

Abstract
Motivation
Most trait-associated genetic variants identified in genome-wide association studies (GWASs) are located in non-coding regions of the genome and thought to act through their regulatory roles.
Results
To account for enriched association signals in DNA regulatory elements, we propose a novel and general gene-based association testing strategy that integrates enhancer-target gene pairs and methylation quantitative trait locus data with GWAS summary results; it aims to both boost statistical power for new discoveries and enhance mechanistic interpretability of any new discovery. By reanalyzing two large-scale schizophrenia GWAS summary datasets, we demonstrate that the proposed method could identify some significant and novel genes (containing no genome-wide significant SNPs nearby) that would have been missed by other competing approaches, including the standard and some integrative gene-based association methods, such as one incorporating enhancer-target gene pairs and one integrating expression quantitative trait loci.
Availability and implementation
Software: wuchong.org/egmethyl.html
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies (GWASs) have identified thousands of loci associated with complex diseases and traits (Visscher et al., 2017). However, the identified genetic variants can only explain a small proportion of the heritability, known as the ‘missing heritability’ problem (Eichler et al., 2010). As an alternative, various gene-based analysis approaches have been adopted (Chen and Wang, 2017; Chen et al., 2017; Pan, 2009; Pan et al., 2014; Wang, 2017; Wu et al., 2011), in which a gene body region is extended up to several kb to cover some regulatory regions, such as promoters. Although appealing, the vast majority (93%) of trait-associated variants are located in non-coding regulatory regions (Maurano et al., 2012) and can affect phenotypes through complex distal genetic regulation (Farh et al., 2015; Wu et al., 2018; Zhu et al., 2016), implying that the usual strategy of mapping the uncovered genetic variants to the nearest genes may be problematic. More importantly, an extension by several kb of a gene body region may not be enough to include all or a majority of its regulatory elements, since some distal regulatory elements are as far as 2 or 3 Mb away from the gene (Krivega and Dean, 2012); on the other hand, a too large extension of a gene region may include too many non-associated SNPs or SNPs from other genes, leading to not only substantial power loss but also difficulties in interpretation.
To boost statistical power and offer biological insights, several integrative gene-based tests (Gamazon et al., 2015; Gusev et al., 2016; Wu and Pan, 2018) have been proposed to incorporate various sources of external information on genetic regulation into GWAS analysis. For example, recent studies show that GWAS risk loci are enriched in enhancers (Ernst et al., 2011), and distal enhancers play important regulatory roles for their target genes through enhancer–promoter interactions in a 3D structure of the chromatin fiber (Ong and Corces, 2014). Accordingly, a new gene-based method, called ‘E + G’ (Wu and Pan, 2018), has been proposed to integrate enhancer-promoter interactions with GWAS summary results. Specifically, when testing on a gene, in addition to its gene body and promoter regions, ‘E + G’ also includes its enhancer regions. Although Hi-C and related technologies have made it possible to experimentally measure enhancer–promoter interactions (Rao et al., 2014), due to their high cost as well as the availability of other (epi-)genomic data, computational methods have emerged to predict enhancers and their target genes (Cao et al., 2017). In the following, we will use ‘enhancer–promoter interactions’ and ‘enhancer–target gene pairs’ exchangeably because either refers to a correspondence between an enhancer and its target gene. In a similar line, inspired by the fact that many genetic variants influence complex traits through transcriptional regulation (Lappalainen et al., 2013; Westra et al., 2013), transcriptome-wide association studies (TWASs) have been proposed (Gamazon et al., 2015; Gusev et al., 2016), which use external expression quantitative trait locus (eQTL) information or databases to select and weight the SNPs associated with gene expression in a largely extended gene region (e.g. up to 1 Mb). As shown previously (Wu and Pan, 2018), either ‘E + G’ or TWAS could identify some significant and novel genes that would be missed by the other and by the standard gene-based testing, indicating their complementary usefulness with possible power gains.
DNA methylation is an extensively studied epigenetic phenomenon, well known to influence gene expression (Wagner et al., 2014) and other genomic functions, including alternative splicing, promoter usage and transcription factor binding (Maunakea et al., 2010; Xu et al., 2015). In turn, DNA methylation may be associated with some genetic variants called methylation quantitative trait loci (mQTLs). To better understand the biological mechanism underlying a disease or phenotype, some association methods have been proposed to integrate genetic and epigenetic information (Freytag et al., 2018; Hannon et al., 2017; So, 2017; Wu et al., 2018). However, these integrative methods are mainly for the purpose of co-localization or mediation analyses, as reviewed in Teschendorff and Relton (2017). For example, Hannon et al. (2017) proposed a modified summary data-based Mendelian randomization to identify pleiotropic genetic variants that are associated with both a complex trait and DNA methylation. By Mendelian randomization, Wu et al. (2018) demonstrated a plausible mechanism by which the effects of genetic variants on a complex trait are mediated through DNA methylation to transcription. Our motivation here is different: by taking advantage of the regulatory roles of DNA methylation and its possibly closer proximity to causal genetic variants than gene expression in causal pathways, we aim to improve statistical power while enhancing the interpretability of any new discoveries. Note that DNA methylation sites associated with gene expression are enriched in enhancers, promoters and gene body regions (including exons and introns) (Gutierrez-Arcelus et al., 2015). In particular, increased DNA methylation in enhancer regions is also known to be associated with gene expression changes of the linked genes (Lu et al., 2014), suggesting the potential usefulness of simultaneous integration of both mQTL and enhancer–promoter interaction information. In particular, as shown in cancer (Aran and Hellman, 2013), for many genes, the correlation between methylation of enhancers and gene expression is much higher than that between SNPs and gene expression. For example, Li et al. (2013) showed that, for a prominent breast cancer oncogene CCND1, although DNA sequence variants of its enhancer region are associated with breast cancer risk, they are not significantly correlated with CCND1 expression level; in contrast, Aran and Hellman (2013) demonstrated a striking correlation of CCND1 expression level with methylation of the same enhancer region. Hence, integrating methylation data may go beyond using only eQTL data to uncover novel risk loci for complex traits. Although integrating either eQTL data or mQTL data alone with GWAS results (Freytag et al., 2018; Gamazon et al., 2015; Gusev et al., 2016; Hannon et al., 2017; So, 2017; Wu et al., 2018; Xu et al., 2017) has become increasingly popular recently, to our knowledge, no previous study has integrated both mQTL data and promoter-enhancer interactions with GWAS summary results. In this article, we propose a new gene-based association testing method, called ‘E + G + Methyl’, that integrates enhancer-target gene maps, mQTL databases, and GWAS summary results to identify significant and novel genes that may be missed by other methods. The main idea is that, when testing on a gene, we search for and then test only on the mQTLs in its gene body (including exons and introns), promoter and enhancer regions. Note that ‘E + G + Methyl’ can be viewed as an extension of ‘E + G’ (Wu and Pan, 2018): instead of using all SNPs in its gene body, promoter and enhancer regions, ‘E + G + Methyl’ uses only mQTLs while excluding other SNPs. In other words, we focus on genetic variants that exert their effects on a trait through some methylation pathways while accounting for enriched association signals in mQTLs and enhancers.
To illustrate the potential usefulness of our new method and better understand the mechanism underlying schizophrenia (SCZ), we reanalyzed two SCZ GWAS summary datasets (Ripke et al., 2013, 2014). These analyses show that, when applied to the smaller SCZ GWAS dataset, our new method ‘E + G + Methyl’ could identify many significant and novel genes that were replicated by the larger GWAS dataset, but would have been missed by other competing methods, such as ‘E + G’, TWAS and the standard gene-based testing. Similarly, when applied to the larger SCZ GWAS data, ‘E + G + Methyl’ could identify 16 significant and novel genes that were missed by competing methods. In summary, we view ‘E + G + Methyl’ as a powerful integrative gene-based association method that is useful and complementary to the existing approaches.
2 Materials and methods
2.1 Enhancer–target and mQTL databases
Enhancers, bound by transcription factors, act independently of the orientation and distance to their target genes, thus it is challenging to determine enhancer–target gene pairs (Shlyueva et al., 2014). We used two publicly available databases as in Wu and Pan (2018) to determine the enhancer regions for each target gene: (i) experimentally measured from the MCF-7 cell line by using genome-wide chromatin interaction analysis with paired-end-tag sequencing (Li et al., 2012), denoted as MCF7 in the following; (ii) computationally predicted for the brain hippocampus region by analyzing ENCODE and Roadmap data with a statistical model (Cao et al., 2017), denoted as Hippo in the following. Note that in (i), for simplicity, we call any DNA fragment interacting with a promoter as an enhancer. Given our focus on SCZ and the relatedness of pathophysiology of SCZ to the hippocampus (Harrison, 2004), we used the computationally predicted enhancer–target gene pairs for the hippocampus as an example of data drawn from a trait-relevant tissue. Since enhancer–promoter interactions are tissue-specific (Andersson et al., 2014), it would be ideal to utilize enhancer–promoter interaction information drawn from a disease- or trait-related tissue. However, because data from some tissues may not be available, and 55–75% DNA interactions are shared among different cell lines (Rao et al., 2014), it might be potentially useful to use the data from a tissue or cell line not necessarily most relevant to the disease or trait, such as from the MCF-7 cell line (Wu and Pan, 2018). Note that in the Hippo data, only enhancers within 1 Mb of each transcription start site (TSS) were considered by the original authors (Cao et al., 2017); for the MCF7 data, there was no such restriction (Li et al., 2012).
The effects of SNPs on complex traits are potentially mediated through some highly dynamic epigenetic processes, such as DNA methylation (Relton and Smith, 2010). To understand how genetic factors (e.g. mQTLs) influence DNA methylation, Gaunt et al. (2016) developed a genome-wide cis- and trans-mQTL database based on analyzing blood samples in the Avon longitudinal study of parents and children. This mQTL database provides mQTL information at five life stages in human blood, and we used middle age mQTL information for the subsequent analysis. It contains 5 421 792 significant SNP–CpG site associations (P-value < 1 × 10–7), covering 1 926 067 SNPs and 45 070 CpG sites.
The genomic coordinates of the SNPs and genes were obtained from the human genome assembly GRCh37 (hg19). Because DNA methylation in enhancer and promoter regions may play some important roles in gene regulation (Lu et al., 2014; Wu et al., 2018), we were motivated to focus on mQTLs located in enhancers, promoters, and gene body (including both exons and introns) regions. Thus we defined a SNP set for each gene to be tested by integrating enhancer–promoter interaction and mQTL data by the following steps.
Gene body: All the introns and exons of a target gene were included and called the gene body. In other words, a gene body region was defined as that flanking its TSS and transcription end site (TES).
Promoters: Two promoter regions of a target gene were defined as a 500-bp extension (Andersson et al., 2014) on either side of the gene body region beyond its TSS and TES respectively. Although most promoters lie immediately upstream of the TSS, a gene might have several proximal promoters scattered around its TES and even introns (Goñi et al., 2007). Hence we extended 500 bp upstream TSS and downstream TES respectively to include its potential cis-acting regulatory elements. For simplicity, we denote the target gene body plus its two promoter regions as region ‘G’.
Enhancers: An enhancer of a target gene was defined as the DNA fragment interacting with a promoter (or as predicted to regulate the target gene). Note that the defined enhancer regions are tissue- or database-dependent, so they may vary with different data sources (e.g. MCF7 and Hippo). We denote the enhancer regions as ‘E’, and further denote the target gene body plus its two promoter regions and its all enhancer regions as ‘E + G’.
mQTL: For each SNP in the ‘E + G’ region of a target gene, we searched the mQTL database: if it was an mQTL for any CpG site in the target gene ‘E + G’ region, we kept it; otherwise, it was excluded.
For simplicity, we denote the set of SNPs in an ‘E + G’ region that could pass the mQTL searching (with pruning) as ‘E + G + Methyl’. We further denote the set of SNPs inside an ‘E + G’ region (with pruning) as ‘E + G’ (Wu and Pan, 2018), while that inside a ‘G’ region (with pruning) as ‘STD’ (for the standard gene-based testing). Note that to reduce the computational burden and minimize the effect of collinearity for the subsequent association testing, we pruned the set of SNPs selected for each gene such that no SNP pairs within the set were highly correlated (with r2 > 0.95).
We point out that the proposed ‘E + G + Methyl’ can be viewed as an extension of ‘E + G’: ‘E + G + Methyl’ selects the subset of SNPs in ‘E + G’ that are mQTLs (while excluding all other SNPs). Because DNA methylation sites in the enhancer and gene body (intron) regions may play vital regulatory roles (Lu et al., 2014; Wu et al., 2018), whereas other SNPs are less likely to be associated with the trait, only using mQTLs inside ‘E + G’ (i.e. ‘E + G + Methyl’) may reduce the number of non-informative SNPs being tested and thus increase statistical power to identify significant and novel genes that are associated with the trait through DNA methylation pathways. Note that because ‘E + G + Methyl’ only selects the mQTL SNPs that pass the mQTL searching, thus including only a subset of SNPs in ‘E + G’ might be more informative for identifying significant genes.
2.2 Statistical tests
For illustration, we applied two representative gene-based tests, a burden test called SPU(1) (i.e. Sum test) and a variance-component test called SPU(2) (i.e. SSU test) (Kwak and Pan, 2016; Pan et al., 2014; Wu et al., 2011), to a set of SNPs for a target gene to test the association between the target gene and a trait. Although not our focus, we also illustrated the application of an adaptive test called aSPU (Kwak and Pan, 2016; Pan et al., 2014). Note that other gene- or SNP set-based tests (Chen and Wang, 2017; Chen et al., 2017; Wang, 2017) can be equally applied.
Here, we focused on analyzing GWAS summary data (i.e. marginal association statistics), which are often publicly available with the estimated marginal effect size, its standard error and P-value of each SNP and some information on each SNP (such as its chromosome position and major allele). We applied SPU(1), SPU(2), and sometimes aSPU, to the ‘E + G + Methyl’ SNP set for each target gene. To save space, a detailed testing procedure was relegated to the Supplementary Material. For comparison, we also applied both SPU(1) and SPU(2) to the ‘E + G’ region and the standard gene regions (denoted ‘STD’) respectively. Since mQTL and eQTL provide orthogonal ways of functionally annotating SNPs for complex traits, and TWAS (Gusev et al., 2016) and its extension (Xu et al., 2017) use eQTL data to weight SNPs (Gamazon et al., 2013), we applied them as well. For notational simplicity, we used (weighted) SPU(1) to represent TWAS and (weighted) SPU(2) to represent its extension. The four sets of eQTL-derived weights (NTR, YFS, METSIM, and CMC) were downloaded from the TWAS/FUSION website (Gusev et al., 2016).
2.3 Application to GWAS
To demonstrate the potential usefulness of our new method ‘E + G + Methyl’ and better understand the genetic basis of SCZ, we applied different gene-based association tests to identify SCZ-associated genes by reanalyzing two SCZ GWAS summary datasets: a meta-analyzed SCZ GWAS dataset with 8832 cases and 12 067 controls (Ripke et al., 2013), denoted as SCZ1, and a larger one with 36 989 cases and 113 075 controls, denoted as SCZ2 (Ripke et al., 2014).
When we focused on the common gene set analyzed by all methods, we used a conservative Bonferroni correction cutoff 0.05/10 000 = 5 × 10−6; otherwise, we used the Bonferroni correction to each method (with possibly different numbers of genes being tested and thus different cutoffs) separately to control for multiple testing. Following Gusev et al. (2016) and Wu and Pan (2018), we evaluated the performance of the methods via the following steps. First, based on the SCZ1 data, we identified the significant and novel genes; a significant and novel gene in one study is defined as a significantly associated gene that does not cover any significant SNP in an extended gene region (with an extension of ±500 kb upstream and downstream its gene body region). Second, we calculated the replication rate of the identified significant and novel genes that also covered one or more genome-wide significant SNPs in the SCZ2 data. Third, the P-value for the replication rate was calculated by a hypergeometric test with the background probability estimated from all the genes being tested.
2.4 Availability of data and materials
The SCZ1 (Ripke et al., 2013) and SCZ2 (Ripke et al., 2014) GWAS summary data are available at the PGC web site https://www.med.unc.edu/pgc/results-and-downloads. TWAS- and eQTL-based weights can be obtained from http://gusevlab.org/projects/fusion/. Our user-friendly software, and some information on the processed mQTLs, enhancer–promoter interactions and the 1000 Genomes reference panel, are available at wuchong.org/egmethyl.html.
3 Results
3.1 Using SCZ1 for discovery
We consider applications to the smaller SCZ1 dataset for discovery. First, with simulated null Z score vectors, we confirmed that ‘E + G + Methyl’ with the standard gene-based tests, such as SPU(1) and SPU(2), yielded well-controlled Type I error rates under various nominal significance levels (Supplementary Fig. S1). Importantly, the well-controlled Type I error rates of SPU(1) and SPU(2) were also established by previous studies (Gusev et al., 2016; Pan, 2009; Pan et al., 2014). Second, we compared the discovery power for different methods. For a fair comparison, we focused on the common set of 3521 genes that could be analyzed by all methods. Figure 1 shows that ‘E + G + Methyl’, ‘E + G’, STD and TWAS identified 6, 4, 3 and 7 significant and novel genes, respectively; a novel gene is defined as one that does not cover any genome-wide significant SNP within a ±500 kb extension upstream and downstream its gene body in the same dataset (i.e. SCZ1 here). Both ‘E + G + Methyl’ and TWAS identified four significant and novel genes that were missed by other methods. Figure 1 displays the combined results from SPU(1) and SPU(2), while the separate results of SPU(1) and SPU(2) showed similar patterns and thus were relegated to Supplementary Figures S2 and S3. Supplementary Table S1 gives the numbers of the significant genes identified by various methods. Note that, although STD identified more significant genes than both ‘E + G + Methyl’ and TWAS, it was likely due to its testing on more genes.
Fig. 1.
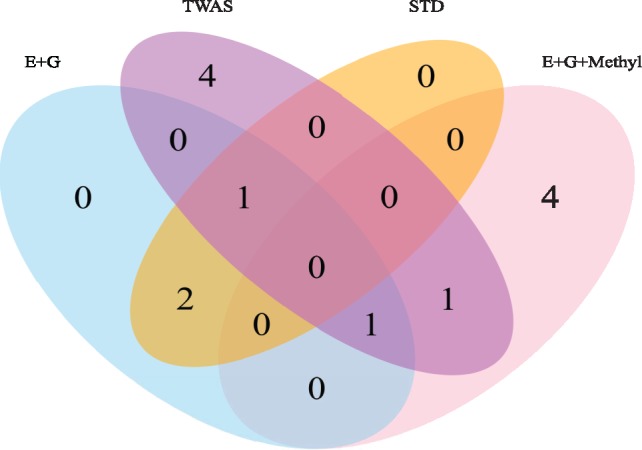
Venn diagrams of the significant and novel genes identified by the different methods applied to the SCZ1 data. ‘E + G’ and ‘E + G+Methyl’ combine the results (i.e. taking the union) of using MCF7 and Hippo data, while TWAS combines the results of using YFS-, NTR-, METSIM- and CMC-based weights. We analyzed the common set of 3521 genes, combined the results from SPU(1) and SPU(2), and used the same significance cutoff (P ≤ 5 × 10−6)
3.2 Using SCZ2 for validation
To further validate our approach, we focused on all significant and novel genes identified from the SCZ1 data that could be confirmed with genome-wide significant SNPs in the larger (but overlapping) SCZ2 data (Table 1). For a fair comparison, we used the Bonferroni correction for each method (with a possibly different number of genes and thus different cutoffs) separately. Supplementary Table S2 shows the replication rates and corresponding statistical significance levels by a hypergeometric test. ‘E + G + Methyl’ with SPU(1) based on MCF7 identified 10 novel genes in the SCZ1 data, of which 6 (60%) contained genome-wide significant SNPs in an extended gene region (±500 kb as before) in the larger SCZ2 data (P-value = 9.6 × 10−6 by a hypergeometric test), constituting a highly significant replication rate of the identified genes by our new method ‘E + G + Methyl’. Both ‘E + G’ and TWAS show similar patterns with highly significant replication rates, confirming the usefulness of incorporating information from other sources.
Table 1.
The numbers of the significant and novel genes identified by analyzing the SCZ1 data
| E + G + Methyl |
E + G |
STD | TWAS |
||||||
|---|---|---|---|---|---|---|---|---|---|
| MCF7 | Hippo | MCF7 | Hippo | (STD) | YFS | NTR | METSIM | CMC | |
| No. genes | 4588 | 2340 | 9127 | 4600 | 22 842 | 4697 | 2452 | 4665 | 5412 |
| SPU(1) | 10/6 | 4/3 | 1/1 | 2/2 | 4/4 | 3/3 | 4/4 | 3/2 | 6/4 |
| SPU(2) | 11/7 | 12/6 | 10/6 | 12/8 | 12/10 | 6/3 | 8/8 | 9/9 | 14/11 |
Note: The numbers a/b in each cell indicate the numbers of (a) the significant and novel genes; and (b) the significant genes that covered one or more GWAS risk variants within ±500 kb in the SCZ2 data.
Furthermore, we searched the GWAS Catalog v1.0 (MacArthur et al., 2017) to identify the genes reported by other studies. Table 2 lists the significant and novel genes identified by ‘E + G + Methyl’, a high proportion of which (12 out of 22, 55%) contained at least one genome-wide significant SNP (P-value < 5 × 10−8) in the (overlapping but larger) SCZ2 data. Importantly, many of them (14 out of 22, 64%) have been reported by other studies. Importantly, the reported genes in the gene list in Table 2 were over-enriched by 14.88 folds (P-value = 1.1 × 10−14 by a hypergeometric test) over that by chance. Overall, these results showcase the power of the ‘E + G + Methyl’ approach in identifying significant and novel genes that were validated by other studies but were missed by the competing methods.
Table 2.
Significant and novel genes (with their P-values) identified by the ‘E + G + Methyl’ approach based on the SCZ1 data
| Gene | CHR | No. mQTL | No. CpG | SPU(1) | SPU(2) | References |
|---|---|---|---|---|---|---|
| Source: Hippo | ||||||
| PLCH2a | 1 | 258 | 49 | [1, 2] | ||
| PLEKHG5 | 1 | 103 | 23 | |||
| NGEFa | 2 | 23 | 3 | [2, 3] | ||
| PSMG4 | 6 | 248 | 24 | |||
| FAM20C | 7 | 1579 | 339 | |||
| TOLLIP | 11 | 559 | 68 | |||
| LRP6 | 12 | 163 | 32 | |||
| MPHOSPH9a | 12 | 7 | 1 | [2, 3] | ||
| C12orf65a | 12 | 4 | 1 | [2] | ||
| CDK2AP1a | 12 | 4 | 2 | [2] | ||
| HMOX2 | 16 | 5 | 4 | |||
| YJEFN3a | 19 | 3 | 4 | |||
| Source:MCF7 | ||||||
| SH3RF1a | 4 | 10 | 2 | |||
| CREB1 | 2 | 3 | 1 | [1] | ||
| CNOT7 | 8 | 5 | 1 | |||
| VPS37A | 8 | 8 | 1 | [3] | ||
| PIK3C2A | 11 | 3 | 1 | [1, 4, 5] | ||
| OGFOD2a | 12 | 4 | 3 | [2, 3] | ||
| PITPNM2a | 12 | 13 | 3 | [2, 3] | ||
| CDK2AP1a | 12 | 6 | 2 | [2] | ||
| XRCC3a | 14 | 5 | 2 | [2] | ||
| SUGP1a | 19 | 3 | 1 | [2] | ||
| NDUFA13a | 19 | 2 | 3 | [2] | ||
| YJEFN3a | 19 | 3 | 4 |
Note: ‘sig SNP’ give the P-value of the most significant SNP within a ± 500 kb extension for each gene in the SCZ2 data; the previously reported gene–SCZ associations appear in ‘References’: [1] Goes et al. (2015); [2] Ripke et al. (2014); [3] Li et al. (2017); [4] Ripke et al. (2011); [5] Ruderfer et al. (2014).
Novel genes, not overlapping with a genome-wide significant SNP within a ± 500 kb extension for each gene in the SCZ2 data.
3.3 New discoveries from the larger SCZ2 data
Having established the usefulness of our new method, we applied the ‘E + G + Methyl’ method to the larger SCZ2 data to identify significant and novel genes. We used a conservative and common Bonferroni cutoff (0.05/10 000 = 5 × 10−6) for all the methods. Overall, ‘E + G + Methyl’ identified 38 significant and novel SCZ-associated genes (Supplementary Table S3), of which 16 were missed by STD, ‘E + G’ based on MCF7 or Hippo, and TWAS with four sets of weights (Table 3). Among these 16 genes, 3 of them have been reported by other studies (Goes et al., 2015). Importantly, there are biological findings supporting our results. For example, the protein encoded by gene CREB1 is a transcription factor involved in regulating gene expression as part of cAMP signaling cascades in the brain (Montminy, 1997), and is a critical component of memory-related synaptic plasticity (Kandel, 2012). Furthermore, some CpG sites inside the introns of CREB1 were reported to be associated with SCZ (Kumar et al., 2015), partially explaining why ‘E + G + Methyl’ could successfully identify CREB1 while the other methods failed. Another example is gene CHRM3: it has been shown that CHRM3 plays a vital role in abnormal thalamo-orbital frontal cortex functional connectivity in SCZ subjects (Wang et al., 2016). Overall, these 16 newly identified genes represent a class of discoveries that would be missed by other competing methods, showcasing the power of our proposed approach to integrating enhancer–promoter interactions, mQTL data and GWAS summary results to gain insights into the genetic basis of complex diseases.
Table 3.
Significant and novel genes identified by both SPU(1) and SPU(2) with the ‘E + G + Methyl’ approach, but not by the STD, ‘E + G,’ and TWAS based on the SCZ2 data
| Gene | CHR | No. mQTL | No. CpG | SPU(1) | SPU(2) | References |
|---|---|---|---|---|---|---|
| Source: Hippo | ||||||
| CHRM3 | 1 | 2 | 2 | |||
| KCNS3 | 2 | 12 | 4 | [1] | ||
| EFR3B | 2 | 3 | 1 | [1] | ||
| CHL1 | 3 | 1340 | 191 | |||
| PSMG4 | 6 | 248 | 24 | |||
| FAM20C | 7 | 1579 | 339 | |||
| C7orf50 | 7 | 805 | 135 | |||
| DRD4 | 11 | 109 | 39 | |||
| TOLLIP | 11 | 559 | 68 | |||
| LRP6 | 12 | 163 | 32 | |||
| FAM177A1 | 14 | 9 | 2 | |||
| MADCAM1 | 19 | 404 | 85 | |||
| CSNK1G2 | 19 | 68 | 17 | |||
| Source:MCF7 | ||||||
| PRADC1 | 2 | 2 | 1 | |||
| CREB1 | 2 | 3 | 1 | [1] | ||
| ZNF623 | 8 | 13 | 3 |
Note: ‘sig SNP’ gives the P-value of the most significant SNP within a ± 500 kb extension for each gene in the SCZ2 data; the previously reported gene-SCZ associations appear in ‘References’: [1] Goes et al. (2015).
3.4 Power gains with an adaptive gene-based test
We also applied aSPU with ‘E + G + Methyl’ to the SCZ1 to show possible power gains from a more powerful and adaptive SNP-set test like aSPU. We used a conservative and common Bonferroni cutoff (0.05/10 000 = 5 × 10–6) in the following. Figure 2 shows that SPU(1), SPU(2) and aSPU identified 6, 9 and 10 novel genes, respectively. Importantly, most novel genes identified by SPU(1) and SPU(2) were recovered by aSPU, showcasing the high power by aSPU. Supplementary Table S4 lists the significant and novel genes identified by aSPU with ‘E + G + Methyl’: a high proportion of them contained at least one genome-wide significant SNP (19 out of 24, 79%) in the SCZ2 data or were reported previously (20 out of 24, 83%). Overall, these results constituted a highly significant replication of the identified genes by aSPU with ‘E + G + Methyl’. Importantly, the Type I error rates of aSPU were well-controlled under the null (Supplementary Fig. S1). To further discover some significant and novel genes that would have been missed by other methods, we applied aSPU with ‘E + G + Methyl’ to the SCZ2 data. Table 4 lists six significant and novel genes uniquely identified by aSPU with ‘E +G + Methyl’. These new findings may further provide biological insights into the mechanism of SCZ.
Fig. 2.
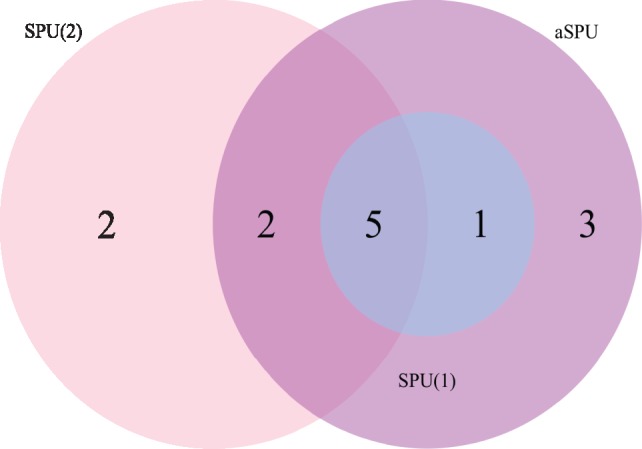
Venn diagrams of the significant and novel genes identified by the ‘E + G+Methyl’ with different methods applied to the SCZ1 data. We combined the results of using MCF7 and Hippo data, and used the same significance cutoff (P ≤ 5 × 10−6)
Table 4.
Significant and novel genes identified by aSPU with the ‘E + G + Methyl’ approach, but not by the SPU(1) and SPU(2) with ‘E + G + Methyl’, STD, ‘E + G’ and TWAS based on the SCZ2 data
| # | # | ||||||
|---|---|---|---|---|---|---|---|
| Gene | CHR | mQTL | CpG | SPU(1) | SPU(2) | aSPU | References |
| Source: Hippo | |||||||
| TCEA3 | 1 | 20 | 4 | ||||
| TRAPPC3 | 1 | 4 | 2 | [1] | |||
| TTLL7 | 1 | 13 | 3 | ||||
| C6orf195 | 6 | 45 | 10 | ||||
| PRH1 | 12 | 183 | 18 | ||||
| FLYWCH1 | 16 | 61 | 13 |
Note: ‘sig SNP’ gives the P-value of the most significant SNP within a ± 500 kb extension for each gene in the SCZ2 data; the previously reported gene-SCZ associations appear in ‘Reference’: [1] Goes et al. (2015).
3.5 Data summary
We provide some basic statistics for ‘E + G + Methyl’ regions. On average, for each target gene, MCF7- and Hippo-based ‘E + G + Methyl’ regions contained 3.1 and 1.6 enhancer regions, respectively (Supplementary Table S5). For MCF7-based ‘E + G + Methyl’, the enhancer (‘E’) regions and ‘G’ regions were 5.2 and 62 kb, respectively (Supplementary Table S5). Because ‘E + G + Methyl’ only selected the mQTLs in the ‘E + G’ regions, for the SCZ1 dataset, MCF7-based’E + G + Methyl’ contained 29.4 SNPs on average while ‘E + G’ contained 34.3 SNPs on average (Supplementary Table S6). Note that ‘E + G + Methyl’ only included a subset of SNPs in ‘E + G’, and the ‘E + G + Methyl’ set might be more informative for identifying significant genes that influence a trait through some methylation pathways.
4 Discussion
Most identified GWAS variants are thought to act by affecting gene regulation, rather than altering protein products, highlighting the importance and thus potential power gains of integrating genetic regulatory information when conducting gene-based association testing. Here we have introduced a new method, called ‘E + G + Methyl’, for identifying trait-associated genes via integrating both mQTL and enhancer–promoter interaction data with GWAS association results. Our approach allows to identify some significant and novel genes that would be missed by other methods, and to gain insights into the genetic mechanisms underlying complex traits. Our method differs from other gene-based tests in how to construct a SNP set for a gene being tested. To the best of our knowledge, ‘E + G + Methyl’ is the first approach to combine mQTL data and enhancer–target gene maps in the gene-based testing framework for GWAS.
To further demonstrate the usefulness of integrating mQTL data, we explored using mQTLs inside the gene body region with a 20-kb extension, denoted as ‘G + Methyl’. As shown in Supplementary Figure S4, ‘G + Methyl’ could identify some significant and novel genes that would be missed by other methods, indicating that mQTLs are indeed informative for GWAS analysis.
We view our method complementary to existing methods, such as TWAS, ‘E + G’ and standard gene-based approaches. Comparatively, we expect that TWAS will be advantageous when one gene contains several cis-eQTLs that are possibly outside annotated enhancers, while ‘E + G’ will be highly powerful when a gene has one or more (far away) enhancers that are enriched with trait-associated SNPs (which may not be either eQTL or mQTL). In contrast, our new method ‘E + G + Methyl’ will be most useful when the enhancers, especially those far away from a gene, contain trait-associated mQTLs. It is noted that, for example in cancer, some known risk variants in enhancers are methylated, but are not easily detectable as eQTLs for their target genes (Aran and Hellman, 2013; Li et al., 2013), suggesting possible gains from integrating methylation data, not just eQTL data, with GWAS.
We note several possible limitations of the proposed ‘E + G + Methyl’ method. First, due to the lack of data, the used ChIA-PET enhancer–promoter interaction data was not from a brain tissue most relevant to SCZ. Although 55%–75% promoter-enhancer interactions are shared among different cell lines (Rao et al., 2014), we expect our method to be more powerful when data drawn from the most relevant tissues are used. There is a similar issue with the mQTL database used (Gutierrez-Arcelus et al., 2015). Second, although we have integrated multiple sources of (epi-)genomic information, some other useful sources of genomic data and/or genome annotations (Chen et al., 2016; Zhang and Hardison, 2017; Zhang et al., 2016) have not been incorporated. Third, ‘E + G + Methyl’ is a GWAS summary data-based method and thus we only focused on common variants, though its extension to including rare variants seems possible. Despite these limitations, ‘E + G + Methyl’ is a powerful new approach to identifying novel genes significantly associated with complex traits.
Supplementary Material
Acknowledgements
We thank the reviewers for helpful comments, and the Minnesota Supercomputing Institute for providing computing resources.
Funding
This work was supported by the National Institutes of Health grants [R21AG057038, R01HL116720, R01GM113250, R01GM126002 and R01HL105397].
Conflict of Interest: none declared.
References
- Andersson R. et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature, 507, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Hellman A. (2013) Dna methylation of transcriptional enhancers and cancer predisposition. Cell, 154, 11–13. [DOI] [PubMed] [Google Scholar]
- Cao Q. et al. (2017) Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet., 49, 1428–1436. [DOI] [PubMed] [Google Scholar]
- Chen L. et al. (2016) DIVAN: accurate identification of non-coding disease-specific risk variants using multi-omics profiles. Genome Biol., 17, 252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Wang K. (2017) A gene-based test of association through an orthogonal decomposition of genotype scores. Hum. Genet., 136, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Chen Z. et al. (2017) A powerful variant-set association test based on chi-square distribution. Genetics, 207, 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler E.E. et al. (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet., 11, 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J. et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh K.K.-H. et al. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag V. et al. (2018) Genetic estimators of DNA methylation provide insights into the molecular basis of polygenic traits. Transl. Psychiatry, 8, 31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon E. et al. (2013) Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol. Psychiatry, 18, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon E.R. et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet., 47, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt T.R. et al. (2016) Systematic identification of genetic influences on methylation across the human life course. Genome Biol., 17, 61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goes F.S. et al. (2015) Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. B Neuropsychiatr. Genet., 168, 649–659. [DOI] [PubMed] [Google Scholar]
- Goñi J.R. et al. (2007) Determining promoter location based on DNA structure first-principles calculations. Genome Biol., 8, R263.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A. et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Arcelus M. et al. (2015) Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet., 11, e1004958.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon E. et al. (2017) Pleiotropic effects of trait-associated genetic variation on DNA methylation: utility for refining GWAS loci. Am. J. Hum. Genet., 100, 954–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P.J. (2004) The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology, 174, 151–162. [DOI] [PubMed] [Google Scholar]
- Kandel E.R. (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain, 5, 14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivega I., Dean A. (2012) Enhancer and promoter interactions-long distance calls. Curr. Opin. Genet. Dev., 22, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G. et al. (2015) Refinement of schizophrenia GWAS loci using methylome-wide association data. Hum. Genet., 134, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak I.-Y., Pan W. (2016) Adaptive gene-and pathway-trait association testing with GWAS summary statistics. Bioinformatics, 32, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. et al. (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell, 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. et al. (2013) Integrative eqtl-based analyses reveal the biology of breast cancer risk loci. Cell, 152, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. et al. (2017) Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet., 49, 1576–1583. [DOI] [PubMed] [Google Scholar]
- Lu F. et al. (2014) Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev., 28, 2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J. et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea A.K. et al. (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature, 466, 253–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M.T. et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy M. (1997) Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem., 66, 807–822. [DOI] [PubMed] [Google Scholar]
- Ong C.-T., Corces V.G. (2014) CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet., 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. (2009) Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol., 33, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. et al. (2014) A powerful and adaptive association test for rare variants. Genetics, 197, 1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S. et al. (2014) A 3d map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton C.L., Smith G.D. (2010) Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med., 7, e1000356.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S. et al. (2011) Genome-wide association study identifies five new schizophrenia loci. Nat. Genet., 43, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S. et al. (2013) Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet., 45, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S. et al. (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer D.M. et al. (2014) Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry, 19, 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D. et al. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet., 15, 272–286. [DOI] [PubMed] [Google Scholar]
- So H.-C. (2017) Epigenome-wide association study and integrative analysis with the transcriptome based on GWAS summary statistics. arXiv Preprint arXiv: 1702.00329.
- Teschendorff A.E., Relton C.L. (2017) Statistical and integrative system-level analysis of dna methylation data. Nat. Rev. Genet., 19, 129–147. [DOI] [PubMed] [Google Scholar]
- Visscher P.M. et al. (2017) 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet., 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.R. et al. (2014) The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol., 15, R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. (2017) Conditional asymptotic inference for the kernel association test. Bioinformatics, 33, 3733–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. et al. (2016) The CHRM3 gene is implicated in abnormal thalamo-orbital frontal cortex functional connectivity in first-episode treatment-naive patients with schizophrenia. Psychol. Med., 46, 1523–1534. [DOI] [PubMed] [Google Scholar]
- Westra H.-J. et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Pan W. (2018) Integration of enhancer-promoter interactions with GWAS summary results identifies novel schizophrenia-associated genes and pathways. Genetics, 209, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.C. et al. (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet., 89, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. et al. (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun., 9, 918.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T. et al. (2015) Base-resolution methylation patterns accurately predict transcription factor bindings in vivo. Nucleic Acids Res., 43, 2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. et al. (2017) A powerful framework for integrating eQTL and GWAS summary data. Genetics, 207, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Hardison R.C. (2017) Accurate and reproducible functional maps in 127 human cell types via 2D genome segmentation. Nucleic Acids Res., 45, 9823–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2016) Jointly characterizing epigenetic dynamics across multiple human cell types. Nucleic Acids Res., 44, 6721–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SCZ1 (Ripke et al., 2013) and SCZ2 (Ripke et al., 2014) GWAS summary data are available at the PGC web site https://www.med.unc.edu/pgc/results-and-downloads. TWAS- and eQTL-based weights can be obtained from http://gusevlab.org/projects/fusion/. Our user-friendly software, and some information on the processed mQTLs, enhancer–promoter interactions and the 1000 Genomes reference panel, are available at wuchong.org/egmethyl.html.