

Abstract
In the present study, we characterized oxidative stress-dependent cellular events in dopaminergic cells after exposure to an organic form of manganese compound, methylcyclopentadienyl manganese tricarbonyl (MMT). In pheochromocytoma cells, MMT exposure resulted in rapid increase in generation of reactive oxygen species (ROS) within 5–15 min, followed by release of mitochondrial cytochrome C into cytoplasm and subsequent activation of cysteine proteases, caspase-9 (twofold to threefold) and caspase-3 (15- to 25-fold), but not caspase-8, in a time- and dose-dependent manner. Interestingly, we also found that MMT exposure induces a time- and dose-dependent proteolytic cleavage of native protein kinase Cδ (PKCδ, 72–74 kDa) to yield 41 kDa catalytically active and 38 kDa regulatory fragments. Pretreatment with caspase inhibitors (Z-DEVD-FMK or Z-VAD-FMK) blocked MMT-induced proteolytic cleavage of PKCδ, indicating that cleavage is mediated by caspase-3. Furthermore, inhibition of PKCδ activity with a specific inhibitor, rottlerin, significantly inhibited caspase-3 activation in a dose-dependent manner along with a reduction in PKCδ cleavage products, indicating a possible positive feedback activation of caspase-3 activity by PKCδ. The presence of such a positive feedback loop was also confirmed by delivering the catalytically active PKCδ fragment. Attenuation of ROS generation, caspase-3 activation, and PKCδ activity before MMT treatment almost completely suppressed DNA fragmentation. Additionally, overexpression of catalytically inactive PKCδK376R(dominant-negative mutant) prevented MMT-induced apoptosis in immortalized mesencephalic dopaminergic cells. For the first time, these data demonstrate that caspase-3-dependent proteolytic activation of PKCδ plays a key role in oxidative stress-mediated apoptosis in dopaminergic cells after exposure to an environmental neurotoxic agent.
Keywords: apoptosis, oxidative stress, Parkinson's disease, environmental factors, manganese, dopaminergic degeneration
Parkinson's disease (PD) is an idiopathic neurodegenerative disorder characterized by profound loss of dopaminergic neurons in the nigrostriatal tract. Although debated, most studies have concluded that aging, environmental neurotoxicant exposures, and genetic alterations are potential risk factors in the development of PD (Oertel and Kupsch, 1993; Langsten and Hill, 1998;Aschner, 2000; Simon et al., 2000). Recently, a study conducted on thousands of twins concluded that genetic factors do not play a role in the pathogenesis of geriatric onset of PD, which further supports the view that environmental factors are dominant risk factors in the etiology of PD (Tanner et al., 1999). Results of several epidemiological studies conducted in rural areas have also suggested that certain pesticides and other environmental factors, including transition metals such as manganese, have a positive association with increased incidences of PD (Seidler et al., 1996; Liou et al., 1997;Gorell et al., 1999). Occupational exposure to manganese during mining was shown to cause a Parkinson's-like syndrome known as Manganism (Mena et al., 1967; Barbeau, 1984; Donaldson, 1987; Gorell et al., 1999). Furthermore, exposure to manganese-containing compounds such as manganese ethylene-bis-dithiocarbamate (a fungicide) and Bazooka (a cocaine-based drug) among farm workers and abusers, respectively, has been shown to result in adverse neurological defects (Roels et al., 1987; Ferraz et al., 1988; Wang et al., 1989; Thiruchelvam et al., 2000).
Methylcyclopentadienyl manganese tricarbonyl (MMT) has been used in Canada as an anti-knock gasoline agent and has been recently legalized for use in the United States as a replacement for tetraethyl lead [(CH3CH2)4Pb] in gasoline (Lynam et al., 1999; Zayed et al., 1999). Because MMT is a manganese-containing compound, its use has raised great a concern regarding increased exposure to the public and its possible adverse health effects (Frumkin and Solomon, 1997; Davis, 1998; Lynam et al., 1999; Zayed et al., 1999). Exposure to MMT produces a prolonged and more pronounced accumulation of manganese in rat brain as compared with manganese derived from an inorganic source, for example, MnCl2 (Zheng et al., 2000). Administration of MMT produces seizures in mice (Fishman et al., 1987) and also results in depletion of dopamine in the mouse striatum (Gianutsos and Murray, 1982). Furthermore, MMT administration has been shown to be an effective inhibitor of complex I in mitochondrial electron transport chain (Autissier et al., 1977), an action similar to the pyridinium metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a Parkinsonian toxin. Recently, we demonstrated that MMT exposure induces reactive oxygen species (ROS) generation, dopamine depletion, and cell death in dopamine-producing rat pheochromocytoma (PC12) cells, which can be protected by pretreatment with antioxidants (Wagner et al., 2000). To further understand the cellular mechanism of MMT-mediated apoptosis, we investigated whether oxidative stress induced by MMT can activate a series of cellular factors associated with apoptotic pathways, which could subsequently lead to programmed cell death in dopaminergic cells. Herein, we report that MMT exposure activates a novel apoptotic pathway in dopaminergic cells through caspase-3-dependent proteolytic cleavage of PKCδ.
MATERIALS AND METHODS
Reagents. MMT was obtained from Sigma-Aldrich (St. Louis, MO); rottlerin was purchased from Calbiochem (San Diego, CA); acetyl-Asp-Glu-Val-Asp-aldehyde (Ac-DEVD-CHO), acetyl-Iso-Glu-Thr-Asp-7-amino-4-methylcoumarin (Ac-IETD-AMC), acetyl-Leu-Glu-His-Asp-7-amino-4-methylcoumarin (Ac-LEHD-AMC), and Z-Asp-Glu-Val-Asp-fluoromethyl ketone (Z-DEVD-FMK) were obtained from Alexis Biochemicals (San Diego, CA); Z-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-FMK) was obtained from Enzyme Systems (Livermore, CA). Acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC) was obtained from Bachem (King of Prussia, PA); fluorescein isothiocyanate conjugated to VAD-FMK (FITC-VAD-FMK) was purchased from Promega(Madison, WI); antibodies to PKCδ, PKCα, PKCβI, and PKCβII were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), cytochrome C (mouse monoclonal) from PharMingen (San Diego, CA), green fluorescent protein (GFP) (mouse monoclonal) from Clontech (Palo Alto, CA), and β-actin (mouse monoclonal) from Sigma (St. Louis, MO). ECL chemiluminescence kit was purchased from Amersham Pharmacia Biotech (Piscataway, NJ). PC12 cells were purchased from American Type Culture Collection (ATCC) (Rockville, MD), and immortalized rat mesencephalic dopaminergic neuronal cell line (1RB3AN27) was a kind gift of Dr. Kedar N. Prasad (University of Colorado Health Sciences Center, Denver, CO). Hydroethidine and Hoechst 33342 were purchased from Molecular Probes (Eugene, OR). Cell Death Detection ELISA Plus assay kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN). PKCδ catalytic fragment, acridine orange, histone H1, β-glycerophosphate, superoxide dismutase (SOD), ATP, Protein-A-Sepharose, phosphatidylserine, and dioleoylglycerol were purchased from Sigma. Mn(III)tetrakis(4-Benzoic acid)porphyrin chloride (MnTBAP) was purchased from Oxis Health Products (Portland, OR). [γ-32P]ATP was purchased from NEN (Boston, MA). Bradford protein assay kit was purchased from Bio-Rad (Hercules, CA). Lipofectamine Plus reagent, Roswell Park Memorial Institute (RPMI)-1640 medium, horse serum, fetal bovine serum,l-glutamine, penicillin, streptomycin, and PCEP4 plasmid were purchased from Invitrogen (Gaithersburg, MD). BioPORTER, protein delivery reagent was purchased from Gene Therapy Systems (San Diego, CA), and plasmids PKCδK376-GFP fusion protein and pEGFP-N1 were kind gifts of Dr. Stuart Yuspa (National Cancer Institute, Bethesda, MD).
Cell culture. PC12 (ATCC CRL1721) cells were grown in RPMI medium supplemented with 10% horse serum, 5% fetal bovine serum, 1%l-glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml) and maintained at 37°C in a humidified atmosphere of 5% CO2. Immortalized rat mesencephalic cells (1RB3AN27) were grown in RPMI medium supplemented with 10% fetal bovine serum, 1%l-glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml), maintained at 37°C in a humidified atmosphere of 5% CO2 (Prasad et al., 1998).
Stable transfection. Plasmid pPKCδK376R-GFP encodes protein kinase Cδ-GFP fusion protein, the number K376R refers to the mutation of lysine residue at position 376 to arginine in the catalytic site of PKCδ rendering it inactive (Li et al., 1999). Plasmid pEGFP-NI encodes the green fluorescent protein alone and used as vector control. pEGFP-N1 and pPKCδK376R were transfected into 1RB3AN27 cells using Lipofectamine Plus reagent according to the procedure recommended by the manufacturer. In brief, 8 μg of DNA, 24 μl of lipid, and 24 μl of Plus reagent were used to transfect 1RB3AN27 cells in 100 mm tissue culture dishes at 50% confluency in 4 ml of culture medium without serum. Fresh medium containing serum was added 3 hr later. For stable cell lines, the 1RB3AN27 cells were selected in 400 μg/ml hygromycin, 48 hr after cotransfection with PCEP4 plasmid, which confers hygromycin resistance. Colonies were isolated with trypsin and glass cloning cylinders, and they were then replated and grown to confluence in T75 flasks. Subsequently, the stable cell lines were maintained in 200 μg/ml hygromycin.
Treatment paradigm. After 2–4 d in culture, PC12 cells and 1RB3AN27 were harvested and resuspended in serum-free growth medium at a cell density of 1–3 × 106/ml. Cell suspensions were treated with varying concentrations of MMT (30–500 μm) over a period of 0.5–5 hr at 37°C. In inhibitor studies SOD (ROS inhibitor, 100 U/ml), MnTBAP (ROS inhibitor, 10 μm), rottlerin (PKCδ inhibitor, 5–20 μm), Ac-DEVD-CHO (caspase-3-specific inhibitor, 100–300 μm), Z-DEVD-FMK (caspase-3-specific inhibitor, 10–50 μm), or Z-VAD-FMK (a broad spectrum caspase inhibitor, 30–100 μm) were added 30–90 min before the addition of MMT. The reaction samples were removed at 0.25, 0.5, 1, 2, 3, and 5 hr, then spun at 200 ×g, and after 5 min, the cell pellets were used for assessing cytochrome C release, caspase-3, caspase-8, and caspase-9 enzymatic activities, extent of PKCδ cleavage, and DNA fragmentation. Dimethylsulfoxide (DMSO) (0.5–1%) was used as a vehicle in control experiments.
Lactate dehydrogenase assay. Lactate dehydrogenase (LDH) activity in the cell-free extracellular supernatant was quantified as an index of cell death (Vassault, 1983). We modified the original method to a 96-well format (Kitazawa et al., 2001). Briefly, PC12 cells were plated in 96-well plate, and after treatment 10 μl of the extracellular supernatant was added to 200 μl of 0.08m Tris buffer, pH 7.2, containing 0.2m NaCl, 0.2 mm NADH, and 1.6 mm sodium pyruvate. LDH activity was measured continuously by monitoring the decrease in the rate of absorbance at 339 nm using a microplate reader (Molecular Devices, Sunnyvale, CA), and the temperature was maintained at 37°C during reading. Changes in absorbance per minute (ΔA/ΔT) were used to calculate LDH activity (U/I), using the following equation: U/I = (ΔA/ΔT) × 9682 × 0.66, where 9682 was a coefficient factor, and 0.66 was a correction factor at 37°C.
Detection of reactive oxygen species and lipid peroxidation by flow cytometry. Flow cytometry analysis was performed on a Becton Dickinson (San Francisco, CA) FACScan flow cytometer. Hydroethidine, a sodium borohydride-reduced derivative of ethidium bromide, is used to detect ROS produced specifically inside the cell (Narayanan et al., 1997). When hydroethidine is loaded in the cells, it binds to cellular macromolecules. Once O is generated, it converts hydroethidine to ethidium bromide and increases red fluorescence (620 nm). A 15 mW air-cooled argon–ion laser was used as an excitation source for hydroethidine at 488 nm, and the optical filter was 585/42 nm bandpass. Cells were detected and distinguished from the background by forward-angle light scattering and orthogonal light scattering characteristics. All the flow cytometric data were analyzed by Cellquest data analysis software to determine the significant increase or decrease of fluorescence intensity.
PC12 cells and engineered 1RB3AN27 cells expressing kinase inactive PKCδ protein were resuspended with HBSS with 2 mm calcium at a density of 0.5 × 106 cells/ml. Cells were then incubated with 10 μm hydroethidine for 15 min at 37°C in the dark to allow dye loading into the cells. After incubation with dye, excess dye was removed, and the cells were resuspended with HBSS. After addition of MMT (30–500 μm) ROS generation was measured at 0, 5, 15, 30, and 45 min after the exposure. In inhibitor studies, cells were incubated with SOD (100 U/ml) and MnTBAP (10 μm) 10–30 min before MMT exposure.
Quantification of cytochrome C release. Cytochrome C release was quantified using a recently developed ELISA kit developed by MBL (Watertown, MA). This is a fast, highly sensitive and reliable assay for the detection of early changes in cytochrome C levels. Briefly, after 2–4 d in culture, PC12 cells were harvested and resuspended in serum-free growth medium at a cell density of 5 × 106/ml. Cell suspensions were exposed to 200 and 500 μm MMT for 15–30 min at 37°C. After treatment the cells were spun at 200 × g, and after 5 min, washed once with 1× ice-cold PBS and resuspended in 1 ml of ice-cold homogenization buffer (10 mm Tris HCl, pH 7.5, 0.3 m sucrose, 1 mm phenylmethylsulfonyl fluoride, 25 μg/ml aprotinin, and 10 μg/ml leupeptin) and homogenized on ice. Cells were then centrifuged for 10,000 × g for 60 min at 4°C. The resulting supernatants were collected as cytoplasmic fraction and used for cytochrome C release measurements. The MBL ELISA kit measures cytochrome C by one-step sandwich ELISA. The assay uses affinity-purified two polyclonal antibodies against cytochrome C. The cytoplasmic fractions were incubated with peroxidase conjugated anti-cytochrome C polyclonal antibody in the 96-well microtiter for 60 min at room temperature (RT). After washing with buffer (provided with the kit), the peroxidase substrate is mixed with the chromogen and allowed to incubate for an additional 15 min. An acid solution provided with the kit is then added to each well to terminate the enzyme reaction and to stabilize the developed color. The optical density of each well is then measured at 450 nm using a microplate reader. The concentration of cytochrome C is calibrated from a standard curve based on reference standards.
Confocal analysis of in situ caspase activity.For this study, we used CaspACE kit (Promega) to label PC12 cells. The kit uses FITC-VAD-FMK, an FITC conjugate of the cell-permeable caspase inhibitor Z-VAD-FMK, which binds to activated caspase and serves as anin situ marker for apoptosis. The experiment was performed as per the manufacturer's protocol with slight modifications. Briefly, PC12 cells were grown on laminin (5 μg/ml)-coated slides for 2–3 d in a 37°C, 5% CO2 incubator. Cells were then exposed to 200 μm MMT for 1 hr in the dark. After exposure, the cells were treated with 10 μm FITC-VAD-FMK for 20 min at 37°C. Cells were then rinsed with 1× PBS and fixed in 10% buffered formalin for 30 min at RT in the dark. After fixing, the cells were washed three times with PBS to remove formalin and then mounted with medium and coverslips, and observed under a Leica TCS-NT confocal microscope (Leica Microsystems Inc., Exton, PA).
Enzymatic assay for caspases. Caspase-3, caspase-8, and caspase-9 activities were performed as previously described byYoshimura et al. (1998). Briefly, after treatment cells were spun and the cell pellets were lysed with Tris buffer, pH 7.4 (50 mm Tris HCl, 1 mm EDTA, and 10 mm EGTA) containing 10 μm digitonin for 20 min at 37°C. Lysates were centrifuged at 900 × g for 3 min, and the resulting supernatants were incubated with specific fluorogenic caspase substrates at 37°C for 1 hr. Ac-DEVD-AMC (50 μm), Ac-IETD-AMC (50 μm), and Ac-LEHD-AMC (50 μm) were used as substrates for determining caspase-3-, caspase-8-, and caspase-9-like protease activities, respectively. Levels of cleaved (active) caspase substrate were monitored at excitation λ 380 nm and emission λ 460 nm using a fluorescence plate reader (model: Fluoroskan-11; Titertek). Caspase activities were expressed as fluorescence units per milligram of protein per hour. The protein concentrations were determined using the Bio-Rad protein assay kit.
Isolation of cytoplasmic fractions. After incubation, the PC12 cells were spun at 200 × g for 5 min. Cell pellets were then washed once with ice-cold Ca2+-free PBS saline and resuspended in 2 ml of homogenization buffer (20 mm Tris-HCl, pH 8.0, 10 mm EGTA, 2 mm EDTA, 2 mm dithiothreitol, 1 mmphenylmethylsulfonyl fluoride, 25 μg/ml aprotinin, and 10 μg/ml leupeptin). The suspensions were sonicated for 10 sec, and centrifuged at 100,000 × g for 1 hr at 4°C. The supernatants were collected as cytosolic fractions. Protein concentration of each sample was determined, and the SDS-gel electrophoresis was performed as described below.
Western blotting. Cytoplasmic fractions containing equal amounts of protein were loaded in each lane and separated on a 10–12% SDS-polyacrylamide gel. Proteins were then transferred to nitrocellulose membrane by electroblotting for 75–90 min at 100 V. Nonspecific binding sites were blocked by treating the nitrocellulose membranes with 5% nonfat dry milk powder for 2 hr before treatment with primary antibodies. The nitrocellulose membranes containing the proteins were incubated with primary antibodies for 1 hr at RT with antibody directed against PKCδ (1:2000 dilution), PKCα (1:2000 dilution), cytochrome C (1:2000 dilution), or GFP (1:1000 dilution). The primary antibody treatments were followed by treatment with secondary HRP-conjugated anti-rabbit or anti-mouse IgG (1:2000 dilution) for 1 hr at RT. Secondary antibody-bound proteins were detected using an ECL chemiluminescence kit (Amersham). To confirm equal protein loading, blots were reprobed with a β-actin antibody (1:5000 dilution). Gel photographs were taken with a gel imaging system and quantification of bands was performed using the imaging software from Scion Corp. (Frederick, MD).
Immunoprecipitation kinase assays. PKCδ enzymatic activity was assayed using an immunoprecipitation kinase assay as described byReyland et al. (1999) and Vancurova et al. (2001). Briefly, after treatment with MMT, PC12 cells were washed once with PBS and resuspended in 1 ml of PKC lysis buffer (25 mmHEPES, pH 7.5, 20 mm β-glycerophosphate, 0.1 mm sodium orthovanadate, 0.1% Triton X-100, 0.3m NaCl, 1.5 mmMgCl2, 0.2 mm EDTA, 0.5 mm DTT, 10 mm NaF, and 4 μg/ml each aprotonin and leupeptin). In inhibition experiments, cells were pretreated with 10 μm rottlerin before the addition of 200 μm MMT. The cell lysates were allowed to sit on ice for 30 min and centrifuged at 13,000 ×g for 5 min, and the supernatants were collected as cytosolic fraction. Protein concentration was determined using a Bradford assay. Cytosolic protein (0.25–0.5 mg) was immunoprecipitated overnight at 4°C using 2 μg of anti-PKCδ, anti-PKCα, anti-PKCβI, or anti-PKCβII antibodies. The immunoprecipitates were then incubated with Protein-A Sepharose (Sigma) for 1 hr at 4°C. The protein A bound antigen–antibody complexes were then washed three times with PKC lysis buffer, three times with 2× kinase buffer (40 mm Tris, pH 7.4, 20 mmMgCl2, 20 μm ATP, and 2.5 mm CaCl2), and resuspended in 20 μl of 2× kinase buffer. Reaction was started by adding 20 μl of reaction buffer containing 0.4 mg Histone H1, 50 μg/ml phosphatidylserine, 4.1 μm dioleoylglycerol, and 5 μCi of [γ-32P] ATP (3000 Ci/mm) to the immunoprecipitated samples and incubated for 10 min at 30°C. SDS gel-loading buffer (2×) was added to terminate the reaction, the samples were boiled for 5 min, and the products were separated on a 12.5% SDS-PAGE gel. For in vitro inhibition of PKCδ kinase activity, 5–20 μm rottlerin was added to 200 μm MMT-treated immunoprecipitated sample 15 min before the addition of 2× reaction buffer containing 5 μCi of [γ-32P] ATP (3000 Ci/mmol). The H1 phosphorylated bands were detected using a Personal Molecular Imager (FX model; Bio-Rad), and quantification was done using Quantity One 4.2.0 software.
Intracellular delivery of PKCδ catalytic fragment.Intracellular delivery of PKCδ fragment was performed using a recently developed lipid-mediated delivery system (BioPORTER; Gene Therapy Systems, San Diego, CA). This is a fast and reliable procedure that delivers proteins in a functionally active form into the cytoplasm of cells (Zelphati et al., 2001). The protein delivery system is composed of a new trifluoroacetylated lipopolyamine (TFA-DODAPL) and dioleoyl phosphatidylethanolamine. This cationic formulation has recently been used for delivery of various bioactive molecules, including antibodies, enzymes (caspase-3, caspase-8, β-galactosidase, and granzyme B), cytochrome C, dextran sulfates, phycobiliproteins, and albumins into the cytoplasm of numerous adherent and suspension cells (Zelphati et al., 2001). Active PKCδ catalytic fragments were delivered into cells using the protein delivery reagent by following the manufacturer's protocol. PC12 cells (∼1–2 × 105 cells/ml) were subcultured in 24-well tissue culture plate for 24 hr. PKCδ catalytic fragment (5 ng) was mixed with 3 μl of protein delivery reagent and 300 μl of serum-free DMEM media and added to each well. The cells were incubated at 37°C for 4 hr. Cells were then lysed, and caspase-3 activity measured as described above. Heat-inactivated PKCδ catalytic fragment was used as negative control and inactivation was performed by incubating the active PKCδ fragment at 95°C for 15 min. The delivery efficiency was ∼70% in PC12 cells as determined using a FITC-tagged antibody control (supplied with the assay kit). Also, the protein delivery system produced no significant cytotoxic response as measured by Trypan blue dye exclusion method.
In situ assessment of apoptosis. To assess nuclear morphology and DNA damage, we stained the cells with fluorescent DNA-binding dyes acridine orange and Hoechst 33342. Acridine orange, a useful probe for detecting apoptotic cells, exhibits metachromatic fluorescence that is sensitive to DNA conformation. Apoptotic cells stained with acridine orange show reduced green and enhanced red fluorescence in comparison with normal cells (Pulliam et al., 1998). Briefly, PC12 cells were grown on laminin (5 μg/ml)-coated slides for 2–3 d in a 37°C, 5% CO2 incubator. Cells were washed twice with PBS and after 1 hr treatment with 200 μm MMT, the cells were incubated with 10 μm acridine orange for 15 min at RT in the dark. The cells were again washed with PBS, mounted with coverslips, and observed under a Nikon DiaPhot microscope, and pictures were captured with a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI).
Morphological changes associated with apoptosis were also assessed by staining with Hoechst 33342. Cells stained with Hoechst 33342 dye fluoresce bright blue after binding to DNA in the nucleus. The nucleus of apoptotic cells exhibit strong blue staining and staining pattern is heterogeneous and occurs in patches, indicative of chromatin condensation, whereas the nucleus of nonapoptotic cells exhibit more diffused, weak and homogenous staining (Shimizu et al., 1996; Du et al., 1997). Briefly, PC12 cells were plated on collagen (6 μg/cm2)-coated cover slides and treated with 200 μm MMT. After 1 hr of exposure, the cells were fixed with 10% buffered formaldehyde for 30 min at room temperature and stained with Hoechst 33342 (10 μg/ml) for 3 min in dark. The cells were again washed three times with PBS, mounted with coverslips, and observed under a Nikon DiaPhot microscope under UV illumination, and pictures were captured with a SPOT digital camera (Diagnostic Instruments).
Quantification assay for DNA fragmentation. DNA fragmentation assay was performed using a recently developed Cell Death Detection ELISA Plus assay kit. This is a fast, highly sensitive and reliable assay for the detection of early changes in apoptotic cell death and measures the appearance and amount of histone-associated low molecular weight DNA in the cytoplasm of cells. This assay has been recently used in quantitation of apoptosis because of its reliability and high sensitivity (Reyland et al., 1999). Briefly, PC12 and engineered 1RB3AN27 cells were exposed to 200–500 μm MMT for 1–3 hr. In inhibitor studies, SOD (100 U/ml), MnTBAP (10 μm), rottlerin (10 μm), Z-DEVD-FMK (50 μm), or Z-VAD-FMK (100 μm) were treated for 30 min at 37°C before the addition of MMT. After MMT treatment, cells were spun down at 200 × g for 5 min and washed once with 1× PBS. Cells were then incubated with a lysis buffer (supplied with the kit) at RT. After 30 min, samples were centrifuged, and 20 μl aliquots of the supernatant were then dispensed into streptavidin-coated 96-well microtiter plates followed by addition of 80 μl of antibody cocktail and incubated for 2 hr at RT with mild shaking. The antibody cocktail consisted of a mixture of anti-histone biotin and anti-DNA-HRP directed against various histones and antibodies to both single-stranded DNA and dsDNA, which are major constituents of the nucleosomes. After incubation, unbound components were removed by washing with the incubation buffer supplied with the kit. Quantitative determination of the amount of nucleosomes retained by anti-DNA-HRP in the immunocomplex was determined spectrophotometrically with 2,2′-azino-di-(3-ethylbenzthiazoline sulfonate (6)) diammonium salt (ABTS) as an HRP substrate (supplied with the kit). Measurements were made at 405 nm against an ABTS solution as a blank (reference wavelength ∼490 nm) using a Molecular Devices Spectramax Microplate Reader.
Data analysis. Data analysis was performed using Prism 3.0 software (GraphPad Software, San Diego, CA). Data from caspase enzymatic activities and DNA fragmentation assays were first analyzed using one-way ANOVA. Neuman–Keuls or Dunnett's post-tests were then performed to compare control with MMT-treated groups, and differences with p < 0.05 were considered significant. For individual comparisons, student t test or Welch's correctedt test where appropriate was used.
RESULTS
MMT-induced cytotoxicity
PC12 cells were exposed to 200 and 500 μm MMT for varying amounts of time. The amount of LDH released into the extracellular media was measured as an index of cytotoxicity (Vassault, 1983; Kanthasamy et al., 1995). Extracellular LDH activity showed a dose- and time-dependent increase with MMT treatment ranging from 2- to 20-fold over the control group (Fig. 1). For example, exposure to 200 μm MMT resulted in 3-, 10-, and 17-fold increase in LDH release over untreated cells at 1, 3, and 5 hr, respectively. To determine the percent cell death, total LDH content of untreated cells (∼2000 U/I) was normalized to 100%. PC12 cells exposed to 200 μm MMT for 3 hr produced ∼50% cell death. Similarly, exposure to 500 μm MMT resulted in a significant (p < 0.05) toxicity in MMT-treated groups as compared with vehicle-treated cells at all time points. There were no significant differences in LDH release between vehicle-treated and untreated PC12 cells during the 5 hr exposure. These data suggest that MMT induces a dose- and time-dependent cell death in dopamine-producing cells.
Fig. 1.

MMT exposure induces cell death. PC12 cells were exposed to 200 and 500 μm of MMT for 0.5–5 hr at 37°C. After the exposure, cell-free extracellular supernatants were collected, and LDH activity was measured by spectrophotometer. Values represent mean ± SEM for three to five separate experiments in triplicate. Significance was determined by ANOVA followed by Dunnett's post-test between the vehicle-treated group and each treatment group (*p < 0.05).
Generation of ROS after MMT treatment
Flow cytometric analysis using the ROS-sensitive fluorescence probe hydroethidine revealed that MMT treatment induces ROS generation. Figure 2A depicts a representative flow cytometric histogram of 200 μm MMT-treated PC12 cells exhibiting time-dependent increases in red fluorescence. MMT treatment increased ROS production in a dose- and time-dependent manner (Fig.2B). For example, a 15 min exposure to 30, 100, and 200 μm MMT resulted in a 10, 41, and 76% increase in ROS production, respectively. Exposure to 200 μm MMT resulted in 40, 76, and 80% increase in ROS production over vehicle treatment at 5, 15, and 30 min, respectively. The time course study also revealed that 500 μm MMT treatment induced a rapid and dramatic increase in ROS generation (>200% of control) within 5 min and then the response rapidly declined over time (data not shown). Pretreatment with SOD (100 U/ml) or MnTBAP (SOD mimetic, 10 μm) significantly (p < 0.05) reduced MMT-induced ROS production, indicating that MMT predominantly generates superoxide species (Fig. 2C).
Fig. 2.
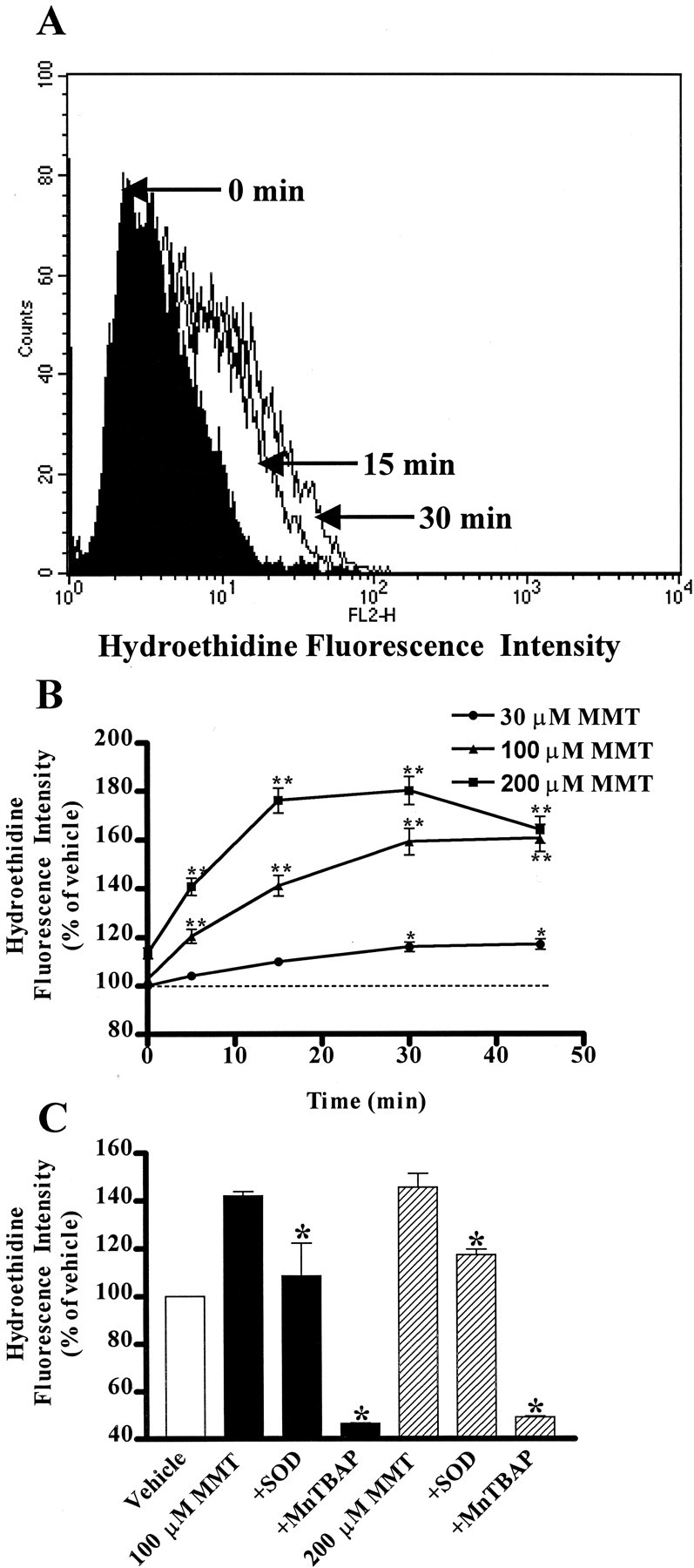
MMT treatment generates ROS in PC12 cells. PC12 cells were suspended in HBSS supplemented with 2 mmCa2+ at a density of 0.5–0.75 × 106 cells/ml. A concentration of 10 μmhydroethidine was added to the cells and incubated for 15 min at 37°C in the dark. A, Time-dependent change in hydroethidine fluorescent intensity in PC12 cells treated with MMT. A concentration of 200 μm MMT was added, and fluorescent intensity was measured at 0, 15, and 30 min by flow cytometry as described in Materials and Methods. The data are a representative flow cytometric histogram of MMT-treated PC12 cells exhibiting a time-dependent increase in red fluorescence. B, Dose- and time-dependent increase in ROS production. Various doses of MMT were added, and fluorescent intensity was measured at 0, 5, 15, and 30 min. Data represent the mean ± SEM of two to five separate experiments in triplicate. Asterisks (*p < 0.5 and **p < 0.01) indicate significant differences compared with the time-matched vehicle-treated cells. C, Effect of SOD and MnTBAP on ROS production. Cells were pretreated with ROS inhibitors, SOD (100 U/ml) and MnTBAP (10 μm), and then exposed to 100 or 200 μm MMT for 15 min. The value of each treatment group is the mean ± SEM from two to three separate experiments performed in triplicate. Asterisks (*p < 0.05) indicate significant differences compared with MMT-treated cells.
Accumulation of cytochrome C in the cytosol after MMT treatment
ROS production in the cells is known to activate many cellular factors including cytochrome C, which subsequently triggers apoptotic cell death (Tan et al., 1998; Cassarino et al., 1999). Release of cytochrome C from the mitochondria into the cytoplasm is an early event that occurs during programmed cell death (Muller-Hocker, 1992;Crompton, 1999), and therefore we determined whether MMT induces release of cytochrome C in PC12 cells. Figure3A shows a time-dependent increase of cytochrome C in the cytoplasmic fractions of PC12 cells treated with MMT. No detectable levels of cytochrome C were detected in the cytosol of vehicle (DMSO)-treated cells up to 3 hr, whereas a profound release of cytochrome C was observed as early as 1 hr in MMT-treated cells. Nitrocellulose membranes were reprobed with β-actin antibody, and the density of 43 kDa β-actin bands was identical in all lanes confirming equal protein loading. To further accurately quantify how soon cytochrome C is released, we used a highly sensitive cytochrome C ELISA sandwich assay. MMT exposure resulted in a dose-dependent increase in cytosolic cytochrome C as early as 15 min (Fig. 3B). A 15 min exposure to 200 and 500 μm MMT resulted in an increase in cytosolic cytochrome C by 40 and 200% over the vehicle-treated group, respectively, and a 30 min exposure resulted in an increase in cytosolic cytochrome C by 70 and 170%, respectively. The reason for the decrease in amount of cytochrome C released at 30 min after 500 μm MMT exposure might be attributed to loss of cellular integrity caused by the observed necrotic cell death (Fig.1).
Fig. 3.

Dose- and time-dependent accumulation of cytosolic cytochrome C in MMT-treated PC12 cells. A, Western blot.B, Cytochrome C ELISA assay. A, Subconfluent cultures of undifferentiated PC12 cells were harvested at 1 and 3 hr after treatment with 200 or 500 μm MMT. The cytosolic fractions were obtained as described in Materials and Methods. Cytosolic fractions were separated by 12% SDS-PAGE, transferred to a nitrocellulose membrane, and cytochrome C (Cyt C) was detected using polyclonal antibody raised against cytochrome C. For β-actin measurements, the membrane used for cytochrome C was reprobed with β-actin antibody to confirm equal protein loading in each lane. The immunoblots were visualized using ECL detection agents from Amersham. B, Subconfluent cultures of undifferentiated PC12 cells were harvested at 15 and 30 min after treatment with 200 or 500 μm MMT. The cytosolic fractions were obtained as described in Materials and Methods. The value of each treatment group is the mean ± SEM from two separate experiments in triplicate. Asterisks (*p < 0.05) indicate significant differences compared with vehicle-treated cells.
Activation of caspase-3 and caspase-9 but not caspase-8 after MMT treatment
Because the release of cytochrome C is known to activate a group of cysteine proteases, namely caspases (Cohen, 1997; Earnshaw et al., 1999; Schultz and Andreasen, 1999; Jellinger, 2000), we examined whether caspase-8, caspase-9, and caspase-3 are activated during MMT exposure. PC12 cells exposed to MMT showed a significant increase in caspase-9 activity, however, no significant increase in caspase-8 enzyme activity was observed (data not shown). A 30 min exposure to 200 and 500 μm MMT produced a threefold and twofold increase in caspase-9 activity, respectively. The lack of dose–response in caspase-9 enzymatic activity at 500 μmMMT concentration is probably caused by acute cytotoxic effects of the toxic compound at higher doses.
MMT treatment in PC12 cells resulted in dramatic increase in caspase-3 enzymatic activity (Fig. 4). After exposure to 200 μm MMT, caspase-3-specific activity was increased 12-, 17-, 7-, and 6-fold over the vehicle-treated groups at 0.5, 1, 2, and 3 hr after treatment, respectively (Fig.4A). Similarly, exposure to 500 μm MMT resulted in an increase in caspase-3 specific activity by 20-, 27-, 20-, and 6-fold over the vehicle-treated groups after 0.5, 1, 2, and 3 hr exposure, respectively. The overall pattern of the time course study of caspase-3 activity revealed a clear pattern of dramatic increase in enzyme activity peaking at 1 hr and then progressively decreasing over time, returning nearly to that of vehicle-treated cells at 3 hr. To further confirm the activation of caspase-3, in situ fluorometric analysis was performed using FITC-VAD-FMK in live cells. In these experiments, we found majority of PC12 cells were labeled within 1 hr of exposure to 200 μm MMT, indicating a profound increase in caspase-3 activity in situ, whereas no labeling was seen in vehicle-treated cells (Fig. 4B).
Fig. 4.
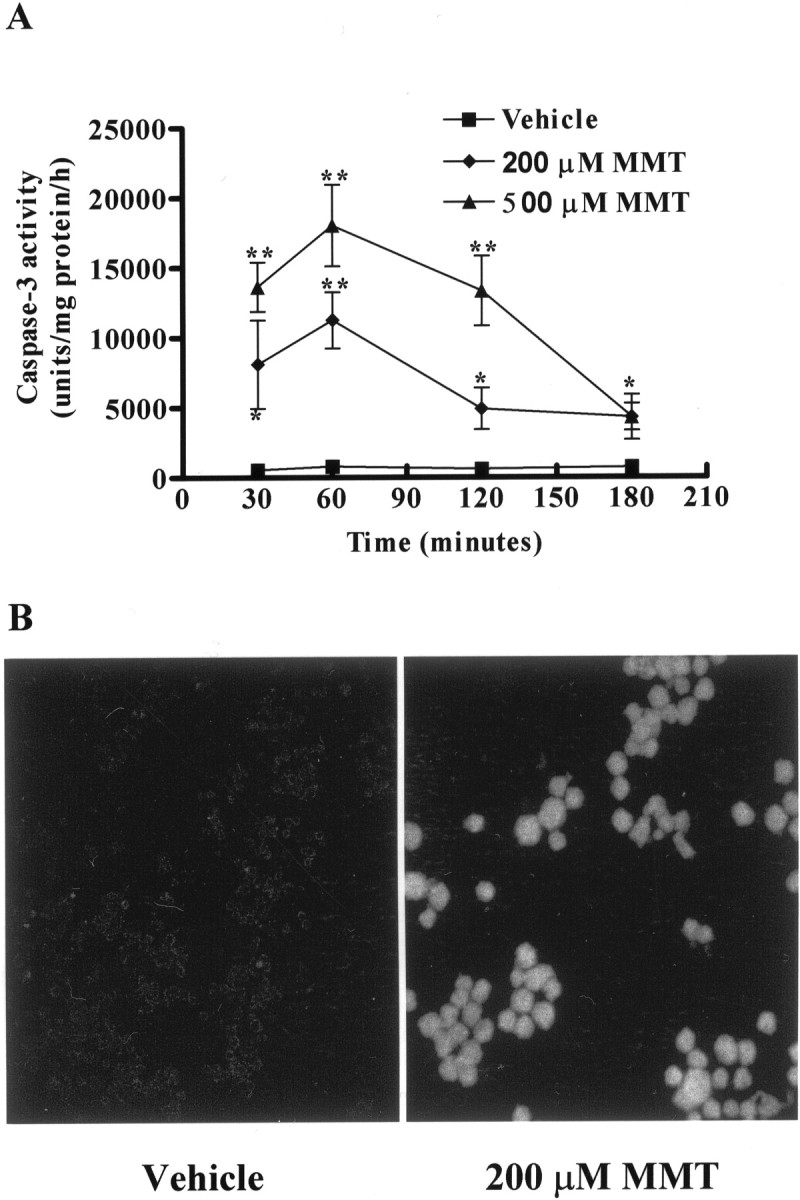
MMT treatment increases caspase-3 activity.A, Caspase-3 enzymatic activity. B, In situ caspase-3 activity. A, Subconfluent cultures of undifferentiated PC12 cells were harvested at 30 min, 1, 2, and 3 hr after MMT treatment. Caspase-3 activity was assayed using specific fluorogenic substrate, Ac-DEVD-AMC (50 μm), as described in Materials and Methods. The data represent mean ± SEM of nine individual measurements from three separate experiments. Asterisks (**p < 0.01; *p < 0.05) indicate significant differences compared with temporally matched vehicle (DMSO)-treated cells. B, PC12 cells were grown on laminin-coated slides for 2–3 d and then exposed to 0.5% DMS0 (vehicle) and 200 μm MMT for 1 hr in the dark. After exposure, cells were treated with 10 μm FITC-VAD-FMK (Promega caspACE, in situ marker for caspase-3 activity) and processed as described in Materials and Methods. Confocal images were obtained using a Leica TCS-NT microscope.
Proteolytic cleavage of protein kinase Cδ but not PKCα by MMT
Recent studies have indicated PKCδ to be one of the endogenous substrates for caspase-3, which cleaves the kinase to yield a 41 kDa catalytically active and a 38 kDa regulatory PKCδ fragments in non-neuronal cell lines, salivary gland acinar cells, (Reyland et al., 1999), rat fibroblasts (Dal Pra et al., 1999), and neutrophils (Pongracz et al., 1999). Because MMT exposure resulted in a profound activation of caspase-3, we decided to examine the proteolytic cleavage of PKCδ in MMT-treated PC12 cells. After treatment of PC12 cells with MMT at 37°C, we observed over a 5 hr period a significant proportion of native PKCδ (72–74 kDa) protein was proteolytically cleaved to yield 38 kDa regulatory and 41 kDa catalytically active fragments when immunoblotted with an antibody raised against PKCδ (Fig.5A). The time course study revealed that almost all of the native PKCδ (72–74 kDa) protein was cleaved within 3 hr of incubation with MMT, evidenced by a reduction in the intensity of the native 72–74 kDa band and a concomitant increase in the catalytically active 41 kDa cleaved fragment. PC12 cells exposed to increasing concentrations of MMT (200 and 500 μm) showed a dose-dependent cleavage of PKCδ. However, no cleavage of PKCδ was observed in vehicle-treated cells during the entire 5 hr experimental time period at the doses tested. Nitrocellulose membranes were reprobed with β-actin antibody, and the density of 43 kDa β-actin band was identical in all lanes confirming equal protein loading.
Fig. 5.

Proteolytic cleavage of PKCδ but not of PKCα in MMT-treated PC12 cells. A, PKCδ; B,PKCα. Subconfluent undifferentiated PC12 cells were harvested at 1, 3, and 5 hr after treatment of 200 or 500 μm MMT. Cytosolic fractions were obtained as described in Materials and Methods, and were separated by 10% SDS-PAGE, transferred to nitrocellulose membrane, and PKCα and PKCδ were detected using antibodies directed against their catalytic subunits. To confirm equal protein loading in each lane, the membranes were reprobed with β-actin antibody. The immunoblots were visualized using ECL detection agents from Amersham. C, PKCδ catalytic subunit;R, PKCδ regulatory subunit.
MMT-induced proteolytic cleavage of PKC was also isoform-specific, because exposure of PC12 cells to 200 or 500 μm MMT for up to 5 hr failed to induce proteolytic cleavage of PKCα (Fig.5B). Membranes were reprobed with β-actin antibody, and the density of 43 kDa β-actin band was identical in all lanes confirming equal protein loading. Additionally, MMT exposure did not result in the translocation of either PKCα or δ from the cytoplasm to the membrane for their activation (data not shown).
MMT-induced proteolytic cleavage of PKCδ is caspase-3-dependent
To further confirm that PKCδ cleavage is mediated by caspase-3, we used caspase-3-specific inhibitors Ac-DEVD-CHO (Fig.6A), Z-DEVD-FMK (Fig.6B) or a broad-spectrum caspase inhibitor, Z-VAD-FMK (Fig. 6A), to block the cleavage. Pretreatment of PC12 cells for 30 min with any of the three inhibitors used here before 3 hr exposure of cells to 200 μm MMT prevented the appearance of the 41 kDa catalytically active PKCδ fragment, and effects of all three inhibitors were dose-dependent. Furthermore, Z-DEVD-FMK and Z-VAD-FMK appeared to be more potent in blocking PKCδ cleavage than Ac-DEVD-CHO. Membranes were reprobed with β-actin antibody (Fig. 6A,B), and the density of 43 kDa β-actin bands was identical in all lanes confirming equal protein loading.
Fig. 6.

Caspase-3 mediates the proteolytic cleavage of PKCδ in MMT-treated PC12 cells. A, Effect of Ac-DEVD-CHO and Z-VAD-FMK on PKCδ cleavage. B, Effect of Z-DEVD-FMK on PKCδ cleavage. Subconfluent undifferentiated PC12 cells were treated with 200 μm MMT, with or without the inclusion of caspase inhibitors Ac-DEVD-CHO, Z-VAD-FMK, or Z-DEVD-FMK. Inhibitors were added 30 min before the addition of MMT. Cells were harvested 3 hr after the addition of MMT. The cytosolic fractions were obtained as described in Materials and Methods, and were analyzed by 10% SDS-PAGE and Western blot. To confirm equal protein loading in each lane, the membranes were reprobed with β-actin antibody.C, PKCδ catalytic subunit; R, PKCδ regulatory subunit.
Rottlerin blocks MMT-induced caspase-3 enzymatic activity: possible feedback activation of caspase-3 by PKCδ
As reported above, we observed a dramatic increase in caspase-3 (6- to 27-fold) (Fig. 4A) enzymatic activity in MMT-treated PC12 cells at 1 hr after treatment. These results prompted us to determine the cause for the dramatic increase in MMT-induced caspase-3 activity, and so we investigated whether PKCδ is capable of activating caspase-3 by a positive feedback mechanism. To address this hypothesis, we tested the ability of PKCδ specific inhibitor rottlerin to modulate caspase-3 activity in MMT-treated PC12 cells by pre- and post-treatment. Pretreatment with rottlerin 30 min before the addition of 200 μm MMT suppressed caspase-3 activity in a dose-dependent manner (Fig.7A). Rottlerin at 5, 10, and 20 μm suppressed MMT-induced caspase-3 activity by 38, 64, and 70%, respectively, whereas the basal caspase-3 activity was not altered by treatment with rottlerin alone. Post-treatment with rottlerin 30 min after the addition of 200 μmMMT also suppressed caspase-3 activity in a dose-dependent manner (Fig.7B). Rottlerin at 5 and 20 μmsuppressed MMT-induced caspase-3 activity by 60 and 98%, respectively, whereas the basal caspase-3 activity was unaltered by post-treatment with rottlerin alone. The extent of MMT-induced caspase-3 inhibition by 5 μm rottlerin was not statistically significant (p > 0.05) between pre- and post-treatments, whereas inhibition with 20 μmrottlerin post-treatment was very significant (p< 0.01) as compared with pre-treatment, reducing the caspase-3 activity to almost the basal level. Overall, these results indicate that there may be a positive feedback activation of caspase-3 by PKCδ, and this activation can be blocked by rottlerin.
Fig. 7.

Suppression of caspase-3 activity by rottlerin in MMT-treated PC12 cells. A, Pre-treatment;B, post-treatment. Subconfluent undifferentiated PC12 cells were treated with 200 μm MMT with or without the inclusion of rottlerin (Rot; 5–20 μm) for 1 hr. Rottlerin was added 30 min before or 30 min after the addition of MMT. Caspase-3 activity was assayed using Ac-DEVD-AMC (50 μm) as substrate, as described in Materials and Methods. The data represent an average of four to nine individual measurements from two or three separate experiments ± SEM. Asterisk (*p < 0.05) indicates significant difference compared with cells exposed to 200 μm MMT.
Rottlerin blocks caspase-3 mediated proteolytic cleavage of PKCδ
Because the pretreatment study with rottlerin blocked caspase-3 enzymatic activity, we further tested whether rottlerin pretreatment attenuates caspase-3-dependent proteolytic cleavage of PKCδ. Pretreatment with rottlerin before the addition of 200 μmMMT prevented the accumulation of PKCδ cleavage product in a dose-dependent manner (Fig. 8). Rottlerin at 20 μm markedly reduced the appearance of PKCδ cleavage product in PC12 cells exposed to 200 μm MMT, indicating that the activation of PKCδ is essential to its cleavage by caspase-3. Membranes were reprobed with β-actin antibody (Fig. 8), and the density of 43 kDa β-actin band was identical in all lanes, confirming equal protein loading.
Fig. 8.

Rottlerin pretreatment blocks proteolytic cleavage of PKCδ in MMT-treated PC12 cells. Subconfluent undifferentiated PC12 cells were treated with 200 μm MMT with or without the inclusion of rottlerin (5–20 μm). Rottlerin was added 90 min before the addition of MMT. Cytosolic fractions were obtained as described in Materials and Methods and were separated by 10% SDS-PAGE, transferred to nitrocellulose membrane, and PKCδ was detected using an antibody directed against its catalytic subunit. The immunoblots were visualized using ECL detection agents from Amersham. To confirm equal protein loading in each lane, the membranes were reprobed with β-actin antibody. C, PKCδ catalytic subunit;R, PKCδ regulatory subunit.
Rottlerin inhibits MMT-induced increases in PKCδ kinase activity in PC12 cells
To determine whether the MMT induced caspase-3- and PKCδ-dependent accumulation of PKCδ cleaved product is attributed to an increase in PKCδ enzyme activity, we performed kinase assays in immunoprecipitated samples from cytosolic fractions using PKCδ specific polyclonal antibody and by examining the ability of PKCδ to phosphorylate histone H1. The enzymatic activity of PKCδ increased after 1 hr exposure to MMT in dose-dependent manner (Fig.9A). Densitometric analysis of phosphorylated histone H1 bands revealed a three-fold and five-fold increase in protein kinase activity in cells exposed to 200 and 500 μm MMT for 1 hr, respectively, and was coincident with generation of PKCδ cleavage. We attribute this increased kinase activity to the persistently active PKCδ catalytic fragment, because activation of intact PKCδ by translocation to the membrane does not occur during MMT treatment (data not shown). There was no increase in kinase activity of PKCα, PKCβI, and PKCβII in immunoprecipitated samples of treated cells (data not shown) suggesting that the MMT-induced increase in kinase activity is isoform specific for PKCδ. Pretreatment with 10 μm rottlerin resulted in 80% reduction in the kinase activity (Fig. 9B), suggesting that the activation of PKCδ is essential for MMT-induced increases in kinase activity, and this may be facilitated via the positive-feedback activation of caspase-3 by the catalytically active PKCδ fragment.
Fig. 9.
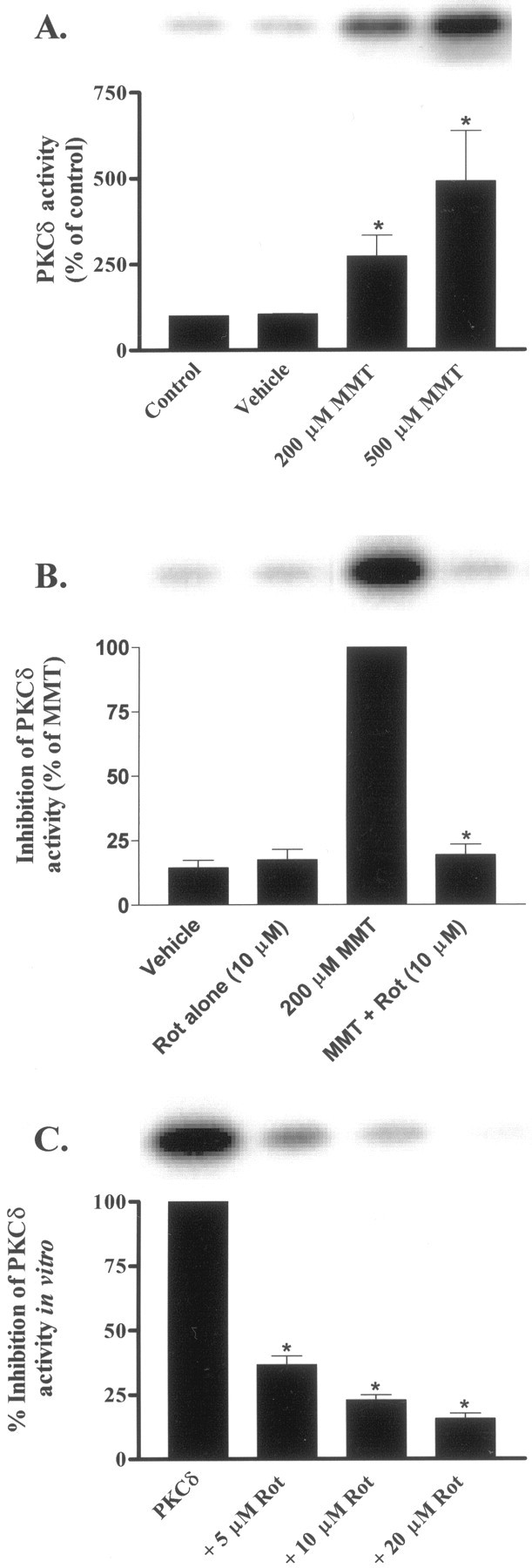
Rottlerin inhibits PKCδ kinase activity in intact cells and in in vitro. A, Dose-dependent increase in PKCδ activity. B, Rottlerin suppresses MMT-induced increase in PKCδ kinase activity in intact cells. C, Rottlerin inhibits PKCδ kinase activityin vitro. Subconfluent undifferentiated PC12 cells were treated with 200 μm MMT for 1 hr at 37°C with or without the inclusion of rottlerin (Rot; 5–20 μm). Rottlerin was added 30 min before the addition of MMT. For in vitro inhibition of PKCδ activity, rottlerin (5–20 μm) was added to the immunoprecipitated samples from MMT-treated cells and incubated for 30 min before the addition of substrate (histone H1) and [γ-32P]ATP. The immunoprecipitation kinase assay was performed as described in Materials and Methods. The bands were quantified by a PhosphoImager after scanning the dried gel and expressed as a percentage of control (untreated cells) (A), percentage of MMT treatment (B), or percentage of PKCδ kinase activity (C). The data represent an average of three individual measurements from two separate experiments ± SEM. Asterisks (*p < 0.05) indicate significant differences compared with control, MMT-treated cells, or PKCδ kinase activity.
Rottlerin directly inhibits PKCδ kinase activityin vitro
Rottlerin was originally reported to inhibit PKCδ kinase activity by competing for the ATP-binding site (Gschwendt et al., 1994). This inhibitor has been used to implicate PKCδ in a variety of cellular events, including apoptosis (Chen et al., 1999; Reyland et al., 1999; Dempsey et al., 2000; Way et al., 2000; Basu et al., 2001;Vancurova et al., 2001). To further confirm the inhibitory potency of rottlerin on PKCδ activity, we tested various concentrations of rottlerin on PKCδ enzyme activity using an in vitro kinase assay. PKCδ was immunoprecipitated from MMT-treated cytosolic fractions using PKCδ specific polyclonal antibody and incubated with rottlerin in vitro for 15 min before the addition of histone H1 and [32P]ATP. For the in vitro reaction, we used same rottlerin concentrations that blocked MMT-stimulated PKCδ kinase activity in intact PC12 cells. Rottlerin at 5, 10, and 20 μm inhibited PKCδ activityin vitro by 63, 77, and 84%, respectively (Fig.9C), and is consistent with rottlerin inhibition of MMT-induced PKCδ activity in intact PC12 cells (Fig. 9B). Our data are also in agreement with previously published values for direct inhibition of PKCδ by rottlerin in vitro kinase assays (Gschwendt et al., 1994; Way et al., 2000; Vancurova et al., 2001).
Activation of caspase-3 after intracellular delivery of PKCδ catalytic fragment
To further confirm the existence of a positive feedback loop between caspase-3 and proteolytic cleavage PKCδ, we investigated the effect of intracellular delivery of PKCδ catalytic fragment on caspase-3 activity in PC12 cells. We used a recently developed lipid-mediated delivery system to introduce the catalytically active PKCδ fragment into the cytoplasm of PC12 cells (Zelphati et al., 2001). The cells were treated with the delivery reagent with or without the PKCδ catalytic fragment for 4 hr at 37°C. As shown in Table1, PC12 cells delivered with PKCδ catalytic fragment showed increases in caspase-3 activity to 341% of reagent control. Neither the intracellular delivery of heat-inactivated PKCδ catalytic fragment nor the delivery reagent alone produced any increases in caspase-3 enzymatic activity. These results strongly suggest that catalytic fragment of PKCδ is capable of mediating caspase-3 activation, further supporting our hypothesis that proteolytic cleavage of PKCδ can augment caspase-3 activity by a positive feedback loop during MMT treatment.
Table 1.
Activation of caspase-3 after intracellular delivery of PKCδ catalytic fragment in PC12 cells
| Treatment | Caspase-3 activity (fluorescence units/mg protein/hr) | % Reagent control |
|---|---|---|
| Reagent control | 25,293 ± 5726 | 100 |
| Catalytic active PKCδ fragment | 86,296 ± 34,116 | 341 ± 135* |
| Heat inactivated PKCδ catalytic fragment | 29,814 ± 6851 | 118 ± 27 |
Asterisk indicates significant difference (p < 0.05) compared with reagent control. The data are given as the mean ± SEM from two separate experiments performed in triplicate.
In situ fluorometric detection of apoptosis
To understand the functional consequence of the activation of many apoptotic factors, we tested whether MMT induces DNA fragmentation. Chromosomal breakdown of DNA into 200 bp nucleosomal fragments and DNA condensation are hallmarks of cells undergoing apoptosis. We usedin situ fluorometric analysis to identify apoptotic cells using acridine orange and Hoechst 33342 to detect nuclear condensation and DNA damage after MMT treatment. In these experiments, we found the majority of PC12 cells exposed to 200 μm MMT for 1 hr showed enhanced red fluorescence and reduced green fluorescence, suggesting that acridine orange dye is bound to single-stranded or highly condensed DNA (Fig.10B), whereas little or no enhanced red fluorescence was seen in vehicle-treated cells (Fig.10A). Similarly, the nucleus of PC12 cells exposed to 200 μm MMT for 1 hr and subsequently stained with Hoechst 33342 dye showed nuclear condensation as the dye bound to the highly condensed DNA (Fig. 10D). The Hoechst 33342 staining of vehicle-treated cells showed a weak and diffused staining, indicating that there is no nuclear condensation in these cells (Fig. 10C).
Fig. 10.
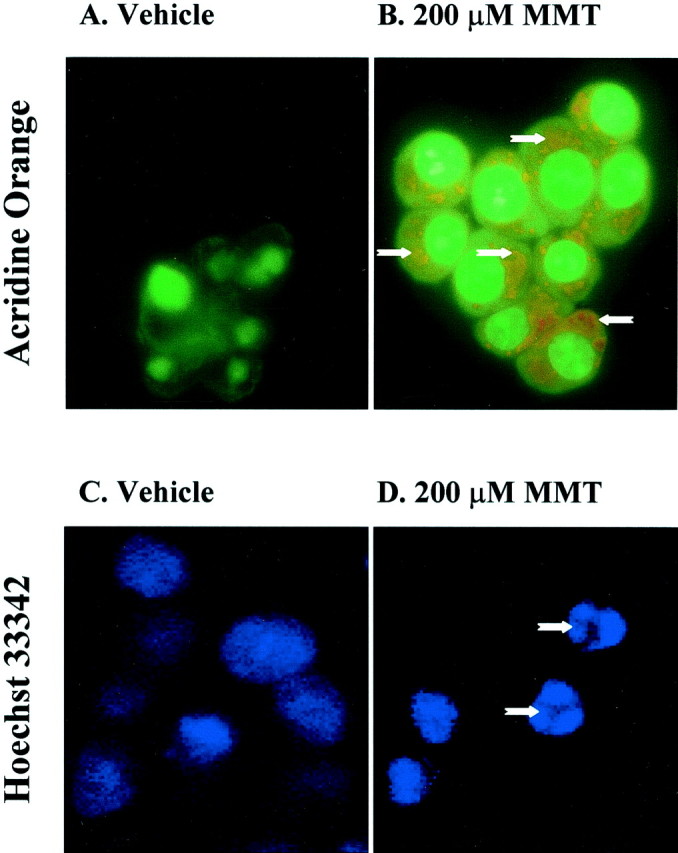
MMT treatment increases apoptosis in situ. A, C, Vehicle-treated cells; B, D, 200 μm MMT-treated cells. PC12 cells were grown on laminin-coated slides for 2–3 d and then exposed to 200 μm MMT for 1 hr. A, B, For acridine orange staining, cells were treated with acridine orange (10 μm) for 15 min in the dark at RT after exposure to MMT.Arrows indicate enhanced red fluorescence and reduced green fluorescence in MMT-treated cells, which are undergoing apoptosis, whereas little or no enhanced redfluorescence was seen in vehicle-treated cells. C, D,For Hoechst 33342 staining, cells were stained with Hoechst 33342 (10 μg/ml) for 3 min in dark after exposure to MMT. Arrowsindicate apoptotic cells containing condensed chromatin.
Oxidative stress, caspase-3, and PKCδ mediate MMT-induced DNA fragmentation
To further confirm the results obtained by in situfluorometric detection of live apoptosis and to assess the involvement of caspases and PKCδ in mediating apoptosis, a quantitative DNA fragmentation assay was performed. PC12 cells treated with 200 μm MMT showed DNA fragmentation within 1 hr of exposure (Fig. 11). MMT treatment resulted in more than a twofold increase over the levels of basal (vehicle-treated) DNA fragmentation. Pretreatment with an ROS inhibitor, SOD (100 U/ml), almost completely blocked MMT-induced DNA fragmentation (Fig. 11A), indicating that SOD is capable of blocking MMT-induced apoptosis. To further confirm the anti-apoptotic effect of SOD in MMT treatment, a cell-permeable SOD mimetic, MnTBAP, was used. Pretreatment with 10 μm MnTBAP also almost completely attenuated MMT-induced apoptosis (Fig. 11A). SOD, but not MnTBAP, when treated alone also significantly attenuated the basal apoptosis in vehicle-treated PC12 cells.
Fig. 11.
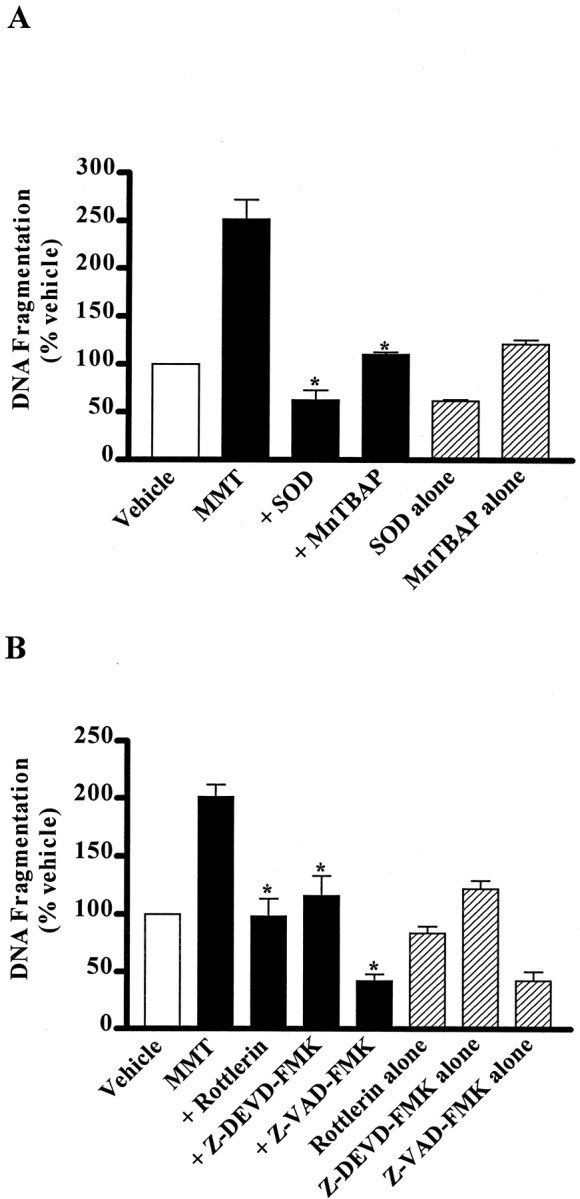
Suppression of MMT-induced apoptosis in PC12 cells. A, ROS inhibitors, SOD and MnTBAP.B, Caspase-3 inhibitors, Z-VAD-FMK and Z-DEVD-FMK and PKCδ inhibitor, rottlerin. Subconfluent cultures of undifferentiated PC12 cells were treated with MMT (200 μm) with or without the inclusion of the following inhibitors: ROS inhibitors SOD (100 U/ml) or MnTBAP (10 μm); caspase inhibitors Z-VAD-FMK (100 μm) or Z-DEVD-FMK (50 μm); and PKCδ inhibitor rottlerin (10 μm). Inhibitors were added 30 min before addition of MMT. Cells were harvested 1 hr after MMT treatment. Apoptosis was assayed using ELISA assay as described in Materials and Methods. The data are expressed as percentage of apoptosis observed in vehicle-treated cells. The data represent the mean ± SEM of six individual measurements from three separate experiments. Asterisks (*p < 0.01) indicate significant differences when compared with cells exposed to 200 μm MMT.
Pretreatment for 30 min with PKCδ inhibitor rottlerin (10 μm) completely prevented 200 μm MMT-induced DNA fragmentation (Fig. 11B). Similarly, pretreatment with caspase inhibitors Z-DEVD-FMK (50 μm) or Z-VAD-FMK (100 μm) almost completely blocked MMT-induced DNA fragmentation (Fig. 11B). Rottlerin, Z-DEVD-FMK, and Z-VAD-FMK when treated alone did not significantly attenuate the basal apoptosis in vehicle-treated PC12 cells, suggesting that caspase-3 and PKCδ are participants specifically in MMT-stimulated, and not basal programmed cell death.
MMT treatment does not induce apoptosis in mesencephalic cells overexpressing mutant PKCδK376R protein
Pretreatment with a PKCδ-specific inhibitor rottlerin significantly reduced MMT-induced DNA fragmentation, supporting the idea that the catalytic activity of PKCδ enzyme is vital for induction of apoptosis. If the kinase activity of PKCδ is essential for apoptosis, then overexpression of a kinase inactive PKCδ mutant protein should suppress MMT-induced DNA fragmentation, which occurs downstream of caspase-3 dependent PKCδ activation. Alternatively, overexpression of a kinase inactive PKCδ mutant protein may not interfere with ROS production, an event that occurs before caspase-3 dependent PKCδ activation. To explore these possibilities, we engineered a rat-immortalized mesencephalic (1RB3AN27) cell line to express a dominant-negative PKCδ mutant by stably transfecting with plasmids pPKCδK376R-GFP (in which a lysine at 376 position is mutated to arginine) and pEGFP-N1 (Fig.12A). The plasmid pPKCδK376R-GFP codes for a catalytically inactive PKCδ mutant fused to GFP and pEGFP-N1 plasmid encodes the green fluorescent protein alone, which was used as a vector control. Figure 12B shows stable GFP expression in cell lines transfected with kinase inactive mutant PKCδK376R-GFP and GFP alone. Antibody directed against GFP detected ∼100 and 27 kDa bands in cell lines expressing kinase inactive mutant PKCδK376R-GFP and GFP alone, respectively. Similarly, antibody directed against PKCδ detected ∼100 and 72 kDa bands in cell line expressing PKCδK376R-GFP fusion, whereas only a 72 kDa band was detected in cells expressing GFP alone. The 100, 72, and 27 kDa bands obtained in Western blots correspond to the expression of intact mutant PKCδK376R-GFP fusion protein, native PKCδ and GFP protein, respectively. In apoptotic measurements, MMT-induced DNA fragmentation was completely abolished in cells stably expressing kinase inactive PKCδ protein but not in GFP-alone (vector) transfected 1RB3AN27 cells (Fig.12C). However, MMT-induced ROS production was not significantly different between the kinase inactive PKCδ-GFP and GFP-alone expressing cell lines. A 15 min exposure of 1RB3AN27 cells stably expressing kinase inactive PKCδ-GFP and GFP alone to 200 μm MMT resulted in a 157 ± 20% and 148 ± 11% increase in ROS production, respectively. These results suggest that the kinase activity of PKCδ is essential for MMT-induced DNA fragmentation. These data also indicate that the suppression of apoptosis in PKCδ dominant-negative cells was not caused by a change in the amount of ROS generated in the PKCδ-GFP overexpressing cells versus GFP-vector cells.
Fig. 12.
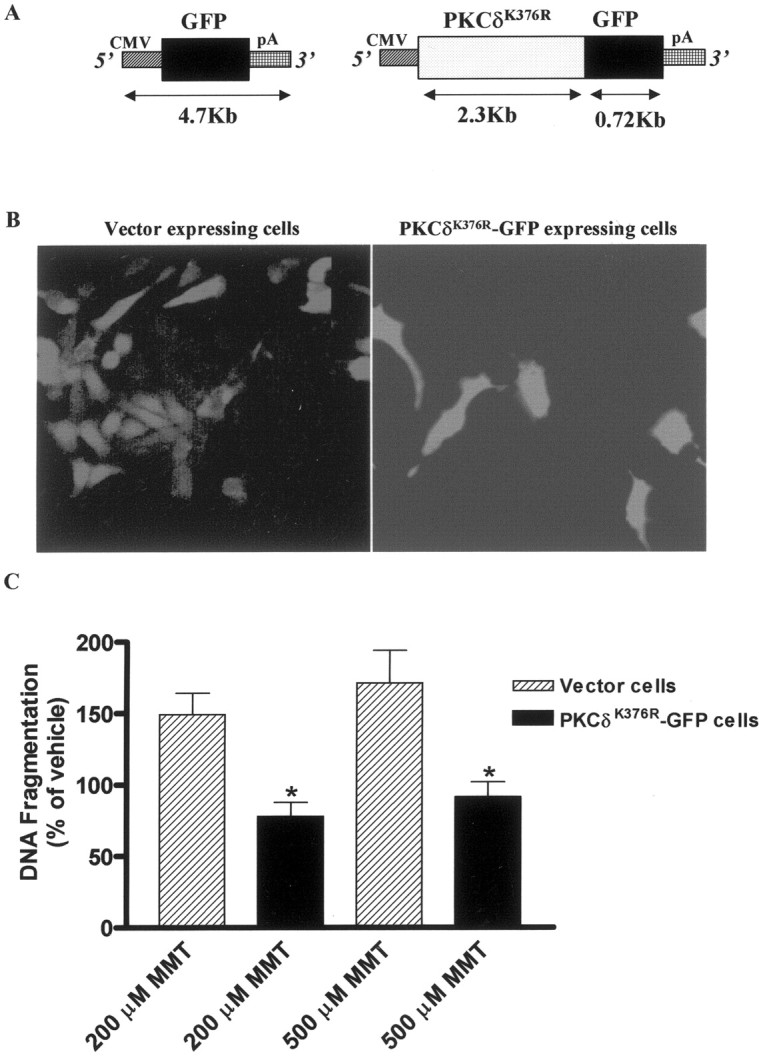
Overexpression of catalytically inactive PKCδ protein blocks MMT-induced apoptosis in immortalized dopaminergic neuronal cell line (1RB3AN27).A, Plasmid description, pEGFP-NI construct codes for the green fluorescent protein (GFP) mRNA transcribed under the 5′ human cytomegalovirus (CMV) immediate early promoter, and the mRNA is stabilized with the 3′ SV40 mRNA polyadenylation signal (pA) and was used as vector control. PKCδK376R-GFP construct codes for the kinase inactive PKCδ-GFP fusion transcript. B, Stable expression of GFP and PKCδK376R-GFP fusion protein in 1RB3AN27 cells. The cells were viewed under a fluorescence microscope, and images were obtained with a SPOT digital camera. C, Subconfluent cultures of undifferentiated 1RB3AN27 cells stably expressing vector or PKCδK376R-GFP fusion protein were treated with MMT (200 and 500 μm) for 3 hr. Apoptosis was assayed using ELISA assay as described in Materials and Methods. The data are expressed as percentage of apoptosis observed in vehicle-treated cells. The data represent a mean ± SEM of four to six individual measurements from two separate experiments. Asterisks (*p < 0.01) indicate significant differences when compared with MMT-treated cells.
DISCUSSION
We recently reported that exposure of PC12 cells to MMT induces dopamine depletion and cytotoxic cell death in a dose- and time-dependent manner (Wagner et al., 2000). The present study extends these observations by demonstrating that MMT induces apoptosis in dopamine-producing cells through ROS production and activation of a series of specific cell death signaling events, including release of cytochrome C into the cytosol, activation of caspase-9 and caspase-3, proteolytic cleavage of PKCδ, and nuclear DNA breakdown. To our knowledge, this is the first report demonstrating that caspase-3-dependent proteolytic cleavage of PKCδ mediates oxidative stress-induced apoptotic cell death in dopaminergic cells after exposure to an environmental neurotoxicant.
In this study, MMT treatment elevated intracellular ROS levels over 45 min in a time- and dose-dependent manner. ROS generation was observed as early as 5 min after MMT exposure, indicating that ROS generation precedes the cytotoxic response. ROS has been shown to induce cytochrome C release from mitochondria in both neuronal and non-neuronal systems by activation of mitochondrial transition pore opening, which results in swelling and rupturing of mitochondrial membrane (Liu et al., 1996; Petit et al., 1996; Blackstone and Green, 1999; Hollensworth et al., 2000; Lee and Wei, 2000). We observed an accumulation of cytosolic cytochrome C in PC12 cells within 15 min after MMT treatment, suggesting that ROS may be an initial signal for the release of cytochrome C. Our data are also consistent with the actions of other dopaminergic toxins, 1-methyl-4-phenylpyridinium (MPP+) (Leist et al., 1998; Cassarino et al., 1999) and 6-hydroxydopamine (6-OHDA) (Dodel et al., 1999) in their ability to induce ROS-mediated cytochrome C release. Cytochrome C, once released into the cytoplasm, forms a complex with apoptotic protease activating factor, and together they activate a series of caspases.
Activation of caspases by cytosolic cytochrome C is an early and essential step in the apoptotic-signaling pathway (Earnshaw et al., 1999; Jellinger, 2000). Several lines of evidence indicate that caspase-3 plays a major role in the regulation and execution phase of both in vitro and in vivo models of apoptosis (Cohen, 1997; Schultz and Andreasen, 1999). In this study, we demonstrate that MMT exposure to PC12 cells results in a dramatic activation of caspase-3, indicating that caspase-3 may play a key role in MMT-induced dopaminergic degeneration. Our data are further supported by a recent study in which caspase-3 activation was observed in neuronal cultures after MPP+ and 6-OHDA treatment (Dodel et al., 1999). The importance of caspase-3 activation as an indicator of apoptosis is further underscored by a recent study from Hartmann et al. (2000), who demonstrated caspase-3 to be a vulnerability factor and a critical effector of apoptotic death in dopaminergic neurons in both MPTP mouse model and in human patients with Parkinson's disease.
Biochemical consequences of caspase-3 activation are proteolytic cleavage of cellular targets associated with apoptosis. Poly (ADP-ribose) polymerase, a DNA cleaving enzyme, has been established as one of the important apoptotic substrates of caspase-3 (Earnshaw et al., 1999; Schultz and Andreasen, 1999). In the present study, we demonstrated that PKCδ is an emerging putative endogenous substrate for caspase-3 and show that MMT exposure induces PKCδ cleavage and increases PKCδ activity in a dose- and time-dependent manner in dopaminergic cells. Additionally, MMT does not induce cleavage of PKCα, suggesting that the cleavage is isoform specific. Proteolytic cleavage of PKCδ in MMT-treated PC12 cells is blocked by specific caspase inhibitors, indicating that the cleavage is mediated by caspase-3. Proteolytic cleavage of PKCδ by caspase-3 results in persistent activation of PKCδ in cytosol, which might initiate a myriad of vital signaling cascades. In a previous study, proteolytic cleavage of PKCδ was observed in KCl-deprived cerebellar granule cell apoptosis, however, this study did not characterize the caspase-3 dependency of PKCδ cleavage (Villalba, 1998). Recent studies have additionally implicated the persistently active catalytic fragment of PKCδ in apoptotic cell death in non-neuronal systems (Earnshaw et al., 1999; Schultz and Andreasen, 1999).
We further determined a possible interaction between PKCδ and caspase-3 activation in MMT-treated PC12 cells using the PKCδ-specific inhibitor rottlerin. Pre- and post-rottlerin treatment effectively blocked MMT-induced caspase-3 activation in PC12 cells in a dose-dependent manner, suggesting a positive feedback modulatory role of PKCδ on caspase-3 activity. Although rottlerin suppressed MMT-induced caspase-3 activity in both pre-and post-treatments (Fig.7), the inhibition was more pronounced in post-treatment at higher concentrations. The reason for the pronounced inhibition is not completely clear at the present time, and we attribute that this might be caused by action of rottlerin on other cellular targets including other kinases (Davies et al., 2000; Way et al., 2000). However, there was no significant difference in caspase activity between pre- and post-treatments of rottlerin at lower dose 5 μm, the concentration at which a pronounced inhibition of PKCδ activityin vitro (Fig. 9C) was observed. Furthermore, delivery of the catalytically active PKCδ fragment alone into PC12 cells increased the caspase-3 activity, confirming the presence of such a positive feedback mechanism. It appears that maximal caspase-3 activity requires the kinase activity of cleaved PKCδ fragment and is made possible by the existence of a positive feedback activation loop. A positive feedback loop between PKCδ and caspase-3 activation has recently been shown to exist in an etoposide-induced salivary cell apoptosis model (Reyland et al., 1999). Thus, the existence of such a positive feedback loop discovered independently by two research groups in two different apoptotic models, MMT-induced PC12 apoptosis (this study) and in etoposide-induced salivary cell apoptosis, suggests that this may be an important regulatory mechanism, allowing for the amplification of apoptotic signaling processes. Further studies are needed to understand the cellular mechanisms of caspase-3 regulation by PKCδ and their role in neuronal apoptosis.
DNA fragmentation and condensation resulting from intranucleosomal cleavage have long been considered biochemical hallmarks of apoptosis and are terminal events in the apoptotic process (Cohen, 1997).In situ fluorometric experiments using two different fluorescent dyes revealed that MMT exposure of PC12 cells induces chromatin condensation in the nucleus. We also took advantage of a recently developed ELISA method that provides a better quantitative measurement of DNA fragmentation. MMT exposure to PC12 cells induced DNA fragmentation, which could be suppressed under conditions where caspase or PKCδ activities were inhibited. Suppression of MMT-induced DNA fragmentation by either caspase inhibitors or rottlerin in the present study indicates that both caspase-3 and PKCδ activities are essential for MMT-induced DNA fragmentation. Furthermore, caspase-3-mediated promotion of DNA fragmentation may be amplified via feedback activation of caspase-3 by the catalytically active PKCδ fragment. To additionally confirm the role of PKCδ in MMT-induced apoptosis, we conducted dominant-negative experiments by stably expressing catalytically inactive PKCδ protein (PKCδK376R) in immortalized rat mesencephalic neurons. MMT treatment produced a significant increase in DNA fragmentation in vector control cells, whereas MMT failed to induce DNA fragmentation in catalytically inactive PKCδ overexpressing cells, thus confirming the key functional role of PKCδ in MMT-induced apoptotic cell death.
Although the events downstream of PKCδ and those that lead to apoptosis remain unclear, recent studies from many research groups have shown that catalytically active PKCδ fragment can regulate the activity of a host of cell signaling molecules such as scrambalase, a membrane phosphatidylserine translocator (Frasch et al., 2000), DNA protein kinase, a DNA repair enzyme (Bharti et al., 1998), heat-shock proteins-25/27 (Maizels et al., 1998), histone H2B (Ajiro, 2000), and lamin kinase (Cross et al., 2000). In addition, PKCδ has been shown to phosphorylate other signaling molecules such as MAP kinases (Chen et al., 1999), Jak2, a tyrosine kinase (Kovanen et al., 2000), and Stat3, signal transducers and activators of transcription (Jain et al., 1999). Most recently, it has been demonstrated that PKCδ activates redox-sensitive transcription factor, NF-κB, and thereby promotes apoptosis in neutrophils (Vancurova et al., 2001). Furthermore, PKCδ has been shown to translocate to cytosol and a variety of cellular organelles to initiate apoptosis (Sawai et al., 1997; Chen et al., 1999; Dal Pra et al., 1999; Li et al., 1999; Dempsey et al., 2000; Majumder et al., 2000). Hence, constitutively active PKCδ fragment can promote loss of cellular regulatory function in many of its substrates, resulting in rapid apoptosis. Currently, our laboratory is focusing on identifying critical cellular targets of PKCδ that might contribute to apoptotic cell death in dopaminergic cells.
In conclusion, we demonstrate for the first time that an environmental neurotoxicant, MMT, induces dopaminergic degeneration by a novel oxidative stress-mediated apoptotic mechanism in which caspase-3-dependent proteolytic cleavage of PKCδ plays a critical role (Fig. 13). Our data also demonstrate a positive feedback amplification loop between PKCδ and capsase-3, which has a regulatory role in the promotion of apoptosis. Further research into identifying molecules that participate in this loop might provide very exciting information regarding cell signaling and neuronal apoptosis. Finally, this study emphasizes the importance of characterizing oxidative stress-induced cell signaling molecules after neurotoxicant exposure to better understand the role of environmental risk factors in the pathogenesis of Parkinson's disease.
Fig. 13.

A model describing the sequence of cell death signaling events in MMT-induced apoptosis. 1, Increased ROS production can be blocked by pretreatment with antioxidants, superoxide dismutase and MnTBAP; 2, cytochrome C is released into the cytosol from the mitochondria; 3,cytosolic cytochrome C activates caspase-9; 4, caspase-9 activates caspase-3; 5, caspase-3 mediates proteolytic cleavage of PKCδ, which can be blocked by pretreatment with the caspase inhibitors Ac-DEVD-CHO, Z-VAD-FMK, and Z-DEVD-FMK;6, pretreatment with rottlerin, a PKCδ inhibitor, reduces caspase-3 activity indicating a possible feedback activation;7, both caspase-3 and PKCδ inhibitors block MMT-induced DNA fragmentation; and 8, dopaminergic cells stably overexpressing catalytically inactive PKCδ [dominant-negative mutant (DNM) PKCδK376RGFP] completely blocked MMT-induced DNA fragmentation. In conclusion, our data suggest that caspase-3-dependent proteolytic activation of PKCδ plays a key role in MMT-induced dopaminergic cell death.
Footnotes
This work was supported by the National Institute of Environmental Health Sciences Grant RO1-ES10586. We acknowledge Dr. Palur Gunasekar (Operational Toxicology, Air Force Research Laboratories, Dayton, OH) for his initial assistance in some experiments. We thank Dr. Michael L. Kirby, Dr. Arthi Kanthasamy, and Mr. Siddharth Ranade in the preparation of this manuscript and Dr. Donghui Cheng for help with flow cytometry.
Correspondence should be addressed to Dr. A. G. Kanthasamy, Parkinson Disorders Research Laboratory, Department of Biomedical Sciences, 2062 Veterinary Medicine Building, Iowa Sate University, Ames, IA 50011. E-mail: akanthas@iastate.edu.
J. Wagner's present address: Department of Chemistry, California State University, Fresno, CA.
REFERENCES
- 1.Ajiro K. Histone H2B phosphorylation in mammalian apoptotic cells An association with DNA fragmentation. J Biol Chem. 2000;275:439–443. doi: 10.1074/jbc.275.1.439. [DOI] [PubMed] [Google Scholar]
- 2.Aschner M. Manganese brain transport and emerging research needs. Environ Health Perspect. 2000;108 [Suppl 3]:429–432. doi: 10.1289/ehp.00108s3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Autissier N, Dumas P, Brosseau J, Loireau A. Effects of methylcyclopentadienyl manganese tricarbonyl (MMT) of rat liver mitochondria. I. Effects, in vitro, on the oxidative phosphorylation. Toxicology. 1977;7:115–122. doi: 10.1016/0300-483x(77)90043-9. [DOI] [PubMed] [Google Scholar]
- 4.Barbeau A. Manganese and extrapyramidal disorders (a critical review and tribute to Dr. George C Cotzias). Neurotoxicology. 1984;5:13–35. [PubMed] [Google Scholar]
- 5.Basu A, Woolard MD, Johnson CL. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. Cell Death Differ. 2001;8:899–908. doi: 10.1038/sj.cdd.4400885. [DOI] [PubMed] [Google Scholar]
- 6.Bharti A, Kraeft SK, Gounder M, Pandey P, Jin S, Yuan ZM, Lees-Miller SP, Weichselbaum R, Weaver D, Chen LB, Kufe D, Kharbanda S. Inactivation of DNA-dependent protein kinase by protein kinase C delta: implications for apoptosis. Mol Cell Biol. 1998;18:6719–6728. doi: 10.1128/mcb.18.11.6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackstone NW, Green DR. The evolution of a mechanism of cell suicide. Bioessays. 1999;21:84–88. doi: 10.1002/(SICI)1521-1878(199901)21:1<84::AID-BIES11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Cassarino DS, Parks JK, Parker WD, Jr, Bennett JP., Jr The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta. 1999;1453:49–62. doi: 10.1016/s0925-4439(98)00083-0. [DOI] [PubMed] [Google Scholar]
- 9.Chen N, Ma W, Huang C, Dong Z. Translocation of protein kinase Cepsilon and protein kinase Cdelta to membrane is required for ultraviolet B-induced activation of mitogen-activated protein kinases and apoptosis. J Biol Chem. 1999;274:15389–15394. doi: 10.1074/jbc.274.22.15389. [DOI] [PubMed] [Google Scholar]
- 10.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 12.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. PKC-delta is an apoptotic lamin kinase. Oncogene. 2000;19:2331–2337. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 13.Dal Pra I, Whitfield JF, Chiarini A, Armato U. Changes in nuclear protein kinase C-delta holoenzyme, its catalytic fragments, and its activity in polyomavirus-transformed pyF111 rat fibroblasts while proliferating and following exposure to apoptogenic topoisomerase-II inhibitors. Exp Cell Res. 1999;249:147–160. doi: 10.1006/excr.1999.4441. [DOI] [PubMed] [Google Scholar]
- 14.Davies PS, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis JM. Methylcyclopentadienyl manganese tricarbonyl: health risk uncertainties and research directions. Environ Health Perspect. 1998;106 [Suppl 1]:191–201. doi: 10.1289/ehp.98106s1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 17.Dodel RC, Du Y, Bales KR, Ling Z, Carvey PM, Paul SM. Caspase-3-like proteases and 6-hydroxydopamine induced neuronal cell death. Brain Res Mol Brain Res. 1999;64:141–148. doi: 10.1016/s0169-328x(98)00318-0. [DOI] [PubMed] [Google Scholar]
- 18.Donaldson J. The physiopathologic significance of manganese in brain: its relation to schizophrenia and neurodegenerative disorders. Neurotoxicology. 1987;8:451–462. [PubMed] [Google Scholar]
- 19.Du Y, Dodel RC, Bales KR, Jemmerson R, Hamilton-Byrd E, Paul SM. Involvement of a caspase-3-like cysteine protease in 1-methyl-4-phenylpyridinium-mediated apoptosis of cultured cerebellar granule neurons. J Neurochem. 1997;69:1382–1388. doi: 10.1046/j.1471-4159.1997.69041382.x. [DOI] [PubMed] [Google Scholar]
- 20.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 21.Ferraz HB, Bertolucci PH, Pereira JS, Lima JG, Andrade LA. Chronic exposure to the fungicide MANEB may produce symptoms and signs of CNS manganese intoxication. Neurology. 1988;38:550–553. doi: 10.1212/wnl.38.4.550. [DOI] [PubMed] [Google Scholar]
- 22.Fishman BE, McGinley PA, Gianutsos G. Neurotoxic effects of methylcyclopentadienyl manganese tricarbonyl (MMT) in the mouse: basis of MMT-induced seizure activity. Toxicology. 1987;45:193–201. doi: 10.1016/0300-483x(87)90105-3. [DOI] [PubMed] [Google Scholar]
- 23.Frasch SC, Henson PM, Kailey JM, Richter DA, Janes MS, Fadok VA, Bratton DL. Regulation of phospholipid scramblase activity during apoptosis and cell activation by protein kinase Cdelta. J Biol Chem. 2000;275:23065–23073. doi: 10.1074/jbc.M003116200. [DOI] [PubMed] [Google Scholar]
- 24.Frumkin H, Solomon G. Manganese in the U.S. gasoline supply. Am J Ind Med. 1997;31:107–115. doi: 10.1002/(sici)1097-0274(199701)31:1<107::aid-ajim16>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Gianutsos G, Murray MT. Alterations in brain dopamine and GABA following inorganic or organic manganese administration. Neurotoxicology. 1982;3:75–81. [PubMed] [Google Scholar]
- 26.Gorell JM, Rybicki BA, Cole Johnson C, Peterson EL. Occupational metal exposures and the risk of Parkinson's disease. Neuroepidemiology. 1999;18:303–308. doi: 10.1159/000026225. [DOI] [PubMed] [Google Scholar]
- 27.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilson GL, LeDoux SP. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med. 2000;28:1161–1174. doi: 10.1016/s0891-5849(00)00214-8. [DOI] [PubMed] [Google Scholar]
- 30.Jain N, Zhang T, Kee WH, Li W, Cao X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J Biol Chem. 1999;274:24392–24400. doi: 10.1074/jbc.274.34.24392. [DOI] [PubMed] [Google Scholar]
- 31.Jellinger KA. Cell death mechanisms in Parkinson's disease. J Neural Transm. 2000;107:1–29. doi: 10.1007/s007020050001. [DOI] [PubMed] [Google Scholar]
- 32.Kanthasamy AG, Isom GE, Borowitz JL. Role of intracellular Cd2+ in catecholamine release and lethality in PC12 cells. Toxicol Lett. 1995;81:151–157. doi: 10.1016/0378-4274(95)03425-0. [DOI] [PubMed] [Google Scholar]
- 33.Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptotic cell death in dopaminergic cells. Free Radic Biol Med. 2001;31:1473–1485. doi: 10.1016/s0891-5849(01)00726-2. [DOI] [PubMed] [Google Scholar]
- 34.Kovanen PE, Junttila I, Takaluoma K, Saharinen P, Valmu L, Li W, Silvennoinen O. Regulation of Jak2 tyrosine kinase by protein kinase C during macrophage differentiation of IL-3-dependent myeloid progenitor cells. Blood. 2000;95:1626–1632. [PubMed] [Google Scholar]
- 35.Langsten R, Hill K. The accuracy of mothers' reports of child vaccination: evidence from rural Egypt. Soc Sci Med. 1998;46:1205–1212. doi: 10.1016/s0277-9536(97)10049-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee HC, Wei YH. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000;7:2–15. doi: 10.1007/BF02255913. [DOI] [PubMed] [Google Scholar]
- 37.Leist M, Volbracht C, Fava E, Nicotera P. 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol Pharmacol. 1998;54:789–801. doi: 10.1124/mol.54.5.789. [DOI] [PubMed] [Google Scholar]
- 38.Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 41.Lynam DR, Roos JW, Pfeifer GD, Fort BF, Pullin TG. Environmental effects and exposures to manganese from use of methylcyclopentadienyl manganese tricarbonyl (MMT) in gasoline. Neurotoxicology. 1999;20:145–150. [PubMed] [Google Scholar]
- 42.Maizels ET, Peters CA, Kline M, Cutler RE, Jr, Shanmugam M, Hunzicker-Dunn M. Heat-shock protein-25/27 phosphorylation by the delta isoform of protein kinase C. Biochem J. 1998;332:703–712. doi: 10.1042/bj3320703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Majumder PK, Pandey P, Sun X, Cheng K, Datta R, Saxena S, Kharbanda S, Kufe D. Mitochondrial translocation of protein kinase C delta in phorbol ester-induced cytochrome c release and apoptosis. J Biol Chem. 2000;275:21793–21796. doi: 10.1074/jbc.C000048200. [DOI] [PubMed] [Google Scholar]
- 44.Mena I, Marin O, Fuenzalida S, Cotzias GC. Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology. 1967;17:128–136. doi: 10.1212/wnl.17.2.128. [DOI] [PubMed] [Google Scholar]
- 45.Muller-Hocker J. Mitochondria and ageing. Brain Pathol. 1992;2:149–158. doi: 10.1111/j.1750-3639.1992.tb00683.x. [DOI] [PubMed] [Google Scholar]
- 46.Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963–3971. [PubMed] [Google Scholar]
- 47.Oertel WH, Kupsch A. Pathogenesis and animal studies of Parkinson's disease. Curr Opin Neurol Neurosurg. 1993;6:323–332. [PubMed] [Google Scholar]
- 48.Petit PX, Susin SA, Zamzami N, Mignotte B, Kroemer G. Mitochondria and programmed cell death: back to the future. FEBS Lett. 1996;396:7–13. doi: 10.1016/0014-5793(96)00988-x. [DOI] [PubMed] [Google Scholar]
- 49.Pongracz J, Webb P, Wang K, Deacon E, Lunn OJ, Lord JM. Spontaneous neutrophil apoptosis involves caspase 3-mediated activation of protein kinase C-delta. J Biol Chem. 1999;274:37329–37334. doi: 10.1074/jbc.274.52.37329. [DOI] [PubMed] [Google Scholar]
- 50.Prasad KN, Clarkson ED, La Rosa FG, Edwards-Prasad J, Freed CR. Efficacy of grafted immortalized dopamine neurons in an animal model of parkinsonism: a review. Mol Genet Metab. 1998;65:1–9. doi: 10.1006/mgme.1998.2726. [DOI] [PubMed] [Google Scholar]
- 51.Pulliam L, Stubblebine M, Hyun W. Quantification of neurotoxicity and identification of cellular subsets in a three-dimensional brain model. Cytometry. 1998;32:66–69. doi: 10.1002/(sici)1097-0320(19980501)32:1<66::aid-cyto9>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 52.Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO. Protein kinase C delta is essential for etoposide-induced apoptosis in salivary gland acinar cells. J Biol Chem. 1999;274:19115–19123. doi: 10.1074/jbc.274.27.19115. [DOI] [PubMed] [Google Scholar]
- 53. Roels H, Lauwerys R, Buchet JP, Genet P, Sarhan MJ, Hanotiau I, de Fays M, Bernard A, Stanescu D. Epidemiological survey among workers exposed to manganese: effects on lung, central nervous system, and some biological indices. Am J Ind Med 11 1987. 307 327 [Erratum (1987) 12:119–120] [DOI] [PubMed] [Google Scholar]
- 54.Sawai H, Okazaki T, Takeda Y, Tashima M, Sawada H, Okuma M, Kishi S, Umehara H, Domae N. Ceramide-induced translocation of protein kinase C-delta and -epsilon to the cytosol. Implications in apoptosis. J Biol Chem. 1997;272:2452–2458. doi: 10.1074/jbc.272.4.2452. [DOI] [PubMed] [Google Scholar]
- 55.Schultz SK, Andreasen NC. Schizophrenia. Lancet. 1999;353:1425–1430. doi: 10.1016/s0140-6736(98)07549-7. [DOI] [PubMed] [Google Scholar]
- 56.Seidler A, Hellenbrand W, Robra BP, Vieregge P, Nischan P, Joerg J, Oertel WH, Ulm G, Schneider E. Possible environmental, occupational, and other etiologic factors for Parkinson's disease: a case-control study in Germany. Neurology. 1996;46:1275–1284. doi: 10.1212/wnl.46.5.1275. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, Tsujimoto Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–2050. [PubMed] [Google Scholar]
- 58.Simon DK, Mayeux R, Marder K, Kowall NW, Beal MF, Johns DR. Mitochondrial DNA mutations in complex I, tRNA genes in Parkinson's disease. Neurology. 2000;54:703–709. doi: 10.1212/wnl.54.3.703. [DOI] [PubMed] [Google Scholar]
- 59.Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem. 1998;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- 60.Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, Langston JW. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341–346. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- 61.Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci. 2000;20:9207–9214. doi: 10.1523/JNEUROSCI.20-24-09207.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vancurova I, Miskolci V, Davidson D. NF-kappa B activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase Cdelta correlation to nuclear Ikappa Balpha. J Biol Chem. 2001;276:19746–19752. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 63.Vassault A. Lactate dehydrogenase. In: Bergmeyer, editor. Methods of enzymatic analysis. Academic; New York: 1983. pp. 118–126. [Google Scholar]
- 64.Villalba M. A possible role for PKC delta in cerebellar granule cells apoptosis. NeuroReport. 1998;9:2381–2385. doi: 10.1097/00001756-199807130-00042. [DOI] [PubMed] [Google Scholar]
- 65.Wagner JR, Anantharam V, Gunasekar PG, Kanthasamy AG. Free radical mediated mechanisms of MMT-induced dopaminergic toxicity in PC12 cells. Toxicol Sci. 2000;54:167. [Google Scholar]
- 66.Wang JD, Huang CC, Hwang YH, Chiang JR, Lin JM, Chen JS. Manganese induced parkinsonism: an outbreak due to an unrepaired ventilation control system in a ferromanganese smelter. Br J Ind Med. 1989;46:856–859. doi: 10.1136/oem.46.12.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- 68.Yoshimura S, Banno Y, Nakashima S, Takenaka K, Sakai H, Nishimura Y, Sakai N, Shimizu S, Eguchi Y, Tsujimoto Y, Nozawa Y. Ceramide formation leads to caspase-3 activation during hypoxic PC12 cell death. Inhibitory effects of Bcl-2 on ceramide formation and caspase-3 activation. J Biol Chem. 1998;273:6921–6927. doi: 10.1074/jbc.273.12.6921. [DOI] [PubMed] [Google Scholar]
- 69.Zayed J, Thibault C, Gareau L, Kennedy G. Airborne manganese particulates and methylcyclopentadienyl manganese tricarbonyl (MMT) at selected outdoor sites in Montreal. Neurotoxicology. 1999;20:151–157. [PubMed] [Google Scholar]
- 70.Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem. 2001;276:35103–35110. doi: 10.1074/jbc.M104920200. [DOI] [PubMed] [Google Scholar]
- 71.Zheng W, Kim H, Zhao Q. Comparative toxicokinetics of manganese chloride and methylcyclopentadienyl manganese tricarbonyl (MMT) in Sprague-Dawley rats. Toxicol Sci. 2000;54:295–301. doi: 10.1093/toxsci/54.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]