

Abstract
Cell cycle regulation, especially faithful DNA replication and mitosis, are crucial to maintain genome stability. Cyclin-dependent kinase (CDK)/cyclin complexes drive most processes in cellular proliferation. In response to DNA damage, cell cycle surveillance mechanisms enable normal cells to arrest and undergo repair processes. Perturbations in genomic stability can lead to tumor development and suggest that cell cycle regulators could be effective targets in anticancer therapy. However, many clinical trials ended in failure due to off-target effects of the inhibitors used. Here, we investigate in vivo the importance of WEE1- and MYT1-dependent inhibitory phosphorylation of mammalian CDK1. We generated Cdk1AF knockin mice, in which two inhibitory phosphorylation sites are replaced by the non-phosphorylatable amino acids T14A/Y15F. We uncovered that monoallelic expression of CDK1AF is early embryonic lethal in mice and induces S phase arrest accompanied by γH2AX and DNA damage checkpoint activation in mouse embryonic fibroblasts (MEFs). The chromosomal fragmentation in Cdk1AF MEFs does not rely on CDK2 and is partly caused by premature activation of MUS81-SLX4 structure-specific endonuclease complexes, as well as untimely onset of chromosome condensation followed by nuclear lamina disassembly. We provide evidence that tumor development in liver expressing CDK1AF is inhibited. Interestingly, the regulatory mechanisms that impede cell proliferation in CDK1AF expressing cells differ partially from the actions of the WEE1 inhibitor, MK-1775, with p53 expression determining the sensitivity of cells to the drug response. Thus, our work highlights the importance of improved therapeutic strategies for patients with various cancer types and may explain why some patients respond better to WEE1 inhibitors.
Subject terms: Cancer models, Mitosis
Introduction
Proliferation of eukaryotic cells requires faithful replication and segregation of the genetic material during cell cycle progression and mitosis. Timely progression through these events is regulated by cyclin-dependent kinases (CDKs). Interestingly, CDK1 alone is sufficient to drive mouse embryogenesis until midgestation in the absence of interphase CDKs (CDK2/3/4/6) [1], whereas CDK1 loss leads to early embryonic lethality due to mitosis entry failure [2]. CDK activity is temporally and spatially regulated through the cell cycle and requires binding to cyclin partners [3], as well as posttranslational modifications. Once associated with cyclin A2 or B1, CDK1 activity is further controlled through its inhibitory phosphorylation on threonine 14 (T14 [4–6]), tyrosine 15 (Y15 [4, 7]), and its activating phosphorylation on threonine 161 (T161[8, 9]). WEE1-dependent phosphorylation on the Y15 residue prevents unscheduled entry into mitosis by keeping CDK1/cyclin B1 complexes in a low activity state [10–13]. In metazoans, MYT1-dependent modification of T14 precludes premature activation of CDK1/cyclin B1 complexes right before their nuclear translocation [4, 6, 13, 14]. The dual-specificity phosphatase CDC25 regulates the mitotic onset by the abrupt dephosphorylation of T14 and Y15, triggering complete activation of CDK1/cyclin B1 complexes [15–17]. Therefore, the dephosphorylation of CDK1 on its inhibitory residues is believed to be a rate-limiting step for its activation and entry into mitosis [18, 19].
As expected, overexpression of CDK1, as well as high expression of its binding partner cyclin B1 [20, 21], has been described in many cancers with poor prognosis. Altered activity of CDK1 due to deranged p53 and the DNA damage-signaling pathway has been reported [22–29]. In response to DNA damage, induction of CHK1/CHK2 controls CDK1 activity via WEE1 and CDC25 regulation to ensure complete DNA repair before entry into mitosis [26, 30]. As the DNA damage response (DDR) is suppressed by elevated CDK1 activity, triggering premature mitotic events in combination with DNA-damaging agents has become an attractive therapeutic strategy for cancer patients [31–33]. Based on this, WEE1 inhibition is an effective approach in clinical trials [29, 33]. However, its off-target effects and cross-inhibition of other kinases (like CDK2) needs further research to establish an effective anticancer strategy [34–38]. The assessment of premature CDK1 activity using diverse approaches has resulted in variable results. WEE1 itself is essential for proliferation and embryogenesis in mice [39] and its inhibition/silencing induces DNA damage but also mitotic catastrophe [29, 34] through premature activation of several substrates including the structure selective endonuclease (SSE) MUS81 [34, 40, 41] in multiple cell lines. In contrast, the ectopic expression of mutant CDK1T14A/Y15F (hereafter referred to as CDK1AF) alone in mammalian cell lines has rather insignificant effects on S phase progression and mitotic timing but when co-expressed with cyclin B1, greatly increases the frequency of premature mitotic events [11, 42, 43]. In addition, the levels of the overexpressed mutant CDK1AF in presence or absence of wild-type CDK1 affects the biological outcome [44].
To fully understand the impact of aberrant CDK1 activity and the importance of the inhibitory phosphorylation on T14 and Y15 at the endogenous level in vivo, we have taken advantage of our recently generated Cdk1AF knockin mouse model, in which both inhibitory phosphorylation sites are replaced by non-phosphorylatable amino acids, T14A and Y15F [45]. We observed that monoallelic expression of Cdk1AF leads to early embryonic lethality and is associated with altered activation of key cell cycle regulators, premature mitotic events, increased levels of DNA damage, replication stress and chromosomal fragmentation leading to S phase failure. We provide evidence of the involvement of MUS81 in these defects, which indicates that inhibitory phosphorylation of CDK1 during S phase safeguards genomic integrity by protecting chromatin from unscheduled endonucleolytic digestion by the mitotic MUS81-SLX4 complexes. Moreover, our work unravels the importance of the p53 status for the sensitivity of cells to CDK1 inhibitory phosphorylation, both in Cdk1AF and control cells treated with the WEE1 inhibitor, MK-1775. Last but not least, we show that liver expressing mutant CDK1AF protein does not develop tumors unlike control mice after induction of tumorigenesis.
Results
The expression of CDK1AF leads to lethality accompanied by DNA damage in mice
To investigate the consequences of CDK1AF expression in vivo, we crossed Cdk1+/SAF (hereafter referred to as Cdk1SAF; SAF stands for Stop-AF [45], whereas AF refers to the AF allele that is actively expressed) animals with mice ubiquitously expressing β-actin-Cre and analyzed the progeny at different developmental stages. Resulting Cdk1AF P21 pups and E13.5 embryos were not viable, whereas mutant blastocysts (E3.5) were obtained at expected frequency (Table 1). Compared with controls, Cdk1AF blastocysts displayed a reduced number of cells accompanied with an increase in the phosphorylation on S139 of the H2AX histone variant (hereafter called γH2AX) (Fig. 1a). To further examine the effects of the ubiquitous CDK1AF expression in adult mice, we injected tamoxifen in Cdk1SAF animals harboring the Rosa26-CreERT2 transgene [46] (hereafter referred to as Rosa-Cre). Similarly to what we previously observed for Cdk1 knockout adult mice [2], animals expressing CDK1AF died within 5–6 days after tamoxifen administration, indicating that CDK1AF expression is also lethal in adult animals. Spleen of control and mutant animals was collected 4 days after tamoxifen injection to evaluate the extent of the DNA damage. Staining for γH2AX of spleen (Fig. 1b) and other tissue sections (data not shown) from Cdk1AF mice revealed a prominent signal increase compared with control mice. Comet assays on splenocytes from Cdk1AF mice confirmed the observed increase of DNA damage since the tail moment was 14 times higher than in the control animals (Fig. 1c, d). In order to assess whether CDK1AF expression could lead to apoptosis, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays on spleen sections from control and CDK1AF-expressing mice. Less than 1% apoptotic cells were observed in wild-type mice, whereas in Cdk1AF mice >8% were detected (Fig. 1e; yellow arrows, Fig. 1f). These in vivo observations suggest that the timely control of CDK1 activity via its inhibitory phosphorylation on T14 and Y15 is essential during the embryogenesis and adult life to prevent the formation of DNA breaks and the onset of apoptosis.
Table 1.
Cdk1+/AF mice are early embryonic lethal
| Age | Total | Control | Cdk1 AF |
|---|---|---|---|
| P21 pups | 39 | 39 | 0 |
| E13.5 embryos | 47 | 47 | 0 |
| E3.5 blastocysts | 33 | 25 (76%) | 8 (24%) |
Fig. 1.

The expression of CDK1AF leads to early embryonic lethality. a Control (Cdk1+/SAF) and β-actin-Cre Cdk1+/AF blastocysts were visualized with Hoechst staining (nuclei). The level of DNA damage was assessed through immunofluorescence staining of phospho-H2AX. b The expression of Cdk1+/AF was induced in all tissues of adult Rosa26-CreERT2 mice upon tamoxifen IP administration. Spleen sections were stained for phospho-γH2AX to visualize DNA damage response. c DNA damage in spleen of control and Rosa26-CreERT2 mutant mice was analyzed by Comet assays using the tail moment as a parameter to determine the extent of DNA breaks. d Quantification of DNA breaks was calculated based on the tail moment. e To investigate chromosomal fragmentation, spleen sections from tamoxifen injected control and Cdk1+/AF Rosa26-CreERT2 mice were analyzed by TUNEL assay to evaluate apoptosis. Yellow arrows indicate apoptotic cells with extensive DNA breaks. f The number of apoptotic cells was quantified in relation to the total number of cells
S phase failure in Cdk1AF MEFs
To unravel the molecular mechanism of Cdk1AF-induced lethality, we used mouse embryonic fibroblasts (MEFs) expressing 4-hydroxytamoxifen (4-OHT) inducible Esr1-CreERT2 to induce CDK1AF expression. As displayed in Fig. 2a, the proliferation of Cdk1AF MEFs over 7 days was impaired once released after synchronization at G0/G1 by serum starvation. Fluorescence-activated cell sorting (FACS) analysis of BrdU-labeled cells after release from serum starvation revealed the appearance of an intermediate BrdU-negative population located between the G1 and G2 population of mutant cells (Fig. 2b and S1A, red arrow). Both control and Cdk1AF MEFs were able to enter S phase and initiated replication at 16 h after release (Figure S1A). Over a period of 30 h, during which control MEFs successfully duplicated their genome and divided (Figure S1A), Cdk1AF cells displayed an increasing population with partially replicated DNA and never completed DNA replication. The percentage of Cdk1AF cells forming the intermediate population, which could be interpreted as unfinished S phase or S phase failure, gradually increased from 4% at 16 h to 42 % at the 30-h time point (Fig. 2c). The lack of proliferation indicated that the S phase cell cycle arrest in Cdk1AF MEFs was permanent.
Fig. 2.

Characterization of Cdk1AF MEFs. Primary MEFs were synchronized at G0/G1 by serum starvation as described in Materials and methods section. The expression of CDK1AF was induced during the starvation period. a The proliferative potential of Cdk1flox/SAF (control) and Cdk1null/AF (Cdk1AF) MEFs was monitored by alamarBlue proliferation assay for seven days. b The distribution of cells at 21-h time point after release from serum starvation was analyzed by BrdU FACS. DNA content was determined by propidium iodide (PI) staining. Based on BrdU incorporation and DNA content cells were classified into four categories: G1, S, G2/M and S phase failure. c Quantitative analysis of S phase failure from (b). Data are represented as mean ± SD from three independent experiments. d BrdU FACS analysis of Cdk1+/SAF (control) and Cdk1+/AF MEFs (Cdk1AF) at 24 h after release in the absence or presence of 10 µM of CDK1 inhibitor, RO-3306. e The abundance of chromatin-bound proteins was determined in Cdk1flox/SAF and Cdk1null/AF MEFs by cellular fractionation followed by western blotting with the indicated antibodies. Histone H3 served as a loading control. f Protein extracts from Cdk1flox/SAF and Cdk1null/AF MEFs were subjected to western blotting using the indicated antibodies. HSP90 served as a loading control. g Protein extracts from (d) were subjected to immunoprecipitation with the indicated antibodies. Kinase activities of immunoprecipitates were measured by in vitro kinase assays, in which radioactive ATP and histone H1 were used as substrates. Results are representative of two (e)/three (f–g) independent experiments. * unspecific band; p – phosphorylated form of Cdk1
Our past work suggests that the loss of either CDK2 or CDK1 activity does not have major effects on S phase progression [2, 47]. To investigate whether a reduction of CDK1 activity could rescue Cdk1AF-induced defects, we treated MEFs with the specific CDK1 inhibitor, RO-3306 [48]. The low dose of RO-3306 used had no effect on S phase progression in control cells but rescued S phase failure in Cdk1AF MEFs (Fig. 2d, S1B). This indicates that the unscheduled CDK1 activity is the cause of the impaired cell cycle progression in mutant fibroblasts.
However, the molecular pathway leading to S phase failure in mutant Cdk1AF cells still remains unknown. To determine its origin, we first investigated the initiation of DNA replication by assessing the formation and loading of the prereplication complex (pre-RC) and its transformation into the preinitiation complex (pre-IC). The pre-RC consists of a double hexameric MCM complex, ORC, CDC6 and CDT1 proteins [49–55]. The pre-RC is loaded at replication origins during G1 phase. Once cells enter S phase, the pre-RC is converted to active helicase/replisome pre-IC in a DDK/CDK-dependent manner by recruiting CDC45 [56] followed by GINS2 loading induced by CDK2/cyclin A activity [57, 58]. We assessed the abundance of different replisome components recruited to chromatin using cellular fractionation followed by western blotting (Fig. 2e). None of the replication factors were detected on chromatin in control and mutant cells in the early G1 phase (6 h). This is likely due to the low expression levels after serum starvation, as these proteins were also undetected in the soluble fraction (Figure S1C). By 16 h, pre-RCs were already converted into active replisomes for both genotypes, as all three replication factors, CDC6, CDC45 and GINS2, were bound to the chromatin. However, by 24 h, the amount of chromatin-bound CDC45 and GINS2 in Cdk1AF fibroblasts was reduced compared with controls. Similarly, the recruitment of proliferating cell nuclear antigen (PCNA), a DNA clamp that stabilizes active replisomes on chromatin and facilitates leading strand synthesis during DNA replication [59, 60], was also reduced in mutant fibroblasts compared with control cells. As shown in Figure S1C, the amount of soluble CDC6 was comparable for both genotypes at any given time, while CDC45, GINS2 and PCNA levels were reduced in mutant cells. Moreover, by 30 h, hardly any replication factors were detected on chromatin in Cdk1AF MEFs (Fig. 2e). Overall, our data indicate that mutant fibroblasts assemble pre-RC and initiate DNA synthesis rather normally. However, impaired DNA replication in Cdk1AF MEFs is likely associated with reduced numbers of active replisomes, which could contribute to the S phase arrest. The lack of replication factors bound to chromatin at 30 h in mutant cells might suggest failure of surveillance mechanisms to stabilize replication forks, which results in DNA breakage [61].
To understand the molecular basis underlying the interrupted S phase, we evaluated the expression levels and the associated kinase activity of key cell cycle regulators including CDK1, CDK2, cyclin B1 and cyclin A2 at different time points after release (Fig. 2f, g). CDK1/cyclin B1 complexes are known to be the main regulators of mitosis [62, 63], whereas CDK2 in complex with cyclin A2 controls S phase progression [64–66]. At the G1/S transition (14 h), the protein expression profiles of control and Cdk1AF MEFs were similar, with only a mild reduction in CDK1 and cyclin A2 levels in mutant cells (Fig. 2f). As the cells progressed into S phase, these differences in protein expression levels between control and mutant cells became more pronounced. In control cells, CDK1 was expressed as early as 14 h after serum starvation and the protein levels gradually increased until it reached a maximum around 24–30 h. Moreover, only in control MEFs we detected the shift in electrophoretic mobility for CDK1 (Fig. 2f), which is likely related to its phosphorylation status [8, 19]. To verify whether the molecular shift was associated with inhibitory phosphorylation of CDK1 on Y15, we performed western blotting using antibodies against phospho-Y15 (pY15). In control MEFs, pY15 was detected between 18 and 30 h when most of the cells progress from S phase to mitosis. In Cdk1AF MEFs, no pY15 was detected, which indicates the lack of WEE1-mediated phosphorylation in both CDK1 and CDK2 (the pY15 antibody recognizes both). In addition, cyclin B1 and A2 were expressed at much lower levels in mutant cells compared with controls (Fig. 2f).
To investigate the kinase activities associated with CDK/cyclin complexes, we performed in vitro kinase assays. During early S phase (16 h), control MEFs exhibited low kinase activities associated with all tested CDKs and cyclins (Fig. 2g), as expected. The kinase activities increased gradually with a peak around 21 h for CDK2/cyclin A2 and at 30 h for CDK1/cyclin B1 complexes. In contrast, the kinase activity associated with CDK1 and cyclin B1 was elevated in Cdk1AF MEFs compared with controls. These were detected as early as 16 h and peaked at 21 h despite that cyclin B1 levels were low (Fig. 2g). This premature activity of CDK1AF was expected because in the absence of WEE1 phosphorylation, CDK1 is activated immediately by binding to the cyclin subunit (assuming that CDK1 is fully phosphorylated on T161 by CDK-activating kinase [CAK] as has been shown before [67]). Furthermore, Cdk1AF MEFs exhibited a significant decrease in kinase activities associated with CDK2 and cyclin A2 compared with controls (Fig. 2g).
Premature mitotic events are observed in S phase in Cdk1AF MEFs
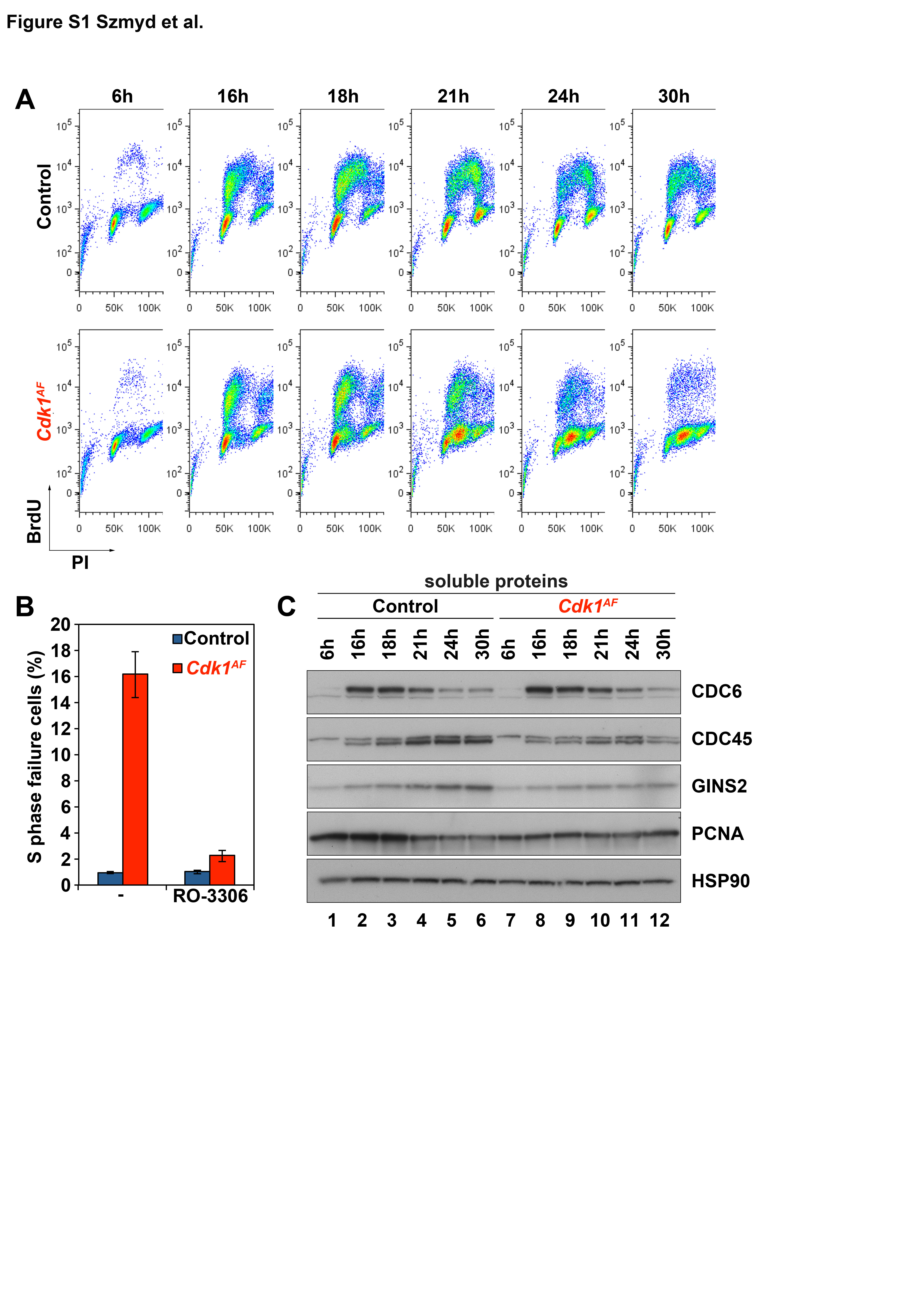
The conserved mechanism leading to mitotic entry is controlled by the WEE1/MYT1 – CDC25 regulatory loop, which regulates CDK1 activity by determining the phosphorylation status of T14 and Y15 [5, 6, 10–12, 14–17, 68]. At the end of G2 phase, the CDC25 phosphatase dephosphorylates CDK1 leading to its immediate activation. Upon CDK1/cyclin A2 activation, the nuclear envelope (NE) breaks down and CDK1/cyclin B1 complexes accumulate in the nucleus causing a sequence of events [69]. CDK1 phosphorylates the nuclear lamina leading to nuclear envelope break down (NEBD) [70, 71]. CDK1 also triggers chromosome condensation due to histone modification including phosphorylation of histone H3 on S10 by Aurora kinase B [72, 73], and induces the phosphorylation of histone H1, which is one of the first known indicators of mitosis [74]. To investigate whether the increase in CDK1 activity during S phase leads to premature mitotic events in Cdk1AF MEFs, we determined the expression of mitotic markers in Cdk1AF MEFs. CDK1-dependent phosphorylation of Lamin A/C on S22 and S392 inhibits the assembly of nuclear lamina and is associated with NEBD [75, 76]. As shown in Fig. 3a, control MEFs displayed elevated phosphorylation of Lamin A/C on S22 only 24 h after release, at a time when cells start to enter mitosis. Interestingly, the increase in phosphorylation on S22 in mutant MEFs was already detected at 18 h when cells were still in S phase and remained at high levels until 24 h. To further investigate the nuclear lamina disassembly in Cdk1AF MEFs, we monitored the cell cycle progression of mutant cells via time-lapse microscopy using stable expression of green fluorescent protein (GFP)-Lamin A/C and 53BP1-mCherry [77, 78]. The GFP-Lamin A/C construct was used to directly monitor NE disassembly, whereas 53BP1-mCherry helped to visualize the accumulation of DNA damage and NE reassembly [79–81]. We determined the mitosis-like state as a time range between NEBD and NE reassembly. NEBD occurred in control MEFs approximately 2.5 h later compared with Cdk1AF MEFs (Fig. 3b, left panel, S2A). Interestingly, Cdk1AF MEFs remained longer in mitotic-like state (55 min) compared with control fibroblasts (43 min) (Fig. 3b, right panel). Moreover, Cdk1AF MEFs were unable to properly reassemble nuclear lamina (Figure S2B) and exited the mitotic-like state with distorted nuclear architecture (Fig. 3c, S2B).
Fig. 3.

Premature mitotic events in Cdk1AF MEFs. MEFs were synchronized at G0/G1 as described in Materials and methods section. a Cells were harvested at different times after release from serum starvation. Protein extracts from Cdk1+/SAF and Cdk1+/AF MEFs were subjected to western blotting using the indicated antibodies. Lamin A/C served as a loading control. b By using live cell imaging in Cdk1+/SAF (n = 27) and Cdk1+/AF (n = 35) MEFs, NEBD was monitored by the disappearance of GFP-lamin A/C expression, while mitosis-like state was calculated by the timing between 53BP1 re-accumulation and NEBD (see Figure S2A). Statistical significance was assessed by unpaired t-test with Welch’s correction. c Cdk1+/SAF and Cdk1+/AF MEFs were fixed 24 h after release from serum starvation. The integrity of nuclear envelope was determined by immunofluorescence staining with antibodies against lamin A/C and Lamin B1. DNA was stained with Hoechst dye. d DNA condensation was determined in Cdk1flox/SAF and Cdk1null/AF MEFs at different time points after serum starvation by pS10-histone H3 FACS. DNA was stained with PI. Red font – premature DNA condensation in S phase; green font – DNA condensation at G2/M; Noco – nocodazole. Nocodazole-treated Cdk1flox/SAF cells served as a positive control. Results are representative of three independent experiments
To further characterize the mitotic phenotype of Cdk1AF MEFs, we determined the presence of mitotic markers including posttranslational modifications of histones H1 and H3. It has been shown that phosphorylation of histone H1 occurs early in the cell cycle and reaches its saturation in mitosis [74]. In line with this study, low levels of phosphorylation of histone H1 in control MEFs were detected when cells entered S phase (16 h) and then gradually increased until 24 h (Fig. 3a, lanes 2–5). In contrast, high levels of phospho-histone H1 persisted in Cdk1AF MEFs through the time course of the experiment. As shown in Fig. 3a, the phosphorylation of histone H3 on S10 in control cells was detected at 24 h after release, which overlapped with increased phosphorylation of Lamin A/C on S22 and histone H1, suggesting entry into mitosis at that time. On the other hand, Cdk1AF MEFs exhibited premature and elevated phosphorylation of histone H3 on S10 as indicated by western blot (Fig. 3a) and FACS (Fig. 3d) at 16 h after release. Importantly, FACS analysis revealed that premature phosphorylation of histone H3 on S10 occurred in cells with 2 n DNA content, suggesting that an early condensation of incompletely replicated DNA might trigger further complications during cell division.
Interrupted S phase in Cdk1AF MEFs causes chromosomal fragmentation and intra-S phase checkpoint activation
So far, our data indicate that CDK1AF expression leads to interruption of DNA replication in MEFs while it causes a prominent increase of γH2AX staining in tissues, indicating DNA damage. In order to elucidate the underlying molecular mechanisms responsible for the observed phenotype, we verified the presence of DNA damage in MEFs by FACS analysis using antibodies against γH2AX (Fig. 4a). γH2AX in control MEFs was barely detectable and never exceeded 1% of the total signal, likely caused by physiological DNA lesions during S phase. In contrast, Cdk1AF MEFs displayed substantial levels of γH2AX as soon as cells entered S phase (Fig. 4b; 12 ± 5% at 16 h) and reached its maximum after 24 h (41 ± 9%). Cdk1AF cells had higher γH2AX levels than control cells treated with Adriamycin, a DNA-damaging agent (15 ± 8% of cells). Importantly, γH2AX-positive cells were mostly restricted to cells with under-replicated DNA (≈3 n DNA content), which may suggest that mutant fibroblasts underwent replication stress.
Fig. 4.

DDR in Cdk1AF MEFs. MEFs were prepared as described in Materials and methods section. a The level of DNA damage was measured in synchronized Cdk1flox/SAF and Cdk1null/AF MEFs by phospho-H2AX FACS. DNA content was determined by PI staining. Red font – DNA damage in S phase; green font – DNA damage at G2/M. Adriamycin-treated (Adr) Cdk1flox/SAF cells served as a positive control. b Quantitative analysis of DNA damage from (a). Data are represented as mean ± SD from at least two independent experiments. c Chromosomal fragmentation was assessed in Cdk1flox/SAF and Cdk1null/AF MEFs by PFGE. The amounts of broken DNA were normalized to intact DNA and control at 6 h using ImageJ.  intact DNA;
intact DNA; 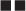 DNA breaks. d Western blots for phosphorylated CHK1 in control and Cdk1AF MEFs. Hsp90 served as a loading control. e Western blots for total (35 kDa) and cleaved CASP3 (• 19 kDa; •• 17 kDa bands) and HSP90 in Cdk1flox/SAF and Cdk1null/AF MEFs at the indicated time points after release from serum starvation. Adriamycin-treated (Adr) Cdk1flox/SAF MEFs served as a positive control
DNA breaks. d Western blots for phosphorylated CHK1 in control and Cdk1AF MEFs. Hsp90 served as a loading control. e Western blots for total (35 kDa) and cleaved CASP3 (• 19 kDa; •• 17 kDa bands) and HSP90 in Cdk1flox/SAF and Cdk1null/AF MEFs at the indicated time points after release from serum starvation. Adriamycin-treated (Adr) Cdk1flox/SAF MEFs served as a positive control
To confirm whether CDK1AF-induced replication stress is associated with chromosomal fragmentation, we analyzed the formation of DNA breaks by pulsed field gel electrophoresis (PFGE) (Fig. 4c). We could not detect any DNA fragmentation in control MEFs. In contrast, Cdk1AF MEFs exhibited a gradual increase in chromosomal breakage with a peak around 21–24 h (lanes 8–11), which was higher than the amount of DNA breaks in Adriamycin-treated fibroblasts (Fig. 4c, lane 13).
To further decipher whether DNA damage induced by CDK1AF activates DDR, we examined the phosphorylation status of the DDR regulator CHK1. In control MEFs, phosphorylated CHK1 was barely detectable (Fig. 4d, lanes 1–6). In contrast, Cdk1AF MEFs displayed significant levels of CHK1 phosphorylated on S345 at 16–24 h compared with control cells (Fig. 4d, lanes 7–12), similar as has been reported for intra-S phase DNA damage induced cells [82].
Chromatin condensation and DNA fragmentation can be triggered by caspase-3, a well-known hallmark of programmed cell death [83], which undergoes autocatalytic cleavage to become fully active [84, 85]. To determine whether the observed DNA breaks in mutant cells are the result of replication stress or apoptosis, we evaluated the expression levels of cleaved caspase-3 at different time points after serum starvation release (Fig. 4e). We detected significant levels of cleaved caspase-3 in Adriamycin-treated (Adr) control cells (19 kDa and 17 kDa bands), as well as in control MEFs at 48 h (19 kDa band). Nevertheless, we did not detect cleaved caspase-3 in mutant cells at any of analyzed time points. Taken together, our data indicate that apoptosis is most likely the long-term consequence of DNA damage triggered by prematurely active CDK1.
Depletion of Mus81 partially rescues DNA damage and S phase arrest in Cdk1AF cells
Controlled processing of joint molecule intermediates of homologous recombination (HR), which often consists of four-way structures known as Holiday Junctions (HJ), is indispensable for proper chromosome segregation in mitosis [86]. The BLM–TopoIIIα–RMI1–RMI2 (BTR complex) is known to dissolve HJs, whereas MUS81–EME1–SLX1–SLX4 and GEN1 endonuclease complexes contribute to the resolution of recombination intermediates [87]. Proliferating cells preferentially use BTR in S phase [88], whereas MUS81–EME1–SLX1–SLX4 and GEN1 cleave DNA at G2/M phase and mitosis, respectively [89]. Importantly, GEN1 functions are restrained by nuclear exclusion [86, 90], whereas MUS81 activation relies on CDK1 activity [91, 92]. CDK1 phosphorylates both MUS81 and its scaffold protein, SLX4, promoting an assembly of active MUS81–SLX4 complex on chromatin [91, 92]. Therefore, we aimed to investigate the status of the MUS81–SLX4 complexes in Cdk1AF MEFs.
In control cells, we were able to detect SLX4-bound MUS81 complexes in mitotic extracts derived from cells arrested by nocodazole (noco) (Fig. 5a, lane 2) but not in S phase (18 h) (Fig. 5a, lane 1), which was expected because CDK1 activity peaks in mitosis and phosphorylates MUS81 and SLX4 [91–93]. However, in Cdk1AF MEFs, MUS81–SLX4 complexes were already present in S phase (Fig. 5a, lane 3). The treatment of mutant cells with nocodazole (Fig. 5a, lane 4) only minimally increased the amount of SLX4-bound MUS81 compared with 18-h time point because Cdk1AF cells do not enter mitosis. Collectively, these data indicate premature formation and activation of MUS81–SLX4 complexes in Cdk1AF MEFs during S phase.
Fig. 5.

Mus81 knockdown partially rescues CDK1AF-induced defects in MEFs. a MEFs were prepared as described in Materials and methods section. Cdk1flox/SAF and Cdk1null/AF cells were harvested in S phase (18 h) or mitosis/mitotic-like state (noco). Noco – nocodazole. Cdk1flox/SAF MEFs were treated with nocodazole from 21 to 26 h to arrest them at metaphase–anaphase transition, while the drug was added to Cdk1null/AF MEFs from 16 to 21 h to capture cells in mitotic-like state. Protein extracts from Cdk1flox/SAF and Cdk1null/AF MEFs were subjected to immunoprecipitation with antibodies against SLX4 (IP: SLX4) followed by immunoblotting using the indicated antibodies. The endogenous levels of MUS81 and SLX4 were verified by western blotting (input). HSP90 served as a loading control. Results are representative of three independent experiments. b S phase failure and c the levels of DNA damage were determined by FACS as previously described (Figs. 2b and 4a, respectively). Striped red column indicates Cdk1AF MEFs infected with an empty vector. Quantitative analyses of FACS data are shown. Results are represented as mean ± SD. d Chromosomal fragmentation was analyzed by PFGE as described in Fig. 4c. Results (b–d) are representative of three independent experiments
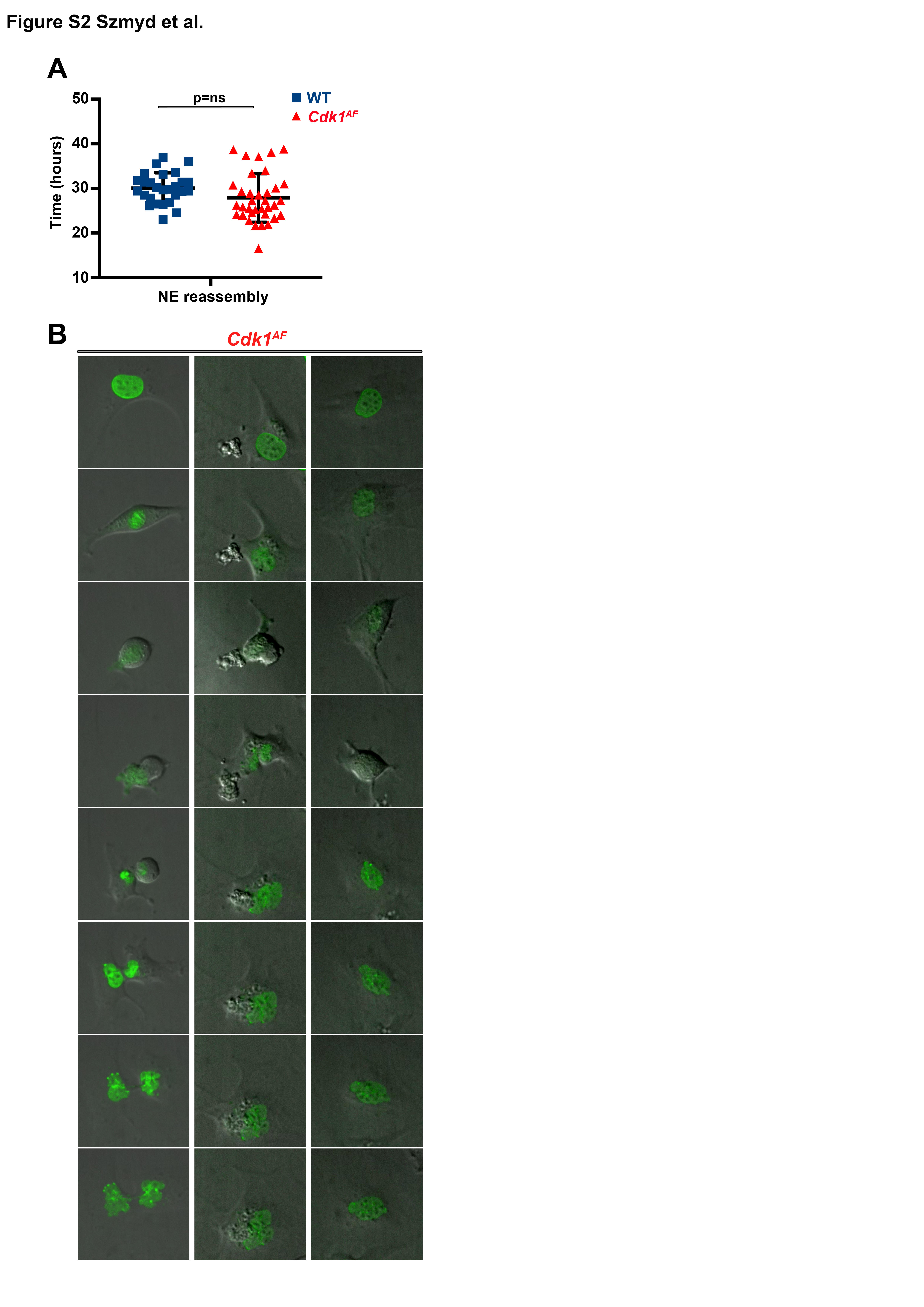
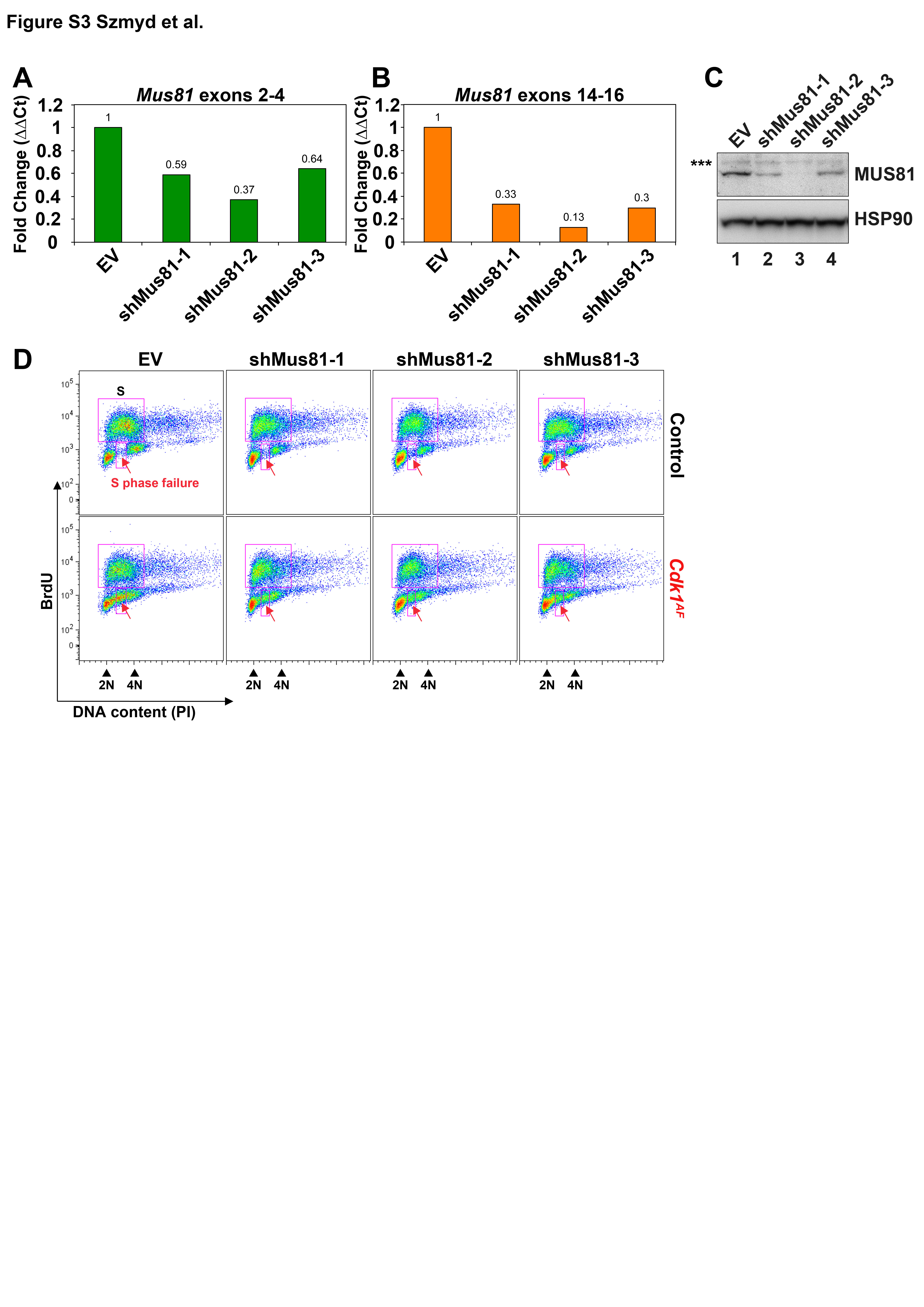
To further examine whether prematurely assembled MUS81–SLX4 complexes are responsible for chromosomal fragmentation in mutant cells, we silenced Mus81 in mutant MEFs. To verify the depletion of Mus81 in control cells after retroviral infection, we used three different short hairpin RNAs (shRNAs) directed against the Mus81 transcript. All infected cells displayed reduced amounts of Mus81 mRNA (Figure S3A-B) and protein (Figure S3C) compared with cells infected with an empty vector. The most efficient knockdown (KD) was achieved in cells treated with shMus81-2. Subsequently, we examined the S phase failure and assessed the levels of DNA damage in Cdk1AF shMus81 cells by BrdU and γH2AX FACS, respectively. We observed an S phase block in Cdk1AF cells (Fig. 5b, S3D; empty vector) with higher percentage of cells positive for γH2AX (Fig. 5c; empty vector). Silencing of Mus81 in Cdk1AF-expressing cells, using any of the tested shRNA against Mus81, reduced the number of cells with under-replicated DNA by around 35% compared with mutant cells infected with an empty vector (Fig. 5b). We observed a similar reduction in γH2AX-positive cells upon Mus81 silencing (Fig. 5c) associated with a lower amount of detected DNA fragmentation by PFGE in Cdk1AF MEFs (Fig. 5d). Taken together, our data suggest that the premature formation of MUS81–SLX4 endonuclease complexes partially contributes to DNA fragmentation during S phase in Cdk1AF MEFs.
p53 status contributes to WEE1 inhibitor sensitivity
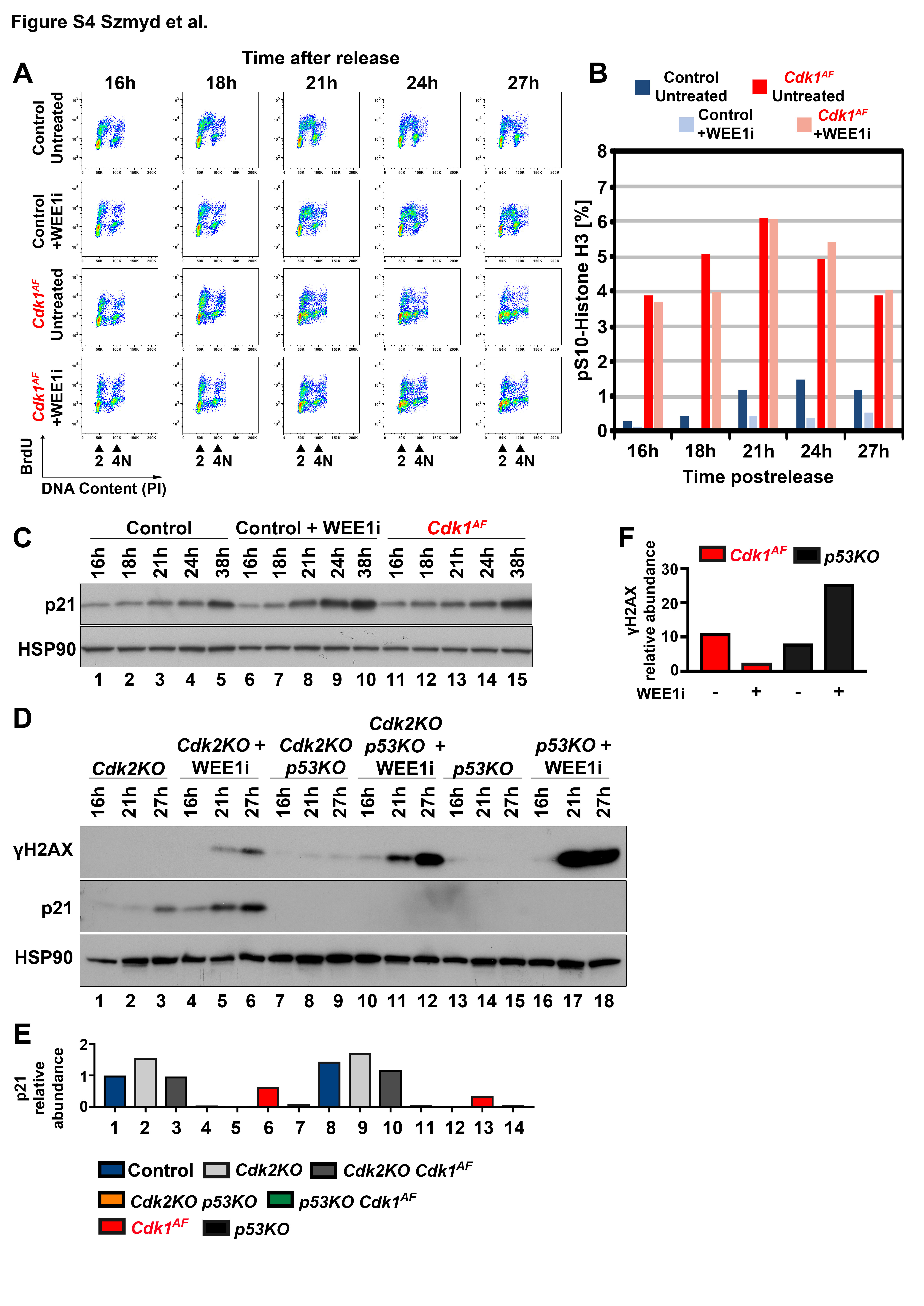
So far, we have demonstrated that the loss of inhibitory phosphorylation on CDK1 results in its premature activation and promotes MUS81-dependent DNA damage in S phase. Previous work established that early activation of CDK1 could be achieved by the inhibition of its negative regulator, the WEE1 kinase [91]. However, WEE1 disruption through silencing or drug inhibition affects the regulation of both CDK1 and CDK2 [35] whereas our genetic model prevents only inhibitory phosphorylation of CDK1 on T14 and Y15. Thus, we investigated whether the genetic ablation of both such sites on CDK1 differs from pharmacological inhibition of the WEE1 kinase. To compare the effects of WEE1 inhibition with CDK1AF expression, we treated control and mutant MEFs with the small-molecule WEE1 inhibitor, MK-1775 (hereafter referred to as WEE1i) [94]. Cells were synchronized at G0/G1 by serum starvation and released into 10% serum medium in the presence of WEE1i. First, we tested the expression levels of CDK1 and Y15 at different time points after release (Fig. 6a). As expected, in control cells CDK1 expression levels gradually increased until reaching a maximum at 24 h. Interestingly, only in control cells, the shift in electrophoretic mobility for CDK1 was detected (Fig. 6a, lanes 1–4). Faint Cdk1 phosphorylation mobility shift in control cells treated with WEE1i (lanes 9–12) might have been due to T161 phosphorylation [8, 9]. Importantly, Y15 phosphorylation remained undetectable in any of analyzed cells but control MEFs (lanes 1–4), which confirms the efficiency of WEE1i in blocking Y15 phosphorylation under these conditions (lanes 9–12). We further assessed the contribution of WEE1i to DNA damage by immunoblotting against γH2AX (Fig. 6a). As expected, γH2AX was only detected in cells either harboring Cdk1AF mutation (lanes 5–9, 13–16) or those treated with WEE1i [9–16]. Interestingly though, Cdk1AF cells treated with the inhibitor (lanes 9–12) displayed DNA damage at later time points compared with untreated mutant MEFs (lanes 5–8). The possible delay in cell cycle progression was likely due to WEE1 inhibition.
Fig. 6.

Different surveillance control mechanisms in WEE1-inhibited and Cdk1AF cells. Primary MEFs were synchronized at G0/G1 by serum starvation and then released into full serum medium in the presence or absence of WEE1i (1 μM). a Protein extracts from control (Cdk1flox/SAF) and Cdk1AF (Cdk1null/AF) MEFs treated with/without WEE1i were subjected to western blotting using the indicated antibodies. HSP90 served as a loading control. b S phase failure was determined by BrdU FACS, whereas in c DNA fragmentation was visualized by PFGE in all four experimental groups. d Protein extracts from (c) were subjected to immunoprecipitation with the indicated antibodies. Kinase activities of the immunoprecipitates were determined by in vitro kinase assays as previously described. e The abundance of p21 bound to different CDKs and cyclins was determined in protein lysates by co-immunoprecipitation with the indicated antibodies followed by western blotting against p21. f WEE1i was added to control MEFs at 21 h after release from serum starvation (time point 0). Cells were collected at the indicated time points (1, 2 and 3 h) in the presence or absence of WEE1i. Protein extracts were isolated and subjected to immunoprecipitation with CDK2 antibodies. Kinase activities associated with CDK2 were measured by in vitro kinase assays. g Protein extracts at the 27-hour time point from control (Cdk1flox/SAF), Cdk2KO, Cdk2KO Cdk1AF (Cdk2KO Cdk1null/AF), Cdk2KO p53KO, p53KO Cdk1AF (p53KO Cdk1null/AF), Cdk1AF (Cdk1null/AF) and p53KO cells. MEFs treated with/without WEE1i were subjected to western blotting using the indicated antibodies. GAPDH served as a loading control. h Relative abundance of γH2AX and in samples presented in panel (g) upon previous normalization to GAPDH
To address whether WEE1 inhibition affects the timing of mitotic entry, we examined phosphorylation of histone H3 on S10. As we have shown before in Fig. 3a, phosphorylation of histone H3 on S10 occurs prematurely in Cdk1AF MEFs compared with control (Fig. 6a, lanes 5–8 vs 1–4). Intriguingly, in control WEE1i-treated cells phospho-S10 signal remained undetectable (lanes 9–12), indicating that WEE1i-treated wild-type MEFs did not enter mitosis at the analyzed time points. Again, the delay in S10 phosphorylation in WEE1i-treated mutant cells (lanes 13–16), in comparison with untreated Cdk1AF MEFs (lanes 5–8), possibly results from slower progression into mitotic-like state in the presence of the inhibitor.
To test whether WEE1-inhibited control cells arrest in S phase, as observed in Cdk1AF MEFs (see Figs. 2b, c), we performed FACS analysis and noted that cells with ≈3 n DNA content (BrdU-negative population with under-replicated DNA) characteristic for Cdk1AF cells, were not observed as a result of WEE1 inhibition in control MEFs during the first 27 h (Fig. 6b, S4A). This unexpected discovery suggests that although both WEE1 inhibition and CDK1AF expression lead to DNA damage, the underlying molecular mechanisms could be distinct. Next, we examined whether DNA breaks are formed in control cells treated with WEE1i using PFGE analysis. We did not detect DNA breakage in control cells treated with WEE1i at the indicated time points, whereas DNA fragmentation was observed in Cdk1AF cells with or without WEE1i treatment (Fig. 6c).
In addition, we examined kinase activities associated with different CDK/cyclin complexes. We carried out in vitro kinase assays after immunoprecipitation in untreated control and Cdk1AF MEFs, as well as control cells treated with WEE1i (Fig. 6d). Untreated control MEFs exhibited low kinase activities at 16-h time point but increased gradually as cells were progressing through S toward G2/M phase (lanes 1–5). Surprisingly, kinase activities in cells treated with WEE1i was barely detectable at 16 and 18 h after release, which might indicate slower replication progression through S phase (lanes 6–10). As expected, Cdk1AF cells displayed elevated kinase activity associated with CDK1 and cyclin B1 (lanes 11–15) compared with untreated control cells (lanes 1–5).
The differences in CDK1 activity between untreated Cdk1AF and WEE1i-treated control MEFs were unexpected, hence we aimed to further investigate this phenotype. The expression levels of p21Cip1/Waf1, a universal inhibitor of multiple CDK/cyclin complexes [95, 96] and a component of the DNA damage checkpoint [97, 98], was similar in untreated control and Cdk1AF MEFs (Figure S4C, lanes 1–5, 11–15). In contrast, in MEFs treated with WEE1i, p21 levels gradually increased from 21-h time point (Figure S4C, lanes 6–10). Next, we determined the abundance of p21 bound to different CDKs and cyclins. At 18-h time point, the levels of p21 bound to CDK1, CDK2, cyclin A2 and cyclin B1 were comparable in control and Cdk1AF MEFs with and without WEE1i treatment (Fig. 6e). Intriguingly though, at the 24-h time point we found elevated p21 bound to all aforementioned CDKs and cyclins in WEE1i-treated control cells (Fig. 6e), which could explain the low kinase activities (see Fig. 6d, lanes 6–10). To investigate the possible impact of CDK2 inhibition in control MEFs treated with WEE1i, at 21 h after serum release, we added WEE1i for 1, 2 or 3 h (Fig. 6f). As expected, at 1 h we observed low CDK2 activity in untreated cells (lane 2), which significantly increased after WEE1i treatment (lane 3). After 2 h WEE1i treatment, CDK2 activity was only slightly elevated in MEFs compared with their untreated equivalents (lanes 4–5), while after 3 h WEE1i treatment, CDK2 activity was lower than in the untreated cells (lanes 6–7). Therefore, CDK2 kinase activity is only transiently induced by WEE1i, followed by its immediate inhibition presumably due to induction of DNA damage and p21 binding.
WEE1i-induced cellular sensitivity in primary MEFs partially differs from its response in cancer cell lines [34, 41, 91]. One of the explanation of the distinct results in previously published reports is the variable p53 status, which is known to affect cell cycle arrest, DNA repair, as well as apoptosis [99]. To address whether p53 deficiency exacerbates DNA damage in cells with prematurely active CDK1, we compared expression levels of γH2AX and p21 in p53KO, Cdk1AF MEFs, and p53KO Cdk1AF MEFs, with and without WEE1i treatment. Interestingly, p53KO MEFs exhibit increased γH2AX phosphorylation (Figs. 6g, h, S4F), which likely was due to the inhibited p21 expression (Figs. 6g, S4D-E). Intriguingly, p53 depletion in Cdk1AF cells aggravate DNA damage when compared with Cdk1AF and p53KO single mutants (Fig. 6g, h) but was reduced by 7.6-fold when double mutant cells were treated with WEE1i (Figs. 6g, h, S4D). The reduction in DNA damage in p53KO Cdk1AF cells could be the consequence of the WEE1i off-target effects.
It has been shown that CDK2 is dispensable for tumorigenesis induced by the loss of p53 in mice [100] but its depletion rescues the DNA damage induced by premature activation of CDK1 due to WEE1 inhibition in breast and ovarian cancer cell lines [35]. In our study, we have presented that the kinase activity associated with CDK2/cyclin A2 complexes is downregulated in Cdk1AF MEFs (see Figs. 2g and 6d). To address whether residual activity of CDK2 could contribute to observed cell cycle defects induced by CDK1AF, we monitored the consequences of CDK2 depletion in Cdk1AF MEFs, with and without WEE1i treatment. Importantly, the Cdk1AF phenotype did not depend on CDK2 but the addition of WEE1i reduced DNA damage in Cdk2KO Cdk1AF (Fig. 6g, h). Interestingly, in comparison with the p53KO single mutant, CDK2 deficiency also contributed do γH2AX reduction in Cdk2KO p53KO cells (Figs. 6g, h, S4D). Taken together, Cdk1AF and WEE1i-treated control MEFs seem to bear partially distinct cell cycle control mechanisms, which rely on the p53 status that sensitizes cells to WEE1i.
The CDK1AF expression inhibits liver tumorigenesis
Our findings indicate that the CDK1AF-induced defects are most likely restricted to proliferating cells. Previously, we had shown that CDK1 is essential for cell division and its depletion inhibits liver tumorigenesis [2]. To further investigate the consequences of Cdk1AF mutation in vivo, we tested whether the expression of CDK1AF would affect liver tumorigenesis. We examined whether liver-specific Cdk1AF mice were able to develop liver tumors by injecting them with tumorigenic mixture of activated oncogene Ras and shRNA against p53, via hydrodynamic tail vein injection (HTVI) technique [2, 101, 102]. Liver tumors were detected in two-thirds of control mice and only in one Cdk1AF mouse at 3 months after HTVI (Fig. 7a). The latter was most likely due to the loss of CDK1AF expression as confirmed by PCR genotyping (data not shown). By 6 months, all control animals developed liver tumors, which differed in size (Figs. 7a, b). Cdk1AF mice did not develop macroscopic liver tumors up to 6 months after induction, at which point all animals were sacrificed. On the microscopic level, in contrast to untreated control livers, samples from Cdk1AF animals displayed signs of liver damage, such as inflammation and fibrosis (Fig. 7c). Hepatocytes, uniform in shape and size, and organized in hepatic plates in healthy control liver, displayed variations in sizes and aberrant nuclear morphology, with small, big or irregular nuclei in the Cdk1AF liver. Liver tumors, encompassing steatotic tumor cells and confined within the boundaries of tumor microenvironment, were detected in control but not in mutant livers. Instead, Cdk1AF mice displayed focal nodular hyperplasia surrounded by fibrous tissues. The altered hyperplastic hepatocytes looked rather normal and seemingly arranged in cords. Hence, liver tumor development was inhibited in Cdk1AF mice, suggesting that the expression of the mutant protein did not allow cancer cells to expand.
Fig. 7.

CDK1AF expression inhibits liver tumorigenesis. Eight weeks old Cdk1+/+ (control) and Cdk1+/SAF Alb-CreERT2 (Cdk1+/AF after injection) mice were IP injected with tamoxifen to induce Cdk1+/AF expression in hepatocytes. Two to three weeks later, tamoxifen-treated mice were subjected to hydrodynamic tail vein injection (HTVI) with tumorigenic mix of activated Ras and shRNA against p53 to initiate liver tumorigenesis. a Quantitative analysis of tumor formation 3 (control n = 6, Cdk1+/AF n = 8) and 6 months (control n = 4, Cdk1+/AF n = 4) after HTVI are presented. b Six months after HTVI, mice (four animals per experimental group) were sacrificed and livers/liver tumors were harvested. c Hematoxylin and eosin (H&E) staining of liver sections from Cdk1+/+ and Cdk1+/AF mice. d TUNEL assay representative images from untreated and subjected to HTVI control and Cdk1+/AF mice (two animals per experimental group). Yellow arrows indicate apoptotic cells. e Quantitative analysis of TUNEL assay presented in panel (d)
We have shown that Cdk1AF blastocysts and Cdk1AF MEFs were not able to proliferate (see Figs. 1a and 2a, respectively). In spleens, 4 days after tamoxifen injection in Rosa-cre Cdk1AF-expressing mice, we detected increased DNA damage (see Fig. 1b, d) and apoptosis (see Fig. 1e, f). Therefore, we tested whether the observed inhibition of tumor formation in Cdk1AF livers after 6 months from HTVI is caused by increased apoptosis of malignant cells. The percentage of apoptotic cells in untreated wild-type liver was <1% of counted cells but after 6 months from HTVI administration in tumors developed in treated controls, the number of apoptotic cells increased up to almost 4% (Fig. 7d, e). In contrast, in Cdk1AF liver the frequency of detected apoptotic cells remained at 1% even after HTVI. Our in vivo data indicate that tumor development was inhibited in Cdk1AF liver, which suggests that the expression of the mutant protein was toxic for cancer cells. Interestingly though, we did not detect apoptosis in Cdk1AF liver, which may be due to effective removal of these cells by macrophages or by other means like autophagy, necroptosis, etc.
Discussion
Using an inducible genetic mouse model, we report that monoallelic expression of CDK1AF in vivo leads to early embryonic lethality in mice (Table 1, Fig. 1a). These data complement our previous study, in which Cdk1KO embryos were unable to develop beyond the blastocyst stage [2], indicating that not just the protein expression but also the timely activation of CDK1 is essential during embryogenesis. Importantly, the regulatory phosphorylation of CDK1 is required for survival of adult animals, where it protects genomic integrity by preventing extensive DNA fragmentation (Fig. 1c, d). Our work highlights the importance of stringent T14/Y15 regulation in governing mammalian cell cycle progression in vivo.
To understand the underlying molecular mechanism of CDK1AF-induced lethality, we performed our work on MEFs isolated from Cdk1AF animals. We found that one copy of endogenously expressed CDK1AF (in the presence or absence of a wild-type Cdk1 allele) is sufficient to trigger premature mitotic events in actively replicating cells leading to replication stress, checkpoint activation, S phase failure and eventually cell cycle arrest (Figs. 2a–c, 4, and S1A). In contrast to previous studies, in which the expression of mutant CDK1 had mild effects on cell cycle progression and mitotic timing in mammalian cell lines [11, 42, 43, 103], Cdk1AF fibroblasts prematurely enter a mitotic-like state characterized by elevated CDK1 kinase activity, incomplete DNA synthesis, premature chromosome condensation and partially disassembled nuclear lamina (Figs. 2f, 3, and S2). The differences in biological outcomes are likely caused by various expression levels of the mutant protein or depend on the experimental model used. Unlike in endogenously expressed CDK1AF, HeLa cells with highly induced CDK1AF expression exhibit cell cycle arrest, whereas cells with moderate levels of mutant CDK1 enter a M-phase-like state prematurely and cycle more rapidly [44]. Moreover, in contrast to our data, survival in CDK1-depleted human cells relies on overexpressed CDK1AF [104]. For aforementioned reasons, in vitro studies in human cell lines with ectopically expressed CDK1AF differ to certain extent from our findings.
The aberrant cell cycle progression of Cdk1AF MEFs is associated with altered kinase activity of both CDK1/cyclin B1 and CDK2/cyclin A2 complexes (Fig. 2g). Premature activation of CDK1/cyclin B1 in mutant fibroblasts triggers the chromatin recruitment of MUS81–SLX4 complexes during S phase (Fig. 5a). We have shown that chromosomal fragmentation is mediated partly by the MUS81–SLX4 endonuclease complex (Figs. 5b–d and S3D), which is activated in a CDK1-dependent manner [91, 92]. In our work, we present that Mus81 KD partially rescues DNA break formation (Figs. 5b–d, S3D), which could be due to the compensating action of other endonuclease(s). In human cells, GEN1 like MUS81, can cleave double-stranded replication or recombination intermediates [105]. Therefore, the combined silencing of GEN1 and MUS81 in Cdk1AF MEFs could potentially inhibit DNA break formation more efficiently.
Our results corroborate the study by Duda et al. [91], where a WEE1 inhibitor was used to block phosphorylation of CDK1 on Y15 to trigger premature activation of the kinase in cancer cell lines. Despite many similarities between these and our findings, the mechanism of WEE1 inhibition and CDK1AF expression do not seem to be exactly the same. In addition to phosphorylation of CDK1 and its role as the gatekeeper of the G2/M transition, the WEE1 kinase also regulates inhibitory phosphorylation of CDK2, therefore controlling the onset of DNA replication [106]. The WEE1 kinase plays a crucial role in maintaining genomic integrity through the control of replication fork progression [41]. Unlike silencing WEE1 in human cancer cell lines, where DDR depends on CDK2 but not CDK1 [41], we demonstrate that DNA damage in Cdk1AF MEFs depends on CDK1 and not on CDK2. First of all, the kinase activity associated with CDK2 is very low in Cdk1AF MEFs (Fig. 2f) likely due to low levels of cyclin A2 (Fig. 2e). Moreover, we have not seen any significant changes in a phenotype of Cdk1AF Cdk2KO MEFs compared with Cdk1AF cells (Fig. 6g, h). Second, S phase failure could be rescued by inhibition of CDK1 kinase activity in mutant fibroblasts treated with RO-3306 (Figs. 2d and S1B). Additionally, homozygous Cdk2AF/AF mice are viable and display only a minimal phenotype [107]. Last but not least, MEFs isolated from those animals exhibit fairly normal cell cycle progression with elevated CDK2/cyclin E kinase activity in the absence of any signs of increased DNA damage.
In our study, we compared WEE1-inhibited control cells with Cdk1AF MEFs. Our data suggest that the underlying molecular mechanisms of WEE1 inhibition and CDK1AF expression differ in certain aspects. Although WEE1i blocks Y15 phosphorylation of CDK1 and CDK2, it does not induce premature mitotic events, such as DNA condensation or may do so only at a much later time point (Figs. 6a and S4B). Despite detected γH2AX phosphorylation in cells treated with WEE1i (Fig. 6a), we did not observe chromosomal breakage (Fig. 6c), which might be caused by stalled, but not yet broken, replication forks [108]. Indeed, the progression of replication in WEE1i-treated cells is possibly delayed comparing with Cdk1AF MEFs due to detected elevated levels of p21 bound to CDK2, cyclin A2, as well as CDK1 and cyclin B1 (Fig. 6e). Intriguingly, CDK2 depletion in Cdk1AF cells does not rescue the mutant phenotype, whereas p53 deficiency aggravates DNA damage in Cdk1AF MEFs (Figs. 6g, h, S4C). Our data support previous findings in various cancer cell lines that p53 mutational status sensitizes cells to WEE1i treatment [109, 110] and here we provide a mechanistic explanation because CDK1/cyclin B1 protein levels and activity are increased in p53KO [100] exacerbating the DNA damage phenotype.
Both WEE1 and MYT1 kinases are needed to inhibit CDK1 activity. Although WEE1 displays a preference for Tyr15, MYT1 specifically phosphorylates Thr-14. Although MYT1 is believed to be more important in meiosis [111], it still is elusive how it contributes to the somatic cell cycle. Despite that MYT1 depletion does not affect entry into mitosis [112], it is not known whether it compensates the lack of WEE1 in cells treated with WEE1i.
The maintenance of genome integrity is crucial for cell viability and for suppression of neoplastic transformation [113]. The challenges to maintain genome integrity arise during S phase, which is jeopardized by multiple impediments affecting replication fork progression [61]. In the present work, we provide a mechanistic view how prematurely activated CDK1 promotes genome instability by triggering premature mitosis-like events in S phase that negatively affects cellular proliferation. Likely, premature condensation in Cdk1AF cells creates harmful topological constraints for replication forks and endonucleytic action of MUS81–SLX4 complexes triggers DNA break formation and DNA damage accumulation.
WEE1 is highly expressed in various cancer types, hence is an attractive target for cancer therapy. WEE1i are currently in clinical trials, covering various cancer types. These clinical studies are still at early stage but already suggest that the action of WEE1i may rely on variety of cell type-dependent mechanisms. Thus, future work should focus on accurate determination of WEE1i selectivity and action, especially in combination with DNA-damaging agents, in a range of cancer types.
Materials and methods
Mouse strains
Cdk1AF mice were generated as a conditional knockin strain as previously described [45]. Cdk1+/AF constitutive expression was obtained by crossing with β-actin-Cre mice [114] (strain name: FVB/N-Tg(ACTB-cre)2Mrt/J; stock no.: 003376; The Jackson Laboratory) or with Rosa26-CreERT2 animals [115] to induce CDK1AF expression in adult animals. Cdk1flox/SAF Rosa26-CreERT2 were obtained by mating the latter with conditional Cdk1flox/flox mice. Primary MEFs were isolated from Cdk1+/SAF or Cdk1flox/SAF mice crossed with inducible Esr1-CreERT2 strain [116] (strain name: B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J; stock no.: 004682; The Jackson Laboratory). Liver-specific Cdk1AF knockin was accomplished by crossing Cdk1+/SAF mice with Alb-Cre [117] and Alb-CreERT2 [118] animals. All Cre-mouse lines were backcrossed to C57Bl6 and all mouse lines used in this study were mostly of C57Bl6 background.
Cre-mediated recombination was induced by three consecutive intraperitoneal injections (24-hourly injection of tamoxifen (1 mg of tamoxifen each) Sigma-Aldrich, T5648). Rosa26-CreERT2 mice were sacrificed 24 h after the last injection. Mice were housed under controlled environmental conditions, 12 h of light/dark cycle (from 7 a.m. to 7 p.m.), and free access to water and food. All experimental procedures were performed in agreement with Institutional Animal Care and Use Committee guidelines.
Cdk1+/AF MEFs were infected with the retroviral constructs such as PKB1927 (pBabe-hygro-GFP-lamin A) and PKB1928 (mCherry-human 53BP1-2pLPC-puro), or PKB1167 (pMSCV-∂3′LTR-H2B-GFP) and PKB1928.
Embryo and blastocyst isolation, culture and synchronization of MEFs
Timed mating between Cdk1+/SAF and β-actin-Cre mice (for blastocysts) or between Cdk1flox/SAF Esr1-CreERT2 was set up and females were monitored for the signs of mating. Pregnant females were sacrificed to isolate blastocysts after 3 days post coitum or 13.5 days for MEFs isolation. Blastocysts preparation and imaging analysis were performed as previously described [2, 119] after releasing them from the uterine wall by vigorous flushing of phosphate-buffered saline (PBS). Primary MEFs were prepared as previously described [47]. Primary MEFs were cultured under humidified atmosphere with 5% CO2 and 3% O2, whereas immortalized p53KO MEFs were maintained in the presence of 21% O2. Cells were synchronized at G0/G1 by serum starvation (Dulbecco’s modified Eagle’s medium, 0.1% fetal bovine serum and 1% penicillin/streptomycin) for 72 h. The expression of Cdk1AF, alone or simultaneously with Cdk1 deletion, was induced by the addition of 4-OHT (40 ng/ml; Sigma-Aldrich, H7904) during the last 48 h of the starvation period. Inhibition of CDK1 was obtained by addition of 10 µM of RO-3306 (Sigma-Aldrich, SML0569) in the medium during the release period.
Immunohistochemistry
For hematoxylin and eosin (H&E) staining, tissues were fixed in 10% neutral buffered formalin (Sigma-Aldrich, HT501128) for 18–24 h, transferred to ice-cold 70% ethanol and embedded in paraffin blocks followed by the staining of tissues sections.
Comet assay
Splenocytes were isolated 48 h after the last (third) tamoxifen injection from spleens of adult Cdk1+/AF Rosa26-CreERT2 mice and their littermate controls (tamoxifen-treated Cdk1+/+ Rosa-Cre mice). The spleens were dissected, incubated in PBS-EDTA (20 mM, 5 min), the cell suspension from the supernatant were centrifuged, and adjusted at 4 × 105 cells/ml. Alkaline and Neutral Comet assay were performed on commercial slides accordingly to manufacturer’s protocol (Trevigen, #4250–050-K). After electrophoresis, slides of cells were stained with SYBR Green solution (Maxima SYBR Green qPCR Master Mix (2 × ), Fermentas, K0252) and dried overnight before images acquisition. Images were taken on Zeiss Axioimager Z1 Epifluorescence microscope at ×20 magnification. At least 100 comets were scored in each sample using TriTek CometScore Freeware v1.5 and the amount of DNA breaks was calculated based on the tail moment (Trevigen Instructions, #4250–050-K).
TUNEL assay
Apoptotic cells were detected by ApopTag Plus Peroxidase In Situ Apoptosis Kit (Millipore #S7101) by labeling and detecting DNA breaks by the indirect TUNEL method.
Proliferation assay
MEFs were seeded in quintuplicate in 96-well plate at the density of 1500 cells/well. The proliferation was monitored daily for a period of 7 days using the alamarBlue assay (AbD Serotec, BUF012B) and the emitted fluorescence signal (excitation: 560 nm; emission: 590 nm) was measured using a microplate reader SPECTRAmax M2 linked to SoftMax Pro V5 software (Molecular Devices).
FACS analysis of MEFs
For BrdU incorporation analysis, MEFs were pulse-labeled for 1 h with 100 μM BrdU (BD Pharmingen, 550891) before collection. Collected cells were fixed in –20 °C cold 70% ethanol and washed in blocking buffer (PBS-bovine serum albumin (BSA) 1%; BSA Sigma-Aldrich, A7906). For non-BrdU analysis, cells were permeabilized with PBS-1% BSA-0.5% Triton X-100, whereas, for BrdU analysis, cells were treated with 2 n HCl/0.5% Triton X-100 and neutralized with 0.1 m sodium tetraborate, pH8.5. Cell staining were performed using anti-BrdU (1:500; BD Pharmingen, 555627, clone 3D4), pS10-histone H3 (1:100; Cell Signaling Technology [CST] #9701) or pS139-histone H2AX (1:500; CST #9718) followed by their incubation with secondary antibody Alexa 647-conjugated goat anti-mouse (1:400; Invitrogen, A21235) or Alexa 647-conjugated donkey anti-rabbit (1:400; Invitrogen, A31573). Before analysis, cells were resuspended in PBS supplemented with 2.5 μg/ml propidium iodide (Merck, 537059) and 20 μg/ml RNase A (Sigma-Aldrich, R6513). Analysis of cells and BrdU incorporation was acquired on BD FACSCalibur or BD LSRII flow cytometers (BD Bioscience) and further analyzed using FlowJo 8.8.7 software.
Protein extraction, immunoblotting, immunoprecipitation and kinase assays
Protein extracts were obtained by lysing cells in CSK/RIPA buffer (see below) or in EBN buffer (80 mM β-glycerophosphate pH7.3, 15 mM MgCl2, 20 mM EGTA, 150 mM NaCl, 0.5% NP-40) supplemented with 1 mM DTT, protease inhibitors cocktail (10 μg/ml each of leupeptin, chymostatin, and pepstatin [Chemicon, E18, E16 and E110]) and phosphatase inhibitors (2 mM NaF, Sigma-Aldrich, 71519; 0.1 mM Na3VO4, Sigma-Aldrich, 450243). Protein concentration was assessed using the BCA assay (Thermo Scientific, 23225). The protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, IPVH0010). The membranes were blocked in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) and 4% milk (Bio-Rad, 1706404), and subsequently probed overnight at 4 °C with the following primary antibodies: CDK1 (Santa Cruz, sc-954, clone C-19), pY15-CDK1 (CST #9111), CDK1 (1:20,000, rabbit anti-mouse [47]), Cyclin A2 (Santa Cruz, sc-596, clone C-19), cyclin B1 (CST #4135), HSP90 (BD Transduction Laboratories, #610419), MUS81 (University of Dundee, DU 37171), SLX4 (University of Dundee, DU7173), CDC6 (Santa Cruz, sc-9964), CDC45 (CST #3673), GINS2 (Proteintech, #16247-1-AP), PCNA (CST #2586, clone PC10), p-histone H1 (Upstate #12D11), Histone H3 (CST #4499, clone DIH2), p-histone H3 (CST #9701), Lamin A/C (CST #4777), p-Lamin A/C (CST #2026), p-CHK1 (CST, #2348, clone 133D3), p21 (Santa Cruz, sc-6246, clone F-5). Washed membranes were probed with the adequate horseradish peroxidase (HRP)-conjugated secondary antibodies and developed using enhanced chemiluminescence (Immobilon western Chemiluminescent HRP substrate, Millipore, WBKLS0500) and visualized using X-ray film (Fujifilm, 47410 19289).
Affinity purification/immunoprecipitation and kinase assay were carried out as previously described [47]. The antibodies used for immunoprecipitation were either covalently conjugated to Sepharose beads, cyclin A2 (Santa Cruz, sc-751, clone H432), cyclin B1 (Santa Cruz, sc-7393, clone D11) and SUC1 (Millipore, 14132), or pre-coupled to protein A agarose beads (Roche, 11719408001) for CDK1 and CDK2 [47] or SLX4 (University of Dundee, DU 37173). In all, 100–1000 µg of protein extracts was incubated with the respective antibody in EBN buffer.
For kinase assays, immunoprecipitated proteins were incubated in 11 µl of EB buffer supplemented with 10 mM DTT, 15 µM ATP, 5µCi [ɣ−32P]-ATP (PerkinElmer, BLU502A) and 1.6 µg of histone H1 (Roche, 11004875001) at room temperature for 30 min, and stopped by addition 5X SDS-sample buffer. Denatured proteins were resolved by SDS–PAGE on 12% gels. The gels were stained with Coomassie Blue and dried. Incorporated radioactivity was determined using a phosphoimager (FLA-7000, Fujifilm).
Chromatin fractionation and immunoblotting
Chromatin fractionation was performed as previously described [120] with modifications. Collected MEFs were resuspended in CSK buffer (10 mM Hepes-KOH pH7.4, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 0.25% Triton-X 100) freshly supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1 mM Na3VO4 and protease inhibitor cocktail. After incubation (on ice, 5 min) and centrifugation (5000 g, 5 min, 4 °C), supernatants containing cytoplasmic and nucleoplasmic soluble proteins were directly snap-frozen in liquid nitrogen, whereas the remaining pellet of chromatin-bound proteins was resuspended and washed twice in CSK buffer. To extract chromatin-bound proteins, pellets were resuspended in RIPA buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 0.5% sodium deoxycholate) freshly supplemented with 1 mM PMSF, 0.1 mM Na3VO4, 0.1 mM NaF and protease inhibitors cocktail.
Pulsed field gel electrophoresis
Plugs were prepared as previously described [121]. Chromosomes were separated by PFGE (CHEF-DR II System, Bio-Rad), and electrophoresis was performed for 12 h at 6 V/cm with 90 sec pulses, followed by 12 h with 60 sec pulses in 0.5x Tris/Borate/EDTA (TBE) buffer at 14 °C. DNA was visualized by ethidium bromide staining.
HTVI and liver tumorigenesis
HTVI was performed as previously described [2, 101, 102]. Briefly, 10-week-old mice were injected within 10 s with a cocktail of plasmids [transposase (15 μg pGK-SleepingBeauty13; PKB1094) and transposon (30 μg pT2-Caggs-NRasV12; PKB1095 and 15 μg pT2-shRNAp53; PKB1096] diluted in lactated Ringer’s solution. Animals were injected with a volume corresponding to 10% of their body weight in the lateral tail vein. Animals were monitored for liver tumors and sacrificed 3 and 6 months after the injection unless stated differently.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Zakiah Talib, June Wang, Vithya Anantaraja and Chloe Sim for animal care; and all past and current members of the Kaldis lab for support and discussions. We thank Jos Jonkers and Anton Berns for providing the Rosa26-CreERT2 mice, Andy McMahon for Esr1-Cre mice, T Jake Liang for Alb-Cre mice and Mark Lewandoski for the β-actin-Cre/Flpe mice. We acknowledge the technical expertise provided by the Advanced Molecular Pathology Laboratory at IMCB. This work was supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore and in part by a grant from the National Medical Research Council Singapore, NMRC (NMRC/CBRG/0091/2015) to PK.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41388-018-0464-0) contains supplementary material, which is available to authorized users.
References
- 1.Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–5. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 2.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA. 2012;109:3826–31. doi: 10.1073/pnas.1115201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–91. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 4.Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- 5.Kornbluth S, Sebastian B, Hunter T, Newport J. Membrane localization of the kinase which phosphorylates p34cdc2 on threonine 14. Mol Biol Cell. 1994;5:273–82. doi: 10.1091/mbc.5.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu F, Stanton JJ, Wu Z, Piwnica-Worms H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol. 1997;17:571–83. doi: 10.1128/mcb.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–7. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 8.Solomon MJ, Lee T, Kirschner MW. Role of phosphorylation in p34cdc2 activation: identification of an activating kinase. Mol Biol Cell. 1992;3:13–27. doi: 10.1091/mbc.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaldis P. The cdk-activating kinase (CAK): from yeast to mammals. Cell Mol Life Sci. 1999;55:284–96. doi: 10.1007/s000180050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker LL, Atherton-Fessler S, Piwnica-Worms H. p107wee1 is a dual-specificity kinase that phosphorylates p34cdc2 on tyrosine 15. Proc Natl Acad Sci USA. 1992;89:2917–21. doi: 10.1073/pnas.89.7.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heald R, McLoughlin M, McKeon F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell. 1993;74:463–74. doi: 10.1016/0092-8674(93)80048-j. [DOI] [PubMed] [Google Scholar]
- 12.McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12:75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coulonval K, Kooken H, Roger PP. Coupling of T161 and T14 phosphorylations protects cyclin B-CDK1 from premature activation. Mol Biol Cell. 2011;22:3971–85. doi: 10.1091/mbc.E11-02-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wells NJ, Watanabe N, Tokusumi T, Jiang W, Verdecia MA, Hunter T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G2/M progression. J Cell Sci. 1999;112(Pt 19):3361–71. doi: 10.1242/jcs.112.19.3361. [DOI] [PubMed] [Google Scholar]
- 15.Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67:197–211. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai A, Dunphy WG. The cdc25 protein controls tyrosine dephosphorylation of the cdc2 protein in a cell-free system. Cell. 1991;64:903–14. doi: 10.1016/0092-8674(91)90315-p. [DOI] [PubMed] [Google Scholar]
- 17.Strausfeld U, Labbe JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, et al. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242–5. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- 18.Krek W, Nigg EA. Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 1991;10:3331–41. doi: 10.1002/j.1460-2075.1991.tb04897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norbury C, Blow J, Nurse P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991;10:3321–9. doi: 10.1002/j.1460-2075.1991.tb04896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aaltonen K, Amini RM, Heikkila P, Aittomaki K, Tamminen A, Nevanlinna H, et al. High cyclin B1 expression is associated with poor survival in breast cancer. Br J Cancer. 2009;100:1055–60. doi: 10.1038/sj.bjc.6604874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao H, Marto JA, Hoffmann TK, Shabanowitz J, Finkelstein SD, Whiteside TL, et al. Identification of cyclin B1 as a shared human epithelial tumor-associated antigen recognized by T cells. J Exp Med. 2001;194:1313–23. doi: 10.1084/jem.194.9.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–9. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 23.Poon RY, Chau MS, Yamashita K, Hunter T. The role of Cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer Res. 1997;57:5168–78. [PubMed] [Google Scholar]
- 24.Bayart E, Grigorieva O, Leibovitch S, Onclercq-Delic R, Amor-Gueret M. A major role for mitotic CDC2 kinase inactivation in the establishment of the mitotic DNA damage checkpoint. Cancer Res. 2004;64:8954–9. doi: 10.1158/0008-5472.CAN-04-1613. [DOI] [PubMed] [Google Scholar]
- 25.Minemoto Y, Uchida S, Ohtsubo M, Shimura M, Sasagawa T, Hirata M, et al. Loss of p53 induces M-phase retardation following G2 DNA damage checkpoint abrogation. Arch Biochem Biophys. 2003;412:13–19. doi: 10.1016/s0003-9861(03)00010-9. [DOI] [PubMed] [Google Scholar]
- 26.Poon RY, Jiang W, Toyoshima H, Hunter T. Cyclin-dependent kinases are inactivated by a combination of p21 and Thr-14/Tyr-15 phosphorylation after UV-induced DNA damage. J Biol Chem. 1996;271:13283–91. doi: 10.1074/jbc.271.22.13283. [DOI] [PubMed] [Google Scholar]
- 27.Vincent F, Deplanque G, Ceraline J, Duclos B, Bergerat JP. p53-independent regulation of cyclin B1 in normal human fibroblasts during UV-induced G2-arrest. Biol Cell. 1999;91:665–74. [PubMed] [Google Scholar]
- 28.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell. 2009;35:327–39. doi: 10.1016/j.molcel.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vriend LE, De Witt Hamer PC, Van Noorden CJ, Wurdinger T. WEE1 inhibition and genomic instability in cancer. Biochim Biophys Acta. 2013;1836:227–35. doi: 10.1016/j.bbcan.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Chow JP, Siu WY, Fung TK, Chan WM, Lau A, Arooz T, et al. DNA damage during the spindle-assembly checkpoint degrades CDC25A, inhibits cyclin-CDC2 complexes, and reverses cells to interphase. Mol Biol Cell. 2003;14:3989–4002. doi: 10.1091/mbc.E03-03-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98:523–8. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194–202. doi: 10.1016/j.drudis.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 33.Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 2016;37:872–81. doi: 10.1016/j.tips.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–36. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc Natl Acad Sci USA. 2015;112:15160–5. doi: 10.1073/pnas.1505283112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chow JP, Siu WY, Ho HT, Ma KH, Ho CC, Poon RY. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J Biol Chem. 2003;278:40815–28. doi: 10.1074/jbc.M306683200. [DOI] [PubMed] [Google Scholar]
- 37.Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell. 2015;28:557–68. doi: 10.1016/j.ccell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia TB, Fosmire SP, Porter CC. Increased activity of both CDK1 and CDK2 is necessary for the combinatorial activity of WEE1 inhibition and cytarabine. Leuk Res. 2018;64:30–33. doi: 10.1016/j.leukres.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tominaga Y, Li C, Wang RH, Deng CX. Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int J Biol Sci. 2006;2:161–70. doi: 10.7150/ijbs.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–8. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- 41.Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol. 1996;134:963–70. doi: 10.1083/jcb.134.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol. 1998;141:875–85. doi: 10.1083/jcb.141.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pomerening JR, Ubersax JA, Ferrell JE., Jr Rapid cycling and precocious termination of G1 phase in cells expressing CDK1AF. Mol Biol Cell. 2008;19:3426–41. doi: 10.1091/mbc.E08-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adhikari D, Busayavalasa K, Zhang J, Hu M, Risal S, Bayazit MB, et al. Inhibitory phosphorylation of Cdk1 mediates prolonged prophase I arrest in female germ cells and is essential for female reproductive lifespan. Cell Res. 2016;26:1212–25. doi: 10.1038/cr.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucl Acids Res. 1999;27:4324–7. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–85. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 48.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA. 2006;103:10660–5. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–34. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 50.Coleman TR, Carpenter PB, Dunphy WG. The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell. 1996;87:53–63. doi: 10.1016/s0092-8674(00)81322-7. [DOI] [PubMed] [Google Scholar]
- 51.Donovan S, Harwood J, Drury LS, Diffley JF. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci USA. 1997;94:5611–6. doi: 10.1073/pnas.94.11.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maiorano D, Moreau J, Mechali M. XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature. 2000;404:622–5. doi: 10.1038/35007104. [DOI] [PubMed] [Google Scholar]
- 53.Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000;404:625–8. doi: 10.1038/35007110. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol. 2002;4:198–207. doi: 10.1038/ncb757. [DOI] [PubMed] [Google Scholar]
- 55.Bowers JL, Randell JC, Chen S, Bell SP. ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Mol Cell. 2004;16:967–78. doi: 10.1016/j.molcel.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 56.Tanaka S, Araki H. Regulation of the initiation step of DNA replication by cyclin-dependent kinases. Chromosoma. 2010;119:565–74. doi: 10.1007/s00412-010-0291-8. [DOI] [PubMed] [Google Scholar]
- 57.Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–5. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci USA. 2006;103:10236–41. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T. Structure-function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J Biol Chem. 1995;270:22527–34. doi: 10.1074/jbc.270.38.22527. [DOI] [PubMed] [Google Scholar]
- 60.Kelman Z, O’Donnell M. Structural and functional similarities of prokaryotic and eukaryotic DNA polymerase sliding clamps. Nucl Acids Res. 1995;23:3613–20. doi: 10.1093/nar/23.18.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 62.Lohka MJ. Nuclear responses to MPF activation and inactivation in Xenopus oocytes and early embryos. Biol Cell. 1998;90:591–9. [PubMed] [Google Scholar]
- 63.Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL. Cyclin is a component of maturation-promoting factor from Xenopus. Cell. 1990;60:487–94. doi: 10.1016/0092-8674(90)90599-a. [DOI] [PubMed] [Google Scholar]
- 64.Fang F, Newport JW. Evidence that the G1-S and G2-M transitions are controlled by different cdc2 proteins in higher eukaryotes. Cell. 1991;66:731–42. doi: 10.1016/0092-8674(91)90117-h. [DOI] [PubMed] [Google Scholar]
- 65.Pagano M, Pepperkok R, Lukas J, Baldin V, Ansorge W, Bartek J, et al. Regulation of the cell cycle by the cdk2 protein kinase in cultured human fibroblasts. J Cell Biol. 1993;121:101–11. doi: 10.1083/jcb.121.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ross KE, Kaldis P, Solomon MJ. Activating phosphorylation of the Saccharomyces cerevisiae cyclin-dependent kinase, cdc28p, precedes cyclin binding. Mol Biol Cell. 2000;11:1597–609. doi: 10.1091/mbc.11.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell. 1991;64:1111–22. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- 69.Gong D, Pomerening JR, Myers JW, Gustavsson C, Jones JT, Hahn AT, et al. Cyclin A2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin B1. Curr Biol. 2007;17:85–91. doi: 10.1016/j.cub.2006.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Enoch T, Peter M, Nurse P, Nigg EA. p34cdc2 acts as a lamin kinase in fission yeast. J Cell Biol. 1991;112:797–807. doi: 10.1083/jcb.112.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA. In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell. 1990;61:591–602. doi: 10.1016/0092-8674(90)90471-p. [DOI] [PubMed] [Google Scholar]
- 72.Monaco L, Kolthur-Seetharam U, Loury R, Murcia JM, de Murcia G, Sassone-Corsi P. Inhibition of Aurora-B kinase activity by poly(ADP-ribosyl)ation in response to DNA damage. Proc Natl Acad Sci USA. 2005;102:14244–8. doi: 10.1073/pnas.0506252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilkins BJ, Rall NA, Ostwal Y, Kruitwagen T, Hiragami-Hamada K, Winkler M, et al. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 2014;343:77–80. doi: 10.1126/science.1244508. [DOI] [PubMed] [Google Scholar]
- 74.Ajiro K, Nishimoto T, Takahashi T. Histone H1 and H3 phosphorylation during premature chromosome condensation in a temperature-sensitive mutant (tsBN2) of baby hamster kidney cells. J Biol Chem. 1983;258:4534–8. [PubMed] [Google Scholar]
- 75.Heald R, McKeon F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell. 1990;61:579–89. doi: 10.1016/0092-8674(90)90470-y. [DOI] [PubMed] [Google Scholar]
- 76.Ward GE, Kirschner MW. Identification of cell cycle-regulated phosphorylation sites on nuclear lamin C. Cell. 1990;61:561–77. doi: 10.1016/0092-8674(90)90469-u. [DOI] [PubMed] [Google Scholar]
- 77.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–8. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–29. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–20. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–90. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem. 2003;278:25207–17. doi: 10.1074/jbc.M300070200. [DOI] [PubMed] [Google Scholar]
- 83.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–19. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–60. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 86.West SC, Blanco MG, Chan YW, Matos J, Sarbajna S, Wyatt HD. Resolution of recombination intermediates: mechanisms and regulation. Cold Spring Harb Symp Quant Biol. 2015;80:103–9. doi: 10.1101/sqb.2015.80.027649. [DOI] [PubMed] [Google Scholar]
- 87.Sarbajna S, Davies D, West SC. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014;28:1124–36. doi: 10.1101/gad.238303.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–4. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 89.Pfander B, Matos J. Control of Mus81 nuclease during the cell cycle. FEBS Lett. 2017;591:2048–56. doi: 10.1002/1873-3468.12727. [DOI] [PubMed] [Google Scholar]
- 90.Matos J, Blanco MG, Maslen S, Skehel JM, West SC. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–72. doi: 10.1016/j.cell.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duda H, Arter M, Gloggnitzer J, Teloni F, Wild P, Blanco MG, et al. A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev Cell. 2016;39:740–55. doi: 10.1016/j.devcel.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 92.Wyatt HD, Sarbajna S, Matos J, West SC. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell. 2013;52:234–47. doi: 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 93.Garner E, Kim Y, Lach FP, Kottemann MC, Smogorzewska A. Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 2013;5:207–15. doi: 10.1016/j.celrep.2013.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 95.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 96.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 97.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 98.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 99.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 100.Padmakumar VC, Aleem E, Berthet C, Hilton MB, Kaldis P. Cdk2 and Cdk4 activities are dispensable for tumorigenesis caused by the loss of p53. Mol Cell Biol. 2009;29:2582–93. doi: 10.1128/MCB.00952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Prot. 2007;2:3153–65. doi: 10.1038/nprot.2007.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ho V, Lim TS, Lee J, Steinberg J, Szmyd R, Tham M, et al. TLR3 agonist and sorafenib combinatorial therapy promotes immune activation and controls hepatocellular carcinoma progression. Oncotarget. 2015;6:27252–66. doi: 10.18632/oncotarget.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blasina A, Paegle ES, McGowan CH. The role of inhibitory phosphorylation of CDC2 following DNA replication block and radiation-induced damage in human cells. Mol Biol Cell. 1997;8:1013–23. doi: 10.1091/mbc.8.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gupta M, Trott D, Porter AC. Rescue of a human cell line from endogenous Cdk1 depletion by Cdk1 lacking inhibitory phosphorylation sites. J Biol Chem. 2007;282:4301–9. doi: 10.1074/jbc.M607910200. [DOI] [PubMed] [Google Scholar]
- 105.Rass U, Compton SA, Matos J, Singleton MR, Ip SC, Blanco MG, et al. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010;24:1559–69. doi: 10.1101/gad.585310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hughes BT, Sidorova J, Swanger J, Monnat RJ, Jr, Clurman BE. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc Natl Acad Sci USA. 2013;110:8954–9. doi: 10.1073/pnas.1302927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao H, Chen X, Gurian-West M, Roberts JM. Loss of cyclin-dependent kinase 2 (CDK2) inhibitory phosphorylation in a CDK2AF knock-in mouse causes misregulation of DNA replication and centrosome duplication. Mol Cell Biol. 2012;32:1421–32. doi: 10.1128/MCB.06721-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–7. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res. 2014;20:4274–88. doi: 10.1158/1078-0432.CCR-13-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–39. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 111.Nakajo N, Yoshitome S, Iwashita J, Iida M, Uto K, Ueno S, et al. Absence of Wee1 ensures the meiotic cell cycle in Xenopus oocytes. Genes Dev. 2000;14:328–38. [PMC free article] [PubMed] [Google Scholar]
- 112.Chow JP, Poon RY. The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene. 2013;32:4778–88. doi: 10.1038/onc.2012.504. [DOI] [PubMed] [Google Scholar]
- 113.Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–68. [PubMed] [Google Scholar]
- 115.Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–7. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 117.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–50. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 118.Schuler M, Dierich A, Chambon P, Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis. 2004;39:167–72. doi: 10.1002/gene.20039. [DOI] [PubMed] [Google Scholar]
- 119.Bonin A, Reid SW, Tessarollo L. Isolation, microinjection, and transfer of mouse blastocysts. Methods Mol Biol. 2001;158:121–34. doi: 10.1385/1-59259-220-1:121. [DOI] [PubMed] [Google Scholar]
- 120.Klotz-Noack K, McIntosh D, Schurch N, Pratt N, Blow JJ. Re-replication induced by geminin depletion occurs from G2 and is enhanced by checkpoint activation. J Cell Sci. 2012;125:2436–45. doi: 10.1242/jcs.100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.