

Abstract
Claviceps purpurea bifunctional Δ12‐hydroxylase/desaturase, CpFAH12, and monofunctional desaturase CpFAD2, share 86% of sequence identity. To identify the underlying determinants of the hydroxylation/desaturation specificity, chimeras of these two enzymes were tested for their fatty acid production in an engineered Yarrowia lipolytica strain. It reveals that transmembrane helices are not involved in the hydroxylation/desaturation specificity whereas all cytosolic domains have an impact on it. Especially, replacing the CpFAH12 cytosolic part near the second histidine‐box by the corresponding CpFAD2 part annihilates all hydroxylation activity. Further mutagenesis experiments within this domain identified isoleucine 198 as the crucial element for the hydroxylation activity of CpFAH12. Monofunctional variants performing the only desaturation were obtained when this position was exchanged by the threonine of CpFAD2. Saturation mutagenesis at this position showed modulation in the hydroxylation/desaturation specificity in the different variants. The WT enzyme was demonstrated as the most efficient for ricinoleic acid production and some variants showed a better desaturation activity. A model based on the recently discovered membrane desaturase structures indicate that these changes in specificity are more likely due to modifications in the di‐iron center geometry rather than changes in the substrate binding mode.
Keywords: hydroxylation‐desaturation specificity, membrane desaturases, modeling, mutagenesis and chimeras, Yarrowia lipolytica
Abbreviation
- %conv
Conversion percentage
- %Hyd
Hydroxylation percentage
- %LA/CpD
Percentage of LA titer compared with the titer of the wild‐type CpFAD2 expressing strain
- %LA/CpH
Percentage of LA titer compared with the titer of the wild‐type CpFAH12 expressing strain
- %RA/CpH
Percentage of RA titer compared with the titer of the wild‐type CpFAH12 expressing strain
- CpFAD2
Claviceps purpurea fatty acid desaturase
- CpFAH12
Claviceps purpurea fatty acid hydroxylase
- FA
Fatty acid
- FAD
Fatty acid desaturase
- FAH
Fatty acid hydroxylase
- FAME
Fatty acid methyl ester
- His‐Box
Histidine‐box
- LA
Linoleic acid (C18:2 Δ9cis,12cis)
- MD(s)
Membrane fatty acid desaturase(s)
- OA
Oleic acid (C18:1 Δ9cis)
- RA
Ricinoleic acid (C18:1 Δ9cis OH12)
- SCD1
Stearoyl‐CoA desaturase
- TFA
Total fatty acids
- TM
Transmembrane
- WT
Wild‐type
1. INTRODUCTION
Vegetable oils are commonly exploited for nutritional, medicinal, and/or energy purposes. Their properties depend on the kind of fatty acids (FAs) esterified on glycerol moieties. The most common FA modification is desaturation, which fulfills diverse physiological needs for the organism such as membrane fluidity. The main enzymes responsible for these modifications belong to two evolutionary distinct families called desaturases (Shanklin & Cahoon, 1998): the soluble fatty acid desaturases (Pfam: PF03405) only found in the stroma of chloroplasts and the cytosol of bacteria, and the membrane fatty acid desaturases (MDs; Pfam: PF00487) that are widespread in all organisms. Both families share an evolutionary convergence like a diiron cluster in their active site, and require an electron transfer chain as well as dioxygen for their catalytic activity.
The MDs are able to perform desaturation on a broad range of substrates (including acyls‐CoA and phospholipids), and on different FAs positions. They have been named according to the localization of the desaturation in the FA chain. For instance, the widespread Δ9 stearoyl‐CoA desaturases 1 (SCD1 family), that belong to the Δ9‐FADs, synthesize oleoyl‐CoA by introducing a double bond between carbons nine and ten on stearoyl‐CoA, whereas the Δ12‐FADs act on the bond between carbons 12 and 13, mostly on oleic acid esterified on the sn2 position of phosphatidylcholine. Some desaturases of the Δ12‐FAD family have evolved to perform modifications other than desaturation (Qiu, Sept, Joseph, Holst, & McCammon, 2001; Shanklin & Cahoon, 1998), such as hydroxylation (Broun, Boddupalli, & Somerville, 1998; Dauk, Lam, Kunst, & Smith, 2007; Meesapyodsuk & Qiu, 2008; Van de Loo, Broun, Turner, & Somerville, 1995; Zhou, Singh, & Green, 2013), epoxidation (Lee et al., 1998; Zhou et al., 2006), or “acetylenization” (Cao et al., 2013; Lee et al., 1998; Minto & Blacklock, 2008; Sperling et al., 2000). The unusual FAs synthesized like the widely used ricinoleic acid (RA; C18:1 Δ9cis OH12R; RA) often present some interesting properties for industrial applications. The hydroxyl group confers some particular properties to the fatty acid that can prove useful in high‐added value fields, such as cosmetics, lubricants, or chemistry synthon. Although MDs are more widespread than soluble desaturases, their structural data are less available due to their localization in the membrane. Only three structures of MDs have been currently described: two SCD1 (Bai et al., 2015; Wang et al., 2015) and a sphingolipid α‐hydroxylase (Zhu et al., 2015). Hence, there is no three‐dimensional structure available for the Δ12‐FAD family and the determinant(s) responsible for their uncommon enzymatic activities are unknown.
The phytopathogenic fungus, Claviceps purpurea, possesses three Δ12‐FAD‐like enzymes: CpFAD2, which preferentially produces linoleic acid (C18:2 Δ9cis‐12cis; LA) from oleic acid (C18:1 Δ9cis; OA), CpFADX, which is a ν + 3 desaturase with a broad range of substrates (Meesapyodsuk, Qiu, Reed, Covello, & Qiu, 2007), and CpFAH12, which is the only characterized fungal Δ12‐hydroxylase and has been used to produce high quantities of RA in different recombinant organisms such as yeasts, microalgae, and plants (Beopoulos et al., 2014; Kajikawa et al., 2016; Meesapyodsuk & Qiu, 2008; Yazawa, Kumagai, & Uemura, 2013). CpFAH12 is a bifunctional Δ12 hydroxylase/desaturase and as such, it produces RA as well as LA. It shares only 38–41% of amino acid identity with other plant hydroxylases (Ricinus communis, Physaria fendleri, Physaria lindheimeri, and Hiptage benghalensis) but up to 86% with CpFAD2. Hence, comparing these two closely related C. purpurea enzymes may lead to the discovery of the determinants(s) responsible for the desaturation/hydroxylation specificity.
To do so, we decided to work with the Yarrowia lipolytica oleaginous yeast which has been shown to efficiently produce RA by expressing the CpFAH12 enzyme (Beopoulos et al., 2014). To identify the determinant(s) involved in the enzyme specificity, we used a chimera strategy, a method that has proved particularly efficient to study MDs substrate or region specificity (Broun et al., 1998; Hoffmann et al., 2007; Libisch, Michaelson, Lewis, Shewry, & Napier, 2000; Lim, Senger, & Vrinten, 2014; Meesapyodsuk et al., 2007; Sasata, Reed, Loewen, &, Covello, 2004). The partition of CpFAH12 sequence was performed according to secondary structure predictions. The analysis of the chimera productions focused our attention on one particular domain where we identified an amino acid which seems to play a major role on the activity of CpFAH12. This position was submitted to a systematic substitution by all possible amino acids to better understand the role of this amino acid within the enzyme activity. We finally confronted the experimental results with those given by a three‐dimensional model to gain deeper insights of the role of this amino acid on the specificity of the enzyme.
2. MATERIAL AND METHODS
2.1. Molecular biology
The Table S1 lists the plasmids used in this study. The shuttle vector was a Jmp62 (Beopoulos et al., 2014), with kanamycin resistance for Escherichia coli amplification and Ura3 selection marker for Yarrowia lipolytica expression. The gene was expressed under the strong constitutive promoter pTEF. Plasmids containing either the wild‐type (WT) CpFAH12 or the WT CpFAD2 or a CpFAH12 chimera, and optimized for Y. lipolytica, were synthetic plasmids ordered either from GenScript (Piscataway) or Synbio Technologies (Monmouth Junction). The accession numbers of both CpFAH12 and CpFAD2 are listed in Table S2.
Site‐directed mutagenesis was performed by polymerase chain reaction (PCR) with primers ordered from Eurogentec (Liège, Belgium) listed in Table S3, on plasmid Jmp62 Ura3ex pTEF CpFAH12 or Cyt2’ _Des and with CloneAmp HiFi PCR Premix (Clontech). The thermocycler used was a T100TM from Bio‐Rad and the amplification conditions followed supplier specifications. Restrictions enzymes were ordered from New England Biolabs (Evry, France).
2.2. Bacterial and yeast strains, transformation and culture
All chemicals, reagents, and substrates were purchased from Sigma (Saint Quentin‐Fallavier, France) except if otherwise stated.
The E. coli XL1‐Blue strain from Invitrogen (Paisley, UK) was used to amplify plasmid DNA. E. coli competent cells were made with Mix & Go!TM Transformation Buffer Set from Zymo Research (Freiburg, Germany) according to the supplier specifications. After transformation, the clones were screened on LBKanamycin (10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl, 40 mg/L kanamycin) plates (with 15 g/L agar). Clones were cultured in LBKanamycin broth at 37°C during 20 hr under agitation. Plasmids extractions were performed with QIAprep Spin Miniprep Kit from Qiagen (Hilden, Germany). All sequences were checked by Sanger sequencing by Eurofins (Ebersberg, Germany) with sequencing primers (Table S3).
The Y. lipolytica chassis was derived from the modified strain JMY2159 (called OleoX) used in previous studies (Beopoulos et al., 2014). The OleoX strain is auxotrophic for leucine and uracil and combines 10 deletions: the two acyl‐CoA:diacylglycerol acyltransferase DGAT1 and DGAT2, the phospholipid:diacylglycerol acyltransferase LRO1, the oleate Δ12‐desaturase Yl.FAD2 and the six genes POX1–POX6. The deletion of POX genes disrupts the β‐oxidation which impairs the consumption of lipids. The deletion of the acyltransferases and the Δ12‐desaturase should lead to an accumulation of sn2 oleoyl‐phosphatidylcholine. The zeta docking platform was added to OleoX strain to obtain the chassis OleoX zeta (OXZ) strain, as described in Bordes, Fudalej, Dossat, Nicaud, & Marty (2007; Figure S1).
OXZ competent cells were made by Frozen‐EZ Yeast Transformation II KitTM from Zymo Research. OXZ strain was transformed with a plasmid digested by NotI‐HF to generate a cassette containing Ura3ex selection marker and the gene of interest surrounded by zeta sequence. After typically 72 hr at 30°C on YNB plates (1.7 g/L YNBwowo, 5 g/L NH4Cl, 10 g/L glucose, 50 mM NaKPO4 buffer at pH 6.8, and 15 g/L agar), transformed OXZ clones were directly picked to start an inoculum for cultures.
2.3. Ricinoleic acid production conditions in OleoX zeta strain
The inoculum used for the production of RA was prepared with freshly transformed cells. Two different clones were grown in 2 ml of YPD broth (10 g/L yeast extract, 10 g/L bactotryptone (Difco, Paris, France) and 10 g/L glucose) at 200 rpm at 28°C during 24–48 hr then the optical density (OD) was measured. The growth was performed in Erlenmeyer flasks of 100 ml containing 9 ml of YT2D5 (10 g/L yeast extract, 20 g/L bactotryptone and 50 g/L glucose, buffered at pH 4 with 100 mM citrate buffer) and 1 ml of decane (weighted). The flasks were shaken at 200 rpm at 28°C during 48 hr.
When cultures were stopped, 400 µl of decane (weighted) with 1 g/L of methyl laurate (mC12) as internal standard was added to the whole 10 ml of media, then OD was measured. The medium was centrifuged at 4,000 g during 10 min and the decane phase was transferred to a glass vial and stored at room temperature until analysis. The supernatant was discarded, and then the cell pellet was freeze‐dried, powdered by vigorous vortexing and conserved at − 20°C until analysis.
2.4. Quantification and analysis of produced fatty acids
Two different quantifications were performed: The free FAs in decane phase were quantified by silylation and the whole FAs contents in cell pellets were quantified by transmethylation.
For the silylation, 200 µl of each sample of decane phase was derivatized with 5 µl of N,O‐Bis(trimethylsilyl)acetamide (BSA, weighted) and mixed thoroughly. The reactions were incubated at 45°C during approximately 1 hr, and the samples were analyzed by gas chromatography. Whenever the concentration of a sample was out of the calibration range of 0.1–1 g/L, a dilution was realized with decane.
The method for the transmethylation of powdered freeze‐dried cells pellet was adapted from Browse, McCourt, & Somerville (1986). A solution of methanol containing 2.5% sulfuric acid (V/V) and 0.3 g/L of myristic acid (C14:0) as internal standard was prepared. About 4 ml of this solution was added to the cell pellets, and the tubes were vortexed to resuspend the powder. After 4 hr of incubation at 80°C with periodic mixing, they were cooled at room temperature. About 3 ml of NaCl at 50 g/L and 3 ml of decane with 0.3 g/L mC12 as internal standard were then added to the samples. After mixing, they were centrifuged during 10 min at 4,000 g. The organic upper phase was analyzed by gas chromatography.
Both analysis were made in GC‐FID‐MS (Thermo Fisher Scientific, Trace 1310‐ISQ LT) with TG‐5MS column (Thermo Fisher Scientific, 30 m x 0.25 mm x 0.25 µm). The injection volume was 1 µl, the SSL injector was put in split mode 1:5 at 250°C and the FID detector was at 270°C. The FID output was used for quantification. Calibration solutions used for quantification were prepared from six FAs (silylated by the previously described protocol or methylated): palmitic acid (C16:0), palmitoleic acid (C16:1 Δ9cis), stearic acid (C18:0), OA, LA, and RA. The temperature programs used for analysis of lipids are shown in Table S4.
2.5. Bioinformatics studies
PSI/TM‐COFFEE (Notredame, Higgins, & Heringa, 2000), PSIPRED (Buchan, Minneci, Nugent, Bryson, & Jones, 2013; Jones, 1999), and Interpro (Finn et al., 2017) were used to make secondary structure prediction (SSP), and Phyre2 (Kelley, Mezulis, Yates, Wass, & Sternberg, 2015) and I‐TASSER (Yang et al., 2015) were used to make predictive models also with SSP. The T‐COFFEE package was used to make alignments between the two SCD1, CpFAH12, and CpFAD2 to infer the localization of secondary structure. The alignment with the 10 MDs were made by PROMALS3D (Pei, Kim, & Grishin, 2008).
2.6. Homology modeling of CpFAH12
The CpFAH12 model was generated using the Rosetta software, which allowed us to combine threading and comparative approaches through RosettaCM protocol (Song et al., 2013). Regarding the threading approach, fragment libraries of three and nine amino residues were built using the Robetta server (Kim et al., 2004). To identify the best template for the comparative approach, we ran the sequence of CpFAH12 through HH server (Söding et al., 2005). This server uses hidden Markov motif to spot similarities within secondary structures even if sequences share a low percentage of identity. Despite sharing only 12% of identity with CpFAH12, the secondary structure of SDC1 from Mus musculus (pdb: 4YMK) (Bai et al., 2015) was identified as the best template with its perfect coverage of the residues 105–399.
Since the chosen template had such a low percentage of identity, to build a likely CpFAH12 model, we had to implement some constraints: (a) Based on the predictions of both Octopus (Viklund & Elofsson, 2008) and the TMHMM2.0 software (Möller, Croning, & Apweiler, 2001), we put a constraint on the TM helices; (b) based on the structure of SCD1, we constrained the eight histidine residues (His155, His159, His191, His194, His195, His391, His394, and His395), which, thanks to sequence alignment, had been previously identified as part of the three histidine‐box (His‐Box) motifs, to keep intact ions coordination sites. All these constraints were added to the RosettaCM protocol and 250 models of the core domain (105–399 residues) were generated. All models were evaluated with ProQM (Ray, Lindahl, & Wallner, 2010) assessment program and the model with the best score and a good His/ion coordination was selected. To mimic the real substrate of CpFAH12, we added an oleic acid by superimposing it on the stearoyl‐CoA of SCD1. The energy of new model was minimized to remove any steric clash.
2.7. Electrostatic potential calculations
Electrostatic potential maps were calculated with the Adaptive Poisson–Boltzmann Solver (APBS; Baker, Sept, Joseph, Holst, & McCammon, 2001) on the model of CpFAH12 and the mutant I198T model, using APBS default parameters (physiological salt concentration of 150 mM, temperature of 298.15 K, solvent dielectric of 78.4, and solute dielectric of 2). The Van der Waals radii and partial charges of both the protein and the Fe2+ ions were those of the AMBER ff99 force field. Solute charges were distributed onto grid points using a cubic B‐spline discretization. The molecular surface was defined by the interface between a 1.4 Å solvent probe, corresponding to the radius of a water molecule, and the solute the van der Waals radii.
3. RESULTS
3.1. Chimeras
3.1.1. Activities of CpFAD2 and CpFAH12 chassis
We checked the functionality of CpFAD2 in a modified strain of Y. lipolytica (Figure S2) and compared it with the one of CpFAH12, an enzyme that had already been successfully expressed in this strain to produce RA (Beopoulos et al., 2014). As RA is largely secreted, cultures were advantageously performed in a biphasic system using decane, thus RA remains trapped in the organic phase which limits its toxic effect. As expected, CpFAD2 did not produce any RA, but its LA titer was greater by two‐fold (220%) than the one of CpFAH12. The conversion percentage (%Conv) was around 47% in this strain, which was lower than the 72% displayed in the strain expressing CpFAH12. Interestingly, the RA produced by the CpFAH12 strain was mainly secreted ( = 93%) and was almost the only FA observed in the decane phase ( = 90%). The CpFAD2 expressing strain presented only traces of FAs in decane ( = 0.63%), probably due to cell lysis.
3.1.2. Dissection of CpFAH12 and CpFAD2 sequences
The secondary structure predictions (SSP) were performed for both CpFAD2 and CpFAH12 enzymes. First, the length and borders for the two pairs of transmembrane (TM) helices, TM1&2 (Ser99 to Ile153) and TM3&4 (Arg295 to His349), were defined from the SSP analysis (Figure 1). The cytosolic N‐term part (Met1 to Lys98) was placed before TM1&2. We chose to split the two remaining large cytosolic parts to reduce their length and to separate the His‐Boxes. The C‐terminal part of two small helices (Phe167–Leu180 and Gly377–His385) were used as borders and led to the cytosolic parts Cyt1 (Gly154 to Leu180) and Cyt3 (Thr350 to His385). The two remaining cytosolic parts were named Cyt2 (Leu181 to Ala294) and C‐term (Gly386 to Gln477). This dissection led to a total of seven main parts which had the same length in CpFAH12 and CpFAD2 (Figure 1).
Figure 1.
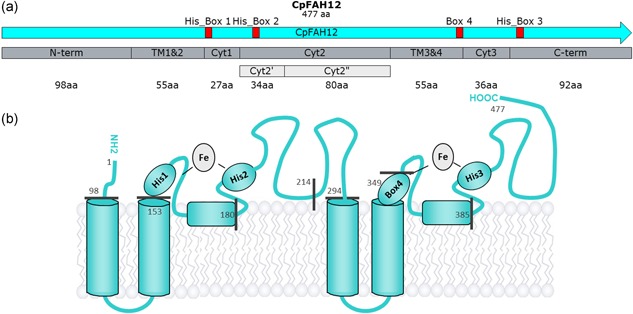
(a) Map of CpFAH12, with the partition determined by SSP in gray. The two lighter gray parts come from subpartitioning the Cyt2 part. Red boxes represent the three His‐Boxes (His_Box 1, 2, and 3) plus the hypothetical fourth box (Box 4) binding the diiron cluster. (b) Topological model. The gray numbers correspond to the positions of amino acids right before the splitting of parts. As a reminder, boxes 1 and 4 coordinate both irons and boxes 2 and 3 bind only one iron [Color figure can be viewed at wileyonlinelibrary.com]
3.1.3. Implication of TM domains in hydroxylation
We first investigated whether the TM regions were involved in the hydroxylation activity. We designed a CpFAH12 TM_Des chimera composed with a sequence of CpFAH12 in which TM1&2 and TM3&4 have been exchanged with the ones found in CpFAD2. TM regions have identical length in both enzymes but differ in their respective sequence of amino acids: The difference touches 10 amino acids in TM1&2 but only one in TM3&4 (Table S5). Switching these TM regions did not affect the FAs concentration, nor the FA profile including RA (Figure 2). Hence, the amino acids found in the TM helices are not involved in catalytic efficiency and hydroxylation/desaturation specificity in CpFAH12.
Figure 2.

Total FAs production in g/L by OXZ strain expressing either wild‐type CpFAH12, wild‐type CpFAD2, or chimeras. All chimeras are based on CpFAH12 chassis, which contains the two pairs of TM helices of CpFAD2 (TM1&2 and TM3&4), in addition to one of the five cytosolic parts of CpFAD2 (N‐term, Cyt1, Cyt2, Cyt3, C‐term, or Cyt2’’). The %Hyd ({[RA])/([RA] + [LA]}) ratio is written above in cyan. Schematic representations of enzymes in topological model are shown below: blue parts correspond to CpFAD2 sequence and cyan parts correspond to CpFAH12 sequences. color code for FA is cyan: RA, blue: LA, violet: OA, light gray: stearic acid, gray: palmitoleic acid and dark gray: palmitic acid [Color figure can be viewed at wileyonlinelibrary.com]
3.1.4. Implication of cytosolic domains in hydroxylation
We investigated the influence of cytosolic parts on hydroxylation activity with five chimeras carrying the CpFAH12 TM_Des chassis with one of the main cytosolic parts interchanged with CpFAD2 homologous regions. FAs analysis of these chimeras (Figure 2 and Table S6A) revealed a decrease of the RA titer for all chimeras. Compared with WT CpFAH12, Cyt1/TM_Des, and C‐term/TM_Des chimeras had the lowest conversion rates and a heavily impacted RA titer. The chimera Cyt2/TM_Des gave the most significant outcome as the activity of OA modification was still high but with a total abolition of the RA production and a huge increase of the LA titer. These results suggest that amino acids involved in hydroxylation specificity and activity are distributed along the whole cytosolic sequences. However, the parts containing the His‐Boxes have the greatest impact on activity, especially the Cyt2 part, which contains His‐Box no.2. Hence, some amino acids essential for the hydroxylation specificity of CpFAH12 must be located in this region.
3.2. Identification of a key residue for hydroxylation activity
We further investigated the role of the rather large Cyt2 part by splitting it in two parts (Figure 1). A predicted helix (Phe207–Gln214) served as border and led to the split in Cyt2’ (Leu181 to Gln214) and Cyt2” (Gln215 to Ala294). Each part differed by only four amino acids between CpFAH12 and CpFAD2 (Figure S3 and Table S5). Compared with WT CpFAH12, the chimera CpFAH12 Cyt2’’/TM_Des showed a decrease in its titer of RA and LA (Figure 2 and Table S6A), albeit less remarkable than the one observed with Cyt2/TM_Des chimera.
To identify which residue(s) were essential for hydroxylation/desaturation specificity, reciprocal‐replacements of one, three or all of the four positions of Cyt2’ in CpFAH12 – namely Gly197, Ile198, Asn200, and Glu213 (GINE) by CpFAD2 Ala197, Thr198, His200, and Ala213 (ATHA) – were made. The FA titer of OXZ strains expressing these mutants are shown in Figure 3 (Table S6B).
Figure 3.

Total FAs production in g/L by OXZ strain expressing either WT CpFAH12, WT CpFAD2, or a CpFAH12 mutant in Cyt2’ part. The four mutations CpFAH12‐to‐CpFAD2 are G197A, I198T, N200H, and E213A. Schematic representations of the mutations in CpFAH12 are shown under the bar charts. Blue spheres correspond to amino acids from CpFAD2 sequence, and cyan spheres, to amino acids from CpFAH12 sequence. The chassis is CpFAH12, as indicated by cyan color. The %Hyd ({[RA])/([RA] + [LA]}) are written above in cyan. color code for FA is cyan: RA, blue: LA, violet: OA, light gray: stearic acid, gray: palmitoleic acid and dark gray: palmitic acid [Color figure can be viewed at wileyonlinelibrary.com]
The mutant CpFAH12 ATHA (corresponding to CpFAH12 Cyt2'_Des), demonstrated no hydroxylation ability but maintained a desaturation capacity. This means that at least one of the four modified amino acids is crucial for the hydroxylation/desaturation specificity.
Comparing the triple mutants with the quadruple mutant CpFAH12 ATHA, only the mutant AIHA which conserved the CpFAH12 Ile198, had a hydroxylase activity, all the mutants that conserved the threonine became monofunctional desaturases. In the simple mutants, the effects of single replacement differed according to the target position. However, the CpFAH12 I198T mutant in which the CpFAH12 Ile198 was switched to a threonine is the only one that completely lost its hydroxylation ability. Hence, the replacement of isoleucine 198 by threonine is exclusively responsible for changing the bifunctional hydroxylase/desaturase CpFAH12 activity into a monofunctional desaturase CpFAD2‐like activity. Such a drastic effect was unexpected, and to better investigate its significance, we tested all the possible amino acids at this specific position.
However, when we tried to incorporate the Isoleucine at the position 198 in the CpFAD2 desaturase to see if it would confer a hydroxylation activity, this mutation resulted in the total loss of the desaturation activity for CpFAD2 mutant. Interestingly, when the four mutations GINE were integrated in CpFAD2, it recovers a weak desaturation activity (9.5% cf. to WT CpFAD2; data not shown). This confirms that hydroxylation activity is driven by multiple determinants that are distributed all along the sequence as we proved in our chimera experiment.
3.3. Systematic exploration of position 198
The 18 other CpFAH12 variants at this position were thus constructed. The FAs titer of OXZ strains expressing these mutants are shown in Figure 4 and Table 1.
Figure 4.

Total FAs production in g/L by OXZ strain expressing either WT CpFAH12, WT CpFAD2, or CpFAH12 variant for I198 position (named I198X, where X is the replaced amino acid). The %Hyd ({[RA])/([RA] + [LA]}) are written in cyan above the RA‐productive strain. The mutants are ordered following their %Hyd. color code for FA is cyan: RA, blue: LA, violet: OA, light gray: stearic acid, gray: palmitoleic acid and dark gray: palmitic acid [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
Overview of the percentages of ricinoleic acid (RA) and linoleic acid (LA) among the total number of fatty acids (TFA) in the strain expressing WT CpFAH12, WT CpFAD2, or a CpFAH12 active variant
| Ratios of RA or LA in total fatty acid | Conversion percentage | Sample RA and LA production compared with respective WT CpFAH12 or CpFAD2 production | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||||||
| CpFAH12 | 33% | 8.7% | 61% | 100% | 100% | 36% | ||||||
| I198L | 19% | 4.7% | 34% | 41% | 39% | 14% | ||||||
| I198V | 23% | 33% | 82% | 61% | 342% | 122% | ||||||
| I198N | 9.7% | 33% | 62% | 22% | 281% | 101% | ||||||
| I198C | 9.8% | 42% | 76% | 23% | 375% | 134% | ||||||
| I198P | 0.4% | 6.9% | 10% | 0.8% | 49% | 18% | ||||||
| I198A | 1.7% | 29% | 44% | 3.3% | 222% | 80% | ||||||
| I198S | 0.7% | 25% | 36% | 1.4% | 182% | 65% | ||||||
| I198H | 0% | 18% | 25% | 0% | 137% | 49% | ||||||
| I198M | 0% | 7.9% | 11% | 0% | 59% | 21% | ||||||
| I198G | 0% | 7.6% | 10% | 0% | 56% | 20% | ||||||
| CpFAD2 | 0% | 35% | 48% | 0% | 279% | 100% | ||||||
Eight variants were found to be totally inactive, giving no traces of LA or RA. Six other variants showed either no hydroxylation activity (I198G, I198M, and I198H) or very little (I198P, I198A, and I198S), and for the former two, the desaturation activity was also really decreased. The four remaining variants, I198C, I198N, I198V, and I198L, kept a significant RA titer but none showed a better hydroxylation performance than the WT CpFAH12 strain that combines the best hydroxylation ratio of 79% and the best RA titer (0.35 g/L). Variant I198L had a similar decrease for both LA and RA titer, and thus a decreased conversion rate, whereas the hydroxylation ratio was maintained (%Hyd ratio 80%). For the three other mutants, a concomitant LA ratio augmentation with a higher (or conserved) conversion rate resulted in around a three‐fold increase in LA production (342%, 281%, and 375% for I198V, I198N, and I198C mutants, respectively) compared with the WT CpFAH12 enzyme. Finally, these three mutants presented a LA production that was higher or equal to CpFAD2. The best LA production was obtained for the I198C variant, in which LA represented 42% of the total amount of fatty acids with a total of 0.34 g/L of culture in 48 hr.
3.4. New insight from the recently resolved 3D structure of SCD
To better understand the crucial effects of position 198 on the hydroxylation/desaturation specificity, we took advantage of the recently resolved structure of the MDs and built a three‐dimensional model of CpFAH12.
3.4.1. Alignments
As CpFAH12 and the SCD1 with solved 3D structures could not be aligned directly by their sequences, the alignments were based on their secondary structures. To improve the alignment quality, we used several Δ12‐FADs of Plantae and Fungi, including CpFAD2: the secondary structure of Δ12‐FADs and SCD1 were then well aligned, and the three His‐Boxes (Figure 1, His‐Box 1–3) coordinating the two iron ions of the active site, were also perfectly aligned. For CpFAH12 and CpFAD2, this corresponds to His155‐His159 (His‐Box no1), His191‐His194‐His195 (His‐Box no2), and His391‐His394‐His395 (His‐Box no3). The alignments also showed a good conservation for a fourth putative di‐iron binding box. Recently, this box composed of a NX3H motif was identified in a SCD1 enzyme (Bai et al., 2015; Wang et al., 2015). The multiple alignments of characterized Δ12‐FADs enzyme and mutagenesis experiments (see Supplementary Results and Figures S4, S5, and S6) on the position 345 and 349 of CpFAH12, lead us to propose that in Δ12‐FADs the fourth diiron binding‐box is composed of a TX3H motif composed of the Thr345 and His349 for CpFAH12 enzyme.
The TM regions that are supposed to accommodate the carbon chain of the FAs were also very well aligned. However, both CpFAH12 and CpFAD2 sequences are longer than the others, especially compared with SCD1, the shortest of the 10 sequences (Figure 5). As SCD1 acts on acyl‐CoA and Δ12‐FADs on phospholipids, it was expected to find differences in the cytosolic regions that should be involved in the accommodation of their lipid head.
Figure 5.

Alignment with PROMALS3D of eight Δ12‐FADs of Fungi (Claviceps purpurea, Yarrowia lipolytica, and Mortierella alpina) and Plantae (Arabidopsis thaliana, Physaria fendleri, and Ricinus communis) and of the two mammalian SCD1 with available tree‐dimensional structures (accession numbers listed in Table S2). The helices are shown in cyan and TM helices are underlined. The italic cyan helix for CpFAH12 and CpFAD2 were obtained by secondary structure prediction whereas cyan helices for SCD1 were obtained directly from the structure. The red letters are the four amino acids that differs between the Cyt2’ of CpFAH12 and CpFAD2. The position Ile198 in CpFAH12 and the equivalent position in other MDs have been put in bold character. The black boxes encompass the His‐Boxes [Color figure can be viewed at wileyonlinelibrary.com]
3.4.2. 3D model
As stipulated in the methods, we generated models of the core structure (positions 105–399) of CpFAH12 by using the structure of stearoyl‐CoA desaturase from Mus musculus (mSCD1) as template. To preserve the integrity of the iron boxes, we added constraints on the three His‐Boxes. We replaced the stearoyl‐CoA (in mSCD1) by superimposing an oleate molecule in the model and the energy of the system was minimized. In this final 3D representation, the four helices are well structured (Figure 6 and Figure S6). Each iron ion is correctly coordinated by the nitrogen of their respective binding histidines (dN‐Fe = 2.0 ± 0.3 Å) and the distance between the iron ions is 8.5 Å, which is 2.1 Å longer than the one observed in SCD1 structure (Bai et al., 2015). Interestingly, the distance between the iron and the C9 or C12 carbon atom of the substrate are in the same range for SCD1 structures and CpFAH12 model (5.2 Å for both mSCD1 and hSCD1 and 4.8 Å for the CpFAH12 model) whereas the distance between the iron ions and the other carbon atom (C10 or C13) is 2 Å longer in the CpFAH12 model (4.7 Å for mSCD1, 4.3 Å for hSCD1, and 6.1 Å for CpFAH12).
Figure 6.

Pymol pictures obtained from the CpFAH12 model. (a) The whole enzyme is shown in ribbon mode in rainbow colors (From N‐term in blue to C‐term in red), the oleic acid is shown in balls representation where light gray stands for the carbon atoms, red for the oxygen, and dark gray for the irons. (b) Closer view of the di‐iron center with histidines that belong to the His‐Boxes shown in green sticks. The Ile198 is displayed in sticks at the top right corner in light gray. In both figures, hydrogen is not represented for clarity [Color figure can be viewed at wileyonlinelibrary.com]
Evaluation of all these parameters confirms that the di‐iron cluster and active site are well organized and allow going further in the understanding of mutation impacts. We thus focused our attention on the environment of the isoleucine 198, located at the end of an amphiphilic helix between two glycine residues (197 and 199, Figure S7). In the model, isoleucine 198 is covered by a loop and interacts tightly with two amino acids within it, Tyr357 and Glu360. On the other side, it is in the close vicinity of three histidines His194, His349, and His395, which coordinate the di‐iron center (3.7 Å, 4.3 Å, and 3.2 Å, respectively). We also generated the electrostatic map of the surface of the protein (Figure S8) and observed a negatively charged surface around this residue.
4. DISCUSSION
The recently obtained structures of SCD1 with stearoyl‐CoA complex (Bai et al., 2015; Wang et al., 2015) confirmed a strong interaction between the substrate and the TM regions which form the binding pocket for the FA carbon chain. In other desaturases chimeras strategies (Libisch et al., 2000; Lim et al., 2014), the authors showed that mutations in TM regions were responsible for changes in regio‐specificity and/or substrate‐length specificity, probably due to different accommodation of the substrate in the binding pocket. On the contrary, in the present study, we found that the TM regions were not involved in the hydroxylation/desaturation specificity of the CpFAH12 bifunctional hydroxylase. This can be explained by the fact the two activities share the same substrate and the same position of modification. However, we demonstrated that all cytosolic regions have an effect on hydroxylation activity, meaning that these regions may interact with one another to sustain good enzyme functionality. This shows that hydroxylation activity is driven by multiple determinants that are distributed all along the sequence.
While working with chimeras to spot the determinant responsible for its bifunctional activity, CpFAH12 Cyt2/TM_Des drew our attention. While its LA titer remained similar to those observed in CpFAD2, the loss of all its hydroxylation capacity indicated that the large Cyt2 region played a major role in the hydroxylation function. When we further investigated the Cyt2 region through mutagenesis experiments to narrow down the crucial sequence responsible for this hydroxylation function, we identified isoleucine 198 as the key residue for this activity and we performed site saturation mutagenesis on this residue. Among the eight amino acids that led to inactive mutants, all were bulky (Phe, Tyr, Trp, and Gln) or charged (Asp, Glu, Lys, and Arg). Substitution by large hydrophobic amino acids like isoleucine, leucine or valine maintained a reasonable ratio of hydroxylation. The smaller hydrophobic amino acid alanine greatly diminished both the hydroxylation ratio and enzyme activity. Interestingly in the Physaria fendleri bifunctional hydroxylase (PfFAH12), this position (which corresponds to Asn149) had been formerly identified as having a role in the hydroxylation specificity of the enzyme (Broun, Boddupalli, & Somerville, 1998). The alignments of Δ12‐FADs at this position reveal that it is always a threonine that is found except for the ones having a hydroxylation activity in which either an isoleucine is found (RcFAH12 and CpFAH12), or an asparagine (PfFAH12 and P. lindheimeri FAH12; Dauk et al., 2007). Since lines of hydroxylases do not share a common ancestor, the factors involved in the desaturation/hydroxylation specificity should differ from one organism to the next. However, the systematic mutation of this position in hydroxylases of different organisms suggests a convergent evolution, which in turn, implies that this position is crucial for the hydroxylation mechanism.
To gain deeper insights into the role of this isoleucine in the hydroxylation, we generated a model of the enzyme based on the recently resolved structure of SCD1 (Bai et al., 2015; Wang et al., 2015). To our knowledge, only two papers have proposed a model for Δ12‐FADs to perform studies on the molecular determinant implicated in the Δ15 desaturase activity for bifunctional fungal Δ12/Δ15 desaturases (Cai, Yu, Liu, Liu, & Shanklin, 2018; Rong et al., 2018). Even if our model gives some pertinent global information on the general fold of the protein, the positioning of the substrate, the position of the iron atoms, and even on the position of the amino acid we identified, we have to keep in mind that it is still a model with a limited accuracy, as it is based on a limited data set of only two structures of distantly related enzymes in terms of sequence, cofactor and activity.
In the model, the OA substrate was docked in the active site with the C12–C13 carbon positioned in a catalytically relevant distance from the two iron ions. It showed that the isoleucine 198 has no contact with the substrate but is located at 4.3 Å from one of the iron ion and in the close vicinity of three histidines that coordinate the two iron atoms. If we refer to the two 3D structures of SCD1, the equivalent position of CpFAH12 Ile198 bears a serine that is buried in a groove on the protein external surface. Bai et al. (2015) suggest that this groove, localized in a predominantly positive surface near the active site, may be the place where the electron transfer occurs with cytochrome b5. An analysis of the electrostatic surface of the CpFAH12 protein does not show a positively charged region near the isoleucine 198. However, this surface of interaction was not found in the related structure of ScS7p sphingolipid α‐hydroxylase (Zhu et al., 2015) and thus the interaction with redox partners could be located elsewhere. Interestingly, the electrostatic surface map shows a positive pocket located 15–20 Å far from Ile198.
We have shown that Ile198 was densely packed with three histidines belonging to His‐Boxes (His194, His349, and His395). Mutation of this residue may modify the location of these histidines and could explain the drastic impact observed on enzyme activity and specificity. It is difficult to go deeper in the reason the hydroxylation/desaturation ratio is modified but the di‐iron center geometry is certainly modified. This could lead to several modifications, such as an effect on the redox potential of iron atoms, the distance between iron atoms, and the substrate or their distance with the dioxygen that could all explain the changes in activity and specificity. Mutation of the adjacent and flexible Gly197 by an alanine (mutant CpFAH12 G197A) has also effect on the activity and specificity. It is possible that modification of this flexible residue will generate displacement in the Ile198 position.
For FAD2 enzymes, it is commonly accepted that the desaturation and hydroxylation reactions share a first common oxidation reaction at the carbon closest to the C1 and that the divergence between these two reactions occurs in a second step (Behrouzian & Buist, 2003; Buist, Behrouzian, Alexopoulos, Dawson, & Black, 1996). Interestingly, our observations fit this mechanism since the same distance is found between this carbon (the C9 or C12 of SCD1 or CpFAH12, respectively) and the iron center for both the CpFAH12 hydroxylase and SCD1 desaturases. The divergence between the two reactions could perhaps be explained by the distance between the other iron ion and the other carbon of the substrate (C10 of SCD1 or C13 of CpFAH12, respectively) that is higher in our model of the bifunctional CpFAH12 than in SCD1 desaturases.
In conclusion, we found that the hydroxylase activity of CpFAH12 does not originate from the transmembrane helices, but is dispatched in the cytosolic parts. The isoleucine at position 198 is essential for the hydroxylation specificity, but substituting it by any other amino acid do not improve the RA titer in any CpFAH12 mutant. The localization of this residue suggests that hydroxylation specificity does not depend on substrate accommodation in the binding pocket, but more likely results from a modification of the geometry of the di‐iron center.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
This study was supported by the INSA of Toulouse, the doctoral school SEVAB and the Occitanie region. Special thanks to ICEO Platform and Nelly MONTIES for the GC‐MS support. We are also grateful to Gilles Truan for the interesting conversation we had about the diiron‐enzymes.
Robin J, Gueroult M, Cheikhrouhou R, et al. Identification of a crucial amino acid implicated in the hydroxylation/desaturation ratio of CpFAH12 bifunctional hydroxylase. Biotechnology and Bioengineering. 2019;116:2451–2462. 10.1002/bit.27102
References
REFERENCES
- Bai, Y. , McCoy, J. G. , Levin, E. J. , Sobrado, P. , Rajashankar, K. R. , Fox, B. G. , & Zhou, M. (2015). X‐ray structure of a mammalian stearoyl‐CoA desaturase. Nature, 524(7564), 252–256. 10.1038/nature14549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, N. A. , Sept, D. , Joseph, S. , Holst, M. J. , & McCammon, J. A. (2001). Electrostatics of nanosystems: Application to microtubules and the ribosome. Proceedings of the National Academy of Sciences, 98(18), 10037–10041. 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrouzian, B. , & Buist, P. H. (2003). Mechanism of fatty acid desaturation: A bioorganic perspective. Prostaglandins, Leukotrienes and Essential Fatty Acids, 68(2), 107–112. 10.1016/S0952-3278(02)00260-0 [DOI] [PubMed] [Google Scholar]
- Beopoulos, A. , Verbeke, J. , Bordes, F. , Guicherd, M. , Bressy, M. , Marty, A. , & Nicaud, J. ‐M. (2014). Metabolic engineering for ricinoleic acid production in the oleaginous yeast Yarrowia lipolytica . Applied Microbiology and Biotechnology, 98(1), 251–262. 10.1007/s00253-013-5295-x [DOI] [PubMed] [Google Scholar]
- Bordes, F. , Fudalej, F. , Dossat, V. , Nicaud, J. ‐M. , & Marty, A. (2007). A new recombinant protein expression system for high‐throughput screening in the yeast Yarrowia lipolytica . Journal of Microbiological Methods, 70(3), 493–502. 10.1016/j.mimet.2007.06.008 [DOI] [PubMed] [Google Scholar]
- Broun, P. , Boddupalli, S. , & Somerville, C. (1998). A bifunctional oleate 12‐hydroxylase: Desaturase from Lesquerella fendleri . The Plant Journal, 13(2), 201–210. 10.1046/j.1365-313X.1998.00023.x [DOI] [PubMed] [Google Scholar]
- Broun, Pierre , Shanklin, J. , Whittle, E. , & Somerville, C. (1998). Catalytic plasticity of fatty acid modification enzymes underlying chemical diversity of plant lipids. Science, 282(5392), 1315–1317. 10.1126/science.282.5392.1315 [DOI] [PubMed] [Google Scholar]
- Browse, J. , McCourt, P. J. , & Somerville, C. R. (1986). Fatty acid composition of leaf lipids determined after combined digestion and fatty acid methyl ester formation from fresh tissue. Analytical Biochemistry, 152(1), 141–145. 10.1016/0003-2697(86)90132-6 [DOI] [PubMed] [Google Scholar]
- Buchan, D. W. A. , Minneci, F. , Nugent, T. C. O. , Bryson, K. , & Jones, D. T. (2013). Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Research, 41(W1), W349–W357. 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buist, P. H. , Behrouzian, B. , Alexopoulos, K. A. , Dawson, B. , & Black, B. (1996). Fluorinated fatty acids: New mechanistic probes for desaturases. Chemical Communications, 23, 2671–2672. 10.1039/cc9960002671 [DOI] [Google Scholar]
- Cai, Y. , Yu, X. ‐H. , Liu, Q. , Liu, C. ‐J. , & Shanklin, J. (2018). Two clusters of residues contribute to the activity and substrate specificity of Fm1, a bifunctional oleate and linoleate desaturase of fungal origin. Journal of Biological Chemistry, 293(51), 19844–19853. 10.1074/jbc.RA118.005972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, S. , Zhou, X. ‐R. , Wood, C. C. , Green, A. G. , Singh, S. P. , Liu, L. , & Liu, Q. (2013). A large and functionally diverse family of Fad2 genes in safflower (Carthamus tinctorius L.). BMC Plant Biology, 13(1), 5 10.1186/1471-2229-13-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauk, M. , Lam, P. , Kunst, L. , & Smith, M. A. (2007). A FAD2 homologue from Lesquerella lindheimeri has predominantly fatty acid hydroxylase activity. Plant Science, 173(1), 43–49. 10.1016/j.plantsci.2007.03.015 [DOI] [Google Scholar]
- Finn, R. D. , Attwood, T. K. , Babbitt, P. C. , Bateman, A. , Bork, P. , Bridge, A. J. , … Mitchell, A. L. (2017). InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Research, 45(D1), D190–D199. 10.1093/nar/gkw1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Hornung, E. , Busch, S. , Kassner, N. , Ternes, P. , Braus, G. H. , & Feussner, I. (2007). A small membrane‐peripheral region close to the active center determines regioselectivity of membrane‐bound fatty acid desaturases from Aspergillus nidulans . Journal of Biological Chemistry, 282(37), 26666–26674. 10.1074/jbc.M705068200 [DOI] [PubMed] [Google Scholar]
- Jones, D. T. (1999). Protein secondary structure prediction based on position‐specific scoring matrices. Journal of Molecular Biology, 292(195–202), 8–202. [DOI] [PubMed] [Google Scholar]
- Kajikawa, M. , Abe, T. , Ifuku, K. , Furutani, K. , Yan, D. , Okuda, T. , … Fukuzawa, H. (2016). Production of ricinoleic acid‐containing monoestolide triacylglycerides in an oleaginous diatom, Chaetoceros gracilis . Scientific Reports, 6(1), 10.1038/srep36809. Article no. 36809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L. A. , Mezulis, S. , Yates, C. M. , Wass, M. N. , & Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10(6), 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. E. , Chivian, D. , & Baker, D. (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Research, 32(Web Server), W526–W531. 10.1093/nar/gkh468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. , Lenman, M. , Banas, A. , Bafor, M. , Singh, S. , Schweizer, M. , … Stymne, S. (1998). Identification of non‐heme diiron proteins that catalyze triple bond and epoxy group formation. Science, 280(5365), 915–918. 10.1126/science.280.5365.915 [DOI] [PubMed] [Google Scholar]
- Libisch, B. , Michaelson, L. V. , Lewis, M. J. , Shewry, P. R. , & Napier, J. A. (2000). Chimeras of Δ6‐fatty acid and Δ8‐sphingolipid desaturases. Biochemical and Biophysical Research Communications, 279(3), 779–785. 10.1006/bbrc.2000.4023 [DOI] [PubMed] [Google Scholar]
- Lim, Z. L. , Senger, T. , & Vrinten, P. (2014). Four amino acid residues influence the substrate chain‐length and regioselectivity of Siganus canaliculatus Δ4 and Δ5/6 desaturases. Lipids, 49(4), 357–367. 10.1007/s11745-014-3880-0 [DOI] [PubMed] [Google Scholar]
- Meesapyodsuk, D. , & Qiu, X. (2008). An oleate hydroxylase from the fungus Claviceps purpurea: Cloning, functional analysis, and expression in Arabidopsis . Plant Physiology, 147(3), 1325–1333. 10.1104/pp.108.117168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meesapyodsuk, D. , Qiu, X. , Reed, D. W. , Covello, P. S. , & Qiu, X. (2007). Primary structure, regioselectivity, and evolution of the membrane‐bound fatty acid desaturases of Claviceps purpurea. Journal of Biological Chemistry, 282(28), 20191–20199. 10.1074/jbc.M702196200 [DOI] [PubMed] [Google Scholar]
- Minto, R. E. , & Blacklock, B. J. (2008). Biosynthesis and function of polyacetylenes and allied natural products. Progress in Lipid Research, 47(4), 233–306. 10.1016/j.plipres.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller, S. , Croning, M. D. R. , & Apweiler, R. (2001). Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics, 17(7), 646–653. [DOI] [PubMed] [Google Scholar]
- Notredame, C. , Higgins, D. G. , & Heringa, J. (2000). T‐coffee: A novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology, 302(1), 205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- Pei, J. , Kim, B. ‐H. , & Grishin, N. V. (2008). PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Research, 36(7), 2295–2300. 10.1093/nar/gkn072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X. , Reed, D. W. , Hong, H. , MacKenzie, S. L. , & Covello, P. S. (2001). Identification and analysis of a gene from Calendula officinalis encoding a fatty acid conjugase. Plant Physiology, 125(2), 847–855. 10.1104/pp.125.2.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, A. , Lindahl, E. , & Wallner, B. (2010). Model quality assessment for membrane proteins. Bioinformatics, 26(24), 3067–3074. 10.1093/bioinformatics/btq581 [DOI] [PubMed] [Google Scholar]
- Rong, C. , Chen, H. , Wang, M. , Gu, Z. , Zhao, J. , Zhang, H. , … Chen, Y. Q. (2018). Molecular mechanism of substrate preference for ω‐3 fatty acid desaturase from Mortierella alpina by mutational analysis and molecular docking. Applied Microbiology and Biotechnology, 102(22), 9679–9689. 10.1007/s00253-018-9321-x [DOI] [PubMed] [Google Scholar]
- Sasata, R. J. , Reed, D. W. , Loewen, M. C. , & Covello, P. S. (2004). Domain swapping localizes the structural determinants of regioselectivity in membrane‐bound fatty acid desaturases of Caenorhabditis elegans . Journal of Biological Chemistry, 279(38), 39296–39302. 10.1074/jbc.M405712200 [DOI] [PubMed] [Google Scholar]
- Shanklin, J. , & Cahoon, E. B. (1998). Desaturation and related modifications of Fatty Acids. Annual Review of Plant Physiology and Plant Molecular Biology, 49, 611–641. Retrieved from Stage. [DOI] [PubMed] [Google Scholar]
- Söding, J. , Biegert, A. , & Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research, 33, W244–W248. 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. , DiMaio, F. , Wang, R. Y. ‐R. , Kim, D. , Miles, C. , Brunette, T. , … Baker, D. (2013). High‐resolution comparative modeling with RosettaCM. Structure, 21(10), 1735–1742. 10.1016/j.str.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling, P. , Lee, M. , Girke, T. , Zähringer, U. , Stymne, S. , & Heinz, E. (2000). A bifunctional Δ6‐fatty acyl acetylenase/desaturase from the moss Ceratodon purpureus: A new member of the cytochrome b5 superfamily. European Journal of Biochemistry, 267(12), 3801–3811. 10.1046/j.1432-1327.2000.01418.x [DOI] [PubMed] [Google Scholar]
- Van de Loo, F. J. , Broun, P. , Turner, S. , & Somerville, C. (1995). An oleate 12‐hydroxylase from Ricinus communis L. is a fatty acyl desaturase homolog. Proceedings of the National Academy of Sciences, 92(15), 6743–6747. 10.1073/pnas.92.15.6743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viklund, H. , & Elofsson, A. (2008). OCTOPUS: Improving topology prediction by two‐track ANN‐based preference scores and an extended topological grammar. Bioinformatics, 24(15), 1662–1668. 10.1093/bioinformatics/btn221 [DOI] [PubMed] [Google Scholar]
- Wang, H. , Klein, M. G. , Zou, H. , Lane, W. , Snell, G. , Levin, I. , … Sang, B. ‐C. (2015). Crystal structure of human stearoyl–coenzyme A desaturase in complex with substrate. Nature Structural & Molecular Biology, 22(7), 581–585. 10.1038/nsmb.3049 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Yan, R. , Roy, A. , Xu, D. , Poisson, J. , & Zhang, Y. (2015). The I‐TASSER Suite: Protein structure and function prediction. Nature Methods, 12(1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa, H. , Kumagai, H. , & Uemura, H. (2013). Secretory production of ricinoleic acid in fission yeast Schizosaccharomyces pombe . Applied Microbiology and Biotechnology, 97(19), 8663–8671. 10.1007/s00253-013-5060-1 [DOI] [PubMed] [Google Scholar]
- Zhou, X. ‐R. , Singh, S. , Liu, Q. , & Green, A. (2006). Combined transgenic expression of Δ12‐desaturase and Δ12‐epoxygenase in high linoleic acid seeds leads to increased accumulation of vernolic acid. Functional Plant Biology, 33, 585–592. 10.1071/FP05297 [DOI] [PubMed] [Google Scholar]
- Zhou, X. ‐R. , Singh, S. P. , & Green, A. G. (2013). Characterisation of the FAD2 gene family from Hiptage benghalensis: A ricinoleic acid accumulating plant. Phytochemistry, 92, 42–48. 10.1016/j.phytochem.2013.05.006 [DOI] [PubMed] [Google Scholar]
- Zhu, G. , Koszelak‐Rosenblum, M. , Connelly, S. M. , Dumont, M. E. , & Malkowski, M. G. (2015). The crystal structure of an integral membrane fatty acid α‐hydroxylase. Journal of Biological Chemistry, 290(50), 29820–29833. 10.1074/jbc.M115.680124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information