

Abstract
Mutations in the Cu/Zn superoxide dismutase (SOD1) gene cause one form of familial amyotrophic lateral sclerosis, a progressive disorder of motor neurons leading to weakness and death of affected individuals. Experiments using both transgenic mice expressing mutant SOD1 and SOD1 knock-out mice have demonstrated that disease is caused by a toxic gain of function and not by a loss of normal SOD1 activity. Precise mechanisms underlying mutant SOD1 toxicity are unclear but may involve abnormal interactions between zinc and SOD1.
The metallothioneins (MTs) represent a family of zinc binding proteins that can function as zinc chaperones for apo-SOD1 in vitro. We hypothesized that manipulation of metallothioneinsin vivo might alter the disease phenotype of transgenic mice expressing G93A SOD1 and therefore crossed this line with MT-I and MT-II or MT-III knock-out mice. G93A SOD1 mice deficient of either MT-I and MT-II or MT-III exhibited significant reductions in survival compared with G93A SOD1 mice. In addition, motor dysfunction was markedly accelerated in G93A SOD1 mice deficient in metallothioneins with regard to onset (MT-I and MT-II) or progression (MT-III).
These results indicate that the disease course in G93A SOD1 mice is dependent on levels of metallothionein expression. Because MT-I and MT-II are expressed in glia whereas MT-III is found in neurons, these results also indicate that primary changes within non-neuronal cells can affect mutant SOD1-induced disease and do so in ways distinct from primary neuronal changes.
Keywords: amyotrophic lateral sclerosis, metallothionein, copper, zinc, glia, transgenic
Although mechanisms underlying mutant superoxide dismutase (SOD1) toxicity remain unclear, abnormalities in zinc binding to SOD1 have been implicated in disease pathogenesis (Elliott, 2001). Zinc is important in maintaining the structural integrity of SOD1, and, when depleted of zinc, SOD1 gains enhanced ability to react with peroxynitrite and catalyze the addition of nitro groups to tyrosine residues (Lyons et al., 1996; Crow et al., 1997a; Goto et al., 2000). Mutant SOD1 exhibits markedly reduced affinity for zinc compared with wild-type SOD1 and has enhanced nitration activity (Crow et al., 1997a,b). Indeed, biochemical hallmarks of protein nitration, including elevated 3-nitrotyrosine products, have been found in spinal cords from amyotrophic lateral sclerosis (ALS) patients and mutant SOD1 (mSOD1) transgenic mice (Beal et al., 1997; Bruijn et al., 1997a; Ferrante et al., 1997; Toghi et al., 1999). Zinc binding may also be important in modulating protein aggregation, another possible mechanism of toxicity (Johnston et al., 2000; Cherney et al., 2001; Curtain et al., 2001; Quaglio et al., 2001). Recent work has addressed biological consequences of zinc-depleted SOD1, finding that it is highly toxic when introduced into motor neurons in vitro (Estevez et al., 1999). However, experiments directly assessing the role of zinc in disease pathogenesis have been limited, partly because of the difficulty in altering zinc binding in live animals. To address the role of zinc in mSOD1 toxicity in vivo, we used an alternative approach involving the manipulation of proteins involved in zinc homeostasis with subsequent analysis of changes in disease phenotype.
The metallothioneins (MTs) represent a family of zinc binding proteins, three of which, MT-I, MT-II, and MT-III, are found in the murine nervous system. Metallothioneins are important in the regulation of zinc bioavailability within cells, by acting as chaperones during the synthesis of metalloenzymes (Jacob et al., 1998; Palmiter, 1998). Metallothioneins are rich in cysteine residues that bind zinc and can be readily oxidized with subsequent release of zinc to accepting proteins, including apo-SOD1 (Suzuki and Kuroda, 1995). Experiments using genetically altered mice with metallothionein overexpression or deletions have shown that these proteins protect the CNS from oxidative injury or heavy metal toxicity (Masters et al., 1994b; Erickson et al., 1997). Metallothionein expression is markedly upregulated in the spinal cords of ALS patients and transgenic mutant SOD1 mice, suggesting that these molecules also serve a protective function in disease potentially related to their zinc binding capacity (Smitt et al., 1992; Blaauwgeers et al., 1996; Gong and Elliott, 2000). Therefore, we decided to use changes in metallothionein expression to study the role of zinc in mutant SOD1 toxicity in vivo. Consequently, we crossed transgenic mice expressing a G93A SOD1 mutation with MT-I and MT-II or MT-III knock-out mice and then studied changes in the disease phenotype.
Motor neuron dysfunction and degeneration are hallmarks of ALS, but recent work using tissue-specific expression of mutant SOD1 in transgenic mice has suggested that interactions between neurons and glia may be necessary to generate a disease phenotype (Gong et al., 2000; Pramatarova et al., 2001). The differential expression of metallothioneins within the CNS can also be used to assess potential neuronal and glial contributions to the disease process. Because MT-I and MT-II are restricted to glia whereas MT-III is neuronal, targeted manipulation of distinct metallothioneins allows change within each cellular compartment (glial or neuronal) to be made directly and independently (Nishimura et al., 1992; Palmiter et al., 1992; Masters et al., 1994a; Gong and Elliott, 2000). Our results indicate that both neuronal and non-neuronal cells contribute to disease progression in G93A SOD1 mice.
MATERIALS AND METHODS
Mouse lines, breeding strategy, and genotyping.Transgenic mice expressing the low copy number human G93A SOD1 mutation (B6SJL-TgNSOD1-G93A; JR2300) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice with targeted deletions of both MT-I and MT-II loci (129S7/SvImJ) were also obtained from The Jackson Laboratory. MT-III knock-out mice (129Sv/C5Bl) were generously provided by Dr. Richard Palmiter (University of Washington, Seattle, WA) and have been well characterized (Erickson et al., 1997). Two differing breeding paradigms were used for MT-I and MT-II or MT-III crossing with G93A SOD1 transgenic mice. For an accelerated breeding program (Ba), G93A SOD1 mice were crossed to either MT-I and MT-II or MT-III knock-out mice. F1 offspring, all heterozygous for metallothionein expression and G93A SOD1 positive, were then backcrossed to metallothionein homozygous founders. Progeny, heterozygous for metallothionein and G93A SOD1 positive, were then bred with metallothionein heterozygous siblings. This cross allows all possible genotypes to be generated with adequate internal controls, and consequently only these mice were included in the study. A prolonged breeding paradigm (Bp) was also used for crossing G93A SOD1 mice with MT-I and MT-II knock-out mice. Using this strategy, G93A SOD1 transgenic mice were crossed with MT-I and MT-II knock-out mice. F1 offspring were then backcrossed for eight generations onto the G93A SOD1 strain. After the eighth generation, metallothionein heterozygotes positive for G93A SOD1 were then bred to metallothionein heterozygous siblings, again allowing all potential genotypes to be generated and analyzed.
Offspring were genotyped using genomic DNA isolated from tails digested overnight with proteinase K at 50°C. The following primer pairs for PCR were used: 5′-CAT CAG CCC TAA TCC ATC TGA-3′ and 5′-CGC GAC TAA CAA TCA AAG TGA-3′. This will amplify a 236 bp fragment from mice carrying the human G93A SOD1 transgenic construct. 5′- CGC GCT CAC TGA CTG CCT TC-3′ and 5′-CTG GGAGCA GCA CTT CGC ACA GC-3′ will amplify a 282 and 299 bp fragment from wild-type or MT-II knock-out alleles, respectively. 5′-ACG TAG CGC ATC CGC TTG-3′ and 5′-CTC TTC TTG CAG TTC GTG C-3′ will amplify a 154 bp fragment from a wild-type MT-III allele. 5′-CTT GGG TGG AGA GGC TAT TC-3′ and 5′-AGG TGA GAT GAC AGG AGA TC-3′ will amplify a 280 bp fragment from the neomycin cassette used to generate the MT-III knock-out.
Motor testing and survival analysis. The stride test was performed with slight modifications from previously published methods (Gong et al., 2000). Mice had their hindpaws dipped in nontoxic ink and were trained to walk across the 1 m white board for recording footprints. Stride lengths were measured in millimeters. Mice unable to move hindlimbs and perform this task were graded as zero. Statistical analysis was performed using the Student's ttest. Forelimb grip testing was done with a grip metertransducer (Ugo Basile, Comerio, Italy) designed especially for mouse. At monthly testing intervals, three independent trials were performed, with the top two being averaged. Animals unable to grip were recorded as zero. All procedures performed on mice were approved by an animal research committee (ARC, University of Texas, Southwestern Medical Center, Dallas, TX) and conform to National Institutes of Health guidelines. Animals unable to correct posture when placed on side were considered end stage and killed. Survival analysis was performed using the Kaplan–Meier method.
Immunochemistry and histology. After overdose with pentobarbital (250 mg/kg, i.p.), animals were killed and then perfused with 4% paraformaldehyde for spinal cord and brain removal. For immunohistochemistry, paraffin-embedded sections (10 μm) were incubated with a rabbit polyclonal GFAP antibody (1:250 dilution; Dako, High Wycombe, UK), visualized using an immunoperoxidase reaction (the one kit; Sternberger Monoclonals, Lutherville, MD), and counterstained with cresyl violet (.5%). For motor neuron counting, 8 μm sections of L5–L6 spinal cord from paraffin-embedded tissue were stained with 0.5% cresyl violet. Random sections through the L5–L6 cord were selected and used for counting. Motor neurons were identified using established criteria (Deckwerth et al., 1996; Reaume et al., 1996;Kostic et al., 1997), and the number of motor neurons per spinal cord section was counted by an examiner unaware of the animal genotype. At least five sections per animal were used, and the average number of motor neurons per slice per animal was assessed.
Western blotting. Animals were overdosed with sodium pentobarbital (250 mg/kg, i.p.). Spinal cords were dissected and homogenized in 20 mm Tris-HCl, pH 7.5, 2 mm dtt, 0.1 mg of leupeptin, 1 mm EDTA, and 1 mm EGTA. The homogenate was then centrifuged at 14,000 × g to pellet debris. Protein concentration was measured using the BCA protein assay (Pierce, Rockford, IL). Protein (25 μg) from each sample was run on a 4–20% Tris-glycine gel (Invitrogen, San Diego, CA). After transfer, membranes were washed in PBS, followed by overnight incubation in blocking buffer [0.2% I-block (Applied Biosystems, Foster City, CA), PBS, and 0.1% Tween 20]. The membrane was then probed with a polyclonal rabbit anti-bovine SOD1 (Chemicon, Temecula, CA) antibody at 1:4000 dilution. After several washes, membranes were incubated with an alkaline phosphatase-conjugated secondary antibody (1:5000 in blocking buffer), and the immunoreactive signals were visualized using an enhanced chemiluminescent reagent, CDP Star (Western Star kit; Tropix). After exposure, films were scanned and then imported into NIH Image for quantitation of band density.
Zinc determination. Mice from 5.5–6 months of age were used for zinc determination. Mice were overdosed with sodium pentobarbital (250 mg) and perfused with PBS before tissue removal. All glassware was first soaked in 5% nitric acid and then rinsed with 18 MΩ deionized water before use. Animal tissue was hydrolyzed overnight in 70% “ultraspec” nitric acid at 102°C. Sample blanks, consisting of nitric acid only heated overnight, were included with tissue samples. Total zinc was determined using a Varian Spectra AA-20 atomic spectrophotometer at 213.9 nm wavelength according to the instructions of the manufacturer. Three to four animals were used per group. Units for zinc are given in micrograms per gram of wet weight of tissue.
RESULTS
Expression of glial metallothioneins influences survival and disease course in G93A SOD1 transgenic mice
To assess a potential role for glial metallothioneins (MT-I and MT-II) in mutant SOD1-induced disease, we crossed G93A SOD1 transgenic mice with knock-out mice lacking both MT-I and MT-II. These double knock-out mice have a normal lifespan and do not exhibit a motor neuron phenotype. Initially, we used an accelerated breeding program (rapid backcross to metallothionein strain) to generate and compare G93A SOD1 mice with the three possible metallothionein genotypes (wild type, heterozygous, and homozygous knock-outs). The survival curves for the accelerated paradigm are shown in Figure1. G93A SOD1 mice deficient in MT-I and MT-II demonstrated a significantly reduced mean survival (204 ± 4 d) compared with G93A SOD1 mice with normal metallothionein expression (240 ± 3 d; p < 0.0001). This 36 d change corresponds to a 15% overall reduction in G93A SOD1 lifespan. G93A SOD1 mice heterozygous for MT-I and MT-II also exhibited a significant reduction in mean survival (221 ± 4 d), although not to the same degree as homozygous knock-outs. Thus, survival in G93A SOD1 mice is dependent on MT-I and MT-II expression in a dose-dependent manner. These results suggest that primary changes within non-neuronal cell populations can significantly affect survival of mutant SOD1 mice.
Fig. 1.

Kaplan–Meier cumulative survival analysis of G93A SOD1 mice with or without MT-I and MT-II expression following the accelerated breeding protocol. Survival in G93A SOD1 mice is dependent on levels of MT-I and MT-II expression (log-rank test;p < 0.0001). MT−/−, MT-I and MT-II knock-outs; MT+/−, mice heterozygous for both MT-I and MT-II.
To ensure that survival changes observed in G93A SOD1 mice lacking MT-I and MT-II were not attributable to artifacts of cross breeding, we repeated the experiment using a prolonged breeding protocol that sought to place the mice on the original G93A transgenic background. The results are shown in Figure2A. G93A SOD1 mice deficient in MT-I and MT-II had a significantly reduced mean survival (229 ± 5 d) compared with G93A SOD1 mice with normal metallothionein expression (261 ± 4 d; p < 0.0001). This 32 d change corresponds to a 12% reduction in overall G93A SOD1 lifespan. As in the accelerated breeding paradigm, G93A SOD1 mice heterozygous for MT-I and MT-II demonstrated a reduction in survival of significant but intermediate proportions (249 ± 4 d). Thus, results obtained from two varying breeding protocols demonstrate a similar dose-dependent effect of MT-I and MT-II expression on the survival of G93A SOD1 mice.
Fig. 2.
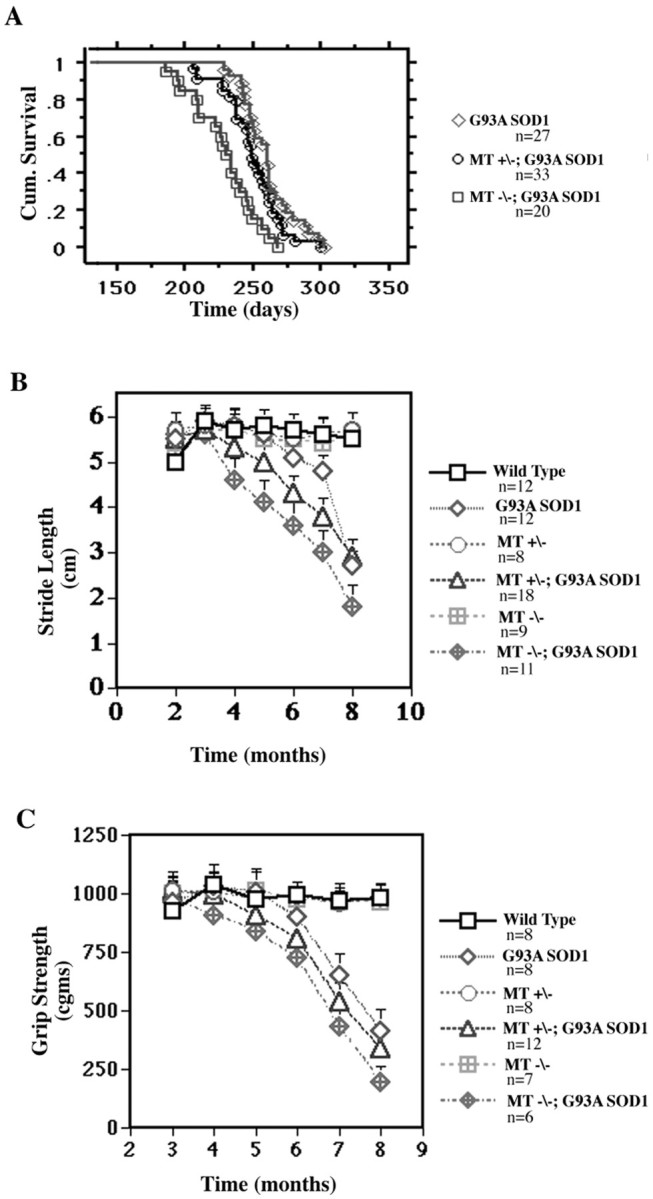
Survival and onset of motor dysfunction is influenced by levels of MT-I and MT-II expression (prolonged breeding protocol). A, Kaplan–Meier cumulative survival analysis (log-rank test; p < 0.0001). B, Stride length analysis; onset of motor dysfunction is significantly influenced by non-neuronal metallothionein expression levels.C, Grip strength analysis; onset of motor dysfunction is significantly affected by non-neuronal metallothionein expression.MT−/−, MT-I and MT-II knock-outs; MT+/−, mice heterozygous for both MT-I and MT-II.
We also performed longitudinal analysis of motor function using both stride length and grip strength to determine the effects of MT-I and MT-II expression on disease onset and progression. Measurement of stride length is primarily an indicator of hindlimb motor function and has been well characterized in mutant SOD1 transgenic mice (Gong et al., 2000). By 6 months of age, G93A SOD1 mice have begun to develop abnormalities in gait characterized by stride shortening, which then progress over the next 2 months at a rate of 1.2 cm/month (Fig.2B). In contrast, G93A SOD1 mice deficient for MT-I and MT-II demonstrated onset of gait abnormalities at a much earlier age. A decrease in stride length was apparent in these mice between 3 and 4 months of age and thereafter continued to decline at a rate of 0.7 cm/month. G93A SOD1 mice heterozygous for MT-I and MT-II demonstrated onset of abnormal gait at 5 months of age, again intermediate between G93A SOD1 and metallothionein-deficient G93A SOD1 mice. We then used grip strength as a primary measure of forelimb motor function in these mice (Fig. 2C). G93A SOD1 mice demonstrated onset of significant grip weakness between 6 and 7 months of age. However, the onset of grip weakness was significantly accelerated in G93A SOD1 mice lacking both MT-I and MT-II, with initial declines observed between 4 and 5 months of age. Again, G93A SOD1 mice heterozygous for the MT-I and MT-II allele first exhibited grip weakness at an intermediate time point between 5 and 6 months.
Results from these tests of motor function reveal a dose-dependent effect of MT-I and MT-II on the course of motor dysfunction in G93A SOD1 similar to what was observed for survival. However, although survival was shifted by ∼1 month in G93A SOD1 mice lacking both MT-I and MT-II, the onset of motor dysfunction was shifted even more robustly. Thus, changes in non-neuronal metallothionein expression appear to alter the onset of motor dysfunction in mutant SOD1 mice to a greater extent than overall survival.
We next investigated whether G93A SOD1 deficient in MT-I and MT-II demonstrated any changes in spinal cord pathology to accompany the observed alterations in motor function. Spinal cords from endstage G93A SOD1 mice (8.5 months) and G93A SOD1 mice lacking MT-I and MT-II (7.5 months) showed extensive astrogliosis and microgliosis throughout the spinal cord and brainstem. Both lines also demonstrated significant and comparable neuronal dropout, with an ∼75% loss in lumbar ventral horn motor neurons. The pathology in presymptomatic (3 month) G93A SOD1 mice and G93A SOD1 mice lacking MT-I and MT-II demonstrated mild astroglial changes and neuronal loss. Examination of tissue from an early symptomatic time point (5.25 months) indicated enhanced astroglial changes in the spinal cords of G93A SOD mice lacking MT-I and MT-II compared with G93A SOD1 mice with normal MT-I and MT-II (Fig.3A,B). In addition, increased GFAP reactivity was observed in white matter tracts of spinal cord and brainstem compared with tissue from age-matched G93A SOD1 mice (Fig. 3C,D). However, despite differences in non-neuronal pathology and motor deficits, neuronal loss in the lumbar ventral horn was similar in G93A SOD1 mice with or without MT-I and MT-II at each examined time point, including presymptomatic, early symptomatic, and end stage (Fig.4). These results indicate that, even if motor neurons are present, they may be dysfunctional and therefore cause a decline in motor function.
Fig. 3.
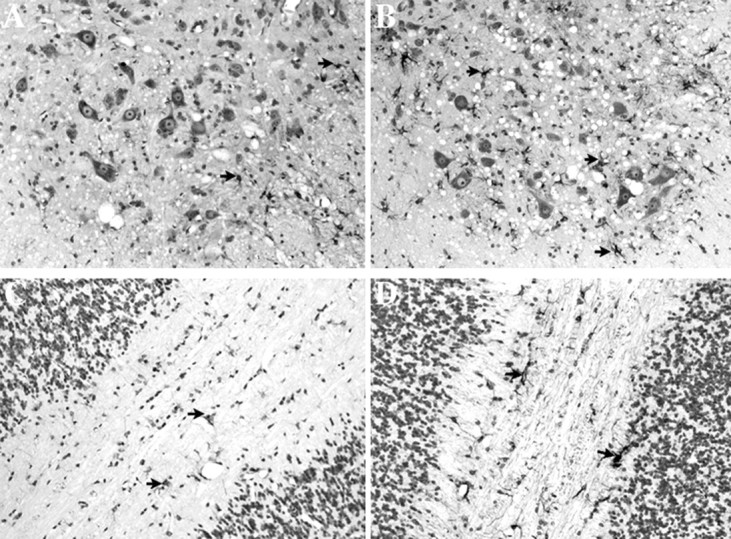
GFAP immunoreactivity is increased in G93A SOD1 mice lacking MT-I and MT-II. A, B, L5–L6 spinal cord ventral horn from 5.25-month-old G93A SOD1 transgenic mice with normal MT-I and MT-II (A) or absent MT-I and MT-II (B). C, D, Cerebellar white matter from 5.25-month-old G93A SOD1 transgenic mice with normal MT-I and MT-II (C) or absent MT-I and MT-II (D). Magnification, 200×.Arrowheads, GFAP reactive cells.
Fig. 4.

Motor neuron counts performed on L5–L6 spinal cords at various functional time points. Motor neuron survival in G93A SOD1 mice is not affected by levels of non-neuronal metallothionein (MT-I and MT-II) expression. n = 4 for each group.
Survival in G93A SOD1 mice is dependent on neuronal MT-III expression
To address whether altering zinc binding proteins in neurons might also influence the course of mSOD1-induced disease in vivo, we crossed G93A SOD1 transgenic mice with MT-III knock-out mice. Because previous experiments using MT-I and MT-II knock-out mice did not demonstrate significant differences between the accelerated and prolonged breeding paradigms, we selected the accelerated breeding paradigm for generating G93A SOD1 mice lacking MT-III.
The effects of reducing MT-III expression on the survival of G93A SOD1 mice are shown in Figure 5. MT-III knock-out mice lacking the G93A SOD1 mutation do not exhibit any change in motor function or survival compared with wild-type mice. G93A SOD1 mice deficient for both MT-III alleles demonstrate a marked reduction in overall survival compared with G93A SOD1 mice possessing both wild-type MT-III alleles. The mean survival for G93A SOD1 mice (253 ± 6 d) was reduced by over 20% to 202 ± 4 d (p < 0.0001 log-rank test) for G93A SOD1 mice lacking both MT-III alleles. No significant change in survival was observed for G93A SOD1 mice heterozygous for MT-III (249 ± 6 d), indicating that one MT-III allele is sufficient to rescue the original phenotype and contrasts to the dose-dependent survival effects described previously for the non-neuronal metallothioneins.
Fig. 5.

Survival and disease course is influenced by MT-III expression. A, Kaplan–Meier cumulative survival analysis (log-rank test; p < 0.0001).B, Stride length analysis; progression but not onset of motor dysfunction is significantly affected by neuronal MT-III expression. C, Grip strength analysis; progression of motor dysfunction is significantly affected by neuronal MT-III expression. MT−/−, MT-III knock-outs;MT+/−, mice heterozygous for MT-III.
If survival in G93A SOD1 mice lacking MT-III is shortened by almost 2 months, we then asked whether a corresponding temporal shift in motor function also occurred in these mice. Longitudinal assessment of stride length in G93A SOD1 mice lacking MT-III revealed significant differences in motor function compared with G93A SOD1 mice (Fig.5B). Although both groups displayed a similar onset of gait abnormality between 5 and 6 months of age, the decline in motor function was much steeper for G93A SOD1 mice lacking MT-III (2 cm) than for G93A SOD1 mice (1.2 cm). Similar changes were observed for grip strength (Fig. 5C). G93A SOD1 mice with and without MT-III exhibit normal grip strength until 5 months of age. After this time point, G93A SOD1 mice lacking MT-III show an accelerated rate of decline in motor function compared with G93A SOD1 mice. Thus, MT-III expression within neurons greatly affects survival of G93A SOD1 mice and markedly influences progression of mutant SOD1-induced disease.
Pathologic examination of spinal cord from presymptomatic and end stage G93A SOD1 mice with or without MT-III expression did not reveal significant differences in neuronal survival. However, at an early symptomatic time point (5.25 months), G93A mice lacking MT-III exhibited a significant increase in motor neuron loss compared with G93A mice with normal MT-III levels (Fig.6). Thus, in mice with alterations in neuronal MT-III, the increased severity of motor dysfunction parallels the more pronounced loss of motor neurons observed. This pattern is in contrast to the knock-out of non-neuronal metallothioneins in which the more severe changes in motor function are not matched by a more significant loss of motor neuron number.
Fig. 6.
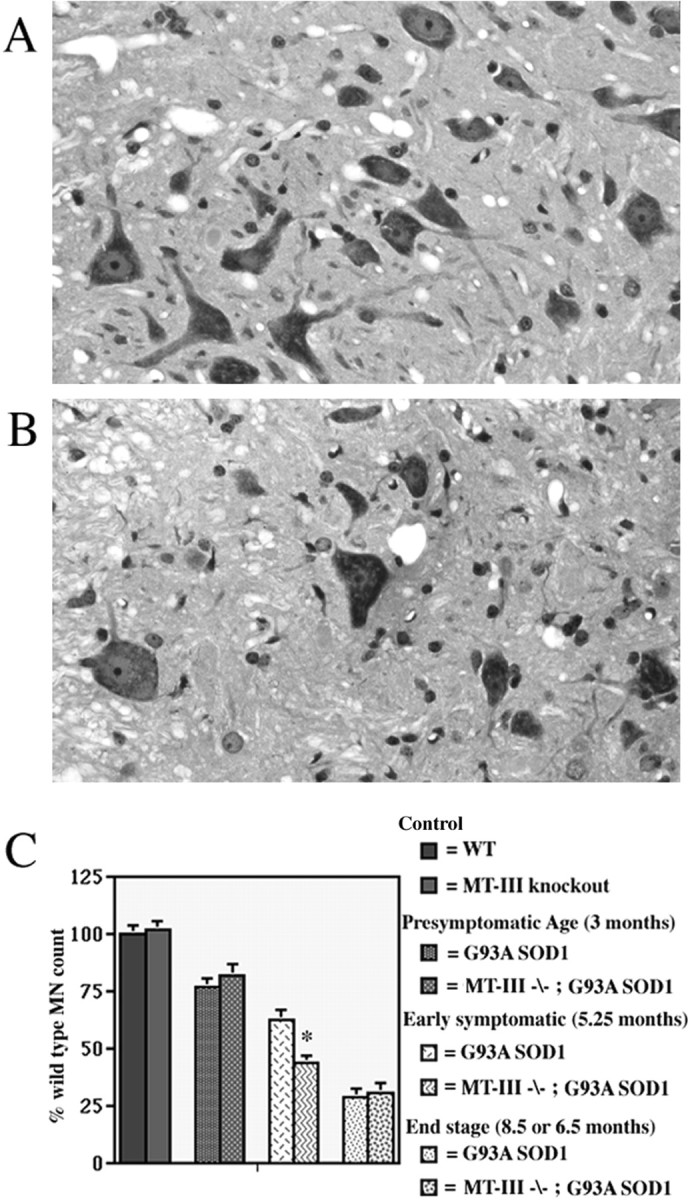
Motor neuron loss is enhanced in G93A SOD1 mice lacking MT-III. A, L5–L6 ventral horn from a 5.25-month-old G93A SOD1 transgenic mouse with normal MT-III.B, L5–L6 ventral horn from a 5.25-month-old G93A SOD1 transgenic mouse lacking MT-III. Magnification, 400×.C, Motor neuron counts performed on L5–L6 spinal cords at various functional time points. Motor neuron survival in G93A SOD1 mice is affected by levels of neuronal MT-III expression at an early symptomatic time point. *p < 0.03.n = 4 for each group.
Because of the importance of metallothioneins in zinc homeostasis, we next asked whether altering MT-III expression within the CNS of G93A SOD1 mice in fact led to corresponding changes in zinc levels. Total zinc levels in brain were determined for adult wild-type, MT-III knock-out, and G93A SOD1 mice with normal and absent MT-III expression. Zinc levels in MT-III knock-out mice were significantly lower than in wild-type mice (11.7 ± 0.1 vs 13.0 ± 0.1 μg/gm tissue;p < 0.01). Zinc levels in G93A SOD1 mice (18.6 ± 1.0 μg/gm tissue; p < 0.002) were significantly higher (>50%) than in wild-type mice. G93A SOD1 mice lacking any MT-III expression also demonstrated significantly increased zinc levels (16.8 ± 0.2 μg/gm tissue; p < 0.004) compared with wild-type mice, although not to the same degree as G93A SOD1 mice with normal MT-III expression. Zinc levels in the brains of G93A SOD1 mice lacking MT-III were reduced compared with G93A SOD1 mice with normal MT-III expression, although results did not reach significance (p < 0.07) because of variance in G93A SOD1 values. These results indicate that the total zinc pool within the CNS is reduced in mice lacking MT-III, raising the possibility that less zinc may be available for binding to SOD1.
Changes in disease phenotype are not related to increases in mutant SOD1 protein expression
Because the severity of mutant SOD1-induced disease in transgenic mice, both in terms of onset and progression, is clearly dependent on levels of mutant SOD1, we wanted to determine whether altering metallothionein synthesis could in fact result in changes of SOD1 protein expression. Western blot analysis of spinal cord and liver extracts from adult G93A SOD1 mice and from G93A SOD1 mice deficient in either MT-III or MT-I and MT-II show equivalent levels of mutant SOD1 protein expression (Fig. 7). Thus, the changes in disease phenotype manifested in the absence of metallothionein expression are not simply attributable to a compensatory increase in mutant SOD1 protein levels.
Fig. 7.

Western blot of adult mouse spinal cord and liver probed with SOD1 antibody. hSOD1, Human SOD1;mSOD1, normal murine SOD1. Spinal cord extract (25 μg) and 15 μg of liver extract were loaded per lane.
DISCUSSION
Precise mechanisms underlying mutant SOD1 toxicity in ALS remain unclear, but there is growing evidence to support a potential role for zinc in the disease process. The mutations in SOD1 linked to familial ALS (FALS) span all five exons of the coding region and encompass virtually all functional domains of the protein, including the copper binding site, active site, dimer interface, and β structural regions (Gaudette et al., 2000). However, to our knowledge, no mutations in the four zinc binding residues in SOD1 have been identified in FALS, suggesting that interactions between zinc and SOD1 may be important for disease expression. Although zinc binding sites may be preserved in SOD1 mutations causing disease, studies have shown that zinc binding to mutant SOD1 is abnormal with consequences both for the enzymatic and structural integrity of the protein. Abnormal zinc binding to SOD1 results in its increased ability to catalyze addition of nitro groups to protein tyrosine residues. Changes in zinc binding also enhance the tendency of certain proteins, including prion protein and β amyloid, to form insoluble complexes, and such mechanisms may contribute to the formation of SOD1 aggregates observed in the disease (Johnston et al., 2000; Cherney et al., 2001; Curtain et al., 2001; Quaglio et al., 2001). Our findings that disease progression in G93A SOD1 transgenic mice is markedly affected by the expression of zinc binding metallothioneins provide one more piece of evidence to implicate interactions of zinc with mutant SOD1 as a contributing factor to the disease phenotype.
There are a number of ways in which changes in metallothionein expression might promote mutant SOD1-induced toxicity. Metallothioneins can function as zinc chaperones, ferrying zinc ions to certain apo-proteins, and in vitro experiments have found that metallothioneins can perform this function for apo-SOD1 (Suzuki and Kuroda, 1995). Without metallothioneins, it may be harder to ensure adequate zinc loading for apo-SOD1, particularly in the context of the decreased affinity for zinc manifested by mutant SOD1 compared with wild-type SOD1 (Crow et al., 1997a). Our findings indicating overall lower zinc levels in mice deficient for MT-III would be consistent with this hypothesis. Experiments have demonstrated that zinc-deficient SOD1 is highly toxic to motor neurons in culture, and similar processes may also occur in vivo (Estevez et al., 1999). Metallothioneins also buffer excess zinc and protect cells from the potentially harmful consequences of excessive intracellular zinc. SOD1 is normally expressed at high levels within the CNS, comprising 1% of total protein, and, with up to a 20-fold decrease in zinc affinity for mutant SOD1 compared with wild-type SOD1, conditions may favor release of zinc from mutant SOD1 (Crow et al., 1997a; Elliott, 2001). Excess zinc may be toxic through a variety of pathways, including modulation of glutamate receptor or transporter function, thus leaving neurons vulnerable to excitotoxicity (Sensi et al., 1999; Trotti et al., 1999;Mitrovic et al., 2001). Recent work has suggested that, at least in yeast, SOD1 itself may play an important role in zinc metabolism (Wei et al., 2001).
It is also possible that the protective effects of metallothioneins in G93A SOD1 mice may be unrelated to specific interactions with SOD1 but rather act through indirect and nonspecific mechanisms. Metallothioneins clearly protect cells against oxidative injury in general and may perform similar roles in mutant SOD1-induced disease. Recently, experiments have demonstrated that metallothioneins may also be found in mitochondria and appear to be important in modulating cellular respiration via changes in zinc levels within mitochondria (Ye et al., 2001). The absence of metallothioneins could potentially lead to abnormal cellular respiration and enhanced mitochondrial dysfunction in cells already subjected to mutant SOD1 toxicity. In fact, studies from mutant SOD1 transgenic mice indicate that early and prominent mitochondrial pathology is a hallmark of mutant SOD1-induced disease (Dal Canto and Gurney, 1994; Wong et al., 1995). Recent evidence suggests that some SOD1 may also localize to mitochondria, allowing for direct interactions between metallothionein and SOD1 in this organelle (Okado-Matsumoto and Fridovich, 2001; Sturtz et al., 2001).
Neuronal and non-neuronal cells in mutant SOD1-induced disease
The restricted expression of distinct metallothioneins in either glia or neurons also allows an assessment of both neuronal and non-neuronal contributions to the disease process. Although motor neuron dysfunction and degeneration are hallmarks of ALS, recent work has demonstrated that mutant SOD1 expression restricted to neurons is not sufficient to cause weakness in transgenic mice, suggesting that expression of mutant SOD1 in other cell types is necessary for the disease phenotype (Pramatarova et al., 2001). Glia cells, particularly astrocytes, may contribute to the generation of disease. Astrocytes develop early pathologic features, including SOD1-positive aggregate formation and selective loss of the principal glutamate transporter EAAT-2 (excitatory amino acid transporter-2) with concomitant alterations in glutamate transport function (Rothstein et al., 1995;Bruijn et al., 1997b; Canton et al., 1998). Indeed, expression of mSOD1 can result in a post-translational inactivation of this glutamate transporter, potentially leading to the increases in extracellular glutamate observed both in familial ALS patients and mSOD1 transgenic mice (Rothstein et al., 1992; Trotti et al., 1999: Alexander et al., 2000). However, targeted expression of mutant SOD1 in astrocytes in transgenic mice, although resulting in astrocytosis, does not cause a motor phenotype or neuronal loss (Gong et al., 2000). Thus, experiments using cell-specific expression of mutant SOD1 suggest that defects in both neuronal and non-neuronal cells may be necessary for disease manifestation.
Our experimental results with metallothioneins indicate that both non-neuronal and neuronal dysfunction contribute to disease progression and survival in this murine model of ALS but do so in disparate ways. Deletion of non-neuronal metallothioneins reduces survival in G93A SOD1 mice by ∼1 month but has a more marked effect on onset of motor deficits. However, this accelerated motor phenotype occurs without significant change in motor neuron number, indicating that extent of motor dysfunction and motor neuron loss may be discordant. In contrast, removal of neuronal MT-III produces a marked change in survival and disease course that is congruent with enhanced neuronal loss. Mechanisms underlying metallothionein-induced changes in either neuronal or non-neuronal populations from mutant SOD1 transgenic mice remain unclear but do not appear to be related to changes in mutant SOD1 expression. We are exploring currently whether those possible mechanisms include an enhanced capacity of SOD1 to catalyze peroxynitrite formation or an increased tendency for mutant SOD1 to form aggregates in the absence of metallothionein expression.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS40911, the Muscular Dystrophy Association, and the Amyotrophic Lateral Sclerosis Association. We thank Dr. Richard Palmiter for the MT-III knock-out mice. We thank Christine Elliott, Yongli Kong, and Jill Marshall for expert technical assistance.
Correspondence should be addressed to Dr. Jeffrey L. Elliott, Department of Neurology, University of Texas, Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390. E-mail:jellio@mednet.swmed.edu.
REFERENCES
- 1.Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Paterson TD. Elevated cortical extracellular glutamate in transgenic mice expressing mutant (G93A) Cu/Zn superoxide dismutase. J Neurochem. 2000;74:1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF, Ferrante RJ, Browne SE, Mattews RT, Kowall NW, Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:646–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 3.Blaauwgeers HGT, Chand MA, van den Berg FM, Vianney de Jong JMB, Troost D. Expression of different metallothionein messenger ribonucleic acids in motor cortex, spinal cord and liver from patients with amyotrophic lateral sclerosis. J Neurol Sci. 1996;142:39–44. doi: 10.1016/0022-510x(96)00013-5. [DOI] [PubMed] [Google Scholar]
- 4.Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, Price DL, Cleveland DW. Elevated free nitrotyrsoine levels but not protein bound or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997a;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997b;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 6.Canton T, Pratt J, Stutzmann JM, Boireau A. Glutamate uptake is decreased tardively in the spinal cord of FALS mice. NeuroReport. 1998;9:775–778. doi: 10.1097/00001756-199803300-00001. [DOI] [PubMed] [Google Scholar]
- 7.Cherney RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim YS, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 8.Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997a;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 9.Crow JP, Ye YZ, Strong M, Kirk M, Barnes S, Beckman JS. Superoxide catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997b;69:1945–1954. doi: 10.1046/j.1471-4159.1997.69051945.x. [DOI] [PubMed] [Google Scholar]
- 10.Curtain CC, Ali F, Volitakis I, Cherney RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer's disease amyloid-β binds to copper and zinc to generate an allosterically ordered membrane penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 11.Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of ALS. Am J Pathol. 1994;145:1271–1280. [PMC free article] [PubMed] [Google Scholar]
- 12.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 13.Elliott JL. Zinc and copper in the pathogenesis of amyotrophic lateral sclerosis. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1169–1185. doi: 10.1016/s0278-5846(01)00185-3. [DOI] [PubMed] [Google Scholar]
- 14.Erickson JC, Hollopeter G, Thomas SA, Froelick GJ, Palmiter RD. Disruption of the metallothionein-III gene in mice: analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J Neurosci. 1997;17:1271–1281. doi: 10.1523/JNEUROSCI.17-04-01271.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide dependent apoptosis in motor neurons by zinc deficient superoxide dismutase. Science. 1999;286:2497–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 16.Ferrante RJ, Shinobu LA, Schulz JB, Mattews RT, Thomas CE, Kowall NW, Gurney ME, Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 17.Gaudette M, Hirano M, Siddique T. Current status of SOD1 mutations in familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:83–89. doi: 10.1080/14660820050515377. [DOI] [PubMed] [Google Scholar]
- 18.Gong YH, Elliott JL. Metallothionein expression is altered in a transgenic model of familial amyotrophic lateral sclerosis. Exp Neurol. 2000;162:735–742. doi: 10.1006/exnr.2000.7323. [DOI] [PubMed] [Google Scholar]
- 19.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goto JJ, Zhu H, Sanchez RJ, Nersissian A, Gralla EB, Valentine JS. Loss of in vitro metal binding in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2000;275:1007–1014. doi: 10.1074/jbc.275.2.1007. [DOI] [PubMed] [Google Scholar]
- 21.Jacob C, Maret W, Vallee BL. Control of zinc transfer between thionein, metallothionein and zinc proteins. Proc Natl Acad Sci USA. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostic V, Jackson-Lewis V, deBilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 24.Lyons TJ, Liu H, Goto JJ, Nersissian A, Roe JA, Graden JA, Cafe C, Ellerby LM, Bredesen DE, Gralla EB, Valentine JS. Mutations in the copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc Natl Acad Sci USA. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masters BA, Quaife CJ, Erickson JC, Kelly EJ, Froelick GJ, Zambrowicz BP, Brinster RL, Palmiter RD. Metallothionein III is expressed in neurons that sequester zinc in synaptic vesicles. J Neurosci. 1994a;14:5844–5857. doi: 10.1523/JNEUROSCI.14-10-05844.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masters BA, Kellly EJ, Quaife CJ, Brinster RL, Palmiter RD. Targeted disruption of metallothionein I and II genes increases sensitivity to cadmium. Proc Natl Acad Sci USA. 1994b;91:584–588. doi: 10.1073/pnas.91.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitrovic AD, Plesko F, Vandenberg RJ. Zn2+ inhibits the anion conductance of the glutamate transporter EAAT4. J Biol Chem. 2001;276:26071–26076. doi: 10.1074/jbc.M011318200. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura N, Nishimura H, Ghaffar A, Tohyama C. Localization of metallothionein in the brain of rat and mouse. J Histochem Cytochem. 1992;40:309–315. doi: 10.1177/40.2.1552172. [DOI] [PubMed] [Google Scholar]
- 29.Okado-Matsumoto A, Fridovich I. Subcellular localization of superoxide dismutases (SOD) in rat liver. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 30.Palmiter RD. The elusive function of metallothioneins. Proc Natl Acad Sci USA. 1998;95:8428–8430. doi: 10.1073/pnas.95.15.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmiter RD, Findley SD, Whitmore TE, Durnham DM. MT-III, a brain-specific member of the metallothionein gene family. Proc Natl Acad Sci USA. 1992;89:6333–6337. doi: 10.1073/pnas.89.14.6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pramatarova A, Laganiere L, Roussel J, Brisebois K, Rouleau GA. Neuron specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quaglio E, Chiesa R, Harris DA. Copper converts the cellular prion protein into a protease resistant species that is distinct from the scrapie isoform. J Biol Chem. 2001;276:11432–11438. doi: 10.1074/jbc.M009666200. [DOI] [PubMed] [Google Scholar]
- 34.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 35.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 36.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 37.Sensi SL, Yin HG, Carriedo SG, Rao SS, Weiss JH. Preferentialz N2+ influx through Ca2+ permeable AMPA channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smitt PAE, Blaauwgeers HGT, Troost D, de Jong JMBV. Metallothionein immunoreactivity is increased in the spinal cord of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1992;144:107–110. doi: 10.1016/0304-3940(92)90727-o. [DOI] [PubMed] [Google Scholar]
- 39.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu, Zn superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki T, Kuroda T. Transfer of copper and zinc from ionic and metallothionein bound forms to Cu, Zn superoxide dismutase. Res Commun Mol Pathol Pharmacol. 1995;87:287–296. [PubMed] [Google Scholar]
- 41.Toghi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 1999;46:129–131. doi: 10.1002/1531-8249(199907)46:1<129::aid-ana21>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 42.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 43.Wei JJ, Srinivasan C, Han H, Valentine JS, Gralla EB. Evidence for a novel role for copper-zinc superoxide dismutase in zinc metabolism. J Biol Chem. 2001;276:44798–44803. doi: 10.1074/jbc.M104708200. [DOI] [PubMed] [Google Scholar]
- 44.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 45.Ye B, Maret W, Vallee BL. Zinc metallothionein imported into liver mitochondria modulates respiration. Proc Natl Acad Sci USA. 2001;98:2317–2322. doi: 10.1073/pnas.041619198. [DOI] [PMC free article] [PubMed] [Google Scholar]