

Abstract
Activation of the transcription factor cAMP response element-binding protein (CREB) by neurotrophins is believed to regulate the survival, differentiation, and maturation of neurons in the CNS and PNS. Although phosphorylation of Ser133 is critical for the expression of CREB-regulated genes, the identity of neurotrophin-regulated Ser133 kinases has remained controversial. We show here that neurotrophin-induced CREB phosphorylation in CNS neurons depends exclusively on the extracellular signal-regulated kinase 1/2-activated kinase mitogen- and stress-activated protein kinase 1 (MSK1). Small interfering RNA directed against ribosomal S6 kinase 1 (RSK1) and RSK2 reduced phosphorylation of a RSK substrate but did not effect CREB-dependent transcription. However, expression of a selective inhibitory MSK1 mutant markedly attenuated BDNF-stimulated CREB phosphorylation and CREB-mediated transcription. Moreover, the ability of neurotrophins to stimulate CREB phosphorylation was abolished in CNS neurons from MSK1 knock-out mice. Consistent with a role for MSK1 in Ser133 phosphorylation, neurotrophin-induced expression of CREB-regulated genes was attenuated in MSK-deficient neurons. These results indicate that MSK1 is the major neurotrophin-activated Ser133 kinase in CNS neurons.
Keywords: differentiation, neurotrophic, nerve growth factor, nucleus, protein kinase, transcription, transduction
Introduction
Activation of the transcription factor cAMP response element-binding protein (CREB) by neurotrophins has been proposed to regulate synaptic plasticity, neuronal survival, neuronal maturation, and developmental plasticity. In particular, recent work suggests that CREB contributes to neurotrophin-mediated survival in the CNS by activating programs of anti-apoptotic gene expression. Although activation of CREB-regulated transcription by neurotrophins requires CREB Ser133 phosphorylation, the identity of the kinase or kinases that trigger CREB phosphorylation has remained obscure. Interestingly, for unknown reasons neurotrophin-stimulated CREB phosphorylation is not always sufficient for transcriptional activation (Ahn et al., 1998; Mayr et al., 2001). Thus, the identification and characterization of the neurotrophin-regulated Ser133 kinase is critical to our understanding of the complex mechanisms that lead to transactivation of CREB.
Neurotrophins bind to receptors composed of Trk tyrosine kinase receptors and/or p75 receptors (Huang and Reichardt, 2001). BDNF and NT4 selectively activate the TrkB receptor, NT3 selectively activates the TrkC receptor, and NGF activates the TrkA receptor. Binding of neurotrophins to Trk receptor subunits results in autophosphorylation of a set of conserved tyrosine residues that recruit activators of the extracellular signal-regulated kinase (ERK) 1/2, PLCγ, and PI3 kinase cascades.
Several studies have suggested that the recruitment of PLCγ to Trk receptors is critical for neurotrophin-stimulated CREB phosphorylation in cortical neurons (Finkbeiner et al., 1997; Minichiello et al., 2002). It has been proposed that PLCγ-mediated release of intracellular Ca2+ triggers CREB phosphorylation via activation of Ca2+–CaM-regulated kinase IV (CaMKIV) (Finkbeiner et al., 1997; Minichiello et al., 2002). In support of this, inhibitors of CaM kinases reduced BDNF-stimulated CREB phosphorylation in primary cortical neurons. PLCγ also plays a role in the activation of ERK1/2 signaling, possibly by activating PKCδ or by recruiting Grb2 (Huang and Reichardt, 2001). Although one study found that ERK1/2 signaling was required for BDNF-induced CREB phosphorylation (Pizzorusso et al., 2000), other studies have suggested that it does not play a major role in cortical neurons (Finkbeiner et al., 1997; Minichiello et al., 2002). Recently ERK5 and p38 mitogen-activated protein kinase (MAPK) have also been proposed to contribute to neurotrophin-stimulated CREB phosphorylation in pheochromocytoma cell line (PC12) cells and sympathetic neurons, respectively (Xing et al., 1998; Watson et al., 2001).
ERK1/2, ERK5, and p38 MAPK cannot directly phosphorylate CREB Ser133 because it is not a proline-directed phosphorylation site. The ribosomal S6 kinase (RSK) family of kinases were proposed to be neurotrophin-regulated CREB kinases because overexpression of inhibitory RSK2 mutants reduced epidermal growth factor (EGF)-induced CREB phosphorylation (Xing et al., 1996), and cells deficient for RSK2 were found to be deficient for EGF-stimulated CREB phosphorylation (Merienne et al., 2001). The mitogen- and stress-activated protein kinase (MSK) family of ERK- and p38 MAPK-activated kinases have also been shown to be important for stress- and mitogen-induced CREB phosphorylation in fibroblasts and embryonic stem cells (Arthur and Cohen, 2000; Wiggin et al., 2002; Schuck et al., 2003) Nevertheless, because mitogen-induced CREB phosphorylation was only partially attenuated in MSK-deficient cells (Wiggin et al., 2002) and is not attenuated in several RSK2-deficient cell types (Bruning et al., 2000; Soloaga et al., 2003), the contribution of MSKs or RSKs to neurotrophin-induced CREB phosphorylation is not clear.
Although the mechanism by which neurotrophins phosphorylate CREB has been extensively studied, the identities of the signaling pathways that mediate CREB phosphorylation remain controversial. Because most attempts to study neurotrophin-mediated CREB phosphorylation have relied on pharmacological approaches that inhibit multiple kinases, the objectives of this study were to rigorously dissect the signaling pathways that mediate neurotrophin-induced CREB phosphorylation in CNS neurons.
Materials and Methods
Pharmacology and reagents. Neurons were pretreated with inhibitors for at least 30 min. Exposure of U0126, PD184352, and KN62 to light was minimized. All inhibitors were purchased from Calbiochem (La Jolla, CA) and were stored at –20°C in single-use aliquots in DMSO. BDNF (Alomone Labs, Jerusalem, Israel; Chemicon, Temecula, CA) and NT-3 (Alomone) were dissolved in dH2O, whereas forskolin (Sigma, St. Louis, MO) was dissolved in DMSO.
Plasmids. The following plasmids have been described previously: DN MEK, cAMP response element (CRE)-luciferase, DN CaM kinase IV, pUB6-GalCREB (Impey et al., 2002), and Δp85 (Poser et al., 2000). MSK1 was cloned from human cDNA, and the D195A and D565A mutations were introduced by PCR-based mutagenesis and verified by automated sequencing (ABI). RSK2 was cloned from rat hippocampal cDNA, and the K100R mutation was introduced using PCR. pCAG-LacZ or pUB6-LacZ were used to normalize for transfection efficiency. RSK1 and RSK2 small interfering RNA (siRNA) oligos were ligated into mU6pro (Yu et al., 2002) and verified by automated sequencing. Details on the subcloning and purification of plasmids will be provided on request.
Cell culture. The neocortex was excised from postnatal day 1–3 pups, finely minced, and digested enzymatically at 37°C for 30 min with papain (100 U/ml; Worthington, Freehold, NJ) in dissociation media (90 mm Na2SO4, 30 mm K2SO4, 16 mm MgCl2, 0.25 mm CaCl2, 32 mm HEPES, and 0.01% phenol red, pH 7.7). The tissue was rinsed by gravity precipitation five times in Opti-MEM (Invitrogen, San Diego, CA) and gently triturated into a single-cell suspension with a 5 ml plastic serological pipette. The cells were resuspended in Neurobasal A (Invitrogen) supplemented with 2% B27, 1 mm l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Invitrogen) and plated on poly-d-lysine-coated plates (100 μg/ml; Sigma). On day 2 in vitro the media was supplemented with 2.5 μm AraC (Sigma). Cultures were maintained for 4–7 d in standard tissue culture incubators at 37°C and 5% CO2.
Transfection. Cortical and hippocampal neurons were transfected with Lipofectamine 2000 (Invitrogen) as described by the manufacturer with the following exceptions: Neurobasal A plus 50 mm HEPES, pH 7.5, was used for lipid-DNA compaction and transfection, a 1:2 ratio was used for DNA:lipofectamine 2000 compaction, and conditioned media was retained and used to replace the transfection media. Electroporation of postnatal day 1 rat cortical neurons was conducted using the rat neuron nucelofection kit according to the manufacturer's instructions (Amaxa, Köln, Germany) except that 5 μg of DNA was used, and cells were plated in Neurobasal A plus B27 (Invitrogen).
Western blotting. We treated 1–5 × 106 hippocampal neurons (day 10–14 in vitro) as indicated and lysed in 50–100 μl of 2× SDS-PAGE buffer and boiled for 10 min. The samples were clarified by centrifugation (10 min at 28,400 × g), and 10–20 μg of protein was subjected to SDS-PAGE electrophoresis. Gels were transferred to Immobilon-P membranes (Millipore, Bedford, MA) and blocked with 5% powdered milk in PBS. The following primary antibodies were used overnight at 4°C in TBST (0.1% Triton X-100, 10 mm NaF, and 5%BSA): rabbit anti-phospho-cAMP response element-binding protein (1:1000; Cell Signaling Technology, Beverly, MA; Upstate Biotechnology, Lake Placid, NY), rabbit anti-phospho-Rsk Thr380 (1:1000; Cell Signaling Technology), rabbit anti-phospho MSK1 Ser376 (1:1000; Cell Signaling Technology), rabbit anti-phospho T308 Akt (1:1000; Cell Signaling Technology), rabbit anti-phospho-ERK5 (1:200; Cell Signaling Technology), mouse anti-phospho-ERK (1:10,000; Sigma), rabbit anti phospho Ser85 HSP27 (1:1000; Cell Signaling Technology), mouse anti-ERK2 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-RSK2 (1:2000; Santa Cruz Biotechnology), goat anti-Rsk1 (1:1000; Santa Cruz Biotechnology), mouse anti-hemagluttinin (HA) epitope (1:2000; Covance, Princeton, NJ), and mouse anti-cAMP response element-binding protein (1:200; Santa Cruz Biotechnology). Secondary antibodies (affinity-purified HRP- and AP-conjugated anti-IgG 1:2500; Jackson ImmunoResearch, West Grove, PA) were used at 22°C for 1–2 hr in 5% milk TBST. Immunoblots were washed 5 × 15 min after antibody (Ab) incubations and were developed using either alkaline phosphatase (Applied Biosystems, Foster City, CA) or HRP chemiluminescence (NEN, Boston, MA; Amersham Biosciences, Arlington Heights, IL).
Immunochemistry. Cortical neurons were fixed in 5% formaldehyde in PBS, pH 7.4, with 50 mm HEPES for 5 min. Neurons and were blocked (1–2hrat22°C) in 5% BSA in PBS with 0.05% Triton X-100 and 10 mm NaF (PBST). All immunolabeling was conducted in 2.5% BSA PBST. The goat anti-RSK (Santa Cruz Biotechnology) Ab was used at 1:500, and the mouse anti-HA Ab was used at 1:200. Both were visualized with 1 μg/ml Alexa 594 anti-rabbit IgG (Molecular Probes, Eugene, OR).
Reporter gene assays. Luciferase and β-galactosidase activity were measured using the Luciferase Assay and Galacoto-Light assay kits as described by the manufacturer (PerkinElmer Life Sciences, Emeryville, CA). A β-galactosidase vector with the same promoter as the other components of the transfection was used to normalize for transfection efficiency.
Immunoprecipitations. We treated 1 × 106 cortical neurons as indicated and lysed in 1 ml of RIPA buffer [TBS, pH 7.5, 1% NP-40, 1% BSA, 0.5% sodium deoxycholate, 0.5% SDS, 10 mm NaF, 1 mm PMSF, 1 mm EDTA, complete protease inhibitor mixture (Roche, East Sussex, UK), and phosphatase inhibitor mixture I (Calbiochem)]. Lysates were sonicated 3 × 30 sec, clarified by centrifugation at 14,000 × g for 30 min, and precleared with 20 μl of protein A or protein A/G (Pierce, Rockford, IL) and 0.5 μg affinity-purified rabbit IgG (Zymed, San Francisco, CA). The supernatant was incubated with 1 μg of anti-RSK2 (Santa Cruz Biotechnology), 1 μg of anti-RSK1, or 8 μg of anti-HA (HA.11; Covance) at 4°C overnight, followed by 20 μl of protein A or A/G agarose (Pierce) for 1 hr. For immunoprecipitation, Western blots were washed 5 × 10 min with RIPA buffer, the beads were pelleted at 8000 × g for 5 min, and denatured by boiling in 2× SDS-PAGE buffer for 5 min. Immunoprecipitates were separated by SDS-PAGE and immunoblotted as described above.
Immunocomplex kinase assays. For MSK1 immunocomplex kinase assays in PC12 cells, the immunoprecipitations were performed as described above and washed twice in kinase assay buffer (25 mm Tris-HCl, pH 7.5, 5 mm β-glycerophosphate, 1 mm dithiothreitol, 0.1 mm Na3VO4, 10 mm MgCl2, Calbiochem phosphatase inhibitor mixture I, and Roche protease inhibitor mixture) and assayed for kinase activity at 30°C for 30 min using 1 μm glutathione S-transferase (GST)-cAMP response element-binding protein in kinase assay buffer containing 0.1 mm ATP. The kinase assays were terminated by the addition of 2× SDS-PAGE buffer and were probed for Ser133 phosphorylation by Western blotting as described above.
For immunocomplex kinase assays in cortical neurons, cells were lysed in 50 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mm EDTA, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 1 mm sodium pyrophosphate, 0.27 m sucrose, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, and complete proteinase inhibitor mixture (Roche). The lysates were centrifuged at 13,000 rpm for 5 min at 4°C, and the supernatants were removed, quick frozen in liquid nitrogen, and stored at –80°C until use. Five hundred micrograms of soluble protein lysate was precleared for use with 5 μlof protein G Sepharose for 30 min an 4°C. MSK1 was then immunoprecipitated using 5 μgofan Ab raised against a peptide corresponding to residues 384–402 of human MSK1 coupled to 5 μg of protein G Sepharose. This Ab has been previously shown to be specific for MSK1 and does not immunoprecipitate MSK2 or RSK (Wiggin et al., 2002). After 2 hr at 4°C, MSK1 immunoprecipitates were pelleted by centrifugation and washed twice in 50 mm Tris-HCl, pH 7.5, 0.1 mm EGTA, 0.5 m NaCl, and 0.1% 2-mercaptoethenol and once in 50 mm Tris-HCl, pH 7.5, 0.1 mm EGTA, and 0.1% 2-mercaptoethenol. MSK1 activity was measured against crosstide (GRPRTSSFAEG, 30 μm) in 50 mm Tris-HCl, pH 7.5, 0.1 mm EGTA, 10 mm MgAc, 0.1 mm 32P-ATP, and 0.1% 2-mercaptoethenol. MSK1 assays were incubated at 30°C for 20 min, and the reaction was stopped by transferring onto P81 paper, which was then washed in orthophosphoric acid. Alternatively GST-cAMP response element-binding protein (5 μg) was used as a substrate in the kinase assay, for these assays the reaction was stopped by the addition SDS sample buffer to give a final SDS concentration of 1%. The assay was then run on polyacrylamide gels. After coomassie staining of the gels, GST-cAMP response element-binding protein bands were cut out from the gel and 32P incorporation was measured by Cerenkov counting. One unit of activity (U) was defined as the phosphorylation of 1 nmol of peptide substrate in 1 min.
Real-time RT-PCR. 2–4 × 106 mouse cortical neurons were treated as indicated, and RNA was isolated using the RNeasy kit (Qiagen, Hilden, Germany) and reverse-transcribed with Moloney murine leukemia virus (Invitrogen) and random decamers (Ambion, Austin, TX) according to the manufacturer's instructions. Levels of cDNA were measured by real-time SYBR green I (Invitrogen) PCR on an Opticon OP346 (MJ). Both Platinum Taq (Invitrogen) or HotStarTaq-based (Qiagen) PCR reactions were used. Details of the PCR reactions will be provided on request. All data were normalized to 18s message levels. Similar results were obtained when Tata-binding protein or glyceraldehyde-3-phosphate dehydrogenase message levels were used for normalization. The following primers were used: 18s-1, ccgcagctaggaataatgga; 18s-2, ccctcttaatcatggcctca; c-fos-1, accatgatgttctcgggtttcaa; c-fos-2, gctggtggagatggctgtcac; Mkp1–1, ggatgctggaggaagggtgttt; Mkp1–2, ggatgctggaggaagggtgttt; Nurr1–1, aagattcctggctttgctg; Nurr1–2, acaggggcatttggtacaag.
Statistical analysis. One-way ANOVA followed by the Tukey–Kramer multiple comparisons post-test was used to assess significance.
Results
An ERK-activated kinase mediates the phosphorylation and activation of CREB by neurotrophins
We used inhibitors and expression of inhibitory mutants to systematically characterize the signaling cascades that mediate neurotrophin-stimulated CREB Ser133 phosphorylation in rat CNS neurons. CaMKIV is widely believed to be the major BDNF-stimulated CREB kinase in cortical neurons (Finkbeiner et al., 1997; Minichiello et al., 2002). However, we were unable to detect a significant rise in cytoplasmic Ca2+ in response to BDNF (data not shown) and the CaM kinase inhibitor, KN62, did not reduce BDNF-stimulated CREB phosphorylation (Fig. 1A), although it attenuated NMDA-stimulated CaMKIV phosphorylation (Fig. 1B). These data are consistent with previous studies (Pizzorusso et al., 2000) and indicate that CaMKIV does not function as a neurotrophin-regulated CREB kinase in primary cortical neurons.
Figure 1.

Neurotrophin-induced CREB phosphorylation is mediated by an ERK1/2-activated kinase. A, B, Cortical neurons were pretreated with 10 μm KN62 or vehicle and stimulated with BDNF or NMDA for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-cAMP response element-binding protein, ERK2, phospho-CaMKIV, or CaMKIV. C, Cortical neurons were pretreated with 10 μm LY294002 or vehicle and stimulated with BDNF for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-cAMP response element-binding protein, phospho-Akt, and total CREB. D, Rat cortical neurons were treated with 50 ng/ml BDNF or vehicle for 5 min. Akt1 was immunoprecipitated and assayed for kinase activity against GST-cAMP response element-binding protein or GST-Bad. The kinase reactions were separated by SDS-PAGE, and the bands corresponding to the substrate were autoradiographed. Similar results were seen in three experiments. E, Cortical neurons were pretreated with 10 μm PD184352 or vehicle and stimulated with BDNF or NT3 for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-CREB, phospho-ERK, phospho-RSK1/2, phospho-Akt, phospho-ERK5, and total ERK2. F,G, Cortical neurons were pretreated with 5 μm SB202190 or vehicle and stimulated with BDNF or sodium arsenite (As) for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-cAMP response element-binding protein, phospho-HSP27, ERK2, and HSP27.
Several studies have suggested that Akt might mediate neurotrophin-induced CREB phosphorylation (Du and Montminy, 1998; Pugazhenthi et al., 2000). To address the involvement of Akt, we used a potent inhibitor of PI3-kinase, LY294002, to inhibit the phosphorylation and activation of Akt. Although BDNF-stimulated Akt phosphorylation was inhibited to below basal levels by LY294002, BDNF-stimulated CREB phosphorylation was not affected by LY294002 (Fig. 1C). Consistent with these results, Akt immunoprecipitated from BDNF-stimulated cortical neurons was not able to phosphorylate CREB in vitro, although it exhibited normal activity toward GSK3 (Fig. 1D). These data demonstrate that Akt is not a physiological Ser133 kinase in cortical neurons.
Although ERK1/2 signaling has been proposed to play a less significant role in neurotrophin-induced CREB phosphorylation (Finkbeiner et al., 1997; Minichiello et al., 2002), these previous studies used inhibitors or mutants that only partially attenuated activation of ERK. Moreover, the small molecule inhibitors of ERK1/2 activation previously used to assess the contribution of this pathway to CREB activation can also perturb activation of ERK5 (Mody et al., 2001). We used the novel MEK1/2 inhibitor, PD184352 (Sebolt-Leopold et al., 1999), to specifically attenuate activation of ERK1/2 without inhibiting ERK5 phosphorylation (Fig. 1E). The slight increase in the intensity and persistence of ERK5 phosphorylation in the presence of PD184352 is likely caused by the previously described negative regulation of ERK5 by the classical ERK1/2 pathway (Mody et al., 2001). Because PD184352 attenuated neurotrophin-stimulated CREB phosphorylation to basal levels, these results suggest that an ERK1/2-activated kinase mediates neurotrophin-induced Ser133 phosphorylation. Similar inhibition of neurotrophin-stimulated CREB phosphorylation was observed in primary hippocampal neurons and cerebellar granule cells (data not shown). Together with the inhibitor data, these results indicate that in CNS neurons an ERK1/2-dependent kinase mediates neurotrophin-induced CREB phosphorylation.
Neurotrophin-induced Ser133 phosphorylation in PC12 cells has also been reported to require coactivation of p38 MAPK and ERK1/2 signaling (Deak et al., 1998; Xing et al., 1998). However, in cortical neurons BDNF is a poor activator of p38 MAPK (Fig. 1F). Moreover, neurotrophin-induced CREB phosphorylation and CREB-mediated transcription were not affected by treatment with SB202190, a potent inhibitor of p38 MAPK α and β kinase activity (Fig. 1G). SB202190 did not inhibit phosphorylation p38 MAPK in vivo, because SB202190 inhibits the kinase activity of p38 MAPK, and not p38 phosphorylation by its upstream activators (Lee et al., 1994; Cuenda et al., 1995). The same dose of SB202190 attenuated HSP27 phosphorylation (a downstream target of p38 MAPK signaling) by arsenite (Fig. 1F). Thus, we conclude that an ERK1/2-regulated kinase is the major neurotrophin-regulated CREB Ser133 kinase in cortical neurons.
Evidence that RSK1 and RSK2 are dispensable for the activation of CREB by neurotrophins
The RSK family of kinases are the most well characterized ERK1/2-regulated kinases, and as expected phosphorylation of RSK1/2 by neurotrophins is dependent on the ERK1/2 cascade (Fig. 1E). Several previous studies used overexpression of kinase-dead RSK2 to suggest that RSK2 is a physiological CREB kinase (Xing et al., 1998; Bonni et al., 1999). However, the specificity of this mutant was never characterized, and we found that expression of K100R RSK2 markedly reduced the phosphorylation of another ERK1/2-activated kinase, MSK1 (Fig. 2A). To characterize the role of RSKs in the activation of CREB more selectively, we developed siRNA expression vectors that target both RSK1 and RSK2. Cotransfection of a pool of four siRNA vectors that target RSK1 and RSK2 attenuated the expression of epitope-tagged RSK1 and RSK2 by ∼90% in cortical neurons (data not shown) and reduced the expression of endogenous RSK2 by >70% (Fig. 2B). We also examined whether expression of siRNAs could deplete neurons for endogenous RSK by immunochemical staining with an Ab that recognizes both RSK1 and RSK2. Cortical neurons contransfected with siRNA constructs no longer expressed detectable RSK, whereas those cotransfected with empty vector had normal levels of RSK (Fig. 2C).
Figure 2.
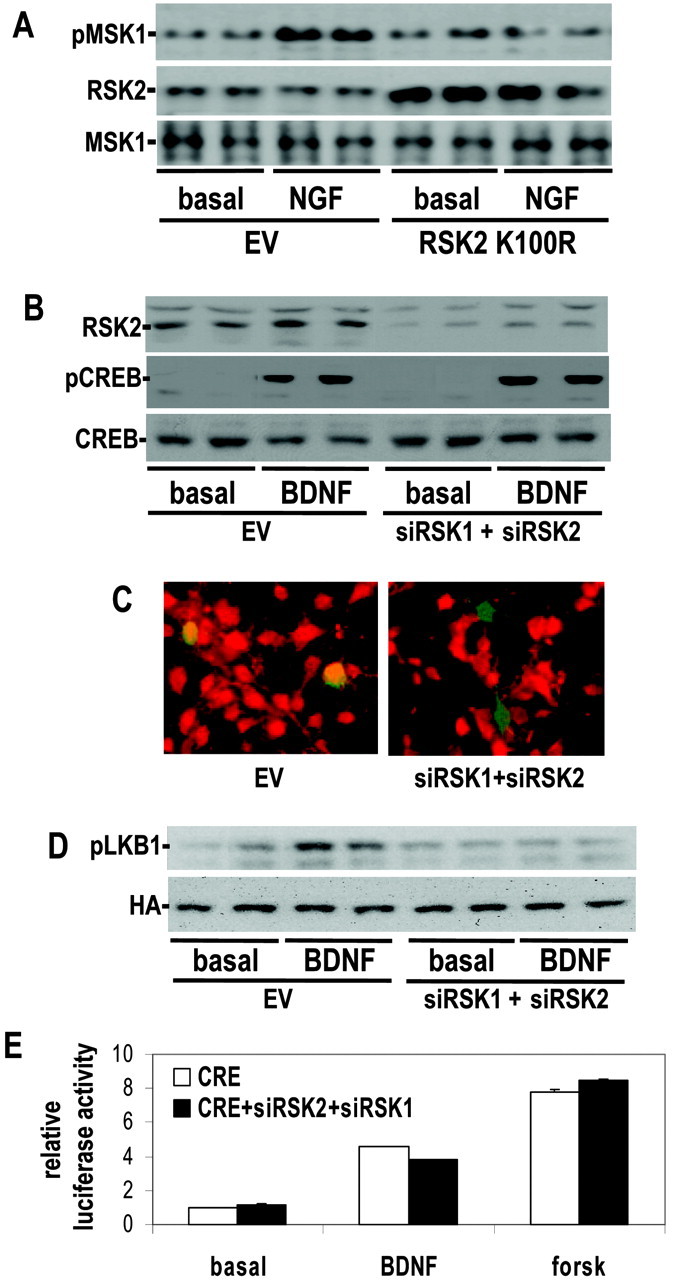
Activation of CREB by neurotrophins does not depend on RSK. A, Rat PC12 cells were transfected with empty vector (EV) or kinase-dead RSK2 mutants (RSK2 K100R) and stimulated with NGF for 15 min. Protein extracts were immunoblotted for phospho-MSK1, RSK2, and MSK1. B, Cortical neurons were electroporated with RSK1 (siRSK1) and RSK2 (siRSK2) siRNA DNAs. The cells were stimulated with BDNF (50 ng/ml), and protein extracts were immunoblotted for RSK2 and pCREB. C, Cortical neurons were transfected with a GFP expression construct and an eightfold excess of RSK1 (siRSK1) and RSK2 siRNA (siRSK2) DNAs. The cells were fixed and immunostained for total Rsk. Green fluorescence is GFP, and red fluorescence is staining for RSK. D, Cortical neurons were transfected with HA-tagged LKB1 and a fourfold excess of RSK1 (siRSK1) and RSK2 siRNA (siRSK2) DNAs. LKB1 was immunoprecipitated with an anti-HA Ab, and immunoprecipitates were immunoblotted for phospho-LKB1 Ser431. E, Cortical neurons were transfected with a CRE-regulated luciferase reporter and an eightfold excess of empty vector, siRNA vector RSK1 (siRSK1), or siRNA vector RSK2 siRNA (siRSK2). Neurons were treated with 50 ng/ml BDNF or 10 μm forskolin (forsk) and assayed for luciferase activity. The data are quadruplicate determinations, and the error is SEM. Unless otherwise noted, all data are representative examples of at least three experiments.
To determine whether the siRNAs were able to inhibit phosphorylation of a RSK target, we transfected neurons with the known RSK substrate, LKB1 (Sapkota et al., 2001), and examined the phosphorylation state of Ser428. Expression of RSK1 and RSK2 siRNA constructs, attenuated neurotrophin-stimulated LKB1 phosphorylation, indicating that these siRNAs effectively downregulated RSK activity (Fig. 2D). Under similar experimental conditions, coexpression of RSK1 and RSK2 siRNAs failed to significantly inhibit BDNF-stimulated CREB phosphorylation or transcription of a CREB-regulated reporter gene (Fig. 2B,E). These results indicate that a novel ERK1/2-activated kinase mediates neurotrophin-induced CREB phosphorylation.
MSK1 is an ERK1/2-regulated kinase that can phosphorylate CREB in response to neurotrophins
MSK1 was initially characterized as an ERK- and p38 MAPK-regulated kinase that based on inhibitor studies was proposed to mediate NGF-induced CREB phosphorylation in PC12 cells (Deak et al., 1998). However, subsequent work showed that the inhibitor used to implicate MSK had activity toward RSKs (Davies et al., 2000), leaving the question of the identity of the ERK1/2-activated CREB kinase unresolved. In contrast to the regulation of MSK1 by NGF, in cortical neurons BDNF-stimulated phosphorylation of MSK1 (at a residue essential for its activation) was completely attenuated by the inhibitor of ERK1/2 signaling, PD184352 (Fig. 3A). Consistent with the idea that it is a physiological CREB kinase, epitope-tagged MSK1 is localized exclusively to the nucleus (Fig. 3B). To test if BDNF could stimulate the catalytic activity of MSK1 and RSK against CREB, both MSK1 and RSK2 were immunoprecipitated and assayed against CREB in vitro. BDNF stimulation of primary cortical neurons markedly stimulated the activity of MSK1 toward full-length CREB in vitro (Fig. 3C). However, in agreement with previous studies (Deak et al., 1998; Swanson et al., 1999) neurotrophin stimulation only marginally increased RSK2 activity toward full-length CREB (Fig. 3C).
Figure 3.

MSK1 is a neurotrophin-regulated CREB kinase. A, Rat cortical neurons were pretreated with 10 μm PD184352 or vehicle and stimulated with 50 ng/ml BDNF (B) or NT3 (N) for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-MSK1 and total MSK1. B, Cortical neurons were transfected with GFP (green) and HA-tagged MSK1. Cells were immunostained for the HA epitope (red) and were also stained with the DNA dye, Hoechst 33258 (blue). C, Mouse cortical neurons were treated with BDNF for 15 min. MSK1 and RSK2 were immunoprecipitated and assayed for kinase activity against full-length CREB. The data are quadruplicate determinations, and the error is SEM.
Expression of kinase-dead MSK1 attenuates neurotrophin-mediated CREB phosphorylation and transcription
We initially explored whether neurotrophin-stimulated transcription depends on MSK1 by developing a selective interfering mutant. Expression of a MSK1 mutant that has inactivating mutations in both kinase domains (MSK1 KD) markedly attenuated NGF-stimulated wild-type MSK1 activity (Fig. 4A). This mutant did not function by nonspecifically inhibiting upstream ERK1/2 activity because under the same conditions it had no effect on the activity of another ERK1/2 substrate, RSK2 (data not shown). Consistent with the idea that MSK1 phosphorylates CREB, expression of MSK1 KD markedly attenuated the ability of BDNF to promote Ser133 phosphorylation and CREB-dependent transcription in cortical neurons (Fig. 4B,C). Expression of MSK1 KD had no detectable effect on forskolin-stimulated transcription or Ser133 phosphorylation, indicating that MSK1 KD does not nonspecifically block Ser133 phosphorylation, but rather that it selectively inhibits the ability of MSK1 to target CREB. Collectively, these data show that expression of MSK1 KD in cortical neurons does not perturb activation of other ERK1/2 targets and strongly suggests that the effect of MSK1 KD on CREB phosphorylation and gene expression is caused by a selective reduction of MSK1 activity.
Figure 4.

Expression of a dominant-negative MSK1 mutant attenuated neurotrophin-induced CREB-dependent transcription. A, PC12 cells were transfected with empty vector (EV) or an HA-tagged MSK1 construct and an excess of either empty vector or kinase-dead MSK1 (MSK1 KD). HA-tagged MSK1 was immunoprecipitated and assayed for kinase activity using purified CREB. Where indicated, PC12 cells were stimulated with 100 ng/ml NGF for 15 min. B, Cortical neurons were electroporated with GFP or kinase-dead MSK1 DNAs and stimulated with BDNF or forskolin (forsk; 10 μm) for the indicated times (in minutes). Protein extracts were immunoblotted for phospho-cAMP response element-binding protein and total CREB. C, Cortical neurons were transfected with a CRE-regulated luciferase reporter and a 10-fold excess of empty vector or kinase-dead MSK1 (MSK1 KD). Neurons were treated with 50 ng/ml BDNF or 10 μm forskolin and assayed for luciferase activity. The data are quadruplicate determinations, the error is SEM, and asterisks denote p < 0.01.
Neurotrophin-stimulated CREB phosphorylation is absent in MSK1-deficient CNS neurons
To address whether MSK1 is a bona fide CREB kinase, we examined neurotrophin-induced CREB phosphorylation in mouse cortical neurons deficient for both MSK1 and MSK2. BDNF-induced CREB phosphorylation was abolished in cortical neurons (Fig. 5A) and hippocampal neurons (data not shown) cultured from MSK1 and MSK2 double knock-out (MSK1/2) mice. Similarly, NT3-induced CREB phosphorylation was also attenuated in MSK1/2-deficient neurons (data not shown). This deficit was not the result of compensatory regulation of ERK signaling because neurotrophin-induced phosphorylation of both ERK1/2 and RSK1/2 was not affected in MSK1/2-deficient neurons (Fig. 5A, B), nor was the deficit in neurotrophin-induced CREB phosphorylation the result of a general impairment in CREB phosphorylation because forskolin-induced Ser133 phosphorylation was normal in MSK1/2-deficient neurons (Fig. 5A).
Figure 5.

Neurotrophin-induced CREB phosphorylation is absent in cortical neurons deficient for MSK1. A, Cortical neurons from wild-type or MSK1/2 double knock-outs were treated with 50 ng/ml BDNF (B) or 10 μm forskolin (F) for the indicated times (in minutes). Extracts were immunoblotted for the indicated epitopes. B, Cortical neurons from wild-type mice, MSK1/2 double knock-outs, MSK1 knock-outs, or MSK2 knock-outs were treated with 50 ng/ml BDNF for the indicated times (in minutes). Extracts were immunoblotted for the indicated epitopes. C, Tissue from the indicated brain regions or cultures was used to prepare cDNA. Real-time quantitative PCR was used to determine mRNA levels for MSK1 and MSK2 in these brain regions.
In murine embryonic fibroblasts, both MSK1 and MSK2 contribute to stress-and 12-O-tetradecanoylphorbol-13-acetate (TPA)-mediated CREB phosphorylation (Wiggin et al., 2002). Consequently, we examined neurotrophin-induced CREB phosphorylation in neurons derived from mice deficient for the individual kinases. BDNF-induced CREB phosphorylation was not detectable in neurons deficient for MSK1, whereas neurons deficient for MSK2 had normal levels of CREB phosphorylation (Fig. 5B). Antibodies that can be used to quantitatively measure endogenous MSK2 protein levels are not available. Therefore, we used real-time quantitative PCR to assess the levels of MSK1 and MSK2 message in cultured cortical neurons and in various adult CNS tissues. Consistent with our results showing that MSK2 is not required for neurotrophin-regulated CREB phosphorylation, MSK2 message was expressed at only 6% of MSK1 message levels in cortical neurons (Fig. 5C). These results suggest that MSK2 may not contribute to CREB phosphorylation in cortical neurons because it is expressed at lower levels. They also raise the possibility that MSK2 may regulate CREB-dependent gene expression in other tissues such as brainstem and striatum. When these results are taken together with our work with MSK1 KD, they strongly suggest that MSK1 is the major neurotrophin-activated Ser133 kinase in CNS neurons.
Neurotrophin-stimulated transcription of endogenous CREB-regulated genes is attenuated in cortical neurons
Neurotrophins are thought to regulate the expression of c-fos and several other immediate early genes by cooperatively activating several transcriptional cascades (Bonni et al., 1995). Although we have shown that MSK1 is critical for neurotrophin-induced CREB phosphorylation, it is conceivable that other pathways predominate at the level of transcription. Therefore, we used quantitative real-time PCR to determine whether neurotrophin-stimulated transcription is affected by the knock-out of MSK1 and MSK2. We chose to study c-fos, Mkp1, and Nurr1 because these genes contain CRE elements proximal to their transcriptional start sites and are believed to be CREB-regulated genes (Lonze et al., 2002). As expected, expression of the inhibitory mutant ACREB attenuated BDNF-induced expression of reporter genes regulated by the c-fos, Mkp1, and Nurr1 promoters (Fig. 6 A). Consistent with the idea that Ser133 phosphorylation is required for transcriptional activation of CREB, BDNF-stimulated transcription of a CREB-regulated reporter was markedly attenuated in MSK-deficient neurons (Fig. 6B). Neurotrophin-stimulated transcription of the prototypical immediate early gene c-fos was reduced by ∼50–60% at early time points in cells deficient for MSK1/2. Because other immediate early gene products and other transcriptional pathways regulate the c-fos promoter, it is not unexpected that c-fos transcription was only modestly reduced in cells that lack MSK1 (Fig. 6C). In contrast to c-fos, both Mkp1 and Nurr1 have promoters in which a CRE is the only well characterized stimulus-regulated enhancer element. Interestingly, neurotrophin-stimulated transcription of both Mkp1 and Nurr1 was markedly attenuated in MSK-deficient cells (Fig. 6 D,E). Importantly, forskolin-induced transcription of all three genes was not significantly different in MSK-deficient cells, indicating that that there was no general impairment in signal-dependent activation of gene expression. Although phosphorylation of ERK1/2, RSK1/2, and Akt was unimpaired in MSK-deficient neurons, it is formally possible that the deficit in neurotrophin-stimulated transcription is the result of a more general impairment in neurotrophin signaling. Therefore, we examined the expression of neurotrophin-regulated genes whose promoters do not contain CREs. BDNF-stimulated transcription of c-jun and SRF was unimpaired in cortical neurons derived from MSK1/2 knock-out mice (data not shown). These data mirror our results at the level of CREB Ser133 phosphorylation and strongly suggest that the genetic lesion of MSK results in the selective impairment of CREB activation by neurotrophins in CNS neurons.
Figure 6.

Neurotrophin-induced expression of CREB-regulated genes is impaired in MSK-deficient cortical neurons. A, Mouse cortical neurons were transfected with fos-luciferase, Mkp1-luciferase, or Nurr1-luciferase, and a 10-fold excess of empty vector or ACREB DNA. The neurons were treated with vehicle or BDNF for 3 hr and assayed for luciferase activity. The data were normalized for CAG-promoter driven β-galactosidase activity. The activity of the CAG promoter is not affected by expression of ACREB. B, Cortical neurons from WT and MSK1 –/– mice were transfected with GalCREB and Gal-luciferase DNAs. The cells were stimulated with forskolin or BDNF for 3 hr and assayed for luciferase activity as in A. C–E, Mouse cortical neurons from WT or MSK1/2 knock-outs were cultured and treated with BDNF, NT3, or forskolin for the indicated times (in minutes). cDNA was prepared, and real-time quantitative PCR was used to determine mRNA levels for c-fos, Mkp1, and Nurr1. The data were normalized to 18s RNA levels also determined by real-time PCR. Error bars denote SEM, and asterisks denote p < 0.01 (n = 4–10).
Discussion
Although neurotrophins are potent activators of CREB-dependent gene expression, the identity of the signaling pathways that mediate neurotrophin-induced CREB phosphorylation and transcriptional activation has remained elusive. In this study, we made use of pharmacological inhibitors, inhibitory mutants, siRNAs, and mouse knock-outs to examine the role of potential CREB kinases in neurotrophin-mediated CREB phosphorylation. Multiple kinases have been proposed to mediate CREB Ser133 phosphorylation in response to neurotrophins, including Akt, MAPKAP-K2, CaMKIV, RSKs, and MSKs (Lonze et al., 2002). Of these, MAPKAP-K2 is the least likely to be an in vivo CREB kinase because studies using small molecule inhibitors and knock-out mice have not supported the original reports that implicated MAPKAP-K2 (Deak et al., 1998; Rolli et al., 1999). Consistent with this, in cortical neurons we find that an inhibitor of p38 MAPK, the upstream activator of MAPKAP-K2, was not able to attenuate BDNF-induced CREB phosphorylation.
CaMKIV has been proposed to play a major role in neurotrophin-mediated CREB phosphorylation (Finkbeiner et al., 1997; Minichiello et al., 2002). However, we found that BDNF did not trigger measurable increases in intracellular Ca2+ in cortical neurons, a result that is consistent with several other reports (Gaiddon et al., 1996; Pizzorusso et al., 2000). Furthermore, we show that inhibitors of CaMKs did not significantly reduce either BDNF-induced CREB phosphorylation or CREB-dependent transcription.
Although previous studies suggested that ERK1/2 signaling does not play a major role in neurotrophin-induced CREB phosphorylation (Finkbeiner et al., 1997; Minichiello et al., 2002), we found that inhibition of ERK1/2 activation blocked neurotrophin-stimulated CREB phosphorylation and CREB-dependent gene expression. One possible reason that our results differ is that we used PD 184352, a novel inhibitor that completely attenuated activation of ERK1/2 in response to neurotrophins, whereas in previous studies ERK1/2 activation was only partially attenuated.
CREB cannot be phosphorylated by ERK1/2, directly indicating that an ERK-activated kinase mediates Ser133 phosphorylation. The RSK family of ERK-activated kinases is widely believed to be major CREB Ser133 kinases. Three main pieces of evidence promote this idea. First, RSK2 was found to be the major CREB kinase in a biochemical purification of NGF-stimulated PC12 cell extracts (Xing et al., 1996). Second, overexpression of RSKs promoted CREB phosphorylation in several cell types (Xing et al., 1996, 1998). Finally, cell lines from Coffin Lowry patients deficient for RSK2 were reported to have impaired EGF-induced CREB phosphorylation (De Cesare et al., 1998).
The initial biochemical characterization of RSK2 as a CREB kinase was not supported by subsequent studies that found that the neurotrophin-regulated CREB kinase was distinct from RSK2 (Deak et al., 1998; Swanson et al., 1999). Although expression of kinase-dead RSK2 has been reported to attenuate the activation of CREB in a number of systems, we found that expression of this RSK2 mutant can also attenuate the activation of MSK1 (another ERK target which had not been identified at the time of the original study). Recent work showing that growth factor-induced CREB phosphorylation was normal in cells derived from RSK2 knock-outs and Coffin Lowry patients reinforces the idea that RSK2 is not a physiological CREB kinase (Bruning et al., 2000; Dufresne et al., 2001; Wiggin et al., 2002; Soloaga et al., 2003). To determine if RSK is a physiological CREB kinase in CNS neurons, we used RNA interference to markedly reduce the expression of RSK1 and RSK2 in cortical neurons. Although phosphorylation of the RSK substrate LKB1 was blocked by duplex siRNA expression, neurotrophin-induced CREB phosphorylation and transcription were not significantly affected. These experiments argue that RSK1 and RSK2 do not play a major role in neurotrophin-mediated CREB phosphorylation.
Because the ERK- and p38 MAPK-activated kinases, MSK1 and MSK2, have been shown to mediate stress-induced CREB phosphorylation in embryonic fibroblasts (Wiggin et al., 2002), we examined the role of these kinases in the activation of CREB. Although MSKs are homologous with p70 S6 kinases and RSKs, they have an entirely different activation mechanism in that they are not phosphorylated by PDK1 and are apparently exclusively nuclear (Deak et al., 1998; Williams et al., 2000). We found that expression of dominant-negative MSK1 inhibited BDNF-induced CREB phosphorylation and CREB-dependent transcription. In addition, we show that in cortical neurons from mice deficient for MSK1 and MSK2, BDNF or NT3 were unable to induce CREB phosphorylation. We also show that MSK1, rather than MSK2, is required for neurotrophin-mediated CREB phosphorylation in cortical neurons. This is in contrast to embryonic fibroblasts in which both MSK1 and MSK2 contribute to CREB phosphorylation by stress signals and TPA (Wiggin et al., 2002). Taken together, the above data provides compelling evidence that MSK1 is the major neurotrophin-induced CREB kinase in CNS neurons.
Although the deficit in CREB phosphorylation in MSK knock-out mice was striking, CREB has been proposed to play a cooperative role in neurotrophin-induced transcription of immediate early genes (Bonni et al., 1995; Lonze et al., 2002). Consequently, it is not clear to what extent a deficit in CREB phosphorylation correlates with a deficit in transcriptional activity. We examined the expression of the CREB-regulated genes, c-fos, Mkp-1, and Nurr1, and found that deletion of MSK1 and MSK2 attenuated neurotrophin-induced transcription in a manner consistent with the dependency of the gene on the CREB pathway. Although previous work has shown that cells deficient for MSK1 and MSK2 have deficits in histone H3 and HMG-14 phosphorylation, these impairments did not correlate with deficits in gene expression (Soloaga et al., 2003). Consistent with this, neurotrophin-stimulated transcription of genes that are not dependent on the CREB pathway was not compromised in MSK-deficient neurons. Thus, our results demonstrate that MSK1 is the major, if not the only, neurotrophin-stimulated CREB kinase in CNS neurons and suggest that MSK1 could mediate CREB phosphorylation in response to other stimuli that activate the ERK1/2 cascade.
Neurotrophins are potent regulators of neuronal survival in both the CNS and the PNS, and recent work suggests that neurotrophins might regulate survival by activating CREB-dependent gene expression (Bonni et al., 1999; Riccio et al., 1999). Nevertheless, CREB also regulates neuronal survival independent of trophic factor stimulation (Herzig et al., 2000; Lonze et al., 2002). Thus, it is unclear whether neurotrophin-mediated survival depends on CREB-regulated gene expression, and this question awaits further analysis of the role that specific CREB-regulated genes play in neuronal survival. In addition to their effects on survival, neurotrophins have been proposed to facilitate neuronal plasticity by triggering programs of de novo gene expression (Schuman, 1999). The finding that MSK1 is the major neurotrophin-activated CREB kinase suggests that MSK1 may play a role in neurotrophin-regulated survival and synaptic plasticity in the CNS. Further study of MSK knock-out mice and MSK-regulated genes should help clarify the role of MSK in these processes.
Footnotes
This work was supported by National Institutes of Health Grant MH64951 to S.I. and by United Kingdom Medical Research Council, Astra-Zeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck and Company, Merck KGaA, and Pfizer grants to J.S.C.A.
Correspondence should be addressed to Soren Impey, Oregon Health Sciences University, The Vollum Institute, 3181 Southwest Sam Jackson Road, Portland, OR 97239. E-mail: impeys@ohsu.edu.
DOI:10.1523/JNEUROSCI.5227-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244324-09$15.00/0
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18: 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JS, Cohen P (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett 482: 44–48. [DOI] [PubMed] [Google Scholar]
- Bonni A, Ginty DD, Dudek H, Greenberg ME (1995) Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci 6: 168–183. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358–1362. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Gillette JA, Zhao Y, Bjorbaeck C, Kotzka J, Knebel B, Avci H, Hanstein B, Lingohr P, Moller DE, Krone W, Kahn CR, Muller-Wieland D (2000) Ribosomal subunit kinase-2 is required for growth factor-stimulated transcription of the c-Fos gene. Proc Natl Acad Sci USA 97: 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364: 229–233. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare D, Jacquot S, Hanauer A, Sassone-Corsi P (1998) Rsk-2 activity is necessary for epidermal growth factor-induced phosphorylation of CREB protein and transcription of c-fos gene. Proc Natl Acad Sci USA 95: 12202–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17: 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Montminy M (1998) CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273: 32377–32379. [DOI] [PubMed] [Google Scholar]
- Dufresne SD, Bjorbaek C, El-Haschimi K, Zhao Y, Aschenbach WG, Moller DE, Goodyear LJ (2001) Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knock-out mice. Mol Cell Biol 21: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031–1047. [DOI] [PubMed] [Google Scholar]
- Gaiddon C, Loeffler JP, Larmet Y (1996) Brain-derived neurotrophic factor stimulates AP-1 and cyclic AMP-responsive element dependent transcriptional activity in central nervous system neurons. J Neurochem 66: 2279–2286. [DOI] [PubMed] [Google Scholar]
- Herzig RP, Scacco S, Scarpulla RC (2000) Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J Biol Chem 275: 13134–13141. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH (2002) Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34: 235–244. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin M, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD (2002) Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34: 371–385. [DOI] [PubMed] [Google Scholar]
- Mayr BM, Canettieri G, Montminy MR (2001) Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci USA 98: 10936–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merienne K, Pannetier S, Harel-Bellan A, Sassone-Corsi P (2001) Mitogen-regulated RSK2-CBP interaction controls their kinase and acetylase activities. Mol Cell Biol 21: 7089–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M (2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36: 121–137. [DOI] [PubMed] [Google Scholar]
- Mody N, Leitch J, Armstrong C, Dixon J, Cohen P (2001) Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett 502: 21–24. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Ratto GM, Putignano E, Maffei L (2000) Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. J Neurosci 20: 2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser S, Impey S, Trinh K, Xia Z, Storm DR (2000) SRF-dependent gene expression is required for PI3-kinase-regulated cell proliferation. EMBO J 19: 4955–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275: 10761–10766. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358–2361. [DOI] [PubMed] [Google Scholar]
- Rolli M, Kotlyarov A, Sakamoto KM, Gaestel M, Neininger A (1999) Stress-induced stimulation of early growth response gene-1 by p38/stress-activated protein kinase 2 is mediated by a cAMP-responsive promoter element in a MAPKAP kinase 2-independent manner. J Biol Chem 274: 19559–19564. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR (2001) Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J Biol Chem 276: 19469–19482. [DOI] [PubMed] [Google Scholar]
- Schuck S, Soloaga A, Schratt G, Arthur JS, Nordheim A (2003) The kinase MSK1 is required for induction of c-fos by lysophosphatidic acid in mouse embryonic stem cells. BMC Mol Biol 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman EM (1999) Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol 9: 105–109. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 5: 810–816. [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS (2003) MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 22: 2788–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KD, Taylor LK, Haung L, Burlingame AL, Landreth GE (1999) Transcription factor phosphorylation by pp90(rsk2). Identification of Fos kinase and NGFI-B kinase I as pp90(rsk2). J Biol Chem 274: 3385–3395. [DOI] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA (2001) Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 4: 981–988. [DOI] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS (2002) MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol 22: 2871–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10: 439–448. [DOI] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273: 959–963. [DOI] [PubMed] [Google Scholar]
- Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 18: 1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL (2002) RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA 99: 6047–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]