

Abstract
In mammals, long-term memory induced by Pavlovian fear conditioning has been shown to be dependent on the amygdala during a protein and mRNA synthesis-dependent phase of memory consolidation. We have used genes identified in a kainic acid model of synaptic plasticity asin situ hybridization probes during the consolidation period after fear conditioning. We found that these genes were transcriptionally regulated in several brain areas only when stimuli were presented in a manner that supported behavioral learning and not after unpaired presentations or footshocks alone. Immediate early genes and neurofilament mRNA peaked ∼30 min after conditioning, as expected. Interestingly, nurr-1, α-actinin, and 16c8 increased ∼2–4 hr later, whereas neurogranin and gephyrin decreased during that time. Our results suggest that fear memory consolidation occurs within a broad neural circuit that includes, but is not limited to, the amygdala. Together, a broad array of transcriptionally regulated genes, encoding transcription factors, cytoskeletal proteins, adhesion molecules, and receptor stabilization molecules, appear to mediate the neural plasticity underlying specific forms of long-term memory in mammals.
Keywords: fear conditioning, learning, consolidation, synaptic plasticity, startle, amygdala
The early, middle, and late temporal phases of memory formation have been well described in invertebrates (Bailey et al., 1996; Dubnau and Tully, 1998). The mechanisms of long-term memory (LTM) in intact mammals have been assumed to have early and late phases of consolidation similar to simpler organisms (McGaugh et al., 1996; Abel et al., 1997; Walker and Davis, 2000; Carew and Sutton, 2001; Schafe et al., 2001). It is thought that activation of cAMP response element-binding protein (CREB) and other immediate early transcription factors [immediate early genes (IEGs)] leads to the transcriptional activation of other genes that presumably play an important role in the structural and functional neuronal changes involved in mammalian LTM (Radulovic et al., 1998; Rosen et al., 1998;Silva et al., 1998; Josselyn et al., 2001). Despite great advances in the understanding of the rapid events in the cellular mechanisms of learning, it remains unclear what many of the later transcribed genes might be. To our knowledge, there has been no systematic study of multiple genes involved in synaptic plasticity during the consolidation phase of a behaviorally relevant learning task.
Pavlovian fear conditioning has provided an excellent model system to study learning that occurs in a very short time frame, so that the temporal relationship between sensory stimuli can be tightly controlled (Davis; 1992; LeDoux, 1998; Fendt and Fanselow, 1999). This model has been studied extensively, and it is known that coincident sensory input coming into the basolateral amygdala (BLA) leads to conditioning. Thus, a previously neutral conditioned stimulus (CS) paired with an aversive unconditioned stimulus (US) now produces a hypothetical state of fear that leads to a fear response in the presence of the CS. This memory has been shown to have a consolidation period that is dependent on NMDA (Miserendino et al., 1990), phosphorylation (Schafe et al., 2000), and protein synthesis (Schafe et al., 1999). There is also some evidence that it is mRNA transcription dependent (Bailey et al., 1999). Although much evidence points to the lateral nucleus of the amygdala as the primary site for coincident learning of the US–CS association (Fanselow and LeDoux, 1999), there is also significant evidence that a broader neural circuitry underlies fear memory that is modulated by amygdala activity (Packard et al., 1994; McGaugh et al., 1996).
Although most studies of gene expression involved in fear conditioning have focused on single genes and generally limited time courses after learning (Campeau et al., 1991; Rosen et al., 1998; Jones et al., 2001), it is probable that a large number of genes will be involved at various stages of synaptic plasticity. To evaluate this possibility, we initially characterized the expression of a relatively large number of transcriptionally activated genes after kainic acid (KA) treatment. This treatment provides a robust model of synaptic plasticity that leads to the apparent induction of new synapse formation and structural reorganization (Ben-Ari and Represa, 1990; Nedivi et al., 1993; Wheal et al., 1998). Using in situ hybridization, we screened the expression of 40 genes initially and then 21 genes in detail that were induced by KA. A significant number of these were also transcriptionally regulated during consolidation in the amygdala and other limbic regions after CS–US pairings but not after unpaired or US alone presentations. Together, these findings suggest that the mechanisms underlying LTM of fear conditioning involve the transcriptional regulation of a broad combination of genes that mediate synaptic plasticity throughout a distributed neural circuit, including, but not limited to, the amygdala.
MATERIALS AND METHODS
Animals
A total of 78 adult male Sprague Dawley rats (Charles River, Raleigh, NC) weighing between 300 and 400 gm were used. Animals were housed in group cages of four rats each in a temperature (24°C)-controlled animal colony, had ad libitum access to food and water, and were maintained on a 12 hr light/dark cycle. All behavioral procedures took place during the light cycle and were approved by the Institutional Animal Care and Use Committee.
Kainic acid treatment
Methods were as described previously (Crispino et al., 1998;Hevroni et al., 1998). Briefly, 10 mg/kg kainic acid (Sigma, St. Louis, MO) was given intraperitoneally 6 hr before animals were killed. Animals were observed for the presence of mild seizure activity, which generally began 30–90 min after KA injection and subsided shortly thereafter. Animals were deeply anesthetized with 100 mg/kg Nembutal intraperitoneally and were perfused intracardially with fresh 4% paraformaldehyde. Eight animals were used (four naive and four KA) in two different experiments with KA.
General behavioral procedures
Animals were trained and tested in identical cages suspended between compression springs within a ventilated, sound-attenuated chamber as described previously (Cassella and Davis, 1986; Walker and Davis, 2000; Paschall and Davis, 2002). Cage movement resulted in displacement of an accelerometer, the analog output of which was amplified and digitized. Startle amplitude (reported in arbitrary linear units) was defined as the peak accelerometer voltage (integrated as velocity) that occurred during the first 300 msec after onset of the startle stimulus. All stimulus input and accelerometer output were automated using a Macintosh G3 computer and custom-designed software. Background white noise was delivered through high-frequency speakers placed 8 cm from the front of each cage. Startle responses were evoked by 50 msec, 95 dB white-noise bursts (0–22 kHz) delivered through the same speakers. The US was a 0.5 sec, 0.4 mA footshock delivered through cage floor bars (Cassella and Davis, 1986; Walker and Davis, 2000;Paschall and Davis, 2002). Visual CS for training and testing was a 4 sec light (82 lux) produced by an 8 W fluorescent bulb (100 μsec rise time) located 10 cm behind each cage. The odor CS was 5% amyl acetate (Sigma) diluted in propylene glycol. This odor was delivered for 4 sec through an olfactometer (model E15-03; Coulbourne Instruments, Allentown, PA) mounted outside the sound-attenuating chamber as described previously (Paschall and Davis, 2002). The olfactometer allowed the odor to be blended with an otherwise steady flow of air at an overall rate of 2 l/min. All stimuli were presented and sequenced under computer control.
Fear conditioning
Animals were pre-exposed to handling and placement in the training/testing chamber for 5 d before fear conditioning. During pre-exposure, baseline startle was measured on each of 2 d by presenting 30 startle stimuli at a 30 sec interstimulus interval (ISI). Animals were then divided into matched groups having equivalent baseline mean startle amplitudes. On the day of fear conditioning, the animal was brought to the room, allowed to habituate, and placed in the chamber as before. The CS–US pairing began after a 5 min acclimation period in the chamber.
Experiment 1. In the light-shock paired group, 15 light-shock pairings were given with an average intertrial interval (ITI) of 2 min (range, 1–3 min), creating a 30 min training period. The shock (US) was delivered during the last 0.5 sec and coterminated with the 4 sec light (CS). The context control group was placed in the chamber as on the previous days for 30 min also, but no stimuli were given. Two cohorts of animals were processed for experiment 1, with a total of 15 animals kept for behavioral testing 24 hr later and 21 animals killed at different time points.
Experiment 2. In the light-shock paired group, 10 light-shock pairings were given with an average ITI of 4 min (range, 3–5 min), over a 40 min training session. The shock (US) was delivered during the last 0.5 sec of the 4 sec light (CS). The unpaired light-shock group received 10 light, 10 odor, and 10 shock stimuli, in a pseudorandom order, with an average ITI of 1.5 min (range, 1–2 min), such that there was no overlap between any stimuli, over a 40 min training session. The shock-only group received 10 shocks, which each lasted 0.5 sec, with an average ITI of 4 min (range, 3–5 min), over a 40 min training session. Animals from all groups (n = 14 total) were returned after training to their home cage and were killed 2 hr later.
Experiment 3. In the odor-shock paired group, five odor-shock pairings were given with an average ITI of 4 min (range, 3–5 min), creating a 20 min training session. The shock (US) was delivered during the last 0.5 sec of the 4 sec odor (CS). The unpaired odor-shock group was placed in the chamber and separately given five shock stimuli and five odor stimuli, with a 2 min ITI (range, 1–3 min), so that there was no overlap between stimuli. The context control group was placed in the chamber for 20 min without stimuli. Eight animals were kept for behavioral testing 24 hr later, and 12 animals were killed at different time points for in situanalysis.
Behavioral testing. Twenty-four hours after fear conditioning, rats were returned to the test chamber for testing for fear-potentiated startle. In experiment 1, 30 initial startle stimuli were presented in darkness at a 30 sec ISI (leader stimuli), followed by 30 startle stimuli presented in darkness (startle alone) and 30 startle stimuli presented 3.5 sec after onset of the 4 sec light (light startle) in a balanced, pseudorandom order at a 30 sec ISI. During the testing for experiment 3, 30 leader stimuli were followed by a total of 40 startle stimuli presented at a 30 sec ISI. The first of every four startle stimuli was presented 3.5 sec after the onset of the 4 sec odor stimulus. The following three stimuli were presented without odor, allowing for the comparison of odor-startle versus startle-alone stimuli within the same session.
Startle amplitude was averaged over all of the startle-alone or light-/odor-startle stimuli within the test session. Data are presented as the absolute level of startle and as a percentage of fear-potentiated startle [100 × (startle amplitude on light or odor startle − startle-alone trials)/startle-alone trials]. Statistical comparisons were made with ANOVA, paired, two-tailedt tests (within-group comparisons) or t tests for independent samples (between-group comparisons).
Preparation of clones and in situ hybridization
cDNA clones of c-fos and corticotrophin-releasing factor (CRF) genes were obtained as indicated. All other genes analyzed were obtained as follows: The rat coding sequence for a gene of interest [obtained from the KA plasticity data (Nedivi et al., 1993;Crispino et al., 1998; Hevroni et al., 1998; Wheal et al., 1998)] was compared against the sequences from all known expressed sequence tags (ESTs) [National Center for Biotechnology Information (NCBI) basic local alignment search tool (BLAST) EST database]. The highest-homology ESTs that were available as IMAGE consortium clones (mouse and human EST databases) were identified and purchased through the Research Genetics IMAGE consortium (www.resgen.com). Individual clones were sequenced to obtain sense/antisense orientation and to verify the identity of the clone. All clones analyzed (Table1) were ≥90% homologous with rat coding sequence as determined by NCBI BLAST.
Table 1.
Synaptic plasticity gene expression after kainic acid and fear conditioning
| Gene | Gene product | GenBank GI number | KA-inducible areas | Changes in fear conditioning | Peak time of change | |
|---|---|---|---|---|---|---|
| 1 | c-fos | Transcription factor | 1-a | Hippocampus, amygdala, striatum, cortex | +++ | 0–60 min |
| 2 | zif268/EGR1 | Transcription factor | 2216697 | Hippocampus, amygdala, striatum, cortex | +++ | 0–60 min |
| 3 | c-jun | Transcription factor | 4058354 | Hippocampus, amygdala, cortex | ++ | 0–60 min |
| 4 | NF-l | Cytoskeletal protein | 813267 | Dentate, amygdala, piriform cortex | +++ | 0–60 min |
| 5 | Gephyrin | GlyR/GABAR anchor | 880230 | Hippocampus, amygdala, piriform cortex | ++ | 30–120 min |
| 6 | RC3/neurogranin | Second messenger modulation | 989352 | Hippocampus, amygdala, piriform cortex | +++ | 30–240 min |
| 7 | nurr-1 | Transcription factor | 5593088 | Hippocampus, amygdala, piriform cortex | +++ | 1–2 hr |
| 8 | 16c8 | Protease inhibitor | 2691513 | Dentate | ++ | 2–4 hr |
| 9 | α-actinin | NMDAR/GluR anchor | 2630541 | Hippocampus | ++ | 2–4 hr |
| 10 | N-cadherin | ECM/cell adhesion | 4198999 | Hippocampus | + | 2–4 hr |
| 11 | ler5/RM5 | Transcription factor | 8597370 | Dentate, amygdala | +/− | 1–4 hr |
| 12 | Tenascin | ECM/cell adhesion | 4061636 | Cortex | +/− | 1–4 hr |
| 13 | VGF | Neuropeptide | 5850546 | Hippocampus | +/− | 1–4 hr |
| 14 | EGR2/Krox20 | Transcription factor | 2158692 | Dentate | − | |
| 15 | CREM | Transcription factor | 2539111 | Hippocampus | − | |
| 16 | EGR4 | Transcription factor | 3988902 | Hippocampus, piriform cortex | − | |
| 17 | ARPP-21 | Signal transduction | 1682348 | Dentate, piriform cortex | − | |
| 18 | Rheb2 | Signal transduction | 2209795 | Dentate, piriform cortex | − | |
| 19 | PAI-2 | Phosphatase inhibitor | 1060156 | Dentate | − | |
| 20 | αB-crystallin | Chaperone protein | 1210687 | Dentate, cortex | − | |
| 21 | CRF/CRH | Neuropeptide | 1-b | Amygdala, hypothalamus | − |
The 21 genes that show robust changes with KA treatment are listed. Columns indicate the common gene name, the putative function of its gene product, the GI accession number of the EST clone used for in situ hybridization, areas of induction (or inhibition) with KA, whether the gene showed changes during consolidation of fear conditioning (qualitative: −, no detectable change; +, equivocal; +, moderate; +++, very robust changes), and the peak period of change during the consolidation of fear learning. CREM, cAMP responsive element modulator; ARPP-21, cAMP regulated phosphoprotein; PAI-2, plasminogen activator inhibitor-2; CRH, corticotrophin releasing hormone.
Gift from T. Curran (St. Jude's Research Hospital, Memphis, TN).
Gift from C. Nemeroff (Emory University School of Medicine, Atlanta, GA).
Animals were given a lethal dose of anesthetic (100 mg/kg Nembutal, i.p.) at various time points after fear conditioning and were perfused with 4% paraformaldehyde in PBS. After overnight fixation, brains were rinsed with PBS and allowed to equilibrate at 4°C in 20% sucrose in PBS. Brains were rapidly frozen in crushed dry ice and stored at −80°C. Brains were sectioned at 12–16 μm thickness on a Leica (Nussloch, Germany) Cryostat at −20°C onto gelatin-coated slides. All sections within the same experiment were of the same thickness. Frontal sections were placed on 25 consecutive slides per brain, such that each slide contained similar sections of brain from three different anatomical areas (anterior commissure, anterior amygdala, and posterior amygdala). In situ hybridization was performed as described previously (Sassoon et al., 1988; Ressler et al., 1993) except for the use of frozen sections instead of paraffin-embedded sections, with the omission of the dewaxing and rehydration steps described in those procedures. 35S-UTP (NEN, Boston, MA)-labeled riboprobes were prepared from linearized receptor clones using T7, T3, or SP6 RNA polymerase. Hybridizations were performed at 52°C overnight as described previously (Ressler et al., 1993) under parafilm. After hybridization, slides were stringently washed as described and dried (Ressler et al., 1993). The slides were placed alongside a linear radiation standard against Kodak (Rochester, NY) magnetic resonance autoradiography film for 12 hr to 7 d.
Films were scanned into a personal computer at 600 dpi, and images were analyzed with Adobe Systems (San Jose, CA) PhotoShop software. Hybridization density quantitation was performed with the mean luminosity histogram feature of Adobe PhotoShop. This measure was shown to produce linear densities with 14C radiation standards with the exposure times and levels used. Within an experiment, slides from all brains hybridized with the same probe were exposed to the same piece of film to ensure equivalent exposure times and conditions between animals and experimental groups. The relative level of mRNA expression (hybridization density) was determined as follows: (1) regions of interest (ROIs) were determined based on qualitative analysis. (2) For each section, density was determined for the ROIs and also for an adjacent area of the same size that did not have hybridization on the same section (background: the corpus callosum or internal capsule). Normalized density for each section was determined as the subtracted difference between the ROI density and background density. (3) The hybridization densities from two different cryostat sections per brain were examined and averaged to give the density for each individual per ROI. (4) For each ROI and experimental group, hybridization density is reported as the average density of all individual animals for that condition ± SEM. Comparisons of means between experimental groups were performed with ANOVA followed by orthogonal contrasts using the SPSS (Chicago, IL) statistics package.
RESULTS
Kainic acid induction of synaptic plasticity genes
Subtractive hybridization screens have described genes previously that are specifically induced in the hippocampus during the neural plasticity phase after KA-induced seizures (Nedivi et al., 1993;Hevroni et al., 1998; Wheal et al., 1998). We hypothesized that a subset of these genes is involved in the synaptic plasticity events underlying fear conditioning, and so we initially characterized the expression of ∼40 of them. Clones of the coding sequences of these genes were prepared as riboprobe templates for in situhybridization studies. Animals were killed 6 hr after the injection of saline or kainic acid (10 mg/kg), and brain sections were hybridized with 35S-labeled antisense riboprobe. Of the initial 40 genes, 21 showed clear changes in the KA-treated animals compared with the control animals (Table 1). The specificity of the antisense probes was confirmed by the similarities in gene expression patterns between the clones and the published patterns of expression for most of the genes, as well as the unique expression patterns with KA induction (Table 1). Figure 1 shows examples of six genes that are upregulated in the dentate gyrus and CA1-CA3 of the hippocampus after KA treatment, as well as two genes, RC3/neurogranin (Fig. 1G,g) and gephyrin (Fig.1H,h), that showed extensive downregulation during KA-induced plasticity. Table 1 also briefly summarizes the temporal changes in expression of these genes during the consolidation phase of fear learning. Interestingly, the anatomical and temporal pattern of gene expression that followed fear conditioning did not correlate in an obvious way with the pattern of gene expression that followed KA-induced seizures.
Fig. 1.

In situ hybridization analyses of genes after kainic acid induction. Magnified bright-field images are shown of hippocampal sections that have been hybridized with35S-labeled antisense riboprobe and exposed to autoradiography film. The brains were from rats that were treated with either saline (A–H) or kainic acid (a–h) and perfused 6 hr later (A, a, c-fos; B, b, zif268; C, c, neurofilament–light chain; D, d, 16c8;E, e, Rheb2; F, f, cAMP responsive element modulator; G, g, RC3/neurogranin; H, h, gephyrin).
Expression of plasticity genes after light-shock associative learning
We initially tested whether we could simply detect differences in plasticity-associated gene expression after fear conditioning versus placement in the context alone without fear conditioning. This would allow us to determine the time course and extent of gene expression change for later, more comprehensive behavioral studies.
Animals were trained and tested as illustrated in Figure2A. On the training day, animals received 15 light-shock pairings given over a 30 min period (light-shock pairing) or no new stimuli (context control) and were returned to the home cage. They were killed at several different time points (0, 1, 4, or 8 hr after training; n = 21 total) or were tested 24 hr later for the presence of fear-potentiated startle (n = 15 total) (Fig. 2A). Animals that had experienced light-shock pairings showed significant fear-potentiated startle as demonstrated by a 60% increase in the acoustic startle reflex in the presence of light (Fig.2B; Light-Startle vs Startle alone; p< 0.05). Animals that had been placed in the context on the day of training but did not receive light-shock pairing showed no appreciable difference between startle in the presence or absence of light, and their difference scores were significantly different from the trained group (t test; p < 0.05).
Fig. 2.
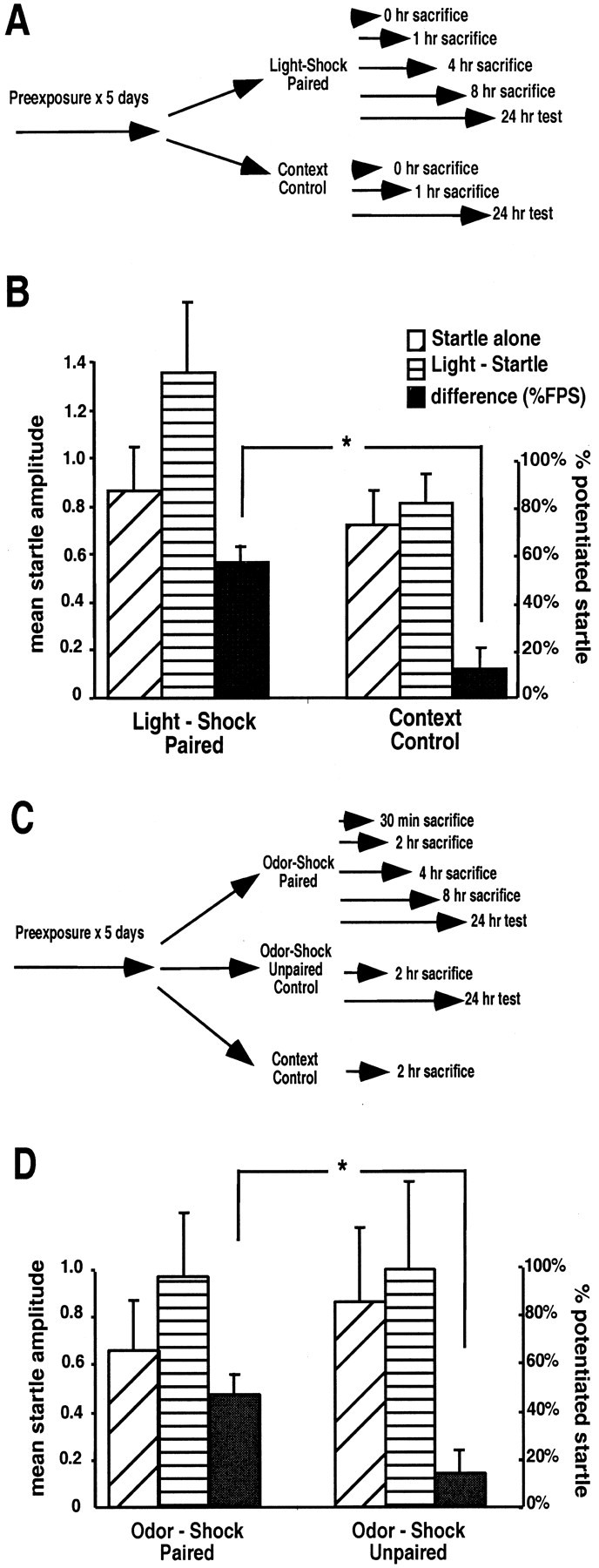
Associative light-shock and odor-shock pairings produce stable fear memory. A, Rats (n = 36) were pre-exposed to handling and the experimental chamber for 5 d. They were then exposed to 15 light-shock pairings over a 30 min period or were simply placed back in the chamber without stimuli for 30 min (Context Control). Animals were killed at time points after fear conditioning or were tested 24 hr later for the presence of fear-potentiated startle. B, Behavioral testing of the remaining animals from A. Mean amplitude ± SEM of startle response is shown on the left axis for startle presentations in the absence (Startle alone) or presence (Light-Startle) of a 4 sec light. The percentage increase (% potentiated startle ± SEM) is indicated by the black bars (scale to theright) for the light-shock paired difference versus the context control difference (∗p < 0.05 between these groups). C, Rats (n = 20) were pre-exposed as in A. They were then exposed to five odor-shock pairings over a 20 min period (Odor-Shock Paired), exposed to five odor and five shock stimuli that were not paired (Odor-Shock Unpaired), or simply placed back in the chamber (Context Control). Animals were then killed or later tested. D, Results of behavioral testing after the odor-shock training experiment inC (∗p < 0.05 between these groups).
Gene expression changes during consolidation
Thirteen of the 21 genes tested showed changes in expression after fear conditioning compared with the control condition. Ten of these genes showed robust expression changes (Table 1), primarily in the piriform cortex (PC) and in the medial and basolateral amygdala. Figure3A illustrates a schematic of the frontal sections analyzed in this experiment. Figure 3Bshows examples of peak changes in the temporal lobe seen with the fear-conditioned animals compared with the context control and KA-treated animals. Notice that the direction of change appears to be similar in both the KA and fear-conditioned animals, although to a different extent with different genes. Indicated are regions that show significant change with fear conditioning (Fig. 3B,arrows) as well as regions that did not change with that gene (Fig. 3B, arrowheads).
Fig. 3.
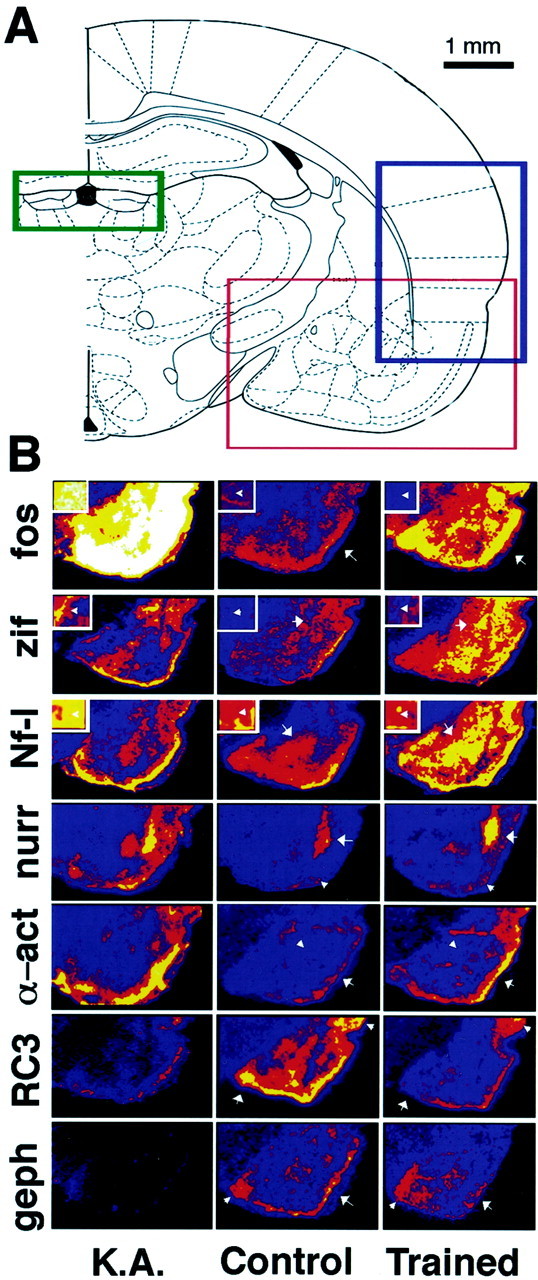
Gene expression changes in the temporal lobe after kainic acid or fear conditioning. A, Schematic diagram from Paxinos and Watson (1986) of regions examined. Areas in thered box were screened for genes indicated inB. Areas in the blue box are represented in Figure 6A, and areas in the green box are represented in Figure 6B. Scale bar, 1 mm. B, Pseudocolor images of in situ hybridization with 35S-radiolabeled probes for the genes indicated. No detectable gene expression is indicated byblack, very low levels are indicated byblue, intermediate levels are indicated byred to orange, and highest levels of expression are indicated by yellow towhite. Brain sections are from animals 6 hr after kainic acid treatment (K.A.), animals receiving context alone (Control), or fear-conditioned animals (Trained) from the experiment in Figure 2. Time points were chosen for the light-paired shock group based on the greatest change of expression for each gene. Arrows, Regions of interest that show significant change (analyzed in Fig. 4).Small arrowheads, Areas in which there are no significant changes in gene expression after fear conditioning. Inset boxes, Thalamic (c-fos, neurofilament–light chain) or cortical (zif268) areas from the same sections to illustrate regions without significant changes in gene expression with fear conditioning.fos, c-fos; zif,zif268; Nf-l, neurofilament–light chain;nurr, nurr-1; α-act, α-actinin; geph, gephyrin.
Figure 4 shows the quantitative time course of expression for a subset of the examined genes. As expected, the immediate early genes (c-fos andzif268) were induced rapidly (0–1 hr) after fear conditioning and returned to baseline within 2 hr (Fig.4A,C). Genes encoding some structural proteins [e.g., neurofilament–light chain (NF-l)] also peaked early (Fig.4B), but other genes, including NMDA receptor (NMDAR) stabilization protein and α-actinin, peaked later (1–4 hr) (Fig.4D). Interestingly, levels of RC3/neurogranin and gephyrin mRNA decreased during this consolidation phase (Fig.4E,F). All genes that were examined appeared to return to baseline by 8 hr after fear conditioning. The mean levels of expression for many of the genes were significantly different for the different time points and control groups even with the small number necessary for these initial broad screening studies. ANOVA between control, 0, 1, 4, and 8 hr groups: c-fos,F(4,11) = 4.3, p < 0.05; zif268, F(4,10) = 4.6, p < 0.05; NF–l,F(4,9) = 10.5, p < 0.01; α-actinin, F(4,11) = 4.9,p < 0.05; RC3/neurogranin,F(4,9) = 5.5, p < 0.05. Subsequent trend analyses showed significant quadratic trends for c-fos (t(7) = 3.3;p < 0.02), zif268(t(6) = 3; p < 0.02), NF-l (t(5) = 4.6; p < 0.01), α-actinin (t(5) = 4;p < 0.01), gephyrin (t(5) = 2.1; p < 0.09), and RC3/neurogranin (t(5) = 3.1; p < 0.03).
Fig. 4.
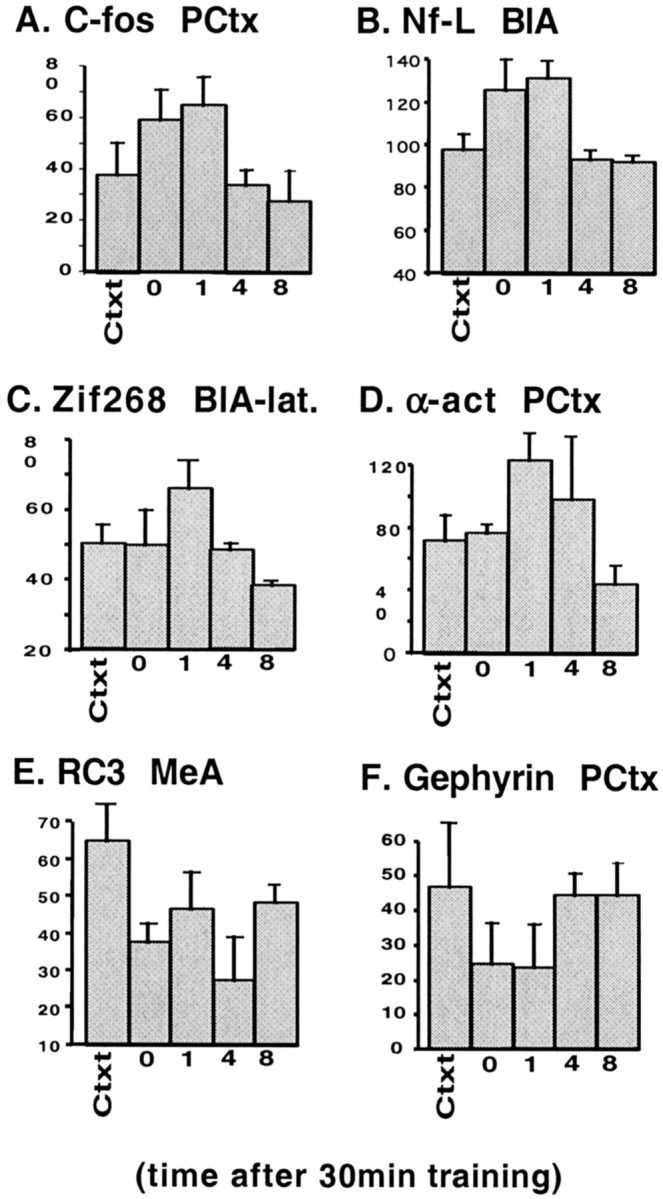
Temporal changes in gene expression after light-shock associative learning. The relative levels of expression of mRNA (in arbitrary units ± SEM) are shown for each of the indicated genes in the indicated region of interest. Levels are shown for the context control at the 1 hr time point compared with the light-shock paired animals that were killed immediately (0 hr) or 1, 4, or 8 hr after the 30 min fear training paradigm described in Figure2A. PCtx, Piriform cortex;Nf-L, neurofilament–light chain;α-act, α-actinin. Please see Results for statistics.
Changes in expression of plasticity genes are caused by associative CS–US pairing
The previous experiments suggested that there were temporally and anatomically specific changes in plasticity gene expression after light-shock pairing. However, it was not clear whether the effects were specific to the associative learning process or caused by general activation or other nonspecific effects. To address this, we performed another set of studies with additional animals at a single time point. The time course experiments suggested that the optimal time to observe changes in most of the genes examined is 2 hr after the fear-conditioning procedure (Fig. 4 and our unpublished results). Therefore, after 5 d of habituation to handling and placement in the experimental chamber, animals were subjected to 10 presentations of footshock alone (n = 4), 10 footshocks explicitly unpaired with light (n = 4), or 10 trials in which light and footshock were paired as in the previous experiments (n = 6). Animals were killed 2 hr after the conditioning or control procedure, and brains were prepared forin situ hybridization analysis with the probes described above.
As expected, the immediate early genes had returned to baseline by 2 hr and were not significantly different in their levels of expression from the control groups (data not shown). However, the other genes examined were significant when comparing the paired light-shock group with the footshock alone or with the unpaired CS –US presentations at the 2 hr time point. Figure 5 illustrates changes that were found to be significant with seven of these genes in several different brain areas. NF-l, nurr-1, α-actinin,N-cadherin, and 16c8 were all found to have significant induction in at least one to two brain areas at 2 hr in the paired group compared with the footshock alone and the unpaired groups (ANOVA: 16c8 piriform cortex, F(2,13) = 4,p < 0.05; 16c8 hippocampus,F(2,13) = 0.7, p < 0.01; NF-l piriform cortex,F(2,13) = 7.5, p< 0.01; NF-l hippocampus, F(2,13) = 4.8, p < 0.05; nurr-1 habenula,F(2,13) = 6.3, p < 0.01; nurr-1 endopiriform nucleus,F(2,13) = 6.4, p < 0.01; N-cadherin piriform cortex,F(2,13) = 9.2, p< 0.01; N-cadherin hypothalamus,F(2,13) = 7.9, p < 0.01; α-actinin BLA, F(2,13) = 5.2,p < 0.05).
Fig. 5.
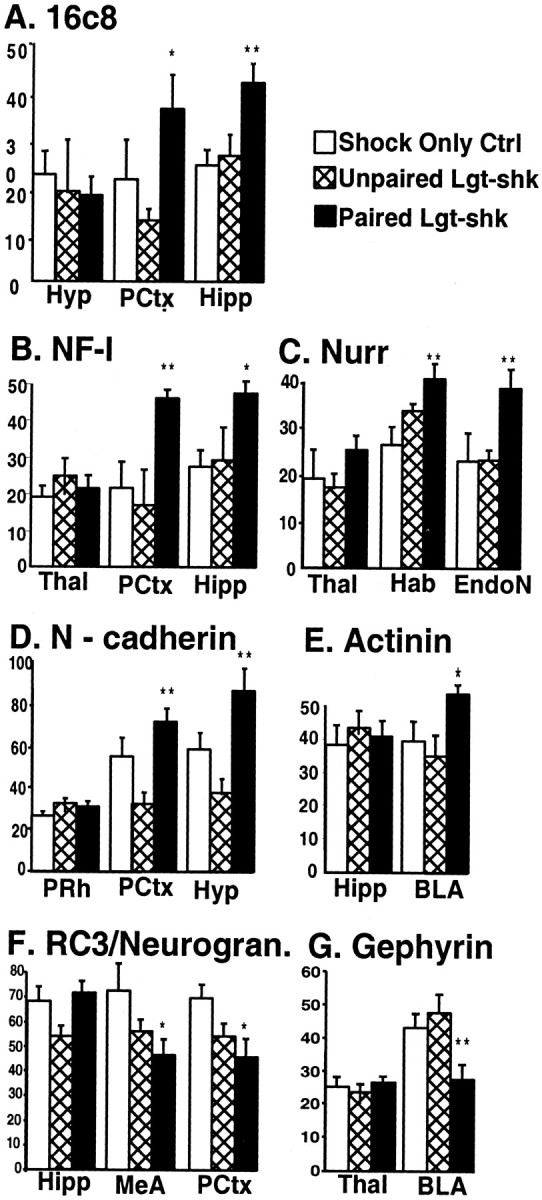
Only associative CS–US pairing alters gene expression. The relative levels of expression of mRNA (in arbitrary units ± SEM) are shown for regions in which there is no change (first region shown) or in regions with significant change for each of the indicated genes. Gene expression was analyzed at the 2 hr time point after light-shock associative learning (Paired Lgt-shk, black bars), unpaired presentations of lights and shocks (Unpaired Lgt-shk, hatched bars), or shock only controls (Shock Only Ctrl,white bars). ∗∗p ≤ 0.01, ∗p ≤ 0.05 with ANOVA. BLA, Basolateral amygdaloid nucleus; EndoN, endopiriform nucleus; Hab, habenula;Hipp, hippocampus; Hyp, hypothalamus; MeA, medial amygdaloid nucleus;PCtx, piriform cortex; PRh, perirhinal cortex; Thal, thalamus; NF-l, neurofilament–light chain; Nurr, nurr-1;Actinin, α-actinin; Neurogran, neurogranin.
Consistent with the previous experiments, RC3/neurogranin and gephyrin were significantly decreased during the same time period (Fig.5E,G) [RC3 medial amygdala (MeA),F(2,13) = 4, p < 0.05; RC3 piriform cortex, F(2,13) = 4.3, p < 0.05; gephyrin BLA,F(2,13) = 7.2, p < 0.01]. This experiment confirms that at a relatively late time period after the presentation of the paired CS and US, there are significant changes in the expression level of genes that may be involved in the long-term storage of conditioned fear memory. These changes occur only if the animals experience the pairing of lights and shocks and not if they receive shocks alone or the same number of lights and shocks in an unpaired manner.
To rule out the possibility of a general increase in multiple areas as opposed to a specific increase in limited areas, we also examined expression levels in regions that showed a basal level of expression that did not seem to change after fear conditioning. The first region shown with each of the genes in Figure 5 illustrates these data. In each of these regions (hypothalamus, thalamus, hippocampus, and perirhinal cortex), no significant change was seen in level of expression with the gene indicated, although significant changes may be seen in some of these areas with other genes. This demonstrates that the increases or decreases in expression described were specific for the time of expression, gene, and region of interest.
Gene expression changes with odor-shock associative learning
The previous experiment suggests that the association between light and shock led to the gene expression changes observed. To rule out the possibility that these findings were related to sensory modality or to the possible aversive properties of light itself, we examined the expression of the same 21 genes during the consolidation phase of an odor-shock training paradigm after paired and unpaired odor-shock presentations. We have shown previously that after only a small number of training trials, a neutral odor paired with mild footshock leads to robust fear conditioning (Paschall and Davis, 2002). Animals were trained and tested as illustrated in Figure 2C. On the training day, different groups of animals received five odor-shock pairings given over a 20 min period (odor-shock paired), five unpaired presentations of odor and shock separately (odor-shock unpaired), or no new stimuli (context control) and were returned to the home cage. They were killed at several different time points (0.5, 2, 4, or 8 hr after training; n = 12 total) or were tested 24 hr later for the presence of fear-potentiated startle (n = 8 total). The odor-shock-conditioned group showed significant fear-potentiated startle as demonstrated by a 58% enhanced startle amplitude when tested in the presence of amyl acetate (Fig.2D) (odor-startle vs startle-alone; pairedt test; p < 0.05), whereas the explicitly unpaired group showed no appreciable fear-potentiated startle (odor-shock paired difference vs odor-shock unpaired difference;p < 0.05).
In situ hybridization revealed that the set of genes induced with odor-shock pairing was similar to that induced with light-shock pairing. In most cases, gene expression at 2 hr after unpaired odor-shock presentation was indistinguishable from the context control condition (t tests; p > 0.5). Furthermore, the time course of induction of the genes that showed robust changes with light-shock pairing was similar to the time course of induction after odor-shock pairing. Again, the immediate early genes (c-fos and zif268) and NF-l showed rapid increases in gene expression at the first time point examined (30 min).nurr-1 and α-actinin were slower to rise and remained elevated for ≥4 hr. Again, the decrease in expression of RC3 and gephyrin lasted for ≥2 hr before returning to baseline. Despite the small number necessary for this multiple time point study, many of these changes were statistically significant or tended toward significance in the piriform cortex and amygdala (ANOVA: c-fos, F(4,9) = 3.85,p < 0.08; NF-l,F(4,9) = 6.4, p < 0.03; nurr-1, F(4,9) = 16.7, p < 0.01; α-actinin,F(4,9) = 3.7, p < 0.09). Subsequent contrasts found significant or near significant quadratic trends (nurr-1,t(5) = 5.04, p < 0.01; α-actinin, t(5) = 2.42,p = 0.06; gephyrin,t(5) = 2.23, p = 0.08; 16c8, t(5) = 2.98, p< 0.03). Therefore, these genes appear to have a delayed onset of transcriptional regulation that correlates specifically with associative learning rather than with the presence of the US alone or with the specific sensory stimulus modality.
Plasticity gene expression changes occur in unexpected brain areas
From studies of immediate early genes in fear conditioning and evidence from amygdala inactivation, we expected to find changes in synaptic plasticity genes within the basolateral amygdala complex. However, we also found that these and other genes examined showed significant changes in expression in several nonamygdala areas. Figure3B illustrates that many of the genes examined showed learning-induced changes within the piriform cortex in addition to the neighboring amygdala. Figure6A shows the time-dependent changes of c-fos and nurr-1expression in the associative somatosensory, perirhinal, and insular cortices along with the endopiriform nucleus (see Fig. 3Afor schematic). These are areas that project extensively to the amygdala, suggesting that much of the synaptic plasticity during the consolidation phase of fear conditioning may occur in the afferent neurons presynaptic to the amygdala.
Fig. 6.
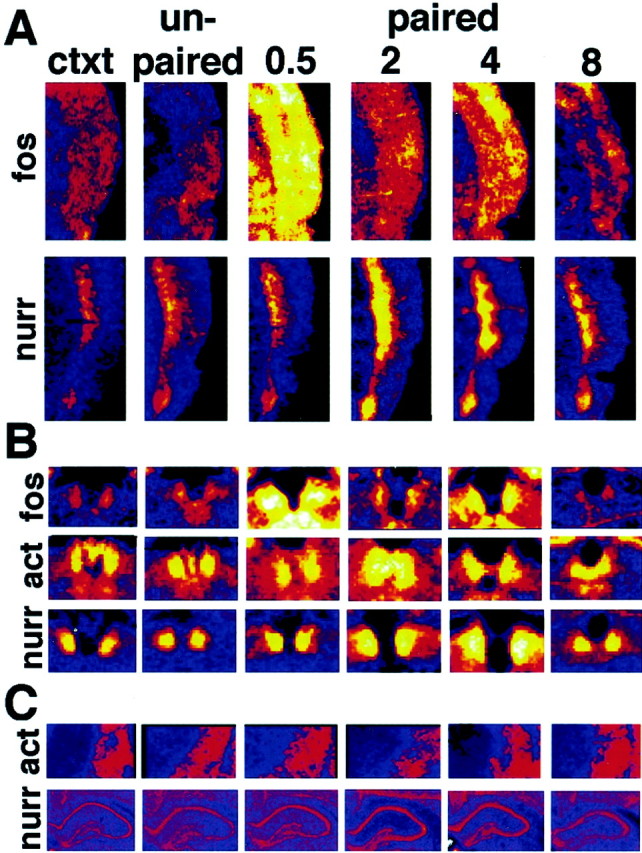
Gene expression changes in the cortex and habenula with fear conditioning. A, Pseudocolor images ofin situ hybridization with the genes indicated in the associative somatosensory (S2), parietal, perirhinal, and insular cortices as outlined in Figure 3A by the blue box. The color palette is the same as in Figure3B, with black showing no signal,blue showing the least mRNA expression, andyellow to white showing the most mRNA expression. Sections are shown after in situhybridization with the indicated genes for the context (ctxt) and unpaired controls and at 30 min and 2, 4, and 8 hr time points after odor-shock pairing. B, Pseudocolor images as in A from the habenula as outlined in Figure 3A by the green box.C, Pseudocolor images as in A from the caudate (act) or hippocampus (nurr) showing that α-actinin and nurr-1 do not show significant changes in these regions after fear conditioning.
Another area found to have extensive levels of synaptic plasticity gene induction is the habenula. Figure 6B illustrates the time-dependent changes in expression of c-fos, α-actinin, and nurr-1 in the habenula. c-fos increased initially and then returned to baseline. α-actinin increased in the same area but with a later peak at 2 hr, whereas nurr-1peaked at 4 hr, and both returned to baseline by 8 hr. There were no obvious qualitative differences between the odor-shock and light-shock groups. It appears that piriform and surrounding medial temporal lobe cortices along with amygdala areas are activated with both CS modalities paired with shock.
Figure 6C illustrates that α-actinin and nurr-1do not show significant changes in the level of expression in the caudate and hippocampus, respectively, in this experiment. This demonstrates the relative temporal and spatial specificity of gene expression during fear consolidation.
For all described experiments, sections of brains from two regions were examined. Anteriorly, brain regions examined were in coronal sections containing the anterior commissure and included areas of frontal and piriform cortices, septum, hypothalamus, striatum, and bed nucleus of stria terminalis. Posteriorly, the coronal sections were focused on the amygdala, including the centromedial, basolateral, and cortical nuclei, hypothalamus, thalamus, hippocampus, habenula and parietal, insular, perirhinal, and piriform cortices (the posterior region is that schematized in Fig. 3A). Table2 describes the brain regions in which altered gene expression was found. This suggests that plasticity after fear conditioning may depend on a broader neural circuit that includes, but is not limited to, the amygdala.
Table 2.
Synaptic plasticity genes are induced in a distributed neuronal circuitry
| Gene | BLA | MeA | CeA | PCtx | PRh | Ins Ctx | EndoN | Hipp | Hab | ThaI | Hyp | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c-fos | + | ++ | +/− | +++ | ++ | +++ | + | +/− | + | + | ++ |
| 2 | zif268 | ++ (LA) | − | − | ++ | ++ | +/− | + | − | + | − | − |
| 3 | c-jun | +/− | − | − | + | − | − | +/− | ++ | − | + | − |
| 4 | NF-l | ++ | + | − | +++ | ++ | +/− | + | ++ | ++ | − | +/− |
| 5 | Gephyrin | + | − | − | ++ | + | + | +/− | − | + | − | − |
| 6 | RC3/neurogranin | +/− | +++ | +/− | ++ | − | − | − | − | − | − | − |
| 7 | nurr-1 | +/− | − | − | − | − | ++ | +++ | − | +++ | − | − |
| 8 | 16c8 | − | − | − | ++ | + | − | − | ++ | − | − | − |
| 9 | α-actinin | ++ | + | − | +++ | +/− | + | − | +/− | + | − | +/− |
| 10 | N-cadherin | − | − | − | + | − | − | − | +/− | − | − | + |
The 10 genes that were found to show robust changes with fear conditioning are listed. The columns indicate the regions of interest examined for levels of gene expression during the consolidation period after fear conditioning. Regions of interest are abbreviated as in Figure 5. Qualitative expression level indicated as in Table 1. CeA, Central amygdaloid nucleus; Ins, insular cortex; LA, lateral amygdala.
DISCUSSION
These studies demonstrate that several genes appear to be regulated at the level of transcription during the consolidation phase of fear memory formation. Twenty-one KA-inducible genes were characterized during the consolidation phase after the presentation of CS–US pairings. These changes in gene expression were shown to occur only when the CS and US were associatively paired, not after US alone or unpaired US presentations. Furthermore, we found that fear conditioning using an olfactory CS activated a similar set of genes in similar areas as fear conditioning using a visual CS. This suggests that the findings are related to the process of associative learning rather than because of the particular CS modality or the US alone. The gene expression changes described occurred with different time courses (Fig. 7A). The IEGs and neurofilament were induced early and decayed within 1–2 hr after fear learning. Genes encoding other transcription factors, structural proteins, and receptor-associated proteins were expressed in similar areas as the IEGs but with later and longer-lasting time courses. Genes whose products are thought to exert a negative regulatory role on neuronal excitability (RC3/neurogranin and gephyrin) showed decreased expression but in a similar temporally restricted manner. Thus, fear conditioning may involve the early and late expression of genes involved in different aspects of the structural and functional plasticity that contributes to LTM formation (schematized in Fig.7B).
Fig. 7.
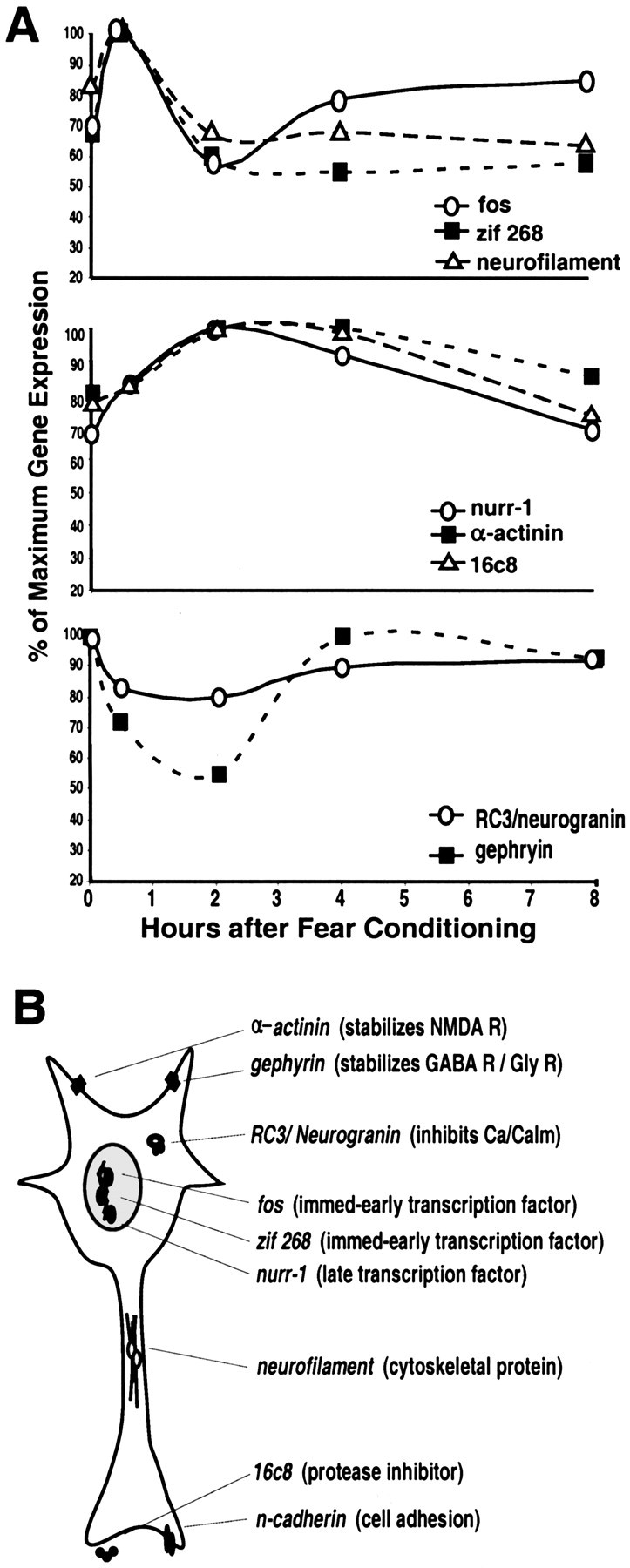
Summary of time course and possible function of gene regulation during the consolidation period of fear conditioning.A, Schematized time courses of the more robust changes in gene expression after fear conditioning in several brain regions, including the amygdala and areas afferent to it. The line graphs represent the percentage of maximal expression (normalized to 100% for each gene) from the odor-shock experiments (0 = context control at 30 min and 2, 4, and 8 hr after conditioning). The top graph represents the genes that show rapid early increases in gene expression. The middle graph represents the genes that show a more delayed and sustained increase. The bottom graph represents the two genes that have a prolonged decrease in expression after fear conditioning. B, The putative cellular location and function of these gene products based on current literature.NMDA R, NMDA receptor; GABA R, GABA receptor; Gly R, glycine receptor;Ca/Calm, calcium/calmodulin; immed-early, immediate early.
Function of transcriptionally regulated genes during consolidation
The genes chosen for this screen were identified previously with the KA model of neuronal plasticity. It is surprising that a large number of these genes are temporally and spatially regulated after fear learning, a much more limited and behaviorally relevant form of synaptic plasticity. The majority of evidence for early transcription regulation after learning has involved invertebrate studies andin vitro studies of long-term potentiation (LTP) (Nguyen et al., 1994; Bailey et al., 1996; Dubnau and Tully, 1998). The only genes that have been examined in detail during the consolidation of fear learning in mammals have been the IEGs zif268/early growth response gene-1 (EGR1) and c-fos (Campeau et al., 1991; Radulovic et al., 1998; Rosen et al., 1998; Malkani and Rosen, 2000; Jones et al., 2001). It is thought that these genes are activated very rapidly after a salient neuronal event has occurred, in part via CREB phosphorylation and transcriptional activation. c-fos,zif268, and c-jun are dynamically induced transcription factors with short half-lives that are thought to activate later transcription events in the activated cell (Herdegen and Leah, 1998). However, most of the important targets of these early transcriptional events have yet to be identified during neuronal plasticity.
NF-l is transcriptionally induced rapidly after associative fear conditioning. Its transcription is regulated by protein kinase A, and it contains a CREB-binding site in its promoter (White et al., 1997). It has also been shown to be involved both in LTP and in long-term depression (Hashimoto et al., 2000). As a structural protein known to be important in cytoskeletal remodeling, its transcriptional regulation during consolidation is consistent with possible presynaptic terminal rearrangements and postsynaptic dendritic spine changes thought to be involved in some forms of learning (Toni et al., 1999).
nurr-1 is a nuclear transcription factor that is involved in regulation of the dopaminergic phenotype (Saucedo-Cardenas et al., 1997) and is induced during neuronal plasticity (Crispino et al., 1998). Hippocampal expression of nurr-1 has been shown to be dynamically regulated with spatial learning (Pena de Ortiz et al., 2000). We observed the induction of nurr-1 slightly later than c-fos or zif268 but within 1 hr after the associative fear learning paradigm. Because nurr-1 has been shown to have a CREB-binding site in its promoter region phenotype (Saucedo-Cardenas et al., 1997), it may be involved in the second wave of transcription factors that mediate LTM.
We found temporal regulation of several genes encoding molecules involved in ECM reorganization. The protease inhibitor 16c8 has been shown to be dynamically regulated with neural plasticity and cell growth (Edwards et al., 1986; Nedivi et al., 1993).N-cadherin is a cell-adhesion molecule known to be involved in developmental axonal connectivity as well as in LTP (Huntley and Benson, 1999). Additionally, we saw some evidence of expression changes with tenascin, an ECM protein expressed principally by astrocytes during axonal growth and plasticity (Faissner, 1997). These findings support the hypothesis that structural changes are occurring during the consolidation of fear-conditioned learning.
α-actinin is a cytoskeletal protein that binds to actin polymers while also mediating the clustering of NMDA and other glutamatergic receptors (Wyszynski et al., 1998). There is evidence that it may play a role in postsynaptic structural events underlying LTP (Lisman and Zhabotinsky, 2001). We find that α-actinin is induced as early as 30 min and remains elevated ≥4 hr after associative US–CS pairing. Thus, its transcriptional regulation may regulate glutamate receptor (GluR) clustering underlying some aspects of fear conditioning.
Two of the genes we examined were actively inhibited with both KA plasticity and after fear learning. RC3/neurogranin is a cytosolic protein that appears to negatively regulate calcium–calmodulin (Ca/Cam)-dependent learning (Pak et al., 2000). When its structure is altered via oxidation by nitric oxide or phosphorylation by protein kinase C, it is thought to be released, thereby activating Ca/Cam. RC3/neurogranin is involved in LTP (Chen et al., 1997) and spatial learning (Pak et al., 2000). The decreased level of RC3/neurogranin via transcriptional regulation may be a longer-term mechanism for enhancing Ca/Cam and synaptic activity after fear learning. Gephyrin is a postsynaptic protein involved in the organization of inhibitory synapses (Sassoe-Pognetto and Fritschy, 2000). It has been shown to actively cluster glycine receptors (GlyRs), and null gephyrin mouse mutants have reduced GABA receptor (GABAR) and glycine receptor clustering in vivo (Fisher et al., 2000). Our finding that gephyrin appears to be actively downregulated suggests that there may be transcriptional regulation leading to decreased glycine and GABA receptor clustering. This could mediate decreased circuitry inhibition during fear consolidation to allow for longer-term mechanisms of memory storage.
Function of brain areas involved in the consolidation of fear
We have demonstrated that multiple brain areas, primarily the amygdala and regions afferent or efferent to it, show significant changes in gene expression during fear consolidation. We have attempted to prove that these expression changes, although complex, are limited in terms of genes, time courses, and regions of interest. Figures 3, 5, and 6 illustrate that there are also regions with basal levels of expression of a gene that do not show temporal changes during fear consolidation. Thus, we believe that the reproducible changes we see with certain genes in certain areas are meaningful and specific for the consolidation of fear learning.
It is generally accepted that the amygdala, specifically the basolateral amygdala, is critically involved in the formation of fear memories (Davis, 1992; Fanselow and LeDoux, 1999). Associative learning has been shown to activate LTP (Rogan et al., 1997) andzif268 gene expression (Rosen et al., 1998) within the amygdala. Furthermore, protein and mRNA synthesis within the amygdala is critical for fear memory consolidation (Bailey et al., 1999; Schafe et al., 1999). The critical role of the BLA is thought to be attributable to the convergence of multimodal sensory input at that site, thus allowing for the pairing of a neutral sensory CS with a US, such as pain (LeDoux et al., 1990; Shi and Davis, 1999; Pitkanen, 2000). The role of the MeA in fear conditioning is less clear. Induction of c-fos has been reported in the MeA after fear conditioning (Campeau et al., 1997; Rosen et al., 1998), although the function of the MeA in fear conditioning remains uncertain.
Multiple plasticity genes were temporally regulated in the piriform cortex in these experiments. Although the piriform cortex receives a large amount of direct olfactory information, it has also been shown to receive inputs from other cortical areas, and it receives multimodal sensory inputs by way of the lateral amygdala (Pitkanen, 2000). Its electrophysiological properties and connectivity suggest that it functions as associative cortex with a large olfactory input (Johnson et al., 2000). Changes in gene expression were also found in the perirhinal, insular, and associative somatosensory (S2) cortices (Fig.6A). The insular cortex and S2 have been shown to play a crucial role in the US pain pathway projection to the BLA (Shi and Davis, 1999). Furthermore, the perirhinal cortex, which projects heavily to the BLA (Shi and Cassell, 1999) and has been shown to be critical for fear memory (Rosen et al., 1992), receives multimodal information and may play a significant role in associative learning and CS–US representation (Pitkanen, 2000). The endopiriform nucleus receives multimodal input from multiple cortical areas and also projects strongly to the BLA. These findings imply that regions projecting to the amygdala, in addition to the amygdala itself, may undergo plasticity-associated changes.
The medial, and to a lesser extent the lateral, habenula were found to have marked changes in gene expression during consolidation (Fig.6B). The habenula receives inputs from a variety of limbic structures, including the central amygdala, bed nucleus of stria terminalis, septum, and cortical areas (Pitkanen, 2000). It projects to several midbrain structures, including the ventral tegmentum and the raphe nuclei (Sutherland, 1982). Our data suggest that plasticity underlying fear learning may occur both at the sensory–input stage of limbic functioning represented by the amygdala and its afferents, as well as in the convergent–output stage as seen with the habenula.
Implications for learning and memory
A debate exists within the literature regarding the storage of fear memories. Connectivity and lesion experiments of the BLA have led to the hypothesis that fear memories are stored within the BLA complex (Fanselow and LeDoux, 1999). Others argue that the amygdala serves primarily to activate salience and vigilance systems, and thus, it modulates the encoding of emotionally salient memories in a distributed cortical network (McGaugh et al., 1996). Our data suggest that both of these hypotheses may be true. Significant changes in expression of plasticity-related genes are found within the amygdala as well as in neurons afferent to it. This implies that plasticity critical for fear consolidation may occur both within neurons of the BLA and within the presynaptic terminals of its afferents. Furthermore, plasticity may be occurring in regions efferent to the amygdala, such as the habenula, suggesting that a broader circuit may underlie the full range of learning and expression of this complex behavior.
Our data imply that several genes that have not been implicated previously in fear conditioning are involved in its consolidation. The data also suggest that brain regions not known previously to be involved in this process may be important. Confirmatory studies, such as region-specific inhibition of gene expression or protein function, will need to be performed to definitively conclude the role of these genes and brain areas in the consolidation of fear conditioning. In summary, our data suggest that the early molecular events necessary for consolidation, the post-translational modification and translational regulation of protein products, are supplemented by complex temporal changes in gene expression. Together, these changes mediate both the early and eventually late phases of long-term memory consolidation in mammals.
Footnotes
This work was supported by National Institute of Mental Health Grants MH 47840 and MH 57250, the Woodruff Foundation, the Science and Technology Center Program (Center for Behavioral Neuroscience) of the National Science Foundation under Agreement IBN-9876754, a Pfizer Postdoctoral Fellowship Award (K.J.R.), and a Rockefeller Brothers Fund Culpeper Scholarship (K.J.R).
Correspondence should be addressed to Kerry J. Ressler, Department of Psychiatry and Behavioral Sciences, Center for Behavioral Neuroscience, Emory University School of Medicine, Atlanta, GA 30322. E-mail:kressle@emory.edu.
REFERENCES
- 1.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey D, Kim J, Sun W, Thompson R, Helmstetter F. Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci. 1999;113:276–282. doi: 10.1037//0735-7044.113.2.276. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Ari Y, Represa A. Brief seizure episodes induce long-term potentiation and mossy fibre sprouting in the hippocampus. Trends Neurosci. 1990;13:312–318. doi: 10.1016/0166-2236(90)90135-w. [DOI] [PubMed] [Google Scholar]
- 5.Campeau S, Hayward MD, Hope BT, Rosen JB, Nestler EJ, Davis M. Induction of the c-fos proto-oncogene in rat amygdala during unconditioned and conditioned fear. Brain Res. 1991;565:349–352. doi: 10.1016/0006-8993(91)91669-r. [DOI] [PubMed] [Google Scholar]
- 6.Campeau S, Falls WA, Cullnan WE, Helmreich DL, Davis M, Watson SJ. Elicitation and reduction of fear: behavioral and neuroendocrine indices and brain induction of the immediate-early gene c-fos. Neuroscience. 1997;78:1087–1104. doi: 10.1016/s0306-4522(96)00632-x. [DOI] [PubMed] [Google Scholar]
- 7.Carew TJ, Sutton MA. Molecular stepping stones in memory consolidation. Nat Neurosci. 2001;4:769–771. doi: 10.1038/90458. [DOI] [PubMed] [Google Scholar]
- 8.Cassella J, Davis M. The design and calibration of a startle measurement system. Physiol Behav. 1986;36:377–383. doi: 10.1016/0031-9384(86)90032-6. [DOI] [PubMed] [Google Scholar]
- 9.Chen SJ, Sweatt JD, Klann E. Enhanced phosphorylation of the postsynaptic protein kinase C substrate RC3/neurogranin during long-term potentiation. Brain Res. 1997;749:181–187. doi: 10.1016/s0006-8993(96)01159-6. [DOI] [PubMed] [Google Scholar]
- 10.Crispino M, Tocco G, Feldman J, Herschman H, Baudry M. Nurr1 mRNA expression in neonatal and adult rat brain following kainic acid-induced seizure activity. Brain Res Mol Brain Res. 1998;59:178–188. doi: 10.1016/s0169-328x(98)00143-0. [DOI] [PubMed] [Google Scholar]
- 11.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 12.Dubnau J, Tully T. Gene discovery in Drosophila: new insights for learning and memory. Annu Rev Neurosci. 1998;21:407–444. doi: 10.1146/annurev.neuro.21.1.407. [DOI] [PubMed] [Google Scholar]
- 13.Edwards DR, Waterhouse P, Holman ML, Denhardt DT. A growth-responsive gene (16C8) in normal mouse fibroblasts homologous to a human collagenase inhibitor with erythroid-potentiating activity: evidence for inducible and constitutive transcripts. Nucleic Acids Res. 1986;14:8863–8878. doi: 10.1093/nar/14.22.8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faissner A. The tenascin gene family in axon growth and guidance. Cell Tissue Res. 1997;290:331–341. doi: 10.1007/s004410050938. [DOI] [PubMed] [Google Scholar]
- 15.Fanselow MS, LeDoux JE. Why we think plasticity underlying pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 16.Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. Neurosci Biobehav Rev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. [DOI] [PubMed] [Google Scholar]
- 17.Fisher F, Kneussel M, Tintrup H, Haverkamp S, Rauen T, Betz H, Wassle H. Reduced synaptic clustering of GABA and glycine receptors in the retina of the gephyrin null mutant mouse. J Comp Neurol. 2000;427:634–648. doi: 10.1002/1096-9861(20001127)427:4<634::aid-cne10>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto R, Nakamura Y, Komai S, Kashiwagi Y, Tamura K, Goto T, Aimoto S, Kaibuchi K, Shiosaka S, Takeda M. Site-specific phosphorylation of neurofilament-L is mediated by calcium/calmodulin-dependent protein kinase II in the apical dendrites during long-term potentiation. J Neurochem. 2000;75:373–382. doi: 10.1046/j.1471-4159.2000.0750373.x. [DOI] [PubMed] [Google Scholar]
- 19.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 20.Hevroni D, Rattner A, Bundman M, Lederfein D, Gabarah A, Mangelus M, Silverman MA, Kedar H, Naor C, Kornuc M, Hanoch T, Seger R, Theill LE, Nedivi E, Richter-Levin G, Citri Y (1998) Hippocampal plasticity involves extensive gene induction and multiple cellular mechanisms. J Mol Neurosci 75–98. [DOI] [PubMed]
- 21.Huntley G, Benson D. Neural (N)-cadherin at developing thalamocortical synapses provides an adhesion mechanism for the formation of somatotopically organized connections. J Comp Neurol. 1999;407:453–471. [PubMed] [Google Scholar]
- 22.Johnson D, Illig K, Behan M, Haberly L. New features of connectivity in piriform cortex visualized by intracellular injection of pyramidal cells suggest that “primary” olfactory cortex functions like “association” cortex in other sensory systems. J Neurosci. 2000;20:6974–6982. doi: 10.1523/JNEUROSCI.20-18-06974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones M, Errington M, French P, Fine A, Bliss T, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- 24.Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeDoux J. Fear and the brain: where have we been, and where are we going? Biol Psychiatry. 1998;44:1229–1238. doi: 10.1016/s0006-3223(98)00282-0. [DOI] [PubMed] [Google Scholar]
- 26.LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisman JE, Zhabotinsky AM. A model of synaptic memory. A CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 28.Malkani S, Rosen JB. Specific induction of early growth response gene 1 in the lateral nucleus of the amygdala following contextual fear conditioning in rats. Neuroscience. 2000;97:693–702. doi: 10.1016/s0306-4522(00)00058-0. [DOI] [PubMed] [Google Scholar]
- 29.McGaugh JL, Cahill L, Roozendaal B. Involvement of the amygdala in memory storage: interaction with other brain systems. Proc Natl Acad Sci USA. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miserendino MJD, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 31.Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363:718–722. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen P, Abel T, Kandel E. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 33.Packard M, Cahill L, McGaugh J. Amygdala modulation of hippocampal-dependent and caudate nucleus-dependent memory processes. Proc Natl Acad Sci USA. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pak J, Huang F, Li J, Balschun D, Reymann K, Chiang C, Westphal H, Huang K. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci USA. 2000;97:11232–11237. doi: 10.1073/pnas.210184697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paschall GY, Davis M. Olfactory-mediated fear-potentiated startle. Behav Neurosci. 2002;116:4–12. doi: 10.1037//0735-7044.116.1.4. [DOI] [PubMed] [Google Scholar]
- 36.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 37.Pena de Ortiz S, Maldonado-Vlaar C, Carrasquillo Y. Hippocampal expression of the orphan nuclear receptor gene hzf-3/nurr1 during spatial discrimination learning. Neurobiol Learn Mem. 2000;74:161–178. doi: 10.1006/nlme.1999.3952. [DOI] [PubMed] [Google Scholar]
- 38.Pitkanen A. Connectivity of the rat amygdaloid complex. In: Aggleton JP, editor. The amygdala, a functional analysis, Ed 2. Oxford UP; New York: 2000. pp. 31–116. [Google Scholar]
- 39.Radulovic J, Kammermeir J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ressler KJ, Sullivan SL, Buck LB. A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell. 1993;73:597–609. doi: 10.1016/0092-8674(93)90145-g. [DOI] [PubMed] [Google Scholar]
- 41.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 42.Rosen JB, Hitchcock JM, Miserendino MJ, Falls WA, Campeau S, Davis M. Lesions of the perirhinal cortex but not of the frontal, medial prefrontal, visual, or insular cortex block fear-potentiated startle using a visual conditioned stimulus. J Neurosci. 1992;12:4624–4633. doi: 10.1523/JNEUROSCI.12-12-04624.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosen JB, Fanselow MS, Young SL, Sitcoske M, Maren S. Immediate-early gene expression in the amygdala following footshock stress and contextual fear conditioning. Brain Res. 1998;796:132–142. doi: 10.1016/s0006-8993(98)00294-7. [DOI] [PubMed] [Google Scholar]
- 44.Sassoe-Pognetto M, Fritschy J. Mini-review: gephyrin, a major postsynaptic protein of GABAergic synapses. Eur J Neurosci. 2000;12:2205–2210. doi: 10.1046/j.1460-9568.2000.00106.x. [DOI] [PubMed] [Google Scholar]
- 45.Sassoon DA, Garner I, Buckingham M. Transcripts of a-cardiac and a-skeletal actins are early markers for myogenesis in the mouse embryo. Development. 1988;104:155–164. doi: 10.1242/dev.104.1.155. [DOI] [PubMed] [Google Scholar]
- 46.Saucedo-Cardenas O, Kardon R, Ediger TR, Lydon JP, Conneely OM. Cloning and structural organization of the gene encoding the murine nuclear receptor transcription factor, NURR1. Gene. 1997;187:135–139. doi: 10.1016/s0378-1119(96)00736-6. [DOI] [PubMed] [Google Scholar]
- 47.Schafe G, Nadel N, Sullivan G, Harris A, LeDoux J. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- 48.Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- 50.Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci. 1999;19:420–430. doi: 10.1523/JNEUROSCI.19-01-00420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi CJ, Cassell MD. Perirhinal cortex projections to the amygdaloid complex and hippocampal formation in the rat. J Comp Neurol. 1999;406:299–328. doi: 10.1002/(sici)1096-9861(19990412)406:3<299::aid-cne2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 52.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 53.Sutherland R. The dorsal diencephalic conduction system: a review of the anatomy and functions of the habenular complex. Neurosci Biobehav Rev. 1982;6:1–13. doi: 10.1016/0149-7634(82)90003-3. [DOI] [PubMed] [Google Scholar]
- 54.Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- 55.Walker DL, Davis M. Involvement of NMDA receptors within the amygdala in short- versus long-term memory for fear conditioning as assessed with fear-potentiated startle. Behav Neurosci. 2000;114:1019–1033. [PubMed] [Google Scholar]
- 56.Wheal HV, Chen Y, Mitchell J, Schachner M, Maerz W, Wieland H, Van Rossum D, Kirsch J. Molecular mechanisms that underlie structural and functional changes at the postsynaptic membrane during synaptic plasticity. Prog Neurobiol. 1998;55:611–640. doi: 10.1016/s0301-0082(98)00026-4. [DOI] [PubMed] [Google Scholar]
- 57.White LA, Reeben M, Saarma M, Whittemore SR. Transcriptional regulation of neurofilament expression by protein kinase A. J Neurosci Res. 1997;47:242–252. doi: 10.1002/(sici)1097-4547(19970201)47:3<242::aid-jnr2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 58.Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs A, Craig A, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]