

Abstract
Motivation
Increasing evidence has shown that nucleotide modifications such as methylation and hydroxymethylation on cytosine would greatly impact the binding of transcription factors (TFs). However, there is a lack of motif finding algorithms with the function to search for motifs with modified bases. In this study, we expand on our previous motif finding pipeline Epigram to provide systematic de novo motif discovery and performance evaluation on methylated DNA motifs.
Results
mEpigram outperforms both MEME and DREME on finding modified motifs in simulated data that mimics various motif enrichment scenarios. Furthermore we were able to identify methylated motifs in Arabidopsis DNA affinity purification sequencing (DAP-seq) data that were previously demonstrated to contain such motifs. When applied to TF ChIP-seq and DNA methylome data in H1 and GM12878, our method successfully identified novel methylated motifs that can be recognized by the TFs or their co-factors. We also observed spacing constraint between the canonical motif of the TF and the newly discovered methylated motifs, which suggests operative recognition of these cis-elements by collaborative proteins.
Availability and implementation
The mEpigram program is available at http://wanglab.ucsd.edu/star/mEpigram.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is a major epigenetic mark that plays crucial roles in many key biological processes. For example, the DNA methylation level at promoters is anti-correlated with gene expression (Smith and Meissner, 2013). DNA methylation often disrupts TF-DNA binding and thus represses transcription (Smith and Meissner, 2013). However, recent studies show that some TFs preferentially bind to methylated sequences that are often different from the canonical motifs they recognize (Hu et al., 2013). For example, Hu et al. have found that Kruppel-like factor 4 (KLF4) can bind to CCmCGCC sequence (mC refers to the methylated cytosine), which is different from its canonical motif (CACACC) (Hu et al., 2013).The protein RBP-J was also demonstrated to bind specifically to a methylated CpG-containing sequence (Bartels et al., 2011) (GmCGGGAA) in a methylation-dependent way. These observations illustrate the importance of identifying methylated motifs (m-motifs).
Currently, aside from Viner et al.’s work (Viner et al., 2016), which was submitted at the same time to BioRxiv as our tool (Ngo and Wang, 2016), there is no other computational method to identify m-motifs. Here we present a new version of our published motif finding method Epigram (Whitaker et al., 2015) to identify sequence motifs containing modifications such as methyl cytosine 5mC, non-CpG methyl cytosine mCpH, hydroxymethyl cytosine 5hmC, formylcytosine 5fC and carboxylcytosine 5caC. Epigram can identify motifs in very large sets of sequences such as in a previous study in 980 465 sequences with a mean length of 1640 bp (Whitaker et al., 2015). Such a large set of sequences would be impractical for other motif finding programs to process. For example, HOMER would simply crash given such a dataset (Heinz et al., 2010; Whitaker et al., 2015); MEME only accepts input size of ≤60 000 characters with sequence lengths of ≤1000 base pairs (Bailey et al., 2006; Tran and Huang, 2014). Epigram’s scaling efficiency is comparable to that of DREME (Bailey, 2011) but it can find motifs longer than 8 base pairs, which is the default motif length in DREME (Bailey, 2011). The program discovers motifs by building position-specific weight matrices (PWMs) from the most enriched k-mers in the positive sequences over the negative sequences as ‘seeds’ and extending the motifs to both directions (Whitaker et al., 2015). We have expanded the alphabet that can in principle represent any modification such as 5mC, 5hmC, 5fC and 5caC. The latest version of MEME and DREME allows quite flexible alphabet but our method shows comparable or superior performance on various scenarios. This new algorithm provides a powerful tool to identify DNA motifs containing any modifications, which would greatly facilitate analyzing the epigenomic data mapping different DNA or RNA modifications.
2 Materials and methods
2.1 Data processing and filtering
2.1.1 Bisulfite sequencing data processing
For H1, the processed whole genome bisulfite sequencing (WGBS) data was obtained from the Roadmap Epigenomics project (GEO: GSM429322); for GM12878, the raw WGBS data was obtained from the ENCODE consortium (GEO: GSM1002650). We used Bismark (Krueger and Andrews, 2011) to process the GM12878 methylome data. We chose bowtie2 (Langmead and Salzberg, 2012) as the option for the process. In addition, we ignored the first 3 nucleotides on read number 2 of each pair-end reads since they have the tendency to be biased. When computing methylation level, we only considered loci covered by at least 2 reads.
Methylated genomes for both H1 and GM12878 were created. Cytosines with beta value ≥0.5 were considered methylated and incorporated into the methylated genomes as the character E instead of C.
2.1.2 ChIP-seq data
TF ChIP-seq peaks for both H1 and GM12878 were downloaded from the ENCODE Consortium. The ChIP-seq datasets contain from 1113 to 75 690 peaks each, with an average of 13 980 peaks; the average length of each peak ranges from 152 to 1426 bp. Peaks were called by the ENCODE consortium (Landt et al., 2012).
2.1.3 DAP-seq data
DAP-seq data for Arabidopsis were downloaded from GEO: GSE60143. Called methylation data was obtained from GEO: GSM1085222. A methylated genome was constructed using methylation calls from the downloaded data.
2.1.4 Data availability
Processed data including methylated genomic sequences of H1 and GM12878 are available in the mEpigram github repository.
2.2 Methods
2.2.1 Motif comparison
To compare our identified motifs with the original PWMs in the simulations, the correlation between the identified PWMs and the original PWMs are calculated. During the comparison, a PWM slides on top of the other and Pearson correlation is calculated for the overlapping segment. The Python function scipy.stats.pearsonr() was used to calculate Pearson correlation. The minimum size of the overlapping segment is 6 bp. Let m1, m2 be two motif PWMs. Let the overlapping segment start at position i for m1 and j for m2. For each overlapping position, a Pearson correlation score is calculated, which compares distribution of bases at m1i and m2j.
These scores were then averaged to get the overall score. The average score per position was calculated as:
, with n being the number of overlapping positions (n ≥ 6).
The similarity scores of all possible alignments, including reverse-complementary, were computed and the smallest one was the distance between m1 and m2.
2.2.2 Motif scanning tool
Because of the introduction of the new bases, a new motif-scanning tool is needed in Epigram to search for matching k-mers using the modified motifs (m-motifs). To scan for the occurrences of a motif of interest in a set of DNA sequences, the program first simulates a score distribution for the motif by dinucleotide-shuffling the input sequences and calculates the scores for all of the k-mers inside the shuffled sequences using the motif’s PWM. The shuffling is repeated several times until an adequate number of k-mers is achieved (set to be 1 million in this study). Motif matches are called by passing a score threshold. This score threshold is defined by given P-values so that only a fraction equaling to the P-value can pass. For example, the score threshold for P-value of 0.01 is the lowest score in the top 1% of the k-mers from the shuffled sequences. The score of a k-mer given a motif (represented as a PWM) is calculated as:
where w is the motif width, and are the probabilities of observing nucleotide xi at position i from the motif and the background distributions, respectively. In this study, we use P-value cutoff of 0.0001.
2.2.3 Choosing appropriate P-value for motif-scanning
For motif scanning, the lower P-value cutoffs gave higher precision but lower sensitivity while the higher cutoffs gave lower precision and higher sensitivity. We calculated the average information content (IC)/position for all known motifs in human (Kulakovskiy et al., 2015). We tested different P-value cutoffs and found that the value of 0.0001 is the most appropriate: for motifs with IC per position of 0.6–1.0, the sensitivity ranges from 0.6 to 0.93 (Supplementary Table S1) while the precision stays above 0.5 for motifs inserted into more than 2% of the sequences. Thus, the default P-value cutoff was set at 0.0001.
2.2.4 Spatial analysis
We carried out functional analyses of the m-motifs using our customized scripts. The approach is based on SpaMo (Whitington et al., 2011). The peak sequences were reformatted into 500 bp-long sequences centered at the center of each original peak. RepeatMasker (Smit et al., 1996) was used to mask repeated sequences with chains of ‘N’ to reduce false positives. We first used our motif scanner to find matches for each of the novel m-motifs in TF peak sequences. The P-value cutoff chosen was 0.001, as the stricter cutoff 0.0001 did not result in many significant matches for our spatial analysis. Our primary interest is the novel methylated motifs; therefore, we used the m-motifs as the primary motifs and look for their significant binding partners. The algorithm assumes that every spacing between the primary and secondary motifs is equally probable if there is no spatial constrain between the two motifs. Then, the number of co-occurrences of the two motifs at a given displacement will follow a binomial distribution with the number of trials as the total number of co-occurrences. The P-values are calculated using this model. Bonferoni’s correction was used to adjust the P-values. The P-value threshold for identifying a significant spacing was chosen at 0.001.
2.2.5 Cross-scan between H1 and GM12878
The motifs found for each TF in H1 were scanned against the peaks for the same TF in GM12878, vice versa. For each TF with replicates, the peak sequences were pooled together to calculate an average enrichment. To determine a significant enrichment level, we generated a distribution of possible enrichment scores using shuffled sets of sequences. The distribution is approximately normal; the enrichment score of 1.5 is approximately 3 standard deviations from the mean (Figure S1). Therefore, we chose 1.5 as the enrichment cutoff for our motifs. An m-motif is considered to have consistent enrichment in both cell types if it has an enrichment score of at least 1.5 in its original cell type, appears in at least 1% of the peaks, and when compared to the original enrichment score (E1), the new enrichment score (E2) has to be similar to it, i.e: E1/E2 < 1.5 and E2/E1 < 1.5. For differentially enriched m-motifs, the enrichment scores have to satisfy: E1/E2 > 1.5 or E2/E1 > 1.5.
2.2.6 Scanning after de-methylation
Similarly to cross-scanning between H1 and GM12878, the m-motif has to have original enrichment of at least 1.5 and appear in at least 1% of the peaks.
2.2.7 Information content calculation
Motifs’ ICs are calculated as:
With pi, j as probability of the DNA sequence having character j at location i of the sequence. The probabilities are provided in the motif’s PWM.
3 Results
3.1 Expansion on de novo motif discovery
In general, mEpigram looks for enriched motifs, both unmethylated and methylated ones, enriched in the input sequences. The program first computes an enrichment score for each k-mer based on how often it appears in the input sequences compared to the shuffled input sequences and a genomic background. K-mers are then ranked based on their final weights:
With W as the k-mer’s enrichment weight, PP as the proportion of sequences that contains the k-mer over the total number of input sequences, Ewg as the k-mer’s enrichment over the genomic background and Esh as its enrichment over the shuffed input. PWMs are then generated by first picking a top k-mer and enriched k-mers similar to it to construct a ‘seed’ PWM, which is then extended by adding more enriched k-mers that are a few base pairs shifted from the original one (for more details, see Whitaker et al., 2015, Supplementary Online Methods). The motifs output by mEpigram are ranked based on their enrichments in the input sequences. The enrichment is calculated as the ratio of number of motif matches in the input sequences over the number of motif matches in the dinucleotide shuffled sequences.
The de novo discovery step requires input of both DNA sequences of interest and the DNA modification data such as DNA methylation data. It has two modes: typeE for finding motifs containing methylated CpG dinucleotides (mCpG), typeEF for other cytosine modifications such as non-CpG methyl cytosine mCpH, hydroxymethyl cytosine 5hmC, formylcytosine 5fC and carboxylcytosine 5caC. The typeE mode only finds symmetrical CpG methylation, i.e mC appears on both strands of the DNA sequence and the reverse complement (the DNA sequence on the other strand of the DNA double helix) of a mCpG dinucleotide is itself, e.g. the reverse complement of ACGTmCG is mCGACGT. In this scenario, there is no need to mark the guanine that pairs with mC and thus we only need to add one base (E) to the conventional four bases (A, C, G, T) to represent mC. In the typeEF mode, we introduce another base F to mark the guanine that pairs with the modified cytosine. This mode creates a more complex alphabet with an increased running time.
The algorithm can reliably identify canonical motifs in ChIP-seq datasets (Fig. 1).
Fig. 1.
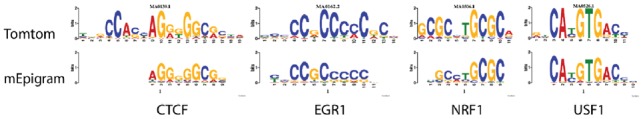
Top ranking non-methylated motifs found by mEpigram are consistent with canonical motifs. For each alignment, the top image is the canonical motif; the bottom image is mEpigram’s result. The top motifs found by mEpigram are compared to other databases using TomTom (Gupta et al., 2007). Matches with P-value lower than 10−6 are reported
3.2 mEpigram outperforms MEME Suite’s algorithms in simulated tests
To evaluate the performance of mEpigram, we conducted several rounds of simulated tests and compared the results with those of MEME Suite’s motif finders. Motifs of different information contents (IC, which measures how conserved a motif is, see Methods) and lengths are inserted to different numbers of sequences at different abundances (Supplementary Table S1). Known motif PWMs were taken from HOCOMOCOv10 (Kulakovskiy et al., 2013) database, with the addition of a constructed PWM to represent our test motif gataEGca. For known motifs, a pair of mCpG was added to each PWM manually at a random site with the probability of mC between 0.7 and 1.0 (Supplementary Table S1). Random sequences of average length 250 and standard deviation of 50 are generated using the human genome as background. The number of sequences is 250, 1000, 5000, 20 000, respectively. For each sequence set, random k-mers from motif PWMs are generated and inserted into 1, 2, 5, 10 and 25% of the sequences.
To determine whether a motif is found, we calculated the Pearson correlation between the original PWM and the output PWMs from each method (see Methods). For each run, only the top three outputs were considered. A motif is considered found in a sequence set if one of the output PWMs has a Pearson correlation of at least 0.85 compared to the original. Default parameters were used for the tests. Since by default, MEME does not allow more than 60 000 base pairs of input, we only performed the test with 250 sequences for MEME. We have also tested MEME by setting the -maxsize parameter to accept larger sequence sets. However, the run time increases quadratically with respect to sequence size, thus making it impractical to use MEME on larger sequence sets. On a computer with a CPU of Xeon E5630 2.53GHz, version 4.12 of MEME took 23.4 min to process 250 sequences of average length of 250 bp, and 353.6 min to process 1000 sequences. For 5000 sequences, the program would take about 6 days. In contrast, our method scales linearly with time. On the same CPU, it takes 19.6 min for 1000 sequences, and 232.2 min for 40 000 sequences.
At large sequence sizes and high ICs (more conserved), mEpigram is able to find motifs in as low as 1% of the sequences (Supplementary Table S1). Compared to the MEME Suite, mEpigram can more reliably retrieve inserted motif in 48.43% of the test cases (Table 1). DREME has the closest performance to mEpigram (44.69%), however the motifs generated by DREME are mostly shorter (6–7 bp, compare to mEpigram’s 8–9 bp), which ignores parts of inserted motifs. MEME has the best performance when the number of input sequences is small. One advantage the MEME suite has over our method is the flexibility in incorporating different alphabets to the motif finder. However, although our method restricts the extension to character E and F, they can still be used to represent other nucleotide modifications (5hmC, 5fC, 5caC, etc.). In this work, we focus on methylated cytosine.
Table 1.
Performance comparison between mEpigram and MEME Suite
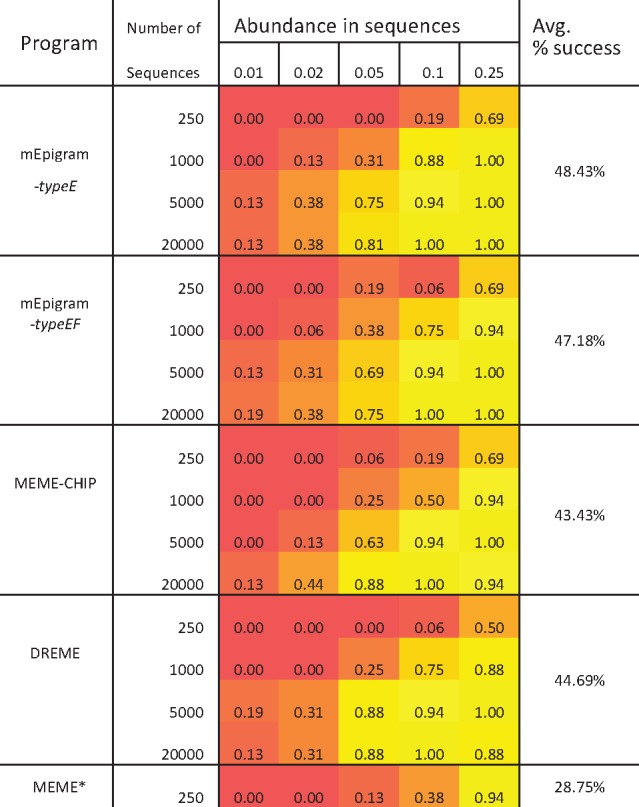
|
Note: mEpigram is able to find inserted m-motifs in more cases than MEME-Suite’s methods.
Since it is impractical for MEME to process large sequence data, the program is only tested on datasets of 250 sequences.
3.3 Identifying m-motifs in known TFs that preferentially bind methylated sites
O’Malley et al. (2016) have demonstrated certain TFs do prefer methylated sequences when bind with DNA. We obtained 270 TF DAP-Seq datasets from this study to test our algorithm’s ability to find m-motifs in validated data. The TFs were categorized based on their sensitivity toward changes in methylation levels. This was calculated as the change in binding affinity after removing methylation from the sequences (O’Malley et al., 2016). Out of 270 TFs, 11 are considered to bind to methylated sequences preferentially, 66 have no preference between methylated and non-methylated DNA, 193 are disrupted by methylation.
We added an additional filter to the program to replicate O’Malley et al.’s method. TF binding specificity was used to simulate TF binding affinity. The binding specificity of a TF motif is defined as the ratio of the density of its loci (number of loci over region length) within the TF-binding regions over the density of its loci found in other regions of the genome. Motif loci are found using our scanning method (see Supplementary) at P-value of 10−4. The importance of methylation in a motif is measured by the logarithm of the fold change in binding specificity before and after removing methylation information from an m-motif (logFC). To remove methylation information from a methylated motif, we simple convert the probabilities for mC in the motif’s PWM to 0.0, and add that probability to the probability of C at the same position. A significant m-motif is thus defined as: i) Appears in at least twice as many regions in the input sequences compared to the dinucleotide-shuffled sequences. ii) When methylation information is removed, m-motif’s binding specificity is reduced by at least 2.83 folds, which corresponds to logFC of 1.5. Since 5-methylcytosine (5mC) in plants, as opposed to mammals, also occur largely in CHG context (Saze et al., 2012), typeEF of the algorithm was used as it does not assume only CpG methylations.
As the result, we were able to find significant m-motifs for all of the 11 TFs reported to bind preferentially to methylated DNA. Out of 66 TFs that were considered insensitive to DNA methylation, seven of them contain significant m-motifs. Only 1 out of 193 TFs known to be disrupted by DNA methylation have m-motifs. Without this additional filter, the numbers are respectively: 11, 40 and 46.
3.4 Application of mEpigram to TF ChIP-seq data
3.4.1 Retrieval of m-motifs
DNA methylation has been believed to disrupt binding of TFs but recent studies suggested that some TFs may prefer methylated motifs [e.g CEBPB (Mann et al., 2013)]. mEpigram provides a tool to study the impact of DNA methylation on TF using the in vivo ChIP-seq binding data. To this end, we applied mEpigram to 55 TF ChIP-seq data in H1 and 44 datasets in GM12878 generated by ENCODE together with the whole genome bisulfite sequencing (WGBS) data. A cytosine is considered methylated when its beta-value is >0.5. In the mEpigram runs, the maximum number of output motifs was set at the default 200. Motifs from the same run were aligned to each other and redundant motifs were removed. Since the data we used only contains CpG methylation information, we took advantage of the typeE mode as it can handle longer k-mers and thus offers higher sensitivity.
First of all, mEpigram successfully found the canonical motifs in majority of the experiments, which indicates the success of the motif-finding algorithm. In H1, 35 out of 40 known canonical motifs were correctly identified by mEpigram in the top 5 most enriched motifs from the corresponding TF ChIP-seq experiments (Supplementary Table S2). For GM12878, 24 out of 31 known canonical motifs are identified (Supplementary Table S2).
The number of m-motifs with enrichment of >1.5 found for each TF ranges from 0 to 16 (Supplementary Table S2). Out of 55 ChIP-seq datasets in H1, 31 show enrichment for m-motifs >1.5 fold (Supplementary Table S2). For GM12878, 24 out of 44 datasets have significantly enriched m-motifs.
The presence of m-motif can have different meanings for each TF. A TF can preferentially bind to methylated sequences or simple tolerate methylations. For example, the top m-motif motif of CEBPB in H1 is highly enriched and very similar to that of the canonical motif. It is identified at the majority of the canonical motif’s loci. For NRF1, the top m-motif does appear at the canonical motif’s loci but less frequently (Supplementary Fig. S2A). There is a sharp increase in methylation level at the center of the motif-matching loci (Supplementary Fig. S3). This suggests that the canonical CEBPB motif binds preferably to methylated sequences. However, the methylation levels around the matching loci for CEBPB’s canonical motif is significantly lower than that of its m-motif, which shows CEBPB does not require methylation to bind with its motif (Supplementary Fig. S2B). For NRF1, the canonical motif prefers regions of lower methylation levels, whereas the m-motif is found in regions with higher methylation levels (Supplementary Fig. S3). In contrast to CEBPB, there is no spike of methylation level in the center of the plot for NRF1’s canonical motif. It can be interpreted that the NRF1 TF does not prefer methylated sequences, but it is insensitive to methylation.
For m-motifs that are different from their TF’s canonical motifs, for example CTCF and EGR1, they do not co-occur in the same ChIP-seq peaks with the TF’s canonical motifs often. CTCF and EGR1’s m-motifs are present in regions with high level of methylation (about 0.8) while their canonical motifs prefer low methylation levels (0.1–0.2, Supplementary Fig. S3). Given that the ChIP-seq peaks tend to occur in low methylation levels, these peaks having high methylation levels suggests that these m-motifs counter-act the DNA methylation to recruit the TF. We hypothesize that removing the methylation of these m-motifs will disrupt the TF’s binding to these loci.
In most of these cases, the m-motif is very different from the canonical one or the most enriched motif present; some examples are shown in Table 2.
Table 2.
Datasets with significant m-motifs: number of motifs and m-motifs of some of the samples with the most enriched m-motifs
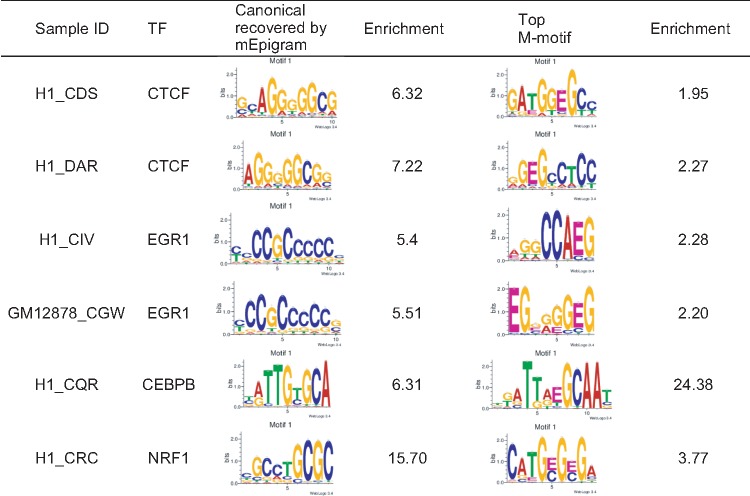
|
Some of the TFs in Table 2 have been previously shown to either have interactions with DNA methylation or bind with specific methylated DNA sequences. For instance, CTCF is known to bind to DNA in a methylation specific manner and CTCF binding is regulated partly by differential DNA methylation (Wang et al., 2012). The top m-motifs for two replicates of CTCF in H1 are similar to each other (Pearson correlation is 0.931 when aligned together with several bp shift). The third CTCF experiment in H1 (H1_CIU) used a different antibody and a different m-motif was found.
For CEBPB, the top motif is the methylated canonical CEBPB motif, which is consistent with the previous observation that CEBPB binds to 39% of the methylated canonical sequence (Mann et al., 2013). We also discovered a strong m-motif for NRF1, present in 3.68% of the peaks, that is the canonical motif methylated at its CG dinucleotide. As a comparison, the canonical motif is present in 25% of the sequences. This finding is consistent with the observation that NRF1 TF exhibits binding with methylated sequences (Hu et al., 2013).
3.4.2 Importance of cytosine
Furthermore, we evaluated the importance of cytosine methylation in the identified m-motifs. For each sample, we first de-methylated the m-motifs identified by mEpigram. In each PWM, at each position i, the probability of E, , was added to and then was set to zero. The resulted PWMs were next scanned against their respective peak regions without the methylation information (containing only A, C, G, T). Some of the m-motifs, when scanned after de-methylation, retain their enrichment (Table 3). This is often because these m-motifs are the methylated canonical motifs. For example, CEBPB and NRF1’s top m-motifs are both methylated canonical motifs. Their enrichments remain relatively unchanged after de-methylation. This further suggests that DNA methylation doesn’t hinder the TFs bindings. In contrast, some m-motifs have their enrichment significantly reduced after de-methylation (Table 3). These motifs generally contain more than 1 methylated cytosine in their sequences. Thus, removing the methylation greatly changes their enrichment. These motifs are likely selectively bound by their TFs.
Table 3.
A) Examples of the m-motifs retaining high enrichments after de-methylation. B) Examples of the m-motifs enriched in their methylated form but not after de-methylation
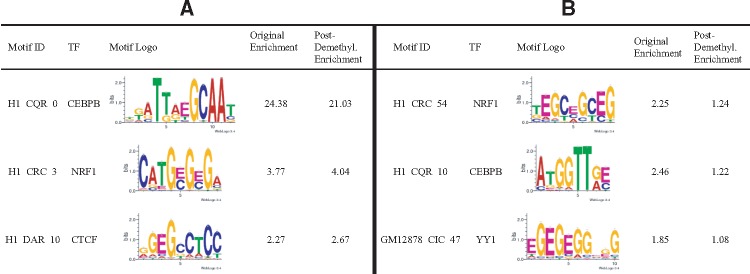
|
3.4.3 Distance constraints
We next searched for distance constraints between methylated motifs and other motifs using the SpaMo algorithm (Whitington et al., 2011) (Table 4). RepeatMasker (Smit et al., 1996, at www.repeatmasker.org) was used to mask repeated sequences with chains of ‘N’ to reduce false positives. Some m-motifs exhibit highly significant spacing constraints with other motifs, most notably the motifs identified from CTCF binding peaks. These CTCF motifs have enrichments over 1.5 and the SpaMo analyses gave the adjusted P-values of less than 0.001.
Table 4.
Most significant spacing pairs between m-motifs and other motifs
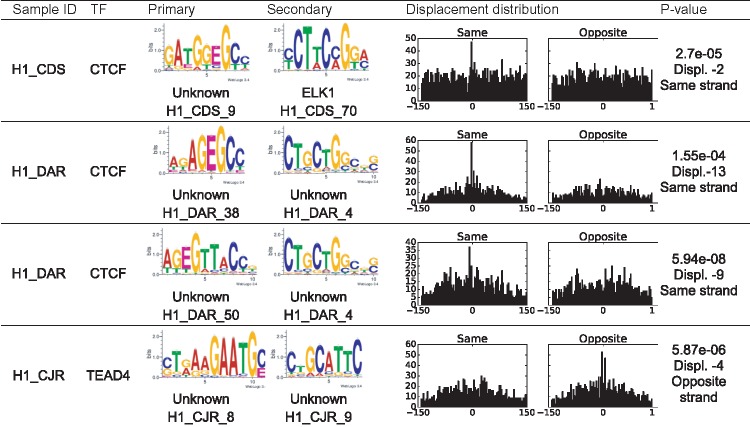
|
Note: The motifs were scanned against HOCOMOCOv10 (Kulakovskiy et al., 2013) database and only matches with E-value less than 0.1 were accepted. The distance is the base pairs between the 3' end of the primary motif and the 5' end of the secondary motif. The P-value cutoff for significant displacement was set at 0.001. The histograms show the distributions of displacements between the primary and secondary motifs. The X-axis is the displacement, the Y-axis is frequency.
3.5 Comparison of m-motifs in H1 and GM12878
To identify m-motifs that are common or unique in H1 and GM12878, we scanned motifs found in H1 peaks against GM12878 peaks and vice versa using our own method (P-value <10−4). The enrichments of the m-motifs in the other cell type were calculated and compared with the enrichment in the cell type where they were discovered. For CTCF, ERG1 and NRF1, several m-motifs are enriched in both GM12878 and H1. These motifs have enrichments of over 1.5 and appear in at least 2% of the peaks in both of the datasets. The top NRF1 m-motifs found in H1 and GM12878 are very similar to each other (Table 5). The top m-motifs for EGR1, on the other hand, are different from one another. The top CEBPB m-motifs found in in H1 were enriched in GM12878. In general, the motifs found in GM12878 for CEBPB appear significantly less often, the maximum enrichment is 4.7 compared to 24.38 in H1 (Table 5). This is unsurprising given the differences between the H1 and GM12878 data.
Table 5.
A) Some of the consistently enriched m-motifs between H1 and GM12878: the enrichments remain significant when scanned in both H1 and GM12878 data. B) Some of the differentially enriched m-motifs between H1 and GM12878
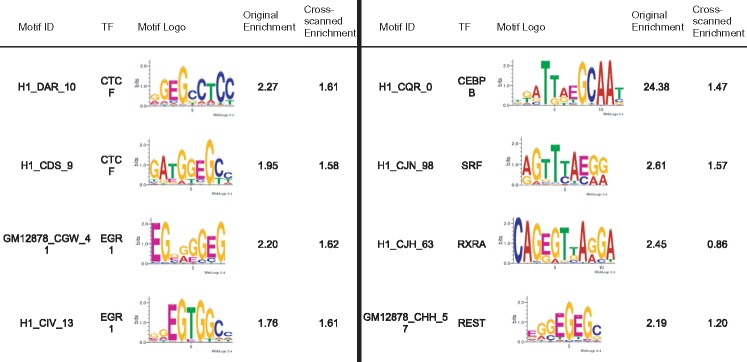
|
3.6 Effect of 5hmC on mEpigram results
Because 5-hydroxymethylcytosine and 5-methylcytosine are undistinguishable by bisulfite sequencing, we examined the effect of 5hmC’s presence on our findings. Using TAB-seq data for H1 (Yu et al., 2012), we found that the large majority of CG loci only contain 5hmC in low probability (95% of the CpG loci in H1 have 5hmC level of less than 0.1, with the mean of 0.045). Removing 5hmC from H1 methylation data did not significantly change the mEpigram results.
4 Discussion
We present here one of the first attempts to expand alphabet in motif search to meet the need of integrative analysis of sequence and epigenomic data. We have demonstrated the power and usefulness of mEpigram in identifying methylated motifs when combining sequence and DNA methylation. mEpigram can readily consider other modifications such as 5hmC, 5fC and 5cacC. When applied to analyzing human TF ChIP-seq data, mEpigram found that several TFs have significantly enriched methylated motifs. The most enriched m-motifs are the methylated canonical motifs (CEBPB, NRF1), which suggests that these TFs may be tolerant or prefer binding to their methylated sequences. Furthermore, additional novel m-motifs, that are not necessarily as enriched as canonical motifs, were also found in many TF binding regions (CTCF, EGR1). Particularly interesting, some of these methylated motifs are significantly enriched in the methylated form compared to the unmethylated form, which suggests possible impact of TF binding by methylation.
Author contributions
Vu Ngo modified the Epigram program, performed the analyses and wrote the manuscript, Mengchi Wang contributed to the data analysis and manuscript preparation, Wei Wang conceived the idea, designed the project, interpreted the data and wrote the manuscript.
Funding
This work was partially supported by NIH (U54HG006997 R01HG009626) and CIRM (RB5-07012).
Conflict of Interest: none declared.
Supplementary Material
References
- Bailey T.L. (2011) DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics, 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. et al. (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res., 34(Web Server issue), W369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels S.J.J. et al. (2011) A SILAC-based screen for methyl-CPG binding proteins identifies RBP-J as a DNA methylation and sequence-specific binding protein. PLoS One, 6, e25884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. et al. (2007) Quantifying similarity between motifs. Genome Biol., 8, R24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S. et al. (2013) DNA methylation presents distinct binding sites for human transcription factors. eLife, 2013, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. (2011) Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy I.V. et al. (2013) HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res., 41 (Database issue), D195–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy I.V. et al. (2015) HOCOMOCO: expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res. 44, D116–D125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt S.G. et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012) Fast Gapped-read Alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann I.K. et al. (2013) CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res., 23, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo V., Wang W. (2016) Finding de novo methylated DNA motifs. doi: 10.1101/043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley R.C. et al. (2016) Erratum: cistrome and epicistrome features shape the regulatory DNA landscape (Cell (2016) 165(5) (1280–1292)). Cell, 166, 1598.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H. et al. (2012) DNA methylation in plants: relationship to small rnas and histone modifications, and functions in transposon inactivation. Plant Cell Physiol., 53, 766–784. [DOI] [PubMed] [Google Scholar]
- Smit A. et al. (1996) RepeatMasker Open-3.0http://www.repeatmasker.org. [Google Scholar]
- Smith Z.D., Meissner A. (2013) DNA methylation: roles in mammalian development. Nat. Rev. Genet., 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Tran N.T.L., Huang C.-H. (2014) A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol. Direct, 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner C. et al. (2016) Modeling methyl-sensitive transcription factor motifs with an expanded epigenetic alphabet. doi: 10.1101/043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. (2012) Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res., 22, 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker J.W. et al. (2015) Predicting the human epigenome from DNA motifs. Nat. Methods, 12, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington T. et al. (2011) Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res., 39, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. et al. (2012) Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell, 149, 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.