

Abstract
The last 40 years have witnessed how p53 rose from a viral binding protein to a central factor in both stress responses and tumor suppression. The exquisite regulation of p53 functions is of vital importance for cell fate decisions. Among the multiple layers of mechanisms controlling p53 function, posttranslational modifications (PTMs) represent an efficient and precise way. Major p53 PTMs include phosphorylation, ubiquitination, acetylation, and methylation. Meanwhile, other PTMs like sumoylation, neddylation, O-GlcNAcylation, adenosine diphosphate (ADP)-ribosylation, hydroxylation, and β-hydroxybutyrylation are also shown to play various roles in p53 regulation. By independent action or interaction, PTMs affect p53 stability, conformation, localization, and binding partners. Deregulation of the PTM-related pathway is among the major causes of p53-associated developmental disorders or diseases, especially in cancers. This review focuses on the roles of different p53 modification types and shows how these modifications are orchestrated to produce various outcomes by modulating p53 activities or targeted to treat different diseases caused by p53 dysregulation.
Keywords: p53, acetylation, deacetylation, phosphorylation, ubiquitination, methylation, transcriptional regulation
Introduction
When referring to tumor suppressors, p53 is an absolutely non-negligible member. Since its discovery in 1979 (DeLeo et al., 1979; Lane and Crawford, 1979; Linzer and Levine, 1979), 40 years have passed, and now p53 has been accepted as the most important tumor suppressor. The significant effect of p53 on tumors has been demonstrated by numerous mouse experiments in which p53 loss of function predisposes cells to permanent damage and neoplastic transformation (Donehower et al., 1992; Jacks et al., 1994; Olive et al., 2004). This is refueled by the clinical data that p53 is mutated in ~ 50% of human tumors, which rank first in all the oncogenes or tumor suppressor genes (Bykov et al., 2018). Even in the p53 wild-type patients, a large portion of them bear a dysfunctional p53 pathway due to various causes (Yue et al., 2017).
p53 exerts its major functions as a homotetrameric transcriptional factor with a multidomain structure. There are six major protein domains in p53, namely two intrinsically disordered N-terminal transactivation domains (TADs), a proline-rich domain (PRD), a central deoxyribonucleic acid (DNA)-binding domain (DBD) followed by a tetramerization domain (TD), and an intrinsically disordered C-terminal regulatory domain (CTD) (Joerger and Fersht, 2008, 2016). By binding the p53-responsive elements located at target genes’ promoters or enhancers, p53 can activate expression of multiple genes. Originally, p53 was called ‘the guardian of the genome’ (Lane, 1992) for it was found to be activated in response to various types of DNA damages. Later research identified that p53 functions as a hub to handle a broad range of cellular stress, both endogenous and exogenous. Besides DNA damage, p53 can respond to multiple upstream stress signals like oncogene activation, telomere erosion, ribosomal stress, and hypoxia (Horn and Vousden, 2007). Once activated, p53 can regulate lots of cellular processes like cell cycle arrest, DNA repair, apoptosis, ferroptosis, senescence, or autophagy to promote cell survival or limit the malignant transformation of cell. Besides its role in cancer suppression, p53 can also participate in the modulation of cell metabolism, pluripotency, epigenetic states, and aging (Kastenhuber and Lowe, 2017). Moreover, increasing data support transcription-independent roles of p53 (Speidel, 2010; Comel et al., 2014). Functions of p53 are far beyond just dealing with DNA damages. Given these facts, p53 should be regarded not as a simple ‘guardian of the genome’, but as an all-powerful ‘guardian of the cell’.
Since p53 has so many vital roles in cell life, its functional regulation is obviously critical. There exist many layers of mechanisms adjusting p53 functions. For example, p53 can be regulated at the genetic level like mutation (Muller and Vousden, 2014) or single-nucleotide polymorphism (Grochola et al., 2010), transcriptional level like epigenetic inhibition of p53 transcription (Saldana-Meyer and Recillas-Targa, 2011), messenger RNA level like alternative splicing (Vieler and Sanyal, 2018), and protein level like protein folding (Walerych et al., 2004) and localization (Liang and Clarke, 2001). Among these mechanisms, p53 posttranslational modifications (PTMs) represent the most widespread and effective type. The multimodular structure of p53 makes it a perfect platform to undergo a multitude of covalent modifications, including phosphorylation, ubiquitination, acetylation, methylation, sumoylation, neddylation, O-GlcNAcylation, ADP-ribosylation, hydroxylation, and β-hydroxybutyrylation (Figure 1).
Figure 1.
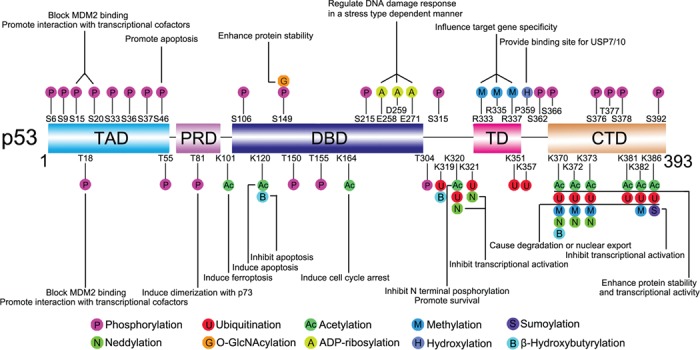
Overview of p53 PTMs. The major sites for p53 modifications (phosphorylation, ubiquitination, sumoylation, neddylation, acetylation, methylation, O-GlcNAcylation, ADP-ribosylation, hydroxylation, and β-hydroxybutyrylation) are plotted. Different colors are used to differentiate distinct modification types. Representative functions of some modifications are indicated. The figure is mainly revised from Dai and Gu (2010) and Gu and Zhu (2012) and not drawn to scale.
These modification types have some common features. (i) Multiple sites: each modification type can occur on many different amino acids; for some amino acids, they can be modified by different chemical groups. (ii) Multiple functions: the functions of the modifications are site-, type-, and context-dependent. The same modification at different sites may have disparate roles; however, different modifications can exert similar functions. (iii) Reversibility: for each modification, there are also one or more corresponding de-modification enzymes. (iv) Widespread crosstalk: modifications can influence the effects of modifications at other sites. The basic working mechanisms of these modifications include affecting p53 stability and localization, causing protein conformational changes, providing interacting partner docking motifs, and altering the local electrostatic forces (Figure 2) (Gu and Zhu, 2012). These features and modes will be reiterated in the sections below.
Figure 2.
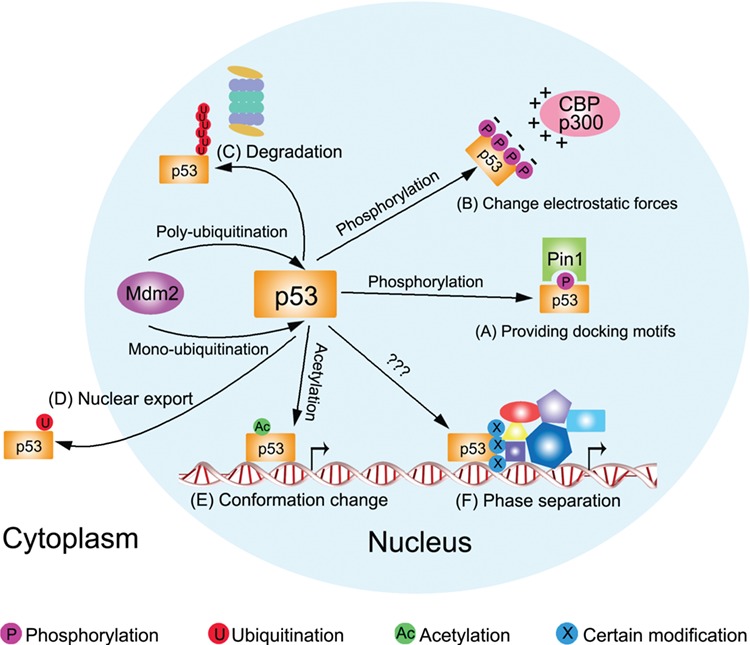
PTMs can regulate p53 in different modes. (A) Phosphorylation at S33, T81, and S315 of p53 provides docking motif for Pin1. (B) Phosphorylations at p53 N-terminal mask it with negative electrostatic forces to facilitate the binding of CBP/p300 with positive electrostatic forces. (C and D) Mdm2 polyubiquitinates p53 for proteasomal degradation whereas monoubiquitinates it for nuclear export. (E) Acetylation at K120 by Tip60 may cause p53 conformational change, making it bind to specific target gene promoter. (F) Certain p53 modifications may help establish phase separation with other regulators.
This review will introduce some typical examples of each modification type. Then the crosstalk between modifications will be emphasized. Additionally, the therapeutic potential of targeting p53 modification pathways will also be discussed. What should be mentioned is that there have been other excellent reviews written previously on similar topics (Bode and Dong, 2004; Meek and Anderson, 2009; Gu and Zhu, 2012; Taira and Yoshida, 2012; Nguyen et al., 2014; Kruiswijk et al., 2015; Meek, 2015; Joerger and Fersht, 2016; Kastenhuber and Lowe, 2017; Hafner et al., 2019). This review includes recent findings.
Phosphorylation
On the p53 protein, there are many serine (S)/threonine (T) sites that can be phosphorylated. The phosphorylation sites span across the whole protein, with an obvious enrichment in the N-terminus. Phosphorylation belongs to the earliest modification type identified on p53. As early as 1992, Hupp et al. (1992) reported that casein kinase II (CK2) can phosphorylate p53 at a C-terminal site to promote its DNA binding.
Some sites (like T55, S376, and S378) are constitutively phosphorylated in unstressed cells (Waterman et al., 1998; Li et al., 2004a, 2007; Wu et al., 2014). Under normal cellular growth conditions, TATA box binding protein-associated factor 1 (TAF1) phosphorylates p53 at T55, promoting degradation of p53 via the proteasome (Li et al., 2004a; Wu et al., 2014). In response to DNA damage, the B56γ subunit of protein phosphatase 2A (PP2A) can dephosphorylate T55, which improves p53 stability and promotes cell cycle arrest. The phosphate group at S376 can also be removed under ionizing radiation (IR) stress (Waterman et al., 1998). This dephosphorylation will create a consensus binding site for 14-3-3, leading to increased DNA binding affinity of p53; conversely, phosphorylation at S33, T81, and S315 of p53 provides docking motif for Pin1 (Figure 2A) (Zacchi et al., 2002; Zheng et al., 2002). Phosphorylation of p53 at S378 by protein kinase C abolishes its reactivity to a carboxyl-terminal antibody PAb421 but activates its DNA binding activity, which can be reversed by protein phosphatases PP1 and PP2A (Takenaka et al., 1995). This provides a model to regulate the transition of p53 from a latent state (PAb421-reactive) to an active (PAb421-negative) form.
Most phosphorylation sites (especially the N-terminal ones) are modified upon meeting cellular stresses to initiate p53 reaction. Once cells are exposed to various types of DNA damage, Ser-Thr kinase VRK1 can phosphorylate p53 at T18 (Teufel et al., 2009). In addition, S15 (mouse S18) and S20 (mouse S23) can be phosphorylated by ataxia–telangiectasia mutated kinase and checkpoint kinase 1/2 (Chk1/2), respectively (Shieh et al., 1997; Shieh et al., 2000). Phosphorylation at these sites protect p53 from its major negative regulator mouse double minute 2 homolog (Mdm2), thus improving its stability and function. Previously, this mechanism was accepted as the first step for p53 activation. However, some in vivo data do not fully support this; for example, Ser18Ala (S18A) or Ser23Ala (S23A) point mutant knockin mice both showed subtle changes on p53 function, unlike the in vitro data (Wu et al., 2002; Chao et al., 2003; MacPherson et al., 2004; Sluss et al., 2004). Although the S18/23A double mutant mice manifest more severe defects on both p53 protein and function, these defects are limited to certain tissues and are mostly limited for DNA damage-induced apoptosis (Chao et al., 2006a). Moreover, in a study by Blattner et al. (1999), the researchers ruled out a role of phosphorylation of some amino acids (S6, S9, S15, S33, S315, S392, and T18 and even several double-site combinations) in the stabilization of p53 after UV and γ irradiation or actinomycin C treatment. These facts, together with some other studies (like the repressive roles of Mdm2/Mdmx on p53), suggest a novel model named ‘antirepression’ for p53 activation, which is highlighted in Kruse and Gu (2009a).
If the DNA damage stress is strong enough, S46 at p53 will be additionally phosphorylated to induce p53-mediated apoptosis. Some pro-apoptotic genes, such as p53-regulated apoptosis-inducing protein 1 (p53AIP1) will be induced, but not those for cell cycle arrest (Olsson et al., 2007; Taira et al., 2007). This is an example revealing the role of p53 modification in p53 target selectivity. Interestingly, phosphorylation of exogenous p53 on S46 was severely impaired in HSC-3 cells. A mutant mimicking S46-phosphorylation (p53S46D) enhanced proapoptotic Noxa promoter activity, overcoming the resistance to p53-mediated apoptosis and growth suppression in HSC-3 cells. These results reveal the possibility that acquisition of resistance to the p53 effect can result from phosphorylation defects (Ichwan et al., 2006).
Beyond the enrichment of phosphorylation sites in the TAD domain, Ser and Thr on other domains can also undergo phosphorylation. S106 of p53 was identified as a novel site phosphorylated by Aurora-A, which inhibited the interaction between p53 and MDM2 (Hsueh et al., 2013). IKKβ phosphorylates p53 at S392 to facilitate its activation, leading to the adaptation of cancer cell to glutamine deprivation (Ishak Gabra et al., 2018). S392 phosphorylation also regulates p53 mitochondrial translocation and transcription-independent apoptosis (Castrogiovanni et al., 2018). Jab1 was reported to stimulate phosphorylation of p53 at T155 to mediate its nuclear export (Lee et al., 2017a). In a recent study, researchers found that downregulation of LATS kinases reduces p53 phosphorylation at S15 and S315 and shifts p53’s conformation and transcriptional output toward a form resembling cancer-associated p53 mutants. This made p53 function as an oncogene to promote migration (Furth et al., 2015). In hepatocellular carcinoma (HCC), overexpression of PAK4 phosphorylated p53 at S215, which not only attenuated its transcriptional transactivation activity but also suppressed the ability of p53 to repress HCC cell invasion (Xu et al., 2016). By computational analysis and experimental validation, Choi et al. (2018) identified NEK2 as a novel kinase that phosphorylates p53 at S315, which represses p53 and promotes cell division.
Phosphorylation of p53 can also create p53 binding protein recognition site. T81 on the p53 PRD domain can be phosphorylated by c-Jun N-terminal kinase (Wolf et al., 2018). T81-phosphorylated p53 gains the ability to dimerize with its homologue p73 to determine cell fate. T18 phosphorylation of p53 specially recruits Pellino1 to the DNA damage site (Dai et al., 2019). Recently, Liao et al., (2017) found that CDK4 can phosphorylate a p53 mutant site R249S . This modification enhances p53 nuclear localization and endows it binding with PIN1. As a result, the p53 mutant interacts with c-Myc and enhances c-Myc-dependent rDNA transcription. IKK2 phosphorylates p53 at S362 and S366 (Xia et al., 2009). E3 ubiquitin ligase β-TrCP1 recognizes these modifications to promote p53 ubiquitination and degradation. Similarly, UBE4B can target S15 and S392 phosphorylated p53 for degradation (Du et al., 2016).
After the damage is relieved, p53 response needs to be terminated, for sustained p53 activation will cause hurt to the cell and is not suitable for cells to resume normal functions. Since phosphorylation plays critical roles in adjusting p53 activity, it is not surprising that dephosphorylation of p53 is very important in reverting p53 activation. All three main types of Ser-Thr phosphatases (PP family, PP type 2 family, and PPM family) regulate the p53 dephosphorylation process (Lazo, 2017). Protein serine/threonine phosphatase 1 (PP1) promotes cell survival by removing the phosphate groups at S15 and S37 of p53 (Li et al., 2006). This causes alteration of p53 activity and downregulation of downstream apoptosis-associated gene expression. The GAS41–PP2Cβ complex was found to be specially required for dephosphorylation of S366 on p53, which increases cell survival upon genotoxic DNA damage (Park et al., 2011). PPM1D (also known as Wip1) is a transcriptional target of p53 (Fiscella et al., 1997) and can reversely inactivate p53 by dephosphorylating it at S15, S33, and S46 (Takekawa et al., 2000; Lu et al., 2005). Once p53 is activated, it induces PPM1D expression. The de novo PPM1D gene expression leaves a time window for p53 to exert its protective roles. Then PPM1D, in conjugation with other phosphatases, dephosphorylates p53 and makes cells restart cell cycle (Shaltiel et al., 2014). Polo-like kinase 1 can also regulate cell cycle re-entry by activating Cdc25C and later will dephosphorylate p53 at S15 to abolish its cell cycle arrest effect (Chen et al., 2006). Moreover, an unknown phosphatase dephosphorylates p53 at S20 to decrease p53 mitochondrial localization during late apoptosis, leading to the attenuation of mitochondrial p53-mediated cell apoptosis (Castrogiovanni et al., 2015).
Taken together, phosphorylation and dephosphorylation are of vital importance in p53 reaction initiation, execution, and termination. When phosphorylation happens (before or after p53 activation) and what effects phosphorylation has on p53 (activation or repression) are highly variable across different conditions.
Ubiquitination, sumoylation, and neddylation
Ubiquitin is a 76-amino acid small protein with a molecular mass of ∼8.5 kDa. After a hierarchical cascade of enzymatic reactions, which are catalyzed by three enzymes—an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin-ligating enzyme, ubiquitins can be transferred to specific substrates, resulting in monoubiquitin- or polyubiquitin-modified substrates. This process is named ubiquitination (Pickart, 2001). The major role of this modification is to target substrates for proteasomal degradation; meanwhile, ubiquitination can also regulate protein localization, protein activity, and protein–protein interaction (Schnell and Hicke, 2003; Mukhopadhyay and Riezman, 2007; Senft et al., 2018). Ubiquitination plays a vital role in p53 regulation. The first report about p53 ubiquitination came from the Howley lab in 1993 that showed that the oncogenic HPV-16 E6 and E6-AP complex could ubiquitinate p53 (Scheffner et al., 1993).
Mdm2 is the major E3 ubiquitin ligase and negative regulator of p53. Mdm2 can modify p53 at six lysine residues within the CTD (K370, K372, K373, K381, K382, and K386) (Rodriguez et al., 2000). High levels of Mdm2 activity promote p53’s polyubiquitination and nuclear degradation (Figure 2C), whereas low levels induce monoubiquitination and nuclear export of p53 (Figure 2D) (Li et al., 2003). However, in the cytoplasm, p53 can perform transcription-independent roles (Green and Kroemer, 2009). So the monoubiquitination-mediated cytoplasmic p53 accumulation may affect these roles. Pivotal evidence to support the important role of Mdm2 on p53 suppression is that the embryonic lethality in Mdm2-null mice can be rescued by the loss of p53 (Brooks and Gu, 2006; Moyer et al., 2017). Interestingly, Mdm2 itself is a transcriptional target of p53. Thus, p53 and Mdm2 can form a double-negative regulatory loop (Brooks and Gu, 2006; Lu, 2017; Zhou et al., 2017). What should be mentioned here is that Mdm2 can also inhibit p53 transcriptional functions by directly binding with p53 at the target DNA site. The E3 ligase activity lacking homologue of Mdm2–Mdmx (or Mdm4) can dimerize with Mdm2 and strengthen this inhibition (Kruse and Gu, 2009a).
Besides Mdm2, there are also other E3 ligases that can target p53. Tripartite motif 69 (TRIM69) can interact with p53 and induce its ubiquitination (Rong et al., 2019). In the cataractogenesis process, TRIM69 expression is inhibited, leading to p53 activation and cataract formation. Another TRIM family member TRIM59 is upregulated in gastric cancer (Zhou et al., 2014). TRIM59 interacts with p53 and induces its ubiquitination and degradation, thus promoting gastric carcinogenesis. In human hepatocellular and colorectal carcinomas, RING1 promotes cancer cell proliferation and survival by directly ubiquitinating p53 to cause its degradation (Shen et al., 2018). Likely, UBE2T, RBCK1, and SMYD3 can ubiquitinate p53 for degradation to support HCC, renal cell carcinoma, and ovarian cancer development, respectively (Liu et al., 2017a; Yu et al., 2019; Zhang et al., 2019). In addition, Arf-BP1, COP1, CHIP, and Pirh2 all ubiquitinate p53 and target it for proteasomal degradation (Leng et al., 2003; Dornan et al., 2004; Chen et al., 2005; Esser et al., 2005). Unlike these E3 ubiquitin ligases, WWP1 and MSL2 ubiquitinate p53 to promote its nuclear export, without proteasomal degradation (Laine and Ronai, 2007; Kruse and Gu, 2009b). Interestingly, an atypical E3 ligase E4F1 ubiquitinates p53 to promote p53-dependent transcriptional program specifically involved in cell cycle arrest, but not apoptosis; similarly, this ubiquitination pattern does not affect its degradation or cellular localization (Le Cam et al., 2006).
Ubiquitin on p53 can also be removed by deubiquitinases (DUBs). Herpesvirus-associated ubiquitin-specific protease (HAUSP, also caller USP7) is an important p53 regulator, which can deubiquitinate p53 (Li et al., 2002a). HAUSP can stabilize p53 both in vitro (even in the presence of excess Mdm2) and in vivo. Interestingly, partial reduction of endogenous HAUSP levels indeed destabilizes endogenous p53; however, total loss of HAUSP stabilizes and activates p53. Further studies reveal that HAUSP can also deubiquitinate Mdm2. Mdm2 will become quite unstable due to its autoubiquitination when HAUSP is inhibited, leading to the stabilization of p53 (Li et al., 2004b). The physiological functions of HAUSP were discovered through generation of HAUSP knockout mice (Kon et al., 2010). These mice die during early embryonic development between E6.5 and E7.5, partly due to p53 activation. Although deletion of p53 did not completely rescue the embryonic lethality caused by the HAUSP knockout, embryonic development was extended in HAUSP and p53 double-knockout embryos (Kon et al., 2010).
Besides HAUSP, a list of other DUBs (including USP3, USP11, USP15, USP49, OTUD1, OTUD5, and ataxin-3) can also deubiquitinate p53 to influence its stability (Luo et al., 2013; Ke et al., 2014; Liu et al., 2016a, 2017b; Fu et al., 2017; Piao et al., 2017; Tu et al., 2018). In U2OS and HEK293 cells, TGF-β promotes the translation of USP15. Upregulated USP15 deubiquitinates p53 to stabilize it (Liu et al., 2017b). In response to DNA damage, p53 promotes the expression of USP49 (Tu et al., 2018). Interestingly, USP49 can in turn stabilize p53 through deubiquitination. Thus, p53 and USP49 form a positive feedback loop upon DNA damage. Unlike these DUBs, which deubiquitinate and stabilize p53 primarily in the nucleus, another deubiquitination enzyme USP10 mainly functions in the cytoplasm. USP10 deubiquitinates p53 to reverse the Mdm2-induced p53 nuclear export and degradation (Yuan et al., 2010).
There are also two other ubiquitin-like proteins, small ubiquitin-like modifier (SUMO) and neural precursor cell expressed developmentally downregulated protein 8 (NEDD8), that can be conjugated to the p53 lysines via a similar mechanism as ubiquitination. These two processes are named sumoylation and neddylation, respectively (Geiss-Friedlander and Melchior, 2007; Carter and Vousden, 2008; Rabut and Peter, 2008). Unlike ubiquitination, sumoylation and neddylation seem unable to affect p53 stability or localization. Protein inhibitor of activated Stat (PIAS) family and TOP1 binding arginine/serine-rich protein (Topors) can sumoylate p53 at K386 to prevent p300 access to the C-terminal lysines. This will inhibit p53 transcriptional activation function (Bischof et al., 2006; Stehmeier and Muller, 2009; Wu and Chiang, 2009). Neddylation of p53 by Mdm2 (at K370, K372, and K373) or FBXO11 (at K320 and K321) inhibits p53 transcriptional activation activity (Xirodimas et al., 2004; Abida et al., 2007).
Acetylation
Histone lysine residue acetylation is a critical epigenetic modification to influence histone structure and gene expression (Verdone et al., 2006). The first example of protein acetylation is reported on histones in 1964 (Allfrey et al., 1964). In the following 30 years, lysine acetylation was also discovered in non-histone proteins, like HMG-1 (Sterner et al., 1979) and tubulin (L'Hernault and Rosenbaum, 1983). However, this field (non-histone protein acetylation) received little attention until the discovery of p53 acetylation in 1997 (Gu et al., 1997; Lill et al., 1997; Verdin and Ott, 2015; Narita et al., 2019).
Co-activators CREB-binding protein (CBP) and its paralog E1A-binding protein (p300) play vital roles in gene transcriptional regulation via various mechanisms, among which is acetylating histones to remold the chromatin conformation (Wang et al., 2013). Initially, we found that CBP/p300 could bind with p53 to promote its transcriptional activity (Gu et al., 1997). Given the inner histone acetyltransferase (HAT) activity of CBP/p300, we speculated that it might also acetylate p53, which was not deemed as a classical acetylation substrate at that time. Strikingly, we demonstrated that CBP/p300 could truly acetylate p53 at several C-terminal lysines (K370, K372, K373, K381, K382, and K386) (Gu and Roeder, 1997). C-terminal acetylation will boost p53 binding to its target gene locus to activate downstream pathways, like cell cycle arrest, senescence, or apoptosis (Reed and Quelle, 2014). Mechanically, there are several explanations for this activation. First, all these six acetylated sites can also be ubiquitinated. Acetylation at these sites may prevent p53 from being ubiquitinated, thus increasing its stability. Second, acetylation of p53 abrogates Mdm2-mediated repression by blocking the recruitment of Mdm2 to p53-responsive promoters (Tang et al., 2008). Third, acetylation of p53 CTD abolishes the binding of the acidic domain of SET, which can inhibit p53 transcriptional activity in unstressed cells (Wang et al., 2016a). Previously, protein acetylation was often recognized to endow proteins with the ability to be bound by some readers (like the bromodomain-containing proteins). However, whether there were some readers that could bind non-acetylated p53 (and if so, what were the biological significances of this binding) was unknown until the discovery of p53 suppression by SET. This finding is of vital importance because it not only identified a new regulator of p53 function, but also revealed a widespread regulatory mode. The same study also identified some other proteins (like VPRBP, DAXX, and PELP1) that may act similarly to SET to regulate p53, which awaits to be demonstrated. Moreover, this mode can also expand to other proteins that can undergo acetylation. However, there are also different effects of p53 acetylation in specific cell types. For example, unlike in many cancer cells, acetylation at K381 and K382 prevents p53 from activating the proapoptotic PUMA gene, decreasing its ability to induce apoptosis in mouse primary cortical neurons (Brochier et al., 2013).
Another extensively studied p53 acetylation site is K120 on the DBD, which is catalyzed by three members (Tip60, MOF, and MOZ) of the MYST HAT family. Tip60 acetylates p53 at K120 to selectively induce the expression of proapoptotic genes (like PUMA and Bax), but not cell cycle arrest genes (like p21). Mechanically, K120 acetylation by Tip60 may change the conformation of the DBD, causing target promoter selectivity (Figure 2E) (Sykes et al., 2006; Tang et al., 2006). K120 is often mutated in cancers. The tumor-derived mutant p53 (K120R) is defective for Tip60-mediated acetylation, thus abrogating p53-dependent activation of apoptosis but having no significant effect on cell growth arrest. Tip60 can also regulate p53 in an acetylation-independent way. For example, Tip60 can block Mdm2-mediated p53 neddylation to improve its transcriptional activity (Dohmesen et al., 2008). Similar to Tip60, MOF also acetylates K120 on p53 to induce apoptosis (Li et al., 2009). However, MOZ-mediated K120 acetylation of p53 specifically enhances its antiproliferative activity. Meanwhile, MOZ can also acetylate K382 (Rokudai et al., 2009, 2013). Recently, NAT10 was reported as a novel regulator for p53 activation (Liu et al., 2016b). Interestingly, NAT10 acts to both acetylate p53 at K120 and ubiquitinate MDM2 for degradation, which synergistically stabilize p53. These results reveal the complexity of p53 modifications. How the target gene selectivity is achieved even when different enzymes add the same modification to the same site needs further investigation.
There are two other sites (K164 and K101) on DBD of p53 that can be acetylated. Both of them are modified by CBP/p300 (Tang et al., 2008; Li et al., 2012; Wang et al., 2016b). K164 acetylation is responsible for the induction of cell cycle arrest. In an in vivo experiment, K117 (K120 in human), K161, and K162 (K164 in human) of mouse p53 were mutated to arginine (3KR). This mutation completely abolished the ability of p53 to mediate cell cycle arrest, apoptosis, and senescence in vivo, verifying the ability of K164 acetylation to induce p21 expression (Li et al., 2012). This result also provides novel evidence to show that the acetylation at the C-terminus of p53 is not a necessary requirement for p53 functions (cell cycle arrest, apoptosis, and senescence), but a regulatory unit (like antagonizing ubiquitination or repressing SET binding). On the other hand, K101 acetylation is critical for regulation of SLC7A11-dependent ferroptosis. p53 with K101R mutation will lose its ability to induce the expression of SLC7A11 (Wang et al., 2016b). Taken together, the acetylation status of the three lysines (K101, K120, and K164) on p53 DBD is critical for p53 target selectivity. Besides these amino acids, there may be other sites on the DBD responsible for different p53 functions (like affecting autophagy and metabolism), which need further investigation. Additionally, the underlining mechanism of this selectivity needs to be solved in the future.
In response to DNA damage, p300–CBP associated factor (PCAF) can acetylate p53 at K320 (Liu et al., 1999). This prevents phosphorylation of crucial serines in the N-terminus of p53, allowing activation of specific target genes to promote cell survival, but not apoptosis (Knights et al., 2006). In a K317R knock-in mouse model (K320 in human), increased p53-dependent apoptosis was observed in cells from various tissues, like thymocytes, small intestine, and retina after IR as well as in E1A/Ras-expressing MEFs after doxorubicin treatment (Chao et al., 2006b). These data support a pro-survival role of p53 K320 acetylation.
Acetylated p53 can be deacetylated by various deacetylases. Previously, we identified that histone deacetylase-1 (HDAC1)-containing complex could deacetylate p53 to strongly repress p53-dependent transcriptional activation, cell growth arrest, and apoptosis (Luo et al., 2000). Interestingly, Mdm2 can promote p53 deacetylation by recruiting a complex containing HDAC1 to p53, thus abrogating the obstruction of acetylation on Mdm2-mediated p53 ubiquitination (Ito et al., 2002; Li et al., 2002b). Sirt1 can repress p53 function through deacetylation (Luo et al., 2001; Vaziri et al., 2001). This process can be inhibited by nicotinamide (Vitamin B3) (Luo et al., 2001) or DBC1 (Zhao et al., 2008). Recently, HDAC6 was reported to deacetylate p53 at K381/382. An HDAC6-selective inhibitor A452 blocked HDAC6 nuclear localization, resulting in increased levels of acetylated p53 at K381/382 and its proapoptotic activity (Ryu et al., 2017). Additionally, other histone deacetylases like SIRT3 and HDAC8 that can deacetylate p53 were also reported (Hua et al., 2017; Xiong et al., 2018).
In summary, acetylation clearly plays an important role in directly or indirectly modulating p53 functions. Targeting this pathway may serve as a promising therapeutic method in various p53-related diseases, which will be discussed later.
Methylation
Like acetylation, methylation is also an important epigenetic mark on the lysine of histone tails. Some lysines on p53 can also be methylated. Chuikov et al. (2004) firstly reported p53 lysine methylation by SET7/9 at K372. This modification can stabilize p53 and restrict it in the nucleus. Meanwhile, it is also associated with enhanced transcription of some target genes like p21. However, monomethylation of p53 at K382 by SET8 attenuates p53-regulated transcription of target genes (Shi et al., 2007). SMYD2 can also methylate p53 at K370 to decrease p53 binding to the promoter of its target genes, thus repressing p53-mediated transcription (Huang et al., 2006). Besides lysine methylation, arginine on p53 can also be methylated. Jansson et al. (2008) showed that protein arginine methyltransferase 5 (PRMT5) is responsible for methylating p53 at R333, R335, and R337. This reaction is regulated during the p53 response to DNA damage and affects p53 target gene specificity.
Methylation sites can also recruit special effectors to p53. SET8-mediated methylation of p53 at K382 promotes the interaction between L3MBTL1 and p53 in p53 target promoters (West et al., 2010). This interaction impedes p53 to transactivate its target genes. On the contrary, PHF20 recognizes p53 dimethylated K370 or K382 to stabilize and activate p53 (Cui et al., 2012). Additionally, a senescence-associated E3 ligase SCF (Fbxo22)-KDM4A can target methylated p53 for degradation (Johmura et al., 2016).
However, only one demethylase of p53 has been conclusively defined to date. Huang et al. (2007) identified that histone lysine-specific demethylase LSD1 could demethylate p53 at K370 to abrogate its interaction with the coactivator 53BP1 (p53-binding protein 1), which repressed p53 function.
Overall, methylation is the simplest (in structure) modification type with complicated effects on p53 functions. Whether there are other sites on p53 that can be methylated or other writers, readers, or especially erasers responsible for p53 methylation regulation remains to be elucidated.
Other modifications
Besides the modification types mentioned above, p53 can also undergo other modifications. p53 S149 is O-GlcNAcylated upon DNA damage (Yang et al., 2006). This modification is associated with decreased T155 phosphorylation and stabilizes p53 in an ubiquitination-dependent manner. When DNA damage happens, poly(ADP-ribose) polymerase-1 (PARP-1) mediates the ADP-ribosylation of p53 to respond to the stress. However, this regulation of p53 by PARP1 is damage type-dependent (Valenzuela et al., 2002; Watanabe et al., 2004). In 2018, p53 was found to be hydroxylated at P359 by PHD3 (Rodriguez et al., 2018). P359 hydroxylation forms a binding site for USP7/10, which can deubiquitinate p53 to increase its stability. Recently, Liu et al. (2019) identified a novel β-hydroxybutyrylation (kbhb) of p53 at K120, K319, and K370, which is catalyzed by CBP. p53 kbhb reduces p53 acetylation and expression of p53 downstream genes p21 and PUMA, thus weakening p53-dependent cell growth arrest and apoptosis.
Crosstalk between modifications
As discussed above, there are so many sites in p53 that can undergo myriad types of modifications, leading to the doubt about the relative significance of a single-site modification in p53 function modulation. Actually, in most cases, the role of a single site modification is hardly decisive. There is widespread crosstalk between modifications. Researchers once differentiated these interplays between modifications into sequential crosstalk and spatial crosstalk (Gu and Zhu, 2012). According to different standards, we can also classify them as homogenous modification crosstalk (one modification influences the other one with the same type at a different site) vs. heterogeneous modification crosstalk (crosstalk between different modification types), nearby modification crosstalk (one modification affects another in its local area) vs. distant modification crosstalk (crosstalk between modifications located at a distance), or cooperative modification crosstalk (one modification promotes or enhances the effect of another) vs. antagonistic modification crosstalk (one modification antagonizes the effect of another). Examples of different crosstalk types are depicted below and in Figure 3.
Figure 3.
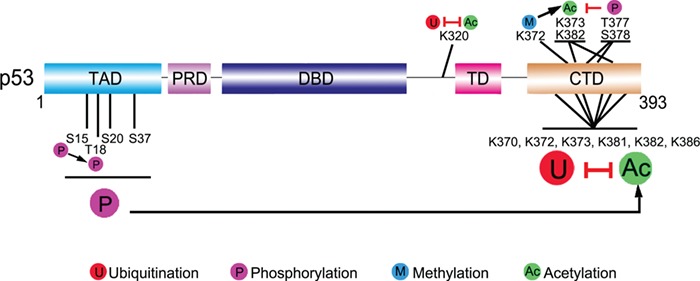
Crosstalk between p53 modifications. There are widespread interplays between p53 modifications. These crosstalks can be divided into different types via diverse standards. For example, homogeneous modification crosstalk (phosphorylation of S15 boosts the latter phosphorylation at T18) vs. heterogeneous modification crosstalk (ubiquitination and acetylation compete for K320 and six C-terminal lysine residues), nearby modification crosstalk (methylation of K372 can promote acetylation of local K373 and K382) vs. distant modification crosstalk (phosphorylation at N-terminus influences the acetylation at C-terminus), or cooperative modification crosstalk (the case that K372 methylation promotes K373 and K382 acetylation can also be categorized as cooperative modification crosstalk) vs. antagonistic modification crosstalk (T377 and S378 phosphorylation inhibits K373 and K382 acetylation). For detailed discussion, see the context. Black arrows indicate positive effects. Red perpendicular bars indicate negative effects.
The simplest case for p53 modification crosstalk is that some amino acids in p53 can be modified by different modification types. This will cause competition between corresponding modifying enzymes to access this site. The six acetylation sites (K370, K372, K373, K381, K382, and K386) at p53 C-terminus can all be ubiquitinated. Thus, acetylation and ubiquitination at those sites can block each other (Nakamura et al., 2000; Ito et al., 2002; Li et al., 2002b). Similarly, E4F1 and PCAF compete at K320 of p53. Ubiquitination of K320 by E4F1 will abrogate acetylation by PCAF at the same site, affecting the downstream transcriptional program specifically involved in cell cycle arrest, but not apoptosis (Le Cam et al., 2006).
Phosphorylation of p53 often performs a cascade behavior, which means that the latter phosphorylations may be dependent on the former ones. In response to DNA damage, T18 is phosphorylated in vivo, which requires the prior S15 phosphorylation (Saito et al., 2003). Different stress signals can induce different phosphorylation cascades. N-terminal phosphorylation of p53 at certain sites (like S15, T18, S20, and S37) or the combination of several phosphorylation sites can promote p53 acetylation through recruiting CBP/p300, likely due to the negative electrostatic forces of phosphate groups (Figure 2B) (Lambert et al., 1998; Kar et al., 2002; Jenkins et al., 2009; Teufel et al., 2009; Lee et al., 2010). After exposing cells to UV light or IR, S33 and S37 of p53 are phosphorylated, which then promote the acetylation of p53 by p300 and PCAF (Sakaguchi et al., 1998). However, T377 and S378 phosphorylations inhibit but not promote p53 acetylation at K373 and K382, without affecting K120 (Ou et al., 2005). Phosphorylation is also influenced by other modifications, such as O-GlcNAcylation and poly(ADP-ribosylation) (Valenzuela et al., 2002; Yang et al., 2006). Methylation of p53 at K372 by Set7/9 is important for the following acetylation of K373 and K382, which leads to the stabilization and activation of p53 (Ivanov et al., 2007). Another study shows that methylation of p53 by Set7/9 is required for Tip60 binding to p53 and subsequent acetylation of p53. Set7/9 mutation fails to methylate p53 K369, thus being unable to induce p53 downstream targets upon DNA damage, and these mutant cells are predisposed to oncogenic transformation (Kurash et al., 2008). However, in vivo studies in other labs challenge the necessity of Set7/9 in p53 activation (Campaner et al., 2011; Lehnertz et al., 2011).
Targeting p53 modification pathway for disease treatment
The p53 pathway is deregulated in various diseases, especially in cancers (Vousden and Lane, 2007). Rescuing the protective function of p53 is a focus in drug discovery. In those patients retaining wild-type p53, the expression, stability, localization, and activity of p53 can also be disrupted due to different mechanisms. As we discussed above, p53 PTMs can influence p53 functions in many aspects, making it an attractive target to recover the p53 functions. Now extensive efforts have been made on targeting the negative modifications of p53 function (Mandinova and Lee, 2011) (Figure 4).
Figure 4.
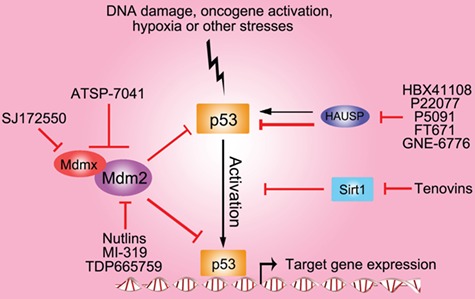
Targeting wild-type p53 modification for disease therapy. Various small-molecular compounds have been developed to target the major enzymes in wild-type p53 modification pathways. These targets and molecules include Mdm2 (Nutlins, TDP665759, and MI-319), Mdmx (SJ172550), Mdm2 and Mdmx (ATSP-7041), HAUSP (P22077, P5049, and HBX41108), and Sirt1 (tenovins). Black arrows indicate positive effects. Red perpendicular bars indicate negative effects.
Mdm2 is the major negative regulator of p53. There have been several small molecules developed to target the Mdm2–p53 interaction, among which are Nutlins. Nutlins are imidazoline derivatives that specially bind MDM2 in the p53-binding pocket and prevent MDM2-mediated p53 degradation, thereby activating the p53 pathway. Nutlin treatment can stabilize and activate p53, improving the effect of p53 to mediate cell cycle arrest, apoptosis, and growth inhibition (Vassilev et al., 2004; Duan et al. 2018). The analog of Nutlin, RG7112, has become the first small-molecule Mdm2 inhibitor in clinical development (Vu et al., 2013). Additionally, other Mdm2 inhibitors like RG7388, MK8242, and AMG232 have entered the clinic (Ding et al., 2013; Sun et al., 2014; Ravandi et al., 2016). MI-319 and TDP665759 are two other types of small-molecule inhibitors of Mdm2, derived from benzodiazepines and spiro-oxindole, respectively (Grasberger et al., 2005; Ding et al., 2006). Both compounds show good antitumor activity in cancer cells. Although Mdmx has no ubiquitin ligase activity, it can dimerize with Mdm2 and strengthen the latter’s function. Thus targeting Mdmx will promote p53 activation. Indeed, the first MDMX inhibitor, SJ172550, has been developed and shows a therapeutic effect (Reed et al., 2010). Moreover, dual inhibitors (like ATSP-7041) targeting both MDM2 and MDMX have also been developed (Chang et al., 2013; Lee et al., 2017b).
Besides inhibiting Mdm2 and Mdmx function on p53, deubiquitinating enzymes that regulate the stability of p53 E3 ligases can also be targeted. For example, our lab has demonstrated that HAUSP inhibition indirectly stabilizes p53 protein levels by destabilizing both MDM2 and MDMX (reviewed in detail by Tavana and Gu, 2017). From these pivotal studies, inhibitors targeting the enzymatic function of HAUSP have been developed (Altun et al., 2011; Chauhan et al., 2012; Chen et al., 2017; Tavana and Gu, 2017). P22077 and P5091, two HAUSP inhibitors discovered by high-throughput screens, were demonstrated to inhibit neuroblastoma or multiple myeloma growth in vivo, respectively (Chauhan et al., 2012; Fan et al., 2013). In 2017, Turnbull et al. (2017) reported two compounds (FT671 and FT827) targeting HAUSP with high potency and a novel binding site on HAUSP. Another group developed two other small-molecule inhibitors GNE-6640 and GNE-6776 for HAUSP, which bound non-covalently with HAUSP to attenuate ubiquitin binding (Kategaya et al., 2017). In a recent study, inhibitors of HAUSP (HBX41108; Colland et al., 2009) and Nutlin-3 were combined (Tavana et al., 2018). Although single treatment with low dose of either agent was unable to induce apoptosis, remarkably, the combination of HAUSP and Mdm2 inhibition can strikingly activate p53 levels and induce apoptosis.
Since acetylation is critical for p53 activity, targeting the deacetylating enzymes may be a good choice to activate p53. In a cell-based screen, researchers discovered a novel compound Tenovins, which inhibits the deacetylating activities of Sirt1 and Sirt2 to activate p53 (Lain et al., 2008). Tenovins can retard growth of tumors derived from a highly aggressive melanoma cell line without significant general toxicity, showing a promising clinical potential.
There have been hundreds of molecules reported to target the p53 modification pathway, among which only limited ones entered the clinical trial stage and even fewer survived into clinical use. One major obstacle in the p53 modification-targeting drug development is the specificity of drugs. There are two dimensions for the specificity. One is that most of the modification enzymes belong to different groups of proteins with a large number of members bearing similar structures (and similar or quite different functions), like the DUB family, SIRT family, and HDAC family. The drug designed for enzyme may also target another family member. For example, HAUSP inhibitor HBX41108 was reported also targeting other USP members (Harrigan et al., 2018). The other aspect for specificity is that most of the modification enzymes have a large list of substrates. For example, inhibiting the HDAC may cause a global epigenetic state and gene expression change (Qin et al., 2017). Therefore, the designs of p53 modification-targeting drugs must take the potential off-target effects into extensive consideration and test. Another question is that these small molecular compounds were developed mainly to activate wild-type p53 activity. In those patients bearing mutant modification sites, like K120 mutation in various cancers or R249S mutation in HCC (Liao et al., 2017), gene therapy (like adenovirus-based or CRISPR-Cas9-based gene therapy) may serve as an ideal choice in the future (Naldini, 2015; Wang et al., 2019; Yin et al., 2019).
Concluding remarks and future directions
The last 40 years’ comprehensive research on p53 exhibits a spectacular scene that a single transcriptional factor can control so many physiological or pathological processes. Meanwhile, the regulator itself is under control of multilayered and complicated regulation, among which PTMs play critical roles. Extensive results about multiple modification sites, modification types, and a myriad of associated enzymes have been shown, which allow for a better understanding about p53 function, mechanism, regulation, and therapeutic application. Promising as this field is, there are some questions calling for attention or solution.
p53 modification is a highly dynamic and context-dependent process. p53 is the hub to orchestrate the multiple responses to various cellular stresses. The final cellular results of p53 activation depend on the types and magnitude of stresses and types of cells. Thus, when studying the p53 modification process in a specific context, the researchers should caution the possible difference between their cases with other studied situations. How exactly do specific p53 modification states help to differentiate different stresses and choose different target genes to activate? The promoter selectivity will remain a central topic in p53 reaction mechanism research in the future. How can the cell characteristics (like the genome structure, epigenetic state, and the endogenous proteome) influence the p53 functions? Crosstalk between modifications expands the functional spectrum of p53 modifications. How is crosstalk precisely regulated? These are still vital and urgent questions to be solved. Novel functions of p53 have been discovered these years, e.g. the role of p53 in stem cell differentiation, autophagy, necrosis, cell epigenetic state regulation, metabolism, and ferroptosis. Are there any other important roles of p53 to be uncovered? In future studies about p53 modifications, these new functions should be considered. What should be specially strengthened is that a large part of the research about p53 modification was performed in vitro. However, these conclusions could not be fully repeated in vivo according to the past research. Future in vitro study results should be validated in vivo.
There are so many proteins (especially modification writers, readers, and erasers) participating in the p53 modification modulation. Are there any other regulators yet to be discovered? How are these proteins regulated in different physiological and pathological processes? Answering these questions not only helps us understand the mechanism of p53 function, but also benefits the clinical application of targeting p53-modifying pathways. At present, there have been some promising small molecular compounds targeting the p53 modification pathway for disease treatment. The complexity of this pathway makes it difficult to target, but also provides many suitable enzymes worth targeting. On the other hand, p53 mutation is quite a common occurrence in many diseases, especially in cancers. Do some mutations disrupt certain normal modifications (like K120 mutation abolishes its acetylation)? Or can some mutations create new modification sites (like the R249S mutation of p53)? Will targeting PTM pathways help regulate the mutant p53 cell stress response? This field is poorly understood and needs more attention. We hope that more candidate drugs targeting both wild-type and mutant p53 modification pathways will be discovered in the future.
The studies about p53 modifications will benefit researches about other transcriptional factors or proteins. The types and mechanisms of the reaction of p53 modifications may extend to other proteins (especially transcriptional factors). Phase separation is an emerging concept to explain a novel mode of protein interaction (Hnisz et al., 2017). Two recent papers revealed that phase separation played an important role in the transcriptional regulation process (Boija et al., 2018; Sabari et al., 2018). Does p53 exert its transcription activation function by phase separation with other proteins? Does p53 modification participate in the regulation of this process? It will be interesting to solve these questions (Figure 2F).
All in all, we are greeting warmly that a new era about p53 research will come in the future. We look forward that in the next decade or 40 years, this powerful guardian will bring us much more surprise and excitement.
Funding
This work was supported by the National Cancer Institute of the National Institutes of Health under Award 5R01CA216884, 5R01CA190477, 5R01CA085533, and 5R01CA193890 to W.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest: none declared.
References
- Abida W.M., Nikolaev A., Zhao W., et al. (2007). FBXO11 promotes the neddylation of p53 and inhibits its transcriptional activity. J. Biol. Chem. 282, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey V.G., Faulkner R., and Mirsky A.E. (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M., Kramer H.B., Willems L.I., et al. (2011). Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 18, 1401–1412. [DOI] [PubMed] [Google Scholar]
- Bischof O., Schwamborn K., Martin N., et al. (2006). The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol. Cell 22, 783–794. [DOI] [PubMed] [Google Scholar]
- Blattner C., Tobiasch E., Litfen M., et al. (1999). DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 18, 1723–1732. [DOI] [PubMed] [Google Scholar]
- Bode A.M., and Dong Z. (2004). Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805. [DOI] [PubMed] [Google Scholar]
- Boija A., Klein I.A., Sabari B.R., et al. (2018). Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C., Dennis G., Rivieccio M.A., et al. (2013). Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 33, 8621–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C.L., and Gu W. (2006). p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykov V.J.N., Eriksson S.E., Bianchi J., et al. (2018). Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102. [DOI] [PubMed] [Google Scholar]
- Campaner S., Spreafico F., Burgold T., et al. (2011). The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol. Cell 43, 681–688. [DOI] [PubMed] [Google Scholar]
- Carter S., and Vousden K.H. (2008). p53-Ubl fusions as models of ubiquitination, sumoylation and neddylation of p53. Cell Cycle 7, 2519–2528. [DOI] [PubMed] [Google Scholar]
- Castrogiovanni C., Vandaudenard M., Waterschoot B., et al. (2015). Decrease of mitochondrial p53 during late apoptosis is linked to its dephosphorylation on serine 20. Cancer Biol. Ther. 16, 1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrogiovanni C., Waterschoot B., De Backer O., et al. (2018). Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 25, 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.S., Graves B., Guerlavais V., et al. (2013). Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA 110, E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C., Hergenhahn M., Kaeser M.D., et al. (2003). Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J. Biol. Chem. 278, 41028–41033. [DOI] [PubMed] [Google Scholar]
- Chao C., Herr D., Chun J., et al. (2006a). Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 25, 2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C., Wu Z., Mazur S.J., et al. (2006b). Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell. Biol. 26, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D., Tian Z., Nicholson B., et al. (2012). A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Song J., Wang J., et al. (2017). Synthesis and biological evaluation of thiazole derivatives as novel USP7 inhibitors. Bioorg. Med. Chem. Lett. 27, 845–849. [DOI] [PubMed] [Google Scholar]
- Chen D., Kon N., Li M., et al. (2005). ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Chen J., Dai G., Wang Y.Q., et al. (2006). Polo-like kinase 1 regulates mitotic arrest after UV irradiation through dephosphorylation of p53 and inducing p53 degradation. FEBS Lett. 580, 3624–3630. [DOI] [PubMed] [Google Scholar]
- Choi B.K., Dayaram T., Parikh N., et al. (2018). Literature-based automated discovery of tumor suppressor p53 phosphorylation and inhibition by NEK2. Proc. Natl Acad. Sci. USA 115, 10666–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S., Kurash J.K., Wilson J.R., et al. (2004). Regulation of p53 activity through lysine methylation. Nature 432, 353–360. [DOI] [PubMed] [Google Scholar]
- Colland F., Formstecher E., Jacq X., et al. (2009). Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 8, 2286–2295. [DOI] [PubMed] [Google Scholar]
- Comel A., Sorrentino G., Capaci V., et al. (2014). The cytoplasmic side of p53's oncosuppressive activities. FEBS Lett. 588, 2600–2609. [DOI] [PubMed] [Google Scholar]
- Cui G., Park S., Badeaux A.I., et al. (2012). PHF20 is an effector protein of p53 double lysine methylation that stabilizes and activates p53. Nat. Struct. Mol. Biol. 19, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C., and Gu W. (2010). p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L., Lin J., Said A.B., et al. (2019). Pellino1 specifically binds to phospho-Thr18 of p53 and is recruited to sites of DNA damage. Biochem. Biophys. Res. Commun. 513, 714–720. [DOI] [PubMed] [Google Scholar]
- DeLeo A.B., Jay G., Appella E., et al. (1979). Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl Acad. Sci. USA 76, 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K., Lu Y., Nikolovska-Coleska Z., et al. (2006). Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 49, 3432–3435. [DOI] [PubMed] [Google Scholar]
- Ding Q., Zhang Z., Liu J.J., et al. (2013). Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 56, 5979–5983. [DOI] [PubMed] [Google Scholar]
- Dohmesen C., Koeppel M., and Dobbelstein M. (2008). Specific inhibition of Mdm2-mediated neddylation by Tip60. Cell Cycle 7, 222–231. [DOI] [PubMed] [Google Scholar]
- Donehower L.A., Harvey M., Slagle B.L., et al. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. [DOI] [PubMed] [Google Scholar]
- Dornan D., Wertz I., Shimizu H., et al. (2004). The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429, 86–92. [DOI] [PubMed] [Google Scholar]
- Du C., Wu H., and Leng R.P. (2016). UBE4B targets phosphorylated p53 at serines 15 and 392 for degradation. Oncotarget 7, 2823–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L., Perez R.E., Chen L., et al. (2018). p53 promotes AKT and SP1-dependent metabolism through the pentose phosphate pathway that inhibits apoptosis in response to Nutlin-3a. J. Mol. Cell Biol. 10, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C., Scheffner M., and Hohfeld J. (2005). The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 280, 27443–27448. [DOI] [PubMed] [Google Scholar]
- Fan Y.H., Cheng J., Vasudevan S.A., et al. (2013). USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 4, e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiscella M., Zhang H., Fan S., et al. (1997). Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl Acad. Sci. USA 94, 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S., Shao S., Wang L., et al. (2017). USP3 stabilizes p53 protein through its deubiquitinase activity. Biochem. Biophys. Res. Commun. 492, 178–183. [DOI] [PubMed] [Google Scholar]
- Furth N., Bossel Ben-Moshe N., Pozniak Y., et al. (2015). Down-regulation of LATS kinases alters p53 to promote cell migration. Genes Dev. 29, 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss-Friedlander R., and Melchior F. (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956. [DOI] [PubMed] [Google Scholar]
- Grasberger B.L., Lu T., Schubert C., et al. (2005). Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 48, 909–912. [DOI] [PubMed] [Google Scholar]
- Green D.R., and Kroemer G. (2009). Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochola L.F., Zeron-Medina J., Meriaux S., et al. (2010). Single-nucleotide polymorphisms in the p53 signaling pathway. Cold Spring Harb. Perspect. Biol. 2, a001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B., and Zhu W.G. (2012). Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., and Roeder R.G. (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Gu W., Shi X.L., and Roeder R.G. (1997). Synergistic activation of transcription by CBP and p53. Nature 387, 819–823. [DOI] [PubMed] [Google Scholar]
- Hafner A., Bulyk M.L., Jambhekar A., et al. (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210. [DOI] [PubMed] [Google Scholar]
- Harrigan J.A., Jacq X., Martin N.M., et al. (2018). Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Shrinivas K., Young R.A., et al. (2017). A phase separation model for transcriptional control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H.F., and Vousden K.H. (2007). Coping with stress: multiple ways to activate p53. Oncogene 26, 1306–1316. [DOI] [PubMed] [Google Scholar]
- Hsueh K.W., Fu S.L., Chang C.B., et al. (2013). A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim. Biophys. Acta 1834, 508–515. [DOI] [PubMed] [Google Scholar]
- Hua W.K., Qi J., Cai Q., et al. (2017). HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 130, 2619–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Perez-Burgos L., Placek B.J., et al. (2006). Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632. [DOI] [PubMed] [Google Scholar]
- Huang J., Sengupta R., Espejo A.B., et al. (2007). p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108. [DOI] [PubMed] [Google Scholar]
- Hupp T.R., Meek D.W., Midgley C.A., et al. (1992). Regulation of the specific DNA binding function of p53. Cell 71, 875–886. [DOI] [PubMed] [Google Scholar]
- Ichwan S.J., Yamada S., Sumrejkanchanakij P., et al. (2006). Defect in serine 46 phosphorylation of p53 contributes to acquisition of p53 resistance in oral squamous cell carcinoma cells. Oncogene 25, 1216–1224. [DOI] [PubMed] [Google Scholar]
- Ishak Gabra M.B., Yang Y., Lowman X.H., et al. (2018). IKKβ activates p53 to promote cancer cell adaptation to glutamine deprivation. Oncogenesis 7, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Kawaguchi Y., Lai C.H., et al. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov G.S., Ivanova T., Kurash J., et al. (2007). Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol. 27, 6756–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T., Remington L., Williams B.O., et al. (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Jansson M., Durant S.T., Cho E.C., et al. (2008). Arginine methylation regulates the p53 response. Nat. Cell Biol. 10, 1431–1439. [DOI] [PubMed] [Google Scholar]
- Jenkins L.M., Yamaguchi H., Hayashi R., et al. (2009). Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry 48, 1244–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A.C., and Fersht A.R. (2008). Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 77, 557–582. [DOI] [PubMed] [Google Scholar]
- Joerger A.C., and Fersht A.R. (2016). The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404. [DOI] [PubMed] [Google Scholar]
- Johmura Y., Sun J., Kitagawa K., et al. (2016). SCF (Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat. Commun. 7, 10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S., Sakaguchi K., Shimohigashi Y., et al. (2002). Effect of phosphorylation on the structure and fold of transactivation domain of p53. J. Biol. Chem. 277, 15579–15585. [DOI] [PubMed] [Google Scholar]
- Kastenhuber E.R., and Lowe S.W. (2017). Putting p53 in context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kategaya L., Di Lello P., Rouge L., et al. (2017). USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538. [DOI] [PubMed] [Google Scholar]
- Ke J.Y., Dai C.J., Wu W.L., et al. (2014). USP11 regulates p53 stability by deubiquitinating p53. J. Zhejiang Univ. Sci. B 15, 1032–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights C.D., Catania J., Di Giovanni S., et al. (2006). Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon N., Kobayashi Y., Li M., et al. (2010). Inactivation of HAUSP in vivo modulates p53 function. Oncogene 29, 1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F., Labuschagne C.F., and Vousden K.H. (2015). p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405. [DOI] [PubMed] [Google Scholar]
- Kruse J.P., and Gu W. (2009a). Modes of p53 regulation. Cell 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse J.P., and Gu W. (2009b). MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 284, 3250–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurash J.K., Lei H., Shen Q., et al. (2008). Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 29, 392–400. [DOI] [PubMed] [Google Scholar]
- Lain S., Hollick J.J., Campbell J., et al. (2008). Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A., and Ronai Z. (2007). Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 26, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert P.F., Kashanchi F., Radonovich M.F., et al. (1998). Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 273, 33048–33053. [DOI] [PubMed] [Google Scholar]
- Lane D.P. (1992). Cancer. p53, guardian of the genome. Nature 358, 15–16. [DOI] [PubMed] [Google Scholar]
- Lane D.P., and Crawford L.V. (1979). T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- Lazo P.A. (2017). Reverting p53 activation after recovery of cellular stress to resume with cell cycle progression. Cell. Signal. 33, 49–58. [DOI] [PubMed] [Google Scholar]
- Le Cam L., Linares L.K., Paul C., et al. (2006). E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 127, 775–788. [DOI] [PubMed] [Google Scholar]
- Lee C.W., Ferreon J.C., Ferreon A.C., et al. (2010). Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl Acad. Sci. USA 107, 19290–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.W., Oh W., Song H.P., et al. (2017a). Phosphorylation of p53 at threonine 155 is required for Jab1-mediated nuclear export of p53. BMB Rep. 50, 373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee X.A., Verma C., and Sim A.Y.L. (2017b). Designing dual inhibitors of Mdm2/MdmX: unexpected coupling of water with gatekeeper Y100/99. Proteins 85, 1493–1506. [DOI] [PubMed] [Google Scholar]
- Lehnertz B., Rogalski J.C., Schulze F.M., et al. (2011). p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol. Cell 43, 673–680. [DOI] [PubMed] [Google Scholar]
- Leng R.P., Lin Y., Ma W., et al. (2003). Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112, 779–791. [DOI] [PubMed] [Google Scholar]
- L'Hernault S.W., and Rosenbaum J.L. (1983). Chlamydomonas α-tubulin is posttranslationally modified in the flagella during flagellar assembly. J. Cell Biol. 97, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.W., Liu J.P., Schmid P.C., et al. (2006). Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 25, 3006–3022. [DOI] [PubMed] [Google Scholar]
- Li H.H., Cai X., Shouse G.P., et al. (2007). A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 26, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.H., Li A.G., Sheppard H.M., et al. (2004a). Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol. Cell 13, 867–878. [DOI] [PubMed] [Google Scholar]
- Li M., Brooks C.L., Kon N., et al. (2004b). A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886. [DOI] [PubMed] [Google Scholar]
- Li M., Brooks C.L., Wu-Baer F., et al. (2003). Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975. [DOI] [PubMed] [Google Scholar]
- Li M., Chen D., Shiloh A., et al. (2002a). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653. [DOI] [PubMed] [Google Scholar]
- Li M., Luo J., Brooks C.L., et al. (2002b). Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277, 50607–50611. [DOI] [PubMed] [Google Scholar]
- Li T., Kon N., Jiang L., et al. (2012). Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wu L., Corsa C.A., et al. (2009). Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol. Cell 36, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S.H., and Clarke M.F. (2001). Regulation of p53 localization. Eur. J. Biochem. 268, 2779–2783. [DOI] [PubMed] [Google Scholar]
- Liao P., Zeng S.X., Zhou X., et al. (2017). Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol. Cell 68, 1134–1146 e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill N.L., Grossman S.R., Ginsberg D., et al. (1997). Binding and modulation of p53 by p300/CBP coactivators. Nature 387, 823–827. [DOI] [PubMed] [Google Scholar]
- Linzer D.I., and Levine A.J. (1979). Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52. [DOI] [PubMed] [Google Scholar]
- Liu H., Li X., Ning G., et al. (2016a). The Machado-Joseph disease deubiquitinase ataxin-3 regulates the stability and apoptotic function of p53. PLoS Biol. 14, e2000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Li F., Sun Q., et al. (2019). p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Scolnick D.M., Trievel R.C., et al. (1999). p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.P., Yang M., Peng Q.Z., et al. (2017a). UBE2T promotes hepatocellular carcinoma cell growth via ubiquitination of p53. Biochem. Biophys. Res. Commun. 493, 20–27. [DOI] [PubMed] [Google Scholar]
- Liu W.T., Huang K.Y., Lu M.C., et al. (2017b). TGF-β upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene 36, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Tan Y., Zhang C., et al. (2016b). NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 17, 349–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. (2017). p53 and MDM2: their Yin-Yang intimacy. J. Mol. Cell Biol. 9, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Nannenga B., and Donehower L.A. (2005). PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 19, 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Lu Z., Lu X., et al. (2013). OTUD5 regulates p53 stability by deubiquitinating p53. PLoS One 8, e77682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Nikolaev A.Y., Imai S., et al. (2001). Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148. [DOI] [PubMed] [Google Scholar]
- Luo J., Su F., Chen D., et al. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381. [DOI] [PubMed] [Google Scholar]
- MacPherson D., Kim J., Kim T., et al. (2004). Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J. 23, 3689–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandinova A., and Lee S.W. (2011). The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci. Transl. Med. 3, 64rv61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek D.W. (2015). Regulation of the p53 response and its relationship to cancer. Biochem. J. 469, 325–346. [DOI] [PubMed] [Google Scholar]
- Meek D.W., and Anderson C.W. (2009). Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer S.M., Larsson C.A., and Lozano G. (2017). Mdm proteins: critical regulators of embryogenesis and homoeostasis. J. Mol. Cell Biol. 9, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D., and Riezman H. (2007). Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205. [DOI] [PubMed] [Google Scholar]
- Muller P.A., and Vousden K.H. (2014). Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Roth J.A., and Mukhopadhyay T. (2000). Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol. Cell. Biol. 20, 9391–9398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L. (2015). Gene therapy returns to Centre stage. Nature 526, 351–360. [DOI] [PubMed] [Google Scholar]
- Narita T., Weinert B.T., and Choudhary C. (2019). Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174. [DOI] [PubMed] [Google Scholar]
- Nguyen T.A., Menendez D., Resnick M.A., et al. (2014). Mutant TP53 posttranslational modifications: challenges and opportunities. Hum. Mutat. 35, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K.P., Tuveson D.A., Ruhe Z.C., et al. (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Olsson A., Manzl C., Strasser A., et al. (2007). How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14, 1561–1575. [DOI] [PubMed] [Google Scholar]
- Ou Y.H., Chung P.H., Sun T.P., et al. (2005). p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol. Biol. Cell 16, 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.H., Smith R.J., Shieh S.Y., et al. (2011). The GAS41-PP2Cβ complex dephosphorylates p53 at serine 366 and regulates its stability. J. Biol. Chem. 286, 10911–10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao S., Pei H.Z., Huang B., et al. (2017). Ovarian tumor domain-containing protein 1 deubiquitinates and stabilizes p53. Cell. Signal. 33, 22–29. [DOI] [PubMed] [Google Scholar]
- Pickart C.M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. [DOI] [PubMed] [Google Scholar]
- Qin H.T., Li H.Q., and Liu F. (2017). Selective histone deacetylase small molecule inhibitors: recent progress and perspectives. Expert Opin. Ther. Pat. 27, 621–636. [DOI] [PubMed] [Google Scholar]
- Rabut G., and Peter M. (2008). Function and regulation of protein neddylation. 'Protein modifications: beyond the usual suspects' review series. EMBO Rep. 9, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravandi F., Gojo I., Patnaik M.M., et al. (2016). A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res. 48, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed D., Shen Y., Shelat A.A., et al. (2010). Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 285, 10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S.M., and Quelle D.E. (2014). p53 acetylation: regulation and consequences. Cancers 7, 30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J., Herrero A., Li S., et al. (2018). PHD3 regulates p53 protein stability by hydroxylating proline 359. Cell Rep. 24, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M.S., Desterro J.M., Lain S., et al. (2000). Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 20, 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokudai S., Aikawa Y., Tagata Y., et al. (2009). Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J. Biol. Chem. 284, 237–244. [DOI] [PubMed] [Google Scholar]
- Rokudai S., Laptenko O., Arnal S.M., et al. (2013). MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl Acad. Sci. USA 110, 3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X., Rao J., Li D., et al. (2019). TRIM69 inhibits cataractogenesis by negatively regulating p53. Redox Biol. 22, 101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H.W., Shin D.H., Lee D.H., et al. (2017). HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 391, 162–171. [DOI] [PubMed] [Google Scholar]
- Sabari B.R., Dall'Agnese A., Boija A., et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S., Yamaguchi H., Higashimoto Y., et al. (2003). Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 278, 37536–37544. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera J.E., Saito S., et al. (1998). DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldana-Meyer R., and Recillas-Targa F. (2011). Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 6, 1068–1077. [DOI] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse J.M., Vierstra R.D., et al. (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Schnell J.D., and Hicke L. (2003). Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J. Biol. Chem. 278, 35857–35860. [DOI] [PubMed] [Google Scholar]
- Senft D., Qi J., and Ronai Z.A. (2018). Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 18, 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel I.A., Aprelia M., Saurin A.T., et al. (2014). Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl Acad. Sci. USA 111, 7313–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J., Li P., Shao X., et al. (2018). The E3 ligase RING1 targets p53 for degradation and promotes cancer cell proliferation and survival. Cancer Res. 78, 359–371. [DOI] [PubMed] [Google Scholar]
- Shi X., Kachirskaia I., Yamaguchi H., et al. (2007). Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh S.Y., Ahn J., Tamai K., et al. (2000). The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Shieh S.Y., Ikeda M., Taya Y., et al. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334. [DOI] [PubMed] [Google Scholar]
- Sluss H.K., Armata H., Gallant J., et al. (2004). Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 24, 976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel D. (2010). Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 20, 14–24. [DOI] [PubMed] [Google Scholar]
- Stehmeier P., and Muller S. (2009). Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair 8, 491–498. [DOI] [PubMed] [Google Scholar]
- Sterner R., Vidali G., and Allfrey V.G. (1979). Studies of acetylation and deacetylation in high mobility group proteins. Identification of the sites of acetylation in HMG-1. J. Biol. Chem. 254, 11577–11583. [PubMed] [Google Scholar]
- Sun D., Li Z., Rew Y., et al. (2014). Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 57, 1454–1472. [DOI] [PubMed] [Google Scholar]
- Sykes S.M., Mellert H.S., Holbert M.A., et al. (2006). Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira N., Nihira K., Yamaguchi T., et al. (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725–738. [DOI] [PubMed] [Google Scholar]
- Taira N., and Yoshida K. (2012). Post-translational modifications of p53 tumor suppressor: determinants of its functional targets. Histol. Histopathol. 27, 437–443. [DOI] [PubMed] [Google Scholar]
- Takekawa M., Adachi M., Nakahata A., et al. (2000). p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka I., Morin F., Seizinger B.R., et al. (1995). Regulation of the sequence-specific DNA binding function of p53 by protein kinase C and protein phosphatases. J. Biol. Chem. 270, 5405–5411. [DOI] [PubMed] [Google Scholar]
- Tang Y., Luo J., Zhang W., et al. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. [DOI] [PubMed] [Google Scholar]
- Tang Y., Zhao W., Chen Y., et al. (2008). Acetylation is indispensable for p53 activation. Cell 133, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavana O., and Gu W. (2017). Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J. Mol. Cell Biol. 9, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavana O., Sun H., and Gu W. (2018). Targeting HAUSP in both p53 wildtype and p53-mutant tumors. Cell Cycle 17, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel D.P., Bycroft M., and Fersht A.R. (2009). Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 28, 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu R., Kang W., Yang X., et al. (2018). USP49 participates in the DNA damage response by forming a positive feedback loop with p53. Cell Death Dis. 9, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull A.P., Ioannidis S., Krajewski W.W., et al. (2017). Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 550, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela M.T., Guerrero R., Nunez M.I., et al. (2002). PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 21, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Vassilev L.T., Vu B.T., Graves B., et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Vaziri H., Dessain S.K., Ng Eaton E., et al. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Verdin E., and Ott M. (2015). 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264. [DOI] [PubMed] [Google Scholar]
- Verdone L., Agricola E., Caserta M., et al. (2006). Histone acetylation in gene regulation. Brief. Funct. Genomic. Proteomic. 5, 209–221. [DOI] [PubMed] [Google Scholar]
- Vieler M., and Sanyal S. (2018). p53 isoforms and their implications in cancer. Cancers 10, E288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden K.H., and Lane D.P. (2007). p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283. [DOI] [PubMed] [Google Scholar]
- Vu B., Wovkulich P., Pizzolato G., et al. (2013). Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walerych D., Kudla G., Gutkowska M., et al. (2004). Hsp90 chaperones wild-type p53 tumor suppressor protein. J. Biol. Chem. 279, 48836–48845. [DOI] [PubMed] [Google Scholar]
- Wang D., Kon N., Lasso G., et al. (2016a). Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 538, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Tai P.W.L., and Gao G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Marshall C.B., and Ikura M. (2013). Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell. Mol. Life Sci. 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.J., Li D., Ou Y., et al. (2016b). Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 17, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe F., Fukazawa H., Masutani M., et al. (2004). Poly(ADP-ribose) polymerase-1 inhibits ATM kinase activity in DNA damage response. Biochem. Biophys. Res. Commun. 319, 596–602. [DOI] [PubMed] [Google Scholar]
- Waterman M.J., Stavridi E.S., Waterman J.L., et al. (1998). ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 19, 175–178. [DOI] [PubMed] [Google Scholar]
- West L.E., Roy S., Lachmi-Weiner K., et al. (2010). The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J. Biol. Chem. 285, 37725–37732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E.R., McAtarsney C.P., Bredhold K.E., et al. (2018). Mutant and wild-type p53 form complexes with p73 upon phosphorylation by the kinase JNK. Sci. Signal. 11, eaao4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.Y., and Chiang C.M. (2009). Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 28, 1246–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Lin J.C., Piluso L.G., et al. (2014). Phosphorylation of p53 by TAF1 inactivates p53-dependent transcription in the DNA damage response. Mol. Cell 53, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Earle J., Saito S., et al. (2002). Mutation of mouse p53 Ser23 and the response to DNA damage. Mol. Cell. Biol. 22, 2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Padre R.C., De Mendoza T.H., et al. (2009). Phosphorylation of p53 by IκB kinase 2 promotes its degradation by β-TrCP. Proc. Natl Acad. Sci. USA 106, 2629–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Wang L., Wang S., et al. (2018). SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 144, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas D.P., Saville M.K., Bourdon J.C., et al. (2004). Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 118, 83–97. [DOI] [PubMed] [Google Scholar]
- Xu H.T., Lai W.L., Liu H.F., et al. (2016). PAK4 phosphorylates p53 at serine 215 to promote liver cancer metastasis. Cancer Res. 76, 5732–5742. [DOI] [PubMed] [Google Scholar]
- Yang W.H., Kim J.E., Nam H.W., et al. (2006). Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. 8, 1074–1083. [DOI] [PubMed] [Google Scholar]
- Yin H., Xue W., and Anderson D.G. (2019). CRISPR-Cas: a tool for cancer research and therapeutics. Nat. Rev. Clin. Oncol. 16, 281–295. [DOI] [PubMed] [Google Scholar]
- Yu S., Dai J., Ma M., et al. (2019). RBCK1 promotes p53 degradation via ubiquitination in renal cell carcinoma. Cell Death Dis. 10, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Luo K., Zhang L., et al. (2010). USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X., Zhao Y., Xu Y., et al. (2017). Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J. Mol. Biol. 429, 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchi P., Gostissa M., Uchida T., et al. (2002). The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419, 853–857. [DOI] [PubMed] [Google Scholar]
- Zhang L., Jin Y., Yang H., et al. (2019). SMYD3 promotes epithelial ovarian cancer metastasis by down-regulating p53 protein stability and promoting p53 ubiquitination. Carcinogenesis, doi: 10.1093/carcin/bgz078. [DOI] [PubMed] [Google Scholar]
- Zhao W., Kruse J.P., Tang Y., et al. (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., You H., Zhou X.Z., et al. (2002). The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419, 849–853. [DOI] [PubMed] [Google Scholar]
- Zhou X., Cao B., Lu H., (2017). Negative auto-regulators trap p53 in their web. J. Mol. Cell Biol. 9, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Ji Z., Wang Y., et al. (2014). TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 147, 1043–1054. [DOI] [PubMed] [Google Scholar]