

Abstract
The synthesis of antibody–drug conjugates (ADCs) using the interchain cysteines of the antibody inherently gives a mixture of proteins with varying drug-to-antibody ratio. The drug distribution profiles of ADCs are routinely characterized by hydrophobic interaction chromatography (HIC). Because HIC is not in-line compatible with mass spectrometry (MS) due to the high salt levels, it is laborious to identify the constituents of HIC peaks. An MS-compatible alternative to HIC is reported here: native reversed phase liquid chromatography (nRPLC). This novel technique employs a mobile phase 50 mM ammonium acetate for high sensitivity in MS and elution with a gradient of water/isopropanol. The key to the enhancement is a bonded phase giving weaker drug–surface interactions compared to the noncovalent interactions holding the antibody–drug conjugates together. The hydrophobicity of the bonded phase is varied, and the least hydrophobic bonded phase in the series, poly(methyl methacrylate), is found to resolve the intact constituents of a model ADC (Ab095-PZ) and a commercial ADC (brentuximab vedotin) under the MS-compatible conditions. The nRPLC-MS data show that all species, ranging from drug-to-antibody ratios of 1 to 8, remained intact in the column. Another desired advantage of the nRPLC is the ability of resolving multiple positional isomers of ADC that are not well-resolved in other chromatographic modes. This supports the premise that lower hydrophobicity of the bonded phase is the key to enabling online nRPLC-MS analysis of antibody–drug conjugates.
Graphical Abstract
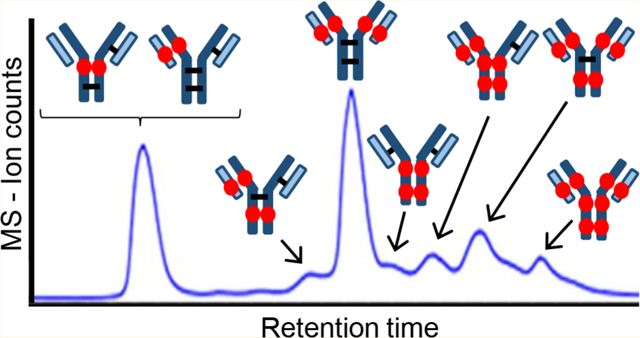
Antibody–drug conjugates (ADCs) are highly selective and potent chemotherapeutics for the treatment of different types of cancer, inspired by Paul Ehrlich.1 An ADC consists of a recombinant monoclonal antibody (mAb) covalently conjugated with a drug via a hydrophilic linker. The mechanism exploits specific binding of tumor-expressed antigens and delivers covalently conjugated cytotoxic payloads to cancer cells selectively over nonmalignant cells, resulting in greater efficacy and minimized systemic toxicity. Four ADCs are currently on the market: Adcetris (brentuximab vedotin) from Seattle Genetics for the treatment of relapsed Hodgkin’s lymphoma and systemic anaplastic large-cell lymphoma, Kadcyla (trastuzumab emtansine) from Genentech for the treatment of metastatic breast cancer,2–4 Mylotarg (gemtuzumab ozogamicin) from Pfizer for acute myeloid leukemia, and Besponsa (inotuzumab ozogamicin) also from Pfizer for acute lymphoblastic leukemia. More than 60 ADCs have been advanced into clinical trials for cancer treatment,3 and there are currently more than 65 ADCs in clinical evaluation to target different hematologic malignancies and solid tumors.3,5 The vast majority of the cytotoxic warheads of the ADCs currently in clinical trials are conjugated to either lysine or cysteine residues on the antibody,6–8 with most using cysteine residues.9 Drug loading in the ADCs is an important design parameter that needs to be characterized.10
Liquid chromatography separation of cysteine-conjugated ADCs to characterize the drug loading distribution is the topic of this paper. Taking IgG1, for example, a common conjugation approach entails partial reduction of four interchain disulfide bonds to generate up to eight reactive thiol groups.11–13 This conjugation scheme yields a mixture of species ranging from 0 to 8 drugs per antibody, which is a broad distribution. The different drug loadings have been reported to affect the pharmacokinetics, stability, and clearance of ADCs.14–18 Native SEC-MS is a rapid technique for determining the distribution of drug loads, where the SEC serves to desalt the sample rather than separate the components and relies solely on MS for characterization and quantitation.19 The technique skews the distribution toward lower drug load due to ion suppression and suboptimal recovery of species with higher drug load.20 Pretreatment by enzymatic cleavage of the hydrophobic drug from the ADC, which leaves the hydrophilic linker attached as a tag, reduces the skewing but does not eliminate it.21 Consequently, chromatographic separations are used for quantitative ADC characterization. Reversed-phase liquid chromatography coupled to mass spectrometry (RPLC-MS) is used to determine the average drug-to-antibody ratio (DAR) by separating the denatured subunits of the reduced ADC,22 but this approach loses information about the drug load distribution.23 Hydrophobic interaction chromatography (HIC) is a nondenaturing separation24–26 that is currently the gold standard for resolving the drug distribution of ADCs.27 A gradient of decreasing salt concentration is used for elution,28,29 and the high initial concentration and low volatility of the salts prevent its direct coupling to mass spectrometry for peak identification.30–34
The Ge and Alpert groups were the first to show that HIC-MS of intact proteins is possible with volatile salts.26,35 In their papers, MS-compatible ammonium acetate salt was used, with a gradient decreasing from 1 M to 20 mM, concurrent with a gradient of increasing acetonitrile in water from 0 to 50%. Because NH4OAc has kosmotropic properties weaker than those of the typical HIC salts of (NH4)2SO4 and Na2HPO4, they used a bonded phase with increased hydrophobicity, and their results demonstrated that the proteins maintained their native forms. HIC-MS has not yet been reported for intact ADCs.
The considerations for HIC-MS of ADCs are different from those of natural proteins. The conjugated drug of an ADC is far more hydrophobic than the solvent-exposed surface of a native protein, as demonstrated by the elution time increasing with increasing drug load in HIC of ADCs. In light of this, the concept behind our work is that a fixed, low concentration of MS-compatible salt, e.g., 50 mM NH4OAc, might give retention of ADCs on hydrophobic columns because less salting-out would be needed. If so, the question then is whether a mild organic additive, isopropanol, can be made to desorb the ADC from the stationary phase without dissociating the noncovalently bound subunits of the antibody. The strategy is to decrease the hydrophobicity of the bonded phase so that less organic component is needed for elution, thereby avoiding the dissociation of the antibody into subunits.36 This is the opposite of the strategy used by Chen and coworkers26,35 because ADCs present a different problem than mAbs, which are more hydrophilic than ADCs. The other difference from the prior work is that the salt concentration is fixed at low level while the organic component is increased, which would make this a reversed-phase separation. Hence, the proposed new method is a nondenaturing version of reversed-phase liquid chromatography (RPLC), and we refer to it as native reversed-phase liquid chromatography (nRPLC).
The purpose of this work is to test the idea that a bonded phase with sufficiently low hydrophobicity would enable a new technique, nRPLC-MS, for separating intact ADCs and determining their molecular weights by in-line coupled mass spectrometry. The method is evaluated using both a model ADC and a commercial ADC, where each ADC has a drug mimic or drug coupled to cysteines of the mAb using a hydrophilic linker.
MATERIALS AND METHODS
Materials.
Nonporous silica particles (1500 nm) were purchased from Superior Silica (Tempe, AZ). Empty stainless-steel columns (2.1 mm ID, 50 mm length), reservoirs (4.6 mm ID, 150 mm), and frits (0.5 μm pore diameter) were purchased from Isolation Technologies (Middleboro, MA). Stainless-steel tubing, ferrules, and internal nuts were all purchased from Valco Instruments (Houston, TX). Silanes, i.e., (chloromethyl)phenyl)dimethylchlorosilane (+99%) and trimethylchlorosilane (+99%), were purchased from Gelest (Morrisville, PA). Methyl methacrylate (MMA, 99%), sodium ascorbate (≥99%), butylamine (99.5%), NH4OAc (99.99%), ammonium sulfate ((NH4)2SO4, ≥99%), sodium phosphate (Na3PO4, 96%), and ammonium hydroxide were purchased from Sigma-Aldrich (St. Louis, MO). Other chemicals used included trifluoroacetic acid (TFA, 99%), difluoroacetic acid (DFA, 98%), and copper(II) chloride (CuCl2, 99%) from Acros Organics (Morris Plains, NJ), tris (2-dimethylaminoethyl)amine (Me6TREN, + 99%) from Alfa Aesar (Haverhill, MA), and formic acid (FA, 99.5%+, LC/MS grade), acetonitrile (ACN), and 2-propanol (IPA) from Fisher Scientific (Hampton, NH). Ultrapure water was obtained from a Milli-Qsystem (MilliporeSigma, Darmstadt, Germany).
IgG1 Ab095 was conjugated with drug-linker mimic PZ in-house at AbbVie (North Chicago, IL) as a model ADC. Brentuximab vedotin was obtained from Seattle Genetics. Both ADCs were prepared at 1 mg/mL in NH4OAc or (NH4)2SO4 with final concentration 0.8–1.0 M.
UHPLC Column Preparation.
The silica particles were modified as described earlier.37 Briefly, the silica particles were calcined at 600 °C for 12 h, then annealed at 1050 °C for 3 h, and rehydroxylated overnight in 1.0 M HNO3. Particles were then rinsed in ultrapure water and dried in a 60 °C vacuum oven. SEM showed that the particles decreased in diameter to 1.2 μm from the heating steps. Freshly rehydroxylated silica particles were suspended in a dry toluene solution containing 2% (v/v) of (chloromethyl)phenyl)dimethylchlorosilane and 0.1% (v/v) of butylamine. The solution was refluxed for 3 h and then rinsed with dry toluene. The particles were then end-capped by suspending in another dry toluene solution containing 2% (v/v) of trimethylchlorosilane and 0.1% (v/v) of butylamine and refluxed for 3 h. The silylated, end-capped particles were then rinsed with dry toluene and allowed to dry in a 60 °C vacuum oven for 2 h.
For polymer growth, each monomer was dissolved in 50:50 H2O/IPA (v/v) in a 50 mL round-bottom flask for a final concentration of 2.5 M. Two other solutions were made: (1) a solution containing 40 mg of CuCl2 and 80 μL of Me6TREN and (2) a solution containing 20 mg of sodium ascorbate. These were also prepared in 2.0 mL of 50:50 H2O/IPA. Afterward, the Cu/Me6TREN solution was added to the round-bottom flask, followed by the sodium ascorbate solution. The resulting solution was poured into a plugged reservoir column of 4.6 × 150 mm. A 2.1 × 50 mm column was packed with 0.24 g of silylated, end-capped particles suspended in acetonitrile. The reservoir and column were connected in series. A high-pressure pump, LabAlliance Series 1500 HPLC Pump (Laboratory Alliance of Central New York, LLC, Syracuse, NY) was used for packing and modification. The reaction solution from the reservoir was pumped into the column starting at 200 μL/min until the reaction mixture dripped from the end of the column. The flow rate was then lowered to 100 μL/min, and the polymerization reaction was allowed to proceed for a range of reaction times from 40 to 85 min for optimization. After reaction, the freshly packed column poly(methyl methacrylate) (PMMA) was rinsed with water for 20 min at 100 μL/min.
UHPLC.
The columns and mobile phases for the various separations are summarized in Table 1.
Table 1.
Summary of HPLC Columns and Their Applications Herein
| column | application | mobile phase A; B |
|---|---|---|
| Thermo MabPac RP | RPLC, reduced ADC | H2O + 0.1% DFA; ACN + 0.1% DFA |
| Supelco Bioshell A400 | RPLC, nonreduced ADC | H2O + 0.1% FA + 0.015% TFA; ACN + 0.1% FA + 0.015% TFA |
| Thermo-HIC butyl | HIC, ADC (Purdue) | 50 mM Na3PO4 + 1 M (NH4)2SO4, pH 7; 50 mM Na3PO4 + 30% IPA, pH 7 |
| Tosoh TSKgel Butyl | HIC, ADC (AbbVie) | 25 mM Na3PO4 + 1.5 M (NH4)2SO4 pH 7; 25 mM Na3PO4 + 25% IPA, pH 7 |
| PMMA, nonporous | nRPLC, ADC | 50 mM NH4OAc, pH 7; 50 mM NH4OAc + 50% IPA, pH 7 |
A Thermo Accela UHPLC system (Thermo-Scientific, Waltham, MA, United States) was used for the development of nRPLC separations at the Purdue lab. Lab-made nRPLC columns (2.1 × 50 mm, 1.2 μm nonporous silica particles coated with various polyalkyl methacrylates were used as the analytical columns. A commercial column, MabPac HIC-Butyl (4.6 × 100 mm, 5 μm nonporous), from Thermo Scientific (Waltham, MA) was used under both HIC and nRPLC conditions for comparison because both have polymeric surfaces. UV absorbance wavelength was set to 280 nm. The column temperature was 30 °C, and the injection volume was 3 μL.
A TSKgel Butyl-NPR column (4.6 × 35 mm, 2.5 μm, Tosoh, King of Prussia, PA) was used for HIC at the AbbVie site, with an Agilent 1200 HPLC (Agilent, Santa Clara, CA). The system is routinely used for HIC separations of ADCs to calculate DAR. With the mobile phase given in Table 1, the gradient started with 90% MPA, decreased to 75% MPA in 2 min followed by a gradient to 0% MPA in 10 min, and was held for 2 min before re-equilibrium. The flow rate was 0.8 mL/min, and column temperature was set to 25 °C.
LC-MS.
For RPLC-MS of the reduced ADC, this was generated in the Purdue lab by adding 1,4-dithiothreitol (DTT), a Thermo Accela UHPLC was used with a Thermo MabPac RP column (2.1 × 50 mm, 4 um, supermacroporous polymer particles) (Thermo-Scientific, Waltham, MA, United States), and the column was coupled to a Thermo LTQ Velos mass spectrometer (Thermo-Scientific, Waltham, MA, United States). With the mobile phase given in Table 1, the gradient started with 27% with MPB2, increased to 43% MPB2 in 15 min, and returned to 27% MPB2 for re-equilibrium. Flow rate was 0.2 mL/min, and column temperature was 80 °C. Peak identities were assigned by matching deconvoluted masses with theoretical masses.
For RPLC-MS of the nonreduced ADC (no DTT), a Supelco Bioshell A400 Protein C4 column (2.1 × 100 mm, 3.4 μm, Sigma-Aldrich, St. Louis, MO) was used in the AbbVie lab. With the mobile phase given in Table 1, the gradient started with 90% MPA3, ramped to 69% MPA3 in 1 min followed by a decrease to 52% MPA3 in 13 min and returned to original condition for re-equilibrium. Flow rate was 0.3 mL/ min, and column temperature was 70 °C. The ADC samples were analyzed using an Acquity UPLC H-Class coupled to a Synapt G2 Si mass spectrometer (Waters, Milford, MA). Peak identities were assigned by matching deconvoluted masses with theoretical masses.
For online nRPLC-MS analysis, the poly(methyl methacrylate) (PMMA) column made in the Purdue lab was used at AbbVie, where the LC-MS system was a Waters Acquity UPLC H-Class coupled to a Xevo G2 qTOF mass spectrometer (Waters, Milford, MA). With the mobile phase given in Table 1, the gradient started with 0% MPB4, held for 2 min, ramped to 15% MPB4 in 3 min followed by a gradient to 50% MPB4 in 15 min and held at 50% MPB4 for 6 min before re-equilibrium. Flow rate was reduced to 0.07 mL/min. Column temperature was 30 °C. For MS condition, capillary voltage was 3.00 kV. Sample cone voltage was 85 V, trap collision energy was set to 60 V, source temperature was 140 °C, and desolvation temperature was 500 °C. The high sampling cone voltage was used to improve resolution and sensitivity in raw MS spectra, but this more prone to cause insource fragmentation
RESULTS AND DISCUSSION
A model ADC was synthesized by AbbVie, with the chemical structure of the linker and drug portions depicted in Figure 1a. The structure is similar to that of the commercial ADC, Brentuximab, also studied here, with its structure depicted in Figure 1b. These are both cysteine-conjugated ADCs using a similar, typical hydrophilic linker with coupling to the mAb cysteines via the maleimide group. The figure shows that the drug mimic for the AbbVie model ADC and the drug for brentuximab vedotin are quite hydrophobic, each with a log P in excess of 3, where P is the partition coefficient for octanol/ water.
Figure 1.
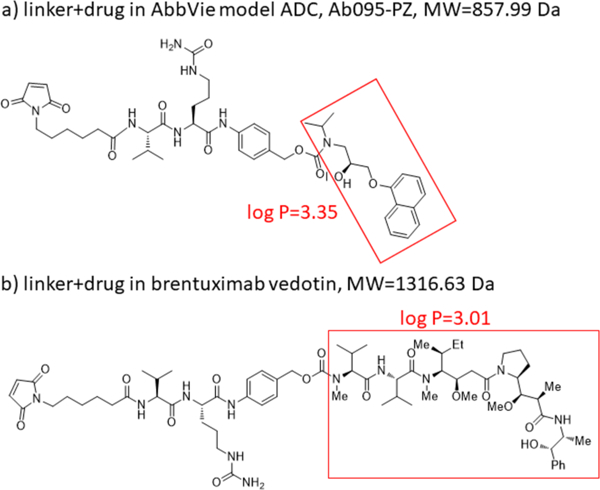
Chemical structures of linker-drug combination for (a) the AbbVie model ADC, Ab095-PZ and (b) brentuximab vedotin. Each drug or drug mimic part is in the red square, and its hydrophobicity is expressed by log P, where P represents the octanol/water partition coefficient.
ADCs can be characterized with respect to their average DAR by fully reducing the ADCs with DTT and then separating the subunits by RPLC. Sketches to indicate labeling for intact ADCs and the various subunits are given in Figure 2a. The RPLC chromatogram for the reduced AbbVie model ADC Ab095-PZ is shown in Figure 2b. The mass spectrum of each peak (Supplementary Figure S1) was used to assign each of the six peaks to the subunit, as labeled in the chromatogram. The first two peaks are light chains without (L0) or with (L1) one drug+linker, and the latter four peaks are heavy chains with 0, 1, 2, and 3 drug+linker attachment(s). The average DAR calculated from the relative peak areas is 3.9. The RPLC chromatogram of Ab095-PZ without DTT reduction (i.e., nonreduced RPLC) is shown in Figure 2c. The peak assignments from MS show that the model ADC was only partially reduced during the conjugation process, as expected. The results demonstrate that in conventional RPLC, without interchain disulfide bond linkages, the ADC dissociates into subunits. The value of this chromatogram is that it can be later compared to that for native RPLC with DTT absent. The HIC chromatogram of Ab095-PZ, using a commercial HIC-Butyl column and typical HIC salt gradient, is shown in Figure 2d. As is common practice, an isopropanol gradient was super-imposed on the salt gradient to attain full elution of the ADC constituents. The species with higher drug loading gave multiple peaks, and this is shown later to be due to partial resolution of positional isomers. Mass spectrometry cannot be used to identify the peaks of Figure 2d because the conventional HIC salts suppress ionization and cause adduct formation, as discussed earlier. One can make tentative peak assignments based on typical drug loading profile for ADCs with an average DAR of 3.9, as indicated in Figure 2d.
Figure 2.
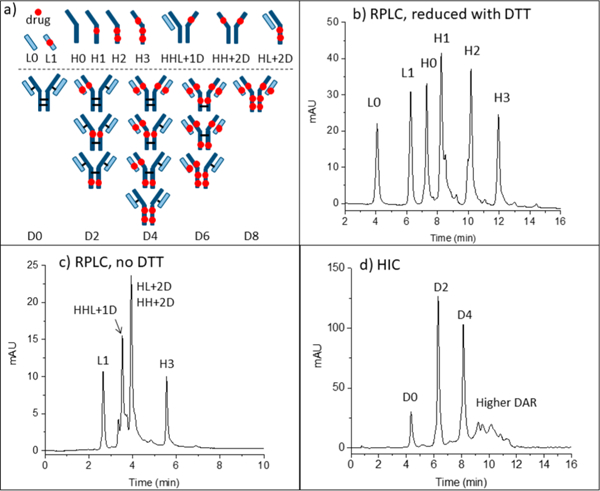
(a) Sketches explain abbreviations for peak assignments. (b) RPLC of the model ADC after reduction with DTT. (c) RPLC of the model ADC without DTT. (d) HIC of the intact ADC with tentative peak assignments. Condition are in the Supporting Information.
The proposed strategy described earlier to enable native RPLC-MS is to use no more than 50 mM NH4OAc. This amount of salt is normally reached at the end of a salt gradient for online HIC-MS;25,26,38 therefore, there is now little value in even running a salt gradient in RPLC mode. Despite this low level of salt, the same commercial HIC column as used for the HIC of Figure 2d (Thermo MabPac HIC-Butyl column) was found to give virtually no elution of the ADC, as shown in Supplemental Figure S2; the retention to the column is too strong for elution. This indicates that the lower kosmotropic power of 50 mM NH4OAc gives more retention than the higher kosmotropic power of 50 mM sodium phosphate of Figure 2d. The strong retention with 50 mM NH4OAc is attributed to irreversible adsorption of the hydrophobic drug rather than to salting out of the intact ADC. The HIC stationary phase, which is said to be made of butyl groups, is thus too hydrophobic for use with isocratic 50 mM NH4OAc, i.e., the hydrophobic interactions between ADC and bonded phase surface are stronger than the intramolecular hydrophobic interactions within the ADC. This inspires the proposed strategy to make the bonded phase less hydrophobic so that the free energy barrier for protein desorption is lower than the free energy barrier for protein denaturation.
Native RPLC chromatograms of model ADC Ab095-PZ using isocratic 50 mM nH4OAc with a gradient of 0–50% isopropanol are shown in Figures 3a–d for a series columns with decreasing bonded phase hydrophobicity, including polymethyl-, polyethyl-, polypropyl-, and poly(butyl methacrylate). The recovery and resolution are progressively higher with lower hydrophobicity, consistent with less denaturation of the ADCs lower mobile phase strength. Poly(methyl methacrylate), with the lowest hydrophobicity, gives a chromatogram similar to that of the native HIC chromatogram of Figure 2d, suggesting that intact ADCs are indeed eluted under mild organic phase content without dissociation. The chromatogram is quite different from than that of the denaturing RPLC case of Figure 2c, again arguing that the ADCs are not dissociated.
Figure 3.
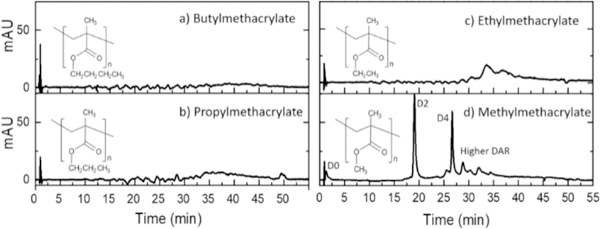
Native RPLC (nRPLC) of AbbVie model ADC with varying hydrophobicity of bonded phase, as denoted by the structures. Gradient conditions are A: 50 mM NH4OAc, pH 7, B: 50 mM NH4OAc, 50% IPA, pH 7, 0–100%B/40 min, 100%B/5 min, 100 μL/min, 30 °C. The same tentative labels as for the HIC chromatogram of Figure 2d are made due to the similarity.
The downside of the nRPLC separation of the ADCs in Figure 3 is that the native antibody, i.e., the species having no conjugated drug, D0, has low retention. This is an inherent outcome of the nRPLC strategy, where the designed retention mechanism is based on the hydrophobic interaction between the exposed/or partially exposed hydrophobic drug with the bonded phase. To increase the retention of D0 species, some mixed-mode copolymer could potentially be used with minimal effect on retention of the hydrophobic drug.
Mass spectrometry is used to test whether the constituents of the ADC peaks are intact vs dissociated under nRPLC conditions for the column with poly(methyl methacrylate) grown for 70 min. Figure 4a shows the nRPLC chromatogram for the AbbVie model ADC using the poly(methyl methacrylate) column, now with the gradient adjusted for faster elution. The peaks are labeled in detail based on the mass of the most prevalent protein for each peak. The raw mass spectral data are given in Figure 4b. By extracting high mass range (extracted in chromatogram not shown), it was confirmed that the unconjugated species (D0) was barely retained and was nearly coeluting with the injection peak. The small peak in the chromatogram of Figure 4a eluting at 10 min was identified as D1. In Figure 4b, the first mass spectrum assigned the peak at 13 min as D2 based on deconvoluted mass, which gives the mass (149 380 Da) corresponding to that expected for D2 (theoretical mass: 147 640 + 2 × 859). There are three peaks for D4, labeled as D4(l), D4(2), and D4(3) in the order of elution, and Figure 2a showed that there are theoretically four positional isomers for D4. Of note, the positional isomers of D4 that result from conjugation of the upper vs lower cysteine pairs in the hinge region likely coelute because they are only subtly different in structure. Similarly, D6 gives two peaks when there are expected to be three, but again, as illustrated in Figure 2a, two cases differ only by position on the heavy chain (upper hinge cysteine conjugation vs lower hinge cysteine conjugation). It is novel for a HIC column to resolve different D4 and D6 isoforms from one another, and the separation on this poly(methyl methacrylate) under nRPLC mode could be advantageous in process understanding and quality control. The mass spectrum of the larger of the two D6 peaks is given in Figure 4b. It shows some peak overlap with D4, as indicated by the light blue lines in Figure 4b. The mass spectrum of the peak labeled D8 indicates it to be mainly D8 with some overlap from D5, D6, and D7. No peaks due to fragments were observed. It is noteworthy that all ADC mass spectra demonstrated a native-like charge envelope distribution with charge state from 24 to 33, which further supports the conclusion of native RPLC.
Figure 4.
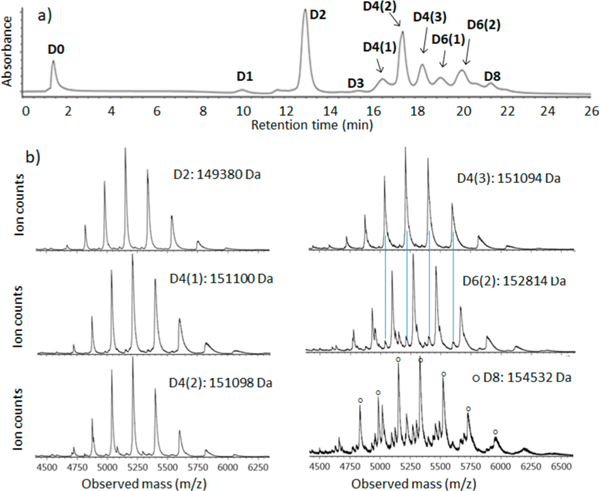
(a) nRPLC of the AbbVie model ADC Ab095-PZ, with peaks labeled based on the mass spectra. Gradient: 0 to 4.5% IPA/water over 3 min, then 4.5 to 50% IPA/water over 20 min. Detection at 280 nm. (b) Raw mass spectra for peaks as labeled, with the molecular weight based on deconvoluted mass spectra for peak ID. The blue lines show that extra peaks are from overlap.
Representative mass spectra of model ADC over a wider range of mass-to-charge ratio (m/z) are given in Figure 5a. All spectra show strong signals from a heretofore unexpected L1 fragment (light chain plus drug), despite the absence of corresponding species (ADC minus L1) in the higher mass range for intact ADC. This at first seems to contradict the claim of intact ADC elution during the discussion of Figure 4 for the higher mass range. Our conclusion is that this L1 signal in Figure 5 arises from two circumstances: (l) in-source fragmentation of the ADC after elution due to the rather high sampling cone voltage and (2) a greater ionization efficiency of the L1 fragment to make its signal appear disproportionately strong compared to that of the intact ADC. If the signal strength were proportional to abundance, there would be a significant amount of ADC-L1 detectable in the higher range of m/z. Therefore, the large peaks for L1 must be due to greater ionization efficiency. In addition, if L1 dissociated from the ADC on the column, the EIC based on L1 would not be correlated with the UV chromatogram. The only reasonable way for the subunits of the ADC to travel together throughout the separation is for the ADC to be intact. An extracted ion chromatogram (EIC) for the L1 fragment is shown in Figure 5b, in comparison with the same chromatogram using UV detection. It is clear that the chromatograms closely track one another for the two different modes of detection. Further, the exceptions prove the rule: the blue arrows in Figure 5b show a D4 peak that is increased and a D4 peak that is decreased for EIC of L1. These are consistent with the expectation that one D4 should have two L1 species and one should have none. The inset images in Figure 5b depict the structures of the positional isomers. D4(2) has twice as many light chains with drug compared to D4(l), which would make its signal increase for EIC of Ll. Likewise, D4(3) has no light chain with drug; hence, signal would decrease for EIC of L1. The EIC supports the conclusion that L1 dissociated postcolumn and all ADCs remained intact throughout the nRPLC separation. It is remarkable that D8, with fully reduced interchain disulfide bonds between subunits, eluted intact.
Figure 5.
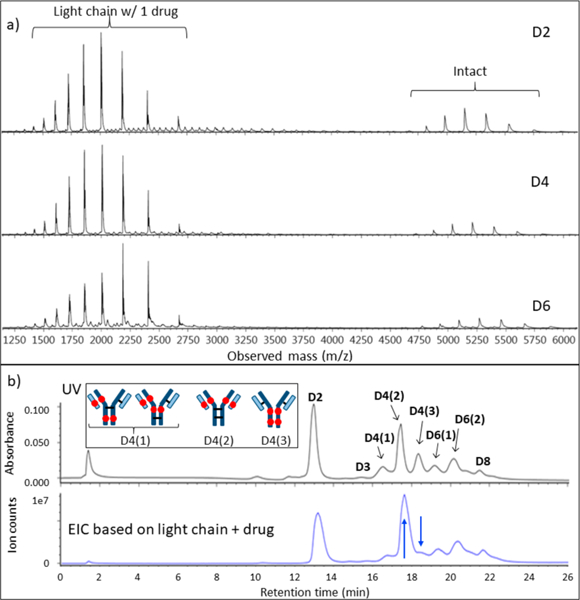
Evidence that light chain dissociates in MS source for AbbVie model ADC. (a) Full-range raw mass spectra show large signals for light chain+drug, but no significant signals for ADC minus light chain+drug. (b) Chromatogram with UV detection (top) and EIC based on light chain+drug (bottom). The blue arrows point to two peaks that changed intensities, and the inset depicts the structures for the isomers consistent with these intensity changes.
The native RPLC-MS strategy was also tested for a commercial ADC, brentuximab vedotin, which is a well-characterized commercial ADC with an average DAR of 4.0, comparable to that of the AbbVie model ADC.39 HIC was performed to compare DAR profiles of the two ADCs. The results are provided in Figure 6, confirming that the DAR profiles of these two ADCs are qualitatively similar. The chromatograms show that the D0 peak elutes later for brentuximab vedotin, indicating that the brentuximab (mAb of brentuximab vedotin) sequence is more hydrophobic than that of the AbbVie model ADC. This is offset by the drug of brentuximab vedotin being less hydrophobic than the drug-linker of the AbbVie model ADC, with its lower octanol/water partition coefficient, log P = 3.01, for MC-VC-MMAE compared to that of the AbbVie model drug, log P = 3.35. The elution times of peaks with higher drug load in HIC are similar in both chromatograms: 10 min.
Figure 6.
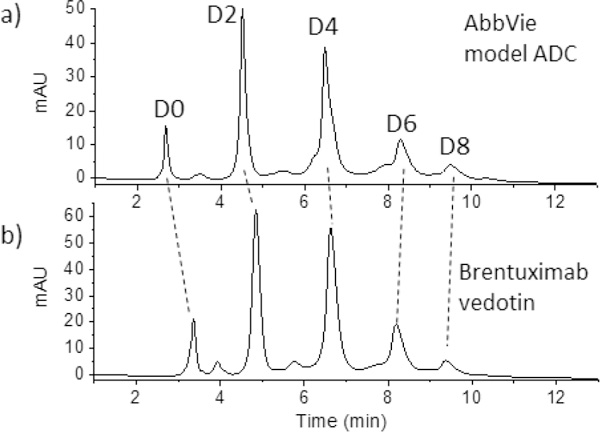
HIC separation of (a) model ADC and (b) commercial ADC Brentuximab vedotin. The dashed lines illustrate that the greater hydrophobicity of the mAb itself for Brentuximab vedotin. Tosoh TSKgel Butyl-NPR, 4.6 × 35 mm, 2.5 μm. MPA: 1.5 M ammonium sulfate, 25 mM sodium phosphate pH 7.0; MPB: 25 mM sodium phosphate pH 7.0 with 25% IPA; flow rate: 0.8 mL/min; column temp: 25 °C.
The nRPLC chromatogram for brentuximab vedotin, using the same poly(methyl methacrylate) column and separation conditions as for the AbbVie model ADC, is given in Figure 7a.
Figure 7.
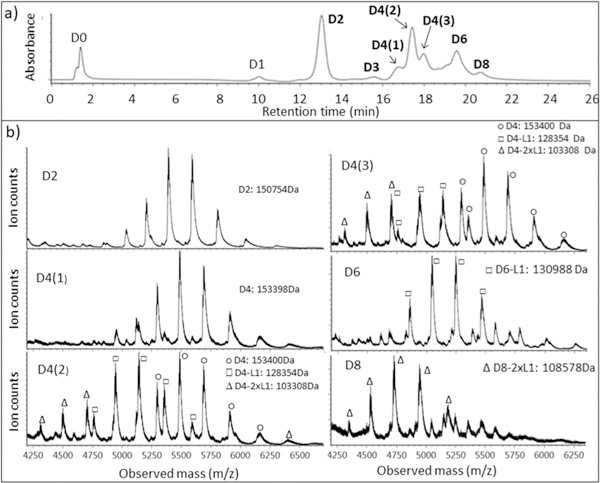
nRPLC and mass spectra for commercial ADC: brentuximab vedotin. (a) nRPLC with detection at 280 nm. Conditions same as those in Figure 4. (b) Raw mass spectra for D2, D4(1,2,3), D6, and D8, with the molecular weight based on deconvoluted mass spectra.
The chromatogram is similar to that for the AbbVie model ADC, with differences in relative peak heights and a small extra peak before D6. The D0 species is now slightly retained in nRPLC, owing to the greater hydrophobicity of the mAb that was noted using HIC. All ADC peaks elute somewhat earlier in nRPLC for brentuximab vedotin, consistent with the lower hydrophobicity of the drug. The mass spectra, detailed in Figure 7b, show that the D2 peak and the first D4 peak, D4(l), are intact, with no loss of Ll. The other two D4 peaks, D4(2) and D4(3), show some loss of one or two light chains with drug (D4–L1 and D4–2xLl), in addition to the intact forms being observed. Overall, the results show that it is much easier to lose L1 from brentuximab vedotin than it is from the AbbVie model ADC.
As was done with the AbbVie model ADC to distinguish on-column vs in-source dissociation, Figure 8a shows representative mass spectra over wider range of m/z for brentuximab vedotin. The relative signals from the L1 fragment are much stronger than those of the AbbVie model ADC, again illustrating the greater ease of loss of L1 for brentuximab vedotin. To determine whether L1 dissociated on-column or in-source, the EIC for the L1 fragment is shown in Figure 8b, in comparison with the same chromatogram using UV detection. As with the case for the AbbVie model ADC, the UV and extracted ion chromatograms closely track one another. The lack of an L1 background across the chromatogram confirms that all of the ADCs remain intact throughout the course of the nRPLC separation. As with the AbbVie model ADC, the L1 fragment signal is more pronounced in the D4(2) position than the latter peak D4(3), indicated by the blue arrows. As with the case for the AbbVie model ADC, the relative abundances of the L1 fragment peaks are likely associated with same two factors: (a) the L1 molar ratio in the positional isomer: 1, 2, and 0, for D4(l), D4(2), and D4(3), respectively; and (b) strength of the noncovalent interaction between L1 and heavy chain in the MS source. These two factors reflect on the L1 fragment peak abundance in the order of D4(2) > D4(l) ≫ D4(3). The comparison of EIC and UV chromatograms again supports the conclusion that nRPLC elutes intact brentuximab vedotin species. The greater loss of L1 in the source for brentuximab vedotin relative to the AbbVie model ADC indicates that the noncovalent interactions between light and heavy chains are weaker for brentuximab vedotin than that of the AbbVie model ADC Ab095-PZ.
Figure 8.
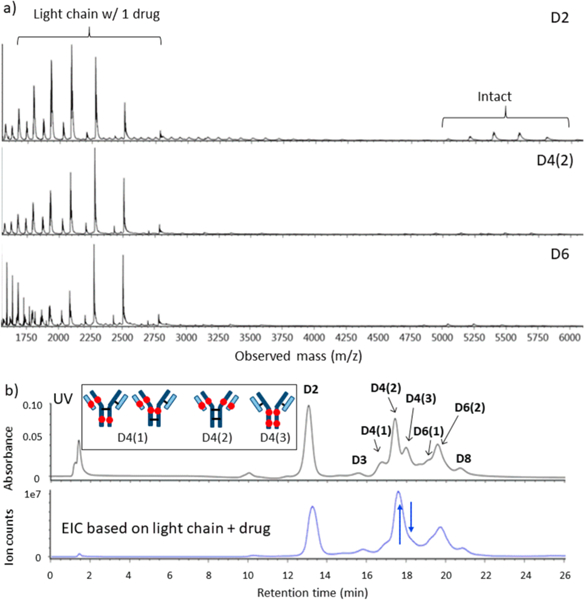
LCMS data for brentuximab vedotin, analogous to Figure 5. (a) Full-range raw mass spectra show even stronger signals for light chain+drug than observed for the AbbVie model ADC. (b) Chromatogram for UV detection (top) approximately tracks that of EIC for signal of light chain+drug, again indicating light chain+drug dissociated after separation. Again, the blue arrows show two peaks that changed intensities, and the inset depicts the structures for the D4 isomers that are consistent with these changes.
CONCLUSIONS
A novel protein chromatography technique intersecting HIC and RPLC modes was developed, termed native reverse-phase liquid chromatography, nRPLC. nRPLC employs the solvent elution model and MS compatibility of RPLC while preserving the native form of protein and ADC as in HIC. This new nRPLC technique is an alternative to HIC for ADCs when inline coupling of MS is desired by virtue of using only 50 mM NH4OAC. The nRPLC method eluted intact ADCs for both a model ADC from AbbVie and a commercial ADC from Seattle Genetics. Key to this chromatographic advance was lower hydrophobicity of the bonded phase to make drug-surface hydrophobic interactions weaker than the intramolecular hydrophobic interactions that maintain the noncovalent complexes. Inherent to this strategy of designing retention only for interactions between the attached drug and chromatographic surface is that the D0 species has little retention at this stage in column development. The column gives partial resolution of positional isomers, thereby providing additional characterization beyond what is typically obtained using HIC. The lesser number of peaks in HIC permits full resolution of the ADC based on drug loading, which enables precise calculation of DAR. With its greater resolution of positional isomers that currently overlap, nRPLC will be a companion rather than a replacement for HIC until resolution is improved. Longer nRPLC columns, refinement of polymer growth conditions, and optimization of separation conditions could lead to sufficient resolution to determine DAR while also characterizing positional isomers. To our knowledge, this is the first time that intact ADCs made from reduced cysteines have been separated based on DAR using an MS-compatible mobile phase.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health, R01GM121910.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.8b04699.
Conditions for conventional chromatography in Figure 2; Figure S1a, RPLC chromatogram for AbbVie model ADC after reduction with DTT; Figure S1b, raw mass spectra and deconvoluted mass spectra for each peak in the chromatograms of part a; Figure S2, nRPLC of model ADC using a commercial HIC column (PDF)
The authors declare the following competing financial interest(s): The corresponding author (M.J.W.) is part owner of a company, bioVidria, that has licensed the technology described in this paper.
REFERENCES
- (1).Hughes B Nat. Rev. Drug Discovery 2010, 9 (9), 665–7. [DOI] [PubMed] [Google Scholar]
- (2).Thomas A; Teicher BA; Hassan R Lancet Oncol. 2016, 17 (6), e254–e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Beck A; Goetsch L; Dumontet C; Corvaia N Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. [DOI] [PubMed] [Google Scholar]
- (4).Polakis P Pharmacol. Rev. 2016, 68 (1), 3–19. [DOI] [PubMed] [Google Scholar]
- (5).Lambert JM; Berkenblit A Annu. Rev. Med. 2018, 69, 191–207. [DOI] [PubMed] [Google Scholar]
- (6).Tsuchikama K; An Z Protein Cell 2018, 9 (1), 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Flygare JA; Pillow TH; Aristoff P Chem. Biol. Drug Des. 2013, 81 (1), 113–21. [DOI] [PubMed] [Google Scholar]
- (8).Peters C; Brown S Biosci. Rep. 2015, 35 (4), No. e00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jain N; Smith SW; Ghone S; Tomczuk B Pharm. Res. 2015, 32 (11), 3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hamblett KJ; Senter PD; Chace DF; Sun MMC; Lenox J; Cerveny CG; Kissler KM; Bernhardt SX; Kopcha AK; Zabinski RF; Meyer DL; Francisco JA Clin. Cancer Res. 2004, 10 (20), 7063–7070. [DOI] [PubMed] [Google Scholar]
- (11).Behrens CR; Ha EH; Chinn LL; Bowers S; Probst G; Fitch-Bruhns M; Monteon J; Valdiosera A; Bermudez A; Liao-Chan S; Wong T; Melnick J; Theunissen JW; Flory MR; Houser D; Venstrom K; Levashova Z; Sauer P; Migone TS; van der Horst EH; Halcomb RL; Jackson DY Mol. Pharmaceutics 2015, 12 (11), 3986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sanderson RJ; Hering MA; James SF; Sun MMC; Doronina SO; Siadak AW; Senter PD; Wahl AF Clin. Cancer Res. 2005, 11, 843–852. [PubMed] [Google Scholar]
- (13).Sun MMC; Beam KS; Cerveny CG; Hamblett KJ; Blackmore RS; Torgov MY; Handley FGM; Ihle NC; Senter PD; Alley SC Bioconjugate Chem. 2005, 16, 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ducry L Antibody-Drug Conjugates; Humana Press: New York City, NY, 2013; Vol. 1045. [Google Scholar]
- (15).Shen BQ; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu SF; Mai E; Li D; Tibbitts J; Baudys J; Saad OM; Scales SJ; McDonald PJ; Hass PE; Eigenbrot C; Nguyen T; Solis WA; Fuji RN; Flagella KM; Patel D; Spencer SD; Khawli LA; Ebens A; Wong WL; Vandlen R; Kaur S; Sliwkowski MX; Scheller RH; Polakis P; Junutula JR Nat. Biotechnol. 2012, 30 (2), 184–9. [DOI] [PubMed] [Google Scholar]
- (16).Adem YT; Schwarz KA; Duenas E; Patapoff TW; Galush WJ; Esue O Bioconjugate Chem. 2014, 25 (4), 656–64. [DOI] [PubMed] [Google Scholar]
- (17).Boswell CA; Mundo EE; Zhang C; Bumbaca D; Valle NR; Kozak KR; Fourie A; Chuh J; Koppada N; Saad O; Gill H; Shen BQ; Rubinfeld B; Tibbitts J; Kaur S; Theil FP; Fielder PJ; Khawli LA; Lin K Bioconjugate Chem. 2011, 22 (10), 1994–2004. [DOI] [PubMed] [Google Scholar]
- (18).Lyon RP; Bovee TD; Doronina SO; Burke PJ; Hunter JH; Neff-LaFord HD; Jonas M; Anderson ME; Setter JR; Senter PD Nat. Biotechnol. 2015, 33 (7), 733–5. [DOI] [PubMed] [Google Scholar]
- (19).Valliere-Douglass JF; McFee WA; Salas-Solano O Anal. Chem. 2012, 84 (6), 2843–2849. [DOI] [PubMed] [Google Scholar]
- (20).Firth D; Bell L; Squires M; Estdale S; McKee C Anal. Biochem. 2015, 485, 34–42. [DOI] [PubMed] [Google Scholar]
- (21).Chen J; Yin S; Wu YJ; Ouyang J Anal. Chem. 2013, 85(3), 1699–1704. [DOI] [PubMed] [Google Scholar]
- (22).Wiggins B; Liu-Shin L; Yamaguchi H; Ratnaswamy GJ Pharm. Sci. 2015, 104 (4), 1362–72. [DOI] [PubMed] [Google Scholar]
- (23).Fekete S; Veuthey JL; Beck A; Guillarme DJ Pharm. Biomed. Anal. 2016, 130, 3–18. [DOI] [PubMed] [Google Scholar]
- (24).Alpert AJ J. Chromatogr. A 1986, 359, 85–97. [Google Scholar]
- (25).Xiu L; Valeja SG; Alpert AJ; Jin S; Ge Y Anal. Chem. 2014, 86 (15), 7899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen B; Peng Y; Valeja SG; Xiu L; Alpert AJ; Ge Y Anal. Chem. 2016, 88 (3), 1885–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wiggins B; Liu-Shin L; Yamaguchi H; Ratnaswamy GJ Pharm. Sci. 2015, 104 (4), 1362–1372. [DOI] [PubMed] [Google Scholar]
- (28).McCue JT Methods Enzymol. 2009, 463, 405–414. [DOI] [PubMed] [Google Scholar]
- (29).Queiroz JA; Tomaz CT; Cabral JM S. J. Biotechnol. 2001, 87, 143–159. [DOI] [PubMed] [Google Scholar]
- (30).Rippel G; Szepesy LJ Chromatogr. A 1994, 664, 27–32. [Google Scholar]
- (31).Machold C; et al. J. Chromatogr. A 2002, 972, 3–19. [DOI] [PubMed] [Google Scholar]
- (32).Xia F; Nagrath D; Garde S; Cramer SM Biotechnol. Bioeng. 2004, 87 (3), 354–63. [DOI] [PubMed] [Google Scholar]
- (33).To BC; Lenhoff AM J. Chromatogr. A 2007, 1141 (2), 235–43. [DOI] [PubMed] [Google Scholar]
- (34).Baca M; De Vos J; Bruylants G; Bartik K; Liu X; Cook K; Eeltink SJ Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 182–188. [DOI] [PubMed] [Google Scholar]
- (35).Chen BF; Lin ZQ; Alpert AJ; Fu CX; Zhang QY; Pritts WA; Ge Y Anal. Chem. 2018, 90 (12), 7135–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bobaly B; Beck A; Veuthey JL; Guillarme D; Fekete SJ Pharm. Biomed. Anal. 2016, 131, 124–132. [DOI] [PubMed] [Google Scholar]
- (37).Huang X; Wirth MJ Anal. Chem. 1997, 69 (22), 4577–4580. [Google Scholar]
- (38).Valeja SG; Xiu L; Gregorich ZR; Guner H; Jin S; Ge Y Anal. Chem. 2015, 87 (10), 5363–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Janin-Bussat MC; Dillenbourg M; Corvaia N; Beck A; Klinguer-Hamour CJ Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2015, 981, 9–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.