

Abstract
G-protein-coupled metabotropic glutamate group I receptors (mGluR1s) mediate synaptic transmission and plasticity in Purkinje cells and, therefore, critically determine cerebellar motor control and learning. Purkinje cells express two members of the G-protein Gq family, namely Gq and G11. Although in vitro coexpression of mGluR1 with either Gα11 or Gαq produces equally well functioning signaling cascades, Gαq- and Gα11-deficient mice exhibit distinct alterations in motor coordination. By using whole-cell recordings and Ca2+ imaging in Purkinje cells, we show that Gαq is required for mGluR-dependent synaptic transmission and for long-term depression (LTD). Gα11 has no detectable contribution for synaptic transmission but also contributes to LTD. Quantitative single-cell RT-PCR analyses in Purkinje cells demonstrate a more than 10-fold stronger expression of Gαq versus Gα11. Our findings suggest an expression level-dependent action of Gαq and Gα11 for Purkinje cell signaling and assign specific roles of these two Gq isoforms for motor coordination.
Keywords: Purkinje cell, mGluR, G-protein, synaptic plasticity, motor control, patch clamp, calcium [Ca] imaging, knock-out, RT-PCR
Introduction
The metabotropic glutamate group I receptor mGluR1 is a key molecule for synaptic signaling and plasticity and thus for motor learning and coordination in the cerebellum (Rose and Konnerth, 2001). Synaptic signaling through mGluR1 involves the activation of guanine nucleotide-binding proteins (G-proteins). On binding of glutamate to mGluR1, the α subunit of the heterotrimeric G-protein activates phospholipase C (PLC), which hydrolyzes phosphatidyl-inositol 4,5-bisphosphate yielding DAG and inositol 1,4,5-trisphosphate (InsP3). InsP3 then mobilizes Ca2+ ions from intracellular stores. Such metabotropic glutamate receptor (mGluR)-mediated Ca2+ release has been shown to be evoked by repetitive stimulation of the synapse between parallel fibers (PFs) and Purkinje cells in rat cerebellum. The resulting transient elevations in the dendritic Ca2+ concentration do not require changes in membrane potential and were, therefore, identified as a new class of postsynaptic response (Finch and Augustine, 1998; Takechi et al., 1998). The importance of the synaptic mGluR-mediated Ca2+ release signals was emphasized by the finding that InsP3-mediated Ca2+release in dendritic spines is essential for the induction of long-term depression (LTD) at PF–Purkinje cell synapses (Miyata et al., 2000). LTD is an activity-dependent form of synaptic plasticity that is presumed to be one of the critical cellular determinants for motor learning in the cerebellum (Ito, 2001). The key role of mGluR1 for cerebellar motor control has found strong support by the demonstration that the functional expression and activation of mGluR1 is required for the induction of LTD (Aiba et al., 1994; Conquet et al., 1994; Ichise et al., 2000). In line with these observations, it was found also that the signals downstream of mGluR1, including G-proteins, PLC, and InsP3, are required for normal LTD and motor coordination. Thus, for example, InsP3 receptor-deficient mice exhibit cerebellar coordination deficits associated with a pronounced ataxia and, on the cellular level, they completely lack LTD (Inoue et al., 1998). It is also necessary to mention here that, besides activating Ca2+ release from intracellular stores, activation of mGluRs under certain conditions evokes a slow EPSP (sEPSP) in Purkinje cells (Batchelor et al., 1994). Recent evidence indicates that this sEPSP is mediated by the TRPC1 cation channel (Kim et al., 2003). Although it was demonstrated that activation of G-proteins is required for the activation of the mGluR-dependent sEPSP (Hirono et al., 1998; Sugiyama et al., 1999; Canepari and Ogden, 2003), to date it is unclear which G-protein subtype is involved.
Group I mGluRs are coupled with the Gq subclass of heterotrimeric G-proteins (Masu et al., 1991; Abe et al., 1992). Of all members of the Gq family, G11 and Gq are the major isoforms in the adult brain (Wilkie et al., 1991; Mailleux et al., 1992). Both Gq and G11 mediate a pertussis toxin-insensitive activation of PLCβ (Rhee, 2001). The α subunits of both isoforms, Gαq and Gα11, have similar amino acid sequences (88%), and Gα11/Gαq immunoreactivity colocalizes with mGluR1 in dendritic spines of Purkinje cells. Therefore, it had been suggested that G11 and Gq are 8the major signal transducers downstream of mGluR1 (Tanaka et al., 2000). This hypothesis was partially supported by the observation that deficiency in Gαq causes coordination deficits and, on the cellular level, a persistent innervation of Purkinje cells by multiple climbing fibers (CFs), instead of the mono-innervation found in normal conditions (Offermanns et al., 1997). However, whereas hippocampal synaptic plasticity is equally impaired in Gαq- and Gα11-deficient mice (Miura et al., 2002), the lack of evidence for behavioral and cellular impairments in cerebellar function in Gα11-deficient mice (Offermanns et al., 1997) remained a puzzle.
Materials and Methods
Tissue preparation
Parasagittal cerebellar slices (300 μm) were prepared from wild-type (C57/BL6) mice at postnatal day (P) 26–P124, Gα11-deficient mice (Stanislaus et al., 1998) (P29–P140), and Gαq-deficient mice (Offermanns et al., 1997) (P36–P63). The animals were decapitated after anesthesia with CO2, and the cerebella were removed rapidly and placed in artificial CSF (ACSF; 0–2°C) containing (in mm) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose, bubbled with 95% O2 and 5% CO2. After cutting, slices were kept for 60 min at 34°C and then for up to 8 hr at 25°C in ACSF.
Patch-clamp recordings
Somatic whole-cell recordings were obtained by using an EPC9 amplifier (HEKA, Lambrecht, Germany). Pipettes (2.5–3.5 MΩ) were pulled from borosilicate glass. The intracellular solution contained (in mm) 148 K-gluconate, 10 HEPES, 10 NaCl, 0.5 MgCl2, 4 Mg-ATP, 0.4 Na3-GTP, and 0.1 Oregon Green BAPTA-1 (Molecular Probes, Eugene, OR), unless stated otherwise, at pH 7.3. During recordings, slices were continuously perfused with ACSF containing 10 μm bicuculline (Sigma, Deisenhofen, Germany). The holding potential was –70 mV, if not stated otherwise. Afferent stimulation was performed by using a pipette filled with 1 m NaCl (1 MΩ resistance) placed in the molecular layer. The stimulus pulse amplitude (100 or 150 μsec duration) was 2–20 V for PF stimulation and 20–55 V for CF stimulation. PF and CF EPSCs were identified by their characteristic features (Konnerth et al., 1990).
For LTD recordings, PFs were stimulated at 0.2 Hz, and EPSCs were recorded in the voltage-clamp mode until a stable baseline amplitude was obtained for 10 min. For synaptic LTD induction, a protocol described previously was used (Barski et al., 2003; Feil et al., 2003). Briefly, while the position of the stimulation pipette remained unchanged, the stimulus intensity was raised to a value of 20% above the CF threshold identified beforehand, and 240 stimuli were delivered at 1 Hz. After pairing, the stimulus intensity was set to the initial value, and the recording of PF EPSCs at 0.2 Hz was resumed for at least 40 min.
Bath-applied 1-amino-cyclopentane-trans-1,3-dicarboxylic acid (ACPD, 50 μm; Tocris Cookson, Bristol, UK) was delivered in the presence of the specific antagonist of mGluR2 receptors, (2S,3S,4S)-2-methyl-(carboxycyclopropyl)glycine (MCCG, 200 μm; Tocris Cookson). In these experiments, the passive membrane properties of Purkinje cells were monitored by applying 5 mV hyperpolarizing pulses. The series resistance was kept constant at 10–20 MΩ.
To measure local responses to ACPD, it was applied by a picospritzer (Parker, Cleveland, OH) coupled with patch pipettes filled with ACSF, to which 200 μm ACPD was added. The pipettes were placed ∼15 μm above the examined dendritic region. Caffeine (40 mm in ACSF; Sigma) was pressure ejected from a fine-tip pipette positioned near (∼10 μm) the soma for the duration of 500–700 msec. The cell was voltage clamped at –50 mV to slightly elevate the resting Ca2+ concentration (Kano et al., 1995).
Flash photolysis of P4(5)-1-(2-nitrophenyl)ethyl ester of myo-d-inositol-1,4,5-trisphosphate (caged InsP3; Calbiochem, La Jolla, CA) was performed by a ∼3 msec UV light pulse from a discharge Xenon lamp, coupled with the epifluorescence port of the microscope via a quartz light guide (T.I.L.L. Photonics, Gräfelfing, Germany). A 470 nm long pass 45° dichroic mirror allowed the UV pulse to follow the same light path as the excitation light. InsP3-induced Ca2+ transients were measured by adding 200 μm Fluo-4 (Molecular Probes) and 150 μm caged InsP3 to the pipette solution (Khodakhah and Ogden, 1995). All patch-clamp experiments were performed at room temperature (22–24°C).
Confocal fluorescence imaging
A confocal laser-scanning microscope (Odyssey; Noran, Middleton, WI), attached to an upright microscope (Axioskop 2; 40× water immersion objective; numerical aperture, 0.75; Zeiss, Oberkochen, Germany) was used to acquire fluorescence images at 60 Hz in parallel to the patch-clamp recordings (Eilers et al., 1995). Full-frame standard video images were recorded at 30 Hz on an optical disk (TQ-FH224; Panasonic, Osaka, Japan) using the Image-1 software (Universal Imaging, Downington, PA) and analyzed offline with custom-made software (FastAnalysis, LABVIEW; National Instruments, Austin, TX). Using FastAnalysis, all images of a sequence were first low-pass filtered to reduce noise in the recording. After that, an average image of 15 or 30 consecutive frames recorded just before stimulation was subtracted from an average image of 45 consecutive frames that followed synaptic stimulation. Pixels in the resulting image were regarded as “active” if their increase in brightness exceeded a threshold of twice the noise level in detected regions outside the cell. The Ca2+-dependent fluorescence signals from dendritic regions consisting of these active pixels were expressed as increases in fluorescence divided by the prestimulus fluorescence values (ΔF/F) and further analyzed using Igor Pro software (Wavemetrics, Lake Oswego, OR). Images displayed in Figures 2 and 3 are averages of 100 consecutive video frames.
Figure 2.
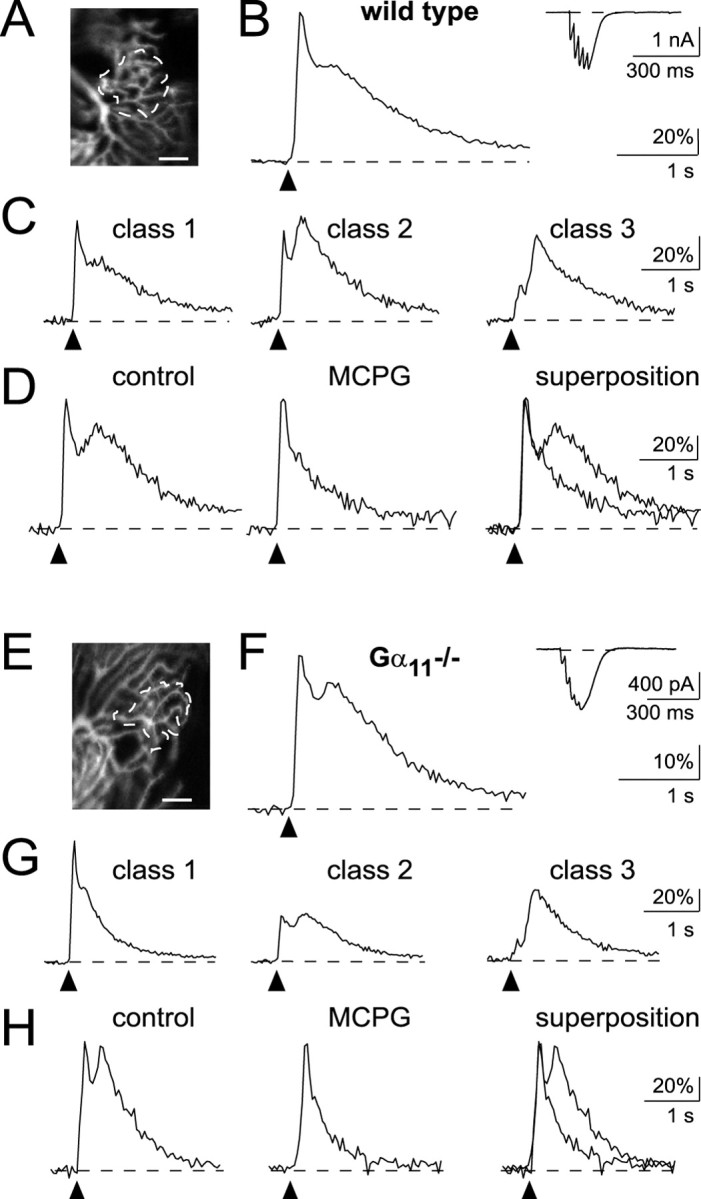
Postsynaptic Ca2+ signals mediated by repetitive PF stimulation in Purkinje cells of wild-type mice and Gα11-deficient mice. A–D, Wild-type mice. A, Confocal image of the active dendritic region of a Purkinje cell (scale bar, 10μm). PFs were stimulated with a train of five pulses (100 μsec, 22 V, at 50 Hz). B, Synaptic Ca2+ signal recorded from the active microdomain in A. The inset shows the corresponding, somatically recorded EPSC. C, Different inputs exhibit different patterns of biphasic Ca2+ responses corresponding to class 1, 2, or 3 (see Results for details). Recordings were obtained in three different Purkinje cells. D, The delayed synaptic Ca2+ signal component is blocked by 2 mm MCPG. E–H, Gα11-deficient mice. E, Confocal image of the active dendritic region of a Purkinje cell (scale bar, 10 μm). PFs were stimulated with a train of five pulses (100 μsec, 6 V, at 50 Hz). F, The traces illustrate the biphasic postsynaptic Ca2+ response recorded from the active microdomain in A and the corresponding EPSC (inset). G, Synaptic Ca2+ responses from different PF synaptic inputs. Recordings were obtained in three different Purkinje cells. H, MCPG sensitivity of the delayed component of the synaptically evoked Ca2+ responses (2 mm MCPG).
Figure 3.
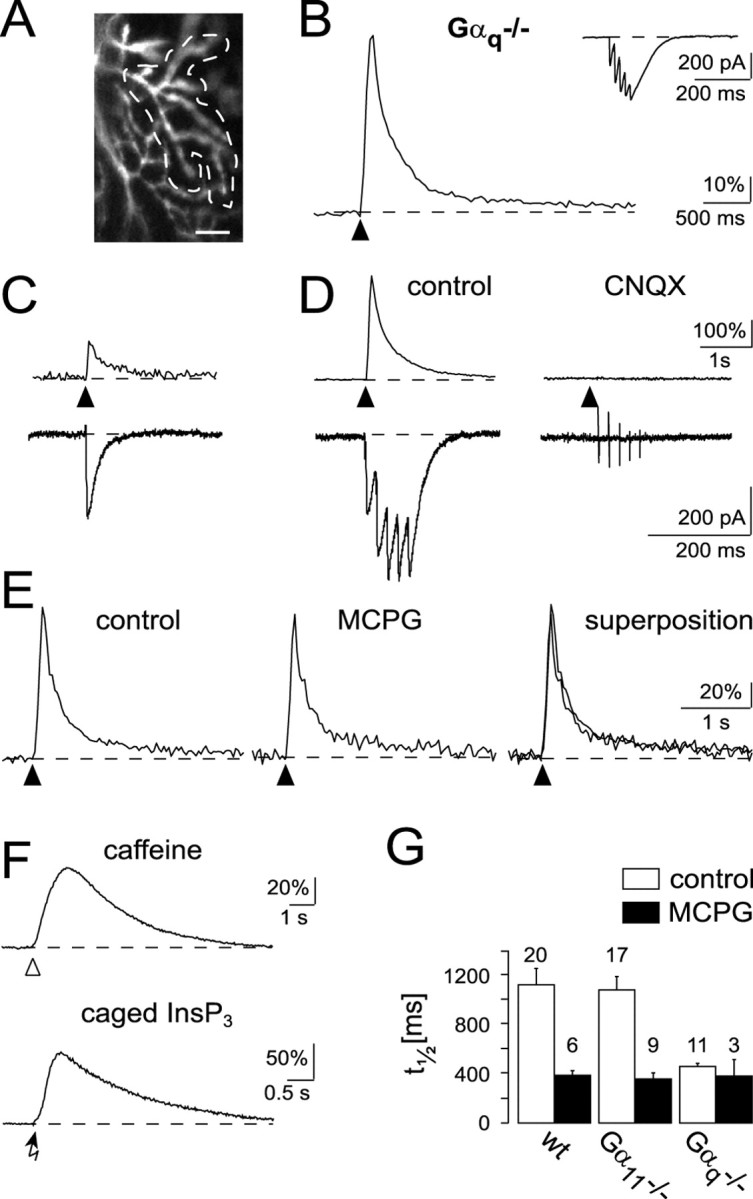
Ca2+ signaling in Purkinje cells of Gαq-deficient mice. A, Confocal image of the active dendritic region of a Purkinje cell responsive to repetitive stimulation of a beam of PFs (5 pulses; 13 V, at 50 Hz; scale bar, 10 μm). B, Ca2+ response recorded from the active microdomain (region within the dashed line) in A. The inset shows the corresponding EPSC. C, Ca2+ transient and EPSC in response to a single synaptic stimulation pulse (10 V). D, Ca2+ transient and EPSC in response to repetitive stimulation (5 pulses of 10 V at 50 Hz) in the same input as in C. Note the larger amplitude but the monotonic decay of the Ca2+ transient (left). The synaptic response was blocked by 10 μm CNQX (right). E, Absence of an MCPG-sensitive Ca2+ response component (2 mm MCPG). F, Ca2+ transients evoked by caffeine application (top, open arrowhead) or by a 3 msec UV pulse (bottom, arrow) applied to a Purkinje cell loaded with caged InsP3 (150 μm). Recordings were obtained from two different Purkinje cells. Each trace is an average of four consecutive responses separated by 3–4 min. The holding potential was –50 mV (caffeine application) and –70 mV (InsP3 uncaging). G, Quantitative comparison of synaptically evoked Ca2+ responses in mutant and in wild-type mice. The time point at which the amplitude of the Ca2+ response was half of the amplitude of the ESCT was defined as t1/2. The bar graph shows the t1/2 values obtained in the three genotypes under control conditions (□) and in the presence of MCPG (▪). The numbers of experiments included in the analyses are indicated on top of each bar.
Behavioral tests
Rotorod test. Tests were performed according to previously published procedures (Offermanns et al., 1997).
Ladder walking test. This test involved a “ladder” that was composed of 38 staves with a diameter of 2 mm placed at intervals of 2 cm. For the creation of irregular patterns, the positions of seven or nine randomly chosen staves were altered. Runs were recorded at 30 Hz, and slips of the forelimbs and hindlimbs were counted in a frame-by-frame analysis.
Elevated beam balancing test. The movement of mice on a round plastic beam (length, 50 cm; diameter, 1 cm) was recorded and analyzed as described above.
Turning ladder test. Mice were placed on a horizontal ladder (38 staves; length, 16 cm; diameter, 2 mm) facing one of the ends. The opposite end was lifted until the ladder reached a vertical position. We measured the time the mice required to turn their body into an upright position.
Quantitative rapid-cycle real-time RT-PCR
Individual Purkinje cells and material for control reactions were harvested under visual control from cerebellar slices as reported previously (Garaschuk et al., 1996; Plant et al., 1998). After solubilization in 0.5% NP-40 (Roche, Basel, Switzerland), reverse transcription (RT) was performed as described (Lambolez et al., 1992). The resulting cDNA was purified by the use of silica matrix. Rapid-cycle PCR reactions were optimized and performed in 20 μl reactions in glass capillaries according to the manufacturer's instructions using the LightCycler DNA Master SYBR Green I kit (Roche). The following primers were selected with the Oligo 6.0 Primer analysis software (MedProbe, Oslo, Norway): (1) Gαq [European Molecular Biology Laboratory (EMBL)/GenBank accession number m55412]: 5′-86-for 5′-AAGCCCGGAGGATCAACGAC-3′, 3′-256-rev 5′-CGCGCTTGTCTTCGTCAGAGT-3′ (single-cell primer set; amplicon size, 171 bp); 5′-724-for 5′-GTAGCGCTTAGCGAATATGAT-3′, 3′-935-rev 5′-CTCTGGGGTCCATCATATTCT-3′ (total brain primer set; amplicon size, 212 bp); and (2) Gα11 (EMBL/GenBank accession number m55411): 5′-119-for 5′-CAACGCGGAGATCGAGAAA-3′, 3′-403-rev 5′-ACCTCCCGGATCAGGAGTG-3′ (total brain and single-cell primer set; amplicon size, 285 bp). The following rapid-cycle PCR conditions were found to be optimal: (1) Gα11 PCR: 1 min denaturation, then 50 cycles with Tdenaturation = 95°C, 0 sec (no hold at 95°C); Tanneal = 59°C, 5 sec; Telongation = 72°C, 12 sec; Tmeasurement = 89°C, 5 sec; primers: each 40 pmol/reaction, 4mm MgCl2; and (2) Gαq PCR: 1 min denaturation, then 50 cycles with Tdenaturation = 95°C, 0 sec; Tanneal = 59°C, 5 sec; Telongation = 72°C, 7 sec; Tmeasurement = 87°C, 5 sec; primers: each 20 pmol/reaction, 5 mm MgCl2. In the case of total brain RNA analysis for Gαq cDNA, the primers were used with the following reaction conditions: 1 min denaturation, then 50 cycles with Tdenaturation = 95°C, 0 sec; Tanneal = 54°C, 5 sec; Telongation = 72°C, 9 sec; Tmeasurement = 81°C, 5 sec; primers: 40 pmol/reaction, 4 mm MgCl2. To be used as standards for absolute cDNA quantification, corresponding amplicons were purified (PCR Purification kit; Qiagen, Hilden, Germany) and quantified using the GeneQuant photometer (Amersham Biosciences, Freiburg, Germany). Then, standards were diluted in 10 mm Tris-HCl, pH 8.5, and 0.1% BSA (w/v). To compare the levels of Gα11 or Gαq expression in individual single cells, the cDNA material of a single cell was divided into five equal parts, of which one part was used for Gαq expression analysis and two parts were used for Gα11 analysis. The remaining material was saved for future tests. Rapid-cycle RT-PCR was performed on the LightCycler (Roche) and analyzed with the LightCycler analysis software (version 3.5.3). To estimate the total amount of Gα11 or Gαq cDNA molecules from single cells or total brain RNA samples, SYBR Green I fluorescence, bound to double-stranded PCR products, was analyzed in the log-linear phase using the so called “fit point” method with standards in the same PCR run that were chosen to represent similar copy numbers (Rasmussen, 2001). Total brain RNA was purified as described (Chomczynski and Sacchi, 1987). PCR products from single cells were identified by melting curve analysis, restriction analysis, and, in some cases, by direct DNA sequencing of PCR products. Sequence analysis was performed at the Genius Network Service (German Cancer Research Center, Heidelberg, Germany).
Transient transfection of COS-7 cells and determination of inositol phosphate levels
COS-7 cells were seeded in 24-well plates at a density of 4 × 104 cells per well and grown overnight. Cells were then washed with PBS, and for cotransfection experiments, 0.2 μg of each DNA mixed with 2 μl of lipofectamine (Invitrogen, Karlsruhe, Germany) in 0.25 ml of Opti-MEM were added to each well. In control experiments, the total amount of DNA was maintained constant by adding DNA from a vector encoding β-galactosidase. After 5 hr at 37°C, 0.25 ml of DMEM containing 20% (v/v) FBS was added to each well. Approximately 24 hr after transfection, cells were labeled for 20–24 hr with 120 pmol of myo-[2-3H]inositol (758.5 Gbq/mmol; NEN, Wilmington, DE), and labeled cells were washed with PBS and were then incubated for 30 min at 37°C with 0.25 ml of inositol–glutamate-free DMEM containing 1 U/ml glutamate pyruvate transaminase and 2 mm pyruvate. Thereafter, the medium was aspirated, cells were washed, and the indicated agents were added in DMEM containing 10 mm LiCl. Inositol phosphate formation was stopped after 20 min by removing the medium and the addition of 0.2 ml of 10 mm ice-cold formic acid. Total inositol phosphates were then extracted, separated, and measured as described (Offermanns and Simon, 1995). Measurements were done in triplicate representing three independently transfected wells.
Immunohistochemistry
Adult mice were perfused with 4% paraformaldehyde, and brains were sectioned at 25 or 50 μm using a vibratome (Leica, Solms, Germany). To block endogenous peroxidase activity, sections were incubated in 1% H2O2 in PBS:methanol (1:1) for 15 min. Background staining was blocked by incubating sections with goat serum for 30 min, and sections were further incubated with the rabbit anti-Gαq/11 antibody (1:1000 or 1:1500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight, followed by biotinylated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA). Incubation with streptavidin and subsequent development of peroxidase staining was done using the Vectastain ABC kit (Vector Laboratories) according to the manufacturer's directions. Images were captured using a CCD camera (Leica) from all mice using identical conditions of light and contrast. Staining intensities were quantified over specific, stained regions with NIH Image software (National Institutes of Health, Bethesda, MD) using nonstained brain regions (white matter) as background controls.
Data analysis
For the analysis of synaptically evoked LTD and ACPD-mediated synaptic depression, all EPSCs collected during the last 10 min of the experiment (30–40 min after stimulation and 20–30 min after ACPD application, respectively) were considered (from every experiment, 10 samples each representing all 12 consecutive test EPSCs per minute, normalized to the control mean value).
Student's t test was used to determine significance between groups of data (if not stated otherwise); p < 0.05 was considered significant. Error bars in bar graphs represent SEM, if not stated otherwise.
Results
To test whether the mGluR is able to interact with both, Gq and G11, we coexpressed mGluR1α receptors and Gαq or Gα11 in COS-7 cells. Figure 1 shows that in this in vitro assay both α subunits were equally effective in augmenting the mGluR1-mediated production of inositol phosphates. In the presence of either Gαq or Gα11, the glutamate-induced inositol phosphate production was about twice as high as in the absence of a cotransfected G-protein α-subunit. This difference is statistically highly significant (p < 0.01). There was no major difference in the potency of glutamate to induce responses through either the mGluR1–Gq or the mGluR1–G11 pathway. This indicates that Gq and G11 indistinguishably couple mGluR1 with the β-isoforms of PLC. In contrast to Gα11-deficient mice, however, mice lacking Gαq have obvious functional cerebellar defects, whereas the general size and anatomy as well as layer structures of the cerebellum appeared to be normal (Offermanns et al., 1997). On the cellular level, we could essentially confirm this observation. Purkinje cells in cerebellar slices obtained from mutant and wild type mice were filled through a patch pipette with the calcium indicator Oregon Green BAPTA-1. The general appearance of the confocal images of the dendritic trees showed no gross abnormalities in both mutants with regard to the size and the apparent complexity of the dendritic ramifications. The number of primary Purkinje cell dendrites was not significantly different in the mutants (wild type, n = 1.23 ± 0.08 in 30 cells; Gα11–/–, n = 1.17 ± 0.05 in 12 cells; Gαq–/– cells, n = 1.16 ± 0.09 in 19 cells). Thus, the single-cell morphology in different mutants did not provide any ostensible clue for the behavioral and cellular alterations observed in Gαq-deficient mice (Offermanns et al., 1997).
Figure 1.

Gαq and Gα11 equally couple mGluR1 activation with inositol phosphate (IP) production. Accumulation of inositol phosphates in COS-7 cells that coexpress mGluR1a and β-galactosidase (lacZ) or Gαq and Gα11 subunits is indicated. Ligand-dependent inositol phosphate formation was determined in the absence or presence of increasing concentrations of glutamate (Glu) for 20 min. Shown are mean values of triplicates ± SD. In the presence of either Gαq or Gα11, the glutamate-induced inositol phosphate production was about twice as high as in the absence of a cotransfected G-protein α subunit. This difference is statistically highly significant (p < 0.01).
PF-mediated synaptic transmission is not detectably altered in Gα11-deficient mice
We next stimulated afferent PFs and recorded the complex synaptic response in Purkinje cells by combining whole-cell voltage-clamp recordings with rapid confocal calcium imaging (Takechi et al., 1998). The electrical component of the synaptic response and the associated local changes of [Ca2+]i in dendrites were recorded simultaneously (Fig. 2A,B,E,F). To compare the characteristics of the Ca2+ response between the different genotypes, we devised an approach that ensured that the entire Ca2+ response was registered within the active dendritic region (see Materials and Methods). Briefly, after PF activation, the active dendritic region was identified by subtracting an averaged image of the resting fluorescence (constructed from a sequence of 15 or 30 images preceding the stimulation) from an average of images taken during the synaptic response. The two-dimensional arrangement of the dendritic tree of Purkinje cells and the strictly laminar organization of the afferent PFs ensured the detection of all responsive dendritic sites. The fluorescence traces represent the integral response of such synaptically “active” sites determined with this approach.
As reported previously for rat cerebellum (Finch and Augustine, 1998; Takechi et al., 1998), in slices of wild-type mouse cerebellum, a brief train of repetitive subthreshold PF stimulation (5 pulses, 50 Hz) evoked a complex Ca2+ signal that was confined to the dendritic target region of the activated PFs (Fig. 2A). The synaptic Ca2+ response consisted of two distinct components: an early response with a fast rising phase and a delayed second component. For consistency, we use here the original terminology (Takechi et al., 1998): early synaptic Ca2+ transient (ESCT) for the initial AMPA receptor (AMPAR)-mediated component and delayed synaptic Ca2+ transient (DSCT) for the mGluR-mediated second component.
Figure 2C shows the different types of PF-mediated complex Ca2+ responses found in control recordings in wild-type mice (n = 28 PF synaptic inputs to Purkinje cells). The responses were grouped into three classes, depending on the ratio of the peak amplitudes of ESCTs versus DSCTs (class 1, ratio ≫1; class 2, ratio ∼1; class 3, ratio ≪1). Both class 1 (left trace; n = 11) and class 2 (middle trace; n = 11) responses were recorded in 39% of the inputs, respectively. In the remaining experiments, class 3 responses were recorded (right trace; 22% of inputs with a complex Ca2+ response; n = 6). Occasionally, no discernable ESCT component was detected (n = 5; data not shown). The reason for the variability of PF-mediated Ca2+ responses is unclear. We assume that the variability reflects either the filling state of the internal stores with Ca2+ and/or a variable distribution of Ca2+-releasing organelles in the dendritic tree. Regardless of their relative amplitudes, DSCTs were always abolished by the presence of the antagonist of mGluR, (R,S)-α-methyl-4-carboxyphenylglycine (MCPG) (n = 6) (Finch and Augustine, 1998; Takechi et al., 1998) (Fig. 2D), whereas the ESCTs were blocked by the antagonist of ionotropic AMPARs, CNQX (data not shown). In mice lacking Gα11, the PF-mediated Ca2+ responses were qualitatively similar to those observed in wild-type mice (Fig. 2E–H). Thus, the size of the active dendritic regions had similar dimensions to those detected in the wild type, when similar stimulation parameters were used (Fig. 2E). As in wild-type mice, in Gα11–/– mice the PF-mediated Ca2+ responses were detected throughout the dendritic tree. The synaptically evoked Ca2+ transients in Gα11-deficient mutants displayed a variability of the ESCT versus DSCT peak amplitude ratios, which is quite similar to that found in wild-type mice (Fig. 2G), although the relative abundance of the different response classes was slightly different. Approximately 30% (6 of 20) of all PF synaptic inputs displaying a complex Ca2+ response belonged to class 1 (left trace), 25% (5 of 20) to class 2 (middle trace), and 45% (9 of 20; right trace) to class 3. Not included in this distribution are three unsuccessful attempts to evoke a clear biphasic postsynaptic Ca2+ response. As in wild-type mice, the DSCTs were abolished by MCPG (n = 9) (Fig. 2H). Taken together, these analyses did not provide support for a major involvement of Gα11 in the process of mGluR-mediated synaptic signaling in cerebellar Purkinje cells. It is, perhaps, also useful to note that AMPAR-mediated signaling seemed to be intact in Gα11-deficient mouse mutants.
Gαq-deficient mice lack mGluR-mediated calcium signaling
The same experimental setting and stimulation procedure yielded entirely different results in Gαq-deficient mice. Although postsynaptic Ca2+ responses with spatial dimensions comparable with those obtained in wild-type and Gα11-deficient mice could be evoked (Fig. 3A), the Ca2+ responses lacked the complexity observed under control conditions. As mentioned above, biphasic Ca2+ signals after repetitive synaptic stimulation were observed at 28 of 33 synaptic inputs (24 cells) and 20 of 23 inputs (17 cells) in wild-type and Gα11-deficient mice, respectively. In contrast, in Gαq-deficient animals, all stimulated inputs (19 of 19 in 15 cells) showed a monophasic Ca2+ transient characterized by a fast rising phase and a mono-exponential decaying phase with an average time constant of 436 ± 179 msec (SD; n = 19 in 15 cells) (Fig. 3B). Thus, the response had a time course that was not distinguishable from that observed with single-shock stimulation (Fig. 3C), a paradigm known to activate exclusively the AMPAR component (Eilers et al., 1995; Takechi et al., 1998). Indeed, in Gαq mice the synaptically evoked calcium response was, in all instances (n = 19), completely blocked by CNQX (Fig. 3D). Furthermore, neither the shape of the synaptic Ca2+ response nor its time course were affected by MCPG (n = 3) (Fig. 3E).
Two types of experiments were performed to test that neither the filling state of the intracellular Ca2+ stores nor the function of InsP3 receptors is impaired in Gαq-deficient mice. First, caffeine, an agonist of ryanodine receptors, was focally applied to Purkinje cells of Gαq–/– mice and was found to produce transient Ca2+ elevations in the soma and dendrites of all four cells tested (Fig. 3F, top trace). Second, photolysis of caged InsP3, which was dialyzed through the patch pipette into Purkinje cells of Gαq–/– mice, evoked Ca2+ transients in all four cells tested (Fig. 3F, bottom trace).
Figure 3G summarizes the results obtained in wild-type mice and the two types of mutant mice and provides a direct comparison of the average synaptically mediated calcium responses obtained in all experiments. As a parameter for the quantification of the different time courses of the Ca2+ transients, we chose t1/2, the time during which the dendritic [Ca2+]i declined to half of the amplitude of ESCT. Figure 3G shows that the t1/2 values were almost equal in the wild-type (1132 ± 139 msec; n = 20 cells; SEM) and Gα11-deficient (1089 ± 110 msec; n = 17 cells; SEM) mice, whereas those found in Gαq-deficient mice (358 ± 23 msec; n = 11 cells; SEM) were similar to those found in MCPG-treated cerebellar slices from wild-type mice (394 ± 46 msec; n = 6 cells; SEM). It is worth mentioning that there was no significant difference between stimulation strengths used in wild-type mice and Gα11–/– and Gαq–/– mice, respectively (p > 0.3). Furthermore, there was no systematic difference of the amplitudes of AMPAR EPSCs in wild-type mice and the two mutants (p > 0.3).
Slow EPSPs require Gαq
In addition to the induction of dendritic Ca2+ signals, synaptic activation of mGluRs at the PF–Purkinje cell synapse produces a mGluR-mediated sEPSP (Batchelor et al., 1994). The sEPSPs, in general, require stronger afferent stimulation than that necessary for the induction of DSCTs, as had been mentioned briefly previously (Takechi et al., 1998). Here, we performed whole-cell voltage-clamp recordings and used, as a standard procedure, 15 stimulation pulses delivered at 50 Hz to afferent PFs to evoke slow EPSCs (sEPSCs). The experiments were performed in the presence of 40 μm CNQX. Under these conditions, sEPSCs were successfully evoked in 5 of 7 inputs (six cells) in wild-type mice and in 6 of 11 inputs (five cells) in Gα11-deficient mice (Fig. 4A). Occasionally, repetitive stimulation evoked a small and rapid, CNQX-resistant inward current. This was possibly a residual AMPAR-mediated current or, perhaps, a postsynaptic glutamate transporter-mediated current (Canepari et al., 2001). We found that sEPSCs were generally accompanied by dendritic Ca2+ signals. These Ca2+ signals as well as the sEPSCs were completely blocked by MCPG (data not shown). In contrast, in Gαq-deficient mice, the sEPSCs could not be evoked at all (Fig. 4A) (n = 7 synaptic inputs in five cells). The summary of these experiments is shown in Figure 4D.
Figure 4.

Analysis of mGluR-mediated currents in wild-type and mutant mice. A, sEPSCs and Ca2+ transients evoked by repetitive stimulation (15 pulses at 50 Hz) are found in wild-type and Gα11-deficient mice but not in Gαq-deficient mice. The recordings were performed in the presence of 10 μm bicuculline and 40 μm CNQX. B, Inward currents (bottom traces) and Ca2+ transients (top traces) in wild-type mice and Gα11- and Gαq-deficient mice are indicated. Responses were evoked by pressure ejecting ACPD (200 μm) from an application pipette placed ∼15 μm above the dendrites of a voltage-clamped Purkinje cell. For the wild-type and Gα11-deficient cells, the Ca2+ transients were obtained from the activated dendritic regions, detected as described in Materials and Methods. In Gαq-deficient cells, the fluorescence was evaluated in the whole dendritic region recorded. The duration of ACPD application was 1 sec. C, Slow inward currents in response to bath application of 50 μm ACPD, which was present for 15 min (indicated by black bars), in the three genotypes are shown. Traces are averages of recordings from five cells in wild-type mice (left), eight cells in Gα11-deficient mice (middle), and eight cells in Gαq-deficient mice (right). Unlike local pressure application of ACPD, the sustained presence of ACPD in the bath induces an increase in the holding current of voltage-clamped Purkinje cells also in Gαq-deficient mice. Compared with Gαq-deficient mice, in wild-type and Gα11-deficient mice the amplitudes were larger (left and middle traces, respectively). In wild-type and Gα11-deficient mice, but not in Gαq-deficient mice, the responses desensitized in the presence of the agonist. D, Summary plot of the sEPSC values. The bar graphs include recordings from five inputs in wild-type mice, six inputs in Gα11-deficient mice, and seven inputs in Gαq-deficient mice. The amplitudes of the sEPSCs are given as ratios of the peak current versus the corresponding voltage value of the stimulation pulse. Values obtained from wild-type and Gα11-deficient mice are not significantly different from each other (p > 0.1). E, Summary plot of the inward current (IACPD) values. The bar graphs include recordings from nine inputs in wild-type mice, six inputs in Gα11-deficient mice, and 12 inputs in Gαq-deficient mice. The difference between wild-type and Gα11-deficient mice is not significant (p > 0.1). F, The corresponding average fluorescence changes of the data shown in B. In the graphs shown in D–F, differences between the respective data obtained from wild-type and Gα11-deficient animals are not significant (p > 0.05). G, Summary plot of the slow ACPD-evoked currents (slow IACPD). The bar graphs include recordings from five cells in wild-type mice and eight cells in Gα11-deficient and Gαq-deficient mice, respectively. The average current amplitude measured in Gαq-deficient mice differs significantly from the corresponding values obtained in wild-type and Gα11-deficient mice (p < 0.01).
An mGluR-mediated current and a corresponding Ca2+ signal can be evoked in Purkinje cells not only by synaptic PF stimulation but also by the direct application of the mGluR agonist ACPD (Vranesic et al., 1991). To study mGluR activation without interference of possible presynaptic changes in the mutants, we locally pressure ejected ACPD to small dendritic regions of Purkinje cells (Fig. 4). ACPD evoked an inward current associated with a marked elevation of the dendritic [Ca2+]i in both wild-type (seven of nine application sites in eight cells) and Gα11-deficient (six of six sites in four cells) mice. In no single instance were any ACPD puff-evoked currents and/or calcium responses detected in Gαq-deficient mice (12 of 12 inputs in six cells). Quantitative results from the ACPD application experiments are given in Figure 4, E and F.
Different results were obtained when ACPD was applied for more prolonged periods of time. Figure 4C shows current traces obtained from Purkinje cells to which 50 μm ACPD was applied for 15 min. In these experiments the group II mGluR inhibitor MCCG (200 μm) was added to the bath to avoid the activation of ACPD-responsive mGluR4 receptors on PFs. In addition, bicuculline in the bath prevented inhibitory activation of Purkinje cells by interneurons expressing mGluR1. Under these conditions, a slowly activating inward current was recorded in cells from all three genotypes, including Gαq-deficient mice. In Gα11-deficient mice, the average amplitude was 181.2 ± 14.7 pA (n = 8; SEM) and thus similar to that obtained in wild-type mice (178.8 ± 29.3 pA; n = 5; SEM). In Gαq-deficient mice, the amplitude, on average, reached only 56.7 ± 12.4 pA (n = 8; SEM).
Impairment of long-term synaptic depression in mutant mice
LTD was induced by conjunctive stimulation of PFs and CFs at 1 Hz for 4 min. In control experiments performed in wild-type mice, the mean amplitude of EPSCs measured 30–40 min after LTD induction was 65.1 ± 1.4% (n = 8 cells; SEM) of the control value (Fig. 5A,D). In Gαq-deficient mice, LTD induction was markedly impaired (Fig. 5C,D), yielding an EPSC amplitude of 85.5 ± 2.4% of the baseline value (n = 9 cells; SEM). This is significantly different from the value obtained in wild-type mice (p < 0.01). Surprisingly, also in Gα11–/– mice, LTD induction was impaired (Fig. 5B,D), although to a lesser extent than in the Gαq–/– mice. On average, the EPSC amplitude values were depressed to 75.9 ± 2.4% of the control (n = 10 cells; SEM). These data are significantly different from both wild-type and Gαq-deficient mice (p < 0.01).
Figure 5.
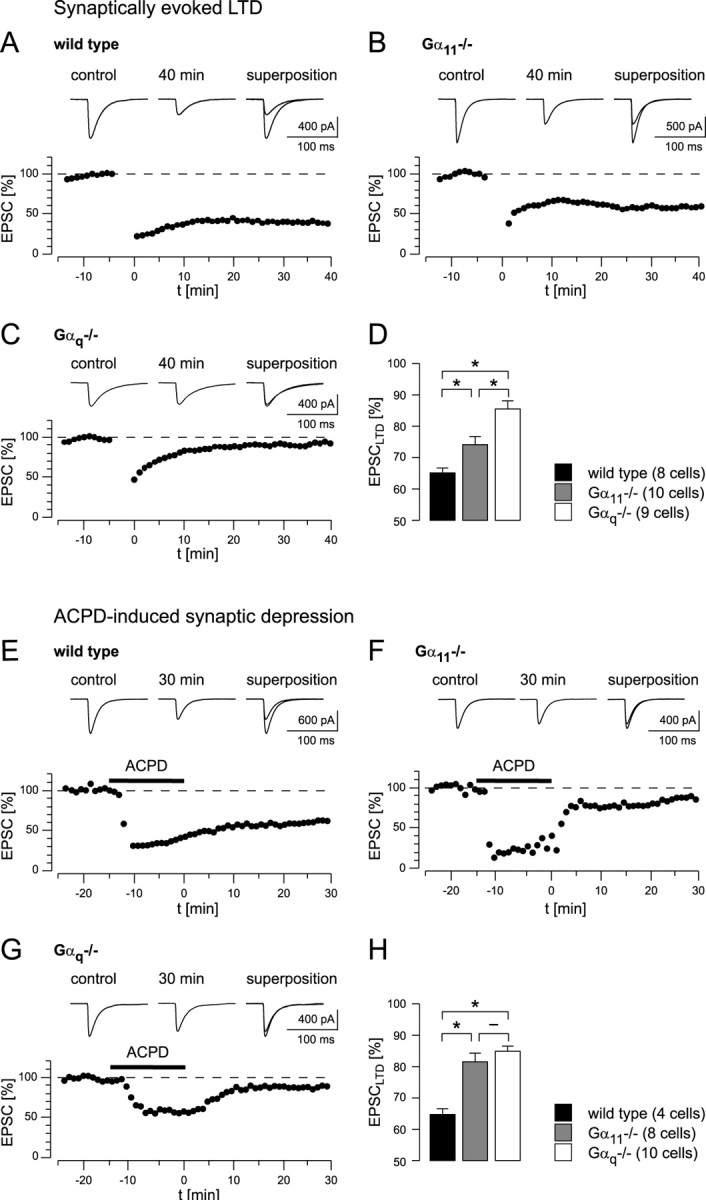
Synaptically evoked LTD and ACPD-induced synaptic depression in wild-type and mutant mice. A, LTD in a wild-type mouse. Top, EPSCs recorded 5 min before the start of LTD induction (control) and 40 min after LTD induction (40 min). Traces are averages of consecutive 10 EPSCs. B, LTD in a Gα11-deficient mouse. Top, Sample traces from the experiment shown below as described in A for wild-type mice. C, Attempt of LTD induction in a Gαq-deficient mouse. Top, Sample traces from a single experiment recorded as described in A for wild-type animals. D, Summary of all LTD experiments. Significant differences are marked with an asterisk (p < 0.01). The bars indicate average values of the remaining EPSC amplitudes (in percentage of the baseline amplitudes). The number of cells included into the analysis is indicated. E, Effect of bath application of ACPD (50 μm for 15 min; marked by the black bar) to PF-mediated synaptic transmission in a wild-type mouse. Top, EPSCs recorded 5 min before the start of ACPD perfusion (control) and 30 min after the beginning of the washout of the agonist (30 min). Traces are averages of 10 consecutive EPSCs recorded in the experiment shown below. In the presence of ACPD, EPSC amplitudes are markedly reduced. During washout of ACPD, they partially recover but remain significantly reduced compared with the baseline after complete washout of ACPD. F, ACPD perfusion experiment, as in E, performed in a Gα11-deficient mouse. G, ACPD perfusion experiment, as in E, performed in a Gαq-deficient mouse. H, Summary of ACPD perfusion experiments (same statistical analysis as that in D). Data points in A–C and E–G represent the average of normalized test EPSCs recorded during 1 min.
Dendritic InsP3 production alone is sufficient to induce a spatially restricted LTD in Purkinje cells (Finch and Augustine, 1998). In hippocampal neurons, group I mGluR- and GTP-dependent plasticity could be induced by bath application of group I mGluR-specific agonists (Fitzjohn et al., 2001). A similar protocol was used previously also in cerebellar Purkinje cells (Crepel et al., 1991), although the synaptic depression after perfusion with ACPD for 5 min was transient in the majority of cells measured. To test for the role of a prolonged InsP3 production, as it may occur during the induction of LTD, we applied ACPD (50 μm) for 15 min while continuing to stimulate the afferent PF input at the normal test frequency (0.2 Hz). Figure 5E shows that in wild-type mice ACPD caused a pronounced depression of EPSCs that persisted in an LTD-like manner for 30 min after the washout of ACPD. Remarkably, an ACPD-evoked EPSC reduction occurred both in Gα11-deficient mice (Fig. 5F) and in Gαq-deficient mice (Fig. 5G), although the persistent depression was much less than in wild-type mice (Fig. 5E). Figure 5H summarizes the results on ACPD-evoked synaptic depression. Taken together, the experiments shown in Figure 5 indicate that prolonged electrical stimulation or prolonged ACPD application are capable of at least partially activating the mGluR1-dependent pathway, even in Gαq-deficient mice.
Behavioral alterations in Gαq- and Gα11-deficient mice
In view of the results obtained in the LTD experiments, especially given that Gα11–/–-deficient mice showed clear impairments, the motor abilities of wild-type and mutant mice were investigated by using various tests (Fig. 6). In the turning rotorod test, Gα11-deficient mice performed similarly well as wild-type mice (Fig. 6A) during the first trials and improved further with increasing trial numbers. In contrast, Gαq-deficient mice immediately fell off the turning rotorod device, independently of the number of previous attempts (Offermanns et al., 1997).
Figure 6.
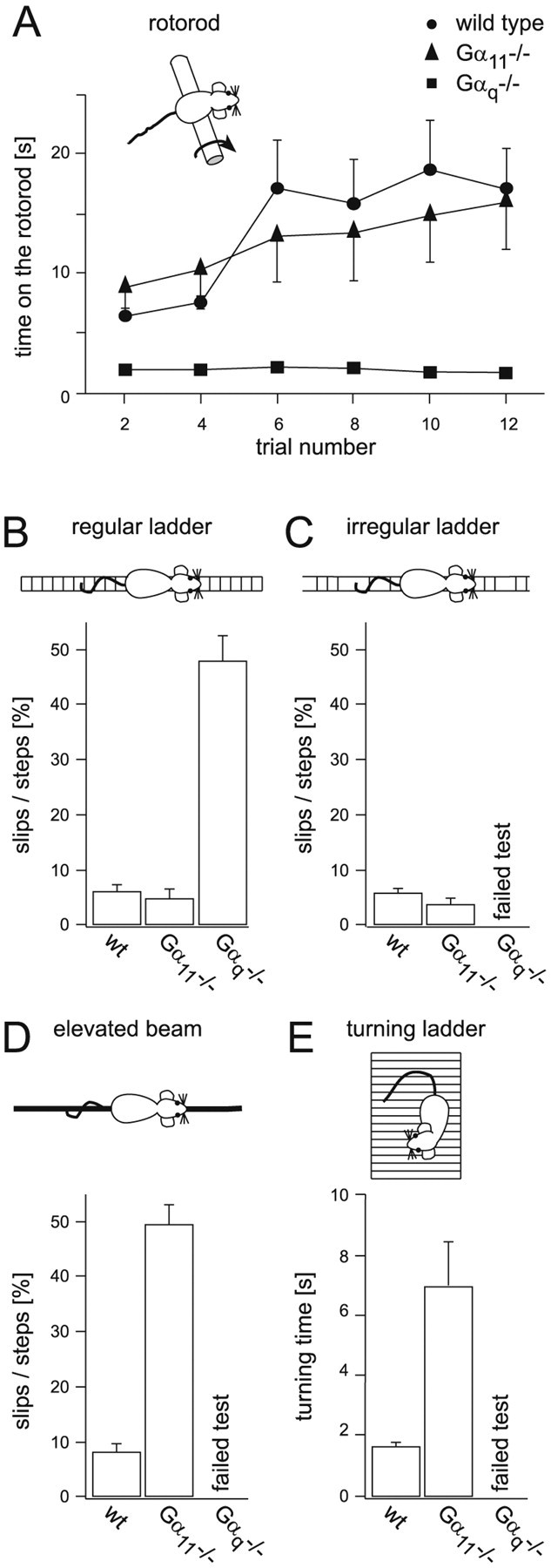
Distinct alterations in motor control in Gαq-deficient and Gα11-deficient mice, respectively. A, Gαq-deficient mice were significantly impaired on the turning rotorod from which they almost immediately fell off (n = 12). Gα11-deficient mice (n = 9), in contrast, displayed a stability very similar to wild-type mice (n = 10). The two data sets from wild-type and Gαq-deficient mice have been published before (Offermanns et al., 1997) but are shown here for a comparison with previously unpublished data from Gα11-deficient mice, which were obtained in the same series of experiments. B, When supposed to walk on a horizontal ladder with a regular stave pattern, Gαq-deficient mice slipped significantly more often than Gα11-deficient mice, which were indistinguishable from wild-type mice in this experiment. C, Gαq-deficient mice completely failed to perform the horizontal ladder test with an irregular stave pattern. Gα11-deficient mice, in contrast, did not differ significantly from wild-type mice in their performance in this test. D, In the elevated beam balancing test, Gα11-deficient mice performed significantly worse than the wild-type mice, as indicated by a sixfold increase in the number of slips. Gαq-deficient mice, again, were not able to pass this test because they could not keep their balance on the beam. The bars in B–D represent averages of two trials of four mice of each genotype. E, In the turning ladder test, the Gα11-deficient mice needed significantly more time to reach an upright position of their body from the reverse starting position than the wild-type mice. Gαq-deficient mice lacked the ability to adhere to the vertical ladder. The bar graphs contain data from three trials of four mice of each genotype. Significance was tested with the ANOVA test (p < 0.01).
A run test on a horizontal ladder (Fig. 6B) yielded comparable results from the three genotypes, consisting of a strong impairment in motor control for Gαq–/– deficiency but not for Gα11 deficiency. When increasing the difficulty of the task, by using a ladder with an irregular spacing of the steps (Fig. 6C), the difference between Gαq-deficient mice versus wild-type and Gα11-deficient mice, respectively, became even more evident. In this test, Gαq-deficient mice were not capable of walking on the ladder at all. Wild-type and Gα11-deficient mice, in contrast, performed similarly well as on the ladder with regularly spaced steps (Fig. 6B,C).
A more challenging test, however, revealed surprising results. The run test on an elevated, narrow beam (see Materials and Methods) exposed a marked impairment in motor control of Gα11-deficient mice. Wild-type mice walked on the bar without any detectable difficulty to reach a platform at the opposite end of the bar. In contrast, Gαq-deficient mice completely failed to walk on the bar. Gα11-deficient mice reached the platform at the end of the bar but had marked difficulties while walking on the bar. Unlike wild-type mice, Gα11-deficient mice walked uncoordinatedly, slipping frequently on their way to the platform. The average relative number of slips during their runs was increased six times compared with wild-type mice (49.7 ± 3.8% for Gα11-deficient mice and 8.18 ± 1.52% for wild-type mice; two trials, n = 4 in both groups) (Fig. 6D). Thus, in this runway test, each genotype had a clearly distinctive behavior that was found in all individuals tested. Thus, by using a challenging motor coordination test, we found, in contrast to previous suggestions (Offermanns et al., 1997), clear behavioral alterations also in Gα11-deficient mice.
Clear behavioral alterations in Gα11–deficient mice were detected also in the “turning ladder” test. In this test, a mouse is placed on a ladder that initially is held in a horizontal position. We determined the time needed for the mice to turn to an upright body position after they unexpectedly found themselves in a vertical position with their heads facing downward. The average turning time of Gα11-deficient mice was 7.05 ± 1.47 sec (n = 9 mice; three trials for each mouse; SEM) compared with 1.74 ± 0.1 sec (n = 9 mice; three trials for each mouse; SEM) for wild-type mice. Gαq-deficient mice entirely failed this test and in no instance managed to turn their body into the upright position (n = 4 mice; repeated trials) (Fig. 6E).
Expression levels of Gαq and Gα11 in Purkinje cells
Because Gαq and Gα11 are activated to the same extent by mGluR1 (see above), the differential effect of the deletion in Gαq or Gα11, respectively, may be attributable to differences in the level of expression. To test this hypothesis, we implemented a quantitative (Liss et al., 2001) real-time PCR approach (Rasmussen, 2001) that we used for total brain and single-cell RT-PCR analyses (Garaschuk et al., 1996; Plant et al., 1998). To investigate the expression of Gαq and Gα11 in total brain RNA, we established external standard curves over a large range of concentrations (Gαq, 1000–400,000 cDNA copies; Gα11, 500–100,000 cDNA copies). In wild-type mice, we detected an average of 1596 ± 68 copies of Gαq cDNA per nanogram of total RNA (n = 6 total brain RNA pools; SEM) and 253 ± 21 copies of Gα11 cDNA per nanogram of total RNA (n = 6 total brain RNA pools; SEM) (Fig. 7A). In Gα11–/– mice, Gαq transcripts were weakly upregulated (1.26-fold) to 2008 ± 106 cDNA copies per nanogram of total RNA (n = 6 pools; p < 0.01). In Gαq–/– mice, the expression level of Gα11 (255 ± 8 cDNA copies per nanogram of total RNA) was similar to that observed in wild-type mice (Fig. 7A).
Figure 7.
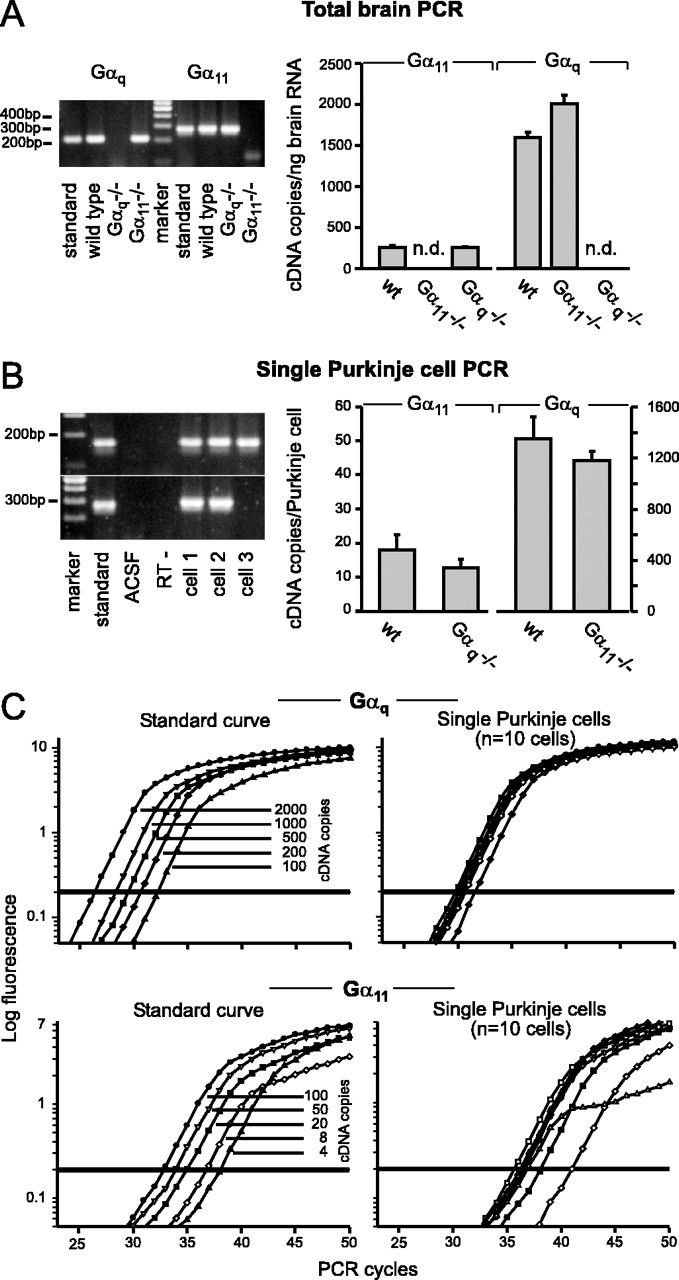
Real-time quantitative RT-PCR of Gαq and Gα11 transcripts in mutant and wild-type mice. A, Quantification of Gαq and Gα11 transcripts in whole brain from wild-type, Gαq-deficient, and Gα11-deficient mice. Left, Agarose gel electrophoresis of representative examples of cDNAs obtained after PCR amplification (Gαq amplicons, 724-for/935-rev, 212 bp; Gα11 amplicons, 119-for/403-rev, 285 bp). Standards: Gαq, 2000 cDNA copies; Gα11, 50 cDNA copies were amplified; the marker is the MassRuler DNA Ladder (Fermentas, St. Leon-Rot, Germany). Right, Bar graph illustrating the average number of cDNA copies per nanogram of total brain RNA. n = 6 mRNA pools from two animals; n.d., Not detectable. B, Quantification of the Gαq and Gα11 isoforms in single Purkinje cells from wild-type, Gαq-deficient, and Gα11-deficient mice. Left, Agarose gel electrophoresis of the cDNA obtained after PCR amplification (above: Gαq amplicons, 86-for/256-rev, 171 bp; below: Gα11 amplicons, 119-for/403-rev, 285 bp). Standards: Gαq, 50 copies; Gα11, 8 copies were amplified; marker (see A); ACSF was harvested directly above the slice; RT–is a Purkinje cell control without reverse transcriptase enzyme added; cells 1–3 are from wild-type animals. Cell 3 shows an example of a single cell that was positive for Gαq but negative for Gα11. Right, Bar graph illustrating the average cDNA copy number per single Purkinje cell; copy numbers are determined based on the standard curves (see C). C, Real-time fluorescence measurements of the amplification of the Gαq (top) and Gα11 cDNA (bottom). Left, For the absolute quantification of cDNAs, known dilutions of the corresponding amplicons were amplified and used to form standard curves. Right, Representative examples of the PCR amplification of single Purkinje cells from wild-type mice (n = 10 cells); only one of five for Gαq or two of five for Gα11 of a Purkinje cell was used for amplification.
For the comparison of the respective expression levels of both transcripts in Purkinje cells, appropriate external standard curves were established for the very low copy numbers expected on the single-cell level (Gαq, 100–2000 cDNA copies; Gα11, 4–100 cDNA copies) (Fig. 7C). In wild-type mice, the number of Gαq cDNA copies/Purkinje cell was 1349 ± 171 (n = 18 cells; SEM) (Fig. 7B). In contrast, the expression level of Gα11 in single Purkinje cells was ∼75-fold lower. Only 18 ± 4 (n = 17 cells; SEM) copies of Gα11 cDNA were detected in the same cDNA material that was used for the Gαq single-cell analysis (Fig. 7B). In Gα11–/– mice, the expression of Gαq was not significantly altered (1180 ± 74 Gαq cDNA molecules/Purkinje cell; n = 25 cells; SEM). It is important to note that the Gα11 expression levels of Gαq–/– mice (13 ± 4.6 cDNA copies/cell; n = 21 cells; SEM) was similar to that found for wild-type mice (see above), indicating the absence of any compensatory upregulation and providing an explanation for the severe behavioral phenotype.
The evaluation of the relative expression of Gαq and Gα11 on the protein level by means of antibody precipitation yielded remarkably similar results. Consistent with previous reports (Tanaka et al., 2000), immunostaining with an anti-Gαq/11 antibody of cerebellar sections of wild-type mice revealed intense staining in the molecular layer, a region densely packed with PF–Purkinje cell synapses, as well as scattered staining in the granule cell layer (Fig. 8A). Parallel staining of cerebellar sections of Gα11–/– mice and Gαq–/– mice with the anti-Gαq/11 antibody enabled investigation of the differential expression of Gαq and Gα11 proteins in the cerebellum. The pattern of staining found in cerebellar sections of wild-type mice was preserved in Gαq–/– and Gα11–/– mice, indicating that Gαq and Gα11 have a similar spatial distribution in the cerebellum. However, the intensity of staining in the molecular layer was severely reduced in Gαq–/– mice compared with wild-type or Gα11–/– mice. The staining intensity in the cerebellar molecular layer of Gαq–/– mice was 57% lower than in wild-type mice and 64% lower than in Gα11–/– mice, respectively (Fig. 8A) (n = 3). In contrast, staining intensity in the granule cell layer was comparable in all mice tested (n = 3). These data indicate that the Gαq protein is the dominant Gq subunit compared with Gα11 in cerebellar Purkinje cells.
Figure 8.

Gαq as the dominating Gq protein in cerebellar Purkinje cells. A, Immunohistochemistry with an anti-Gαq/Gα11 antibody in cerebellar sections in mice of three genotypes, as indicated. In wild-type and Gα11-deficient mice, the molecular layer, in which the dendritic trees of the Purkinje cells are located, is intensely stained, whereas in Gαq-deficient mice, only very weak staining is present in the molecular layer. Mo, Molecular layer; Pu, Purkinje cell layer; Gr, granule cell layer. Scale bar, 50 μm. B, Model for the mGluR1-dependent signal transduction through Gαq and Gα11 in cerebellar Purkinje cells. Both Gαq and Gα11 are involved in signal transduction pathways leading from activation of mGluR1 to release of Ca2+ from dendritic endoplasmic reticulum and/or to a sEPSP. However, the dominating Gq protein in cerebellar Purkinje cells is Gαq, according to its prevalent expression level compared with Gα11. The mGluR-mediated slow depolarization in Purkinje cells is G-protein dependent.
Discussion
Our results demonstrate that both Gq isoforms, Gαq and Gα11, contribute to PF–Purkinje cell synaptic signaling. Both mediate the coupling of mGluR1 to PLCβ and thus to the cytosolic production of InsP3 and DAG and, furthermore, both are involved in the activation of the mGluR-mediated slow depolarization. However, their specific contributions to Purkinje cell synaptic signaling as well as to, presumably, cerebellum-controlled motor behavior display distinct differences.
Synaptic Ca2+ signaling evoked by activation of mGluRs
In cerebellar Purkinje cells, repetitive PF stimulation evokes a complex postsynaptic dendritic Ca2+ signal consisting of an early and a delayed component (termed ESCT and DSCT, respectively) (Takechi et al., 1998). The signal cascade underlying the occurrence of DSCTs involves activation of a G-protein, followed by the activation of PLCβ, and, finally, InsP3-mediated Ca2+ release from the dendritic endoplasmic reticulum (Finch and Augustine, 1998; Takechi et al., 1998). It has been reported previously that mGluR1, the mGluR subtype found in Purkinje cells, is coupled with a G-protein of the Gq family, which includes Gα11 and Gαq as its only members expressed in the brain (Wilkie et al., 1991; Mailleux et al., 1992).
The results of the present study indicate that Gα11-deficient mice are similar to wild-type mice with respect to mGluR-mediated synaptic Ca2+ signaling. Thus, the time course of the dendritic Ca2+ transients after repetitive PF stimulation as well as the composition of these complex responses resembled those found in control animals (Fig. 2). In contrast to wild-type and Gα11-deficient mice, in which DSCTs were found in 28 of 33 and in 20 of 23 inputs, respectively, in Gαq-deficient mice a complex Ca2+ response was never observed (19 of 19 inputs).
sEPSPs evoked by activation of mGluRs
The first piece of evidence for mGluR-mediated synaptic transmission in Purkinje cells was a slow depolarizing postsynaptic potential (Batchelor et al., 1994). New evidence indicates that the membrane conductance change responsible for this sEPSP occurs because of the activation of the cation channel TRPC1 (Kim et al., 2003). As for the DSCTs, the induction of sEPSPs requires repetitive PF stimulation. In our experimental conditions, the generation of sEPSCs needed an approximate three times stronger afferent stimulation (15 pulses at 50 Hz) than that used for the induction of DSCTs.
The involvement of G-proteins for the activation of the sEPSCs had been suggested by previous studies (Hirono et al., 1998; Tempia et al., 1998; Sugiyama et al., 1999; Canepari and Ogden, 2003), showing that in cerebellar Purkinje cells the sEPSCs are abolished by GDPβS, a general inhibitor of G-proteins, and by GTPγS, an unhydrolyzable GTP analog. Our new results suggest that Gαq and, to a lesser extent, Gα11 mediate the coupling of mGluR1 with TRPC1 (Kim et al., 2003).
It is important to note that in CA3 pyramidal cells of rat hippocampus, mossy fiber stimulation evokes an mGluR1-dependent excitatory postsynaptic response independent of G-protein activation. This mGluR–EPSC was neither affected by N-ethylmaleimide, an inhibitor of pertussis toxin-sensitive G-proteins, nor by GDPβSorGTPγS. Instead, it was found that in CA3 hippocampal pyramidal cells mGluR1 activation is linked to a transient cationic conductance increase through the activation of a Src family tyrosine kinase (Heuss et al., 1999).
Role of Gα11 and Gαq for synaptic signaling and motor control
Thus far, it had been assumed that the receptors in mammalian systems do not discriminate between Gαq and Gα11 (Wange et al., 1991; Wu et al., 1992; Offermanns et al., 1994; Yule et al., 1999). Figure 1 shows, indeed, that both G-proteins can couple with mGluR1 with very similar efficiency to inositol phosphate production when expressed in COS cells. Furthermore, there is experimental evidence that Gαq and Gα11 activate the same PLCβ isoforms with the same efficiency (Taylor and Exton, 1991; Wu et al., 1992; Hepler et al., 1993). Nevertheless, we found that only Gαq-deficient mice had altered mGluR responsiveness in Purkinje cells. The immunohistochemical and the quantitative PCR results resolve this dilemma by demonstrating that in Purkinje cells the expression levels of Gαq are markedly higher than those of Gα11. The single-cell PCR results demonstrate that in individual Purkinje cells the expression of Gαq is up to 75-fold higher than that of Gα11. Thus, our data provide strong evidence for an expression level-dependent action of Gαq and Gα11 in PF-to-Purkinje cell synaptic signaling and LTD as well as in cerebellar motor behavior.
Figure 8B summarizes our results. We provide for the first time evidence that Gα11 is directly involved in Purkinje cell synaptic signaling and in distinct aspects of motor control. Both the LTD measurements and the behavioral examination revealed alterations of Gα11-deficient mice. These were, especially on the behavioral level, by far not as severe as those encountered in Gαq-deficient mice, but nonetheless significant. Whereas Gαq exerts a powerful control of mGluR-evoked synaptic Ca2+ signaling, activation of Gα11 comes into play during the prolonged trains of PF stimulation, like those required for the induction of LTD. Thus, we propose that Gαq and Gα11 control PF-to-Purkinje cell transmission during distinct functional states and may, therefore, enhance the tuning range of cerebellar synaptic plasticity.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft. We are grateful to Dr. Hajime Takechi for help and advice in the initial experiments and to Dr. Detlef Hof for technical support with imaging data analysis. We thank Reiko Trautmann, Irene Schneider, Susanne Schickle, Roswitha Maul, Ines Mühlhahn, Ursula Brandt, and Barbara Wallenwein for excellent technical help.
Correspondence should be addressed to Prof. Arthur Konnerth, Institut für Physiologie, Ludwig-Maximilians-Universität München, Pettenkoferstr. 12, 80336 Munich, Germany. E-mail: konnerth@lrz.uni-muenchen.de.
DOI:10.1523/JNEUROSCI.4193-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/245119-12$15.00/0
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S (1992) Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem 267: 13361–13368. [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994) Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79: 377–388. [PubMed] [Google Scholar]
- Barski JJ, Hartmann J, Rose CR, Hoebeek F, Mörl K, Noll-Hussong M, De Zeeuw CI, Konnerth A, Meyer M (2003) Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J Neurosci 23: 3469–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ, Garthwaite J (1994) Synaptic activation of metabotropic glutamate receptors in the parallel fibre–Purkinje cell pathway in rat cerebellar slices. Neuroscience 63: 911–915. [DOI] [PubMed] [Google Scholar]
- Canepari M, Ogden D (2003) Evidence for protein tyrosine phosphatase, tyrosine kinase, and G-protein regulation of the parallel fiber metabotropic slow EPSC of rat cerebellar Purkinje neurons. J Neurosci 23: 4066–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Papageorgiou G, Corrie JE, Watkins C, Ogden D (2001) The conductance underlying the parallel fibre slow EPSP in rat cerebellar Purkinje neurones studied with photolytic release of l-glutamate. J Physiol (Lond) 533: 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crépel F (1994) Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372: 237–243. [DOI] [PubMed] [Google Scholar]
- Crepel F, Daniel H, Hemart N, Jaillard D (1991) Effects of ACPD and AP3 on parallel-fibre-mediated EPSPs of Purkinje cells in cerebellar slices in vitro. Exp Brain Res 86: 402–406. [DOI] [PubMed] [Google Scholar]
- Eilers J, Augustine GJ, Konnerth A (1995) Subthreshold synaptic Ca2+ signalling in fine dendrites and spines of cerebellar Purkinje neurons. Nature 373: 155–158. [DOI] [PubMed] [Google Scholar]
- Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M, Konnerth A, De Zeeuw CI, Hofmann F (2003) Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol 163: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ (1998) Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature 396: 753–756. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL (2001) A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol (Lond) 537: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Schneggenburger R, Schirra C, Tempia F, Konnerth A (1996) Fractional Ca2+ currents through somatic and dendritic glutamate receptor channels of rat hippocampal CA1 pyramidal neurones. J Physiol (Lond) 491: 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, Kozasa T, Smrcka AV, Simon MI, Rhee SG, Sternweis PC, Gilman AG (1993) Purification from Sf9 cells and characterization of recombinant Gqα and G11α. Activation of purified phospholipase C isozymes by Gα subunits. J Biol Chem 268: 14367–14375. [PubMed] [Google Scholar]
- Heuss C, Scanziani M, Gähwiler BH, Gerber U (1999) G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci 2: 1070–1077. [DOI] [PubMed] [Google Scholar]
- Hirono M, Konishi S, Yoshioka T (1998) Phospholipase C-independent group I metabotropic glutamate receptor-mediated inward current in mouse Purkinje cells. Biochem Biophys Res Commun 251: 753–758. [DOI] [PubMed] [Google Scholar]
- Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, Katsuki M, Aiba A (2000) mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288: 1832–1835. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K (1998) Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18: 5366–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M (2001) Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev 81: 1143–1195. [DOI] [PubMed] [Google Scholar]
- Kano M, Garaschuk O, Verkhratsky A, Konnerth A (1995) Ryanodine-receptor-mediated intracellular calcium release in rat cerebellar Purkinje neurons. J Physiol (Lond) 487: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodakhah K, Ogden D (1995) Fast activation and inactivation of inositol trisphosphate-evoked Ca2+ release in rat cerebellar Purkinje neurones. J Physiol (Lond) 487: 343–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ (2003) Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426: 285–291. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM (1990) Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA 87: 2662–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crepel F, Rossier J (1992) AMPA receptor subunits expressed by single Purkinje cells. Neuron 9: 247–258. [DOI] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J (2001) Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J 20: 5715–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux P, Mitchell F, Vanderhaeghen JJ, Milligan G, Erneux C (1992) Immunohistochemical distribution of neurons containing the G-proteins Gqα/G11α in the adult rat brain. Neuroscience 51: 311–316. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S (1991) Sequence and expression of a metabotropic glutamate receptor. Nature 349: 760–765. [DOI] [PubMed] [Google Scholar]
- Miura M, Watanabe M, Offermanns S, Simon MI, Kano M (2002) Group I metabotropic glutamate receptor signaling via Gαq/Gα11 secures the induction of long-term potentiation in the hippocampal area CA1. J Neurosci 22: 8379–8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Finch EA, Khiroug L, Hashimoto K, Hayasaka S, Oda SI, Inouye M, Takagishi Y, Augustine GJ, Kano M (2000) Local calcium release in dendritic spines required for long-term synaptic depression. Neuron 28: 233–244. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Simon MI (1995) Gα15 and Gα16 couple a wide variety of receptors to phospholipase C. J Biol Chem 270: 15175–15180. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Heiler E, Spicher K, Schultz G (1994) Gq and G11 are concurrently activated by bombesin and vasopressin in Swiss 3T3 cells. FEBS Lett 349: 201–204. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI (1997) Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Gαq Proc Natl Acad Sci USA 94: 14089–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TD, Schirra C, Katz E, Uchitel OD, Konnerth A (1998) Single-cell RT-PCR and functional characterization of Ca2+ channels in motoneurons of the rat facial nucleus. J Neurosci 18: 9573–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen R (2001) Quantification on the LightCycler. In: Rapid-cycle real-time PCR. Methods and applications (Meurer S, Wittwer C, Nakawara K, eds), pp 21–34. Berlin: Springer.
- Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C*. Annu Rev Biochem 70: 281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Konnerth A (2001) Stores not just for storage: intracellular calcium release and synaptic plasticity. Neuron 31: 519–522. [DOI] [PubMed] [Google Scholar]
- Stanislaus D, Janovick JA, Ji T, Wilkie TM, Offermanns S, Conn PM (1998) Gonadotropin and gonadal steroid release in response to a gonadotropin-releasing hormone agonist in Gqα and G11α knockout mice. Endocrinology 139: 2710–2717. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Hirono M, Suzuki K, Nakamura Y, Aiba A, Nakamura K, Nakao K, Katsuki M, Yoshioka T (1999) Localization of phospholipase Cβ isozymes in the mouse cerebellum. Biochem Biophys Res Commun 265: 473–478. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A (1998) A new class of synaptic response involving calcium release in dendritic spines. Nature 396: 757–760. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Nakagawa S, Kushiya E, Yamasaki M, Fukaya M, Iwanaga T, Simon MI, Sakimura K, Kano M, Watanabe M (2000) Gq protein α subunits Gαq and Gα11 are localized at postsynaptic extra-junctional membrane of cerebellar Purkinje cells and hippocampal pyramidal cells. Eur J Neurosci 12: 781–792. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, Exton JH (1991) Two alpha subunits of the Gq class of G proteins stimulate phosphoinositide phospholipase C-β 1 activity. FEBS Lett 286: 214–216. [DOI] [PubMed] [Google Scholar]
- Tempia F, Miniaci MC, Anchisi D, Strata P (1998) Postsynaptic current mediated by metabotropic glutamate receptors in cerebellar Purkinje cells. J Neurophysiol 80: 520–528. [DOI] [PubMed] [Google Scholar]
- Vranesic I, Batchelor A, Gähwiler BH, Garthwaite J, Staub C, Knöpfel T (1991) Trans-ACPD-induced Ca2+ signals in cerebellar Purkinje cells. NeuroReport 2: 759–762. [DOI] [PubMed] [Google Scholar]
- Wange RL, Smrcka AV, Sternweis PC, Exton JH (1991) Photoaffinity labeling of two rat liver plasma membrane proteins with [32P]γ-azidoanilido GTP in response to vasopressin. Immunologic identification as α subunits of the Gq class of G proteins. J Biol Chem 266: 11409–11412. [PubMed] [Google Scholar]
- Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI (1991) Characterization of G-protein α subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci USA 88: 10049–10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI (1992) Activation of phospholipase C by α1-adrenergic receptors is mediated by the α subunits of Gq family. J Biol Chem 267: 25798–25802. [PubMed] [Google Scholar]
- Yule DI, Baker CW, Williams JA (1999) Calcium signaling in rat pancreatic acinar cells: a role for Gαq, Gα11, and Gα14 Am J Physiol 276: G271–G279. [DOI] [PubMed] [Google Scholar]