

ABSTRACT
Site-specific antibody-drug conjugates (ADCs) are designed to overcome the heterogeneity observed with first-generation ADCs that use random conjugation to surface-exposed lysine residues or conjugation to interchain disulfide bonds. Despite significantly enhanced homogeneity, however, the production of site-specific ADCs yields some process-related species heterogeneity, including stereoisomers, unconjugated antibody, underconjugated species, and overconjugated species. An elevated level of size variants, such as heavy chain-light chain species (half ADC), heavy chain-heavy chain-light chain species, and light chain species, is also observed with the final site-specific ADC product. To understand the root cause of heterogeneity generated during the ADC conjugation process, we designed time-course studies for each conjugation step, including reduction, oxidation, conjugation, and quenching. We developed both non-reduced peptide map and LabChip-based capillary electrophoresis sodium dodecyl sulfate methods for time-course sample analysis. On the basis of our time-course data, the half ADC and unconjugated antibody were generated during oxidation as a result of alternative disulfide bond arrangements. During oxidation, two hinge cysteines formed an intra-chain disulfide bond in the half ADC, and three inter-chain hinge disulfide bonds were formed in the unconjugated antibody. Time-course data also showed that the elevated level of size variants, especially heavy chain-heavy chain-light chain species and light chain species, resulted from the quenching step, where the quenching reagent engaged in a disulfide bond exchange reaction with the ADC and broke the disulfide bonds connecting the heavy chain and light chain. Underconjugated and overconjugated species arose from the equilibrium established during the conjugation reaction.
KEYWORDS: Antibody-drug conjugate, ADC, conjugation heterogeneity, conjugation time-course study, stereoisomer, underconjugated species, overconjugated species, unconjugated antibody, disulfide bond exchange reaction
Introduction
Although chemotherapy has historically been used as the primary treatment against cancer, its efficacy is hampered by a lack of specificity. Antibody-drug conjugates (ADCs) improve specificity and widen the therapeutic window by combining the specificity of antibodies with the high potency of cytotoxic drugs. Since their introduction in the mid-1990s, ADCs have undergone substantial improvements in molecular designs and manufacturing processes, and, as a consequence, the number of ADCs assessed in clinical studies has increased.1–5 As of 2017, approximately 90 ADCs were under assessment in clinical studies,1 and four were approved: gemtuzumab ozogamicin,6,7 brentuximab vedotin,8 trastuzumab emtansine,9 and inotuzumab ozogamicin.10
Lysine modification, one of the conjugation methods used to produce first-generation ADCs such as gemtuzumab ozogamicin, trastuzumab emtansine, and inotuzumab ozogamicin, generates a complex heterogeneous mixture of species with variable drug-to-antibody ratios (DARs). For example, species with DARs of 0 to 7 have been observed with trastuzumab emtansine,11 and approximately 50% unconjugated antibody and species with DARs of 4 to 6 were detected with gemtuzumab ozogamicin.7 In addition, due to the availability of many conjugatable lysine sites, any two conjugates with the same DAR are likely regioisomers. One study estimated that there were 4.5 million unique molecules for a lysine-conjugated ADC, with DARs ranging from 0 to 6 and 40 of 86 lysine residues conjugated to some degree.12 Such heterogeneity might have contributed partially to the low efficacy and possible toxicity observed in Phase 3 studies of gemtuzumab ozogamicin, which led Pfizer to remove this agent voluntarily from the market 10 years after it was first approved.13
Because fewer conjugable cysteines are available in an antibody, ADC manufacturers have employed cysteine modification to reduce the heterogeneity observed with lysine-based ADCs. In the conventional cysteine conjugation process used to produce the first-generation ADC brentuximab vedotin, the four interchain disulfide bonds in the immunoglobulin G1 molecule were reduced selectively, generating eight potential conjugation sites.14 The intrachain disulfide bonds remained untouched during the optimized reducing condition because they were buried and inaccessible to solvent. Despite the significantly reduced heterogeneity for the cysteine-conjugated ADC, the final product still contained variants with DARs between 0 and 8, and there were several possible structures with the same DAR.
Despite the success of some first-generation ADCs, their complex heterogeneity has been associated with several problems, including suboptimal therapeutic index, higher clearance rates for high-DAR species, narrower therapeutic windows, and poor stability.15–17 To overcome these challenges, the biotechnology and pharmaceutical industries have designed and developed second-generation, site-specific ADCs. The production of site-specific ADCs precisely controls not only the average DARs but also the number of unique conjugation sites. Compared with the production of first-generation ADCs, which yields thousands to millions of different structures, the production of second-generation ADCs yields only a few unique structures, which presumably can be optimized, developed, and manufactured more easily.
Numerous approaches have been developed to generate site-specific ADCs.18–21 One approach achieves site-specific conjugation by incorporating unnatural amino acids.22,23 Although this approach could use a wide variety of bioorthogonal ligation reactions with minimal structure perturbation, the technology is complicated. A second approach takes advantage of enzymatic or chemoenzymatic reactions to generate site-specific ADCs,24–26 but the enzymatic conversion efficiency is site dependent. A third approach, which achieves conjugation through inter-chain disulfide bonds or engineered cysteines, has been adopted widely by the pharmaceutical industry because of its simplicity, fast conjugation reactions, and minimal structure perturbation.
Three conjugation strategies have been used to produce cysteine-based, site-specific ADCs. One approach generates site-specific ADCs by replacing four inter-chain disulfide bonds with disulfide-bridging reagents.27,28 This approach has reduced the heterogeneity of the final products significantly by reducing the maximum number of attachable payloads from 8 to 4. Another approach generates site-specific ADCs by using site-directed mutagenesis to introduce reactive, surface-accessible cysteines at specific residues.29 This method yields site-specific ADCs with a DAR of 2 (vc-PAB-MMAE linker/payload) and high purity, providing comparable efficacy but significantly lower toxicity in vivo. An alternative approach, which inserts reactive cysteines into the hinge region rather than relying on cysteine mutagenesis, is an efficient method to prepare site-specific ADCs.30 The third approach, which was reported recently, selectively conjugates π-clamp, the unique amino acid sequence FCPF within the antibody, with perfluoro-aromatic reagents containing the drug payload.31
Second-generation, site-specific ADCs display significantly enhanced homogeneity, compared with first-generation ADCs. For example, ADC-A, the site-specific ADC used in this study, has one additional cysteine inserted into the hinge region of each heavy chain, resulting in two additional cysteines in the hinge region.30 Thus, the hinge region contains three pairs of cysteines: two pairs, which originated from the initial immunoglobulin G1 antibody and form inter-chain disulfide bonds between two heavy chains; and the pair of cysteines inserted as the site for drug conjugation. Because the inserted cysteines are the only conjugation sites, this design is expected to generate only one kind of desired ADC molecule with a DAR of 2.
ADC conjugation is a complex process that includes multiple steps: reduction, oxidation, conjugation, and quenching.19 With ADC-A, the reduction step removes the cysteinylation capping of the two inserted cysteines and makes them available for conjugation. The oxidation step is needed to re-establish the disulfide bond between the two heavy chains and the disulfide bond between the light and heavy chain. At the same time, the two inserted cysteines are maintained as free thiols and remain available for thiol maleimide conjugation. The conjugation step attaches the drug payload to the two free-inserted cysteines. The quenching reaction consumes any unreacted payload. As a result of this complex process, some heterogeneous species are still observed with site-specific ADCs.
Here, we report the heterogeneous species, such as unconjugated antibody, underconjugated and overconjugated species, stereoisomers, and size variants, that we observed during production of ADC-A. We also report the results of time-course studies we conducted to explore the potential causes of heterogeneity at each conjugation step.
Results
Conjugation heterogeneity observed for the site-specific ADC
ADC structures with a target DAR of 2
In the hydrophobic interaction chromatography (HIC) profile for ADC-A and its corresponding monoclonal antibody (mAb) intermediate, clear differences were observed between the mAb intermediate, which appeared as one main peak, and the ADC, which appeared as multiple peaks (Figure. 1A, B). Because the ADC was distributed among multiple peaks in a broad retention time, the intensity for each ADC peak was significantly reduced, compared with the intensity of the peak for the mAb intermediate. However, the total peak areas for ADC (after subtracting payload absorbance) and the mAb intermediate were comparable, indicating that no ADC sample was lost during HIC. The change in the HIC profile from a single peak for the mAb intermediate to multiple peaks for the ADC indicated that heterogeneous species were generated during the conjugation process.
Figure 1.
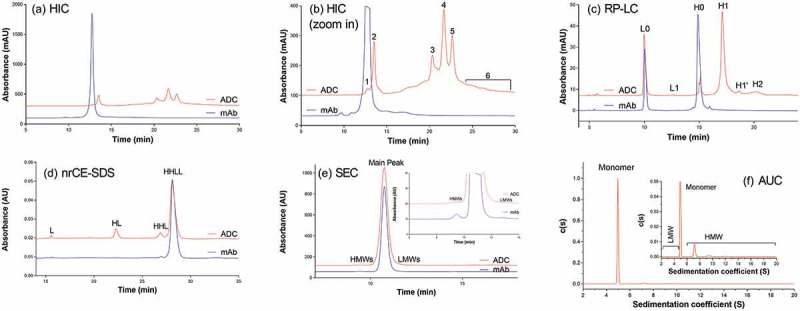
Analytical method profiles for ADC and its corresponding mAb intermediate: (A) Hydrophobic Interaction Chromatograph (HIC); (B) Zoom in HIC profile (Peak # 1–6 are indicated); (C) Reverse Phase Liquid Chromatograph (RP-LC); (D) non-reduced CE-SDS; (E) Size Exclusion Chromatograph (SEC); and (F) Analytical Ultracentrifuge (AUC).
Peaks 3, 4, and 5 were three major peaks that eluted close to each other in the chromatogram. Intact mass measurement indicated that the masses for those peaks were similar, and all these masses matched the theoretical mass of ADC-A, with a DAR of 2, within the mass error range of intact mass measurement (Table 1). Nonreduced peptide mapping analysis showed the same hinge peptides, H15-H15 (2SS, 2Drug), for the peaks 3, 4, and 5 fractions (Table 1). The appearance of peptide H15-H15 (2SS, 2Drug) was consistent with the design of the ADC-A structure, with its target DAR of 2, and with the antibody-drug conjugation through the two cysteines inserted into the hinge region (Table 1, Designed DAR2 ADC). This agreement suggested that peak 3, 4, and 5 could be structural variants for ADC-A. No noticeable differences in structure were observed among the peak 3, 4, and 5 fractions as determined by multiple physicochemical analyses such as DAR measurement by reverse-phase high-performance liquid chromatography (RP-HPLC), aggregation analysis by size-exclusion chromatography (SEC), charge profile analysis by ion-exchange chromatography, fragment analysis by capillary electrophoresis sodium dodecyl sulfate with nonreduced (nrCE-SDS) and reduced (rCE-SDS) sample preparations, and post-translational modification analysis by peptide mapping. Potency assay data revealed similar antigen binding activities of 102%, 108%, and 101% for peaks 3, 4, and 5, respectively. The cytotoxicity assay data also showed similar cytotoxicity, at 102%, 99%, and 100% for peaks 3, 4, and 5, respectively. The fractions eluted from these peaks differed only in their retention times, indicating differences in hydrophobicity.
Table 1.
Intact mass and nonreduced peptide map data for HIC fractions.
| HIC PK# | Intact Structure | Intact Mass (Da) |
Peptide Name | Peptide Structure |
Peptide Mass (Da) |
||
|---|---|---|---|---|---|---|---|
| Theor | Exper | Theor | Exper | ||||
| 1 |
 Unconjugated/Tri SS bonds |
146,242 | 146,242 | H15-H15 (3SS) | 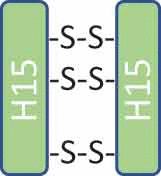 |
5661.8 (a) | 5661.9 (a) |
| 2 |
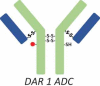 DAR 1 ADC |
147,741 | 147,742 | H15-H15 (2SS, 1Drug, 1SH) | 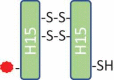 |
7285.6 (a) | 7285.8 (a) |
| 3 |
 (S,S), (R,S), (R,R) Designed DAR2 ADC |
149,237 | 149,236 | H15-H15 (2SS, 2Drug) | 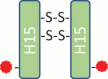 |
8657.2 (a) | 8657.5 (a) |
| 4 | 149,237 | 8657.5 (a) | |||||
| 5 | 149,234 | 8657.5 (a) | |||||
| 6 |
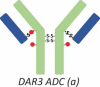 DAR3 ADC (a) |
150,734 | 150,748 | H14-Drug |  |
1946.9 (m) | 1946.9 (m) |
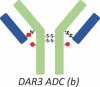 DAR3 ADC (b) |
L16-Drug |  |
2307.0 (m) | 2307.1 (m) | |||
 LC-Drug LC-Drug |
25,116 | 25,115 | |||||
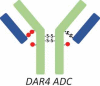 DAR4 ADC |
152,231 | 152,249 | L16-H14 (1SS) |  |
1260.5 (m) | 1260.5 (m) | |
| Crossing HIC peaks |
 Half ADC |
74,619 | 74,620 | H15 (1SS, 1Drug) |  |
4326.1 (m) | 4326.2 (m) |
 LC-QuechCap |
23,781 | 23,780 | L16-QC (Quench Cap) |  |
972.3 (m) | 972.4 (m) | |
(a) Average mass; (m) mono isotopic mass.
The differing hydrophobicity of these three peaks despite the similarities in other properties might arise from the stereochemistry of maleimide-thiol conjugation. Following the maleimide-thiol conjugation reaction for ADC-A, two stereoisomers, R and S, were formed (Scheme I). The presence of two conjugation sites yielded three possible stereoisomer combinations (R, R), (R, S), and (S, S). (R, S), the combination with the highest probability, likely corresponded to peak 4, which was the most intense. The three-dimensional orientation differences among these stereoisomer combinations may have led to different arrangements and interactions with the two payloads on the mAb surface, and thus to differences in surface hydrophobicity and separation by HIC. Otherwise, the three stereoisomer combinations would be expected to have similar properties such as aggregation, charge profiles, fragmentation, and bioactivities. Thus, no obvious differences would have been observed for these stereoisomer combinations by other analytical methods. Indeed, the results were compared with respect to aggregation, charge profiles, fragmentation, and bioactivities among the three major peaks, and we concluded that the coexistence of the three stereoisomer combinations would not be expected to affect the stability, efficacy, or safety of ADC-A.
Scheme I.
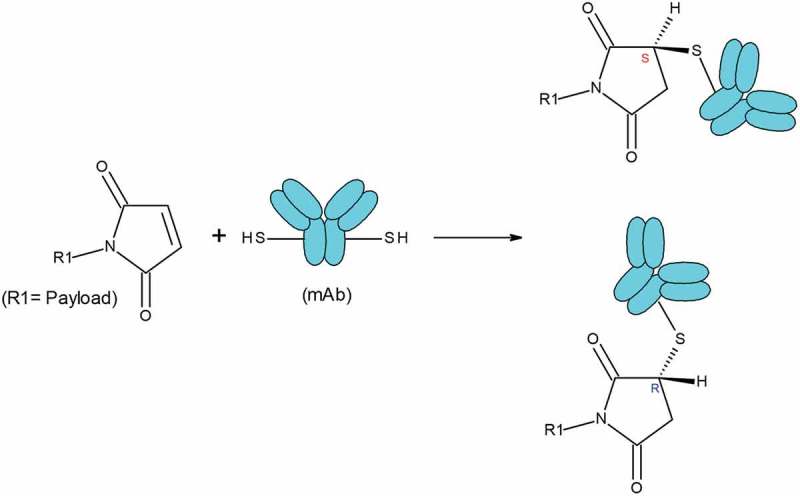
Maleimide-thiol conjugation reaction. After conjugation reaction, one chiral center is generated, which leads to two stereoisomers R and S.
Unconjugated antibody and underconjugated ADC species
The mass observed for peak 1 matched with the expected mass of unconjugated antibody. In the unconjugated antibody species, the two inserted cysteines could exist as free thiols or by forming additional disulfide bonds. As observed with non-reduced peptide mapping, the main hinge peptide from the peak 1 fraction was H15-H15 (3SS), which contained three disulfide bonds between two H15 peptides (Table 1, Unconjugated/Tri SS Bonds). This observation indicated that the two inserted cysteines in the unconjugated antibody species formed a disulfide bond. The peptide H15-H15 (2SS, 2SH), which would contain two disulfide bonds and two free cysteines and correspond to the unconjugated antibody structure with the inserted cysteine existing as free thiols, was not observed in the non-reduced peptide map. Thus, the inserted cysteine pair in the unconjugated antibody formed a disulfide bond, resulting in lack of free cysteines available for conjugation.
The mass measured for peak 2 suggested that this fraction contained underconjugated ADC-A with a DAR of 1 (Table 1, DAR 1 ADC). Non-reduced peptide mapping data for the peak 2 fraction showed that the hinge peptide, H15-H15 (2SS, 1Drug, 1SH), contained two interchain disulfide bonds between the H15 peptides, one inserted cysteine that was conjugated, and the second-inserted cysteine unconjugated. This was consistent with a structure in which one inserted cysteine was conjugated with the drug payload and the other inserted cysteine existed as a free thiol. Potency assay data showed that the antigen binding of the underconjugated species in the peak 2 fraction was comparable with that seen with the species in the peak 3, 4, and 5 fractions, which each had a DAR of 2. Thus, payload conjugation did not affect antigen binding. However, the cytotoxicity for peak 2 was significantly reduced compared with that for peaks 3, 4, or 5 due to less payload per molecule.
We used RP-HPLC (Figure 1C) to characterize the underconjugated species further. The ADC was reduced into heavy chain species (H) and light chain species (L), which were separated from each other by reverse-phase chromatography based on differing hydrophobicity. Unconjugated light chain (L0) and conjugated heavy chain with only one drug payload (H1) were the major species for L and H, respectively. This result confirmed that conjugation occurred through the cysteines inserted into the heavy chain of the antibody. Approximately 9% of heavy chain were unconjugated H0, which was consistent with the approximate levels of underconjugated species (13%) and unconjugated species (2%) detected by HIC. Following reduction, underconjugated ADC generated one unconjugated H0 and one conjugated H1. Thus, the unconjugated heavy chain accounted for half the level of underconjugated ADC. Unconjugated antibody produced two unconjugated heavy chains H0 after reduction. This consistency illustrated that both HIC and RP-HPLC are suitable for monitoring the levels of unconjugated antibody and underconjugated ADC species.
Overconjugated ADC species
RP-HPLC also revealed that overconjugated species, such as heavy chain with two drug payloads (H2) or light chain with one drug payload (L1), were generated during the conjugation process for the site-specific ADC-A (Figure 1C). Although HIC yielded no distinguished peaks for overconjugated species (Figure 1A and B), the peak 6 fraction appeared to be enriched for ADC species with DARs of 3 and 4 ADC (Table 1, DAR3 and DAR4 ADC). However, ADCs with a DAR of 2 were still the major species observed in this fraction, and the overconjugated ADC species were the minor species. The intact masses observed for the overconjugated ADC species were not as accurate as those measured for other species, probably because of low intensity and interference from high-intensity DAR2 species.
The presence of overconjugated ADC species raised questions as to where the additional conjugation sites were located. Peptide mapping revealed several conjugation sites. For example, the two cysteines connecting H and L appeared to be conjugated to some extent, as illustrated by the detection of the peptides H14-Drug and L16-Drug. However, the extent of conjugation for these undesired conjugation sites was very low, which might explain why no distinct overconjugated species appeared in the HIC profile. In this respect, RP-HPLC, which could detect overconjugated species, was advantageous. Thus, the RP-HPLC method was more suitable for monitoring the conjugation because it could measure both underconjugated and overconjugated species.
Size variants observed for ADC
The nrCE-SDS profiles for the ADC-A and the corresponding mAb intermediate showed elevated levels of fragments, such as heavy chain-heavy chain-light chain species (HHL), heavy chain-light chain species (HL) corresponding to half-ADC species, and light chain species (L), for the ADC (Figure 1D). Intact mass measurement also revealed masses corresponding to fragments (Table 1). A mass corresponding to half the size of ADC-A was detected across all HIC fractions and starting materials. Additional nonreduced peptide map studies uncovered H15 (1SS, 1Drug), the peptide structure of the half-ADC. In this structure, an intrachain disulfide bond was formed within a single H15 peptide, preventing the formation of an interchain disulfide bond between two H15 peptides. The half-ADC structure might have contributed to the level of HL detected by nrCE-SDS in the ADC-A samples.
Intact mass measurement and nonreduced peptide mapping also revealed conjugated light chain and light chain capped with quench reagent (Table 1). The conjugated light-chain L1 peak was also detectable by RP-HPLC (Figure 1C). The presence of both these fragments explain the elevated level of L detected by nrCE-SDS. Despite the appearance of HHL in the nrCE-SDS profile, intact mass measurement showed no direct evidence of these species. The absence of an HHL peak in intact mass measurement might arise from ion suppression by major heavy chain-heavy chain-light chain-light chain species (HHLL) species: HHL and HHLL may have similar hydrophobicity, and therefore coelute during intact mass measurement.
It is worth noting that the elevated level of ADC fragments was detectable only by nrCE-SDS, which relies on samples prepared under denaturing conditions. Other methods used to monitor size variants, such as SEC (Figure 1E) and analytical ultracentrifugation (AUC) (Figure 1F), did not show an elevated level of fragments. The monomer peak percentages observed with both SEC and AUC were greater than 99%. Only 0.1% and 0.6% of the ADC fragments were detected by SEC and AUC, respectively. Because these methods used samples prepared under native conditions, it is likely that under these conditions, HL, HHL, and L were held together by noncovalent interactions, and that these fragments would not have a functional impact. Indeed, when bioactivity was evaluated for ADC-A samples with fragment levels ranging from approximately 15% to more than 90%, there were no significant differences in antigen binding or cytotoxicity across these samples.
Time-course studies
The heterogeneity observed with ADC-A most likely arises from the complex conjugation process. We thus needed to determine where in the process heterogeneity was introduced and the rate at which it builds up to optimize the conjugation process and minimize the formation of undesired species. To achieve this, we conducted time-course studies for each step in the conjugation workflow (see supplementary material for conjugation scheme). Analyses of the samples generated from the time-course studies used the LabChip GX Protein Characterization system (GX), which provided overall pictures of species linked by interchain disulfide bonds, and nonreduced peptide mapping, which provided detailed information for each disulfide bond.
Reduction time-course study
As shown by the GX data (Figure 2A), the starting material, HHLL or whole antibody species, was consumed quickly during the reduction reaction. HHL, heavy chain-heavy chain species (HH), and HL were the intermediates generated during reduction. Their concentrations increased initially, then decreased eventually to low levels. H and L were the final products. Significant levels of HHL, HH, and HL were present at the end of reduction under “Red” reduction condition, but not under “Green” or “Blue” reduction conditions (Figure 2A) (the reducing agent concentration: “Red” < “Blue” < “Green”). Different reduction conditions yield different reaction rates for starting material, intermediates, and final products.
Figure 2.
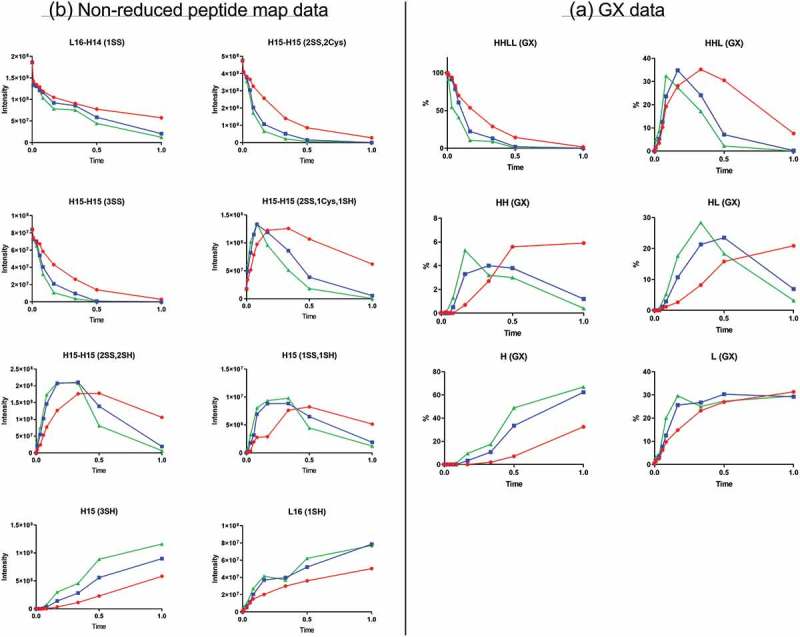
Reduction time-course data. Red, blue, and green represent three different reduction conditions. (A) GX data. H, heavy chain; L, light chain; HL, combination of heavy chain-light chain; HH, combination of heavy chain-heavy chain; HHL, combination of heavy chain-heavy chain-light chain; HHLL, heavy chain-heavy chain-light chain-light chain combination (whole antibody). (B) Nonreduced peptide map data. H15 is the hinge peptide containing three cysteines: two that form interchain disulfide bonds between the two heavy chains, and another engineered cysteine designed for conjugation. H14 and L16 are peptides containing cysteines that connect the antibody heavy chain and light chain. H15 (3SH) and L16 (1SH) are the final products of the reduction reaction. L16-H14 (1SS), peptide structure with disulfide bond link heavy chain and light chain; H15-H15 (2SS, 2Cys), peptide structure with disulfide bonds linking two heavy chains and two inserted cysteine capped with cysteinylation in the starting material; H15-H15 (3SS), structure with three disulfide bonds between two H15 peptides; H15-H15 (2SS, 1Cys, 1SH), intermediate with cysteinylation removed from one inserted cysteine; H15-H15 (2SS, 2SH), intermediate with cysteinylation removed from both inserted cysteines; H15 (1SS, 1SH), half-antibody structure intermediate.
Generally, the nonreduced peptide map data agreed well with the GX data (Figure 2B). The interchain disulfide bonds from the starting material, namely the disulfide bond linking light chain and heavy chain (peptide L16-H14(1SS)) and the disulfide bond between two heavy chains (peptide H15-H15(2SS, 2Cys)), decreased during reduction. At the same time, peptides representing the final light chain and heavy chain products, L16(1SH) and H15(3SH), increased as reduction progressed. Nonreduced peptide map data of the intermediate peptides, H15-H15(2SS, 1Cys, 1SH) and H15-H15(2SS, 2SH), showed the sequence by which cysteinylation was removed from the two inserted cysteines. By the end of reduction, the level of cysteinylation declined substantially under “Blue” and “Green” reduction conditions (Figure 2B). However, a substantial level of cysteinylated peptides remained by the end of reduction reaction under “Red” conditions, consistent with the GX data showing a high level of HHL, HH, and HL intermediate species under these conditions.
Nonreduced peptide mapping data also indicated the presence of peptide H15-H15(3SS), the unconjugated antibody structure containing three disulfide bonds (Figure 2B, Table 1), and its disappearance during reduction reaction. These data also suggested that a half-antibody structure, peptide H15(1SS, 1SH), was generated during reduction and further reduced eventually to H and L by the end of reaction. Although unconjugated antibody and half antibody did exist during the reduction process, they were eventually reduced to H and L, which indicated that the reduction reaction did not contribute to the heterogeneity seen with the final ADC product.
Oxidation time-course study
As expected, the H and L products from the reduction step were consumed quickly during the oxidation reaction (Figure 3A). The level of HHLL, the final product from oxidation, reached a plateau by the end of the reaction. The levels of HH and HHL, the oxidation reaction intermediates, increased initially and were consumed eventually to very low levels. The level of another intermediate, HL, also increased initially and decreased after it reached its maximum, but the remaining level of 10% was higher than that for HH and HHL and was steady through the end of the reaction. The HL intermediate generated during oxidation is likely the main source for the conjugated HL species (half-ADC) found in the final product.
Figure 3.
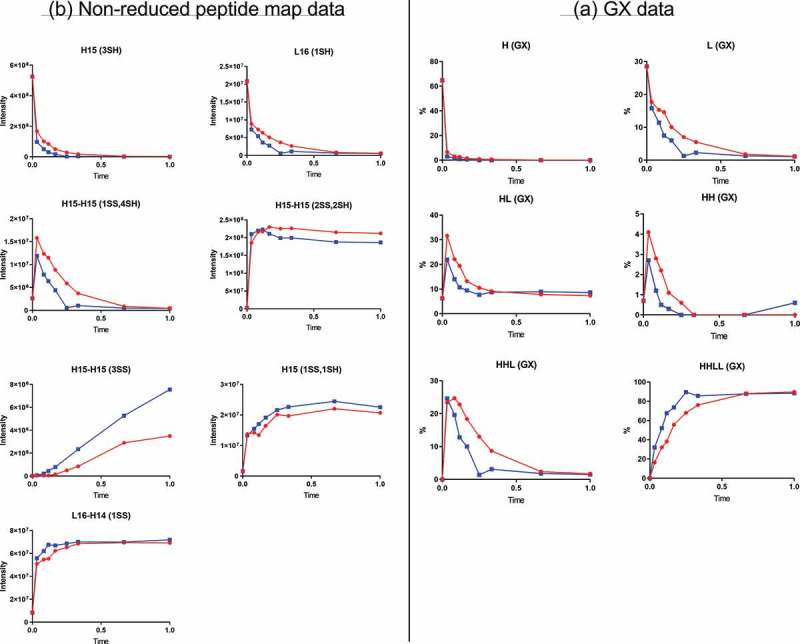
Oxidation time-course data. Red and blue represent two different oxidation conditions. (A) GX data. H, heavy chain; L, light chain; HL, combination of heavy chain-light chain; HH, combination of heavy chain-heavy chain; HHL, combination of heavy chain-heavy chain-light chain; HHLL, heavy chain-heavy chain-light chain-light chain. (B) Nonreduced peptide map data. H15 is the hinge peptide containing three cysteines: two that form interchain disulfide bonds between the two heavy chains, and another engineered cysteine designed for conjugation. H14 and L16 are peptides containing cysteines that connect the antibody heavy chain and light chain. H15 (3SH) and L16 (1SH) are the starting products for the oxidation reaction. H15-H15 (2SS, 2SH) and L16-H14 (1SS) are the desired final products of the oxidation reaction and contain disulfide bonds between the two heavy chains (H15-H15 (2SS, 2SH)) or the heavy and light chain (L16-H14 (1SS)). H15-H15 (1SS, 4SH), intermediate with one disulfide bond between two H15 peptide and four free cysteine; H15-H15 (3SS), undesired product containing three disulfide bonds, which correspond to the unconjugated antibody in the final ADC product; H15 (1SS, 1SH), undesired product corresponding to half-antibody, which is converted to half-ADC in the final ADC product.
Nonreduced peptide map data showed two peptides related to the desired product: peptide L16-H14(1SS), which contained a disulfide bond connecting the unconjugated heavy and light chains, and peptide H15-H15(2SS,2SH), which contained disulfide bonds connecting the two unconjugated heavy chains and two free-inserted cysteines (Figure 3B). The levels for both peptides reached a plateau early in the oxidation reaction, around 40% of total reaction time, indicating that the disulfide bonds were rebuilt successfully. Of note, the level of peptide H15-H15(2SS,2SH) decreased slightly after it reached its maximum. This decrease resulted from further oxidation of peptide H15-H15(2SS,2SH) to triple-disulfide-bond peptide structure H15-H15(3SS) (corresponding to unconjugated antibody in final ADC product). This finding was confirmed by the time-course profile of peptide H15-H15(3SS), where the level of this peptide increased continuously during oxidation. Different oxidation conditions generated different time-course profiles. As shown in Figure 3B, for example, the level of H15-H15(2SS,2SH) decreased faster from its maximum level, and the level of H15-H15(3SS) increased faster, under the “Blue” oxidation condition than under the “Red” oxidation condition (oxidizing agent concentration: “Red” < “Blue”).
The level of peptide H15 (1SS, 1SH), which corresponded to unconjugated HL, reached a plateau during oxidation. This plateau resulted from the formation of an intrachain disulfide bond between the two hinge cysteines in same heavy chain and their subsequent unavailability to form interchain disulfide bonds between the two heavy chains. Thus, the peptide map data were consistent with the GX profile for HL, which declined to 10% and remained there through the remainder of the oxidation reaction.
Conjugation time-course study
The GX data for the conjugation time course study revealed four peaks corresponding to HHLL, HHL, HL, and L (Figure 4A). These peaks remained fairly consistent during conjugation. Slight increases in HHLL and decreases in HL were observed, but those differences fell within the variation associated with the GX assay itself, so no unambiguous conclusions could be made about them. The slight increases of HHLL and decreases of HL might be explained by differences in the absorbance associated with the payload. After conjugation, two drug payloads were attached to HHLL, and only one payload was attached to HL. Because of this difference, HHLL showed more absorbance compared with HL.
Figure 4.
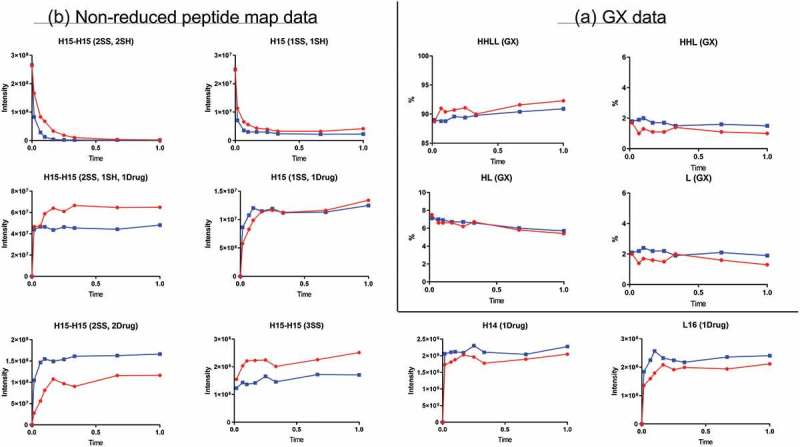
Conjugation time-course data. Red and blue represent two different conjugation conditions. (A) GX data. H, heavy chain; L, light chain; HL, combination of heavy chain-light chain; HHL, combination of heavy chain-heavy chain-light chain; HHLL, heavy chain-heavy chain-light chain-light chain. (B) Nonreduced peptide map data. H15 is the hinge peptide containing three cysteines: two that form interchain disulfide bonds between the two heavy chains, and another engineered cysteine designed for conjugation. H14 and L16 are peptides containing cysteines that connect the antibody heavy chain and light chain. H15-H15 (2SS, 2SH) is the desired starting product for conjugation, and H15 (1SS, 1SH) is the undesired, half-antibody starting product. H15-H15 (2SS, 2Drug) is the desired final product of conjugation. H15-H15 (2SS, 1SH, 1Drug), underconjugated product with a DAR of 1; H15 (1SS, 1Drug), half-ADC; H15-H15 (3SS), triple-disulfide bond structure (unconjugated antibody) generated during the oxidation step; H14 (1Drug), undesired overconjugated heavy-chain species; L16 (1Drug), undesired overconjugated light-chain species.
On the nonreduced peptide map for the conjugation time-course study (Figure 4B), levels of peptide H15-H15(2SS,2Drug), the peptide related to the desired conjugated product, reached its plateau very quickly and remained almost unchanged throughout the rest of the reaction. Undesired underconjugated species (related peptide: H15-H15(2SS,1SH,1Drug)) and overconjugated species (related peptide H14(1Drug) and L16(1Drug)) were also detected. Like the level of the desired peptide, the levels of these undesired species quickly reached a plateau and stayed almost unchanged through the rest of the reaction. These profiles indicate that longer conjugation times would not have changed the final composition of conjugation product and suggest that the conjugation reaction quickly reached equilibrium. However, the state of equilibrium differed based on conjugation conditions. More of the DAR2 product and less of the underconjugated species were generated under “Blue” conjugation conditions than under “Red” conjugation condition (payload concentration: “Red” < “Blue”). However, more overconjugated species were also generated under “Blue” conditions. Thus, conjugation reaction conditions should be optimized to balance the levels of underconjugated and overconjugated species.
The half-antibody species generated during oxidation was converted to half-ADC peptides during conjugation. The level of the unconjugated antibody generated during the oxidation step increased slightly during the conjugation reaction, most likely as a result of continuous oxidation during conjugation step.
Quenching time-course study
Surprisingly, the GX data showed that the level of intact ADC, represented here by HHLL, decreased initially and then stabilized by a certain time point (Figure 5A). The decrease in intact ADC occurred more rapidly under “Blue” quenching conditions than under “Red” quenching conditions (quenching agent concentration: “Red” < “Blue”), but it was significant under both conditions. The level of ADC fragments, as represented by HHL, HH, HL, and L, increased as the quenching reaction progressed, until a plateau was reached. Consistent with the rate at which intact ADC declined, fragment-generation occurred more rapidly under “blue” conditions than under “red” conditions.
Figure 5.
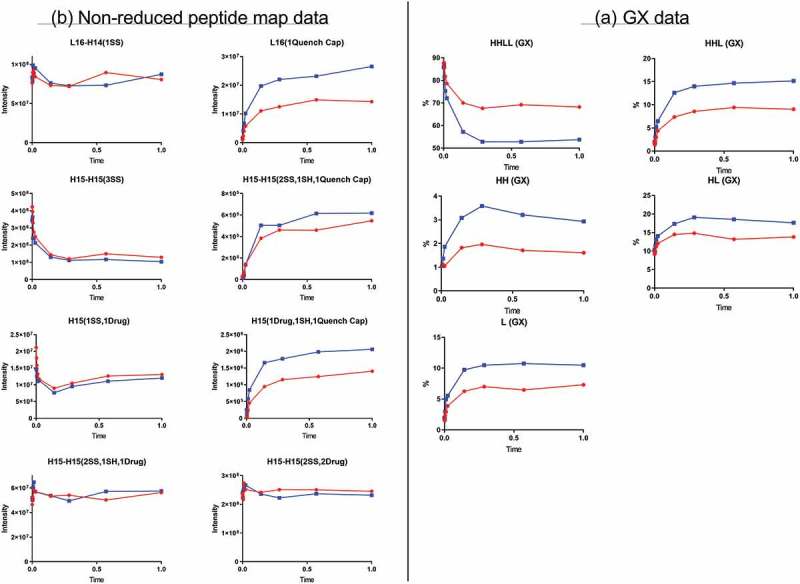
Quench time-course data. Red and blue represent two different quench conditions. (A) GX data. H, heavy chain; L, light chain; HL, combination of heavy chain-light chain; HH, combination of heavy chain-heavy chain; HHL, combination of heavy chain-heavy chain-light chain; HHLL, heavy chain-heavy chain-light chain-light chain. (B) Nonreduced peptide map data. H15 is the hinge peptide containing three cysteines: two that form interchain disulfide bonds between the two heavy chains, and another engineered cysteine designed for conjugation. H14 and L16 are peptides containing cysteines that connect the heavy chain and light chain. H15-H15 (2SS, 2Drug) is the desired product from the conjugation reaction. L16-H14 (1SS), peptide containing a disulfide bond between the light chain and heavy chain; L16 (1 Quench Cap), L16 attached to a molecule of quenching reagent; H15-H15 (3SS), triple-disulfide bond peptide structure (unconjugated antibody); H15-H15 (2SS, 1SH, 1Quench Cap), triple-disulfide bond structure with a molecule of quenching reagent attached; H15 (1SS, 1Drug), half-ADC; H15 (1Drug, 1SH, 1Quench Cap), half-ADC with a molecule of quenching reagent attached; H15-H15 (2SS, 1SH, 1Drug), underconjugated ADC.
Nonreduced peptide map data revealed the generation of new peptides during quenching (Figure 5B), which were the results of disulfide bond exchange reaction occurred during this step. The peptide L16 (1 Quench Cap), in which a molecule of quenching reagent was connected to a light-chain peptide through a disulfide bond, was not detected before the quenching step. The level of L16(1 Quench Cap) increased and reached a plateau during the quenching step. Consistent with this change, the level of corresponding peptide L16-H14(1SS) in which a disulfide bond connected L and H decreased slightly in the early stages of the quenching reaction. These changes indicated that the disulfide bond between L and H was reduced during the quenching reaction and that part of L16-H14(1SS) was converted into L16(1 Quench Cap). The possible mechanism was a disulfide bond exchange reaction between L16-H14(1SS) and the quench reagent (Scheme II). According to this mechanism, new peptides L16 (1 Quench Cap) and H14 (1 Quench Cap) were generated, and peptide L16-H14(1SS) was consumed. H14 (1 Quench Cap) was not detected by our peptide map method because it was too small and too hydrophilic to be retained on column.
Scheme II.
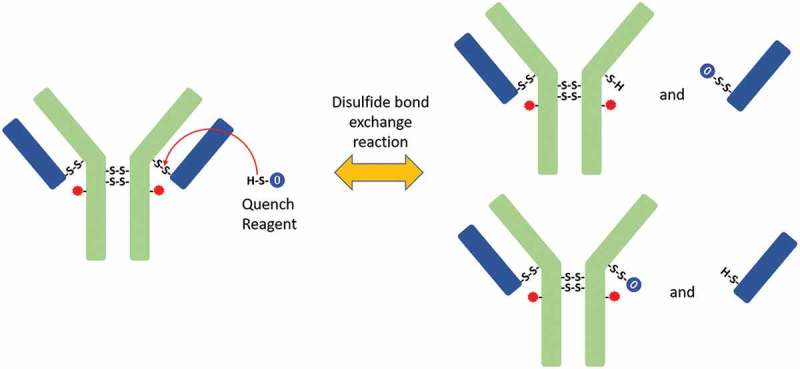
Disulfide bond exchange reaction. The free thiol of quenching reagent reacts with disulfide bond in ADC, which breaks the disulfide bond in ADC and attaches quenching reagent to heavy chain or light chain.
The quench reagent also appeared to react with the hinge disulfide bonds between the two heavy chains in the ADC. A new peptide, H15-H15 (2SS, 1SH, 1 Quench Cap), appeared and reached a plateau during quenching, whereas the level of the corresponding triple-disulfide bond peptide H15-H15 (3SS) decreased initially, then reached equilibrium, indicating that one disulfide bond between two H was exchanged with quenching reagent. The quenching reagent also appeared to react with the intradisulfide bond in the half-ADC, as illustrated by an increase in a new peptide, H15(1Drug, 1SH, 1 Quench Cap), and a decrease in peptide H15 (1SS, 1Drug). No significant changes were observed for peptides related to other species such as underconjugated or overconjugated species, indicating that the quenching reaction did not change those heterogeneities.
Disulfide bond exchange reaction also explained our GX data. The decrease in intact ADC (HHLL) resulted from the consumption of the disulfide bond between H and L or between two H by disulfide bond exchange with molecules of quenching reagent. Likewise, the increase in HHL, HL, HH, and L fragments resulted from disulfide bond exchange reaction.
Discussion
Overall, the heterogeneous species observed for our model ADC-A molecule included stereoisomers for the target ADC, which all had DARs of 2; unconjugated antibody; underconjugated species, which had a DAR of 1; overconjugated species, which had DARs of 3 and 4; and the size variants half-ADC, HHL, and L. The impact of this heterogeneity on ADC-A bioactivities varied among species. The stereoisomers of the target ADC showed similar bioactivity because the stereoisomers were not expected to affect the functionality of ADC. Size variants (half ADC, HHL, L) also did not show an impact on ADC-mediated bioactivity, which was expected because these fragment species bind together through noncovalent interactions under native conditions. On the other hand, the underconjugated and overconjugated species showed noticeable differences in ADC-mediated bioactivities, primarily due to payload differences.
To understand the root cause of the heterogeneity generated during the ADC conjugation process, we conducted time-course studies for each step in the conjugation workflow. These studies provided more detail about the conjugation process. For example, the distinct differences we observed between the different reduction conditions illustrated the power of the time-course study in process optimization. Reduction time-course data indicated that certain reduction conditions were required to successfully remove cysteinylation from two inserted cysteines. These data also indicated that undesired species, such as half-antibody and unconjugated antibody, existed but were eventually reduced to low levels during the reduction reaction. Thus, the reduction reaction did not contribute to the heterogeneity seen with the final ADC product.
Our data showed that the oxidation reaction successfully rebuilt the disulfide bonds between the two H and the disulfide bond between H and L. However, the oxidation step also generated undesired species such as half antibody, which accounted for approximately 10% of the final product from oxidation, and antibody with triple-disulfide bonds in the hinge region. During oxidation, an intrachain disulfide bond was formed in a single heavy chain, leading to the production of half-antibody. The half-antibody generated during oxidation was the main source of the half-ADC observed in the final ADC product. At the same time, the additional disulfide bond formed between the two inserted cysteines in the triple-disulfide bond structure generated during oxidation led to the generation of the unconjugated antibody observed alongside the final ADC at the end of the overall conjugation process. Different oxidation conditions produced different heterogeneity profiles, indicating the need to optimize these conditions carefully.
The time-course studies showed that the conjugation reaction successfully attached two drug payloads to the two inserted cysteines. However, this step also produced undesirable species in the form of underconjugated and overconjugated species, with DARs of 1 for the underconjugated species and 3 or 4 for the overconjugated ones. Because these species arose from conjugation equilibrium, increasing conjugation time would not change the final composition. At the same time, the unconjugated antibody generated during the oxidation step increased slightly during the conjugation process due to continuous oxidation.
The quench step contributed to the elevated level of fragments observed with the final ADC product. These elevated levels were the result of disulfide bond exchange reactions that occurred during the quench step, and this was confirmed by the appearance of additional peptide species and the consumption of corresponding peptides (Scheme II). Both GX data and nonreduced peptide map data showed the levels of these fragments reaching plateau, indicating that the disulfide bond exchange reaction eventually reached equilibrium. Generation of these fragments can be minimized by shortening the quenching reaction time.
We note that the heterogeneity discussed above and the impact on ADC activities are project-specific. Thus, the production of different ADCs may yield different types of heterogeneous species, and their impact on ADC biological activities should be evaluated for each molecule. However, the methodologies used to analyze ADC-associated heterogeneity and the use of time-course studies to understand the overall conjugation process will benefit all ADC developers. Understanding the chemical properties and reactions, such as stereoisomer structure, conjugation equilibrium, intradisulfide bond formatting, and disulfide bond exchange reactions, that contribute to ADC-associated heterogeneity will help ADC developers optimize their manufacturing processes, regardless of ADC project.
Materials and methods
Reagents and materials
Engineered antibody intermediate and the ADCs were produced by MedImmune (Gaithersburg, MD). Ammonium sulfate, sodium acetate N-Ethylmaleimide, sodium phosphate dibasic, sodium phosphate monobasic monohydrate, sodium chloride, formic acid, and trifluoroacetic acid (TFA) were obtained from Sigma-Aldrich. Urea (OmniPur), water (OmniSolv, HPLC, and spectrophotometry grade), acetonitrile (OmniSolv, HPLC and spectrophotometry grade), and isopropyl alcohol (OmniSolv, HPLC, and spectrophotometry grade) were obtained from EMD Serono. LysC was obtained from Promega. Dithiothreitol (no-weight format) was obtained from Pierce Protein Biology.
HIC
A Thermo MabPac HIC-10, 5 µm, 1000 A column (4.6 mm x 250 mm) was used for HIC chromatography. Mobile phase A consisted of 1.5 M ammonium sulfate and 20 mM sodium acetate, pH 5.0, and mobile phase B consisted of 20 mM sodium acetate, pH 5.0. ADC or mAb samples were diluted to 1 mg/mL in mobile phase A, and 50 µL were injected onto the HIC column. The column temperature was set at 30ºC. The sample was eluted in a gradient of 30% to 100% mobile phase B over 35 min at a flow rate of 1 mL/min. Eluted protein was detected by UV absorbance at a wavelength of 220 nm.
The challenge of developing HIC method comes from the separating of low-level unconjugated mAb (<2%) from the underconjugated ADC (peak 1 and 2, respectively, in the HIC method). These two species have similar hydrophobicity; hence, it is very challenging to separate them completely. Optimized separation was achieved by using appropriate salt concentration in mobile phase A.
DAR measurement by RP-HPLC
Each ADC or mAb sample was normalized to 2 mg/mL in HPLC water. Fifty μL of denaturing buffer (8 M guanidine HCl, 160 mM Tris, 1 mM EDTA, pH 7.6) and 2 μL of dithiothreitol (DTT) were added to 50 μL of each sample and incubated at 37ºC for 30 min. 10μL of each sample were injected onto a Waters BioResolve RP mAb polyphenyl column (2.1*150 mm, 2.7 µm, 450Å). Mobile phase A consisted of 0.1% TFA and water, and mobile phase B consisted of 0.1% TFA in acetonitrile. A gradient of 32.5% to 46.5% mobile phase B was run from 2 to 30 min at a flow rate of 0.5 mL/min. The eluted protein was detected by UV absorbance at a wavelength of 280 nm. DAR and drug-load distribution were calculated based on peak areas.
Since a RP-LC method is used to measure the DAR, a critical quality attribute for ADC, the precision and accuracy are very important for this method. The peak purity of H0, H1, H1’, and H2 has a significant impact on the accuracy and precision of the final DAR value calculation. Incomplete reduction will result in peaks splitting and overlapping, leading to inaccurate DAR values. Denatured condition was used to ensure complete reduction. Good separation is critical for this method, which was achieved by appropriate RP column and mobile phase and gradient combination.
nrCE-SDS analysis
ADC or mAb samples were denatured in a prepared sample buffer with 100 mM sodium phosphate and 4% SDS pH 6.0. Samples were diluted to 0.5 mg/mL in 50% v/v sample buffer, 5% v/v N-ethylmaleimide (300 mM stock), and water. Samples were then denatured on a heating block at 65°C for 5 min. Following denaturation, samples were spun at 13200 rpm to cool and collect condensate. Samples were loaded into microvials and placed in a Sciex PA800plus Capillary Electrophoresis system. The autosampler was set to 15°C for sample storage before testing. Data were collected through the 32 Karat Software, and then exported to Empower for data analysis.
SEC analysis
SEC analysis was performed by loading 200 μg of each ADC sample onto a Tosoh TSK-gel G3000SWxL column (7.8 mm x 30 cm) at 30ºC column temperature. The sample was eluted isocratically with a mobile phase composed of 0.1 M sodium phosphate, 0.1 M sodium sulfate and 0.05% sodium azide, pH 6.8, containing 10% isopropanol, at a flow rate of 0.8 mL/min over 22 min. Eluted protein was detected by UV absorbance at 280 nm.
AUC analysis
Sedimentation velocity experiments were performed on a Beckman Optima XL-I analytical ultracentrifuge. ADC samples were diluted to 0.5 mg/mL with reference buffer, and the resulting protein solution was loaded in a 12 mm centrifuge cell in the sample channel. Additionally, reference buffer was loaded into the reference channel of each cell. Loaded cells were placed into an AN-50Ti analytical rotor and equilibrated to 25°C. The samples were scanned under a rotor speed of 42,000 rpm at full vacuum while the optical density was measured at 280 nm. A total of 100 scans of each cell were collected for data analysis. The first scan of each sample was excluded to avoid artifacts caused by the meniscus.
GX analysis
Samples were first diluted to 2.0 mg/mL in 1 × phosphate-buffered saline containing N-ethylmaleimide. All samples and the protein ladder were heated on a heating block at 100°C for 2 min. Following denaturation, samples and the ladder were diluted with ultrapure water and loaded on a 96-well plate. The plate and a chip that contains the gel dye, the destain solution, and the protein express lower marker were placed into the LabChip GX System for analysis.
Non-reduced peptide mapping analysis
Samples were denatured in 100 mM phosphate buffer and 7 M guanidine, pH 7.0, at 37ºC for 30 min. The denatured protein solutions were diluted about fourfold in 100 mM phosphate buffer and 0.1 mM EDTA, and endoproteinase Lys-C was added at a 1:10 enzyme: protein ratio. The reaction mixtures were incubated at 37ºC overnight. The same amount of Lys-C was added, and the samples were incubated at 37ºC for another 4 to 6 h. Following Lys-C digestion, half of each reaction mixture was reduced by adding 500 mM DTT. The Lys-C digests were separated by an Acquity UPLC CSH C18 column (1.7 µm, 2.1 × 150 mm) and analyzed by a tunable ultraviolet detector and an online Orbitrap Fusion mass spectrometer. Mobile phase A consisted of 0.02% TFA in water, and mobile phase B contained 0.02% TFA in acetonitrile. Peptides were eluted at a flow rate of 0.2 mL/min with a gradient of 0% to 95% mobile phase B over 90 min.
The main challenge for non-reduced peptide mapping method is how to ensure consistent digestion across a wide dynamic extent of reduction, re-oxidation, conjugation reactions. The challenge was overcome by choosing an appropriate combination of denaturing condition and enzyme. Lys-C is the enzyme chosen due to its reliable activities at current denaturing condition.
Abbreviations
- ADC
antibody-drug conjugate
- AUC
analytical ultracentrifugation
- DAR
drug-to-antibody ratio
- GX
LabChip GX Protein Characterization System
- H
heavy chain species (antibody or ADC)
- H0
unconjugated heavy chain
- H1
heavy chain with one drug payload
- H2
heavy chain with two drug payloads
- HH
heavy chain-heavy chain species (antibody or ADC)
- HHL
heavy chain-heavy chain-light chain species (antibody or ADC)
- HHLL
heavy chain-heavy chain-light chain-light chain species (antibody or ADC)
- HIC
hydrophobic interaction chromatography
- HL
heavy chain-light chain species (antibody or ADC)
- IEC
ion-exchange chromatography
- L
light chain species (antibody or ADC)
- L0
unconjugated light chain
- L1
light chain with one drug payload
- nrCE-SDS
capillary electrophoresis sodium dodecyl sulfate with non-reduced sample preparation
- rCE-SDS
capillary electrophoresis sodium dodecyl sulfate with reduced sample preparation
- RP-HPLC
reverse-phase high-performance liquid chromatography
- SEC
size-exclusion chromatography
- TFA
trifluoroacetic acid
Acknowledgments
The authors thank Neetha Thomas, Anthony Shannon, and Douglas Johnson for GX analysis. Editorial support was provided by Frances McFarland (funded by MedImmune).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2017. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM.. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–81. doi: 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 4.Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53(15):3796–827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 5.Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016;68(1):3–19. doi: 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- 6.Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, Hallett W, Tsou HR, Upeslacis J, Shochat D, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13(1):47–58. [DOI] [PubMed] [Google Scholar]
- 7.Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–96. [PubMed] [Google Scholar]
- 8.Katz J, Janik JE, Younes A. Brentuximab Vedotin (SGN-35). Clin Cancer Res. 2011;17(20):6428–36. doi: 10.1158/1078-0432.CCR-11-0488. [DOI] [PubMed] [Google Scholar]
- 9.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17(20):6437–47. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 10.Kantarjian HM, Vandendries E, Advani AS. Inotuzumab ozogamicin for acute lymphoblastic leukemia. N Engl J Med. 2016;375(21):2100–01. doi: 10.1056/NEJMc1612040. [DOI] [PubMed] [Google Scholar]
- 11.Kim MT, Chen Y, Marhoul J, Jacobson F. Statistical modeling of the drug load distribution on trastuzumab emtansine (Kadcyla), a lysine-linked antibody drug conjugate. Bioconjug Chem. 2014;25(7):1223–32. doi: 10.1021/bc5000109. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005;14(9):2436–46. doi: 10.1110/ps.051478705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17(20):6417–27. doi: 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- 14.Sun MM, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FG, Ihle NC, Senter PD, Alley SC. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem. 2005;16(5):1282–90. doi: 10.1021/bc050201y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 16.Beckley NS, Lazzareschi KP, Chih HW, Sharma VK, Flores HL. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjug Chem. 2013;24(10):1674–83. doi: 10.1021/bc400182x. [DOI] [PubMed] [Google Scholar]
- 17.Adem YT, Schwarz KA, Duenas E, Patapoff TW, Galush WJ, Esue O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug Chem. 2014;25(4):656–64. doi: 10.1021/bc400439x. [DOI] [PubMed] [Google Scholar]
- 18.Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem. 2015;26(2):176–92. doi: 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sochaj AM, Swiderska KW, Otlewski J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol Adv. 2015;33(6 Pt 1):775–84. doi: 10.1016/j.biotechadv.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Akkapeddi P, Azizi SA, Freedy AM, Cal P, Gois PMP, Bernardes GJL. Construction of homogeneous antibody-drug conjugates using site-selective protein chemistry. Chem Sci. 2016;7(5):2954–63. doi: 10.1039/c6sc00170j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher D, Hackenberger CP, Leonhardt H, Helma J. Current status: site-specific antibody drug conjugates. J Clin Immunol. 2016;36(Suppl 1):100–07. doi: 10.1007/s10875-016-0265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian F, Lu Y, Manibusan A, Sellers A, Tran H, Sun Y, Phuong T, Barnett R, Hehli B, Song F, et al. A general approach to site-specific antibody drug conjugates. Proc Natl Acad Sci USA. 2014;111(5):1766–71. doi: 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, Rivers PJ, et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem. 2014;25(2):351–61. doi: 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]
- 24.Kornberger P, Skerra A. Sortase-catalyzed in vitro functionalization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. MAbs. 2014;6(2):354–66. doi: 10.4161/mabs.27444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, Jones LC, et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug Chem. 2014;25(7):1331–41. doi: 10.1021/bc500189z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dennler P, Chiotellis A, Fischer E, Bregeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug Chem. 2014;25(3):569–78. doi: 10.1021/bc400574z. [DOI] [PubMed] [Google Scholar]
- 27.Jones MW, Strickland RA, Schumacher FF, Caddick S, Baker JR, Gibson MI, Haddleton DM. Polymeric dibromomaleimides as extremely efficient disulfide bridging bioconjugation and pegylation agents. J Am Chem Soc. 2012;134(3):1847–52. doi: 10.1021/ja210335f. [DOI] [PubMed] [Google Scholar]
- 28.Badescu G, Bryant P, Bird M, Henseleit K, Swierkosz J, Parekh V, Tommasi R, Pawlisz E, Jurlewicz K, Farys M, et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem. 2014;25(6):1124–36. doi: 10.1021/bc500148x. [DOI] [PubMed] [Google Scholar]
- 29.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 30.Dimasi N, Fleming R, Zhong H, Bezabeh B, Kinneer K, Christie RJ, Fazenbaker C, Wu H, Gao C. Efficient preparation of site-specific antibody-drug conjugates using cysteine insertion. Mol Pharm. 2017;14(5):1501–16. doi: 10.1021/acs.molpharmaceut.6b00995. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, Welborn M, Zhu T, Yang NJ, Santos MS, Van Voorhis T, Pentelute BL. Pi-Clamp-mediated cysteine conjugation. Nat Chem. 2016;8(2):120–28. doi: 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.