

Abstract
Invasion of the nervous system and neuronal spread of infection are critical, but poorly understood, steps in the pathogenesis of transmissible spongiform encephalopathies or prion diseases. To characterize pathways for the uptake and intraneuronal trafficking of infectious, protease-resistant prion protein (PrP-res), fluorescent-labeled PrP-res was used to infect a neuronally derived murine cell line (SN56) and adult hamster cortical neurons in primary culture. Concurrent with the establishment of persistent scrapie infection, SN56 cells internalized PrP-res aggregates into vesicles positive for markers for late endosomes and/or lysosomes but not synaptic, early endocytic, or raft-derived vesicles. Internalized PrP-res was then transported along neurites to points of contact with other cells. Similar trafficking was observed with dextran, Alzheimer's Aβ1-42 fibrils and noninfectious recombinant PrP fibrils, suggesting that PrP-res is internalized by a relatively nonspecific pinocytosis or transcytosis mechanism. Hamster cortical neurons were also capable of internalizing and disseminating exogenous PrP-res. Similar trafficking of exogenous PrP-res by cortical neurons cultured from the brains of PrP knock-out mice showed that uptake and neuritic transport did not require the presence of endogenous cellular PrP. These experiments visualize and characterize the initial steps associated with prion infection and transport within neuronal cells.
Keywords: prion, trafficking, transport, neurites, endosomes, lysosomes
Introduction
Most transmissible spongiform encephalopathy (TSE) or prion infections penetrate peripheral tissues and follow neuroanatomical pathways to the CNS where the devastating neuropathology of these diseases occurs (Kimberlin et al., 1983; for review, see Glatzel et al., 2004). For this to happen, the infection must invade neurons and be transported from the point of infection to cellular extremities where transfer to other cells presumably occurs. The abnormal, partially protease-resistant form of prion protein (PrP-res or PrPSc) is a primary component of the infectious agent (or prion), and its spread can be detected in peripheral nerves and the brain coincident with the axonal transport of infectivity and the appearance of neuropathological signs (Bruce et al., 1989; Taraboulos et al., 1992; McBride and Beekes, 1999; McBride et al., 2001; Bartz et al., 2002; Glatzel et al., 2004). However, the cellular mechanisms by which PrP-res and TSE infections are transported between and within neural cells are unclear.
Studies in scrapie-infected neuroblastoma cells have shown that once PrP-res is formed on the cell surface or in endosomes, it can be sequestered in lysosomes (Caughey and Raymond, 1991; Caughey et al., 1991; McKinley et al., 1991; Borchelt et al., 1992). In the brains of TSE-infected hosts, PrP-res usually accumulates in both plasmalemmal and extracellular deposits but can also be seen occasionally in late endosome/lysosome-like structures in neurons (Laszlo et al., 1992; Arnold et al., 1995; Grigoriev et al., 1999) and microglia (Jeffrey et al., 1994). These results raise the possibility that neuronal cells might similarly internalize exogenously derived PrP-res when infected for the first time. The normal protease-sensitive precursor of PrP-res, PrP-sen or PrPC, is also endocytosed from the cell surface in neuroblastoma cells and either recycled to the plasma membrane or degraded (Shyng et al., 1993; Magalhaes et al., 2002; Prado et al., 2004). In PNS and CNS neurons, fast anterograde and retrograde axonal transport of PrP-sen also has been detected (Borchelt et al., 1994; Moya et al., 2004), facilitating movement to and from neural extremities. However, PrP-sen differs dramatically from PrP-res in its biophysical properties, turnover, and subcellular distribution and might not be trafficked by neurons in the same way. In epithelial cells, PrP-res appears to be cotransported with ferritin by a receptor- or transporter-mediated pathway (Mishra et al., 2004). Other recent work shows that heparan sulfate molecules are involved but not always essential for PrP-res uptake by several cell types (Hijazi et al., 2005; Horonchik et al., 2005).
To understand how exogenous PrP-res is handled and transported by neural cells during initial infection, we examined the acute interactions between fluorescent-labeled PrP-res and cultured neuronal cells concurrent with the establishment of chronic scrapie infections. Large aggregates of PrP-res bound initially to the surface of cells were broken up and internalized into small vesicles that, in turn, could be seen in real time to move along neuritic projections and congregate at points of contact with other cells. To our knowledge, this represents the first visualization and characterization of the acute process by which TSE agents initially invade neural cells and spread along neuritic pathways.
Materials and Methods
SN56 cell cultures. SN56 cell cultures are neuronal cells derived from mouse septum neurons (Hammond et al., 1990) that, after differentiation with cAMP, present several cholinergic neuronal features (Barbosa et al., 1999; Kushmerick et al., 2001). Cells were passed at 1:10 every 4 d and maintained at 37°C in a 5% CO2 atmosphere in OptiMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were provided by Dr. Bruce Wainer (Department of Pathology, Emory University School of Medicine, Atlanta, GA).
Plasmids. The constitutively activated green fluorescent protein (GFP)-Rab7 mutant Q67L was a gift from Dr. Stephen S. G. Ferguson (J. P. Robarts Research Institute and Department of Physiology, University of Western Ontario, London, Ontario, Canada). A GFP mouse vesicular acetylcholine transporter (VAChT) has been described previously (Santos et al., 2001).
Cell transfection. SN56 cells were plated at 1:20 on Delta T dishes (Bioptechs, Butler, PA) and differentiated for 1 d with cAMP (1 mm) in the absence of serum before transfection. Cell transfection was performed with Effectene (Qiagen, Hilden, Germany), according to the instructions of the manufacturer. A total of 500 ng of GFP-Rab7 mutant or GFP-VAChT plasmid and 12.5 μl of Effectene were used for each dish.
Labeling of organelles. Labeling of endosomes was performed by incubating cells with 40 μg/ml Alexa Fluor 488-labeled transferrin (Molecular Probes, Eugene, OR) at 37°C in 5% CO2 for 20-60 min. After incubation, cells were washed three times in OptiMEM at room temperature without serum and then imaged. Labeling of lysosomes was done by incubating cells with 100 nm LysoTracker Green DND-26 (Molecular Probes) at 37°C in 5% CO2 for 40 min and then cells were washed as described above and imaged.
Cholera toxin subunit B-Alexa Fluor 488 was used as a marker for lipid rafts at 5 μg/ml for 30 min. Cells were washed three times in OptiMEM at room temperature and visualized by confocal microscopy. Dextran-Alexa Fluor 488 (molecular weight, 10,000) was used as a fluid-phase endocytic marker. Cells were labeled at 500 μg/ml for 12 h at 37°C in 5% CO2 in OptiMEM. Cells were washed and imaged using a confocal microscope as described below.
Fluorescent labeling of proteins. PrP-res was purified from the brains of scrapie-infected animals using a series of detergent extractions, differential centrifugations, and treatments with nucleases, salts, and proteinase K (PK) (Raymond and Chabry, 2004). Samples of PK-treated PrP-res or in vitro aggregated amyloid fibrils of recombinant mouse PrP23-231 or Alzheimer's β-amyloid (Aβ) 1-42 peptide (all at ∼0.5 mg/ml in PBS, pH 7.4) were incubated with 1 mg/ml Alexa Fluor 568-succinimidyl ester (Molecular Probes) for 1 h at room temperature and then overnight at 4°C. The aggregated proteins were recovered by centrifugation, washed in PBS containing 10 mm glycine to quench and remove unreacted dye, and resuspended in PBS. Alexa Fluor-labeled material was diluted into sample buffer without running dye for SDS-PAGE analysis. Fluorescent bands were imaged by scanning gels on a Typhoon scanner (Molecular Dynamics, Sunnyvale, CA).
Fluorescence imaging. SN56 cells plated at a 1:20 dilution and differentiated with cAMP (1 mm) and the absence of serum for 1 d were treated with various fluorescent protein aggregates. A total of 16 ng of PrP-resA568 or amounts of fluorescent PrP23-231 or Aβ normalized for total fluorescence were added to the cells in a 4.2 cm2 well. After incubation for 1 h, live cells were visualized by confocal microscopy, and after 4 h, the medium was replaced. Then, cells were returned to the incubator and analyzed every day thereafter for 5 d. Images were obtained with a PerkinElmer (Wellesley, MA) UltraView spinning-disk confocal system connected to a Nikon (Tokyo, Japan) Eclipse TE2000-S microscope with an oil-immersion objective [60×; 1.4 numerical aperture (NA)] or a Bio-Rad (Hercules, CA) MRC 1024 laser-scanning confocal system coupled to a Zeiss (Thornwood, NY) microscope with a water-immersion objective (40×; 1.2 NA). Image processing and analysis were performed with Lasersharp (Bio-Rad), Confocal Assistant, Adobe Photoshop (Adobe Systems, San Jose, CA), and NIH Image J software.
Primary cultures. Primary neuronal cultures were prepared as described previously (Brewer, 1997) from adult hamster brain and PrP knock-out mice. The latter were generated originally on a 129/Ola background (Manson et al., 1994) and then bred onto a mixed 129/Ola and C57BL/10 background while maintaining the PrP knock-out genotype (R. Race and B. Chesebro, unpublished data). Briefly, the cortex was dissected from the adult brain tissue in HAB27 (Hibernate A, B27, 0.5 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin), minced, and digested with 6 ml of 2 mg/ml papain for 30 min at 30°C. After digestion, the cells were washed once with HAB27 and dissociated mechanically by pipetting 10 times with a 1 ml pipette tip. The resultant cell suspension was loaded onto an OptiPrep density gradient with four steps (17.5, 12.5, 10.0, and 7.5%) and centrifuged at 800 × g for 15 min. Fractions corresponding to 10 and 12.5% OptiPrep were collected and washed twice with HAB27. Cells were resuspended in 2 ml of NAB27 (Neurobasal A, B27, 0.5 mm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin). The cells in suspension were counted, and 40,000 cells were seeded in a glass bottom culture dish (diameter, 10 mm) previously coated with 150 μl of 50 μg/ml poly-d-lysine. After 1 h of incubation at 37°C, unattached cells and cell debris were removed by rinsing the culture dish four times with HAB27, and then NAB27 containing 5 ng/ml FGF and 5 ng/ml BDNF was added. After 4 d in culture, cells were exposed to 10 ng of hPrP-resA568 or m(Ch)PrP-resA568 and observed 2-5 d after infection by confocal microscopy.
Immunofluorescence. To stain for microtubule-associated protein 2 (MAP2), cells were washed once with PBS and fixed with 3.7% paraformaldehyde in PBS for 5 min at room temperature followed by incubation with MeOH at -20°C for 1 min. To block nonspecific antibody binding, cells were incubated with 1% nonfat milk in PBS for 30 min at room temperature. Chicken antibody against MAP2 (EnCor Biotechnology, Alachua, FL) was diluted in PBS (1:2000) and added to the cells. After incubation overnight at 4°C, the cells were washed three times with PBS and incubated with secondary anti-chicken IgG antibody (1:500) for 1 h at room temperature. Cells were washed three times and observed by confocal microscopy.
To immunostain for PrP-res, cells infected with PrP-resA568 were fixed with 4% paraformaldehyde in PBS containing 4% sucrose for 10 min at room temperature. Then, the cells were permeabilized with saponin (0.01% in PBS) for 5 min at room temperature and washed once with PBS containing 0.1 m glycine. To reveal the D13 or 3F4 antibody epitopes, cells were treated with 3 m guanidinium-HCl in PBS containing 0.01% saponin for 5 min at room temperature. After two washes with PBS, cells were preincubated with 10% normal goat serum in PBS containing 0.01% saponin for 30 min at room temperature and then with primary antibody D13 (1:500) diluted in blocking solution overnight at 4°C. After washing three times with PBS containing 0.005% saponin, secondary antibody anti-human Fab conjugated with FITC (1:100; Sigma, St. Louis, MO) was added to the cells and incubated for 1 h at room temperature. Cells were washed three times and observed by confocal microscopy.
Immunoblotting. To assay for cell-associated PrP-res, cells were washed twice with phosphate-buffered balanced saline and then lysed in PBS/0.5% (v/v), pH 7.4, Triton X-100/0.5% (w/v) deoxycholate. Nuclei and cell debris were removed by centrifugation at 2700 × g for 5 min at 4°C. The postnuclear supernatant fractions were assayed for total protein using a BCA assay (Pierce, Rockford, IL). Samples normalized for total protein were digested with 20 μg/ml PK for 30 min. PK digestion was terminated by the addition of 2 mm Pefabloc SC and incubation on ice for 5 min. PrP-res was recovered by phosphotungstate (PTA) precipitation essentially as described previously (Wadsworth et al., 2001), with the exception that the benzonase digestion step was omitted. The PK digestion step was omitted for PK- samples. Pellets were resuspended in SDS-PAGE sample buffer. Samples were separated on 10% Bis-Tris Nu-PAGE gels in MES running buffer (Invitrogen). Immunoblot detection of PrP was performed as described previously (Horiuchi et al., 1999) using a mouse/human recombinant anti-PrP monoclonal antibody Fab (D13; InPro Biotechnology, South San Francisco, CA) followed by an alkaline phosphatase-conjugated goat anti-human IgG Fab secondary antibody (Sigma).
Results
Fluorescent labeling of PrP-res
PrP-res was purified from the brains of mice or hamsters infected with various strains of scrapie and treated with PK; thus, the term PrP-res will refer to its PK-treated form. To generate forms of PrP-res that can be monitored visually after exposure to cells, PrP-res was covalently linked to a primary amine-reactive analog of Alexa Fluor 568 (A568). Conditions were selected to add approximately one A568 molecule per PrP molecule. A568-labeled PrP-res preparations from mice infected with the Chandler and 22L strains [m(Ch)PrP-resA568 and m(22L)PrP-resA568, respectively] and hamsters infected with the 263K strain (hPrP-resA568) were analyzed by SDS-PAGE. Fluorescence scanning and immunoblotting of the gels showed that the major fluorescent-labeled bands (∼19-33 kDa and oligomers thereof) reacted with anti-PrP antibodies (Fig. 1a,b). However, in some preparations, most notably with m(Ch)PrP-resA568 and m(22L)PrP-resA568, 10-15 kDa bands were also detected that did not react with anti-PrP antibodies. Thus, in the trafficking experiments described below, additional steps were taken to confirm the cellular colocalization of intact PrP-res and A568 fluorescence (vide infra).
Figure 1.
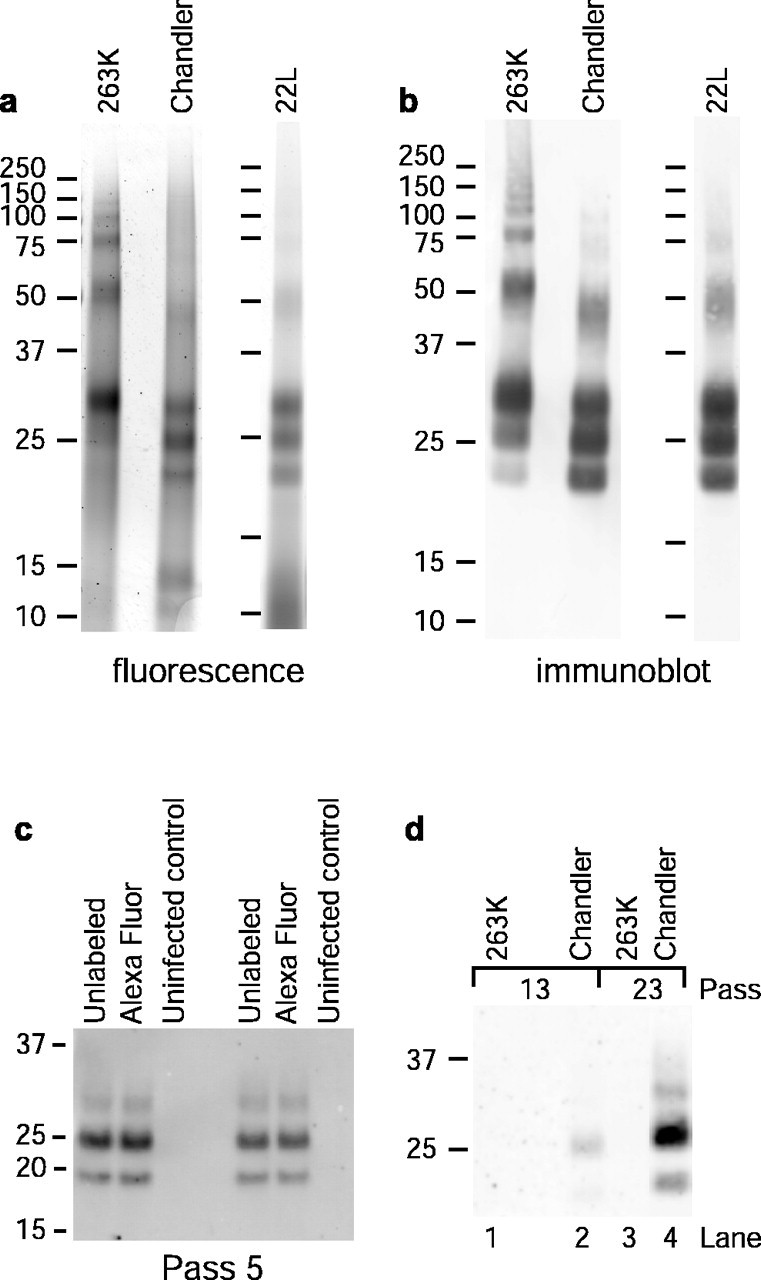
Fluorescence labeling of PrP-res and chronic infection of SN56 cells. a, SDS-PAGE and fluorescence detection of PK-treated PrP-resA568 preparations. b, Immunoblots of the samples shown in a using anti-PrP antibody D13. c, Comparison of long-term PrP-res production in cells infected with A568-labeled or unlabeled PrP-res. Cells at ∼30-40% confluency in a 96-well plate were exposed to 20 ng of PrP-res or PrP-resA568 in 30 μl of medium (0.67 ng/μl PrP-res), incubated for 5 h, fed with an additional 150 μl of medium, incubated for 2 d, and passed into a T-24 well for another 4 d of growth (pass 1). Cells were passed at a 1:10 dilution from this point. At pass 5, PrP-res was precipitated from cell lysates and assayed by immunoblot with D13. Each sample was derived from lysate aliquots containing 224 μg of total protein. d, Long-term PrP-res production in cells treated with the designated types of PrP-resA568 under conditions identical to those used in the confocal microscopy experiments described below and in Materials and Methods. Compared with the protocol used in c, the concentration of PrP-resA568 (0.032 ng/μl) was much lower, and the time of exposure before the change of medium (4 h) was much shorter, resulting in lower overall levels of PrP-res in the cultures. A total of 500 μg of total lysate protein equivalents was loaded per lane. The results in c and d show that A568-labeled PrP-res remained infectious for SN56 cells.
Infection of SN56 cells by fluorescent PrP-res
SN56 cells are transformed murine cells of septal neuronal origin that can be differentiated into neuron-like cells (Blusztajn et al., 1992; Kushmerick et al., 2001). These changes include reduced cell division and elaboration of numerous neuritic projections (Barbosa et al., 1999). Separate experiments have shown that undifferentiated SN56 cells are readily susceptible to infection by Chandler and 22L PrP-res and will remain infected, as evidenced by stable PrP-res production, for at least 60 in vitro passages (G. S. Baron and B. Caughey, unpublished observations).
To verify that A568 labeling of PrP-res did not eliminate its infectivity, SN56 cells were exposed to m(Ch)PrP-resA568, passaged multiple times in the absence of any additional m(Ch)PrP-resA568, and analyzed for PrP-res content by immunoblot. Although the original PrP-resA568 inoculum would be diluted well below detectable limits within four passages (data not shown), immunoreactive PrP-res remained consistently detectable after higher numbers of passages (Fig. 1c,d). Furthermore, there was no difference in the PrP-res production in cells infected with A568-labeled versus unlabeled PrP-res (Fig. 1c). Thus, exposure to m(Ch)PrP-resA568 caused sustained PrP-res formation and, by inference, scrapie infection of the SN56 cells. In contrast, no evidence for sustained PrP-res formation was detected after similar exposures to hPrP-resA568 (Fig. 1d, lanes 1 and 3). Exposures of undifferentiated SN56 cells, to even sixfold-higher amounts of unlabeled 263K PrP-res than was used in the previous experiment, failed to induce persistent infection (Baron and Caughey, unpublished observations), consistent with the low transmissibility of 263K scrapie into mice.
Internalization and trafficking of fluorescent PrP-res by SN56 cells
To visualize events occurring after acute exposure of cells to PrP-res, m(Ch)PrP-resA568 was added to cultures of SN56 cells. The fate of m(Ch)PrP-resA568 was followed in SN56 cells that had reduced cell division and multiple neuritic projections attributable to a 1 d exposure to low-serum differentiation conditions. To facilitate determination of the sources of infection of individual cells, low-dose-exposure conditions were selected so that only a minority of cells were in contact with single m(Ch)PrP-resA568 aggregates, with surrounding cells being devoid of fluorescent particles. Indeed, confocal fluorescence microscopy of live SN56 cells showed that 1-3 h after exposure to m(Ch)PrP-resA568, single large fluorescent aggregates were associated with a subset of cells (Fig. 2a,a1). Over the next few days, progressively fewer large aggregates were observed, and dramatically increasing numbers of much smaller particles of fluorescence were observed on the cell surface, within cell bodies, and along neuritic projections of 70-96% of the cells in contact with m(Ch)PrP-resA568 (Fig. 2b). Frequently, m(Ch)PrP-resA568 aggregates associated with cell bodies were transported in a net anterograde direction to neuritic extremities contacting adjacent cells. The intracellular localization of the small fluorescent particles within cell bodies and neurites was indicated by analyses of individual optical slices (data not shown); however, additional evidence of internalization of PrP-res was provided by colocalization studies with organelle markers (see below). After 10 d and a single passage of the cells, virtually no m(Ch)PrP-resA568 aggregates of the original size remained, yet the small intracellular particles were still readily apparent (data not shown). Similar internalization and trafficking was observed with m(22L)PrP-resA568 (Fig. 2c,c1,d). Thus, it appeared that these PrP-res aggregates were disassembled and disseminated widely within individual cells.
Figure 2.

PrP-resA568 disaggregation and intracellular trafficking in SN56 cells. Confocal fluorescence microscopy 2-4 h (a, a1, c, c1, e, e1) or 3-4 d (b, d, f, f1) after exposure to PrP-resA568 aggregates of various strains. a, a1, b, m(Ch) PrP-resA568. c, c1, d, m(22L)PrP-resA568. e, e1, f, f1, hPrP-resA568. PrP-resA568 of the mouse Chandler and 22L strains was much more readily disaggregated and disseminated throughout the cell bodies and cellular projections than the PrP-resA568 of the hamster 263K strain. Scale bars, 20 μm. Phase-contrast or differential interference contrast images of the cells are shown in combination with the fluorescence images. Arrows point to initial PrP-resA568 aggregates bound to cells.
Several observations suggested that the punctate intracellular fluorescence remaining in the cells after several days was associated with largely intact PrP-res molecules. First, SDS-PAGE analysis of the cell extracts revealed that a majority of the input m(22L)PrP-resA568 (Fig. 3) and m(Ch)PrP-resA568 (data not shown) could be recovered after 3-4 d in SN56 cultures. Second, immunofluorescent staining of fixed and permeabilized cultures after exposure to m(22L)PrP-resA568 showed that the punctate A568 fluorescence colocalized with PrP molecules containing the D13 antibody epitope (i.e., residues 95-104) (Peretz et al., 1997) near the N terminus of the PK-resistant portion of PrP-res molecules (Fig. 4). Although D13, which can also bind PrP-sen, and to a lesser extent the secondary antibody, gave widespread diffuse staining of uninfected control SN56 cells, bright punctate staining that colocalized with A568 was only seen in infected cultures. Furthermore, as is typical in PrP-res rather than PrP-sen (Peretz et al., 1997), this staining required exposure of the D13 epitope by pretreatment with 3 m guanidine hydrochloride (Gdn). Finally, treatments of the cells with a 1000-fold higher concentration of prereacted (with glycine) A568 alone was virtually undetectable with the settings used to acquire confocal images of m(Ch)PrP-resA568 or m(22L)PrP-resA568. Thus, A568 itself would be unlikely to accumulate in intracellular compartments if PrP-resA568 molecules were fully degraded.
Figure 3.
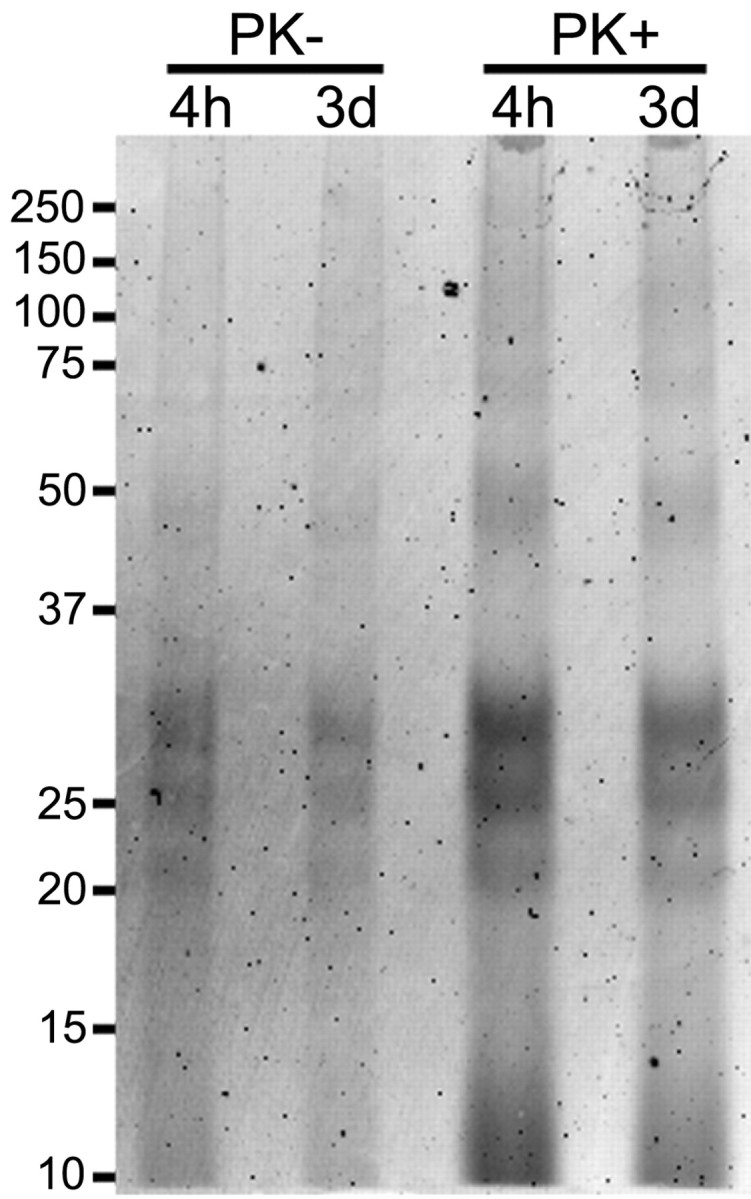
Detection of intact PrP-resA568 in SN56 cells 3 d after infection. SN56 cells were grown in six-well plates and incubated with 100 ng of PrP-res for 4 h or 3 d. After incubation, the cells were washed and lysed. Seventy percent of each lysate was digested with PK, and PrP-res was recovered by PTA precipitation (PK+). Twenty percent of the cell lysates was precipitated with PTA without PK digestion (PK-). The precipitates were analyzed by SDS-PAGE and A568 fluorescence detection.
Figure 4.
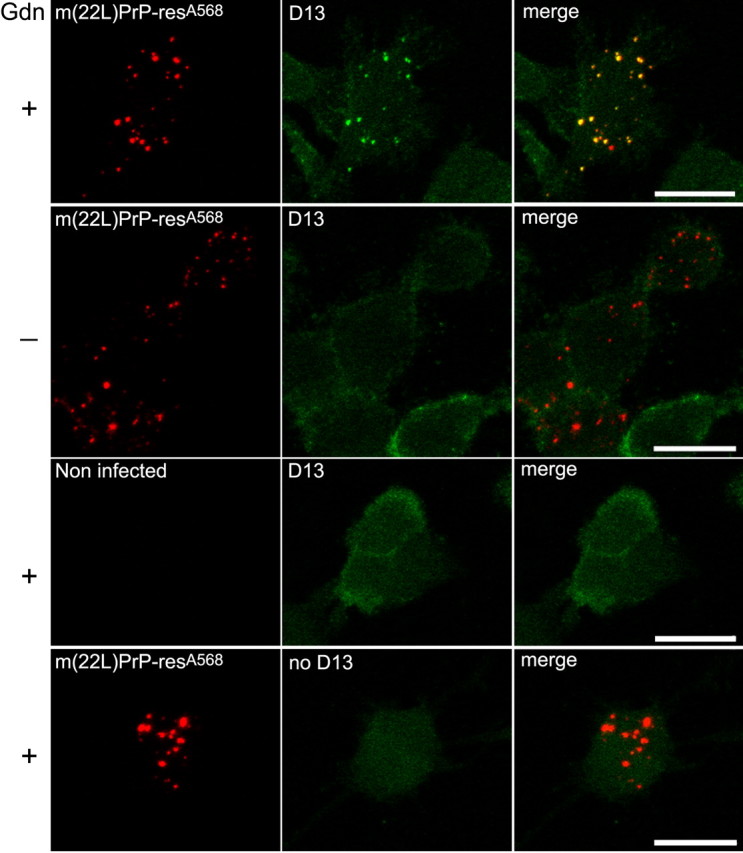
Immunofluorescent labeling of PrP-res in m(22L)PrP-resA568-positive vesicles. First row, Four days after exposure of SN56 cells to m(22L)PrP-resA568, the cells were fixed, permeabilized, treated with 3 m guanidine HCl (Gdn) to expose the D13 epitope in PrP-res, and stained with anti-PrP antibody D13. The left column shows red fluorescence from the Alexa 568 label. The middle column shows the green fluorescence from the FITC-labeled secondary antibody. The right column shows merged images of the left and middle panels. Note the extensive colocalization of the punctate fluorescence from m(22L)PrP-resA568 and D13 immunostaining. Second row, The same treatment as the first row except for the omission of the Gdn treatment. In this case, punctate D13-mediated staining was lost (middle). Third row, The same treatment as the first row except for omission of the m(22L)PrP-resA568 exposure. The lack of m(22L)PrP-resA568 eliminated both the Alexa 568- and D13-mediated intense punctate fluorescence in the cells. Fourth row, The same treatment as the first row except for omission of D13 antibody. Although faint and diffuse green fluorescence was apparent from the secondary antibody alone, no punctate staining and less cell surface staining was observed (middle). Scale bars, 20 μm.
Videos (data not shown) of the labeled cells revealed rapid movement (0.3-5 μm/s; median, 1.4 μm/s) of small fluorescent particles within cell bodies and along neurites. Interestingly, on occasion, we observed vesicles formed from large aggregates indicating how these aggregates were processed by SN56 cells. Some vesicles moved first in one direction, paused, and then reversed direction. Some of the neuritic transport particles appeared to be approximately spherical, whereas others were rod shaped. These observations visualize for the first time the uptake, intracellular dissemination, and neuritic transport of PrP-res coincident with events leading to the sustained infection of cells with a TSE/prion agent.
Species dependence of uptake of PrP-res by SN56 cells
To compare the above observations to the behavior of fluorescent PrP-res of another species that is incapable of producing chronic infection of SN56 cells, similar experiments were performed using hPrP-resA568. On a majority of cells, the large aggregates of hPrP-resA568 mostly remained intact over several days (Fig. 2e,e1,f,f1). However, ∼20% of the cells showed punctuate staining in both the cell body and neuritic projections similar to that observed with m(Ch)PrP-resA568 and m(22L)PrP-resA568. Thus, the internalization and neuritic trafficking of hPrP-resA568 appeared to be much less efficient in the SN56 cells than the mouse 22L and Chandler PrP-res preparations, despite the fact that the population of initial hPrP-resA568 aggregate sizes were similar to m(Ch)PrP-resA568 (data not shown). Nonetheless, the fact that some internalization of hPrP-resA568 occurred suggested that the resistance of SN56 cells to persistent 263K infection is not a result of a total lack of internalization and intracellular trafficking of hPrP-resA568.
Uptake and trafficking of recombinant PrP fibrils and Aβ1-42 fibrils
To determine whether other protein aggregates are internalized and trafficked like m(Ch)PrP-resA568 in SN56 cells, amyloid fibrils were prepared from recombinant mouse PrP23-231 and Alzheimer's β1-42 peptide, conjugated to A568, and added to the cell cultures. Both of these types of fibrils were taken into what appeared to be cytoplasmic vesicles and distributed along neuritic projections within a couple of days in a manner apparently similar to that described for m(Ch)PrP-resA568 (data not shown). With the PrP23-231A568 fibrils, occasional perinuclear localization was also seen. With Alzheimer's β1-42A568 fibrils, colocalization with the acidic organelle marker LysoTracker was seen (data not shown), which was similar to that for PrP-resA568 shown below. Thus, a variety of protein aggregates may be handled by the pathway that is responsible for the uptake and neuritic transport of PrP-res by SN56 cells.
Intracellular localization of m(Ch)PrP-resA568
To identify the subcellular compartments that accumulate and transport exogenous PrP-res, costaining with a variety of organelle markers was performed. Figure 5 shows that m(Ch)PrP-resA568 colocalized with the acidic organelle marker LysoTracker in cell bodies and neuritic locations (Fig. 5b). In contrast, colocalization with markers of lipid rafts (cholera toxin, a ganglioside GM1 ligand) (Nichols et al., 2001) and sorting/early and recycling endosomes (transferrin) (Yamashiro and Maxfield, 1984) was not apparent (Fig. 5c,d). Indeed, internalized cholera toxin, which accumulates in the Golgi apparatus (Nichols et al., 2001), was never seen in the same vesicles as m(Ch)PrP-resA568. Observation of vesicle movement in videos of double-labeled cells indicated that vesicles filled with m(Ch)PrP-resA568 did not seem to fuse to vesicles with fluorescent cholera toxin, although they sometimes appeared to contact each other (data not shown). Moreover, transfection of cells with GFP-VAChT, a cholinergic synaptic vesicle protein fused to green fluorescent protein that labels early and recycling endosomes and synaptic-like vesicles in SN56 cells (Santos et al., 2001; Barbosa et al., 2002), also showed that organelles containing m(Ch)PrP-resA568 did not contain this synaptic vesicle marker (Fig. 6). In contrast, cells labeled overnight with fluorescent dextran, as a marker of fluid-phase endocytosis, showed extensive colocalization between dextran and m(Ch)PrP-resA568 (Fig. 5a). In time-lapse movies of double-labeled cells, almost all vesicles with fluorescent dextran and m(Ch)PrP-resA568 trafficked together (data not shown). Fluid-phase markers accumulate in late endosomes and lysosomes (Swanson and Watts, 1995; Ellinger et al., 1998), which agrees with the observed colocalization of m(Ch)PrP-resA568 with LysoTracker. To confirm the localization of m(Ch)PrP-resA568 in these organelles, cells were transfected with GFP-Rab7 Q67L, a mutant of Rab7 that remains constitutively activated in the GTP-bound form and accumulates preferentially in the membranes of late endosomes and lysosomes (Bucci et al., 2000). After infection of GFP-Rab7 Q67L-transfected cells, most m(Ch)PrP-resA568 particles were enclosed in the lumen of vesicles with GFP-Rab7 Q67L-positive membranes (Fig. 7). Collectively, these results demonstrated that, like the fluid-phase endocytosis marker dextran, exogenous PrP-res was internalized and delivered to late endosomes and/or lysosomes, which in turn distributed the PrP-res along neuritic processes.
Figure 5.
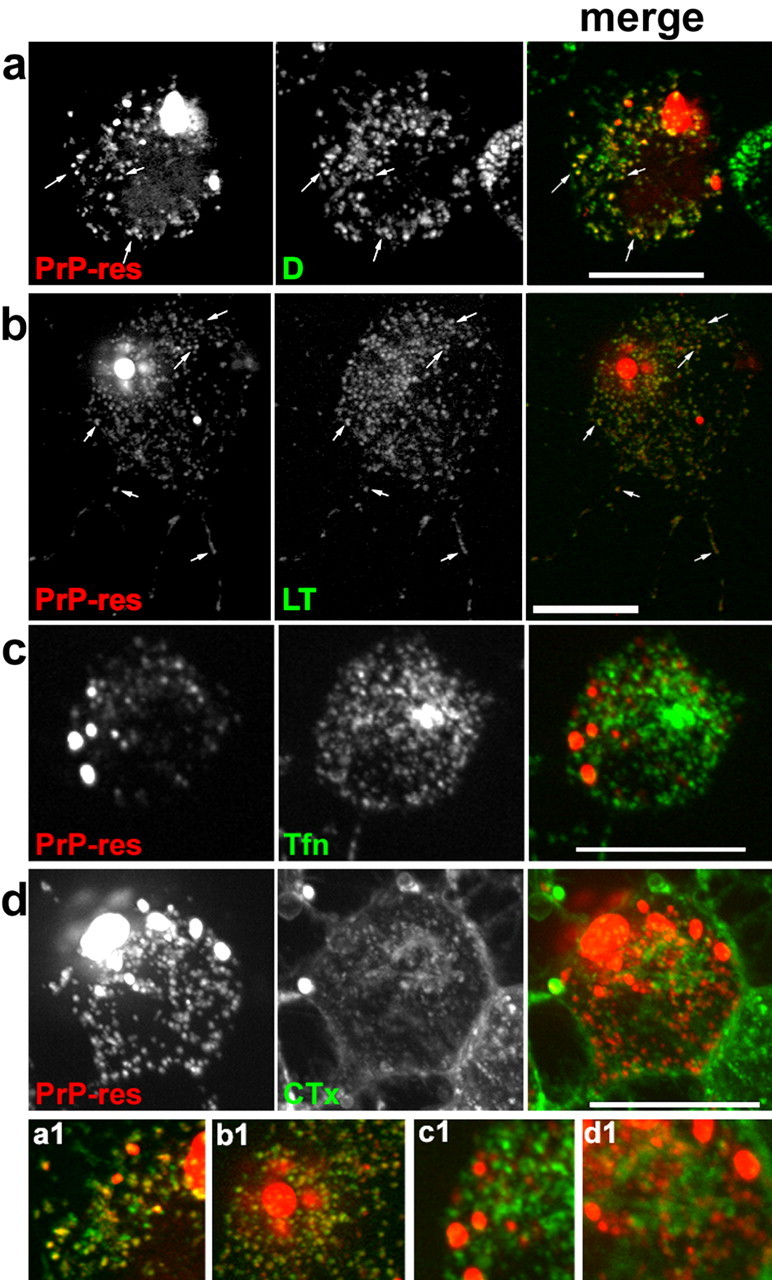
Targeting of m(Ch)PrP-resA568 to late endosomes/lysosomes after infection. SN56 cells with dispersed m(Ch)PrP-resA568 (left, rows a-d) were incubated with the following markers shown in the middle panels: a, dextran-Alexa Fluor 488 (D) for fluid-phase endocytosis (consistent with results of 22 cells from 2 independent experiments); b, LysoTracker Green DND-26 (LT) for acidic organelles and lysosomes (67 cells from 4 experiments); c, transferrin-Alexa 488 (Tfn) for early/sorting and recycling endosomes (84 cells from 8 experiments); d, cholera toxin subunit B-Alexa 488 (CTx) for raft membranes (52 cells from 6 experiments). The right panels in a-d show merged images of the left and middle panels with m(Ch)PrP-resA568 in red and the markers shown in green. Arrows indicate some of the colocalizing vesicles (yellow fluorescence), which were observed with dextran (a) and LysoTracker (b) but not with transferrin (c) or cholera toxin (d). a1-d1 show magnified regions of the merged images of a-d, respectively. Scale bars, 20 μm.
Figure 6.

m(Ch)PrP-resA568-labeled vesicles do not colocalize with GFP-Vacht, a synaptic vesicle marker. a, m(Ch)PrP-resA568 distribution in the cytosol. b, GFP-Vacht accumulates in a perinuclear region and neuritic processes. c, Merged image (GFP-Vacht, green; m(Ch)PrP-resA568, red). d, e, Magnifications of the merged images of neuritic processes (d, e) in c. Images are consistent with 26 cells in four independent experiments. Scale bars, 20 μm.
Figure 7.

Effect of expression of Rab7 Q67L mutant on PrP-res trafficking. SN56 cells expressing GFP-Rab7 Q67L (Q67L) were infected with m(Ch)PrP-resA568 (PrP-res). a, Cells with large GFP-Rab7 Q67L-positive endosomes/lysosomes that also accumulated m(Ch)PrP-resA568 (arrows). b, Large m(Ch)PrP-resA568 aggregate internalized and trafficked to a GFP-Rab7 Q67L-positive endosome/lysosome (arrow). c, m(Ch)PrP-resA568 and GFP-Rab7 Q67L distributed and colocalized in neurites (arrow). Data are representative of 41 cells visualized 3 d after infection in four independent experiments. Scale bars, 20 μm.
Internalization and trafficking of PrP-resA568 in primary cultured neurons
To test whether neurons cultured directly from the brain also have the ability to dissociate and internalize PrP-resA568, we prepared primary cell cultures from cortex tissue of adult hamsters using a method described previously (Brewer, 1997). The cultures contained >70% of neuronal cells as judged by staining with an antibody to the neuronal marker neuron-specific MAP2 (Fig. 8a). After 4 d in culture, cells were incubated with hPrP-resA568, and the distribution of fluorescence was examined by confocal microscopy for 5 d. After 6 h of incubation, most of the PrP-res was associated with cell surfaces as large aggregates (data not shown). However, a minor subset of neurons had already broken up the aggregates and internalized the PrP-res into small vesicles. During the next 4 d, increasing numbers of cells had dissociated the PrP-res aggregate. By the fifth day of incubation with hPrP-resA568, >50% of neuronal cells had PrP-res-positive intracellular vesicles that were distributed through their cell body and processes as observed in SN56 cells infected with m(Ch)PrP-resA568 and m(22L)PrP-resA568 (Fig. 8c). To confirm that relatively intact PrP-res was present in the A568-positive vesicles, immunofluorescent staining was performed with monoclonal antibody 3F4 (Kascsak et al., 1987), which, like antibody D13, binds to PrP-res only after partial denaturation (Peretz et al., 1997). Punctate, Gdn-HCl-dependent staining with 3F4 colocalized with hPrP-resA568 fluorescence in the cell bodies and neuritic projections of the hamster neurons, providing evidence that at least some of the hPrP-resA568 remained intact in the intracellular vesicles (data not shown). These results indicate that, like the mouse SN56 cells, neurons cultured from the adult brain can internalize PrP-res and transport it along neuritic processes.
Figure 8.

Internalization and trafficking of PrP-resA568 in primary cultured neurons. Cultures derived from the adult hamster brain contained a majority of neuronal cells that were stained with anti-MAP2 (a) with its corresponding differential interference contrast image in b. After 4 d of culture, hPrP-resA568 was added to the hamster cells and imaged at 2 d after exposure to hPrP-resA568. A representative neuron that internalized hPrP-resA568 is shown in c and d. e, m(Ch)PrP-resA568 uptake and trafficking after 2 d of exposure to a neuron cultured from knock-out mice lacking PrP gene expression, with its corresponding differential interference contrast image in f. Scale bars, 20 μm.
To test whether endogenous PrP-sen expression is required for exogenous PrP-res uptake, experiments were performed with primary cultures of neurons isolated from knock-out mice lacking PrP expression. Internalization and neuritic trafficking of both m(Ch)PrP-resA568 (Fig. 8 e) and hPrP-resA568 (data not shown) were observed, which appeared to be similar to that observed with the hamster neurons, indicating that PrP-sen-PrP-res interactions are not necessary for these events.
Effect of PrP-res aggregate size
Although the uptake of large aggregates of PrP-res was observed occasionally (Fig. 7b), it was much more common for large cell-surface aggregates to be broken up extensively before internalization. Additional experiments indicated that sonication of certain highly aggregated preparations of hPrP-resA568 before treatment of both primary hamster cortical neurons and SN56 cells markedly expedited internalization and neuritic trafficking (data not shown). Collectively, these results suggest that the size of exogenous PrP-res aggregates can strongly influence their internalization rate. However, particle size does not appear to be the sole determinant of internalization, because hPrP-resA568 was broken up and internalized much less efficiently than m(Ch)PrP-resA568 by SN56 cells, despite being of similar input particle size (data not shown).
Discussion
For clinical symptoms to arise in a TSE-infected host, it is necessary for infections originating in peripheral tissues to invade the nervous system and spread within the brain. Here, we visualize a process by which PrP-res aggregates are reduced to smaller aggregates, internalized, and widely disseminated via late endosome or lysosome-like acidic vesicles within two neuronal cell types. Although we cannot yet define which aspects of the observed trafficking of exogenous PrP-res in the recipient cells are required for initiation of new PrP-res formation, the results show that these PrP-res trafficking events accompany the establishment of a persistent infection and chronic PrP-res formation in SN56 cells. Thus, it is plausible that such an uptake and neuritic transport pathway is a means by which TSE infections are spread within the nervous system.
The PrP-res uptake pathway
A spectrum of endocytotic pathways has been distinguished in various cell types depending on the involvement, or lack thereof, of clathrin, caveolin, dynamin, and lipid rafts (for review, see Pelkmans and Helenius, 2003). The internalization of labeled transferrin and cholera toxin probes that we observed in SN56 cells provides evidence that receptor- and lipid raft-mediated endocytotic pathways are operative, but the lack of colocalization of these markers with PrP-res suggests that these pathways may not be responsible for most exogenous PrP-res internalization. However, it remains possible that if receptor- and lipid raft-mediated transport between the cell surface and late endosomes and lysosomes is rapid relative to the initial dispersion and uptake of large cell-surface PrP-res aggregates, detection of internalized m(Ch)PrP-resA568 in the initial transport endosomes as opposed to late endosomes and lysosomes might be difficult. However, another possibility consistent with the lack of an absolute requirement for the presence of endogenous PrP-sen (Fig. 8e) is that exogenous PrP-res can be taken up by a relatively nonselective process such as macropinocytosis or absorptive pinocytosis. Macropinocytosis occurs in many cell types and results in the delivery of internalized substances to acidic late endosomal or lysosomal organelles that, like the m(Ch)PrP-resA568-positive organelles in SN56 cells, label with LysoTracker and the fluid phase endocytosis marker dextran (Swanson and Watts, 1995; Ellinger et al., 1998).
Consistent with both of these possibilities is the sequestration of m(Ch)PrP-resA568 in large late endosomal or lysosomal vesicles induced and labeled by GFP-Rab7 Q67L (Fig. 7). Rab proteins are involved with different aspects of vesicular trafficking in cells by switching between GTP- and GDP-bound forms (Zerial and McBride, 2001) and can influence PrP-res formation (Beranger et al., 2002). GTP-bound Rab proteins are located on the cytoplasmic face of distinct intracellular compartments in which they regulate vesicle motility, docking, and fusion (Zerial and McBride, 2001). Rab7 has been shown to regulate early-to-late endosomal trafficking (Feng et al., 1995) and also late endosome-to-lysosome trafficking (Meresse et al., 1995; Bucci et al., 2000). The GTP-bound form of Rab7 associates with late endosomal/lysosomal membranes (Cantalupo et al., 2001; Mizuno et al., 2003). The fact that most, if not all, intracellular deposits of m(Ch)PrP-resA568 were surrounded by membranes positive for the constitutively GTP-bound Rab7 mutant GFP-Rab7 Q67L confirms that these aggregates reside and are processed in the late endosomal/lysosomal system.
The lack of colocalization of m(Ch)PrP-resA568 with the raft membrane marker cholera toxin is surprising, because endogenous PrP-res can be copurified with raft membranes from scrapie-infected cells (Vey et al., 1996). One possible explanation for these results is that the exogenous PrP-res aggregates were not efficiently incorporated into raft or other membranes, but endogenously converted PrP-res is already anchored to rafts via its glycophosphatidylinositol moiety before and after conversion. The fact that much of the m(Ch)PrP-resA568 in Figure 7 occupied the lumen of GFP-Rab7 Q67L-labeled vesicles rather than the surrounding membrane is consistent with this possibility. The lack of raft association of m(Ch)PrP-resA568 is also compatible with a proposal that PrP-res formation occurs outside raft domains (Sanghera and Pinheiro, 2002; Sunyach et al., 2003) as well as observations of the accumulation of endogenous PrP-res in lysosomes (McKinley et al., 1991; Laszlo et al., 1992), a compartment not believed to contain detergent-resistant membranes (Hao et al., 2004). However, it is worth considering also that in vivo, some nonamyloid forms of exogenous PrP-res might be membrane associated in a way that alters its interaction with host cell membranes.
Although the m(Ch)PrP-resA568-positive vesicles were transported to the neuritic extremities, little or no colocalization with the synaptic vesicle marker GFP-VAChT was apparent. This agrees with the lack of colocalization of m(Ch)PrP-resA568 with markers of the early endocytic pathway, which is thought to participate in the genesis of synaptic vesicles (de Wit et al., 2001).
The apparent lack of selectivity of the pathway by which PrP-res and other aggregates are internalized and trafficked via acidic organelles does not negate its potential physiological relevance to TSE pathogenesis. If exogenous PrP-res can be taken up by a pinocytotic or transcytotic mechanism and transported to cellular extremities via acidic vesicles, that might be sufficient to account for the spread of infection within neurons even if the pathway is relatively nonselective and shared by other aggregates. This would suggest that other factors are important for the unique transmissible nature of TSEs versus other amyloid diseases.
PrP-res uptake versus establishing a sustained infection
Another prerequisite for the sustained infection of cells is the initiation of continuous new PrP-res formation. Although we have demonstrated that this occurs in conjunction with exposure of the SN56 cells to m(Ch)PrP-res and m(22L)PrP-res, it is unclear where and when within the cells this occurs. In neuroblastoma cells, PrP-sen is known to reside in the Golgi apparatus, on the plasma membrane, and in early and recycling endosomes (Borchelt et al., 1990; Shyng et al., 1993; Prado et al., 2004). In scrapie-infected cells, PrP-sen is converted to PrP-res after transport to the cell surface but before degradation in lysosomes (Caughey and Raymond, 1991; Caughey et al., 1991; Borchelt et al., 1992). Thus, it is likely that contact between PrP-sen and exogenously added PrP-res might first occur on the plasma membrane and/or along an endocytic pathway to lysosomes. This scenario is consistent with our current experiments showing that exogenous PrP-res is transported to late endosomes and lysosomes. Attempts to localize new PrP-res formation during acute infection of cells expressing epitope-tagged PrP-sen have failed, perhaps because of insufficient sensitivity (data not shown). Because striking amino-acid sequence dependence has been observed in cell-free conversion reactions between PrP-res and PrP-sen (Horiuchi et al., 2000), it may be sequence specificity in the interactions between the exogenous PrP-res and the endogenous PrP-sen, rather than the internalization and neuritic trafficking of the invading PrP-res, that determines whether sustained new PrP-res formation can be established. This most likely contributes to the lack of infection of murine SN56 cells by hamster 263K scrapie. However, acute new PrP-res formation can be initiated after exposure of cells to scrapie without leading to sustainable PrP-res formation (Vorberg et al., 2004). Thus, the establishment of persistent cellular scrapie infections would appear to have requirements beyond simple exposure of cells to exogenous PrP-res and acute conversion of endogenous PrP-sen. Regardless of the subcellular site(s) of new PrP-res formation, the neuritic transport of PrP-res described in the present study provides a pathway for the intraneuronal dissemination of TSE/prion infections.
Intercellular transfer of PrP-res
Having observed the uptake and neuritic trafficking of PrP-res in SN56 cells and primary cultures of neurons, the question arises as to whether cell-to-cell transfer of fluorescent PrP-res was observed. On rare occasions, apparent transfer of fluorescent particles from one cell to an adjacent cell was observed in the SN56 cell cultures, but additional study will be required to establish the relevance and reproducibility of such observations. In the vast majority of cases in which m(Ch)PrP-resA568 accumulated at the neuritic extremities close to adjacent cells, no evidence of intercellular transfer was seen. Therefore, it would appear that in the SN56 cell cultures at least, the direct intercellular transfer of PrP-res via neuritic contacts is inefficient. Intercellular transfers of infection in vivo might be aided by other cell types, such as glial cells, or by structures that are not fully recapitulated in these cell cultures. Alternatively, transport between cells might occur primarily via particles that are too small or dilute to remain readily visible in recipient cells. Fevrier et al. (2004) recently reported evidence that scrapie infectivity can be released from cells in small (30-100 nm) membranous vesicles called exosomes. We detected release of small infectious, PrP-res-containing particles into the medium of scrapie-infected SN56 cells (Baron and Caughey, unpublished observations). Whether these particles are visible by light microscopy is not known, but typical exosomes cannot be resolved. An intriguing possibility is that intercellular transfer of PrP-res and TSE infectivity occurs via tunneling nanotubes. Tunneling nanotubes have been described recently as extremely thin (diameter, 50-200 nm) and barely visible, cellular projections that can make contact with nearby cells and mediate the transfer of small LysoTracker-positive vesicles (Rustom et al., 2004). The fact that we have seen extensive colocalization of internalized PrP-res with LysoTracker in small neuritic vesicles makes it tempting to speculate that tunneling nanotubes may mediate the spread of infection between adjacent cells.
Footnotes
A.C.M. received a fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brazil) in an exchange Ph.D. program. M.A.M.P. is a fellow of the John Simon Guggenheim Memorial Foundation (2004-2005) and was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnológico, Fundação de Amparo à Pesquisa de Minas Gerais, and the American Health Assistance Foundation (Alzheimer's program). We are indebted to Drs. Jean Manson, Richard Race, and Bruce Chesebro for providing the PrP knock-out mice, Dr. Ravindra Kodali for providing the fibrils of recombinant mouse PrP23-231, Andrew Hughson and Gregory Raymond for purifying PrP-res, Dr. John Portis for critical review of this manuscript, and Anita Mora and Gary Hettrick for graphics assistance.
Correspondence should be addressed to either of the following: Byron Caughey, Rocky Mountain Laboratories, National Institutes of Health, 903 South Fourth Street, Hamilton, MT 59840, E-mail: bcaughey@nih.gov; or Marco A. M. Prado, Department of Pharmacology, Federal University of Minas Gerais, Avenue Antonio Carlos 6627, Belo Horizonte, MG 31270-901 Brazil, E-mail: mprado@mono.icb.ufmg.br.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255207-10$15.00/0
References
- Arnold JE, Tipler C, Laszlo L, Hope J, Landon M, Mayer RJ (1995) The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J Pathol 176: 403-411. [DOI] [PubMed] [Google Scholar]
- Barbosa Jr J, Massensini AR, Santos MS, Meireles SI, Gomez RS, Gomez MV, Romano-Silva MA, Prado VF, Prado MA (1999) Expression of the vesicular acetylcholine transporter, proteins involved in exocytosis, and functional calcium signaling in varicosities and soma of a murine septal cell line. J Neurochem 73: 1881-1893. [PubMed] [Google Scholar]
- Barbosa Jr J, Ferreira LT, Martins-Silva C, Santos MS, Torres GE, Caron MG, Gomez MV, Ferguson SS, Prado MA, Prado VF (2002) Trafficking of the vesicular acetylcholine transporter in SN56 cells: a dynamin-sensitive step and interaction with the AP-2 adaptor complex. J Neurochem 82: 1221-1228. [DOI] [PubMed] [Google Scholar]
- Bartz JC, Kincaid AE, Bessen RA (2002) Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J Virol 76: 5759-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranger F, Mange A, Goud B, Lehmann S (2002) Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J Biol Chem 277: 38972-38977. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Venturini A, Jackson DA, Lee HJ, Wainer BH (1992) Acetylcholine synthesis and release is enhanced by dibutyryl cyclic AMP in a neuronal cell line derived from mouse septum. J Neurosci 12: 793-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB (1990) Scrapie and cellular prion proteins differ in the kinetics of synthesis and topology in cultured cells. J Cell Biol 110: 743-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Taraboulos A, Prusiner SB (1992) Evidence for synthesis of scrapie prion protein in the endocytic pathway. J Biol Chem 267: 16188-16199. [PubMed] [Google Scholar]
- Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL (1994) Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem 269: 14711-14714. [PubMed] [Google Scholar]
- Brewer GJ (1997) Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 71: 143-155. [DOI] [PubMed] [Google Scholar]
- Bruce ME, McBride PA, Farquhar CF (1989) Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett 102: 1-6. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell 11: 467-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C (2001) Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J 20: 683-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ (1991) The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 266: 18217-18223. [PubMed] [Google Scholar]
- Caughey B, Raymond GJ, Ernst D, Race RE (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol 65: 6597-6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Lichtenstein Y, Kelly RB, Geuze HJ, Klumperman J, van der SP (2001) Rab4 regulates formation of synaptic-like microvesicles from early endosomes in PC12 cells. Mol Biol Cell 12: 3703-3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger I, Klapper H, Fuchs R (1998) Fluid-phase marker transport in rat liver: free-flow electrophoresis separates distinct endosome subpopulations. Electrophoresis 19: 1154-1161. [DOI] [PubMed] [Google Scholar]
- Feng Y, Press B, Wandinger-Ness A (1995) Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol 131: 1435-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101: 9683-9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzel M, Giger O, Braun N, Aguzzi A (2004) The peripheral nervous system and the pathogenesis of prion diseases. Curr Mol Med 4: 355-359. [DOI] [PubMed] [Google Scholar]
- Grigoriev V, Escaig-Haye F, Streichenberger N, Kopp N, Langeveld J, Brown P, Fournier JG (1999) Submicroscopic immunodetection of PrP in the brain of a patient with a new-variant of Creutzfeldt-Jakob disease. Neurosci Lett 264: 57-60. [DOI] [PubMed] [Google Scholar]
- Hammond DN, Lee HJ, Tonsgard JH, Wainer BH (1990) Development and characterization of clonal cell lines derived from septal cholinergic neurons. Brain Res 512: 190-200. [DOI] [PubMed] [Google Scholar]
- Hao M, Mukherjee S, Sun Y, Maxfield FR (2004) Effects of cholesterol depletion and increased lipid unsaturation on the properties of endocytic membranes. J Biol Chem 279: 14171-14178. [DOI] [PubMed] [Google Scholar]
- Hijazi N, Kariv-Inbal Z, Gasset M, Gabizon R (2005) PrPSc incorporation to cells requires endogenous GAGs expression. J Biol Chem 280: 17057-17061. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Chabry J, Caughey B (1999) Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J 18: 3193-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Priola SA, Chabry J, Caughey B (2000) Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci USA 97: 5836-5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horonchik L, Tzaban S, Ben Zaken O, Yedidia Y, Rouvinski A, Papy-Garcia D, Barritault D, Vlodavsky I, Taraboulos A (2005) Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem 280: 17062-17067. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce M, McBride PA, Scott JR, Halliday WG (1994) Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res 656: 329-343. [DOI] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61: 3688-3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin RH, Field HJ, Walker CA (1983) Pathogenesis of mouse scrapie: evidence for spread of infection from central to peripheral nervous system. J Gen Virol 64: 713-716. [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Romano-Silva MA, Gomez MV, Prado MA (2001) Changes in Ca2+ channel expression upon differentiation of SN56 cholinergic cells. Brain Res 916: 199-210. [DOI] [PubMed] [Google Scholar]
- Laszlo L, Lowe J, Self T, Kenward N, Landon M, McBride T, Farquhar C, McConnell I, Brown J, Hope J, Mayer RJ (1992) Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J Pathol 166: 333-341. [DOI] [PubMed] [Google Scholar]
- Magalhaes AC, Silva JA, Lee KS, Martins VR, Prado VF, Ferguson SS, Gomez MV, Brentani RR, Prado MA (2002) Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J Biol Chem 277: 33311-33318. [DOI] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8: 121-127. [DOI] [PubMed] [Google Scholar]
- McBride PA, Beekes M (1999) Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci Lett 265: 135-138. [DOI] [PubMed] [Google Scholar]
- McBride PA, Schulz-Schaeffer WJ, Donaldson M, Bruce M, Diringer H, Kretzschmar HA, Beekes M (2001) Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol 75: 9320-9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, Prusiner SB, Gonatas N (1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab Invest 65: 622-630. [PubMed] [Google Scholar]
- Meresse S, Gorvel JP, Chavrier P (1995) The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J Cell Sci 108: 3349-3358. [DOI] [PubMed] [Google Scholar]
- Mishra RS, Basu S, Gu Y, Luo X, Zou WQ, Mishra R, Li R, Chen SG, Gambetti P, Fujioka H, Singh N (2004) Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: implications for species barrier in prion uptake from the intestine. J Neurosci 24: 11280-11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Kitamura A, Sasaki T (2003) Rabring7, a novel Rab7 target protein with a RING finger motif. Mol Biol Cell 14: 3741-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya KL, Hassig R, Creminon C, Laffont I, Di Giamberardino L (2004) Enhanced detection and retrograde axonal transport of PrPc in peripheral nerve. J Neurochem 88: 155-160. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, Lodge R, Roberts TH, Hirschberg K, Phair RD, Lippincott-Schwartz J (2001) Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol 153: 529-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L, Helenius A (2003) Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol 15: 414-422. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273: 614-622. [DOI] [PubMed] [Google Scholar]
- Prado MA, Alves-Silva J, Magalhaes AC, Prado VF, Linden R, Martins VR, Brentani RR (2004) PrPc on the road: trafficking of the cellular prion protein. J Neurochem 88: 769-781. [DOI] [PubMed] [Google Scholar]
- Raymond GJ, Chabry J (2004) Purification of the pathological isoform of prion protein (PrPSc or PrP-res) from transmissible spongiform encephalopathy-affected brain tissue. In: Techniques in prion research (Lehmann S, Grassi J, eds), pp 16-26. Basel: Birkhauser Verlag.
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH (2004) Nanotubular highways for intercellular organelle transport. Science 303: 1007-1010. [DOI] [PubMed] [Google Scholar]
- Sanghera N, Pinheiro TJ (2002) Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol 315: 1241-1256. [DOI] [PubMed] [Google Scholar]
- Santos MS, Barbosa Jr J, Veloso GS, Ribeiro F, Kushmerick C, Gomez MV, Ferguson SS, Prado VF, Prado MA (2001) Trafficking of green fluorescent protein tagged-vesicular acetylcholine transporter to varicosities in a cholinergic cell line. J Neurochem 78: 1104-1113. [DOI] [PubMed] [Google Scholar]
- Shyng S-L, Huber MT, Harris DA (1993) A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem 268: 15922-15928. [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R (2003) The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 22: 3591-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, Watts C (1995) Macropinocytosis. Trends Cell Biol 5: 424-428. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Jendroska K, Serban D, Yang S, DeArmond SJ, Prusiner SB (1992) Regional mapping of prion proteins in brain. Proc Natl Acad Sci USA 89: 7620-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB (1996) Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA 93: 14945-14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorberg I, Raines A, Priola SA (2004) Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro J Biol Chem 279: 29218-29225. [DOI] [PubMed] [Google Scholar]
- Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358: 171-180. [DOI] [PubMed] [Google Scholar]
- Yamashiro DJ, Maxfield FR (1984) Acidification of endocytic compartments and the intracellular pathways of ligands and receptors. J Cell Biochem 26: 231-246. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107-117. [DOI] [PubMed] [Google Scholar]