

FCM data indicated that cells conventionally considered to be dead and which would not give rise to CFU in a plate count assay, e.g., cells heated to 80°C, were labeled by antibody staining. This finding suggests that without the inclusion of a live/dead discriminating dye, these cells would be erroneously detected as viable within an FCM assay. Reductions in antibody staining due to physicochemical treatment were strain related, reflecting the complexity of the phenomenon under study and illustrating that substantial validation of any new antibody detection-based method, including physiological staining and cell sorting, should be undertaken. Researchers should be aware of physicochemical treatments causing false-negative results: in this study, freeze-thawing severely reduced antibody binding without affecting the viability of a substantial percentage of cells. Scanning electron microscopy carried out on treated cells revealed a range of morphological changes resulting from physicochemical treatments which may have hindered antibody binding.
KEYWORDS: Staphylococcus aureus, antibody-based detection, cell viability, flow cytometry, food-borne pathogens, morphology, physiology
ABSTRACT
The effects of heat and chemical treatments on Staphylococcus aureus viability and physiology and their subsequent effects on antibody binding ability and cell morphology were measured. Treatments included lethal and sublethal heat; exposure to organic acids, salt, and sodium hydroxide; and freeze-thawing. Strain-related differences in viability were noted depending on treatment and were reflected in changes in physiology as monitored by flow cytometry (FCM) using three different staining protocols: SYTO 9/propidium iodide (PI), DiOC2(3), or calcein acetoxymethyl ester (calcein-AM)/PI. Treatments that resulted in significant losses in viability as measured by plate counting were reflected better by the first two staining combinations, as intracellular calcein-AM uptake may have been impaired by certain treatments. FCM analysis using labeling by commercial anti-S. aureus antibodies indicated that differences in cell physiology as a result of treatments influenced immunofluorescence detection. The ratio of the mean fluorescence intensities of stained cells to those of unstained cells [MFI/MFI(us)] varied with treatment, five of these treatments, including freeze-thaw, citric acid, oxalic acid, NaCl, and NaOH treatments, resulted in significantly lower fluorescence values compared to controls.
IMPORTANCE FCM data indicated that cells conventionally considered to be dead and which would not give rise to CFU in a plate count assay, e.g., cells heated to 80°C, were labeled by antibody staining. This finding suggests that without the inclusion of a live/dead discriminating dye, these cells would be erroneously detected as viable within an FCM assay. Reductions in antibody staining due to physicochemical treatment were strain related, reflecting the complexity of the phenomenon under study and illustrating that substantial validation of any new antibody detection-based method, including physiological staining and cell sorting, should be undertaken. Researchers should be aware of physicochemical treatments causing false-negative results: in this study, freeze-thawing severely reduced antibody binding without affecting the viability of a substantial percentage of cells. Scanning electron microscopy carried out on treated cells revealed a range of morphological changes resulting from physicochemical treatments which may have hindered antibody binding.
INTRODUCTION
Flow cytometry (FCM) combines direct multiplex staining, rapid analysis, and high throughput with the ability to analyze individual cells and moves beyond traditional plate count methods which can only provide information regarding viability under controlled growth conditions (1). In combination with antibody staining, FCM can specifically label target cells within a sample, rendering it one of the most rapid and potentially sensitive microbial detection and enumeration techniques (2–4). Despite this, little work has been undertaken to understand the factors affecting the antibody labeling of bacteria for subsequent immunofluorescence FCM analysis (compared to a growing interest in antibody performance in other fields) (5).
As well as a long-standing recognition within the wider field of microbial immunology that cell physiological state can have an effect on antigenicity (6), anomalies in the antibody staining of bacterial cells in the context of FCM analysis have long been reported. Previous authors generated a number of antibodies against synthetic peptides derived from the outer membrane protein F molecule of Pseudomonas aeruginosa and found that the maximum median percentage of antibody-binding cells was only 36.6% and proposed that the surface accessibility of protein F epitopes must have varied during the cell cycle (7). This contrasted with the reaction of antisera directed toward the homologous lipopolysaccharide of P. aeruginosa (which is constitutively expressed), where 98.8% of the cell population was positively stained. These authors also suggested that the microagglutination of cells or the partial disruption of their outer membranes may also have contributed to this variation in binding. It has also been reported that the percentage of Helicobacter pylori cells displaying the HSP60 antigen on the cell surface, as detected by FCM, differed between strains and culture conditions despite immunoblotting demonstrating similar expression levels of the protein (8). Recently, other authors took advantage of the measurable loss of attachment between antibodies and inactivated cells of their target, Escherichia coli O157:H7, in order to generate differential counts of live and dead cells in a water treatment system using a novel microelectrical detection system (9). These authors ascribed the loss of antibody binding of cells to outer membrane damage.
One well-characterized case of a lethal treatment causing alterations in the reaction of a bacterial species to antibody staining is that of Neisseria meningitidis (10). It was reported that live cells of this organism presented poor binding of antibody to its PorB3 proteins (whereas other outer membrane proteins, PorB2 and PorA, displayed much stronger binding). After lethal treatment with ethanol, strong antibody binding to PorB3 was detected and ascribed to “epitope exposure.” Treatment with ethanol appeared to negate the shielding effect of the carbohydrate chains of lipopolysaccharides. A direct example of inactivating treatments causing a loss of antigenicity was demonstrated for a number of anti-Bacillus anthracis antibodies, with labeling being reduced after autoclaving or irradiation treatment of spores (11).
Labeling of target cells is dependent on the affinity or strength of the reaction between a single antigenic determinant and a single combining site on the antibody (12). Changes in the physiology or morphology of cells as affected by factors such as exposure to stressor treatments such as heat or cold shock, extremes of pH, or other challenges could, in theory, impact antibody affinity and avidity (the overall strength of binding) through alterations in antigen conformation or availability (13, 14). Many of the above stressors are experienced by bacteria during food processing unit operations, e.g., spray drying or pasteurization, and result in the generation of a heterogeneous mixture of various subpopulations in different physiological and morphological states (15). Hence, for pathogens such as Staphylococcus aureus, exposure to such treatments may generate a range of subpopulations, from viable to viable but nonculturable to nonviable (1). Not only do such treatments provide challenges for detection using traditional plate counting techniques, but alterations in antibody binding may lead to anomalies in the context of rapid immunofluorescence FCM detection methodologies.
Therefore, we investigated whether antibody-based staining procedures for the detection of S. aureus by FCM were affected by the physiological and morphological status of the cells induced by physical and chemical treatments. Labeling and staining of S. aureus strains was subsequently evaluated using two commercially available primary antibodies directed toward surface antigens and measured by FCM in tandem with three physiological staining regimes, which were used to gain detailed insight into the extent of cellular physiology and their effects on antibody binding.
The combination of SYTO 9/propidium iodide (PI) was used to assess membrane integrity based on dye exclusion. The permeant nucleic acid dye SYTO 9 enters all cells, while only membrane-damaged cells are stained red by the impermeant nucleic acid dye PI (16). In the case of highly damaged cells, PI will fully displace SYTO 9 (1). The dye DiOC2(3) was utilized to evaluate membrane potential, since this parameter has previously been correlated with viability in Gram-positive bacteria such as S. aureus and Micrococcus luteus (17). Positive membrane potential indicates the ability of the cell to generate electrochemical gradients which assist viability. Cells with damaged membranes cannot sustain an electrochemical gradient and are normally classified as dead (18), although neither integrity nor active membrane potential necessarily reflect the ability of the cell to replicate and grow (1).
Intracellular esterase activity, which is probably the most common form of metabolic activity detected by FCM (19), was measured using the dye calcein acetoxymethyl ester (calcein-AM), a nonfluorescent substrate that is converted by intracellular esterase activity into the fluorescent calcein and which is subsequently retained intracellularly. Calcein-AM has been used as a viability marker because of its high cell retention and pH-insensitive fluorescence. However, in some cases both calcein-AM and calcein may rapidly leak out from dead or damaged cells with compromised membranes even if the cell retains some residual esterase activity (20). Esterase activity may be classified as energy independent, since the enzyme will remain functional in cells provided it is retained by the intact membrane and protected from the environment (18, 21).
Scanning electron microscopy (SEM) was used to visualize changes in cell morphology as induced by the stressor treatments. It was expected that alterations in cell integrity detectable by SEM, including alterations to cell size and shape, light scattering ability, the cell matrix, and evidence of autolysis would yield an extra insight into FCM and plate count data and contribute to an integrated understanding of the variability in antibody staining measured.
RESULTS
Enumeration of CFU using plate counting.
Data for plate counts on nutrient agar (NA) for the various treatments were converted to their log10 values, and the effects on viability are presented in Fig. 1. Log reductions for both strains are plotted, with the reduction for each being compared to individual controls.
FIG 1.
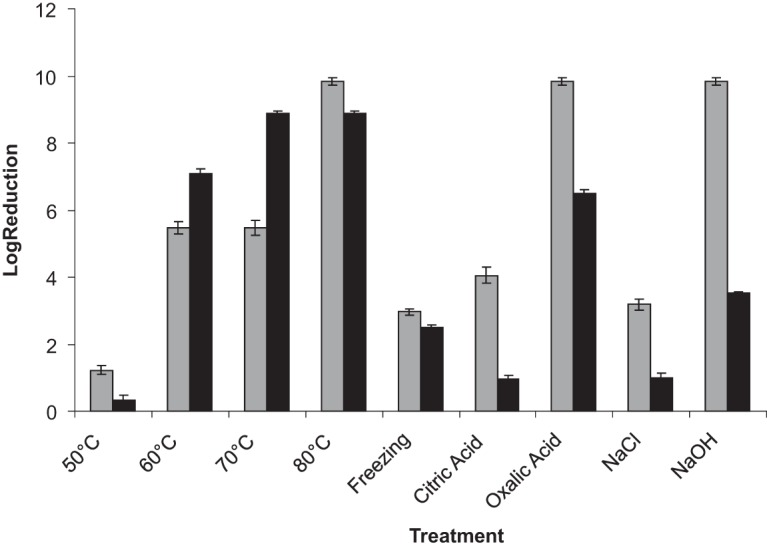
Mean log10 reduction expressed as CFU/ml for S. aureus strain 1 (light gray) or strain 2 (dark gray) after the following treatments (all for 10 min): 50°C, heated to 50°C; 60°C, heated to 60°C; 70°C, heated to 70°C; 80°C, heated to 80°C; citric acid, exposure to 1% (wt/vol) citric acid solution; oxalic acid, exposure to 1% (wt/vol) oxalic acid solution; NaCl, exposure to 1% (wt/vol) NaCl solution; and NaOH, exposure to 2.5% (wt/vol) NaOH solution. Freezing, cells frozen overnight and thawed at room temperature for 1 h before plating.
Flow cytometric analysis of bacterial cells.
(i) Staining protocol 1 and SYTO 9/PI. Results were expressed as the percentage of the total population present as PI-positive “dead” or SYTO 9-positive “live” cells (Table 1). A typical cytograph of these gated regions is presented in Fig. 2. The percentages of PI-positive cells increased substantially as heating temperature increased beyond 60°C, with up to 98% of cells becoming PI positive at 80°C for both strains. Strain 2 showed a more substantial increase in PI permeability compared to strain 1 at 70°C (76 versus 46%) and appeared to be more susceptible to heat damage over the range from 60 to 70°C. Exposure to oxalic acid indicated some strain-related differences for permeability to PI, with strain 2 having ∼15% more PI-positive cells compared to strain 1. CTAB (cetyltrimethylammonium bromide) treatment generated a specific highly permeabilized population outside the typical “live/dead” regions and appeared to generate low levels of cells positive for PI only, with a complete absence of regions positive for SYTO 9 only for both strains.
TABLE 1.
Mean percentages of populations of a pair of S. aureus strains falling into PI-positive or SYTO 9-positive regions following various physicochemical treatments, staining, and FCM analysisa
| Sample | Mean ± SEM (%) |
|||
|---|---|---|---|---|
|
S. aureus strain 1 |
S. aureus strain 2 |
|||
| PI positive | SYTO 9 positive | PI positive | SYTO 9 positive | |
| Live control | 0.01 ± 0.02 | 99.07 ± 0.36 | 0.00 ± 0.00 | 98.85 ± 0.58 |
| Heat-killed control | 96.63 ± 1.56 | 2.36 ± 1.36 | 98.26 ± 0.61 | 0.07 ± 0.13 |
| 50°C | 0.60 ± 0.56 | 96.67 ± 2.73 | 0.43 ± 0.60 | 96.13 ± 3.15 |
| 60°C | 0.47 ± 0.64 | 97.62 ± 1.84 | 0.79 ± 1.23 | 93.44 ± 3.69 |
| 70°C | 45.48 ± 2.10 | 39.51 ± 4.27 | 76.21 ± 9.71 | 5.66 ± 2.06 |
| 80°C | 94.17 ± 3.39 | 3.11 ± 1.88 | 98.23 ± 1.32 | 0.63 ± 0.57 |
| Freezing | 44.99 ± 7.12 | 46.37 ± 2.69 | 60.04 ± 3.72 | 22.30 ± 3.15 |
| Citric acid | 3.37 ± 2.66 | 92.35 ± 1.83 | 9.82 ± 1.22 | 74.19 ± 3.85 |
| Oxalic acid | 63.72 ± 5.89 | 27.28 ± 2.57 | 78.84 ± 4.68 | 13.91 ± 1.76 |
| NaCl | 0.97 ± 0.60 | 96.50 ± 1.91 | 2.75 ± 0.42 | 95.68 ± 1.49 |
| NaOH | 90.98 ± 2.14 | 6.43 ± 3.63 | 97.15 ± 2.95 | 1.47 ± 1.11 |
| CTAB | 24.04 ± 4.71 | 0.00 ± 0.00 | 11.05 ± 0.89 | 0.00 ± 0.00 |
Treatments: live control, untreated stained cells; heat-killed control, cells exposed to temperatures of >95°C for 30 min to ensure complete loss of viability; freezing, cells frozen overnight and thawed at room temperature for 1 h before analysis. All of the following treatments were for 10 min: 50°C, heated to 50°C; 60°C, heated to 60°C; 70°C, heated to 70°C; 80°C, heated to 80°C; citric acid, exposure to 1% (wt/vol) citric acid solution; oxalic acid, exposure to 1% (wt/vol) oxalic acid solution; NaCl, exposure to 1% (wt/vol) NaCl solution; NaOH, exposure to 2.5% (wt/vol) NaOH solution; and CTAB, exposure to 0.5 mM CTAB.
FIG 2.

Cytographs of S. aureus strain 1 exponential-phase live control cells stained with SYTO 9 and PI (a) and DiOC2(3) (b) and of heat-treated dead control cells (95°C for 30 min) stained with SYTO 9 and PI (c) and CCCP-treated cells (depolarized) stained with DiOC2(3) (d).
(ii) Staining protocol 2 and DiOC2(3). A range of depolarized, dead, and live controls were used to construct gates (Fig. 2), which allowed the discrimination of polarized from depolarized cells. From a ratiometric histogram showing FL3/FL1, the percentage of depolarized or polarized cells for each of the treatments was determined (Table 2). Increasing depolarization of both strains was evident with increasing temperature, in particular over the range 50 to 60°C, where levels increased beyond 80% of the total. Freeze-thawing resulted in a greater degree of depolarization for strain 2 (∼70%) compared to strain 1 (∼43%). Exposure to oxalic acid or NaCl generally resulted in a 20% increase in depolarization for strain 2. In contrast, strain 1 had substantially higher levels of depolarization after exposure to sodium hydroxide. For both strains, CTAB treatment resulted in extensive depolarization (∼80%).
TABLE 2.
Mean percentages of cells of a pair of S. aureus strains identified as polarized or depolarized following physicochemical treatments, staining with DiOC2(3), and analysis by FCMa
| Sample | Mean ± SEM (%) |
|||
|---|---|---|---|---|
|
S. aureus strain 1 |
S. aureus strain 2 |
|||
| Depolarized | Polarized | Depolarized | Polarized | |
| Live control | 12.05 ± 2.14 | 84.05 ± 3.78 | 10.47 ± 3.74 | 87.60 ± 3.08 |
| Heat-killed control | 89.04 ± 4.48 | 7.01 ± 2.37 | 94.73 ± 1.18 | 7.46 ± 2.22 |
| 50°C | 47.96 ± 2.08 | 46.90 ± 2.34 | 24.00 ± 2.39 | 67.98 ± 3.65 |
| 60°C | 80.90 ± 3.71 | 14.26 ± 3.21 | 83.79 ± 3.24 | 7.21 ± 4.15 |
| 70°C | 87.79 ± 3.11 | 7.43 ± 1.01 | 93.09 ± 3.02 | 10.18 ± 3.59 |
| 80°C | 91.88 ± 0.62 | 2.26 ± 0.71 | 93.71 ± 0.59 | 2.92 ± 0.65 |
| Freezing | 43.01 ± 1.52 | 34.06 ± 2.26 | 69.97 ± 6.40 | 22.63 ± 3.90 |
| Citric acid | 80.96 ± 2.86 | 4.86 ± 3.02 | 83.03 ± 1.89 | 9.59 ± 0.73 |
| Oxalic acid | 49.35 ± 4.56 | 33.77 ± 4.20 | 77.43 ± 3.76 | 15.51 ± 1.84 |
| NaCl | 7.92 ± 2.87 | 89.45 ± 2.27 | 26.51 ± 8.14 | 63.29 ± 1.03 |
| NaOH | 85.07 ± 5.01 | 8.41 ± 3.56 | 50.46 ± 8.10 | 37.68 ± 0.60 |
| CTAB | 77.12 ± 4.18 | 0.00 ± 0.00 | 81.11 ± 1.24 | 0.00 ± 0.00 |
Treatments: live control, untreated stained cells; heat-killed control, cells exposed to temperatures of >95°C for 30 min to ensure complete loss of viability; freezing, cells frozen overnight and thawed at room temperature for 1 h before analysis. All of the following treatments were for 10 min: 50°C, heated to 50°C; 60°C, heated to 60°C; 70°C, heated to 70°C; 80°C, heated to 80°C; citric acid, exposure to 1% (wt/vol) citric acid solution; oxalic acid, exposure to 1% (wt/vol) oxalic acid solution; NaCl, exposure to 1% (wt/vol) NaCl solution; NaOH, exposure to 2.5% (wt/vol) NaOH solution; and CTAB, exposure to 0.5 mM CTAB.
(iii) Staining protocol 3 and calcein-AM/PI. Data for this staining protocol are expressed as the percentages of total cells present in either PI-positive or esterase-positive gated regions (Fig. 3). Clear, distinguishable regions were evident and used to evaluate all treatments for both strains (Table 3). As noted previously for both strains, PI permeability increased as temperature increased. Intracellular esterase activity was initially higher for strain 1 at 50 or 60°C, where ∼90% of cells were esterase positive. In contrast, for strain 2 only 30% of the cells possessed this activity at 50°C. For both strains, increasing temperature from 50 to 60°C resulted in decreases in the intracellular esterase activity on the order of 43 to 54%. Exposure to higher temperatures (70 or 80°C) resulted in an almost complete loss of intracellular esterase activity for both strains. Intracellular esterase activity was higher for strain 2 after freeze-thawing, where ∼30% cells were esterase positive, while only 13% exhibited this activity in strain 1. In the case of treatment with organic acids, the esterase activity was minimal for both strains after exposure to oxalic acid, whereas after exposure to citric acid ∼20 to 28% of the cells of either strain were esterase positive. Both strains maintained very high levels of esterase activity following treatment with NaCl (on the order of 90 to 97% of the total profile). Treatment with sodium hydroxide greatly affected intracellular esterase activity, with only ∼7% of strain 1 and ∼3% of strain 2 maintaining this activity. Following CTAB treatment, esterase activity was wholly eliminated in both strains, and large increases in membrane permeability were detected, with 60 to 70% of the cells becoming PI positive and the remainder showing extremely high PI fluorescence outside this region.
FIG 3.

Cytographs of S. aureus strain 1 exponential-phase live control cells (a) and heat-killed control cells (95°C for 30 min) (b) stained with calcein-AM and PI. (c) Overlay of green fluorescence distributions of calcein-AM- and PI-stained cells subjected to various treatments. For an explanation of the labels, see Fig. 1.
TABLE 3.
Mean percentages of cells of a pair of S. aureus strains identified as esterase or PI positive following various physicochemical treatments, staining with calcein-AM and PI, and analysis by FCMa
| Sample | Mean ± SEM (%) |
|||
|---|---|---|---|---|
|
S. aureus strain 1 |
S. aureus strain 2 |
|||
| Esterase positive | PI positive | Esterase positive | PI positive | |
| Live control | 97.76 ± 1.60 | 1.12 ± 0.18 | 98.46 ± 1.89 | 0.07 ± 0.11 |
| Heat-killed control | 0.42 ± 0.59 | 96.56 ± 2.02 | 0.07 ± 0.06 | 98.30 ± 0.86 |
| 50°C | 91.86 ± 1.45 | 0.47 ± 0.72 | 30.73 ± 5.33 | 7.49 ± 2.79 |
| 60°C | 56.44 ± 0.83 | 20.73 ± 3.93 | 13.89 ± 3.22 | 50.46 ± 3.86 |
| 70°C | 1.17 ± 0.95 | 68.57 ± 8.40 | 0.77 ± 0.89 | 88.67 ± 2.12 |
| 80°C | 0.33 ± 0.56 | 95.61 ± 2.38 | 0.00 ± 0.00 | 98.21 ± 1.20 |
| Freezing | 13.49 ± 3.59 | 41.11 ± 4.40 | 30.35 ± 2.53 | 44.65 ± 0.87 |
| Citric acid | 21.88 ± 1.92 | 20.02 ± 3.76 | 28.52 ± 4.62 | 18.54 ± 0.96 |
| Oxalic acid | 1.94 ± 0.56 | 58.46 ± 2.43 | 1.28 ± 1.85 | 45.64 ± 3.41 |
| NaCl | 90.86 ± 1.58 | 0.51 ± 0.38 | 96.95 ± 1.67 | 0.84 ± 0.77 |
| NaOH | 7.24 ± 0.55 | 70.39 ± 1.92 | 2.83 ± 1.44 | 77.54 ± 1.07 |
| CTAB | 0.00 ± 0.00 | 59.46 ± 3.20 | 0.00 ± 0.00 | 67.32 ± 0.67 |
Treatments: live control, untreated stained cells; heat-killed control, cells exposed to temperatures of >95°C for 30 min to ensure complete loss of viability; freezing, cells frozen overnight and thawed at room temperature for 1 h before analysis. All of the following treatments were for 10 min: 50°C, heated to 50°C; 60°C, heated to 60°C; 70°C, heated to 70°C; 80°C, heated to 80°C; citric acid, exposure to 1% (wt/vol) citric acid solution; oxalic acid, exposure to 1% (wt/vol) oxalic acid solution; NaCl, exposure to 1% (wt/vol) NaCl solution; NaOH, exposure to 2.5% (wt/vol) NaOH solution; and CTAB, exposure to 0.5 mM CTAB.
Antibody staining.
Figure 4 shows the overlaid fluorescence profiles of antibody-labeled cells subjected to the various treatments, as well as the ratio of the mean fluorescence intensity (MFI) of the stained sample to the MFI of its respective unstained control [MFI(us)]. The MFI/MFI(us) values allow for the measurement of differences in the fluorescence intensities which reflect differences in antibody binding as a result of the various treatments. The MFI/MFI(us) values were similar to those for the controls for all heat treatments for both strains (Fig. 5). These values decreased substantially for the following treatments for both strains: freeze-thawing and exposure to the organic acids and 1% NaCl. However, some strain-related differences were noted for these treatments. Generally, strain 1 had higher MFI/MFI(us) values after exposure to freeze-thawing. In contrast, strain 2 exhibited higher MFI/MFI(us) values following exposure to both acids or sodium hydroxide. MFI/MFI(us) varied from ∼12 to 40 for both strains when primary antibody A was used and from ∼14 to 41 when primary antibody B was used. Both strains had similar lower MFI/MFI(us) values compared to controls following exposure to 1% NaCl. Overall, there were no significant differences when we compared the fluorescence values for strain 1 and strain 2 (P > 0.05 [unpaired t test], two-tailed df = 19) and no significant differences between antibody A and antibody B labeling (P > 0.05). When we compared the treatments, a one-way analysis of variance using Dunnett’s multiple-comparison test showed that, overall, five treatments differed significantly from the control (P < 0.05). These were the freeze-thaw, citric acid, oxalic acid, NaCl, and sodium hydroxide treatments. Exposure of both strains to CTAB resulted in unusually high MFI/MFI(us) intensities, which were orders of magnitude greater than those noted for controls and the various treatments (data not shown). Fluorescence arising from antibody labeling and staining following CTAB treatment was detectable in all channels and did not follow the normal Gaussian distribution required to calculate a statistically valid fluorescence intensity ratio.
FIG 4.

Overlays of the PE fluorescence distributions of S. aureus exposed to various physical or chemical treatments (for an explanation of the labels, see Fig. 1) and stained with one of a pair of commercial anti-S. aureus antibodies, followed by staining with R-PE-conjugated secondary antibody. The left panel shows the average MFI/MFI(us) PE fluorescence values for the various treatments for both pairs of strains and antibodies.
FIG 5.

Comparison of the MFI/MFI(us) of the PE fluorescence of S. aureus strains subjected to various physicochemical treatments (for an explanation of the labels, see Fig. 1, and for an explanation of the bars' color coding, see Fig. 4) and stained with primary antibody A or B, followed by staining with R-PE-conjugated secondary antibody, with log10 plate counts obtained after each treatment. The red line is an illustration of the log10 counts for strain 1, while the orange line represents strain 2.
Scanning electron microscopy.
In general, SEM revealed at a microstructural level the effects of the various stressor treatments on cell morphologies (Fig. 6). Control exponential-phase cells showed walls that appeared to be intact. Cells were also examined after they reached the stationary phase and appeared to be similar to those in the exponential phase. Cultures of stationary-phase cells appeared to be homogenous, with a more uniform distribution of groups of cells.
FIG 6.

Scanning electron micrographs of S. aureus cells. (A) Exponential-phase live control cells; (B) stationary-phase cells; (C) cells after freeze-thaw treatment; (D) cells after heating at 80°C for 10 min; (E) cells exposed to 2.5% (wt/vol) sodium hydroxide for 10 min; (F) cells exposed to 1% (wt/vol) citric acid for 10 min; (G) cells exposed to 1% (wt/vol) oxalic acid for 10 min; (H) cells exposed to 1% (wt/vol) sodium chloride for 10 min.
Cells subjected to freeze-thawing revealed some degree of cell autolysis or permeabilization and also the presence of “ghost” cells devoid of interior cell material. Cells heated to 80°C showed extensive membrane damage with cratering present, together with some cell lysis and extruded cell debris evident. Surface topography was clearly more uneven than in control samples for this treatment. Exposure to sodium hydroxide had extensive effects on cells, with cell lysis and ruptured cell membranes evident. The remaining whole cells appeared to be highly cratered. Cell debris and clumped structures were evident following this treatment.
When analyzing cells treated with citric acid, some unusual results were noted. The majority of cells appeared unaffected and intact. However, some breakaway material and small “bubble”-like structures were present surrounding the cells. It also appeared that exposure to citric acid reduced the extent of cell clumping.
Exposure to oxalic acid had a more severe effect on the S. aureus cells. Substantially more cell debris was evident, and a majority of cells appeared to be lysed. The presence of “bubble”-like structures, also seen following exposure to citric acid, was also evident. “Blebbing” was also obvious in cells subjected to oxalic acid treatment, as was the case with cells exposed to sodium chloride. Somewhat surprisingly, this exposure to low levels of sodium chloride (1%) appeared to greatly affect the cell surface and cell membrane. Cell walls appeared uneven with cratering and irregular variation in cell shape which may have arisen as a result of plasmolysis caused by increased osmotic pressure. Some “ghost” cells and external debris were again evident in the micrographs.
DISCUSSION
In the development of antibody-based detection methods for microorganisms, an issue which has often been overlooked is the effect of cell physiological status on antibody binding, in terms of the intensity of the fluorescence signal generated and the resolution between positive and negative cells. This study sought to address this topic in the case of a pair of commercial monoclonal antibodies and a pair of S. aureus strains, one a food isolate (strain 1) and the other a type strain (strain 2). Previous studies by our group used flow cytometry (FCM) combined with cell sorting to elucidate the mechanisms underlying bacterial responses to measures commonly employed in food manufacturing (1, 15, 22). This study showed that following processing, bacterial cells in a foodstuff or food production environment may exist in a variety of physiological states, ranging from damaged, stressed, or viable to actively reproducing. In the present study, subjecting strains to typical physicochemical treatments encountered during food processing showed that alterations in viability, physiology, and morphology also gave rise to changes in antibody binding for FCM analysis. Indeed, clear treatment and strain-related differences in antibody binding were observed.
Changes induced in bacterial strains by the treatments described here can potentially compromise cell integrity or morphology with consequent effects on binding of antibody conjugates. Two components of membrane functionality were measured using FCM: permeability (using PI and calcein-AM retention [20]) and polarization [using DiOC2(3)]. The physicochemical treatments used in this study were shown to permeabilize cells, which could result in nonspecific binding of antibodies to exposed internal components of cells. This is regarded as problematic when staining mammalian cells and explains the standard use of viability dyes in mouse and human immunological staining panels (23), whereby membrane-compromised and presumably dead cells are removed at an early stage of gating to eliminate artefacts such as false positives, false negatives, and excessive autofluorescence. As in reports (24), the use of electron microscopy in the present study revealed further information about the external morphology of treated cells. Most of the treatments gave rise to observable structural alterations ranging from ruffling of the outer wall to lysis, “blebbing” and the presence of ghost cells.
It is interesting to note that while all the treatments gave rise to significant reductions in viability, as measured by plate counting, these nonviable cells bound antibodies to varying extents depending on the treatment. Heat treatments resulted in losses in viability as reflected by plate counts, with SEM revealing severe structural damage to cells. However, heat treatments to simulate mild to more severe heat processing treatments (from 50 to 80°C) resulted in only small differences recorded for the MFI values between treatments (P > 0.05). Generally, heat treatments had similar log reductions for both strains at all temperatures, with the exception of the treatment of strain 2 at 70°C, where a high loss of viability was noted. In the case of strain 1, complete nonviability was achieved at 80°C. This strain-related difference in response to heat was also evident from FCM SYTO 9/PI staining. At 70°C, ∼93% of the population of strain 2 became PI positive, while for strain 1 ∼91% became PI positive at 80°C. According to DiOC2(3) staining data, it appeared that cell membranes were almost completely depolarized above 60°C, with >80% cells appearing in depolarized regions for both strains. When staining with calcein-AM, esterase-positive events were higher for strain 1 at 50 or 60°C compared to strain 2. However, for both strains, increasing the temperature beyond 60°C resulted in an almost complete loss of intracellular esterase activity. When calcein-AM was counterstained with PI, similar profiles were found for SYTO 9/PI staining, which would indicate that heating generated extensive adverse cellular effects.
While freezing and thawing of cells resulted in a less considerable loss of viability as reflected in plate counts compared to other treatments (an ∼3-log reduction), antibody binding was greatly reduced by this treatment, particularly in the case of strain 2. This strain exhibited higher levels of damage at the cell membrane level, as evidenced by FCM data, and it is possible that the nature of this structural damage may have caused a rapid decrease in fluorescence intensity due to the occlusion or absence of cell surface antigens, as may have been the case with the variability in the staining of P. aeruginosa that was previously reported (7). Electron microscopy revealed the presence of autolysis, permeabilization, and “ghost” cells among freeze-thawed cells. FCM data following SYTO 9/PI staining indicated that the majority of cells sustained membrane damage, with 40 to 60% of the populations of each strain becoming PI positive, with somewhat higher levels for strain 2. Depolarization was also evident, particularly for strain 2, with ∼70% gated in the depolarized region. Calcein-AM staining indicated higher intracellular esterase activity after freeze-thawing for strain 2. Strain-related differences in the stability of intracellular esterase activity and the ability of the cell membrane to retain calcein may account for strain-related differences noted for calcein-AM staining.
Exposure to citric acid or oxalic also resulted in a significant decrease in antibody binding for both strains, particularly for strain 1, and may have arisen from damage to the antibody binding sites for both primary antibodies. Interestingly, citric acid-treated cells appeared less damaged than oxalic acid-treated cells upon examination using electron microscopy. Exposure to citric acid resulted in a 4-log reduction in plate counts for strain 1 and in only a 1-log reduction for strain 2. The majority of cells of both strains were SYTO 9 positive, indicating little effect on membrane permeability from this treatment. With regard to membrane depolarization, 84 to 85% of both strains became depolarized following this treatment. Calcein-AM data indicated low intracellular esterase activity for both strains. However, a small percentage of both strains became PI positive (10 to 17%), indicating that cell membranes may have been mildly damaged through the action of citric acid and that the treatment either reduced esterase activity or decreased the ability of the cells to retain the calcein-AM stain. Indeed, the majority of cells for both strains were present in unstained regions (data not shown), giving further indication of a lack of dye retention. Exposure to oxalic acid resulted in a substantial loss in viability for both strains. FCM profile data indicated that 60 to 80% of cells became PI positive, with the remainder SYTO 9 positive (13 to 27%). Extensive membrane depolarization was also evident for both strains; however, strain 2 had a higher proportion of depolarized cells (∼77%) compared to strain 1 (∼49%). For this particular treatment, it is interesting to note the loss in viability suggested by the plate count data, but the presence of significant SYTO 9-positive populations shown by FCM analysis, and this may indicate the possible presence of a viable but nonculturable subpopulation. Calcein-AM staining showed the almost complete absence of esterase-positive subpopulations for both strains (∼2%) and, as outlined previously, may reflect a lack of retention of this stain. The damage caused by exposure to organic acids may therefore arise from a combination of membrane damage and damage to intracellular enzymes. Overall, exposure to oxalic acid had a more detrimental effect on cells than citric acid. Oxalic acid is present in foods such as fruit and leafy vegetables (25), but it is not used as a preservative or additive in the manner of citric acid.
Exposure to 1% (wt/vol) NaCl caused only relatively small percentages of cells to be damaged, as reflected by FCM analysis (92 to 97% of the cells of either strain remained SYTO 9 positive, and ∼90% of the cell membrane potential was retained, while 90 to 97% of the populations were esterase positive) and a moderate loss in viability, but the extent to which it appeared to affect antibody binding was surprising. MFI values were reduced by 64 to 80% compared to untreated antibody-stained controls. It is possible that NaCl even at low concentrations may alter the cell surface morphology of S. aureus strains through ionic effects which could significantly reduce the number of exposed cell surface antibody binding sites for the two commercially available primary antibodies. Salt concentrations in excess of 1% are frequently added to products such as processed meats, cheese, and ready meals and could therefore present a problem with immunofluorescence-based FCM analysis of these foods. Our data would also support the recognized nonlethal effects of NaCl against S. aureus at such a low concentration (26, 27).
After treatment with sodium hydroxide, a high reduction in cell viability (assessed using plate counting), as well as a reduced MFI when labeled with either primary antibody, was observed for strain 1. In contrast, strain 2 did not appear to be similarly affected in terms of either viability or MFI ratio. These data again illustrate strain-related differences in responses to stressors or treatments which subsequently affected antibody binding conditions. FCM profiling also revealed extensive cell damage following exposure to sodium hydroxide with 90 to 97% of cells of both strains becoming PI positive. Strain-related differences in membrane depolarization were noted, with 85 and 50% of populations becoming depolarized for strain 1 or 2, respectively. Low levels of intracellular esterase activity were evident (3 to 7%) for both strains. It would appear that the presence of a significant subpopulation with a functioning membrane potential for strain 2 may have allowed a higher degree of survival, as noted from viable plate counts. This functioning membrane potential may allow diffusion or active transport of essential nutrients into the cell to maintain viability (17, 28). CTAB treatment, as previously described (1), generated unusual staining profiles which indicated extensive cell damage, particularly through high permeabilization levels.
Given the alterations in antibody binding to cells subjected to simulated food processing treatments recorded in this study, a cautious approach to the use of antibodies to detect bacteria in food systems in advised. Significant discrepancies between FCM counts and traditional plate counts (a feature of comparative studies between the two techniques [19]) may arise from the inclusion of nonviable but nonetheless antibody-positive cells in an FCM-derived count (false positives). In this study, we have demonstrated the presence of nonviable cells which give a strong reaction to the antibody (e.g., heat-treated cells), as well as catastrophically damaged cells which showed dramatic, generalized increases in autofluorescence in all channels (e.g., CTAB-treated cells). Flow cytometric staining assays should therefore include a step to remove false-positive events arising from the final antibody-positive “count gate.” The solution is the inclusion of viability dyes such as those used in this study. As in the case of the majority of flow cytometric studies of mammalian cells (23, 29), we recommend the early gating out of dead cells from any analysis. Given that we have shown that in the case of citric acid-treated cells, permeability was not an indicator of nonviability, whereas membrane polarization was, it would be prudent to include both a permeability dye such as PI and a membrane potential dye such as DiOC2(3). It would be of further benefit to include a vitality dye such as calcein AM, and in this study, for oxalic acid treatment of cells, esterase activity and calcein retention better reflected plate count data than the other staining protocols. Indeed, the recent ISO method for the quantification of lactic acid bacteria by FCM includes the application of three staining protocols similar to those used here (30).
A key question raised by this study is how to deal with cells that viability dyes have marked as alive or vital but which concurrently display reduced antibody binding (as shown for NaCl-treated cells here). A first step in overcoming this problem would involve challenging pure cultures of the target organism with physicochemical treatments, followed by a study of the organism’s reaction to antibody staining. The trial testing of various commercial antibodies to aid in the selection of an antibody which generated the least amount of false negatives would then form a subsequent step. As a form of validation (31), during assay development, cells from subpopulations such as those found in the present study could be sorted directly by fluorescence-activated cell sorting onto selective media to arrive at the best estimate of the incidence of false negatives (and indeed false positives [1]). The availability of a well-characterized monoclonal antibody against a defined epitope, as advocated by other workers, could also reduce this problem (32).
In conclusion, the development of immunofluorescence FCM detection and enumeration methods for bacterial pathogens such as S. aureus in food systems is highly complex. Immuno-FCM is highly promising for quality applications in the food industry. However, its adoption as a routine quality control method will require improvements in current commercial antibodies and the use of multiparameter viability/vitality staining of samples and cell sorting to develop detailed information on how dead and sublethally damaged cells react to antibody staining and can be discriminated and excluded from any assay.
MATERIALS AND METHODS
Preparation of pure suspensions of bacterial cells.
Two strains of Staphylococcus aureus were used for all experiments. The first was a food isolate from the University of Limerick culture collection (strain 1), and the second was S. aureus ATCC 8325 (strain 2). Cultures in exponential phase having similar CFU/ml levels and grown under the same conditions were used in all experiments. Monthly working stocks of cultures were maintained on nutrient agar (NA; Thermo Fisher, Bio Sciences, Ltd., Dún Laoghaire, Ireland) slants, while weekly stocks were also maintained on NA. From the weekly stocks, a single pure colony was inoculated into 10 ml of nutrient broth (NB; Thermo Fisher) and incubated overnight at 37°C. Next, 5 ml of the suspension was added to 95 ml of NB in a conical flask, followed by incubation at 37°C with shaking at 200 rpm. The suspension was left to grow for 1 to 2 h or until cells reached an optical density at 600 nm of ∼0.2 with a cell population of 1.0 × 107 CFU ml−1.
Stressors applied to bacterial cell suspensions.
The following stressor treatments were applied to cell suspensions: exposure to 50, 60, 70, or 80°C for 10 min; overnight freezing at –20°C, followed by thawing; exposure to an organic acid (1% citric or oxalic acid); exposure to 1% salt solution (NaCl); and exposure to 2.5% sodium hydroxide solution.
Heat treatments were carried out using an Eppendorf Thermomixer Comfort (Eppendorf, Ltd., Vienna, Austria) using 1-ml aliquots of cell suspensions. For freeze-thawing, cell suspensions were stored at –20°C overnight and thawed at room temperature for 1 h before use. For all other treatments, suspensions were exposed to substances for 10 min at room temperature. This was done by centrifuging 1-ml aliquots of the cell suspensions in a microcentrifuge (Beckman Coulter, High Wycombe, UK) at 2,500 × g for 5 min and then resuspending the pellet in the various stressor solutions. Suspensions were next centrifuged and resuspended in sterile phosphate-buffered solution (PBS) at pH 7.2 (Thermo Fisher) prior to plate counting, antibody staining, and FCM analysis.
Enumeration of CFU using plate counting.
Plate counts were carried out on NA. Serial dilutions of each treated sample and control were performed in 0.1% peptone water (Thermo Fisher; pH 7.0). Portions (100 μl) of each relevant dilution were spread onto NA plates and allowed to dry. Plates were then inverted in an incubator overnight at 37°C. Colony counts were performed on plates containing colony numbers in the range 30 to 300.
Flow cytometric analysis of bacterial cells.
Three staining regimes were used to analyze by FCM cells subjected to the stressor treatments described above. The first regime involved staining cells with the dyes SYTO 9 (Thermo Fisher) and PI (Sigma-Aldrich, Dublin, Ireland). SYTO 9 (excitation, 485 nm; emission, 498 nm) solution (in dimethyl sulfoxide [DMSO]) was diluted in sterile water and used at a final concentration of 10 μM, and PI (excitation, 493 nm; emission, 636 nm) was first dissolved in DMSO and thereafter in sterile water and used at a final concentration of 5 μM.
The second staining regime involved using a Molecular Probes BacLight bacterial membrane potential kit (Thermo Fisher). The kit was used according to manufacturer’s instructions, where a final concentration of 5 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was used to create a depolarized control, and 30 μM DiOC2(3) was added to all samples except the unstained control. In many Gram-positive species, including S. aureus, the ratio of red to green fluorescence (excitation, 490 nm; emission, 515 [green] and >660 nm [far red]) produced by the dye has been shown to vary with the intensity of the proton gradient (33). A ratiometric histogram of red to green fluorescence was set up according to the manufacturer’s guidelines, and polarized and depolarized regions were gated based on live and CCCP-treated depolarized cells, respectively. Dot plots of green versus yellow fluorescence (the MoFlo FL1 and FL2 channels, respectively [see below]) were also used to distinguish polarized and depolarized cells.
The third staining regime involved using calcein-AM (Thermo Fisher; excitation, 495 nm; emission, 515 nm). Cells retaining esterase activity and whose membranes were intact would be expected to exhibit strong green fluorescence, whereas those lacking esterase activity and/or with compromised membranes would exhibit no fluorescence. Calcein-AM solution (in dry DMSO) was diluted in sterile water and used at a final concentration of 10 μM. In this regime, the cells were also counterstained with PI as previously described to differentiate permeabilized cells. As a positive control for permeabilization, the cells were treated with 0.5 mM CTAB.
For all regimes, samples were vortexed after addition of dye and incubated at room temperature in the dark for 15 min, with the exception of calcein-AM, where samples were incubated at 37°C for 30 min. Samples were vortexed prior to analysis on a MoFlo cell sorter having a 50-mW argon 488-nm blue laser and a 15-mW 635-nm red diode laser (Beckman Coulter, High Wycombe, UK). Data were acquired and analyzed using Summit v4.3 software (Beckman Coulter). Acquiring samples, the trigger was set on the forward-scatter small particle photomultiplier tube (PMT) detector. Prior to analysis, compensation and voltage settings were optimized by running unstained, single-stained, and dual-stained exponential-phase cells. Green fluorescence was captured through a 530/30 band-pass filter using the instrument’s FL1 detector, while red fluorescence was captured through a 670/30 band-pass filter using the FL3 detector. Instrument calibration involved daily laser alignment with Flow-Check beads (Beckman Coulter) and particle size calibration using 1-μm beads (Sysmex, Lincolnshire, IL). A total of 10,000 events were collected for each sample.
Antibody staining.
Two commercially available primary antibodies were used (referred to as antibody A or B). Both antibodies A (catalog no. OBT1530; Bio-Rad, Oxford, UK) and B (catalog no. ab23568; Abcam, Cambridge, UK) were mouse monoclonal antibodies and species specific to S. aureus, belonged to isotype IgM, and had an approximate protein concentration of 19.0 mg/ml. Both antibodies’ immunogens were, according to their suppliers, UV-inactivated S. aureus cells, while a single commercial fluorochrome-conjugated secondary antibody was used against the primary antibodies, namely, goat anti-mouse IgM conjugated to R-phycoerythrin (PE; Thermo Fisher). In order to optimize the concentrations for the secondary antibody, the manufacturer’s protocol was followed with respect to the concentration of the primary antibody. The optimal concentration of primary antibody was initially determined by a series of titrations against the target bacterial strains. The procedure was further optimized by evaluating the following variables: the number of wash steps, the types and concentration of blocking reagents, and analysis of single-stained controls on the flow cytometer. The final labeling procedure involved growth of the cells as described above, centrifugation at 2,500 × g for 5 min to generate cell pellets, resuspension of the pellets in 1 ml of sterile PBS (pH 7.2), with two further washing steps at 2,500 × g for 5 min in 1 ml of buffer. Primary antibody was diluted in PBS with 0.1% bovine serum albumin (Sigma-Aldrich) and added to resuspended washed cells at various concentrations before the samples were placed on ice in the dark for 30 min. Next, primary antibody-labeled cells were washed three times in PBS (as described above), after which the secondary antibody was added. The primary and secondary antibody-labeled cells were then held on ice for a further 30 min and then washed three more times before analysis by FCM. PE fluorescence was collected using the MoFlo FL2 detector using a 580/30-nm band-pass filter. The flow cytometer settings for physiological staining regimes and antibody staining are outlined in Table 4.
TABLE 4.
Typical settings used for the analysis of S. aureus strains on the MoFlo cell sorter for three physiological protocols and one antibody staining protocola
| Parameter | Settings |
|||
|---|---|---|---|---|
| Antibody staining | SYTO 9/PI | DiOC2(3) | Calcein-AM/PI | |
| FSC | Log A | Log A | Log A | Log A |
| SSC | Log A | Log A | Log A | Log A |
| FL1 | Log A; 500 V | Log A; 500 V | Log A; 520 V | |
| FL2 | Log A; 500 V | Log A; 500 V | ||
| FL3 | Log A; 400 V | Log A; 469 V | Log A; 460 V | |
Abbreviations: FSC, forward-scattered light; SSC, side-scattered light; FL1, detector for green fluorescence (530/30 nm); FL2, detector for orange fluorescence (580/30 nm); FL3, detector for red fluorescence (670/30 nm); log A, logarithmic amplification of pulse area signal.
Scanning electron microscopy of cells.
After the cells were subjected to various treatments, they were washed three times in PBS by centrifugation at 2,500 × g for 2 min, and the pellet resuspended in 0.2 ml of sterile-distilled water. The preparation of samples for analysis by SEM was as follows: 0.1% (wt/vol) poly-l-lysine solution (Sigma-Aldrich) was diluted 1:2 with sterile water, and drops were placed onto clean-cut microscope slides. When the slides were almost dry, ∼100-μl portions of the cell suspensions were placed onto the slides, which were then left to dry for ∼1 h. The cells were then fixed by placing the slides in 2% (vol/vol) glutaraldehyde in 0.1 M sodium cacodylate buffer (Electron Microscopy Sciences, Hatfield, PA) for 1 h. The slides were washed by immersion in PBS (pH 7.2) for 10 min, a process repeated twice. Slides were then dried by stepwise placement in graded ethanol solutions (10, 25, 40, 60, 75, and 90%) for approximately 10 min or until completely dried.
The slides were fixed to stubs for gold sputter coating, which was carried out using a Polaron E5100 (Quorum Technologies, Ltd., Laughton, UK) SEM sputter coater. Samples were sputter coated at a vacuum of 9.21 Pa for 3 min at 2,500 V in argon gas at a power of 18 to 20 mA. A JEOL Carryscope JCM-5700 (Jeol, Nieuw-Vennep, The Netherlands) scanning electron microscope was used to obtain the electron images. Stubs containing samples on glass slides were attached to the center of the chamber holder and then placed under high-vacuum conditions and analyzed using magnifications of ×1,800 to ×33,000. Acceleration voltages were ranged from 19 to 20 kV.
Statistical analysis and presentation of data.
Following acquisition, recording, and analysis, FCM data were exported and analyzed using Microsoft Excel 2000 9.0.2720 (Microsoft Corporation, Redmond, WA) and Minitab 15 English (Minitab, Inc., State College, PA). Cytograms and FCM histogram overlays for publication were prepared in FCSExpress Lite Standalone Research v6.06.0014 (32-bit; DeNovo Software, Glendale, CA).
ACKNOWLEDGMENTS
Funding for this research was provided under the National Development Plan through the Food Institutional Research Measure, administered by the Department of Agriculture, Fisheries, and Food, Ireland. U.P.C. is a Marie Skłodowska-Curie Career-FIT Fellow and acknowledges the support of Enterprise Ireland and Marie Skłodowska-Curie Actions. We also acknowledge the support of the Materials and Surface Science Institute, University of Limerick, where the electron microscopy work was carried out.
REFERENCES
- 1.Kennedy D, Cronin UP, Wilkinson MG. 2011. The response of Escherichia coli, Listeria monocytogenes and Staphylococcus aureus to simulated food processing treatments using fluorescence activated cell sorting and plate counting. Appl Environ Microbiol 77:4657–4668. doi: 10.1128/AEM.00323-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy D, Wilkinson MG. 2017. Application of flow cytometry to the detection of pathogenic bacteria. Curr Issues Mol Biol 23:21–38. doi: 10.21775/cimb.023.021. [DOI] [PubMed] [Google Scholar]
- 3.McClelland RG, Pinder AC. 1994. Detection of Salmonella typhimurium in dairy products with flow cytometry and monoclonal antibodies. Appl Environ Microbiol 60:4255–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller S, Nebe-von-Caron G. 2010. Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev 34:554–587. doi: 10.1111/j.1574-6976.2010.00214.x. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury A, Plückthun A. 2015. Reproducibility: standardize antibodies used in research. Nature 518:27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- 6.Dietrich MA, Truax RE, French DD, Lea DF, Stear MJ, Newman MJ. 1991. Measurement of antibody binding to intact bacteria using flow cytometric techniques. J Microbiol Methods 13:281–291. doi: 10.1016/0167-7012(91)90065-X. [DOI] [Google Scholar]
- 7.Hughes EE, Gilleland HE Jr, Matthews-Greer JM. 1996. Analysis by flow cytometry of surface-exposed epitopes of outer membrane protein F of Pseudomonas aeruginosa. Can J Microbiol 42:859–862. doi: 10.1139/m96-109. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi H, Osaki T, Taguchi H, Hanawa T, Yamamoto T, Kamiya S. 1996. Flow cytometric analysis of the heat shock protein 60 expressed on the cell surface of Helicobacter pylori. J Med Microbiol 45:270–277. doi: 10.1099/00222615-45-4-270. [DOI] [PubMed] [Google Scholar]
- 9.Beeman MG, Nze UC, Sant HJ, Malik H, Mohanty S, Gale BK, Carlson K. 2018. Electrochemical detection of E. coli O157:H7 in water after electrocatalytic and ultraviolet treatments using a polyguanine-labeled secondary bead sensor. Sensors (Basel) 18:1497. doi: 10.3390/s18051497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michaelsen TE, Aase A, Kolberg J, Wedge E, Rosenqvist E. 2001. PorB3 outer membrane protein on Neisseria meningitidis is poorly accessible for antibody binding on live bacteria. Vaccine 19:1526–1533. doi: 10.1016/S0264-410X(00)00324-8. [DOI] [PubMed] [Google Scholar]
- 11.Dang JL, Heroux K, Kearney J, Arasteh A, Gostomski M, Emanuel PE. 2001. Bacillus spore inactivation methods affect detection assays. Appl Environ Microbiol 67:3665–3670. doi: 10.1128/AEM.67.8.3665-3670.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies DR, Cohen GH. 1996. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A 93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Li Y. 2002. Detection of Escherichia coli O157:H7 using immunomagnetic separation and absorbance measurement. J Microbiol Methods 51:369–377. doi: 10.1016/S0167-7012(02)00107-0. [DOI] [PubMed] [Google Scholar]
- 14.Lydyard P, Whelan A, Fanger MW. 2004. Immunology. W. W. Norton and Company, New York. NY. [Google Scholar]
- 15.Cronin UP, Wilkinson MG. 2008. Physiological response of Bacillus cereus vegetative cells to simulated food processing treatments. J Food Prot 71:2168–2176. doi: 10.4315/0362-028X-71.11.2168. [DOI] [PubMed] [Google Scholar]
- 16.Stiefel P, Schmidt-Emrich S, Maniura-Weber K, Ren Q. 2015. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiol 15:36. doi: 10.1186/s12866-015-0376-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novo DJ, Perlmutter NG, Hunt RH, Shapiro HM. 2000. Multiparameter flow cytometric analysis of antibiotic effects on membrane potential, membrane permeability, and bacterial counts of Staphylococcus aureus and Micrococcus luteus. Antimicrob Agents Chemother 44:827–834. doi: 10.1128/aac.44.4.827-834.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vives-Rego J, Lebaron P, Nebe-von Caron G. 2000. Current and future applications of flow cytometry in aquatic microbiology. FEMS Microbiol Rev 24:429–448. doi: 10.1111/j.1574-6976.2000.tb00549.x. [DOI] [PubMed] [Google Scholar]
- 19.Wilkinson MG. 2018. Flow cytometry as a potential method of measuring bacterial viability in probiotic products: a review. Trends Food Sci Technol 78:1–10. doi: 10.1016/j.tifs.2018.05.006. [DOI] [Google Scholar]
- 20.Comas-Riu J, Vives-Rego J. 1999. Use of calcein and SYTO-13 to assess cell cycle phases and osmotic shock effects on Escherichia coli and Staphylococcus aureus by flow cytometry. J Microbiol Methods 34:215–221. doi: 10.1016/S0167-7012(98)00091-8. [DOI] [Google Scholar]
- 21.Ueckert J, Breeuwer P, Abee T, Stephens P, Nebe-von-Caron G, ter Steeg PF. 1995. Flow cytometry applications in physiological study and detection of foodborne microorganisms. Int J Food Microbiol 28:317–326. doi: 10.1016/0168-1605(95)00066-6. [DOI] [PubMed] [Google Scholar]
- 22.Cronin UP, Wilkinson MG. 2008. Bacillus cereus endospores exhibit a heterogeneous response to heat treatment and low-temperature storage. Food Microbiol 25:235–243. doi: 10.1016/j.fm.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Chattopadhyay PK, Melenhorst JJ, Ladell K, Gostick E, Scheinberg P, Barrett AJ, Wooldridge L, Roederer M, Sewell AK, Price DA. 2008. Techniques to improve the eirect ex vivo detection of low frequency antigen-specific CD81 T cells with peptide-major histocompatibility complex class I tetramers. Cytometry 73A:1001–1009. doi: 10.1002/cyto.a.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergmans L, Moisiadis P, Van Meerbeek B, Quirynen M, Lambrechts P. 2005. Microscopic observation of bacteria: review highlighting the use of environmental SEM. Int Endod J 38:775–788. doi: 10.1111/j.1365-2591.2005.00999.x. [DOI] [PubMed] [Google Scholar]
- 25.Wu F, Zhang D, Zhang H, Jiang G, Su X, Qu H, Jiang Y, Duan X. 2011. Physiological and biochemical response of harvested plum fruit to oxalic acid during ripening or shelf-life. Food Res Int 44:1299–1305. doi: 10.1016/j.foodres.2010.12.027. [DOI] [Google Scholar]
- 26.Hajmeer M, Ceylan E, Marsden JL, Fung D. 2006. Impact of sodium chloride on Escherichia coli O157:H7 and Staphylococcus aureus analyzed using transmission electron microscopy. Food Microbiol 23:446–452. doi: 10.1016/j.fm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Bruins MJ, Juffer P, Wolfhagen M, Ruijs G. 2007. Salt tolerance of methicillin-resistant and methicillin-susceptible Staphylococcus aureus. J Clin Microbiol 45:682–683. doi: 10.1128/JCM.02417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petit JM, Denis-Gay M, Ratinaud MH. 1993. Assessment of fluorochromes for cellular structure and function studies by flow cytometry. Biol Cell 78:1–13. doi: 10.1016/0248-4900(93)90109-R. [DOI] [PubMed] [Google Scholar]
- 29.Perfetto SP, Chattopadhyay PK, Lamoreaux L, Nguyen R, Ambrozak D, Koup RA, Roederer M. 2010. Amine-reactive dyes for dead cells. Current Protoc Cytom Chapter 9:Unit 9.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ISO. 2015. Milk and milk products—starter cultures, probiotics and fermented products—quantification of lactic acid bacteria by flow cytometry. ISO 19344:2015(E); IDF 232:2015(E). ISO copyright office, Vernier, Geneva, Switzerland. International Dairy Federation, Brussels, Belgium. [Google Scholar]
- 31.Bordeaux J, Welsh AW, Agarwal S, Killiam E, Baquero MT, Hanna JA, Anagnostou VK, Rimm DL. 2010. Antibody validation. Biotechniques 48:197–209. doi: 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiron C, Tompkins T, Burguière P. 2018. Flow cytometry: a versatile technology for specific quantification and viability assessment of micro-organisms in multistrain probiotic products. J Appl Microbiol 124:572–584. doi: 10.1111/jam.13666. [DOI] [PubMed] [Google Scholar]
- 33.Thermo Fisher Scientific. 2019. BacLight™ bacterial membrane potential kit: product information. Thermo Fisher Scientific, Waltham, MA. [Google Scholar]