

Abstract
Dopaminergic midbrain (DA) neurons display two principal activity patterns in vivo, single-spike and burst firing, the latter coding for reward-related events. We have shown recently that the small-conductance calcium-activated potassium channel SK3 controls pacemaker frequency and precision in DA neurons of the substantia nigra (SN), and previous studies have implicated SK channels in the transition to burst firing. To identify the upstream calcium sources for SK channel activation in DA SN neurons, we studied the sensitivity of SK channel-mediated afterhyperpolarization (AHP) currents to inhibitors of different types of voltage-gated calcium channels in perforated patch-clamp recordings. Cobalt-sensitive AHP currents were not affected by L-type and P/Q-type calcium channel inhibitors and were reduced slightly (26%) by the N-type channel inhibitor ω-conotoxin-GVIA. In contrast, AHP currents were blocked substantially (85–94%) by micromolar concentrations of nickel (IC50, 33.75 μm) and mibefradil (IC50, 4.83 μm), indistinguishable from the nickel and mibefradil sensitivities of T-type calcium currents (IC50 values, 33.86 and 4.59 μm, respectively). These results indicate that SK channels are activated selectively via T-type calcium channels in DA SN neurons. Consequently, SK currents displayed use-dependent inactivation with similar time constants when compared with those of T-type calcium currents and generated a transient rebound inhibition. Both SK and T-type channels were essential for the stability of spontaneous pacemaker activity, and, in some DA SN neurons, T-type channel inhibition was sufficient to induce intrinsic burst firing. The functional coupling of SK to T-type channels has important implications for the temporal integration of synaptic input and might help to understand how DA neurons switch between pacemaker and burst-firing modes in vivo.
Keywords: dopamine, substantia nigra (A9), electrophysiology, apamin, nifedipine, agatoxin-TK, FTX-3.3, amphotericin, cyclopiazonic acid (CPA)
Dopaminergic midbrain (DA) neurons are important for voluntary movement, cognition, and reward and are implicated in major disorders such as schizophrenia and Parkinson's disease (Dunnett and Bjorklund, 1999; Goldman-Rakic, 1999; Carlsson et al., 2000; Wise, 2000). In vivo, DA neurons show two principal patterns of activity: they either fire in regular or irregular single-spike mode or discharge bursts of action potentials (Wilson et al., 1977; Grace and Bunney, 1984a,b; Sanghera et al., 1984;Freeman et al., 1985). Burst-firing episodes code for the unpredicted rewarding aspects of environmental stimuli and thus might constitute a mechanism for reward-based learning (Schultz, 2000; Reynolds et al., 2001). During burst firing, dopamine release is increased phasically in striatal (Gonon and Buda, 1985) and cortical (Bean and Roth, 1991) target areas of DA neurons, whereas tonic release during pacemaker firing controls the background of dopamine levels that, among other functions, regulates the intensity of the phasic burst-firing signal (Grace, 1991; Overton and Clark, 1997). Because changes in the degree and/or pattern of dopamine signaling have been implicated in the pathophysiology of Parkinson's disease and schizophrenia (Dunnett and Bjorklund, 1999; Grace, 2000; Svensson, 2000), it is critical to understand better the cellular mechanisms that control the transition between pacemaker and burst firing in DA neurons.
Unlike thalamic neurons (Huguenard, 1998), DA neurons show only pacemaker spiking and no spontaneous burst firing in in vitro preparations (Sanghera et al., 1984; Kita et al., 1986;Grace and Onn, 1989; Wolfart et al., 2001). Thus, it generally is assumed that a particular type of synaptic activity, which is present only in vivo, is necessary for DA neurons to switch into burst mode. In this context NMDA receptor activation (Johnson et al., 1992), GABAA receptor-mediated disinhibitory processes (Paladini and Tepper, 1999), and modulation of postsynaptic conductances (Kitai et al., 1999) have been proposed as candidate mechanisms. In particular, apamin-sensitive, small-conductance, calcium-activated potassium (SK) channels (Blatz and Magleby, 1987;Kohler et al., 1996; Sah, 1996) have been reported to facilitate synaptically mediated burst induction (Seutin et al., 1993; Johnson and Seutin, 1997) or, in some cases, to be sufficient to induce burstingin vitro (Shepard and Bunney, 1988, 1991; Gu et al., 1992;Ping and Shepard, 1996). Furthermore, we have shown recently that SK3 channels control the frequency and precision of pacemaker spiking in DA neurons of the substantia nigra (SN), but not in a subpopulation of DA neurons in the ventral tegmental area (Wolfart et al., 2001).
SK channels form a signaling complex with calmodulin as a calcium detector, and channel opening depends solely on submembrane changes of the intracellular calcium concentration (Xia et al., 1998). All major classes of voltage-gated calcium (Cav) channels (Nowycky et al., 1985; Llinas et al., 1989; Ertel et al., 2000) are present in DA SN cells (Kang and Kitai, 1993b; Stea et al., 1994;Williams et al., 1994; Cardozo and Bean, 1995; Craig et al., 1999;Talley et al., 1999; Takada et al., 2001) and could, in principle, contribute to SK channel activation. However, preferential coupling of SK channels to particular Cav channels has been reported to be present in other neurons (Wisgirda and Dryer, 1994;Marrion and Tavalin, 1998; Sah and Davies, 2000; Bowden et al., 2001). In addition, calcium signals generated by Cavchannels might be amplified by secondary calcium release from intracellular stores, which also has been shown to activate SK channels in various cell types (Yoshizaki et al., 1995; Davies et al., 1996;Tanabe et al., 1998; Cordoba-Rodriguez et al., 1999). Indeed, in DA neurons SK channels can be activated by intracellular calcium release evoked via metabotropic glutamate receptors (Morikawa et al., 2000;Seutin et al., 2000). Because there is increasing evidence that the functional pool of SK channels in DA neurons controls pacemaker stability and potentially is involved in the still elusive burst transition (Shepard and Bunney, 1988, 1991; Gu et al., 1992; Ping and Shepard, 1996; Wolfart et al., 2001), in the present study we aimed to characterize the upstream regulation of SK channel activity in DA SN neurons.
MATERIALS AND METHODS
Slice preparation. Procedures involving animals were conducted in accordance with the Animals (Scientific Procedures) Act, 1986 (UK) and with the Society for Neuroscience policy on the use of animals in research. C57BL/6J mice (Charles River, Margate, UK; 10–14 postnatal d old) were killed by cervical dislocation. Brains were removed quickly, immersed in ice-cold artificial CSF (ACSF), and then blocked for sectioning. Thin (250 μm) coronal midbrain slices were collected with a Vibroslice (Campden Instruments, London, UK) in ice-cold ACSF containing (in mm) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, and 25 glucose, oxygenated with a mixture of 95% O2/5% CO2. After sectioning, midbrain slices were maintained submerged in oxygenated ACSF and were allowed to recover for >30 min before the experiment. Midbrain slices containing a clearly defined substantia nigra pars compacta (SN) at the level of the rostral interpeduncularis nucleus and the caudal mamillary nucleus were used for the experiments.
Electrophysiological recordings. For patch-clamp recordings the midbrain slices were transferred to a recording chamber and perfused continuously at 2–4 ml/min with oxygenated ACSF at room temperature (22–24°C, except see below). Recordings were made from SN neurons visualized by infrared differential interference contrast video microscopy with a Newvicon camera (C2400; Hamamatsu, Hamamatsu City, Japan) mounted to an upright microscope (Axioskop FS; Zeiss, Oberkochen, Germany). Records were digitized at 2–5 kHz and low-pass filtered before acquisition (Bessel characteristic of 1 kHz cutoff frequency). Patch pipettes were pulled from borosilicate glass (GC150TF/F; Harvard Apparatus, UK) with tip resistances between 2 and 5 MΩ when filled with patch solution. For perforated patch-clamp recordings the patch pipettes were tip filled with a solution containing (in mm) 140 KMeSO4, 5 KCl, 10 HEPES, 0.1 EGTA, and 2 MgCl2, pH 7.35, and backfilled with the same solution containing amphotericin B (0.4 mg/ml). For current- and hybrid-clamp (Pennefather et al., 1985) recordings the perforated whole-cell configuration was used (except see below). After GΩ seal formation the perforation was monitored until a stable level of action potential (AP) amplitudes was reached. To elicit and record afterhyperpolarization (AHP) currents under voltage-clamp conditions, we used short (20 msec) unclamped (“hybrid”) depolarizations (from −10 to +100 mV; holding and recording potential, −60 mV). For the AHP current recordings shown in Figure 3A, the conditions described previously were used (Wolfart et al., 2001). Current-clamp recordings were conducted either at 22–24°C or at 36–37°C, and no difference in spiking pattern was found, except that AP frequencies were increased (see Table 1). Therefore, data recorded in both temperatures were pooled for the apamin plus nickel treatment shown in Figure 5. For extracellular local (<50 μm) application of drugs, the cells were perfused at a flow rate of 50–100 μl/min under visual control with the use of a quartz pipette (inner tip diameter, 0.25 mm) attached to a second manipulator and a syringe pump system (World Precision Instruments, Sarasota, FL). Switching between control and drug-containing solutions was controlled by an automated application system (AutoMate Scientific, Oakland, CA). The application solution for perforated patch recordings contained (in mm) 145 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2, 2 MgCl2, and 25 glucose plus 50 μm picrotoxin and 50 μm kynurenic acid, pH 7.4. For the recording of low voltage-activated calcium (LVA) currents the standard whole-cell configuration was used, and patch pipettes were filled with a solution containing (in mm) 140 TEA-Cl, 10 HEPES, 10 EGTA, and 2 MgCl, pH 7.35. Uncompensated series resistances were 8–20 MΩ in recordings for kinetic analysis. Dopaminergic SN neurons were identified by their characteristic low frequency firing by using either the cell-attached or the whole-cell patch-clamp configuration. The application solution for LVA calcium current recordings contained (in mm) 145 TEA-Cl, 2.5 CsCl, 10 HEPES, 2 CaCl2, 2 MgCl2, 25 glucose, and 4 4-aminopyridine plus (in μm) 50 picrotoxin, 50 kynurenic acid, 0.5 tetrodotoxin, 10 nifedipine, and 10 nm ω-conotoxin-GVIA. The holding potential was −100 mV, and the test pulse for drug applications was −50 mV. No leak subtraction was used in these recordings. The steady-state membrane potential of half-maximal activation (V½act) was determined by 1-sec-long voltage steps (5 mV) from −80 to −30 mV from a holding potential of −100 mV. The steady-state membrane potential of half-maximal inactivation (V½inact) was determined by a 3-sec-long conditioning voltage step from −120 to −40 mV and a test pulse to −50 mV. The tail current protocol used to characterize deactivation contained a prepulse pulse to −50 mV and test potentials from −120 to −80 mV. Lipophilic substances (cyclopiazonic acid, amphotericin, picrotoxin, kynurenic acid, and nifedipine) were dissolved in DMSO and diluted 1:1000 to final concentrations. Nifedipine, ω-conotoxin-GVIA, agatoxin-TK, FTX-3.3, and cyclopiazonic acid were obtained from Alomone Labs (Jerusalem, Israel). Mibefradil was a gift from Roche (Basel, Switzerland). All other substances were obtained from Sigma (Dorset, UK). The EPC-9 patch-clamp amplifier and program package PULSE+PULSEFIT (HEKA Electronics, Lambrecht, Germany) were used for data acquisition.
Fig. 3.
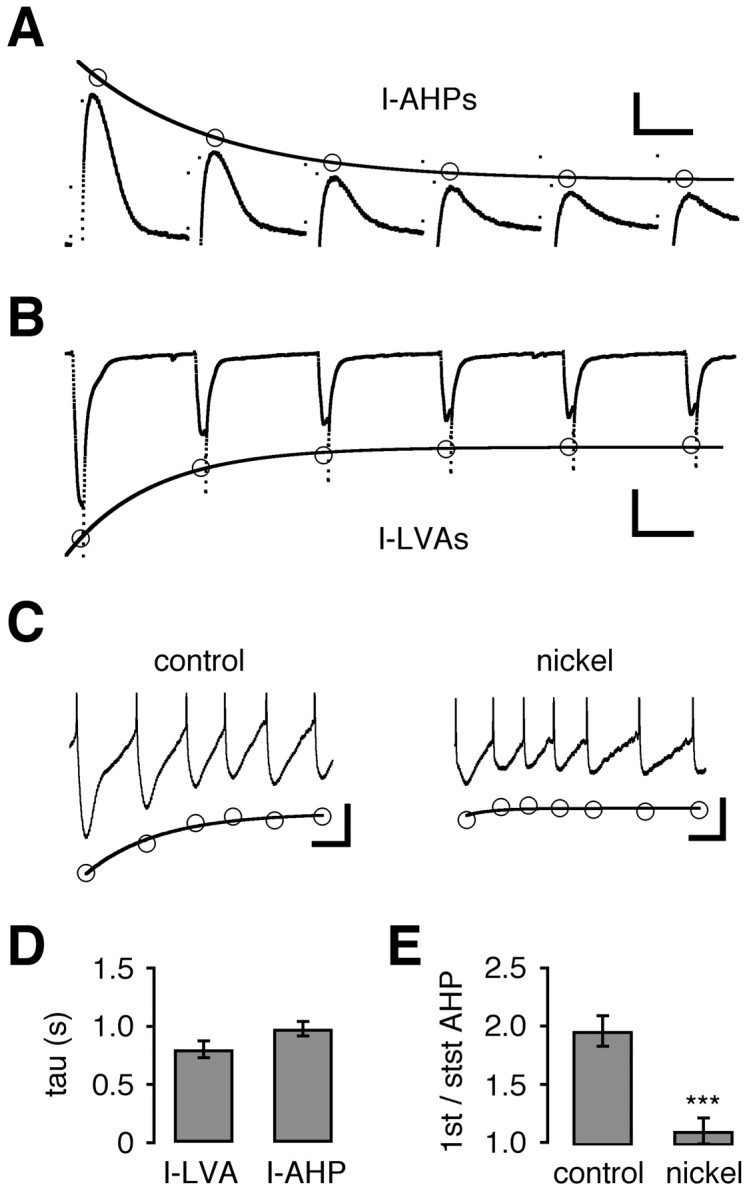
Use-dependent inactivation of SK and T-type currents displayed similar kinetics. A, AHP currents (I-AHPs) evoked with hybrid-clamp depolarizations (100 msec, +60 mV) at a frequency of 1 Hz by using the standard whole-cell configuration (recording potential, −80 mV). Successive AHP currents decreased, reaching a steady-state level at 38% of the initial amplitude. The time constant of cumulative inactivation was 1.26 sec.B, Recording of T-type-mediated low voltage-activated calcium currents (I-LVA; see Results) evoked by the same voltage pulse protocol as in A, using the standard whole-cell configuration and calcium channel recording solutions. Successive activation at 1 Hz led to a decrease of I-LVAs, reaching a steady-state level of 42%. The time constant of use-dependent inactivation was 0.77 sec. C, Perforated current-clamp recording of a train of APs evoked by injections of 10 pA for 4 sec from a hyperpolarized membrane potential of −80 mV. At the onset of depolarization the AHPs were large but decreased with successive APs. Application of nickel (250 μm) decreased the AHP amplitudes and abolished the effect of cumulative inactivation. Note that the control rate of cumulative AHP inactivation (τ = 1.10 sec) was similar to the time constants determined for I-AHPs and I-LVAs. D, Time constants (τ) of cumulative inactivation determined by experiments in A andB. LVA and AHP currents both had cumulative inactivation time constants in the range of 1 sec: I-LVA, 0.80 ± 0.07 sec (n = 10); I-AHP, 0.98 ± 0.06 sec (n = 10; p > 0.05).E, The summary of experiments in D shows that AHPs were reduced twofold under control conditions (2.0 ± 0.1; n = 8), whereas the effect was abolished by nickel application (1.1 ± 0.1; n = 8; ∗∗∗p < 0.0005). Calibration:A, 0.5 sec, 50 pA; B, 0.5 sec, 200 pA;C, 0.5 sec, 10 mV.
Table 1.
Different forms of burst firing in DA neurons obtained in present and previous studies
| Burst induction | Recording method | Reference | APs/burst (bursting cells) | Intra-burst ISI (msec) | Inter-burst ISI (msec) | AP red |
|---|---|---|---|---|---|---|
| Nickel | m, vitro(23°), pp | Present study | 2.3 ± 0.1 (3 of 27) | 151 ± 5 | 2.4 ± 0.3 | No |
| Ni + apamin | m, vitro (37°), pp | Present study | 2.6 ± 0.1 (5 of 16) | 117 ± 13 | 1.5 ± 0.4 | No |
| Ni + apamin | m, vitro (23°), pp | Present study | 2.6 ± 0.1 (3 of 9) | 166 ± 44 | 1.9 ± 0.3 | No |
| Spontaneous | r, vivo, ec, ic | Grace and Bunney, 1984 | 2.9 (50 of 75) | 73 ± 13 | — | Yes |
| Apamin | r,vitro (36°), ic | Shepard and Bunney, 1991 | 3–12 (3 of 10) | — | — | Yes |
| NMDA | r, vitro (35°), ic | Johnson et al., 1992 | 2–10 | 50–200 | 1–5 | No |
| Picrotoxin | r, vivo, ec | Paladini and Tepper, 1999 | 3.8 ± 0.4 | — | — | Yes |
Summary of burst firing characteristics induced by various inhibitors and/or recording techniques obtained in previous studies and the present work. Inhibition of T-type channels (nickel, ni) alone (3 of 27 cells) or in combination with inhibition of SK channels (apamin, 5 of 16 cells in 37°C, 3 of 9 cells in 23°C) induced burst firing consisting mainly of AP doublets or triplets that had interspike intervals (ISIs) of ∼150 msec (Intra-burst ISI), reoccurring every 1–3 sec (Inter-burst ISI) and had similar amplitudes (no AP reduction within bursts). Recording methods: m, mouse; r, rat; vitro,in vitro; vivo, in vivo; pp, perforated patch-clamp recordings; ic/ec, intracellular or extracellular recordings.
Fig. 5.
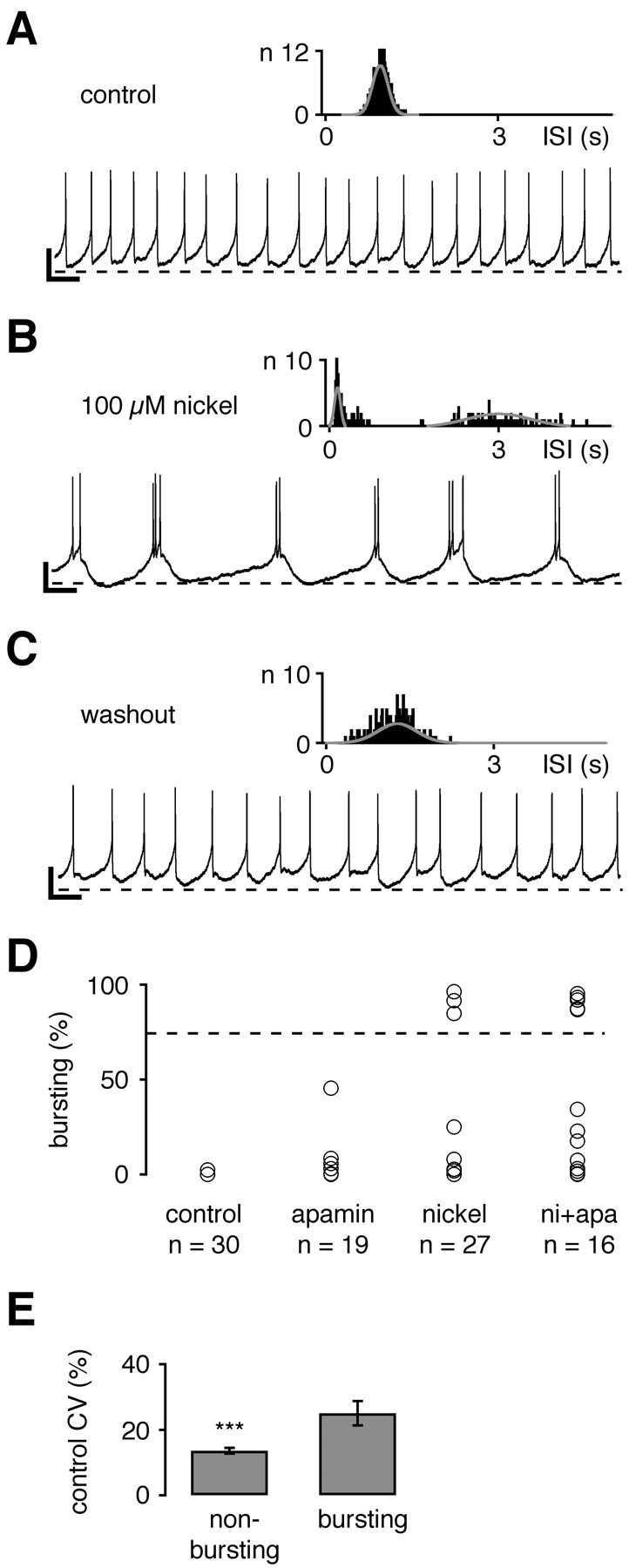
Inhibition of T-type channels evoked bursting in a subpopulation of dopaminergic midbrain neurons. A–C, Perforated current-clamp recordings during control (A), nickel (B), and washout (C) conditions. A 20 sec recording trace representative of a 5 min recording is shown for each condition. As a measure of pacemaker precision the coefficient of variation (CV) was calculated from Gaussian fits of ISI histograms (insets).A, No bursting (0%) was detected during control application. Note that this neuron showed pacemaker spiking at the lower end of firing precision (CV of 20%; compare with Fig. 4).B, Application of 100 μm nickel switched the firing pattern from pacemaker to bursting, with two to three closely spaced APs alternating with long inter-burst intervals. C, With the washout of nickel the firing pattern returned to (irregular) pacemaker spiking.D, Firing patterns during perforated patch-clamp recordings were assessed by a burst evaluation program (spikes/burst per trace = bursting in percentage values; e.g., 85% bursting for the recording shown in B). Under control conditions the bursting value was 0% (n = 30). Application of 300 nm apamin did not change the bursting value (3 ± 2%;n = 19) significantly, although one cell showed an increased value (45%) because of short periods of “burst-like” pattern. Inhibition of T-type channels with 100 μm nickel significantly increased the bursting value to 12 ± 6% (n = 27; p < 0.05), and three cells displayed bursting values above 84%. The combination of nickel and apamin application (ni+apa) was most effective in switching from pacemaker to bursting behavior, increasing the mean bursting value to 34 ± 10% (n = 16;p < 0.0005), with five neurons reaching bursting values of >86%. E, Differential effects of T-type channel inhibition on firing patterns were associated to pacemaker precision under control conditions. Control CV values were correlated with the effect of nickel and apamin application on firing patterns of respective cells. Neurons that were converted to bursting had significantly higher CV values (25 ± 4%; n = 6) compared with cells that became irregular with nickel (or nickel + apamin) application (14 ± 1%; n = 22; ∗∗∗p < 0.0005). Calibration:A–C, 1 sec, 10 mV. Dotted lines inA–C, −50 mV. Dotted line inD, 75% bursting.
Data analysis. For analysis and plotting, the software IgorPro (WaveMetrics, Lake Oswego, OR) was used. Time constants of AHP and LVA current inactivation were determined by fitting mono- or double-exponential functions to 1- to 5-sec-long current traces, respectively. Steady-state activation and inactivation parameters were obtained by fitting Boltzmann functions to the data. Coefficients of variation (CVs) were obtained by dividing the SD of the interspike interval (ISI) distribution (fit to a Gaussian function) by the mean ISI and expressed as a percentage. Drug sensitivities of AHP currents were determined as the average response of three to five steady-state traces in comparison to the average control amplitudes (3–5 traces). Drug sensitivities of transient LVA currents were determined either as above (stable recordings) or by prediction of rundown via linear regression fits (LVA currents with linear rundown kinetics). To determine the dose–response relationships for nickel and mibefradil as well as the degrees of residual drug-insensitive components, we fit the respective mean data to Hill equations. For spiking pattern analysis a burst detecting algorithm was programmed that compared all ISIs of a 5-min-long recording trace with its mean spiking rate and detected the coincidence of a short ISI (<0.5 × mean rate) with a long ISI (>1.25 × mean rate) within two to seven consecutive spikes and marked it as a “burst” (Grace and Bunney, 1984b). Intervals within the burst were validated additionally by a Poisson surprise mechanism that compared ISIs with the Poisson distribution of all ISIs of a recording (Legendy and Salcman, 1985). Spikes within bursts were summed up and normalized to the total number of spikes in the trace (bursting in percentage values). The outcome of the burst analysis was optimized until only unambiguous burst-firing patterns resulted in values above 75% bursting and thus were well separated from irregular firing with occasional burst-like events. AHP current traces (see Figs. 1-3) represent averages from three to five filtered (300 Hz) traces. To evaluate statistical significance (∗p < 0.05; ∗∗p < 0.05; ∗∗∗p < 0.0005), we subjected the data to paired or unpaired Student's t tests in Microsoft Excel. Numbers, symbols, andcolumns with error bars represent means ± SEM.
Fig. 1.
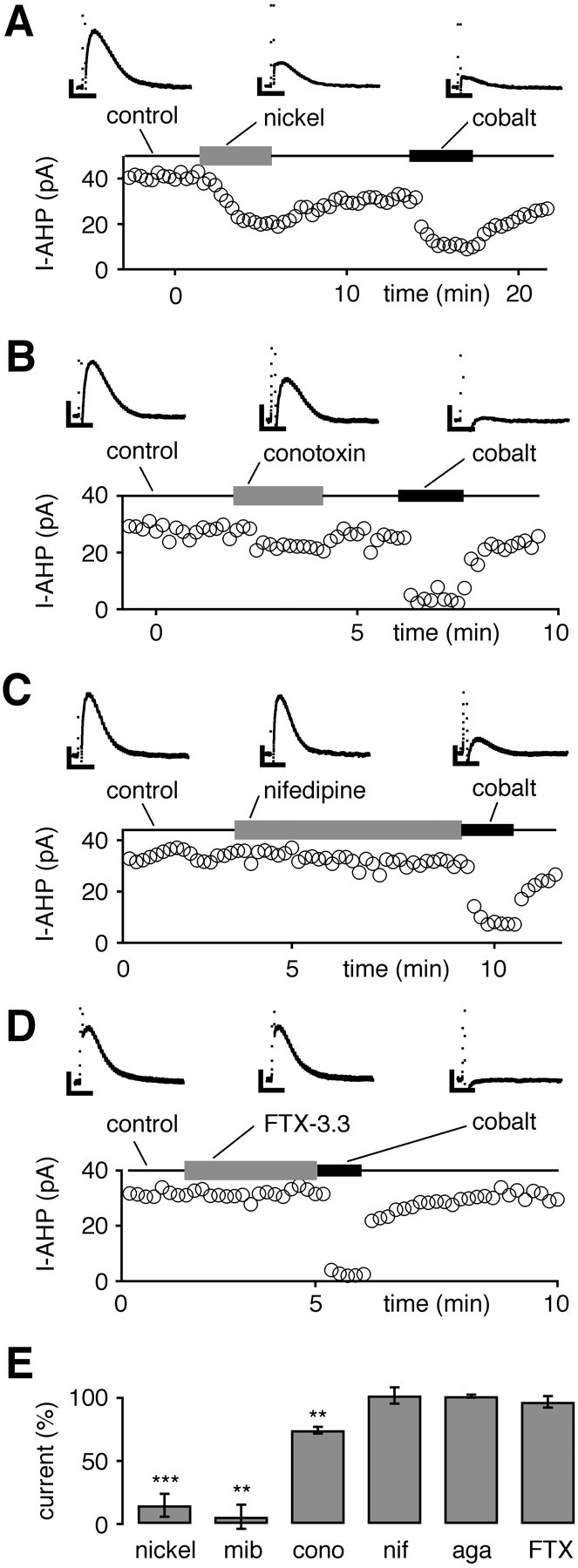
Sensitivity of hybrid-clamp-evoked SK channel-mediated AHP currents (I-AHP) to inhibitors of Cav channels recorded in the perforated whole-cell configuration. A, Low micromolar concentrations (100 μm) of nickel (T-type) reversibly inhibited most of the cobalt-sensitive I-AHP. B, ω-Conotoxin-GVIA (conotoxin; 1 μm) reversibly reduced a minor part of the cobalt-sensitive I-AHP. C, Nifedipine (10 μm) did not affect I-AHPs.D, FTX-3.3 (1 μm) had no effect on I-AHPs.E, The summary of experiments in A–Dshows that cobalt-sensitive I-AHPs were activated preferentially via calcium channels sensitive to low micromolar nickel (100 μm; 85 ± 9%; n = 13; ∗∗∗p < 0.0005) and mibefradil (mib; 10 μm; 94 ± 10%; n = 6; ∗∗p < 0.005), whereas only a small component was sensitive to 1 μm ω-conotoxin-GVIA (cono; 26 ± 3%; n = 4; ∗∗p < 0.005). Nifedipine (nif; 10 μm) did not affect I-AHPs (residual current, 102 ± 6%; n = 6; p > 0.05). Similarly, 1 μm FTX-3.3 (FTX; residual current, 97 ± 5%; n = 5; p > 0.05) and 0.1 μm agatoxin-TK (aga; residual current, 101 ± 1%; n = 3; p > 0.05) had no effect on I-AHPs. Current amplitudes were normalized to cobalt-sensitive I-AHP in each individual experiment except for mibefradil, in which the mean value of cobalt block was used (1 mm; residual current, 29 ± 4%; n= 29). Calibration: A–D, 0.2 sec, 10 pA.
Fig. 2.
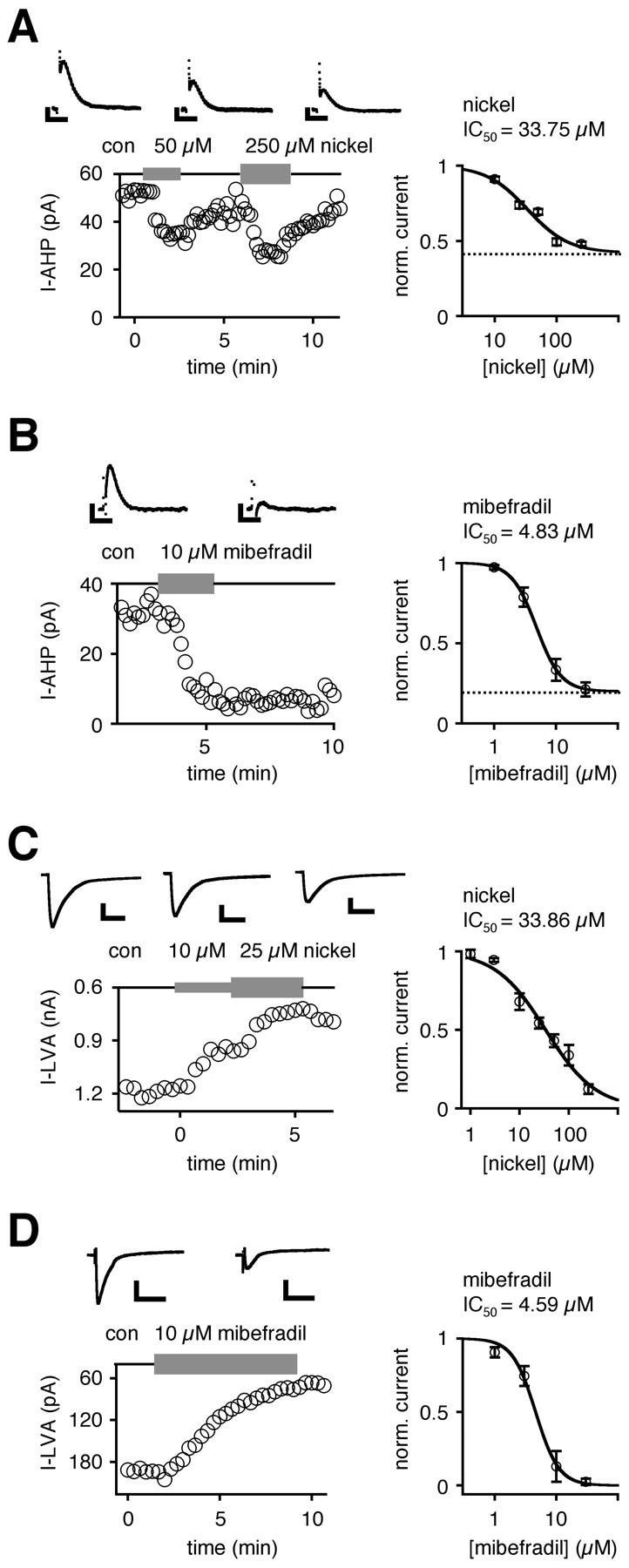
SK-mediated AHP and T-type-mediated low voltage-activated calcium currents (I-LVA; see Results) possessed almost identical nickel and mibefradil sensitivities in dopaminergic neurons. A, Nickel reduced AHP currents (I-AHP; evoked by 20 msec hybrid-clamp depolarizations) in a concentration-dependent manner. The mean dose–response relationship for nickel I-AHP inhibition was described by a Hill function, with an IC50 of 33.75 μm, a Hill coefficient of 1.30 (n = 19), and a relative fitted residual I-AHP component (42%; dotted line).B, Mibefradil inhibited a major component of the I-AHP irreversibly. The mean dose–response relationship for mibefradil I-AHP inhibition was described by a Hill function, with an IC50 of 4.83 μm(Hill coefficient, 2.16; n = 10) and a relative fitted residual I-AHP component of 20% (dotted line).C, T-type-mediated LVA currents evoked by depolarizations to −50 mV from a holding potential of −100 mV were recorded by using standard whole-cell recordings (see Results and Materials and Methods). Nickel reduced the I-LVA in a concentration-dependent manner. The mean dose–response relationship for nickel I-LVA inhibition was described by a Hill function, with an IC50 of 33.86 μm (Hill coefficient, 0.85;n = 16). D, Mibefradil inhibited the I-LVA irreversibly. The mean dose–response for mibefradil I-LVA inhibition was described by a Hill function, with an IC50of 4.59 μm (Hill coefficient, 2.27; n= 14). Calibration: A, B, 0.2 sec, 10 pA;C, 0.2 sec, 300 pA; D, 0.2 sec, 60 pA.
RESULTS
SK channel-mediated AHP currents are activated preferentially by calcium channels with high nickel and mibefradil sensitivities
DA neurons recorded in the SN showed slow pacemaker firing (1–3.5 Hz) with single APs followed by large AHPs, consistent with our previous recordings from tyrosine hydroxylase-immunopositive SN cells (Wolfart et al., 2001) and the biophysical fingerprint of DA SN neurons (Grace and Onn, 1989; Richards et al., 1997). Similar to our previous study, SK-mediated AHP currents were evoked by hybrid-clamp depolarizations (Wolfart et al., 2001), using the perforated patch-clamp configuration to preserve physiological calcium handling. AHP currents evoked by 20 msec hybrid pulses to approximate single APs were small sized (Fig. 1; 27 ± 1.8 pA; range, 9–80 pA; n = 58) but stable throughout the experiment and decayed monoexponentially with a time constant of 75–208 msec (123 ± 4 msec; n = 58). As shown previously, these AHP currents were inhibited by the bee venom toxin apamin, which is a selective blocker of SK channels (Blatz and Magleby, 1987; Kohler et al., 1996; Wolfart et al., 2001) (residual current in 300 nm apamin, 12 ± 3%;n = 8; data not shown). In contrast, slow AHPs after 5-sec-long depolarizations (to mean potentials between −15 and +6 mV) that included multiple APs were not affected by 300 nm apamin (n = 6; data not shown).
We used a panel of established selective and nonselective inhibitors of Cav channels to define which of the different types (L-, P/Q-, N-, R-, and T-type) of neuronal Cav channels were involved in SK channel activation (Nowycky et al., 1985; Llinas et al., 1989; Zhang et al., 1993; Tsien et al., 1995; Randall, 1998; Miller, 2001). AHP currents evoked by 20 msec hybrid pulses were inhibited by 1 mmcobalt (Fig. 1A–D; residual current, 29 ± 4%;n = 29), indicating that activation of Cav channels is essential for the recruitment of SK channels in DA SN neurons. Consistent with our previous results (Wolfart et al., 2001), the presence of a residual cobalt-insensitive AHP current suggests that a minor part of the AHP current is independent of hybrid pulse-mediated calcium influx via voltage-activated calcium channels. To focus on the role of voltage-activated calcium influx for SK channel activation, we normalized the effects of Cav inhibitors to the cobalt-sensitive component of the AHP current amplitude in each individual experiment. Nickel is an nonselective inhibitor of calcium channels, but transient low voltage-activated (LVA) T-type (Perez-Reyes et al., 1998; Perchenet et al., 2000) and high voltage-activated (HVA) R-type (Soong et al., 1993; Schneider et al., 1994) Cav channels are particularly sensitive (IC50, <50 μm), whereas other HVA Cav channels (L-, P/Q-, and N-type) are less sensitive (IC50, >90 μm) (Zhang et al., 1993; Randall, 1998). Figure1A shows a recording of hybrid-evoked AHP currents during control and nickel (100 μm) application and subsequent cobalt (1 mm) application. The main component of the cobalt-sensitive AHP current was blocked by low micromolar concentrations of nickel. Most of the AHP current was also sensitive to mibefradil (Fig. 1E; 10 μm), a drug that inhibits T-type calcium channels in low micromolar concentrations (Martin et al., 2000;Perchenet et al., 2000). Thus, cobalt-sensitive AHP currents were blocked almost completely either by 100 μmnickel (Fig. 1E; residual current, 15 ± 4%;n = 13) or 10 μm mibefradil (Fig. 1E; residual current, 6 ± 10%;n = 6). In contrast, the snail toxin ω-conotoxin-GVIA (ctx-GVIA; 1 μm), an N-type calcium channel inhibitor (Williams et al., 1992a; Randall, 1998), reduced the AHP current only to a small degree (Fig. 1B,E; residual current, 77 ± 2%; residual cobalt-sensitive current, 74 ± 3%; n = 4), indicating a possible minor role for N-type channels in SK channel activation. AHP currents were completely unaffected by 10 μm nifedipine, a dihydropyridine that inhibits L-type channels (Tanabe et al., 1987;Williams et al., 1992b; Randall, 1998) (Fig. 1C,E; residual current, 102 ± 6%; n = 6). Previous studies have shown that nifedipine indeed does inhibit L-type currents and affects AP discharge or high voltage-activated depolarizations in DA neurons (Kang and Kitai, 1993a,b; Nedergaard et al., 1993; Mercuri et al., 1994; Cardozo and Bean, 1995; Ping and Shepard, 1999; Shepard and Stump, 1999). Funnel web spider toxins that block P/Q-type channels (Mori et al., 1991; Randall, 1998), such as FTX-3.3 (Dupere et al., 1996) (Fig. 1D,E; 1 μm; residual current, 97 ± 5%; n = 5), as well as agatoxin-TK (Teramoto et al., 1993) also had no effect on AHP currents (Fig. 1E; residual current, 101 ± 1%;n = 3).
Because there is evidence that receptor-mediated calcium release from intracellular stores activates SK channels in DA neurons (Morikawa et al., 2000; Seutin et al., 2000) and other types of cells (Yoshizaki et al., 1995; Davies et al., 1996; Tanabe et al., 1998; Cordoba-Rodriguez et al., 1999), we assessed the contribution of calcium released from intracellular stores to AHP currents evoked by hybrid pulse-mediated activation of plasmalemmal Cav channels. Cyclopiazonic acid (CPA; 10 μm), an agent used to block intracellular calcium release by inhibiting endoplasmatic reticulum calcium ATPases (Taylor and Broad, 1998), reduced AHP currents by 77 ± 7% (n = 4; data not shown). Thus the inhibition of intracellular calcium release and the inhibition of Cav channels reduced AHP currents to a similar degree. These results suggest that intracellular calcium release acts downstream of plasmalemmal Cav channels and amplifies their calcium signal. In summary, our data demonstrate a selective role for calcium channels with high nickel and mibefradil sensitivity in SK channel activation in DA SN neurons.
SK channel-mediated AHP currents and T-type calcium channels possess almost identical nickel and mibefradil sensitivities in DA SN neurons
The pharmacological profile of AHP currents indicated that T-type Cav channels might be the primary calcium source for SK channel activation after an action potential. Consequently, T-type calcium currents should possess similar nickel and mibefradil sensitivities compared with those of SK currents. To elicit T-type currents, we applied voltage step protocols from hyperpolarized holding potentials in the standard whole-cell configuration while blocking other calcium and potassium channels (see Materials and Methods). These protocols elicited typical LVA T-type calcium currents (Figs.2C,D,3B) (Kang and Kitai, 1993b; Perez-Reyes et al., 1998) that activated at negative membrane potentials (rise time, 23.3 ± 0.9 msec, n = 51; V½act, −56.3 mV, slope = 4.9,n = 18; V½inact, −78.3 mV, slope = 5.3, n = 12; data not shown), had amplitudes in the range of 50–750 pA (268 ± 23 pA;n = 51), and inactivated with major fast (45.0 ± 2.0 msec; n = 51) and minor slow time constants (30.0 ± 2.6%; 242 ± 14 msec; n = 51). Deactivation time constants were in the range of 4–7 msec, as determined by tail current protocols (−120 mV, 4.7 ± 0.6 msec; −100 mV, 5.9 ± 0.6 mV; −80 mV, 7.0 ± 0.9 msec;n = 9; data not shown) (see Materials and Methods). These results are consistent with the transient LVA calcium currents previously reported in rat DA neurons (Kang and Kitai, 1993b) and the biophysical properties of native and recombinant T-type channels (Huguenard, 1996; Perez-Reyes et al., 1998; Lee et al., 1999; McRory et al., 2001).
The mean dose–response relationship for nickel inhibition was almost identical between AHP currents (Fig. 2A; IC50, 33.75 μm; Hill coefficient, 1.30; n = 19) and T-type currents (Fig.2C; IC50, 33.86 μm; Hill coefficient, 0.85; n = 16). Likewise, the mean dose–response curve for mibefradil inhibition was also very similar between AHP currents (Fig. 2B; IC50, 4.83 μm; Hill coefficient, 2.16; n = 10) and T-type currents (Fig.2D; IC50, 4.59; Hill coefficient, 2.27; n = 14). The fitted nickel- or mibefradil-insensitive components of the AHP current (Fig.2A,B, dotted line; nickel, 42%; mibefradil, 20%) were in a range comparable with the cobalt-insensitive AHP current component (29 ± 4%; Fig. 1). This suggests that most of the flux through Cavchannels involved in SK channel activation was indeed nickel- and mibefradil-sensitive. The pharmacological profile of AHP currents was not sufficient to rule out a contribution of R-type calcium channels. However, the striking similarity of quantitative nickel and mibefradil sensitivities between biophysically identified native T-type currents and AHP currents strongly suggests that T-type calcium channels are coupled selectively to SK channels in DA SN neurons.
SK currents closely follow the temporal profile of T-type currents
One predicted consequence of the functional coupling of SK channels to T-type channels was that SK currents should follow the characteristic gating behavior of T-type channels such as use-dependent inactivation (Huguenard, 1996; Perez-Reyes et al., 1998). In agreement with this prediction, we noted that hybrid-clamp-evoked AHP currents showed a use-dependent inactivation when successive voltage steps were applied (Fig. 3A). In contrast, apamin-sensitive AHP currents in SN neurons with an electrophysiological profile typical of GABAergic neurons (Richards et al., 1997; Liss et al., 1999) did not exhibit this use dependency (first/sixth AHP amplitude, 0.93 ± 0.04; n = 15; data not shown). Use-dependent inactivation is a typical feature of channels that possess inactivation gating, including T-type channels (Huguenard, 1996; Perez-Reyes et al., 1998), but has not been reported to be an intrinsic property of SK channels (Kohler et al., 1996; Xia et al., 1998; Hirschberg et al., 1999). Accordingly, SK channels can be activated tonically in DA SN neurons by 1-EBIO (>0.5 mm), a compound that increases the open probability of SK channels (Xia et al., 1998), inducing long-lasting hyperpolarizations (Wolfart et al., 2001). We tested use-dependent inactivation of AHP and T-type currents by eliciting trains of step potentials (holding potential, −80 mV; 100 msec pulses to +60 mV at 1 Hz) in both AHP and T-type current recording conditions (see Materials and Methods). The time constants of use-dependent inactivation for AHP and T-type currents were not significantly different [AHP current (Fig. 3A,D): 0.98 ± 0.06 sec, n = 10; T-type (Fig. 3B,D): 0.80 ± 0.07 sec, n= 10; p > 0.05]. We studied the functional implications of this cumulative inactivation for the temporal integration of neuronal activity by using perforated patch-clamp recordings in current-clamp mode (Fig. 3C). Consistent with our voltage-clamp data, AHP amplitudes cumulatively decreased in response to prolonged positive current injections from hyperpolarized membrane potentials. This cumulative decrease of AHP amplitudes had similar time constants when compared with those of AHP currents and T-type currents (Fig. 3C; 1.08 ± 0.12 sec,n = 8; p > 0.05) and was blocked by nickel (Fig. 3C,E; initial/steady-state AHP: control, 2.0 ± 0.1, n = 8; 250 μmnickel, 1.1 ± 0.1, n = 8; p < 0.0005). Thus in DA SN neurons the SK currents inactivated cumulatively in a physiologically relevant frequency range and closely followed the inactivation kinetics of their major upstream calcium source, the T-type calcium channel. This affects the rebound behavior of DA SN neurons because the functional coupling of T-type to SK channels generates a transient rebound inhibition.
The functional coupling of T-type and SK channels maintains pacemaker precision in DA SN neurons
Next we studied whether the functional coupling of T-type channels to SK channels is operative during spontaneous pacemaker activity. In particular, we were interested in how the functional pairing of T-type and SK channels is involved in the regulation of pacemaker spiking, because we have shown previously that pacemaker frequency and precision strongly depend on SK channel activity in DA SN neurons (Wolfart et al., 2001). To quantify the role of T-type channels in pacemaker precision, we analyzed the CV of ISIs during continuous perforated patch-clamp recordings in the current-clamp configuration (Fig.4). Consistent with previous studies, pacemaker spiking was highly precise at 22–24°C (Fig.4A,D; CV, 14 ± 2%, n = 11) (Wolfart et al., 2001) and at 36–37°C (14 ± 1%,n = 15; p > 0.4; data not shown) (Shepard and Bunney, 1988). Application of 100 μm nickel slowed the spiking frequency (control: 2.13 ± 0.14 Hz, n = 11; nickel: 1.79 ± 0.19 Hz, n = 11; p < 0.005) and rendered the pacemaker more irregular (Fig.4B,D; CV, 27 ± 5%, n = 11;p < 0.005) in such a way that was similar to the effect of apamin application on spiking regularity (Fig.4C,D; CV, 26 ± 4%, n = 11;p < 0.005). Thus, inhibition of T-type and SK channels reduced the precision of the intrinsic pacemaker to a similar extent (p > 0.8), demonstrating that T-type channels are the essential calcium source for SK channels also during spontaneous pacemaker activity in DA SN neurons.
Fig. 4.
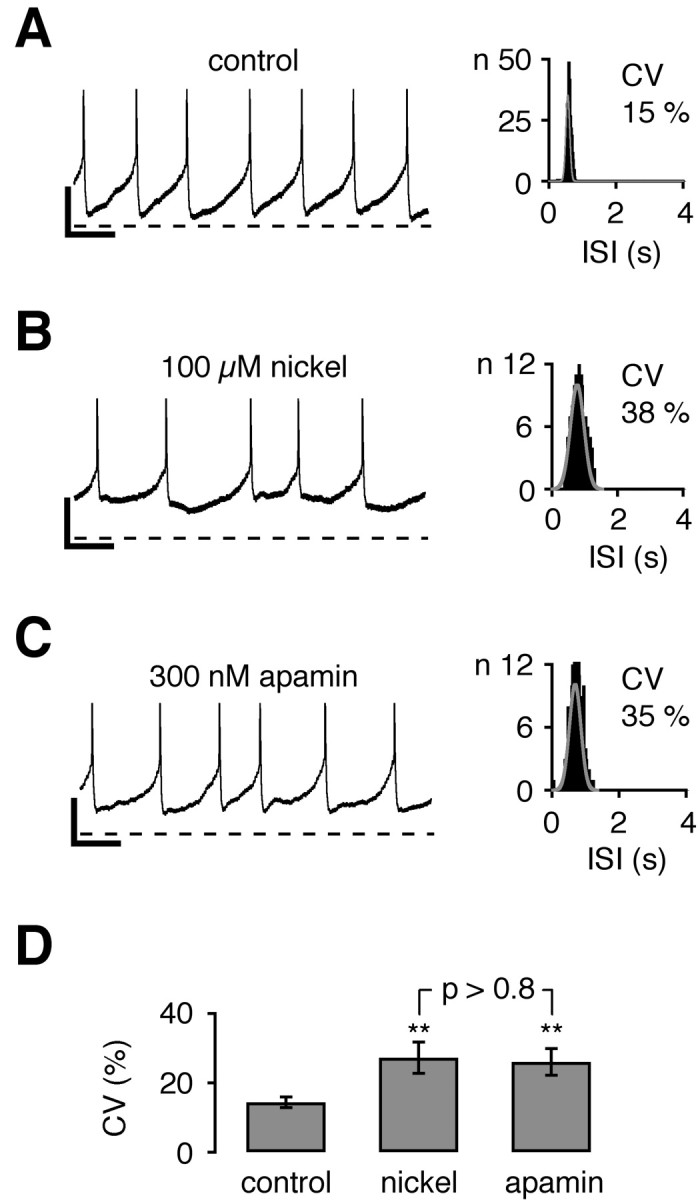
Nickel-sensitive T-type and apamin-sensitive SK channels maintained the high precision of pacemaker spiking in dopaminergic neurons. A–C, Perforated current-clamp recordings during control (A), 100 μm nickel (B), and 300 nm apamin (C) application.Left panels show a 4 sec recording trace representative of a 5 min recording for each condition. Interspike interval (ISI) frequency distributions are displayed in the right panels for each recording. As a measure of pacemaker precision the coefficient of variation (CV) was calculated from the Gaussian fit of ISI histograms. APs were truncated at −20 mV. A, During control conditions pacemaker spiking was relatively regular, with a CV of 15%. B, Application of 100 μm nickel reversibly rendered pacemaker spiking to be more irregular (CV of 38%). C, Application of 300 nm apamin did increase the irregularity of pacemaker spiking to a similar degree (CV of 35%). D, The summary of experiments inA–C shows that nickel (100 μm) and apamin (300 nm) decreased the pacemaker precision (∗∗p < 0.005, respectively) to a similar extent (p > 0.8). Mean CVs: control, 14 ± 2% (n = 11); nickel, 27 ± 5% (n = 11); apamin, 26 ± 4% (n = 11). Calibration: A–C, 0.5 sec, 10 mV. Dotted line in A–C, −50 mV.
Coupling of T-type and SK channels prevents intrinsic burst firing in DA SN neurons
T-type currents are known to promote bursting in many central neurons (Huguenard, 1996, 1998). However, the present results indicate that, in DA SN neurons, T-type channels might have an opposite role because of their functional coupling to SK channels. As shown in Figure4, the activity of T-type channels stabilizes single-spike pacemaker firing. Indeed, application of 100 μm nickel alone led to an unequivocal burst-like pattern in a minor subset (3 of 27) of DA SN neurons recorded via the perforated patch-clamp technique (Fig. 5B; see Table 1 for comparison of burst parameters with those described in the literature). The bursting pattern consisted mainly of AP doublet bursts (range, 2–6; mean, 2.3 ± 0.1 APs/burst; mean intra-burst interval, 151 ± 5 msec) followed by prolonged, large hyperpolarizations (mean inter-burst interval, 2.39 ± 0.31 sec), which were not inhibited by the additional application of 300 nm apamin (n = 2; data not shown). A comparable switch in spiking pattern was never observed during apamin application in this study nor in our previous 10-min-long perforated patch recordings in apamin (Wolfart et al., 2001). As described by Grace and Bunney (1984b), the degree of bursting was quantified as APs involved in bursts per treatment (percentage of bursting; see Materials and Methods). No significant degree of bursting was detected in control and apamin recordings in this study (Fig. 5D; control: 0% bursting,n = 30; 300 nm apamin: 3 ± 2%, n = 19) as well as in recordings of previously published data (control: 0% bursting, n = 11; 300 nm apamin: 5 ± 3% bursting,n = 10; data not shown) (Wolfart et al., 2001). In contrast, 100 μm nickel was sufficient to increase the mean bursting values significantly (Fig. 5D; 12 ± 7% bursting, n = 27; p < 0.05) and, indeed, induced robust bursting (>84% bursting) in three cells. Most effective in the induction of bursting was the combination of T-type and SK channel inhibition. It strongly increased the degree of bursting (Fig. 5D; 34 ± 10%, n = 16; p < 0.0005) and induced robust bursting in five cells (>86% bursting).
We also noted that the degree of bursting was predicted by the degree of pacemaker precision under control conditions. Cells that fired in bursts after nickel (100 μm) or after nickel and apamin (300 nm) application had significantly higher control CV values (Fig. 5E; 25 ± 4%, n = 6) compared with cells that fired irregularly only with the inhibition of T-type channels or T-type and SK channels (Fig. 5E; 14 ± 1%, n = 22; p < 0.0005). In summary, T-type calcium channels stabilized pacemaker firing and in addition prevented the switch to an intrinsic burst-firing mode in DA SN neurons.
DISCUSSION
On the basis of our pharmacological and biophysical analysis of AHP and T-type currents, we conclude that SK channels are activated almost exclusively via T-type channels in mouse DA SN neurons. Although various subtypes of Cav channels are known to be present at the mRNA (Soong et al., 1993; Stea et al., 1994; Craig et al., 1999; Talley et al., 1999), protein (Williams et al., 1994; Craig et al., 1999; Takada et al., 2001), and functional levels (Kang and Kitai, 1993a; Nedergaard et al., 1993; Mercuri et al., 1994; Cardozo and Bean, 1995; Ping and Shepard, 1999; Shepard and Stump, 1999), SK currents were not affected by established L-type and P/Q-type channel blockers (Zhang et al., 1993; Tsien et al., 1995; Randall, 1998;Miller, 2001). In contrast, between 85 and 94% of SK currents activated by voltage-activated calcium influx were blocked selectively in a dose-dependent manner by nickel and mibefradil, respectively, in a concentration range that is likely to block T-type channels (Martin et al., 2000; Perchenet et al., 2000). Although we cannot exclude a minor role for N-type channels in SK channel activation, the reversible inhibition by ω-conotoxin-GVIA also might be an effect on native T-type channels (McCleskey et al., 1987). Although nickel and mibefradil also have been reported to affect HVA R-type channels (Bezprozvanny and Tsien, 1995; Randall and Tsien, 1997), the identical quantitative pharmacological profiles of SK and biophysically identified T-type currents, as well as their very similar use-dependent inactivation, strongly suggest that T-type channels constitute the selective calcium source for SK channel activation in DA SN neurons. In comparison to previous studies in other cell types (Wisgirda and Dryer, 1994; Davies et al., 1996; Marrion and Tavalin, 1998; Tanabe et al., 1998; Sah and Davies, 2000; Shah and Haylett, 2000; Bowden et al., 2001), this selective coupling of SK to T-type channels in SN DA neurons appears to be unique and might have important functional implications.
During phasic activation from relatively hyperpolarized membrane potentials, T-type channel activation does not lead to rebound excitation, as seen in thalamocortical neurons (Huguenard, 1996, 1998). Instead, T-type channel function is inverted by activating SK channels, thereby generating a transient rebound inhibition. The coupling of SK channels to T-type channel gating might explain why SK channels do not contribute to frequency adaptation in DA SN neurons (Shepard and Bunney, 1991), which is an important function of SK channels in many other types of neurons (Sah, 1996; Bond et al., 1999; Sah and Davies, 2000). Similar to sino-atrial pacemaker cells (Hagiwara et al., 1988;Huser et al., 2000), T-type channels in DA SN neurons were active during spontaneous pacemaker activity. Our data provide evidence that the functional pairing of T-type and SK channels maintains the precision and stability of the single-spike pacemaker. Previous studies have showed that the inhibition of SK channels alone can induce intrinsic bursting in rat DA neurons recorded in vitro(Shepard and Bunney, 1988, 1991; Gu et al., 1992; Ping and Shepard, 1996). We did not detect robust bursting in apamin alone, but bursting occurrence in most previous studies was low (<50% of neurons), and other studies also have failed to evoke intrinsic bursting by SK channel inhibition alone (Seutin et al., 1993; Johnson and Seutin, 1997). In marked contrast to the role of T-type channels in other types of neurons (Huguenard, 1996, 1998), the inhibition of T-type channels alone switched the firing pattern of some DA SN neurons to an intrinsic burst-firing mode. Thus in DA SN neurons the role of T-type channels corresponds to that described for SK channels alone (Shepard and Bunney, 1988, 1991; Gu et al., 1992; Ping and Shepard, 1996), because T-type currents are translated into SK currents by their functional coupling. Although burst firing was not evoked by SK channel inhibition alone, burst occurrence was increased significantly when SK channels were blocked in addition to T-type channels. This indicates that the burst switch is regulated by several converging conductances as suggested by a recent burst-modeling study (Goldman et al., 2001).
Our results support the notion that DA SN neurons possess an intrinsic bursting mechanism (Shepard and Bunney, 1988) that might be “unmasked” synaptically (Kitai et al., 1999) and sustained by a combination of intrinsic and synaptic mechanisms (Johnson et al., 1992;Kitai et al., 1999; Paladini and Tepper, 1999). Differences betweenin vivo bursting (Grace and Bunney, 1984b) and in vitro bursting induced by NMDA (Johnson et al., 1992), apamin (Shepard and Bunney, 1991), and/or nickel further suggest that a combination of intrinsic and synaptic mechanisms is responsible for the transition from pacemaker to burst firing in DA SN neurons in vivo (see Table 1).
We can only speculate about the molecular organization of the functional coupling between SK and T-type channels. We have reported previously that hybrid pulse-evoked AHP currents are reduced significantly by millimolar concentrations (10 mm) of the slow calcium buffer EGTA, arguing against a very close spatial coupling of T-type and SK channels in DA neurons (Wolfart et al., 2001). This is in contrast to the tight spatial coupling of SK channels to nicotinic acetylcholine receptors in outer hair cells (<10 nm) (Oliver et al., 2000). Our results also show that release from intracellular stores significantly amplifies the calcium signal for SK channel activation. As described previously for other cell types (Yoshizaki et al., 1995;Tanabe et al., 1998; Cordoba-Rodriguez et al., 1999; Huser et al., 2000), our data suggest that T-type channel-mediated calcium influx during pacemaker firing acts as a “calcium spark,” triggering secondary calcium-induced calcium release that subsequently activates SK channels in DA SN neurons. We did not detect SK channel activation during nickel-induced bursting or after long depolarizations, conditions that are likely to lead to the recruitment of HVA calcium channels in DA neurons (Kang and Kitai, 1993b; Nedergaard et al., 1993;Mercuri et al., 1994; Cardozo and Bean, 1995). Consequently, we hypothesize that T-type channels, calcium stores, and SK channels might be colocalized selectively, possibly forming a specialized calcium signaling complex (Marrion and Tavalin, 1998; Husi et al., 2000; Bowden et al., 2001) in DA neurons. This functional signaling complex could provide a new framework for the temporal integration of synaptic input in DA SN neurons, which might help us to understand how and why DA neurons switch between pacemaker and burst-firing mode in vivo, thus contributing to reward-based learning (Schultz, 2000;Reynolds et al., 2001).
Footnotes
This work was supported by the Medical Research Council. J.R. holds the Monsanto Senior Research Fellowship at Exeter College, Oxford University. We thank Dr. Ian Jones and Dr. Peter Magill for critically reading this manuscript.
Correspondence should be addressed to Dr. Jochen Roeper, Medial Research Council Anatomical Neuropharmacology Unit, Oxford University, Mansfield Road, Oxford OX1 3TH, UK. E-mail:jochen.roeper@pharm.ox.ac.uk.
REFERENCES
- 1.Bean AJ, Roth RH. Extracellular dopamine and neurotensin in rat prefrontal cortex in vivo: effects of median forebrain bundle stimulation frequency, stimulation pattern, and dopamine autoreceptors. J Neurosci. 1991;11:2694–2702. doi: 10.1523/JNEUROSCI.11-09-02694.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967). Mol Pharmacol. 1995;48:540–549. [PubMed] [Google Scholar]
- 3.Blatz A, Magleby K (1987) Calcium-activated potassium channels. Trends Neurosci 463–467.
- 4.Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann NY Acad Sci. 1999;868:370–378. doi: 10.1111/j.1749-6632.1999.tb11298.x. [DOI] [PubMed] [Google Scholar]
- 5. Bowden SE, Fletcher S, Loane DJ, Marrion NV. Somatic colocalization of rat SK1 and D class (Cav 1.2) L-type calcium channels in rat CA1 hippocampal pyramidal neurons. J Neurosci 21 2001. RC175:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol. 1995;74:1137–1148. doi: 10.1152/jn.1995.74.3.1137. [DOI] [PubMed] [Google Scholar]
- 7.Carlsson A, Waters N, Waters S, Carlsson ML. Network interactions in schizophrenia—therapeutic implications. Brain Res Brain Res Rev. 2000;31:342–349. doi: 10.1016/s0165-0173(99)00050-8. [DOI] [PubMed] [Google Scholar]
- 8.Cordoba-Rodriguez R, Moore KA, Kao JP, Weinreich D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proc Natl Acad Sci USA. 1999;96:7650–7657. doi: 10.1073/pnas.96.14.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig PJ, Beattie RE, Folly EA, Banerjee MD, Reeves MB, Priestley JV, Carney SL, Sher E, Perez-Reyes E, Volsen SG. Distribution of the voltage-dependent calcium channel α1G subunit mRNA and protein throughout the mature rat brain. Eur J Neurosci. 1999;11:2949–2964. doi: 10.1046/j.1460-9568.1999.00711.x. [DOI] [PubMed] [Google Scholar]
- 10.Davies PJ, Ireland DR, McLachlan EM. Sources of Ca2+ for different Ca2+-activated K+ conductances in neurones of the rat superior cervical ganglion. J Physiol (Lond) 1996;495:353–366. doi: 10.1113/jphysiol.1996.sp021599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunnett SB, Bjorklund A. Prospects for new restorative and neuroprotective treatments in Parkinson's disease. Nature. 1999;399:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 12.Dupere JR, Moya E, Blagbrough IS, Usowicz MM. Differential inhibition of Ca2+ channels in mature rat cerebellar Purkinje cells by sFTX-3.3 and FTX-3.3. Neuropharmacology. 1996;35:1–11. doi: 10.1016/0028-3908(95)00156-5. [DOI] [PubMed] [Google Scholar]
- 13.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 14.Freeman AS, Meltzer LT, Bunney BS. Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci. 1985;36:1983–1994. doi: 10.1016/0024-3205(85)90448-5. [DOI] [PubMed] [Google Scholar]
- 15.Goldman MS, Golowasch J, Marder E, Abbott LF. Global structure, robustness, and modulation of neuronal models. J Neurosci. 2001;21:5229–5238. doi: 10.1523/JNEUROSCI.21-14-05229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldman-Rakic PS. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol Psychiatry. 1999;46:650–661. doi: 10.1016/s0006-3223(99)00130-4. [DOI] [PubMed] [Google Scholar]
- 17.Gonon FG, Buda MJ. Regulation of dopamine release by impulse flow and by autoreceptors as studied by in vivo voltammetry in the rat striatum. Neuroscience. 1985;14:765–774. doi: 10.1016/0306-4522(85)90141-1. [DOI] [PubMed] [Google Scholar]
- 18.Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- 19.Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 20.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984a;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984b;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu X, Blatz AL, German DC. Subtypes of substantia nigra dopaminergic neurons revealed by apamin: autoradiographic and electrophysiological studies. Brain Res Bull. 1992;28:435–440. doi: 10.1016/0361-9230(92)90044-x. [DOI] [PubMed] [Google Scholar]
- 24.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol (Lond) 1988;395:233–253. doi: 10.1113/jphysiol.1988.sp016916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating properties of single SK channels in hippocampal CA1 pyramidal neurons. Biophys J. 1999;77:1905–1913. doi: 10.1016/S0006-3495(99)77032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 27.Huguenard JR. Low-voltage-activated (T-type) calcium channel genes identified. Trends Neurosci. 1998;21:451–452. doi: 10.1016/s0166-2236(98)01331-9. [DOI] [PubMed] [Google Scholar]
- 28.Huser J, Blatter LA, Lipsius SL (2000) Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol (Lond) 524[Pt 2]:415–422. [DOI] [PMC free article] [PubMed]
- 29.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor–adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 30.Johnson SW, Seutin V. Bicuculline methiodide potentiates NMDA-dependent burst firing in rat dopamine neurons by blocking apamin-sensitive Ca2+-activated K+ currents. Neurosci Lett. 1997;231:13–16. doi: 10.1016/s0304-3940(97)00508-9. [DOI] [PubMed] [Google Scholar]
- 31.Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-d-aspartate: role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- 32.Kang Y, Kitai ST. Calcium spike underlying rhythmic firing in dopaminergic neurons of the rat substantia nigra. Neurosci Res. 1993a;18:195–207. doi: 10.1016/0168-0102(93)90055-u. [DOI] [PubMed] [Google Scholar]
- 33.Kang Y, Kitai ST. A whole cell patch-clamp study on the pacemaker potential in dopaminergic neurons of rat substantia nigra compacta. Neurosci Res. 1993b;18:209–221. doi: 10.1016/0168-0102(93)90056-v. [DOI] [PubMed] [Google Scholar]
- 34.Kita T, Kita H, Kitai ST. Electrical membrane properties of rat substantia nigra compacta neurons in an in vitro slice preparation. Brain Res. 1986;372:21–30. doi: 10.1016/0006-8993(86)91454-x. [DOI] [PubMed] [Google Scholar]
- 35.Kitai ST, Shepard PD, Callaway JC, Scroggs R. Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol. 1999;9:690–697. doi: 10.1016/s0959-4388(99)00040-9. [DOI] [PubMed] [Google Scholar]
- 36.Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 37.Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Legendy CR, Salcman M. Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J Neurophysiol. 1985;53:926–939. doi: 10.1152/jn.1985.53.4.926. [DOI] [PubMed] [Google Scholar]
- 39.Liss B, Bruns R, Roeper J. Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO J. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llinas R, Sugimori M, Lin JW, Cherksey B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl Acad Sci USA. 1989;86:1689–1693. doi: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by colocalized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 42.Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther. 2000;295:302–308. [PubMed] [Google Scholar]
- 43.McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW, Yoshikami D. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons, but not muscle. Proc Natl Acad Sci USA. 1987;84:4327–4331. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McRory JE, Santi CM, Hamming KS, Mezeyova J, Sutton KG, Baillie DL, Stea A, Snutch TP. Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem. 2001;276:3999–4011. doi: 10.1074/jbc.M008215200. [DOI] [PubMed] [Google Scholar]
- 45.Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol. 1994;113:831–838. doi: 10.1111/j.1476-5381.1994.tb17068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller RJ. Rocking and rolling with Ca2+ channels. Trends Neurosci. 2001;24:445–449. doi: 10.1016/s0166-2236(00)01859-2. [DOI] [PubMed] [Google Scholar]
- 47.Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 48. Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci 20 2000. RC103:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nedergaard S, Flatman JA, Engberg I. Nifedipine- and ω-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol (Lond) 1993;466:727–747. [PMC free article] [PubMed] [Google Scholar]
- 50.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 51.Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- 52.Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- 53.Paladini CA, Tepper JM. GABAA and GABAB antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse. 1999;32:165–176. doi: 10.1002/(SICI)1098-2396(19990601)32:3<165::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 54.Pennefather P, Lancaster B, Adams PR, Nicoll RA. Two distinct Ca-dependent K currents in bullfrog sympathetic ganglion cells. Proc Natl Acad Sci USA. 1985;82:3040–3044. doi: 10.1073/pnas.82.9.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perchenet L, Benardeau A, Ertel EA. Pharmacological properties of Cav 3.2, a low voltage-activated Ca2+ channel cloned from human heart. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:590–599. doi: 10.1007/s002100000238. [DOI] [PubMed] [Google Scholar]
- 56.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 57.Ping HX, Shepard PD. Apamin-sensitive Ca2+-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. NeuroReport. 1996;7:809–814. doi: 10.1097/00001756-199602290-00031. [DOI] [PubMed] [Google Scholar]
- 58.Ping HX, Shepard PD. Blockade of SK-type Ca2+-activated K+ channels uncovers a Ca2+-dependent slow afterdepolarization in nigral dopamine neurons. J Neurophysiol. 1999;81:977–984. doi: 10.1152/jn.1999.81.3.977. [DOI] [PubMed] [Google Scholar]
- 59.Randall AD. The molecular basis of voltage-gated Ca2+ channel diversity: is it time for T? J Membr Biol. 1998;161:207–213. doi: 10.1007/s002329900327. [DOI] [PubMed] [Google Scholar]
- 60.Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology. 1997;36:879–893. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- 61.Reynolds JN, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67–70. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- 62.Richards CD, Shiroyama T, Kitai ST. Electrophysiological and immunocytochemical characterization of GABA and dopamine neurons in the substantia nigra of the rat. Neuroscience. 1997;80:545–557. doi: 10.1016/s0306-4522(97)00093-6. [DOI] [PubMed] [Google Scholar]
- 63.Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles, and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 64.Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- 65.Sanghera MK, Trulson ME, German DC. Electrophysiological properties of mouse dopamine neurons: in vivo and in vitro studies. Neuroscience. 1984;12:793–801. doi: 10.1016/0306-4522(84)90171-4. [DOI] [PubMed] [Google Scholar]
- 66.Schneider T, Wei X, Olcese R, Costantin JL, Neely A, Palade P, Perez-Reyes E, Qin N, Zhou J, Crawford GD. Molecular analysis and functional expression of the human type E neuronal Ca2+ channel α1 subunit. Receptors Channels. 1994;2:255–270. [PubMed] [Google Scholar]
- 67.Schultz W. Multiple reward signals in the brain. Nat Rev Neurosci. 2000;1:199–207. doi: 10.1038/35044563. [DOI] [PubMed] [Google Scholar]
- 68.Seutin V, Johnson SW, North RA. Apamin increases NMDA-induced burst firing of rat mesencephalic dopamine neurons. Brain Res. 1993;630:341–344. doi: 10.1016/0006-8993(93)90675-d. [DOI] [PubMed] [Google Scholar]
- 69.Seutin V, Mkahli F, Massotte L, Dresse A. Calcium release from internal stores is required for the generation of spontaneous hyperpolarizations in dopaminergic neurons of neonatal rats. J Neurophysiol. 2000;83:192–197. doi: 10.1152/jn.2000.83.1.192. [DOI] [PubMed] [Google Scholar]
- 70.Shah M, Haylett DG. Ca2+ channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2554–2561. doi: 10.1152/jn.2000.83.5.2554. [DOI] [PubMed] [Google Scholar]
- 71.Shepard PD, Bunney BS. Effects of apamin on the discharge properties of putative dopamine-containing neurons in vitro. Brain Res. 1988;463:380–384. doi: 10.1016/0006-8993(88)90414-3. [DOI] [PubMed] [Google Scholar]
- 72.Shepard PD, Bunney BS. Repetitive firing properties of putative dopamine-containing neurons in vitro: regulation by an apamin-sensitive Ca2+-activated K+ conductance. Exp Brain Res. 1991;86:141–150. doi: 10.1007/BF00231048. [DOI] [PubMed] [Google Scholar]
- 73.Shepard PD, Stump D. Nifedipine blocks apamin-induced bursting activity in nigral dopamine-containing neurons. Brain Res. 1999;817:104–109. doi: 10.1016/s0006-8993(98)01231-1. [DOI] [PubMed] [Google Scholar]
- 74.Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- 75.Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Svensson TH. Dysfunctional brain dopamine systems induced by psychotomimetic NMDA receptor antagonists and the effects of antipsychotic drugs. Brain Res Brain Res Rev. 2000;31:320–329. doi: 10.1016/s0165-0173(99)00048-x. [DOI] [PubMed] [Google Scholar]
- 77.Takada M, Kang Y, Imanishi M. Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Eur J Neurosci. 2001;13:757–762. doi: 10.1046/j.1460-9568.2001.01435.x. [DOI] [PubMed] [Google Scholar]
- 78.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanabe M, Gahwiler BH, Gerber U. L-type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. J Neurophysiol. 1998;80:2268–2273. doi: 10.1152/jn.1998.80.5.2268. [DOI] [PubMed] [Google Scholar]
- 80.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 81.Taylor CW, Broad LM. Pharmacological analysis of intracellular Ca2+ signaling: problems and pitfalls. Trends Pharmacol Sci. 1998;19:370–375. doi: 10.1016/s0165-6147(98)01243-7. [DOI] [PubMed] [Google Scholar]
- 82.Teramoto T, Kuwada M, Niidome T, Sawada K, Nishizawa Y, Katayama K. A novel peptide from funnel web spider venom, omega-Aga-TK, selectively blocks P-type calcium channels. Biochem Biophys Res Commun. 1993;196:134–140. doi: 10.1006/bbrc.1993.2225. [DOI] [PubMed] [Google Scholar]
- 83.Tsien RW, Lipscombe D, Madison D, Bley K, Fox A. Reflections on Ca2+ channel diversity, 1988-1994. Trends Neurosci. 1995;18:52–54. [PubMed] [Google Scholar]
- 84.Williams ME, Brust PF, Feldman DH, Patthi S, Simerson S, Maroufi A, McCue AF, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of an omega-conotoxin-sensitive human N-type calcium channel. Science. 1992a;257:389–395. doi: 10.1126/science.1321501. [DOI] [PubMed] [Google Scholar]
- 85.Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 1992b;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 86.Williams ME, Marubio LM, Deal CR, Hans M, Brust PF, Philipson LH, Miller RJ, Johnson EC, Harpold MM, Ellis SB. Structure and functional characterization of neuronal α1E calcium channel subtypes. J Biol Chem. 1994;269:22347–22357. [PubMed] [Google Scholar]
- 87.Wilson CJ, Young SJ, Groves PM. Statistical properties of neuronal spike trains in the substantia nigra: cell types and their interactions. Brain Res. 1977;136:243–260. doi: 10.1016/0006-8993(77)90801-0. [DOI] [PubMed] [Google Scholar]
- 88.Wise RA. Addiction becomes a brain disease. Neuron. 2000;26:27–33. doi: 10.1016/s0896-6273(00)81134-4. [DOI] [PubMed] [Google Scholar]
- 89.Wisgirda ME, Dryer SE. Functional dependence of Ca2+-activated K+ current on L- and N-type Ca2+ channels: differences between chicken sympathetic and parasympathetic neurons suggest different regulatory mechanisms. Proc Natl Acad Sci USA. 1994;91:2858–2862. doi: 10.1073/pnas.91.7.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 92.Yoshizaki K, Hoshino T, Sato M, Koyano H, Nohmi M, Hua SY, Kuba K. Ca2+-induced Ca2+ release and its activation in response to a single action potential in rabbit otic ganglion cells. J Physiol (Lond) 1995;486:177–187. doi: 10.1113/jphysiol.1995.sp020801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang JF, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]