

Abstract
Total genetic map length and local recombination landscapes typically vary within and across populations. As a first step to understanding the recombination landscape in the domestic horse, we calculated population recombination rates and identified likely recombination hotspots using approximately 1.8 million SNP genotypes for 485 horses from 32 distinct breeds. The resulting breed-averaged recombination map spans 2.36 Gb and accounts for 2939.07 cM. Recombination hotspots occur once per 23.8 Mb on average and account for ∼9% of the physical map length. Regions with elevated recombination rates in the entire cohort were enriched for genes in pathways involving interaction with the environment: immune system processes (specifically, MHC class I and class II genes), responses to stimuli, and serotonin receptor pathways. We found significant correlations between differences in local recombination rates and population differentiation quantified by FST. Analysis of breed-specific maps revealed thousands of hotspot regions unique to particular breeds, as well as unique “coldspots,” regions where a particular breed showed below-average recombination, whereas all other breeds had evidence of a hotspot. Finally, we identified relative enrichment (P = 5.88 × 10−27) for the in silico–predicted recognition motif for equine PR/SET domain 9 (PRDM9) in recombination hotspots. These results indicate that selective pressures and PRDM9 function contribute to variation in recombination rates across the domestic horse genome.
Meiotic recombination is a major driving force in shaping genetic variation and haplotype structure in sexually reproducing eukaryotes. In most species studied to date, recombination events are not uniformly distributed along chromosomes but tend to cluster in small regions termed “hotspots” (Petes 2001; Paigen and Petkov 2010). Fine-scale recombination rates and hotspots are poorly conserved across mammalian species and can differ significantly between even closely related species (Winckler et al. 2005; Smagulova et al. 2016; Stevison et al. 2016), a phenomenon primarily attributed to variations in the DNA-binding specificity of the zinc finger array present in the PR/SET domain 9 (PRDM9) protein (Baudat et al. 2010; Grey et al. 2011). PRDM9 binding results in histone H3 lysine 4 trimethylation, a modification associated with recombination initiation via formation of a double-strand break (DSB) (Buard et al. 2009). In species with functional copies of the PRDM9 gene, total genetic map length and local recombination rates vary between individuals, as well as across populations (Crawford et al. 2004; Coop et al. 2008; Keinan and Reich 2010; Ma et al. 2015; Petit et al. 2017), indicating that recombination landscapes are rapidly evolving in these species and may be subject to selective pressures.
Several factors are associated with a reduction in recombination. In humans, rates tend to be lower within transcribed regions of genes (Myers et al. 2005) and in regulatory domains (Liu et al. 2017), possibly because of increased mutation frequency associated with DSBs. It is also well known that chromosomal inversions result in suppressed regional recombination when heterozygous (Sturtevant 1921; Jaarola et al. 1998), a proposed mechanism by which speciation arises in the presence of gene flow (Noor et al. 2001; Rieseberg 2001). Regions showing little or no recombination (“coldspots”) also occur in association with copy number variants (CNVs) in mice (Morgan et al. 2017). Finally, although population-based estimates of recombination rates are limited by the fact that genetic variation is necessary to detect historical recombination events, a downward bias in these rate estimates is often present in regions of the genome under selection (O'Reilly et al. 2008), an area of particular interest in the study of domestication and selective breeding of livestock.
The horse is a unique species for which domestication has resulted in numerous breeds specialized for the purpose of performing work or participating in athletic competition, and genomic resources have been under development to enable investigation of the genetic bases for such traits. Low-resolution linkage maps for the domestic horse were first created using microsatellite markers (Guérin et al. 1999; Swinburne et al. 2000, 2006; Penedo et al. 2005), paving the way for mapping coat color traits and several diseases of economic importance to the horse industry (Swinburne et al. 2002; Pielberg et al. 2005; Brunberg et al. 2006; Tryon et al. 2007; McCue et al. 2008). Since that time, linkage maps and microsatellite markers have given way to genome-wide SNP chip data and high-throughput sequencing, allowing an even closer look at genetic variation in the horse. Very little is known about fine-scale recombination and its determinants in the domestic horse, although several studies have reported relevant findings. Steiner and Ryder (2013) found evidence for positive selection in the zinc fingers of the PRDM9 gene across several species of equids. They also identified multiple PRDM9 alleles in the domestic horse and hybrids thereof, indicating that there may be population-specific differences in recombination rates and usage of recombination hotspots in this species. In addition, structural variation in the equine genome has long been a subject of interest to the equine genetics community, leading to the identification of thousands of CNVs (Doan et al. 2012; Metzger et al. 2013; Ghosh et al. 2014) with possible ties to phenotypes such as body size and sexual development, as well as a chromosomal inversion on equine Chromosome (ECA) 3 responsible for the Tobiano coat color phenotype (Brooks et al. 2007). Because the horse is a domestic species with several breeds possessing unique distinguishing phenotypes such as gaitedness and athletic performance, investigations of the genetic targets of natural and artificial selection in the horse have resulted in several discoveries providing new insights into selective breeding's functional consequences (Petersen et al. 2013b, 2014; McCoy et al. 2014; Avila et al. 2018). The impacts of the PRDM9 gene, structural variants, and selective pressures on the local recombination landscape in the horse remain unknown.
The recent development of a 2-million-marker SNP array (Schaefer et al. 2017) allows for fine-scale investigation of recombination rate variation across the equine genome, as historical recombination rates can be estimated by coalescent analysis of linkage disequilibrium (LD) patterns in high-density SNP data (Auton and McVean 2007). These estimates can help identify putative recombination hotspots and compare local recombination rate differences between populations, as well as improve genotype imputation accuracy from lower- to higher-density SNP arrays. Here we report creation of fine-scale recombination maps and predicted hotspot locations for the domestic horse as well as 12 individual breeds. We assess relative enrichment for the in silico predicted DNA recognition motif for equine PRDM9 in predicted recombination hotspots compared with genomic regions without evidence of hotspot activity. Finally, we investigate recombination rate distribution across the equine genome as it relates to genes and gene function and compare recombination rates across breeds.
Results
Recombination rate distribution across the equine genome
We estimated fine-scale population recombination rates (4Ner) with the LDhat program rhomap, which uses a coalescent model-based approach to infer historical recombination rates between SNP markers from LD patterns (Auton and McVean 2007). Our data set consisted of 1,931,359 SNP genotypes for 485 horses from 32 distinct breeds (Schaefer et al. 2017). We constructed a breed- and sex-averaged recombination map, as well as 12 breed-specific maps for breeds in which we had genotype data for at least 18 individuals (i.e., Arabian, Belgian, Franches-Montagnes, French Trotter, Icelandic, Lusitano, Maremmano, Morgan, Quarter Horse, Standardbred, Thoroughbred, and Welsh Pony). To convert 4Ner estimates to centimorgans (cM) for all maps, we estimated contemporary effective population sizes (Ne) for all equine chromosomes via gradient boosting regression using coalescent simulations modeling the demographic history of the horse. True and estimated Ne values had a Pearson's correlation coefficient of 0.85 (95% CI: 0.83–0.87; P < 2.2 × 10−16) (Supplemental Fig. S1) based on 1000-fold cross-validation. Ne estimates for the domestic horse data set ranged from 6843 for ECA11 to 9836 for ECA22, with a genome-wide average of 8460. Individual breed estimates varied from 1784 in the Thoroughbred to 6516 in the Quarter Horse (Supplemental Table S1).
Our fine-scale breed- and sex-averaged recombination map spans ∼2.36 Gb and accounts for 2939.07 cM, making the average genome-wide recombination rate 1.24 cM/Mb (Table 1). Chromosomal recombination rates range from 1.06 cM/Mb on ECA15 to 2.13 cM/Mb on ECA12 (Table 1), with smaller autosomes generally showing higher rates. The Pearson's correlation coefficient for log of autosome size in megabases versus recombination rate (in cM/Mb) was −0.61 (P = 0.0002972). Approximately 75% of recombination occurs in 40% of the genome (Fig. 1A), illustrating the nonuniform nature of recombination distribution. Further, the probability of recombination events within transcribed regions of genes is relatively low, with a symmetrical increase in variability in recombination estimates as distance increases from the nearest start codon (Fig. 1B). We identified a significant weak correlation (linear regression R2 = 0.00314; P < 2.2 × 10−16) between recombination rate and per-site nucleotide diversity. Supplemental Figure S2 shows that recombination rates along each equine chromosome vary considerably across chromosomes on a fine scale.
Table 1.
Physical and genetic distance, mean recombination rate, and number of hotspots identified by chromosome
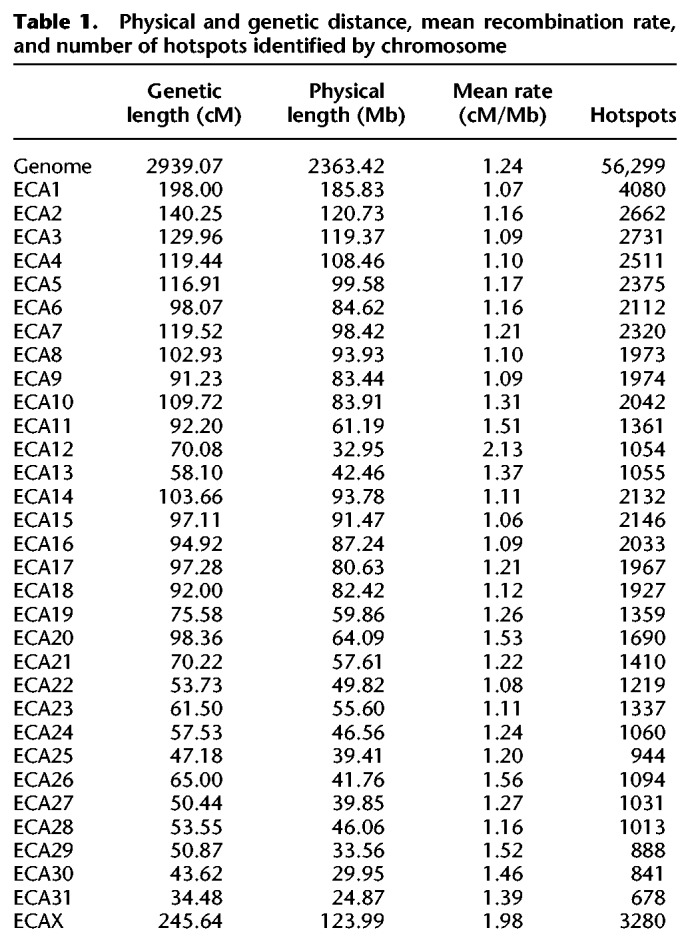
Figure 1.

Recombination rate distribution. (A) Proportion of total recombination versus proportion of sequence covered, sorted by highest inter-recombination rate estimates. (B) Recombination rate as a function of distance from the nearest start codon in 1-kb bins.
We used the g:GOSt function of g:Profiler (Reimand et al. 2016) to assess functional enrichment of genes associated with Gene Ontology (GO) terms (The Gene Ontology Consortium 2000) in regions with both high and low recombination rates (Supplemental Table S2). The top 500 genes by recombination rate ranged from 4.77 to 15.70 cM/Mb with an average rate of 6.28 cM/Mb, whereas the bottom 500 genes ranged from 0.14 to 0.20 cM/Mb with an average rate of 0.19 cM/Mb. Genes with elevated recombination rates were enriched for immune system process terms, specifically major histocompatibility class I and class II complex genes (Fig. 2; Supplemental Table S3). Other significant GO terms with high recombination rates included immunoglobulin production, antigen processing and presentation, immune response, serotonin receptor signaling pathway, amide binding (peptide antigen binding), G-protein-coupled serotonin receptor activity, sulfuric ester hydrolase activity, and G-protein-coupled receptor activity (Supplemental Table S3). Significant GO terms with low recombination rates included the intrinsic apoptotic signaling pathway in response to oxidative stress, regulation of membrane lipid distribution, H3 histone acetyltransferase complex (MOZ/MORF histone acetyltransferase complex), and intracellular organelle/intracellular membrane-bound organelle (Supplemental Table S4).
Figure 2.

Recombination rates across ECA20 with zoom window including predicted hotspots and protein-coding genes located in the region spanning the MHC class I and MHC class II loci.
Recombination hotspots and PRDM9
We used 4Ner estimates to predict recombination hotspots using the LDhot program (Auton et al. 2014), which defines hotspots based on likelihood ratio tests comparing a constant recombination rate model to a model with a 1-kb hotspot in the center of 100-kb sliding windows. Hotspots found within the entire data set range in width from 1.0 to 185.0 kb with an average of 3.8 kb (Supplemental Table S5). These regions occur once per 23.8 Mb on average and account for ∼217.1 Mb in total distance, ∼9% of the physical map length of the equine genome. In contrast, hotspot regions account for 705.6 cM, or 24.0% of the genetic map length. Recombination rates in predicted hotspot regions averaged 3.4 cM/Mb, 2.74 times the genome-wide average.
To assess the involvement of PRDM9 in equine recombination hotspots, we extracted EquCab2 (Wade et al. 2009) DNA reference sequence from predicted hotspot regions and regions of similar size with below-average recombination rates. We compared these regions for the presence of PRDM9's Cys2His2 zinc finger domain DNA recognition motif (Fig. 3) using analysis of motif enrichment (AME) (McLeay and Bailey 2010), revealing a relative enrichment (P = 5.88 × 10−27) for the in silico predicted equine PRDM9 recognition motif in hotspot regions.
Figure 3.

Zinc finger array for the equine reference PRDM9 protein (top) and corresponding in silico–predicted 31-mer DNA recognition sequence (bottom). Each colored box represents a single zinc finger, with distinct zinc fingers represented in different colors. Letters within the boxes signify DNA-contacting amino acids at the −1, 2, 3, and 6 positions. Dotted lines lead to the portion of the DNA sequence motif recognized by each zinc finger.
Breed-specific maps
Breed-specific maps varied in length from 2539.56 cM in the Arabian (genome-wide rate = 1.07 cM/Mb) to 5243.97 cM in the Icelandic (genome-wide rate = 2.22 cM/Mb) and showed significant variation in local recombination rates across breeds (Fig. 4A). We compared recombination dissimilarity matrices for each chromosome with mean pairwise fixation indices (FST) by a Mantel test, establishing a significant positive correlation (r = 0.67–0.84; P = 0.0001–0.0016) between local recombination rates and population differentiation quantified by FST (Fig. 4B; Supplemental Fig. S3).
Figure 4.

Fine-scale recombination rate variation on ECA7. (A) Recombination rate variation along ECA7 in five breeds. (B) Pairwise FST versus recombination dissimilarity (1 − Spearman's rank correlation coefficient) across ECA7.
Analysis of breed-specific recombination hotspots revealed thousands of hotspot regions unique to a particular breed, as well as unique “coldspots,” or regions where one breed showed below-average recombination, whereas all other breeds showed evidence of hotspot activity (Supplemental Table S6; Supplemental Figs. S6–S17).
We calculated and compared breed-specific genic recombination rates (Supplemental Table S2) for functional enrichment using g:Profiler (Reimand et al. 2016). Most enriched GO terms are shared across all or many breeds for genes with both high and low recombination rates and reflect findings from the breed- and sex-averaged map. However, there were several GO enrichments that were unique to a particular breed (Supplemental Figs. S4, S5). Notable examples include enrichment of organic acid and anion transmembrane transporter activity in regions of elevated recombination rates in the Standardbred and negative regulation of metabolic processes in regions of low recombination rates in the Quarter Horse. We also found several breed-specific recombination hotspots and coldspots that overlap with regions previously reported as signatures of selection based on genetic divergence from other breeds in work by Petersen et al. (2013b) for nine breeds included in both studies (Supplemental Figs. S6–S17).
Discussion
Here we report the first look at fine-scale recombination rate variation in the domestic horse, a species for which genetic bases of health, disease, and performance traits are being increasingly discovered as new resources become available to study genome structure and function. We used SNP data from 1,931,359 loci in 485 horses to estimate effective population sizes, calculate population recombination rates, and predict recombination hotspots across the equine genome. To investigate these results further, we analyzed known functions of the genes with the highest and lowest recombination rates across the genome. Finally, we identified differences in recombination rates and hotspot usage across 12 different breeds.
We estimate contemporary effective population size for the domestic horse at 8460. Our breed-specific Ne estimates are much higher than previously reported in many of the same breeds (Poncet et al. 2006; Corbin et al. 2010; Janssens et al. 2010; Campana et al. 2012; Vicente et al. 2012; Petersen et al. 2013a; Lee et al. 2014). This discrepancy is likely because of differences in methods used, which ranged from pedigree-based analyses to using genome-wide SNP array data with varying sample sizes. Ne is a difficult parameter to estimate, sometimes leading to confidence intervals spanning multiple orders of magnitude, and estimates based on genetic markers are relative (Wang 2005; Waples 2006). Previous SNP arrays for the horse had much lower SNP density than the array used in this study and were developed with strong bias in favor of common SNPs with the overall goal of high informativeness (minor allele frequency [MAF] ≥0.05) in many breeds (McCue et al. 2012). Further, the SNPs included in these arrays have relatively poor informativeness in draft and pony breeds (McCue et al. 2012; Schaefer et al. 2017). These features are likely to lead to underestimation of genetic diversity across and within breeds and downward biases in SNP-based estimates of Ne. Similar discrepancies in Ne estimates between methods and marker sets for an animal breed can be seen in other agricultural species, in particular, Holstein cattle (Kim and Kirkpatrick 2009; Boitard et al. 2016) and Lacaune sheep (Kijas et al. 2012; Petit et al. 2017). Further, correction for Ne in this study results in reasonable genome-wide recombination rate figures both within and across breeds that are largely in agreement with existing equine linkage maps, as described below.
On the genome-wide scale, our recombination map length of 2939.07 cM falls between that of the two most recent existing linkage maps for the horse (2772 cM by Swinburne et al. [2006] and 3740 cM by Penedo et al. [2005]), an indication of relative agreement between these different methods despite the differences between studies in populations used and density of marker sets genotyped. The map created by Penedo et al. (2005) is a male-specific map, but it is longer than both the sex-averaged map created by Swinburne et al. (2006) and the map created as a part of this study. In mammals, genome-wide recombination rates are generally higher in females than in males (Kong et al. 2010; Stapley et al. 2017), with the notable exceptions of cattle (Ma et al. 2015) and sheep (Johnston et al. 2016). Although the biological significance of this discrepancy is unknown, one proposed mechanism is intense artificial selection on males of these species leading to indirect selection for higher recombination rates in males (Ma et al. 2015). Collectively, our results, in conjunction with the previous linkage maps, suggest that this explanation could also extend to the horse, a species subjected to strong sex bias during domestication and traditional focus on stallions in selective breeding programs (Lindgren et al. 2004; Wallner et al. 2013). Because our recombination rate estimates are based on LD patterns in SNP data from unrelated individuals, we are unable to estimate differences between male and female recombination rates in the horse using this data set.
Our average genome-wide recombination rate of 1.24 cM/Mb is comparable to findings in other mammals (Stapley et al. 2017). At the chromosome level, we found a significant correlation between autosome size and recombination rate, a result that has been reported in other species as well (Kaback et al. 1992; Jensen-Seaman et al. 2004; Johnston et al. 2016; Stapley et al. 2017). In the equine genome, ∼75% of recombination occurs in 40% of sequence, resembling figures found in other mammals (Myers et al. 2005; Petit et al. 2017).
We identified 56,299 putative recombination hotspot regions in the equine genome. This is similar to the number of hotspots detected in other mammals with functional PRDM9 genes (The International HapMap Consortium 2007; Brunschwig et al. 2012; Petit et al. 2017). We also found that hotspot regions account for 9% of the physical length of the equine genome. This proportion falls between that found in humans at 6% (The International HapMap Consortium 2007) and mice at ∼13% (Brunschwig et al. 2012). Within these hotspot regions, we found significant enrichment for the in silico predicted DNA recognition motif for PRDM9 compared with regions with below-average recombination rates, suggesting the possibility that PRDM9 may play a role in hotspot determination in the horse.
The GO term analysis in regions with elevated recombination shows enrichment for genes associated with pathways involving interactions with the environment; in particular, immune system processes, responses to stimuli, and serotonin receptor signaling pathways. This is consistent with previous findings suggestive of historical diversifying selection in immune-related and olfactory receptor genes in the horse (Orlando et al. 2013). Genomic regions containing MHC complex genes have been shown in other species to be more genetically diverse and have relatively higher recombination rates and hotspot densities than the genome average (Jeffreys et al. 2001; Schaschl et al. 2006; The 1000 Genomes Project Consortium 2010; Fulton et al. 2016), reflecting the vital importance of maintaining within-species variability at these loci for antigen recognition and pathogen resistance. An animal's sense of smell serves a similar purpose to its immune system, helping it to recognize and distinguish thousands of different odorant molecules as a means of gathering information (Young and Trask 2002). Although analysis of our breed-averaged map did not show enrichment for olfactory receptors in highly recombining regions, the majority of individual breeds showed significant enrichment for genes responsible for sensory perception of smell. These results echo those found in a study of the genetic determinants of recombination in sheep (Petit et al. 2017). In contrast, genes with historically low recombination rates in the horse had fewer enriched GO terms but were primarily involved in intracellular processes and as parts of organelles. A similar pattern exists in humans (The International HapMap Consortium 2007). These findings may correspond to selection against recombination in regions of the genome containing more highly conserved genes. However, it is important to consider that our approach is limited by the fact that genetic variation and heterozygosity within a population are required in order to detect recombination events, so these regions of low recombination could potentially represent regions of low variation rather than regions with truly suppressed recombination.
We were able to identify trends in local recombination rate estimates across 12 different horse breeds. However, given the number of breeds and the complexity of the data presented here, a full detailed description of breed-specific recombination maps is beyond the scope of this study. Individual breeds showed positive correlations between recombination rate differences and population differentiation quantified by FST, similar to results found in humans (Keinan and Reich 2010; Laayouni et al. 2011). This variability may be in part a result of variation in demographic history between populations, as 4Ner estimates are sensitive to these differences (Dapper and Payseur 2018). Icelandic horses had a much higher genome-wide recombination rate in this study than any of the other breeds analyzed. It is important to note that we generated both 4Ner and Ne estimates in this breed based on SNP data from the smallest sample size (n = 18) of all breeds for which a breed-specific map was made. As such, it is likely that the discrepancy is in part because of errors in the Ne estimate used to normalize recombination rates. We identified thousands of hotspots unique to a breed, illustrating significant within-species variation in hotspot usage in the horse. Significant GO terms associated with genes with low recombination rates unique to a breed as well as breed-specific coldspots may represent targets of selection resulting from downward bias in 4Ner estimates because of local reductions in Ne.
Overall, our findings add to the growing body of knowledge surrounding fine-scale recombination rate variability within and across species. We have identified several areas warranting further investigation. Recombination rate estimates based on LD patterns represent historical recombination events and are influenced by demographic history, fluctuations in SNP density, and variability in Ne on a finer scale than just by chromosome. Large-scale pedigree-based recombination analyses using dense SNP data are necessary to resolve these possible confounding factors. This approach would also allow for the investigation of total map length and fine-scale recombination differences between male and female horses. Additionally, hotspots and coldspots presented here are computationally predicted and have not yet been experimentally validated to rule out false positives. We show that PRDM9 is likely involved in recombination hotspot determination in the horse and that local recombination landscape varies considerably by breed, but it is not yet known whether population-specific PRDM9 alleles exist that may influence hotspot usage at a breed level. Further characterization of this gene and its effects on recombination in a larger population would help resolve this question.
Methods
Data set
Genotype data from 485 horses (235 male; 281 female) representing 32 distinct breeds were previously generated as a part of the development of a high-density SNP array for the horse (Schaefer et al. 2017). Briefly, whole-genome sequence data compiled from 153 horses representing 24 separate breeds were used to discover approximately 23 million biallelic candidate SNPs. After quality control and filtration based on breed representation, even spacing across the genome, and probe design considerations, 2,001,826 SNPs were selected for the Affymetrix equine MNEc2M SNP array. A cohort of 332 horses from 20 breeds was genotyped using this array to serve as a reference population for genotype imputation and creation of a recombination map (Schaefer et al. 2017). Of these SNPs, 70,467 were located on unplaced contigs and therefore excluded from this study, leaving 1,931,359 SNPs for analysis. Whole-genome sequence SNPs from the variant discovery cohort (NCBI BioProjects PRJEB14779, PRJNA273402, and PRJEB10098) were masked to the MNEc2M SNP loci and combined with the array data (genotypes available at https://www.animalgenome.org/cgi-bin/util/vcf) to create a uniform set of SNPs on Chromosomes 1 through 31 and X from 485 total horses. Genotypes were phased with Beagle version 4.0 (Browning and Browning 2007) to fill in missing genotypes and predict haplotypes.
Recombination rate estimation and hotspot detection
One hundred random samples of 40 chromosomes from the data set described above were generated to estimate population recombination rates (4Ner) using the rhomap program in the LDhat package (Auton and McVean 2007). Likelihood lookup tables for all analyses were generated from the table included with the software package, assuming a mutation rate (θ) of 0.001 per site per generation. Chromosomes were split into intervals of 2000 SNPs with 200-SNP overlaps between adjacent windows. Each analysis was run for 10,100,000 total iterations, sampling every 2000 iterations, with a 100,000-iteration burn-in period. To generate a breed- and sex-averaged recombination map, rates were averaged across the random samples. Breed-specific maps were generated following the above procedure in individual breeds with genotypes for 18 or more individuals (Arabian, Belgian, Franches-Montagnes, French Trotter, Icelandic, Lusitano, Maremmano, Morgan, Quarter Horse, Standardbred, Thoroughbred, and Welsh Pony). Rate estimates from the first 100 SNPs on the 5′ end and last 100 SNPs on the 3′ end of overlapping intervals were discarded to achieve continual progression between consecutive windows. LDhot (Auton et al. 2014) was used to perform likelihood ratio tests over 100-kb sliding windows, comparing a constant recombination rate model to a model with a 1-kb hotspot in the center of the window, with the null distribution for each window coming from up to 1000 coalescent simulations matched for sample size, SNP density, and background recombination rate. Hotspots were considered significant at P < 0.001, with a cutoff of 0.1 for joining adjacent windows.
Effective population size estimation
Effective population size (Ne) was calculated by gradient boosting regression for the domestic horse as well as for each of the 12 breeds for which breed-specific maps were made. Gradient boosting regression is an ensemble machine learning method that uses random sampling of classifiers with replacement over weighted data to train a predictive model. Coalescent simulations modeling the demographic history of the horse were performed using cosi2 (Shlyakhter et al. 2014). Demographic parameters were randomly selected from uniform prior distributions ranging from a 100 to 20,000 contemporary effective population size (Ne), 10 to 1000 generations since domestication or breed formation bottleneck, and a 100 to 50,000 ancestral population size (Na), including a rule that Na must be greater than Ne. Recombination maps were simulated for chromosomes of random lengths matching those of equine chromosomes (i.e., 25–186 Mb), featuring varying recombination rates with gamma-distributed hotspots. SNPs generated from cosi2 were used to calculate MAF proportions in bins (MAF ≤ 0.02, 0.02–0.04, 0.04–0.06, 0.06–0.08, 0.08–0.10, 0.10–0.15, 0.15–0.20, 0.20–0.25, 0.25–0.30, 0.30–0.35, and 0.35–0.40) and pairwise LD measurements in the form of r2 in 25-kb bins up to 200 kb for each simulation. Gradient boosting regression was performed using the Python scikit-learn machine learning module (Pedregosa et al. 2011). Hyperparameters were tuned via grid search, with min_samples_leaf = 100, max_features = “sqrt,” subsample = 0.8, learning_rate = 0.1, n_estimators = 575 identified as the best combination based on mean accuracy for this data set. The effectiveness of this method for estimating Ne from our simulated data was tested via 1000-fold cross-validation. Finally, Ne was estimated for the entire population and each breed 100 times per chromosome with different random seeds, and the mean of these estimates was used to convert 4Ner estimates to centimorgans. An average genome-wide Ne estimate was determined for each breed and for the entire population by back-calculating Ne from the resulting total genetic map lengths in centimorgans and 4Ner.
Recombination rate distribution
A list of the 18,129 annotated equine genes with associated GO terms (The Gene Ontology Consortium 2000) was extracted using BioMart (Smedley et al. 2015). To prevent biased estimates of recombination rates because of differences in gene size, gene size was controlled for as in phase II of the human HapMap project (The International HapMap Consortium 2007). An average recombination rate was estimated for each gene over a region with 10 kb flanking either side of the midpoint of the transcribed region of the gene. Genes were subsequently ranked by recombination rate, and the top and bottom 500 genes were analyzed using the g:GOSt function of g:Profiler (Reimand et al. 2016) as ordered queries.
PRDM9 motif analysis
Motif enrichment analysis was used to assess relative enrichment for the DNA recognition sequence for equine PRDM9. The amino acid sequence for horse PRDM9 (Steiner and Ryder 2013) was used for de novo prediction of the DNA-binding specificity position weight matrix (PWM) of its Cys2His2 zinc finger array. This was achieved with software developed by Persikov and Singh (2014), which predicts DNA-binding motifs by calculating contact energies between the amino acids of each Cys2His2 zinc finger and all 256 possible 4-bp sequence combinations using support vector machines trained with an experimental database derived from known examples of zinc finger protein–DNA interactions. Sequences from hotspot regions (n = 56,299) predicted from the recombination map were extracted from the EquCab2 equine reference genome (Wade et al. 2009). These sequences were matched with “coldspot” sequences of identical length from the same chromosome extracted from regions with recombination rates less than half the mean rate across the chromosome. AME (McLeay and Bailey 2010) was used to determine whether the in silico predicted zinc finger DNA recognition motif for equine PRDM9 was enriched in hotspot DNA sequences relative to regions with low recombination. Sequences were scored using AME's average odds score method, and a Fisher's exact test was used to determine motif enrichment.
Breed-specific comparisons
Recombination rates were compared between the 12 breed-specific maps to identify breed-level variation. Mean pairwise fixation index (FST) values (Weir and Cockerham 1984) were calculated with VCFtools (Danecek et al. 2011) for each breed pair on each chromosome. Recombination dissimilarity was calculated by subtracting the Spearman's rank correlation coefficient of recombination rates across each chromosome from one as described by Laayouni et al. (2011). For each chromosome, symmetric FST and recombination dissimilarity distance matrices containing comparisons of each breed to all other breeds were tested for correlation using a Mantel test (Mantel 1967) as implemented in the Python package scikit-bio (scikit-bio.org) using the Pearson method with P-values determined based on 10,000 matrix permutations. Hotspots were considered unique to a breed if there was no evidence of boundary overlap with a hotspot predicted in another breed. Unique coldspots were defined as regions of below-average recombination in one breed where hotspots occur in all other breeds tested. Recombination rates were averaged for genes using the method described above. The top and bottom 500 genes by recombination rate per breed were analyzed using the g:Cocoa function of g:Profiler (Reimand et al. 2016) as ordered queries for across-breed comparison of biological function of genes in high and low recombination regions.
Data access
Recombination maps and hotspots from this study are available in the Supplemental Material and have been submitted to https://www.animalgenome.org/repository/pub/UMN2019.0219/.
Supplementary Material
Acknowledgments
We thank Dr. Kent Reed for his thoughtful suggestions during preparation of this manuscript. Research support was provided by the U.S. Department of Agriculture (USDA) NIFA project 2012-67,015-19,432, Minnesota Agricultural Experiment Station Multistate project MIN-62-090, and the National Animal Genome Project (NRSP8) through the equine genome coordinator: USDA-NRSP8 (2013–2018) horse-technical-committee coordinator funds. Salary support for S.K.B. was provided by the National Institutes of Health NRSA institutional training grant 2T32AR007612 and fellowship 5F30OD023369.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.243311.118.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, McVean G. 2007. Recombination rate estimation in the presence of hotspots. Genome Res 17: 1219–1227. 10.1101/gr.6386707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Myers S, McVean G. 2014. Identifying recombination hotspots using population genetic data. arXiv:1403.4264 [q-bio.QM].
- Avila F, Mickelson JR, Schaefer RJ, McCue ME. 2018. Genome-wide signatures of selection reveal genes associated with performance in American Quarter Horse subpopulations. Front Genet 9: 249 10.3389/fgene.2018.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. 10.1126/science.1183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard S, Rodríguez W, Jay F, Mona S, Austerlitz F. 2016. Inferring population size history from large samples of genome-wide molecular data: an approximate Bayesian computation approach. PLoS Genet 12: e1005877 10.1371/journal.pgen.1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks S, Lear T, Adelson D, Bailey E. 2007. A chromosome inversion near the KIT gene and the Tobiano spotting pattern in horses. Cytogenet Genome Res 119: 225–230. 10.1159/000112065 [DOI] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. 10.1086/521987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunberg E, Andersson L, Cothran G, Sandberg K, Mikko S, Lindgren G. 2006. A missense mutation in PMEL17 is associated with the Silver coat color in the horse. BMC Genet 7: 46 10.1186/1471-2156-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunschwig H, Levi L, Ben-David E, Williams RW, Yakir B, Shifman S. 2012. Fine-scale maps of recombination rates and hotspots in the mouse genome. Genetics 191: 757–764. 10.1534/genetics.112.141036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard J, Barthès P, Grey C, de Massy B. 2009. Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J 28: 2616–2624. 10.1038/emboj.2009.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana MG, Stock F, Barrett E, Benecke N, Barker GWW, Seetah K, Bower MA. 2012. Genetic stability in the Icelandic horse breed. Anim Genet 43: 447–449. 10.1111/j.1365-2052.2011.02266.x [DOI] [PubMed] [Google Scholar]
- Coop G, Wen X, Ober C, Pritchard JK, Przeworski M. 2008. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science 319: 1395–1398. 10.1126/science.1151851 [DOI] [PubMed] [Google Scholar]
- Corbin LJ, Blott SC, Swinburne JE, Vaudin M, Bishop SC, Woolliams JA. 2010. Linkage disequilibrium and historical effective population size in the Thoroughbred horse. Anim Genet 41: 8–15. 10.1111/j.1365-2052.2010.02092.x [DOI] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M. 2004. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet 36: 700–706. 10.1038/ng1376 [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapper AL, Payseur BA. 2018. Effects of demographic history on the detection of recombination hotspots from linkage disequilibrium. Mol Biol Evol 35: 335–353. 10.1093/molbev/msx272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan R, Cohen N, Harrington J, Veazy K, Juras R, Cothran G, McCue ME, Skow L, Dindot SV. 2012. Identification of copy number variants in horses. Genome Res 22: 899–907. 10.1101/gr.128991.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton JE, McCarron AM, Lund AR, Pinegar KN, Wolc A, Chazara O, Bed'Hom B, Berres M, Miller MM. 2016. A high-density SNP panel reveals extensive diversity, frequent recombination and multiple recombination hotspots within the chicken major histocompatibility complex B region between BG2 and CD1A1. Genet Sel Evol 48: 1 10.1186/s12711-015-0181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Qu Z, Das PJ, Fang E, Juras R, Cothran EG, McDonell S, Kenney DG, Lear TL, Adelson DL, et al. 2014. Copy number variation in the horse genome. PLoS Genet 10: e1004712 10.1371/journal.pgen.1004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Barthès P, Chauveau-Le Friec G, Langa F, Baudat F, de Massy B. 2011. Mouse PRDM9 DNA-binding specificity determines sites of histone H3 lysine 4 trimethylation for initiation of meiotic recombination. PLoS Biol 9: e1001176 10.1371/journal.pbio.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin G, Bailey E, Bernoco D, Anderson I, Antczak DF, Bell K, Binns MM, Bowling AT, Brandon R, Cholewinski G, et al. 1999. Report of the international equine gene mapping workshop: male linkage map. Anim Genet 30: 341–354. 10.1046/j.1365-2052.1999.00510.x [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. 10.1038/nature06258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarola M, Martin H, Ashley T. 1998. Direct evidence for suppression of recombination within two pericentric inversions in humans: a new sperm-FISH technique. Am J Hum Genet 63: 218–224. 10.1086/301900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Stinckens A, Schroyen M, Peeters L, De Keyser K, De Wael R, Lamberigts C, Luyten T, Ons E, Buys N. 2010. Genetic diversity in the Belgian Draught Horse breed as revealed by pedigree analysis and molecular marker data. Anim Genet 41: 205–206. 10.1111/j.1365-2052.2010.02114.x [DOI] [Google Scholar]
- Jeffreys A, Kauppi L, Neumann R. 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29: 217–222. 10.1038/ng1001-217 [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Furey TS, Payseur BA, Lu Y, Roskin KM, Chen C, Thomas MA, Haussler D, Jacob HJ. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res 14: 528–538. 10.1101/gr.1970304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SE, Bérénos C, Slate J, Pemberton JM. 2016. Conserved genetic architecture underlying individual recombination rate variation in a wild population of Soay sheep (Ovis aries). Genetics 203: 583–598. 10.1534/genetics.115.185553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback DB, Guacci V, Barber D, Mahon JW. 1992. Chromosome size-dependent control of meiotic recombination. Science 256: 228–232. 10.1126/science.1566070 [DOI] [PubMed] [Google Scholar]
- Keinan A, Reich D. 2010. Human population differentiation is strongly correlated with local recombination rate. PLoS Genet 6: e1000886 10.1371/journal.pgen.1000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, et al. 2012. Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol 10: e1001258 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kirkpatrick BW. 2009. Linkage disequilibrium in the North American Holstein population. Anim Genet 40: 279–288. 10.1111/j.1365-2052.2008.01831.x [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103. 10.1038/nature09525 [DOI] [PubMed] [Google Scholar]
- Laayouni H, Montanucci L, Sikora M, Melé M, Dall'Olio GM, Lorente-Galdos B, McGee KM, Graffelman J, Awadalla P, Bosch E, et al. 2011. Similarity in recombination rate estimates highly correlates with genetic differentiation in humans. PLoS One 6: e17913 10.1371/journal.pone.0017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Woo Lee J, Kim H. 2014. Estimating effective population size of thoroughbred horses using linkage disequilibrium and theta (4 Nμ) value. Livest Sci 168: 32–37. 10.1016/j.livsci.2014.08.008 [DOI] [Google Scholar]
- Lindgren G, Backström N, Swinburne J, Hellborg L, Einarsson A, Sandberg K, Cothran G, Vilà C, Binns M, Ellegren H. 2004. Limited number of patrilines in horse domestication. Nat Genet 36: 335–336. 10.1038/ng1326 [DOI] [PubMed] [Google Scholar]
- Liu Y, Sarkar A, Kheradpour P, Ernst J, Kellis M. 2017. Evidence of reduced recombination rate in human regulatory domains. Genome Biol 18: 193 10.1186/s13059-017-1308-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, O'Connell JR, VanRaden PM, Shen B, Padhi A, Sun C, Bickhart DM, Cole JB, Null DJ, Liu GE, et al. 2015. Cattle sex-specific recombination and genetic control from a large pedigree analysis. PLoS Genet 11: e1005387 10.1371/journal.pgen.1005387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N. 1967. The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220. [PubMed] [Google Scholar]
- McCoy AM, Schaefer J, Petersen JL, Morrell PL, Slamka MA, Mickelson JR, Valberg SJ, McCue ME. 2014. Evidence of positive selection for a glycogen synthase (GYS1) mutation in domestic horse populations. J Hered 105: 163–172. 10.1093/jhered/est075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue ME, Valberg SJ, Lucio M, Mickelson JR. 2008. Glycogen synthase 1 (GYS1) mutation in diverse breeds with polysaccharide storage myopathy. J Vet Intern Med 22: 1228–1233. 10.1111/j.1939-1676.2008.0167.x [DOI] [PubMed] [Google Scholar]
- McCue ME, Bannasch DL, Petersen JL, Gurr J, Bailey E, Binns MM, Distl O, Guérin G, Hasegawa T, Hill EW, et al. 2012. A high density SNP array for the domestic horse and extant Perissodactyla: utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet 8: e1002451 10.1371/journal.pgen.1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, Bailey TL. 2010. Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11: 165 10.1186/1471-2105-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger J, Philipp U, Lopes MS, da Camara Machado A, Felicetti M, Silvestrelli M, Distl O. 2013. Analysis of copy number variants by three detection algorithms and their association with body size in horses. BMC Genomics 14: 487 10.1186/1471-2164-14-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Gatti DM, Najarian ML, Keane TM, Galante RJ, Pack AI, Mott R, Churchill GA, de Villena FPM, et al. 2017. Structural variation shapes the landscape of recombination in mouse. Genetics 206: 603–619. 10.1534/genetics.116.197988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. 10.1126/science.1117196 [DOI] [PubMed] [Google Scholar]
- Noor MAF, Grams KL, Bertucci LA, Reiland J. 2001. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci 98: 12084–12088. 10.1073/pnas.221274498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly PFO, Birney E, Balding DJ. 2008. Confounding between recombination and selection, and the Ped/Pop method for detecting selection. Genome Res 18: 1304–1313. 10.1101/gr.067181.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando L, Ginolhac A, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, et al. 2013. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499: 74–78. 10.1038/nature12323 [DOI] [PubMed] [Google Scholar]
- Paigen K, Petkov P. 2010. Mammalian recombination hot spots: properties, control and evolution. Nat Rev Genet 11: 221–233. 10.1038/nrg2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. 2011. Scikit-learn: machine learning in Python. J Mach Learn Res 12: 2825–2830. [Google Scholar]
- Penedo MCT, Millon LV, Bernoco D, Bailey E, Binns M, Cholewinski G, Ellis N, Flynn J, Gralak B, Guthrie A, et al. 2005. International equine gene mapping workshop report: a comprehensive linkage map constructed with data from new markers and by merging four mapping resources. Cytogenet Genome Res 111: 5–15. 10.1159/000085664 [DOI] [PubMed] [Google Scholar]
- Persikov AV, Singh M. 2014. De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res 42: 97–108. 10.1093/nar/gkt890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JL, Mickelson JR, Cothran EG, Andersson LS, Axelsson J, Bailey E, Bannasch D, Binns MM, Borges AS, Brama P, et al. 2013a. Genetic diversity in the modern horse illustrated from genome-wide SNP data. PLoS One 8: e54997 10.1371/journal.pone.0054997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, Bailey E, Bannasch D, Binns MM, Borges AS, et al. 2013b. Genome -wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet 9: e1003211 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JL, Valberg SJ, Mickelson JR, McCue ME. 2014. Haplotype diversity in the equine myostatin gene with focus on variants associated with race distance propensity and muscle fiber type proportions. Anim Genet 45: 827–835. 10.1111/age.12205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD. 2001. Meiotic recombination hot spots and cold spots. Nat Rev Genet 2: 360–369. 10.1038/35072078 [DOI] [PubMed] [Google Scholar]
- Petit M, Astruc JM, Sarry J, Drouilhet L, Fabre S, Moreno CR, Servin B. 2017. Variation in recombination rate and its genetic determinism in sheep populations. Genetics 207: 767–784. 10.1534/genetics.117.300123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielberg G, Mikko S, Sandberg K, Andersson L. 2005. Comparative linkage mapping of the Grey coat colour gene in horses. Anim Genet 36: 390–395. 10.1111/j.1365-2052.2005.01334.x [DOI] [PubMed] [Google Scholar]
- Poncet PA, Pfister W, Muntwyler J, Glowatzki-Mullis ML, Gaillard C. 2006. Analysis of pedigree and conformation data to explain genetic variability of the horse breed Franches-Montagnes. J Anim Breed Genet 123: 114–121. 10.1111/j.1439-0388.2006.00569.x [DOI] [PubMed] [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. 2016. g:Profiler: a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44: W83–W89. 10.1093/nar/gkw199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol Evol 16: 351–358. 10.1016/S0169-5347(01)02187-5 [DOI] [PubMed] [Google Scholar]
- Schaefer RJ, Schubert M, Bailey E, Bannasch DL, Barrey E, Bar-Gal GK, Brem G, Brooks SA, Distl O, Fries R, et al. 2017. Developing a 670 k genotyping array to tag ∼2M SNPs across 24 horse breeds. BMC Genomics 18: 565 10.1186/s12864-017-3943-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaschl H, Wandeler P, Suchentrunk F, Obexer-Ruff G, Goodman SJ. 2006. Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 97: 427–437. 10.1038/sj.hdy.6800892 [DOI] [PubMed] [Google Scholar]
- Shlyakhter I, Sabeti PC, Schaffner SF. 2014. Cosi2: an efficient simulator of exact and approximate coalescent with selection. Bioinformatics 30: 3427–3429. 10.1093/bioinformatics/btu562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero D, Petukhova GV. 2016. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30: 266–280. 10.1101/gad.270009.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, et al. 2015. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 43: W589–W598. 10.1093/nar/gkv350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. 2017. Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Phil Trans R Soc B 372: 20160455 10.1098/rstb/2016.0455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner CC, Ryder OA. 2013. Characterization of Prdm9 in equids and sterility in mules. PLoS One 8: e61746 10.1371/journal.pone.0061746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevison LS, Woerner AE, Kidd JM, Kelley JL, Veeramah KR, McManus KF, Great Ape Genome Project, Bustamante CD, Hammer MF, Wall JD. 2016. The time scale of recombination rate evolution in great apes. Mol Biol Evol 33: 928–945. 10.1093/molbev/msv331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant A. 1921. A case of rearrangement of genes in Drosophila. Proc Natl Acad Sci 7: 235–237. 10.1073/pnas.7.8.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinburne J, Gerstenberg C, Breen M, Aldridge V, Lockhart L, Marti E, Antczak D, Eggleston-Stott M, Bailey E, Mickelson J, et al. 2000. First comprehensive low-density horse linkage map based on two three-generation, full-sibling, cross-bred horse reference families. Genomics 66: 123–134. 10.1006/geno.2000.6207 [DOI] [PubMed] [Google Scholar]
- Swinburne JE, Hopkins A, Binns MM. 2002. Assignment of the horse grey coat colour gene to ECA25 using whole genome scanning. Anim Genet 33: 338–342. 10.1046/j.1365-2052.2002.00895.x [DOI] [PubMed] [Google Scholar]
- Swinburne JE, Boursnell M, Hill G, Pettitt L, Allen T, Chowdhary B, Hasegawa T, Kurosawa M, Leeb T, Mashima S, et al. 2006. Single linkage group per chromosome genetic linkage map for the horse, based on two three-generation, full-sibling, crossbred horse reference families. Genomics 87: 1–29. 10.1016/j.ygeno.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Tryon RC, White SD, Bannasch DL. 2007. Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse. Genomics 90: 93–102. 10.1016/j.ygeno.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Vicente AA, Carolino N, Gama LT. 2012. Genetic diversity in the Lusitano horse breed assessed by pedigree analysis. Livest Sci 148: 16–25. 10.1016/j.livsci.2012.05.002 [DOI] [Google Scholar]
- Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR, et al. 2009. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 326: 865–867. 10.1126/science.1178158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner B, Vogl C, Shukla P, Burgstaller JP, Druml T, Brem G. 2013. Identification of genetic variation on the horse Y chromosome and the tracing of male founder lineages in modern breeds. PLoS One 8: e60015 10.1371/journal.pone.0060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. 2005. Estimation of effective population sizes from data on genetic markers. Phil Trans R Soc B 360: 1395–1409. 10.1098/rstb.2005.1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS. 2006. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7: 167–184. 10.1007/s10592-005-9100-y [DOI] [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, Bontrop RE, McVean GAT, Gabriel SB, Reich D, Donnelly P, et al. 2005. Comparison of fine-scale recombination rates in humans and chimpanzees. Science 308: 107–111. 10.1126/science.1105322 [DOI] [PubMed] [Google Scholar]
- Young JM, Trask BJ. 2002. The sense of smell: genomics of vertebrate odorant receptors. Hum Mol Genet 11: 1153–1160. 10.1093/hmg/11.10.1153 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.