

The phosphoproteome of the endothelial receptor type tyrosine phosphatase VE-PTP has been identified for mouse endothelial cells by a substrate trapping approach and a second anti-phosphotyrosine affinity isolation approach. In agreement with the central role of VE-PTP as regulator of endothelial junctions, 29% of the identified substrate candidates were junction related. Besides the well characterized substrates Tie-2 and FGD5, the tyrosine kinase receptor EPHB4 was identified as novel substrate, which formed a ternary complex with VE-PTP and Tie-2.
Keywords: substrate identification, cell adhesion, cell-cell interactions, tyrosine kinases, phosphorylation, immunoaffinity, protein complex analysis, protein phosphatases, substrate trapping
Graphical Abstract

Highlights
Identification of the substrates profile of the endothelial phosphatase VE-PTP.
A large fraction of VE-PTP substrate candidates (29%) is cell junction related.
Tie-2 and EPHB are substrates which associate as ternary complex with VE-PTP.
Abstract
Vascular endothelial protein tyrosine phosphatase (VE-PTP, PTPRB) is a receptor type phosphatase that is crucial for the regulation of endothelial junctions and blood vessel development. We and others have shown recently that VE-PTP regulates vascular integrity by dephosphorylating substrates that are key players in endothelial junction stability, such as the angiopoietin receptor TIE2, the endothelial adherens junction protein VE-cadherin and the vascular endothelial growth factor receptor VEGFR2. Here, we have systematically searched for novel substrates of VE-PTP in endothelial cells by utilizing two approaches. First, we studied changes in the endothelial phosphoproteome on exposing cells to a highly VE-PTP-specific phosphatase inhibitor followed by affinity isolation and mass-spectrometric analysis of phosphorylated proteins by phosphotyrosine-specific antibodies. Second, we used a substrate trapping mutant of VE-PTP to pull down phosphorylated substrates in combination with SILAC-based quantitative mass spectrometry measurements. We identified a set of substrate candidates of VE-PTP, of which a remarkably large fraction (29%) is related to cell junctions. Several of those were found in both screens and displayed very high connectivity in predicted functional interaction networks. The receptor protein tyrosine kinase EPHB4 was the most prominently phosphorylated protein on VE-PTP inhibition among those VE-PTP targets that were identified by both proteomic approaches. Further analysis revealed that EPHB4 forms a ternary complex with VE-PTP and TIE2 in endothelial cells. VE-PTP controls the phosphorylation of each of these two tyrosine kinase receptors. Despite their simultaneous presence in a ternary complex, stimulating each of the receptors with their own specific ligand did not cross-activate the respective partner receptor. Our systematic approach has led to the identification of novel substrates of VE-PTP, of which many are relevant for the control of cellular junctions further promoting the importance of VE-PTP as a key player of junctional signaling.
Reversible protein phosphorylation on tyrosine residues is an essential signaling mechanism in eukaryotic cells. It is controlled by the antagonistic activities of protein-tyrosine kinases (PTKs)1 and protein-tyrosine phosphatases (PTPs). Our current knowledge of molecular activities and targets of PTKs in cell signaling exceeds by far that of PTPs; and, for many PTPs, their cellular substrates remain unidentified (1, 2). A recent large-scale comprehensive screening approach focusing on protein-protein interactions within the membrane could map receptor tyrosine kinase - phosphatase interactions on a genome-wide level (3). However, for a true, holistic understanding of cellular signaling processes, a deeper and more detailed knowledge of the substrate repertoire of phosphatases would be highly desirable.
We are particularly interested in the receptor-type protein-tyrosine phosphatase VE-PTP, which is specifically expressed in vascular endothelium and is essential for proper blood vessel formation and the integrity of endothelial junctions. It is a member of the R3-subclass of R-PTPs and composed of 17 extracellular fibronectin type III-like domains, a transmembrane (TM) domain and a cytoplasmic PTP-domain (4, 5). Mice carrying a mutation in VE-PTP leading to the deletion of the transmembrane and cytoplasmic domains suffer from strongly enlarged blood vessels in the yolk sac, impeded heart development, and early embryonic lethality shortly before 10 days of gestation (6). The same defects also occur in mice carrying a null allele of the VE-PTP gene (7). A molecular substrate for VE-PTP implicated in these developmental defects is the endothelial tyrosine kinase receptor TIE2. VE-PTP interacts with TIE2 (4) and negatively regulates its signaling activity in angiogenic processes by dephosphorylation (8). TIE2 was demonstrated to be responsible for embryonic vessel enlargement caused by the lack of VE-PTP phosphatase activity (8). Another interaction partner of VE-PTP is VEGFR-2, which is negatively regulated by VE-PTP under resting conditions. Both proteins were described to dissociate subsequent to the binding of VEGF to its receptor leading to enhanced VEGFR-2 phosphorylation and thus activation (9). This interaction with a growth factor receptor is also likely to be of importance for the proper formation of blood vessels during embryonic development. Further, VE-PTP interacts with the major adhesion molecule of endothelial adherens junctions, VE-cadherin (5). This interaction depends on the extracellular domains of both proteins and is crucial for the regulation of endothelial cell-cell contact stability, because suppression of VE-PTP expression in endothelial cells greatly reduces the adhesive function of VE-cadherin (10). The VE-cadherin-VE-PTP complex is rapidly dissociated by adhesion of leukocytes to activated endothelium or by VEGF (10). This dissociation is triggered via a RAC1/NOX/PYK2 signaling pathway and a yet undefined substrate of VE-PTP (11). VE-PTP also directly dephosphorylates plakoglobin, another component of the VE-cadherin-catenin complex (10).
The development of a potent VE-PTP inhibitor (AKB-9778) allowed further insights into the signaling of this phosphatase (12, 13). This sulfamic acid-based inhibitor is highly selective for VE-PTP compared with other closely related phosphatases except for DEP-1 (VE-PTP IC50 = 17 pm and DEP-1 IC50 = 36 pm) (14). It was shown that pharmacological VE-PTP inhibition strongly activates TIE2 signaling in endothelial cells. In tumor models, the inhibitor delays tumor growth and metastatic progression through TIE2-dependent stabilization of tumor vessels (15). Further, the activation of TIE2 in ocular vasculature via VE-PTP inhibition blocks ischemia and VEGF-induced retinal neovascularization and suppresses macular edema formation (14). Interestingly, this TIE2-mediated junction-stabilizing effect of the VE-PTP inhibitor in vivo overrides the negative effect of VE-PTP inhibition on the adhesive function of VE-cadherin (16). At present, the identified biological effects of the VE-PTP inhibitor AKB-9778 can all be ascribed to the activation of TIE2.
To understand more about the role of VE-PTP in endothelial signaling, we have conducted two mass spectrometry-based proteomic approaches to identify new substrates. First, we monitored the abundance of proteins harboring phosphotyrosine residues subsequent to exposure of cells to the VE-PTP inhibitor AKB-9778 by label-free quantification of proteins immunoprecipitated by a pY-specific antibody. Second, we identified potential VE-PTP substrates using a catalytically inactive VE-PTP substrate trapping mutant in combination with SILAC quantification. We found several new substrate candidates for VE-PTP with a surprisingly clear preference for junction related proteins. Further, one of the most prominently phosphorylated substrates on VE-PTP inhibition was the tyrosine kinase receptor EPHB4, which was found in a ternary complex with TIE2 and VE-PTP.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
VE-PTP inhibitor AKB-9778 was a kind gift of Aerpio (Cincinnati, OH) and. Comp-Ang1 was a gift from G.Y. Koh. Complete EDTA-free protease inhibitor as well as PhosStop phosphatase inhibitor cocktails were obtained from Roche Applied Science (Merck, Darmstadt, Germany), gelatin was purchased from Sigma-Aldrich (Schnelldorf, Germany) and angiopoietin-1 (923AN) as well as EphrinB2 Fc (496EB) from R&D Systems (Wiesbaden, Germany). DMA and DST were obtained from Pierce (Thermo Scientific, Dreieich, Germany). The following antibodies were used: mAb 4G10 directed against phosphotyrosine (Millipore, Darmstadt, Germany), pAb Z-5 against GST (Santa Cruz Biotechnology, Heidelberg; Germany), pAb AF446 against EPHB4 (R&D Systems), mAb 3G1 against TIE2 (17), pAb VE-PTP-C against VE-PTP (5), pAb VE-PTP-1–8 against human VE-PTP extracellular domains (unpublished), pAb 9102 against ERK1/2 and pAb against phospho-ERK1/2-T202/Y204 (Cell Signalling Technology, Frankfurt, Germany).
DNA Constructs
GST-VE-PTP and the trapping mutant GST-VE-PTP C/S were described previously (4). The D/A mutation was introduced into GST-VE-PTP and GST-VE-PTP C/S by PCR using the following primers (Eurogentec): antisense (5′-CTCTGGGACCCCATGGGCTGGCCACACCGTG TAG-3′) and sense (5′-CTACACGGTGTGGCCAGCCCATGGGGTCCCAGAG-3′). The Q/A mutation was introduced into GST-VE-PTP D/A by PCR using the following primers: antisense (5′-CATATTGACACTCGGTCGCGACCATGTGAACCCTG-3′) and sense (5′-CAGGGTTCACAT GGTCGCGACCGAGTGTCAATATG-3′). GST, GST-VE-PTP, GST-VE-PTP C/S, GST-VE-PTP D/A, GST-VE-PTP C/S D/A and GST-VE-PTP D/A Q/A fusion proteins were expressed in Escherichia coli (strain BL21).
Cell Culture and SILAC Labeling
HUVECs were cultured in EBM-2 medium supplemented with SingleQuots (Lonza) and the following cells were cultivated as described: bEnd.3 cells (18) and bEnd.5 cells (19). For SILAC measurements, two isotopically distinct populations of bEnd.3 cells were created by a serial passage (five times 1:3) in arginine- and lysine-deficient Dulbecco′s modified Eagle′s medium containing 10% dialyzed fetal bovine serum supplemented with l-lysine (0.67 mm) and l-arginine (0.24 mm) either “medium” labeled (Lys4/Arg6) or “heavy” labeled (Lys8/Arg10) Labeling efficiencies for medium and heavy labeled cells were better than 95% (data not shown).
SILAC VE-PTP Substrate Trapping
For SILAC substrate trapping pull-down experiments, nine 15-cm plates of confluent bEnd.3 cells were pretreated with 1 mm pervanadate for 30 min and lysed (20 mm Tris/HCl pH 7.5, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, 10% glycerine, 5 mm iodoacetic acid [IAA], 1× Complete EDTA-free protease inhibitor mixture) for 30 min at 4 °C. Subsequently, 10 mm DTT was added to inactivate IAA. After centrifugation at 14,000 × g for 30 min at 4 °C, cleared lysates were incubated with glutathione-Sepharose for 90 min at 4 °C to remove proteins binding nonspecifically to the Sepharose. After centrifugation at 500 × g for 5 min at 4 °C, cleared lysates were incubated overnight with 20 μg GST-VE-PTP or GST-VE-PTP C/S D/A, as previously described (n = 4) (20). Beads were washed five times with 5 ml and three times with 1 ml of lysis buffer. GST-VE-PTP and GST-VE-PTP C/S D/A beads were then combined, boiled in Laemmli sample buffer to elute and denature bound proteins, followed by 1D-separation by SDS-PAGE and LC-MS/MS analysis.
For immunoblotting analysis of trapping mutants three 15-cm plates of confluent bEnd.5 cells were treated as described above except that cleared lysates were incubated overnight with 7 μg GST, GST-VE-PTP, GST-VE-PTP C/S, GST-VE-PTP D/A, GST-VE-PTP C/S D/A or GST-VE-PTP D/A Q/A, bound to glutathione-Sepharose and subjected to SDS-PAGE and immunoblotting.
Immunoprecipitation of Tyrosine Phosphorylated Proteins
Following treatment with AKB-9778 (50 μm; Aerpio, Cincinnati, OH) for 30 min at 37 °C bEnd.5 cells were lysed in lysis buffer (20 mm Tris/HCl pH 7.4, 150 mm NaCl, 2 mm CaCl2, 1 mm Na3VO4, 1% Triton X-100, 0.04% NaN3, 1× Complete EDTA-free protease inhibitor mixture) for 30 min at 4 °C. Lysates were centrifuged at 4 °C for 30 min at 20,000 × g. Cleared lysates were incubated for 2 h at 4 °C with 4G10 anti-phosphotyrosine antibody. Protein G Sepharose was used to precipitate immunocomplexes, which were eluted using 0.1 m glycine pH 2.5 and neutralized by the addition of 1 m Tris buffer. Eluted proteins were then acetone precipitated before GelC-MS/MS carried out as described below. For Western blotting, immunocomplexes were washed five times with lysis buffer, size-separated by reducing SDS-PAGE and transferred to nitrocellulose. Blots were analyzed as previously described (21). For detection of phosphotyrosine, milk powder in the blocking buffer was replaced by 2% BSA, and 200 μm Na3VO4 was added.
Tryptic Digest
Proteins were subjected to SDS-PAGE using BioRad Ready-Gels (4–15%) according to the manufacturer′s instructions. Following electrophoretic separation and visualization of proteins by Coomassie Blue R250 staining, the resulting lanes were cut into 13–15 slices. In gel digests were performed according to Shevchenko et al. (22). Proteins co-immunoprecipitated from bEnd.5 cells treated or untreated with the phosphotyrosine - specific 4G10 Ab were eluted from the Ab-beads using glycine buffer pH 2.5 the peptide fractions were desalted and stored on StageTips (22) until analysis by LC-MS/MS.
Nano-LC-MS/MS Analysis
Using a Proxeon EasyLC nanoflow system each peptide fraction was injected onto an in-house packed fused silica capillary column (length 15 cm; ID 75 μm; ReproSil-Pur C18-AQ, 3 μm) that was online coupled via a Proxeon electrospray ion source to an LTQ-Orbitrap Velos mass spectrometer. Bound peptides were eluted using a linear 120 min gradient from 5–35% B (80% ACN, 0.5% acetic acid) followed by a gradient from 35 to 98% B in 15 min. After washing at 98% B the column was re-equilibrated at starting conditions. The mass spectrometer was operated in the positive ion mode, switching in a data-dependent fashion between survey scans in the orbitrap (mass range m/z = 300–1650 (300–1750 in pY-specific pulldowns); resolution R = 60,000; target value = 1E6; lockmass set to 445.120025) and collision induced fragmentation and MS/MS acquisition in the LTQ part. MS/MS spectra of the 15 most intense ion peaks (10 in pY-specific pulldowns) detected in the MS were recorded. Raw MS data were processed using MaxQuant (v. 1.5.3.8) with the built-in Andromeda search engine.
Tandem mass spectra were searched against the mouse uniprotKB database (UP000000589_10090.fasta; version from 12/2015, containing 20997 searchable entries) concatenated with reversed sequence versions of all entries and containing common contaminants. Carbamidomethylation on cysteine residues was set as fixed modification for the search in the database, whereas oxidation at methionine, acetylation of the protein N termini were set as variable modifications. For the identification of phosphopeptides, phosphorylation at Ser, Thr and Tyr were allowed as variable modifications in addition. Trypsin was defined as the digesting enzyme, allowing a maximum of two missed cleavages and requiring a minimum length of 7 amino acids. The maximum allowed mass deviation was 20 ppm for MS and 0.5 Da for MS/MS scans. Protein groups were regarded as being unequivocally identified with a false discovery rate (FDR) of 1% for both the peptide and protein identifications. To further increase the stringency of the identification process, proteins had to be identified with at least 2 different peptides, one of which being unique to the protein group. Phosphosites were accepted when they were identified with a localization probability of >0.75 and a score difference >5 (class I phosphosites). Data transformation as well as evaluation was performed using Perseus software (version 1.5.5.3). Common lab contaminants and proteins containing reverse sequences that were derived from the decoy database were filtered out from the dataa set before any further analysis. For label free quantification of immunoprecipitated proteins on inhibitor treatment, normalized intensity (LFQ) values were first log2 transformed, followed by filtering for at least 2 valid values in one of both experimental groups (inhibitor-treatment; control). Missing values were replaced by imputation (width 0.3; down shift 1.8). Significant differences between control and inhibitor-treated samples were determined using Student′s t test with a threshold p value set to 0.05. Only proteins, which were > 2-fold enriched were considered for further evaluation. For quantification of SILAC trapping results significance B was calculated in Perseus on log2 transformed average H/M ratios (n = 4) using a p value set to 0.05 for truncation. Only proteins determined as significant outliers were considered for further evaluation.
Bioinformatic Analysis
Network construction and functional gene ontology (GO) enrichment analysis of the proteins identified from the immunoprecipitation experiment involving the phosphotyrosine specific 4G10 antibody and the SILAC trapping approach were performed using Cytoscape (v3.6.0) in combination with the stringApp (v1.3.0) (24) and the enhancedGraphics plugin (v1.2.0). The lists of significantly changed proteins obtained from either the substrate trapping, or the AKB-9778 inhibitor experiment were searched against the STRING database to obtain the network of interacting proteins (confidence score cutoff at the default value of 0.4). Proteins without interaction partners within the corresponding network (singletons) were omitted from the visualized network. Participating proteins in the network are represented as nodes size proportional to their relative abundance, and are linked by gray lines (edges), when directly interacting with each other. The confidence of the protein-protein interaction is reflected by the STRING confidence score, which is proportional to edge thickness/intensity. False discovery rate values for functional GO term enrichments were calculated using the stringapp with a FDR value of 0.05 as cutoff (Figs. 1, 2). The network in Fig. 3 was constructed using the same tools as described above including all significantly changed proteins from both approaches (FDR = 0.05) albeit with the focus on the degree of connectivity between the protein nodes. Node diameter thus is directly proportional to the number of links with other proteins in the network. This allows for the visualization of potential network hubs. As above, singleton proteins were excluded from the network visualization. The Venn diagram was generated using the BioVenn web application (25).
Fig. 1.

Identification of potential VE-PTP substrates by anti-phosphotyrosine affinity purification after VE-PTP inhibition. A, Schematic representation of label free quantification of immunoprecipitated pY-containing and associated proteins on VE-PTP inhibitor treatment. Phosphotyrosine-containing proteins in untreated or AKB-9778-treated cells were precipitated by 4G10-antibody, separated by SDS-PAGE, in gel - digested and analyzed by label free quantitative LC-MS/MS. B, Functional interaction network of significantly enriched proteins from the immunoprecipitation experiment. Only proteins that are part of the network are displayed (40 out of 54). The size of the protein nodes reflects quantitative differences in enrichment based on label free quantification of proteins. Red label: Proteins associated with GO process signal transduction; green label: proteins associated with GO compartment cell junction.
Fig. 2.

Identification of potential VE-PTP substrates by substrate trapping. A, Domain structure and catalytic domains of wildtype VE-PTP (FN III: extracellular fibronectin type III-like repeat; TM: transmembrane domain) and schematic representation of the engineered GST-VE-PTP fusion proteins. B, Mouse bEnd.5 endothelioma cells, pretreated with 1 mm pervanadate for 30 min, were subjected to substrate trapping pull downs with GST (GST only), GST-VE-PTP wt, GST-VE-PTP C/S, GST-VE-PTP D/A, GST-VE-PTP C/S D/A or GST-VE-PTP D/A Q/A, followed by immunoblotting for phospho-tyrosine (upper, middle panel) and GST (lower panel). C, Synopsis of the SILAC-based substrate trapping strategy. bEnd.3 endothelioma cells labeled with either Lys4/Arg6 (medium condition) or Lys8/Arg10 (heavy condition) were treated with 100 μm pervanadate for 30 min to achieve maximal protein-tyrosine phosphorylation. The heavy lysate was incubated with GST-VE-PTP C/S D/A double mutant and the medium lysate was incubated with GST-VE-PTP wt. Enzyme-substrate complexes were purified by GST-pulldowns, combined and analyzed by GeLC-MS/MS. D, Functional interaction network of significantly enriched proteins from the substrate trapping experiment. Only those proteins that are part of the network are displayed. VE-PTP (PTPRB) was added to the network as additional node, as well as 5 nearest neighbors. The size of the protein nodes reflects quantitative differences in enrichment. Proteins in this network associated with signal transduction are labeled in red and proteins associated with cell junction in green.
Fig. 3.
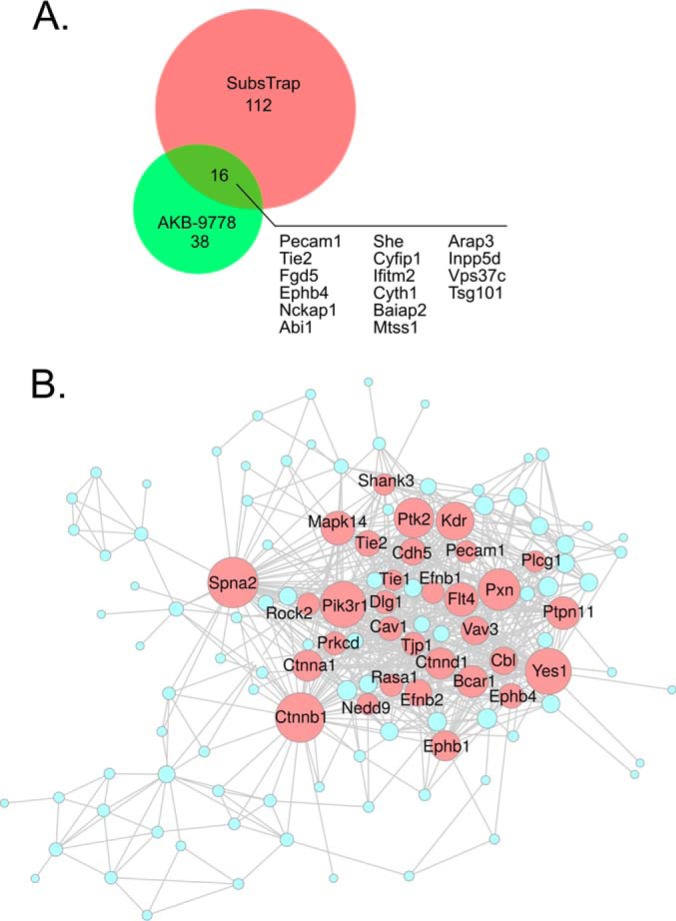
Comparison of Co-immunoprecipitations and Trapping SILAC approach. A, Size proportional Venn diagram to show the overlap of enriched proteins from both experimental approaches (SILAC substrate trapping and AKB-9778 inhibitor treatment/4G10-mediated CoIP). B, Combined functional interaction network of all significantly regulated proteins from both exerimental approaches. Node sizes are based on the degree of connectivity within the network, which allows for the visualization of likely network hubs. Highlighting arbitrarily proteins with 15 or more connections to neighboring protein nodes (red circles) reveals the impact of VE-PTP on signal transduction by transmembrane receptors at cell junctions and on elements regulating the actin cytoskeleton.
Analysis of the VE-PTP-TIE2-EPHB4 Complex
Detection of the ternary VE-PTP-TIE2-EPHB4 complex was performed based on a method described in (26), except for using a different cleavable crosslinker. bEnd.5 or bEnd.3 cells were incubated with the periodate-cleavable crosslinker DST (Disuccinimidyl tartrate) at 500 μm for 30 min. Following washing and lysis, cleared lysates were incubated for 3 h with VE-PTP (or control IgG) antibodies coupled to protein G-Sepharose beads by DMA (dimethyl adipimidate). Bound complexes were eluted with 100 mm glycine (pH 2.5), followed by a second precipitation with anti-TIE2 antibodies overnight. Finally, crosslinks were cleaved with 15 mm sodium meta-periodate and binding partners were analyzed by Western blotting. For stimulation of signaling through TIE2 or EPHB4, bEnd5 cells were either incubated for 30 min with 200 ng/ml Comp-Ang1 or with 10 μg/ml EphrinB2-Fc plus 2 μg/ml anti-human Fc-gamma, respectively.
Experimental Design and Statistical Rationale
Immunoprecipitations of tyrosine phosphorylated proteins by the 4G10 antibody were designed as label free experiments. Biological triplicates of DMSO- and AKB-9778 - treated bEnd.5 cells enabled us to ascertain target proteins by using a t test. SILAC substrate trapping experiments were carried out with a total of 4 biological replicates. In each replicate experiment, potential substrates were trapped from nine 15-cm plates of bEnd.3 cells per labeling state (H, M). Significant outliers from the total population of quantified proteins were identified by using the intensity dependent significance B algorithm of MaxQuant based on log2 transformed average H/M ratios (p = 0.05).
RESULTS
Substrate Identification by MS-based Analysis of Phosphoproteins
Based on the rationale that VE-PTP may constitutively remove phosphate groups from tyrosine-phosphorylated substrate proteins, we expected that inhibition of VE-PTP phosphatase activity would cause a marked increase in the extent of tyrosine phosphorylation of corresponding substrate proteins. Therefore, we treated bEnd.5 endothelioma cells with AKB-9778, a potent, and highly selective inhibitor of VE-PTP catalytic activity (12, 14, 16, 27), followed by immunoprecipitation of tyrosine-phosphorylated proteins (Fig. 1A). Label free quantitative LC-MS/MS of these proteins (n = 3 biological replicates, each for untreated and inhibitor treated cells) revealed that among all 2769 identified proteins (supplemental Table S1), 54 proteins were significantly enriched on VE-PTP inhibition between 2 to 26 fold (p < 0.05, Table I). Bioinformatic analysis suggested that of these 54 proteins, 40 proteins clustered in a functional interaction network (Fig. 1B). Gene ontology enrichment analysis revealed that proteins related to cell junctions (p = 3.5E-9), adherens junction (p = 1.39E-8), regulation of actin cytoskeleton organization (p = 5.24E-7) and signal transduction (p = 5.46e-8) were strongly enriched among these proteins. Proteins related to cell junctions were 5.4 times more prominently enriched within the group of potential VE-PTP substrates than would be expected among a randomly picked group of proteins. Among the 10 phospho-tyrosine proteins that were most strongly enriched on VE-PTP inhibition (9.4 to 26-fold), we found the known substrate TEK/TIE2 (9.4 fold) (8, 14, 16) and the Cdc42 GEF FGD5 (11.5 fold). We have recently identified FGD5 as a direct VE-PTP substrate essential for the regulation of endothelial junctions by VE-PTP and TIE2 (28). Also Tie-1 (8.6-fold), and VEGFR3 (Flt4, 7 fold) and the ephrin type B receptor 4 (EPHB4, 18.4 fold) were highly enriched. The adaptor protein Shb, which was reported to mediate VEGF-induced vascular permeability by binding to VEGFR2 (29), was the most prominently enriched substrate candidate (26-fold). Collectively, cell junction-related proteins seem to be preferred substrates for VE-PTP.
Table I. VE-PTP inhibitor experiment; significantly enriched proteins upon AKB-9778 inhibition and 4G10 immunoaffinity enrichment (p < 0.05).
| Protein names | Gene names | Peptides | Score | MS/MS count | −log10 p value | log2 difference | fold difference |
|---|---|---|---|---|---|---|---|
| SH2 domain-containing adapter protein B | Shb | 8 | 17.04 | 19 | 2.37 | 4.69 | 25.74 |
| Bone morphogenetic protein receptor type-2 | Bmpr2 | 9 | 15.19 | 21 | 2.46 | 4.50 | 22.65 |
| Ephrin type-B receptor 4 | Ephb4 | 37 | 257.22 | 249 | 1.79 | 4.20 | 18.41 |
| Vacuolar protein sorting-associated protein 37C | Vps37c | 6 | 50.15 | 30 | 2.16 | 3.80 | 13.97 |
| Cytohesin-1 | Cyth1 | 12 | 22.98 | 51 | 1.74 | 3.72 | 13.15 |
| FYVE, RhoGEF and PH domain-containing protein 5 | Fgd5 | 28 | 136.07 | 157 | 2.05 | 3.52 | 11.46 |
| SH3 and multiple ankyrin repeat domains protein 3 | Shank3 | 47 | 323.31 | 565 | 1.80 | 3.35 | 10.16 |
| Tensin-like C1 domain-containing phosphatase | Tenc1 | 33 | 68.97 | 147 | 2.84 | 3.31 | 9.92 |
| Roundabout homolog 2 | Robo2 | 4 | 9.83 | 12 | 1.78 | 3.25 | 9.52 |
| Angiopoietin-1 receptor | Tie2/Tek | 50 | 323.31 | 901 | 3.73 | 3.22 | 9.35 |
| Beta-chimaerin | Chn2 | 5 | 9.66 | 15 | 1.56 | 3.18 | 9.05 |
| Probable E3 ubiquitin-protein ligase MID2 | Mid2 | 9 | 43.57 | 24 | 1.37 | 3.12 | 8.71 |
| Tyrosine-protein kinase receptor Tie-1 | Tie1 | 40 | 323.31 | 557 | 3.41 | 3.10 | 8.55 |
| Metastasis suppressor protein 1 | Mtss1 | 8 | 16.08 | 28 | 1.43 | 3.08 | 8.43 |
| Abl interactor 1 | Abi1 | 15 | 40.08 | 79 | 1.35 | 3.05 | 8.27 |
| Mesoderm-specific transcript protein | Mest | 4 | 19.70 | 8 | 1.46 | 2.91 | 7.51 |
| Lipid phosphate phosphohydrolase 3 | Ppap2b | 6 | 13.16 | 49 | 2.68 | 2.87 | 7.31 |
| Vascular endothelial growth factor receptor 3 | Flt4 | 30 | 127.44 | 195 | 1.85 | 2.80 | 6.98 |
| Flrt2 | 14 | 32.61 | 64 | 1.51 | 2.73 | 6.65 | |
| Tumor susceptibility gene 101 protein | Tsg101 | 16 | 87.37 | 81 | 3.00 | 2.59 | 6.03 |
| Activated CDC42 kinase 1 | Tnk2 | 6 | 9.25 | 12 | 1.62 | 2.55 | 5.86 |
| Vacuolar protein sorting-associated protein 28 homolog | Vps28 | 10 | 41.14 | 53 | 2.52 | 2.51 | 5.68 |
| Interferon-induced transmembrane protein 2 | Ifitm2 | 3 | 23.99 | 22 | 2.95 | 2.43 | 5.37 |
| Multivesicular body subunit 12B | Mvb12b | 8 | 19.59 | 24 | 1.53 | 2.42 | 5.35 |
| General receptor for phosphoinositides 1-associated scaffold protein | Grasp | 13 | 48.40 | 100 | 1.71 | 2.23 | 4.71 |
| Anthrax toxin receptor 2 | Antxr2 | 7 | 15.38 | 22 | 1.47 | 2.12 | 4.36 |
| ABI gene family member 3 | Abi3 | 6 | 11.29 | 16 | 1.54 | 2.12 | 4.35 |
| Caskin-2 | Caskin2 | 53 | 323.31 | 1066 | 1.85 | 2.10 | 4.29 |
| Mitogen-activated protein kinase 14 | Mapk14 | 9 | 36.65 | 55 | 2.23 | 2.07 | 4.21 |
| SH2 domain-containing adapter protein E | She | 7 | 26.80 | 59 | 1.60 | 2.07 | 4.19 |
| Nck-associated protein 1 | Nckap1 | 50 | 323.31 | 410 | 1.61 | 1.93 | 3.80 |
| Growth arrest-specific protein 7 | Gas7 | 6 | 8.81 | 11 | 1.62 | 1.85 | 3.61 |
| Arf-GAP with Rho-GAP domain, ANK repeat and PH domain-containing protein 3 | Arap3 | 27 | 66.58 | 148 | 1.95 | 1.84 | 3.58 |
| Cytoplasmic FMR1-interacting protein 1 | Cyfip1 | 55 | 323.31 | 351 | 1.48 | 1.82 | 3.53 |
| Homer protein homolog 3 | Homer3 | 17 | 195.55 | 225 | 2.89 | 1.81 | 3.50 |
| Wiskott-Aldrich syndrome protein family member 2 | Wasf2 | 14 | 99.56 | 123 | 2.39 | 1.77 | 3.42 |
| Tyrosine-protein kinase Yes | Yes1 | 18 | 79.11 | 132 | 1.31 | 1.60 | 3.03 |
| NCK-interacting protein with SH3 domain | Nckipsd | 11 | 19.20 | 27 | 1.60 | 1.59 | 3.02 |
| Slc25a1 | 10 | 23.81 | 53 | 1.38 | 1.55 | 2.92 | |
| Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 | Inpp5d | 41 | 166.04 | 266 | 3.00 | 1.52 | 2.86 |
| KN motif and ankyrin repeat domain-containing protein 2 | Kank2 | 9 | 26.94 | 44 | 1.43 | 1.45 | 2.74 |
| 60S ribosomal protein L9 | Rpl9 | 7 | 20.51 | 34 | 1.57 | 1.42 | 2.67 |
| Brain-specific angiogenesis inhibitor 1-associated protein 2 | Baiap2 | 8 | 13.92 | 18 | 1.47 | 1.35 | 2.55 |
| Ephrin-B1 | Efnb1 | 4 | 6.03 | 8 | 1.45 | 1.33 | 2.52 |
| Lipoma-preferred partner homolog | Lpp | 13 | 66.64 | 77 | 1.47 | 1.33 | 2.51 |
| Programmed cell death protein 6 | Pdcd6 | 6 | 35.84 | 46 | 1.75 | 1.30 | 2.46 |
| NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial | Ndufs4 | 4 | 12.28 | 23 | 1.45 | 1.25 | 2.38 |
| Monoacylglycerol lipase ABHD6 | Abhd6 | 2 | 6.90 | 2 | 1.53 | 1.22 | 2.33 |
| Gm8730 | 10 | 13.33 | 12 | 1.84 | 1.16 | 2.23 | |
| Coiled-coil domain-containing protein 124 | Ccdc124 | 5 | 5.84 | 10 | 1.96 | 1.11 | 2.16 |
| Protein phosphatase 1 regulatory subunit 21 | Ppp1r21 | 13 | 27.59 | 23 | 1.72 | 1.08 | 2.12 |
| Transmembrane protein 33 | Tmem33 | 5 | 24.17 | 42 | 1.33 | 1.08 | 2.12 |
| Platelet endothelial cell adhesion molecule | Pecam1 | 25 | 228.69 | 366 | 2.12 | 1.04 | 2.06 |
| 40S ribosomal protein S16 | Rps16 | 8 | 27.46 | 68 | 2.03 | 1.01 | 2.01 |
Because the enrichment of VE-PTP substrate candidates in this screen was based on affinity isolation of the complete proteins with an anti pY-antibody, most identified peptides were not phosphorylated. However, for some substrate proteins, certain sites were so strongly phosphorylated that we could even detect them as phosphopeptides without prior phosphopeptide enrichment. This way, we found 179 class I phosphotyrosine sites that were enriched on cell treatment with VE-PTP inhibitor out of a total of 649 detectable class I phosphosites (27,6%; localization probabilities > 0.75; Suppl. Table II). Of these, 79 class I sites were identified exclusively in proteins of inhibitor-treated but not of control cells. Among them, we found phosphopeptides from the tyrosine kinase receptors EPHB4, Tek/TIE2, and Tie1, as well as from PECAM1 (Table II).
Table II. Phosphotyrosine containing peptides, identified subsequent to VE-PTP inhibition by AKB-9778.
| Protein names | Gene names | pY position | Sequence window |
|---|---|---|---|
| Ephrin type-B receptor 4 | Ephb4 | 774 | SRFLEENSSDPTpYTSSLGGKIPIRW |
| Ephrin type-B receptor 4 | Ephb4 | 590 | HGQYLIGHGTKVpYIDPFTYEDPNEA |
| Platelet endothelial cell adhesion molecule | Pecam1 | 702 | HQALGTRATETVpYSEIRKVDPNLME |
| SH3 and multiple ankyrin repeat domains protein 3 | Shank3 | 930 | KRRPRPSGPDSPpYANLGAFSASLFA |
| Tyrosine-protein phosphatase non-receptor type substrate 1 | Sirpa | 208 | ETTVNPSGKNVSpYNISSTVRVVLNS |
| Angiopoietin-1 receptor | Tie2/Tek | 1106 | LEERKTYVNTTLpYEKFTYAGIDCSA |
| Angiopoietin-1 receptor | Tie2/Tek | 895 | IINLLGACEHRGpYLYLAIEYAPHGN |
| Angiopoietin-1 receptor | Tie2/Tek | 814 | NRKAKNNPDPTIpYPVLDWNDIKFQD |
| Tensin-like C1 domain-containing phosphatase | Tenc1 | 483 | LAHTRGPLDGSPpYAQVQRVPRQTPP |
| Tyrosine-protein kinase receptor Tie-1 | Tie1 | 1023 | LPVRWMAIESLNpYSVYTTKSDVWSF |
SILAC-based Substrate Trapping Reveals Multiple Putative Substrates
To complement the screen described above, which was based on the use of the VE-PTP inhibitor AKB-9778, we applied an inhibitor-independent approach by using a substrate-trapping construct of VE-PTP as bait to directly pull down substrates. It was shown before, that the active center of tyrosine phosphatases can be mutated in a way that its dephosphorylation activity is ablated, yet specific binding to substrates is still intact and even stabilized because of the lack of dephosphorylation (hence “substrate-trapping”) (20, 30). Based on this concept, we generated various VE-PTP substrate-trapping mutants (Fig. 2A) and tested these constructs for efficient entrapment of proteins containing phosphorylated tyrosine residues in bEnd.5 endothelioma cell lysates. We found that the C/S D/A double mutant was most effective in binding phosphotyrosine-containing proteins, whereas the C/S and the D/A single mutants as well as the D/A Q/A double mutant showed reduced trapping capacity in comparison (Fig. 2B). For the SILAC trapping experiment, the lysates from heavy (Lys8/Arg10) SILAC labeled bEnd.3 endothelioma cells were then incubated with the C/S D/A GST-VE-PTP double mutant as bait to trap its corresponding substrate proteins, whereas the lysate labeled with Lys4/Arg6 was incubated with the catalytically active GST-VE-PTP fusion protein as negative control. Eluates of both incubations were combined, separated by SDS-PAGE and subjected to in-gel tryptic digestion and LC-MS/MS analysis (Fig. 2C).
In four independent substrate trapping experiments, 18,496 peptides were identified that could be matched to 1809 proteins (ignoring splice variants) and quantified on the basis of their H/M ratios (supplemental Table S3). Of these, 128 proteins were significantly enriched by the VE-PTP trapping construct when compared with the wt form of VE-PTP (p < 0.05; Table III). Investigating the potential functional association of these proteins again demonstrated that a majority of the enriched proteins appear to interact with each other in networks that are related to signal transduction (red; p = 1.63E-12) and cell junctions (green; p = 1.3E-19) among others (Fig. 2D).
Table III. Substrate trapping experiment; significant binders (p < 0.05).
| Protein names | Gene names | Peptides | Q-value | Score | Ratio H/M normalized | Significance B |
|---|---|---|---|---|---|---|
| Sodium bicarbonate cotransporter 3 | Slc4a7 | 17 | 0 | 47.966 | 7.82 | 1.94E-15 |
| Hematopoietic lineage cell-specific protein | Hcls1 | 12 | 0 | 78.89 | 6.86 | 3.61E-12 |
| Src substrate cortactin | Cttn | 21 | 0 | 261.7 | 6.83 | 1.91E-19 |
| Platelet endothelial cell adhesion molecule | Pecam1 | 32 | 0 | 323.31 | 6.65 | 1.82E-18 |
| 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-2 | Plcg2 | 42 | 0 | 323.31 | 6.46 | 1.51E-17 |
| Focal adhesion kinase 1 | Ptk2 | 43 | 0 | 323.31 | 6.03 | 1.84E-15 |
| Receptor-type tyrosine-protein phosphatase S | Ptprs | 2 | 0.0072 | 2.4766 | 5.57 | 2.59E-12 |
| Docking protein 1 | Dok1 | 4 | 0 | 7.1982 | 5.29 | 3.18E-09 |
| Rho GTPase-activating protein 27 | Arhgap27 | 24 | 0 | 309.22 | 5.13 | 1.52E-11 |
| Prelamin-A/C;Lamin-A/C | Lmna | 5 | 0 | 12.329 | 4.98 | 2.59E-08 |
| Cell surface glycoprotein MUC18 | Mcam | 8 | 0 | 32.322 | 4.96 | 5.50E-07 |
| Breast cancer anti-estrogen resistance protein 1 | Bcar1 | 26 | 0 | 119.98 | 4.93 | 8.90E-11 |
| Sorting nexin-9 | Snx9 | 11 | 0 | 96.837 | 4.90 | 7.65E-07 |
| Leucyl-cystinyl aminopeptidase | Lnpep | 29 | 0 | 245.59 | 4.69 | 7.08E-10 |
| Rab GDP dissociation inhibitor beta | Gdi2 | 8 | 0 | 49.985 | 4.45 | 7.47E-06 |
| Ephrin type-B receptor 1 | Ephb1 | 9 | 0 | 7.834 | 4.19 | 3.11E-06 |
| Protein Rasa1 | Rasa1 | 30 | 0 | 240.95 | 4.14 | 5.37E-08 |
| Afadin | Mllt4 | 67 | 0 | 323.31 | 4.11 | 6.75E-08 |
| Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 | Inpp5d | 33 | 0 | 323.31 | 4.10 | 7.37E-08 |
| Protein Pcdh1 (Fragment) | Pcdh1 | 21 | 0 | 199.52 | 4.06 | 1.00E-07 |
| Tight junction protein ZO-2 | Tjp2 | 30 | 0 | 323.31 | 3.93 | 2.57E-07 |
| Casein kinase II subunit beta | Csnk2b | 6 | 0 | 28.667 | 3.76 | 8.21E-07 |
| Tight junction protein ZO-1 | Tjp1 | 47 | 0 | 323.31 | 3.75 | 8.90E-07 |
| Casein kinase II subunit alpha | Csnk2a2 | 11 | 0 | 154.96 | 3.72 | 1.12E-06 |
| Bleomycin hydrolase | Blmh | 2 | 0.0011 | 3.8154 | 3.55 | 1.14E-05 |
| NSFL1 cofactor p47 | Nsfl1c | 4 | 0 | 13.487 | 3.53 | 9.19E-05 |
| Tyrosine-protein phosphatase non-receptor type 11 | Ptpn11 | 5 | 0 | 11.752 | 3.53 | 9.25E-05 |
| Zinc finger protein 593 | Znf593 | 2 | 0 | 7.3828 | 3.49 | 4.85E-04 |
| Eukaryotic translation initiation factor 4H | Eif4h | 2 | 0 | 10.833 | 3.48 | 5.03E-04 |
| Ephrin-B2 | Efnb2 | 5 | 0 | 16.658 | 3.37 | 1.99E-04 |
| Spectrin alpha chain, non-erythrocytic 1 | Sptan1 | 92 | 0 | 323.31 | 3.34 | 1.25E-05 |
| Casein kinase II subunit alpha | Csnk2a1 | 14 | 0 | 279.56 | 3.30 | 1.58E-05 |
| Serine/threonine-protein kinase N2 | Pkn2 | 37 | 0 | 298.51 | 3.27 | 1.93E-05 |
| Catenin delta-1 | Ctnnd1 | 44 | 0 | 323.31 | 3.23 | 2.35E-05 |
| Spectrin beta chain, non-erythrocytic 1 | Sptbn1 | 82 | 0 | 323.31 | 3.22 | 2.59E-05 |
| Brain-specific angiogenesis inhibitor 1-associated protein 2 | Baiap2 | 3 | 0 | 11.512 | 3.13 | 1.79E-03 |
| Heterogeneous nuclear ribonucleoproteins C1/C2 | Hnrnpc | 3 | 0 | 28.859 | 3.12 | 1.22E-04 |
| USP6 N-terminal-like protein | Usp6 nl | 12 | 0 | 70.455 | 3.08 | 2.09E-03 |
| Serpin B5 | Serpinb5 | 5 | 0 | 70.589 | 3.07 | 2.21E-03 |
| Actin filament-associated protein 1-like 2 | Afap1l2 | 2 | 0.0025 | 2.9245 | 2.98 | 2.51E-04 |
| Complement component C1q receptor | Cd93 | 5 | 0 | 20.157 | 2.94 | 1.24E-03 |
| Protein Crocc2 | E030010N08Rik | 2 | 0.0081 | 2.4245 | 2.92 | 3.53E-04 |
| ELKS/Rab6-interacting/CAST family member 1 | Erc1 | 26 | 0 | 283.34 | 2.88 | 1.69E-04 |
| Annexin A1 | Anxa1 | 4 | 0 | 23.892 | 2.84 | 4.68E-03 |
| Phosphatidylinositol 4-phosphate 5-kinase type-1 gamma | Pip5k1c | 3 | 0 | 31.856 | 2.84 | 5.14E-04 |
| E3 ubiquitin-protein ligase CBL | Cbl | 10 | 0 | 44.378 | 2.83 | 1.89E-03 |
| Nck-associated protein 1 | Nckap1 | 29 | 0 | 234.21 | 2.82 | 2.41E-04 |
| Platelet endothelial aggregation receptor 1 | Pear1 | 2 | 0 | 17.647 | 2.76 | 7.42E-04 |
| 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase beta-3 | Plcb3 | 15 | 0 | 127.01 | 2.74 | 6.38E-03 |
| Abl interactor 1 | Abi1 | 3 | 0 | 37.773 | 2.74 | 2.73E-03 |
| Scavenger receptor class F member 1 | Scarf1 | 8 | 0 | 34.326 | 2.72 | 2.95E-03 |
| Tyrosine-protein kinase Fes/Fps | Fes | 16 | 0 | 147.24 | 2.71 | 7.09E-03 |
| Guanine nucleotide exchange factor VAV3 | Vav3 | 25 | 0 | 223.37 | 2.69 | 7.54E-03 |
| Mitogen-activated protein kinase kinase kinase kinase 3 | Map4k3 | 17 | 0 | 239.14 | 2.67 | 8.03E-03 |
| Solute carrier family 12 member 4 | Slc12a4 | 10 | 0 | 45.778 | 2.64 | 3.91E-03 |
| Cytoplasmic FMR1-interacting protein 1 | Cyfip1 | 42 | 0 | 318.55 | 2.58 | 7.79E-04 |
| Apoptosis-stimulating of p53 protein 2 | Tp53bp2 | 4 | 0 | 13.437 | 2.54 | 1.99E-03 |
| Rho-associated protein kinase 2 | Rock2 | 28 | 0 | 242.99 | 2.54 | 9.77E-04 |
| Adenomatous polyposis coli protein 2 | Apc2 | 2 | 0.0025 | 2.998 | 2.53 | 5.82E-03 |
| Interferon-induced transmembrane protein 2 | Ifitm2 | 4 | 0 | 75.342 | 2.45 | 1.51E-02 |
| Pleckstrin homology-like domain family B member 1 | Phldb1 | 10 | 0 | 42.854 | 2.45 | 7.75E-03 |
| 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 | Plcg1 | 8 | 0 | 91.185 | 2.43 | 1.63E-02 |
| THUMP domain-containing protein 1 | Thumpd1 | 3 | 0 | 9.9834 | 2.41 | 8.67E-03 |
| Nucleolin | Ncl | 44 | 0 | 323.31 | 2.40 | 1.82E-03 |
| Ankycorbin | Rai14 | 14 | 0 | 214.61 | 2.40 | 1.78E-02 |
| Enhancer of filamentation 1 | Nedd9 | 6 | 0 | 7.278 | 2.38 | 9.82E-03 |
| SH2 domain-containing adapter protein E | She | 4 | 0 | 87.672 | 2.37 | 1.90E-02 |
| Tyrosine-protein kinase Mer | Mertk | 4 | 0 | 21.03 | 2.33 | 4.83E-03 |
| Tyrosine-protein kinase Fer | Fer | 10 | 0 | 14.036 | 2.32 | 1.19E-02 |
| 26S proteasome non-ATPase regulatory subunit 1 | Psmd1 | 13 | 0 | 25.012 | 2.31 | 1.23E-02 |
| Cytohesin-3;Cytohesin-2 | Cyth3;Cyth2 | 8 | 0 | 28.802 | 2.31 | 2.26E-02 |
| Nardilysin | Nrd1 | 3 | 0 | 5.7932 | 2.29 | 5.69E-03 |
| Interferon-induced transmembrane protein 3 | Ifitm3 | 6 | 0 | 104.77 | 2.27 | 3.28E-03 |
| Kin of IRRE-like protein 1 | Kirrel | 12 | 0 | 285.18 | 2.27 | 2.53E-02 |
| Protein IWS1 homolog | Iws1 | 3 | 0.0006 | 4.7896 | 2.27 | 6.20E-03 |
| Guanine nucleotide-binding protein subunit alpha-11 | Gna11 | 7 | 0 | 27.664 | 2.26 | 2.61E-02 |
| Protein Sec24b | Sec24b | 10 | 0 | 17.687 | 2.24 | 1.54E-02 |
| RNA-binding protein 25 | Rbm25 | 2 | 0.0006 | 4.887 | 2.23 | 7.06E-03 |
| Ubiquitin carboxyl-terminal hydrolase 15 | Usp15 | 10 | 0 | 23.853 | 2.22 | 1.65E-02 |
| STE20-like serine/threonine-protein kinase | Slk | 8 | 0 | 94.699 | 2.20 | 1.72E-02 |
| Intersectin-2 | Itsn2 | 17 | 0 | 144.9 | 2.20 | 3.04E-02 |
| Cytohesin-1 | Cyth1 | 7 | 0 | 35.718 | 2.18 | 8.57E-03 |
| Small nuclear ribonucleoprotein Sm D2 | Snrpd2 | 4 | 0 | 11.107 | 2.16 | 1.94E-02 |
| ERC protein 2 | Erc2 | 4 | 0.0063 | 2.5821 | 2.16 | 9.31E-03 |
| Ephrin type-B receptor 4 | Ephb4 | 18 | 0 | 58.562 | 2.15 | 3.40E-02 |
| Metastasis suppressor protein 1 | Mtss1 | 10 | 0 | 105.81 | 2.15 | 2.03E-02 |
| Putative RNA-binding protein 3 | Rbm3 | 5 | 0 | 27.446 | 2.14 | 5.66E-03 |
| Vacuolar protein sorting-associated protein 37C | Vps37c | 4 | 0 | 44.041 | 2.14 | 3.56E-02 |
| Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 2 | Inppl1 | 7 | 0 | 9.4336 | 2.11 | 2.26E-02 |
| Paxillin | Pxn | 3 | 0 | 8.9379 | 2.11 | 1.14E-02 |
| Ras-related protein Rab-7a | Rab7a | 7 | 0 | 23.962 | 2.08 | 2.49E-02 |
| TATA element modulatory factor | Tmf1 | 2 | 0.0011 | 3.8652 | 2.07 | 1.28E-02 |
| Wee1-like protein kinase | Wee1 | 6 | 0 | 36.904 | 2.07 | 1.29E-02 |
| Ezrin | Ezr | 7 | 0 | 15.673 | 2.06 | 2.63E-02 |
| Matrin-3 | Matr3 | 2 | 0 | 6.0982 | 2.06 | 1.35E-02 |
| Arf-GAP with Rho-GAP domain, ANK repeat and PH domain-containing protein 3 | Arap3 | 34 | 0 | 323.31 | 2.05 | 8.07E-03 |
| Protein kinase C delta type | Prkcd | 21 | 0 | 155.33 | 2.05 | 4.43E-02 |
| 26S proteasome non-ATPase regulatory subunit 7 | Psmd7 | 3 | 0 | 14.014 | 2.04 | 2.80E-02 |
| Membrane-associated guanylate kinase, WW and PDZ domain-containing protein 1 | Magi1 | 17 | 0 | 188.01 | 2.02 | 4.77E-02 |
| Sorting nexin-1 | Snx1 | 7 | 0 | 106.72 | 2.01 | 1.61E-02 |
| E3 ubiquitin-protein ligase HECTD1 | Hectd1 | 4 | 0 | 6.6585 | 2.00 | 1.68E-02 |
| Angiopoietin-1 receptor | Tie-2/Tek | 11 | 0 | 59.25 | 2.00 | 4.99E-02 |
| 28S ribosomal protein S2, mitochondrial | Mrps2 | 3 | 0 | 37.502 | 1.98 | 3.34E-02 |
| 26S protease regulatory subunit 6A | Psmc3 | 2 | 0 | 17.228 | 1.98 | 1.81E-02 |
| NHS-like protein 1 | Nhsl1 | 7 | 0 | 30.79 | 1.95 | 3.58E-02 |
| Charged multivesicular body protein 4b | Chmp4b | 2 | 0 | 7.4329 | 1.94 | 3.78E-02 |
| Disks large homolog 1 | Dlg1 | 6 | 0 | 10.473 | 1.93 | 2.09E-02 |
| cGMP-dependent 3,5-cyclic phosphodiesterase | Pde2a | 2 | 0.0025 | 2.7977 | 1.92 | 2.23E-02 |
| FYVE, RhoGEF and PH domain-containing protein 5 | Fgd5 | 39 | 0 | 323.31 | 1.91 | 1.38E-02 |
| Nectin-2 | Pvrl2 | 7 | 0 | 18.197 | 1.90 | 4.15E-02 |
| Fatty acid synthase | Fasn | 11 | 0 | 35.125 | 1.90 | 4.24E-02 |
| 26S proteasome non-ATPase regulatory subunit 3 | Psmd3 | 2 | 0.0021 | 3.2938 | 1.87 | 4.52E-02 |
| Probable ATP-dependent RNA helicase DDX58 | Ddx58 | 10 | 0 | 21.358 | 1.84 | 4.91E-02 |
| Protein Slc4a1ap | Slc4a1ap | 2 | 0.0006 | 4.4031 | 1.76 | 3.73E-02 |
| Phosphatidylinositol 3-kinase regulatory subunit alpha | Pik3r1 | 3 | 0.0006 | 5.086 | 1.75 | 3.82E-02 |
| Tumor susceptibility gene 101 protein | Tsg101 | 12 | 0 | 59.347 | 1.75 | 2.45E-02 |
| 39S ribosomal protein L37, mitochondrial | Mrpl37 | 2 | 0 | 9.7657 | 1.74 | 3.95E-02 |
| Apoptosis-stimulating of p53 protein 1 | Ppp1r13b | 4 | 0 | 25.745 | 1.73 | 4.04E-02 |
| Eukaryotic translation initiation factor 3 subunit L | Eif3l | 5 | 0 | 19.961 | 1.71 | 2.77E-02 |
| Reticulon-4 | Rtn4 | 25 | 0 | 143.96 | 1.68 | 3.12E-02 |
| Catenin alpha-1 | Ctnna1 | 44 | 0 | 323.31 | 1.67 | 3.16E-02 |
| Catenin beta-1 | Ctnnb1 | 31 | 0 | 323.31 | 1.59 | 4.12E-02 |
| Rab11 family-interacting protein 5 | Rab11fip5 | 24 | 0 | 164.59 | 1.58 | 4.27E-02 |
| Cadherin-5/VE-cadherin | Cdh5 | 16 | 0 | 259.78 | 1.57 | 4.38E-02 |
| Caveolin-1 | Cav1 | 5 | 0 | 20.233 | 1.57 | 4.40E-02 |
| Protein DEK | Dek | 15 | 0 | 242.01 | 1.57 | 4.41E-02 |
| Vascular endothelial growth factor receptor 2 | Kdr | 27 | 0 | 274.11 | 1.55 | 4.64E-02 |
| Eukaryotic translation initiation factor 3 subunit F | Eif3f | 11 | 0 | 196.76 | 1.54 | 4.83E-02 |
Several of the candidates identified with this approach were characterized before as functionally relevant substrates for the regulation of endothelial junctions such as VE-cadherin (5, 10), TIE2 (8, 16), VEGFR2 (9) and FGD5 (28), which supported the validity of our SILAC based substrate trapping approach. Other junction related molecules enriched by this method were the catenins, known to associate with VE-cadherin. In addition, junctional components such as the tight junction proteins ZO-1 (Tjp1) and ZO-2 (Tjp2) and the junctional scaffold protein MAGI1 were enriched in our screen. Further, the kinase ROCK2 turned up in this screen, which regulates baseline junctional tension of endothelial cells (31). PECAM-1, an adhesion molecule at endothelial cell contacts involved in shear sensing (32) and leukocyte extravasation (33, 34), and the focal adhesion kinase (FAK/Ptk2), which regulates integrin mediated adhesion at focal contacts as well as stability of junctions via the VE-cadherin-catenin complex (35), were each found more than 6 fold enhanced by VE-PTP substrate trapping. The tyrosine phosphatase SHP2 (Ptpn11), 3.5-fold enriched in this screen, is another regulator of VE-cadherin dependent junction stability (36, 37). Finally, cortactin (CTTN), a prominent Src substrate that supports Arp2/3 nucleation of actin branches and plays an important role in various actin based processes (38)—among them again the control of endothelial junctions and of leukocyte extravasation (39)—was the third most strongly enriched VE-PTP substrate candidate. Collectively, also this second screen for potential VE-PTP substrates identified prominent regulators of endothelial junction stability, highlighting the importance of this phosphatase for the regulation of vascular integrity.
Among the 128 candidates identified in the substrate trapping approach, there were 20 receptor and nonreceptor kinases and 4 other phosphatases (e.g. protein tyrosine phosphatases PTPRS and PTPN11), suggesting that VE-PTP could be involved in multiple signaling events. Two of the identified nonreceptor kinases, FES and FER, belonging to a small kinase subfamily, have multiple roles in cell adhesion and cell migration. Fes kinase has been implicated in endothelial migration stimulated by various angiogenic factors such as FGF2, VEGF-A, and Angiopoietin-2 (40, 41, 42) possibly acting by promoting microtubule assembly (43). Fer kinase was shown to interact with and phosphorylate the cadherin binding partner p120 (CTNND1) (44), cortactin (CTTN) (45) (46), the kinase FAK1 (Ptk2) (47), as well as PECAM1 and the nonreceptor type tyrosine-phosphatase SHP2 (Ptpn11) (48), all of which are involved in the control of endothelial junctions.
Each of our large screens identified a set of substrate candidates, which were unique for this screen. However, a fraction of 16 proteins was common to both screens (Fig. 3A), and contained the well characterized VE-PTP substrate TIE2. Interesting novel substrates of this group were PECAM-1 and the tyrosine kinase receptor EPHB4. Network analysis of all enriched proteins from both approaches, which was based on the degree of connectivity between individual protein nodes, revealed potential network hubs as targets of VE-PTP, which included numerous proteins that regulate endothelial cell adhesive behavior (Fig. 3B). Among these were VE-cadherin (CDH5) and the associated α-catenin (CTNNA), β-catenin (CTNNB), and p120 (CTNND1), which are essential components of endothelial junctions. Other members of this network comprise proteins typical for focal adhesions such as the signaling adaptor paxillin (Pxn) and Bcar1. The latter is a member of the Crk associated substrate (CAS) family of scaffold proteins, which forms an adaptor at focal adhesions for the kinase FAK (Ptk2).
The proto-oncogene CBL is also highlighted in this network, a RING finger E3 ubiquitin ligase, involved in the negative regulation of other signaling pathways, because it recognizes activated receptor tyrosine kinases (TIE2/TEK, KDR, FLT4, EPHB4, EPHB1, PTK2; compare with Fig. 3B) and membrane bound members of the SRC family of kinases (YES) and can terminate signaling by these proteins by promoting their proteasomal degradation (49). Thus, VE-PTP appears to impact multiple processes within endothelial cells related to signal transduction by receptor tyrosine kinases, eventually modulating adhesive properties of endothelial cells as well as cytoskeletal reorganization processes.
VE-PTP Interacts Constitutively with EPHB4 and TIE2 in a Ternary Complex
Because our knowledge on the potential regulation of EPH receptors by phosphatases in general and specifically by VE-PTP is limited, we decided to focus on the potential role of VE-PTP for the regulation of EPHB4, because EPHB4 was detected as being a VE-PTP target in both the pY-specific pulldown as well as the SILAC-based substrate trapping approach. To confirm that EPHB4 is a substrate of VE-PTP whose phosphorylation status is affected by VE-PTP activity, we again treated endothelial cells with 50 μm VE-PTP inhibitor AKB-9778 and immune-precipitated tyrosine phosphorylated proteins from bEnd.5 cell lysates followed by Western blotting with specific antibodies against EPHB4 and TIE2. Both proteins could be identified as prominent bands (at 120 and 150 kDa), apparently highly phosphorylated on VE-PTP inhibition (Fig. 4A). A minor band at ∼140 kDa corresponded to the phosphorylated kinase receptor Tie-1, which had also been identified as a putative VE-PTP interactor in our substrate trapping screen. Importantly, VE-PTP inhibitor concentrations below 1 μm were already enough to induce extensive EPHB4 phosphorylation in bEnd.5 cells (data not shown). Antibody depletion of EPHB4 from bEnd.5 cells eliminated the 120 kDa phosphotyrosine protein band, indicating that this phospho-tyrosine protein was indeed EPHB4 (Fig. 4B). In addition, specific down-regulation of VE-PTP from the cell surface of HUVEC (8) by inducing its endocytosis with a polyclonal antibody against the extracellular part of VE-PTP increased phosphorylation of EPHB4 as well as of TIE2 to a similar extent as treatment with AKB-9778, emphasizing that tyrosine phosphorylation of EPHB4 was indeed specifically induced by interference with VE-PTP (Fig. 4C).
Fig. 4.

EPHB4 phosphorylation is enhanced on VE-PTP inhibition. A, bEnd.5 cells were treated with (+) or without (−) VE-PTP inhibitor AKB9778, phosphorylated proteins were then immunoprecipitated with 4G10 antibody and immunoblotted for EPHB4, TIE2 and Tie1 (3 top panels). Total cell lysates were botted with indicated antibodies (bottom four panels). B, bEnd.5 cells were treated without (−) or with (+) VE-PTP inhibitor and pY-containing proteins were immunoprecipitated either after control-depletion or after depletion of cell lysates for EPHB4 (as indicated above). Immunoprecipitates were blotted for anti pY (4G10) or for EPHB4. Bottom panel: Immunoblot of total cell lysate for EPHB4. C, HUVEC were either untreated (−) or pre-treated with polyclonal Ab against the extracellular part of VE-PTP (to induce endocytosis) or with AKB9778, followed by immunoprecipitation for either TIE2 or EPHB4 and immunoblotting with anti-pY (4G10). Bottom panels: Immunoblots of total cell lysates.
Using a polyclonal antibody directed against VE-PTP (VE-PTP-C) (5) for immunoprecipitations, we could co-precipitate EPHB4, demonstrating a clear association of VE-PTP with EPHB4 (Fig. 5A). The stoichiometry of this interaction with a novel binding partner was comparable to the known interaction between VE-PTP and Tie2 (Fig. 5A). To show that the interaction between EPHB4 and VE-PTP is direct and not mediated by a bridging TIE2 molecule, we depleted first TIE2 and could still observe that EPHB4 was co-precipitated together with VE-PTP (Fig. 5B).
Fig. 5.

EPHB4 is a direct interactor of both VE-PTP and TIE2. A, bEnd5 cell lysates were immunopreciptated with control antibodies (IgG) or with antibodies against TIE2 or VE-PTP, followed by immunoblotting for EPHB4 (top panel) or TIE2 or VE-PTP (second panel). Bottom panels: Immunoblots of total cell lysates for the indicated antigens. B, bEnd5 cell lysates were either mock depleted (left lane) or depleted for TIE2 (two lanes on the right), followed by immunoprecipitation of either TIE2 or VE-PTP (as indicated above) and immunoblotting for EPHB4, TIE2 or VE-PTP (as indicated below). Note that EPHB4 co-precipitates with VE-PTP in the absence of TIE2. Bottom panels: Immunoblots of total lysates for the indicated antigens. C, Similar as for B, except that VE-PTP was depleted instead of TIE2. Note that EPHB4 co-precipitates with TIE2 in the absence of VE-PTP.
Because developmental studies on vascular morphogenesis have suggested a link between EPHB4 and TIE2 (50), we tested whether EPHB4 might interact with TIE2. Indeed, we found that EPHB4 was co-precipitated together with TIE2 in immunoprecipitations with anti-TIE2 antibodies (Fig. 5C). The interaction between EPHB4 and TIE2 was direct because depletion of VE-PTP did not interfere with the efficiency of the co-precipitation of EPHB4 with TIE2 (Fig. 5C). These results demonstrate that EPHB4 can directly associate with VE-PTP and with TIE2. Because TIE2 can also associate with VE-PTP (8), it is possible that a ternary complex exists that contains EPHB4, TIE2 and VE-PTP. However, it could not be excluded at this point that three independent complexes exist, each one consisting of only two of the three membrane proteins.
To distinguish between these two possibilities, we tested the existence of a ternary complex by performing sequential immunoprecipitations of VE-PTP and TIE2. This approach was facilitated by reversible in-cell crosslinking in bEnd.5 cells to prevent dissociation of the complexes. Cells were then lysed and subjected to a first immunoprecipitation with anti VE-PTP antibodies, followed by elution of enriched proteins and a second immunoprecipitation with anti-TIE2 antibodies and subsequent crosslink cleavage and Western blotting for EPHB4. As shown in Fig. 6A and 6B, EPHB4 was specifically enriched by this sequential immunoprecipitation from bEnd.5 or bEnd.3 cell lysates, respectively. Interestingly, only minute amounts of the VE-PTP binding partner VE-cadherin were detected in such sequential VE-PTP/TIE2 immunoprecipitations, indicating that VE-cadherin, in contrast to EPHB4, may not be a constitutive member of the novel ternary receptor complex consisting of TIE2, VE-PTP and EPHB4 (Fig. 6C) and may associate with VE-PTP in a separate pool of receptor molecules.
Fig. 6.

Biochemical characterization of the VE-PTP-TIE2-EPHB4 complex. Sequential immunoprecipitations from bEnd.5 cell lysates (A) or bEnd.3 cell lysates (B) with either first (1.) anti VE-PTP antibodies followed by (2.) anti-TIE2 antibodies after elution (left lane in A, right lane in B) or first (1.) with control antibodies (IgG), followed by (2.) anti-TIE2 antibodies (right lane in A, left lane in B). Cells have been pre-treated with cleavable DST crosslinker (+) to stabilize complexes. Immunoprecipitates were immunoblotted for the antigens indicated on the right. Bottom panels. Immunoblots of total cell lysates for the indicated antigens. C, Sequential immunoprecipitations of bEnd.5 cell lysates were done as in A. followed by immunoblotting for the indicated antigens. Cells had been either pre-treated with DST crosslinker (+), to stabilize complexes or were left untreated (−). Panels on the right depict immunoblots of total cell lysates. Note that VE-cadherin was not found associated with the ternary VE-PTP/EPHB4/TIE2 complex.
Lack of Transactivation Between Receptor Tyrosine Kinases EPHB4 and TIE2 in the Ternary Complex
The direct association of EPHB4 and TIE2 suggested a possible (VE-PTP-modulated) receptor kinase crosstalk. Therefore, we investigated whether EPHB4 could be trans-activated by the TIE2 ligand angiopoetin-1 or whether TIE2 could be activated by the EPHB4 ligand ephrinB2. Although activation of TIE2 by comp-Ang-1 (a recombinant form of Ang1) resulted in robust TIE2 phosphorylation as expected (Fig. 7A), a simultaneous stimulation of EPHB4 phosphorylation could not be detected (Fig. 7B). Likewise, EPHB4 activation by receptor crosslinking did not increase TIE2 phosphorylation (Fig. 7A, 7B). Although VE-PTP inhibition leads to enhanced phosphorylation of both kinases (Fig. 7B), specific activation of one of the two receptor tyrosine kinases had no obvious effect on the corresponding other receptor tyrosine kinase (Fig. 7B). Pushing receptor activation even further by receptor stimulation with specific ligands in the presence of the VE-PTP inhibitor, again indicated that there is no clear transactivation between EPHB4 and TIE2 transmembrane receptors that might be modulated by VE-PTP (Fig. 7C). These results suggest that EPHB4 and TIE2 trigger independently distinct signaling pathways even though they are co-associated with VE-PTP in a ternary complex. Further, inhibiting VE-PTP enhances the signaling of each receptor, but does not induce sensitivity for trans-activation of the receptors.
Fig. 7.

Activation of EPHB4 or TIE2 does not lead to receptor transactivation. A, bEnd5 cells were either stimulated with Angiopoietin1 (Ang1) or by ephrinB2-Fc crosslinking (EphrinXL) followed by immunoprecipitation of either TIE2 or EPHB4 and subsequent immunoblotting for the antigens indicated below. Bottom panel: immunoblot of total cell lysate for the indicated antigens. B, and C, Principally similar as in A. Note that transactivation - such as activation of TIE2 by ephrin crosslinking or activation of EPHB4 by stimulation with Ang1 - was not detectable neither in comparison (B), nor in combination (C) with VE-PTP inhibitor.
DISCUSSION
To learn more about the regulatory potential of VE-PTP in endothelial cells, we undertook two complementary proteomic approaches to identify potential phosphatase substrates. One approach was based on the use of a highly specific inhibitor of VE-PTP, which enhanced tyrosine phosphorylation of potential substrate candidates. The other approach was independent of the use of the VE-PTP inhibitor and relied on the substrate binding specificity of a trapping mutant of VE-PTP, which still recognizes substrates but can no longer dephosphorylate them and therefore bind with much enhanced stability. Both approaches identified large sets of significantly enriched candidates: 54 in the first approach and 128 in the second, of which 16 were identified in both screens. Two VE-PTP substrates, namely TIE2 and Fgd5, which we confirmed previously, and which are important for the effects of VE-PTP on junction-integrity (16, 28), were identified in both approaches, supporting the validity of these screens. In addition, other substrates previously identified, such as VE-cadherin (5, 10) and VEGFR2 (9), were only found in the substrate trapping approach.
Remarkably, proteins related to the regulation of cell junctions were strongly represented among the identified VE-PTP substrate candidates. Among those, the most prominently enriched newly identified phosphoproteins in the VE-PTP-inhibitor based approach were the adaptor protein Shb known to associate with VEGFR2 and participate in junction destabilizing signaling (29), the tyrosine kinase receptor EPHB4 (51, 52), the orphan receptor Tie-1 known to associate with TIE2 (53), VEGFR3 (54, 55) and bone morphogenetic protein receptor 2 (BMPR2) (56). Proteins with well documented relevance for the regulation of endothelial junctions and most prominently enriched by the VE-PTP substrate trapping mutant were PECAM-1 (34), cortactin (38), Focal adhesion kinase 1 (FAK 1/PTK2) (57), and the tyrosine phosphatase SHP-2 (PTPN11). In line with a proposed role of VE-PTP in cell spreading and migration (58), several strong VE-PTP-substrate candidates are related to the function of focal contacts/adhesions, such as FAK1/PTK2, the signaling adaptor paxillin, the scaffold protein BCAR1, and Tensin2/TENC1 (59). Many of these proteins plus additional substrate candidates involved in signal transduction (adapter proteins SHE, SHB, SHANK, ABI1; nonreceptor tyrosine kinases YES1, ACK1), regulation of cell morphology and cytoskeletal reorganization (e.g. WAVE complex proteins WASF2, NCKAP1, NCKIPSD, CYFIP1) (60), are all known to impact on the modulation of endothelial proliferation, inter-endothelial junctions and endothelial adhesive properties (61).
Although our two different screening approaches identified several VE-PTP substrate candidates, which were common to both searches, a considerable number of proteins were exclusively found by only one of the two approaches. This discrepancy has technical but also principal reasons. Although the VE-PTP inhibitor-based approach had been performed with bEnd.5 mouse endothelioma cells, the VE-PTP trapping mutant approach was done with bEnd3 cells. This could account for some of the differences. More important is the fact that the specificity of each approach is based on different criteria. The first approach we used was based on the use of the VE-PTP inhibitor AKB-9778. This reagent shows excellent selectivity for VE-PTP (IC50 = 17 pm) versus a variety of phosphatases with IC50 values 1000 to 10,000 times higher or more for many other receptor type and nonreceptor type tyrosine phosphatases, with the exception of HPTPη (DEP-1, PTPRj; IC50 = 36 pm) and HPTPγ (PTPRG; (IC50 = 100 pm). Because HPTPη is also expressed in endothelial cells, we cannot exclude that some of the substrates we identified in our AKB-9778 based approach might also be substrates of HPTPη. However, the use of the direct VE-PTP trapping mutant approach circumvents this ambiguity. In addition, TIE2 is not a substrate of HPTPη in endothelial cells (14). A second important difference between both approaches is because the use of AKB-9778 will only enhance tyrosine phosphorylation of those substrates that are in direct vicinity of an active kinase specific for this substrate. This is probably the reason why a substrate such as the receptor kinase TIE2 is strongly phosphorylated on VE-PTP inhibition, because it is thought to have some baseline activity even in the absence of the agonist Ang1. A similar situation may be the case for EPHB4 and, also, the phosphorylation of the VE-PTP substrate FGD5 is strongly enhanced by AKB-9778, because FGD5 phosphorylation depends on TIE2 (28). Other substrates such as VE-cadherin or the VEGFR2 require active kinase stimulation and, therefore, inhibition of VE-PTP alone may not necessarily be enough to achieve strong phosphorylation of such substrates. A third important difference between both proteomics approaches is based on the peroxyvanadate treatment of endothelial cells in the substrate trapping approach. Peroxyvanadate is a strong nonselective inhibitor of all tyrosine phosphatases, which leads to near maximal tyrosine phosphorylation level of all potential tyrosine phosphatase substrates. Hence, this approach probably allowed the detection of substrates that were missed in the phosphoprotein affinity enrichment experiment because of a lack of sensitivity. Fourth, differences in the repertoire of detectable substrates by both approaches may be because antiphospho-tyrosine antibodies do not bind to all tyrosine phosphorylation sites with the same affinity. Different binding affinities are equally expected for a phosphatase trapping mutant possibly leading to the preferential identification of some substrates in one approach but not in the other approach. Finally, the VE-PTP inhibitor-based approach would also detect proteins, which might be indirectly upregulated in tyrosine phosphorylation because of being a downstream signaling target of VE-PTP. Such targets are excluded in the VE-PTP trapping approach.
Recently, we characterized FGD5, a GTPase exchange factor for CDC42, as a direct substrate of VE-PTP in a global phosphoproteomics experiment, in which we had enriched for phosphopeptides from AKB-9778-treated endothelioma cells (28). Interestingly, when we perform a meta-analysis and integrate the AKB-9778 inhibitor-specific phosphotyrosine containing proteins from this dataa set into another Venn diagram analogous to Fig. 3A, EPHB4 as well as TIE2 are still identified as targets next to FGD5 (supplemental Fig. S1). This observation further validates our decision to focus on EPHB4 and TIE2 as substrates of VE-PTP in the present study.
The identification of EPHB4 as a direct interactor and substrate of VE-PTP is a novel finding, that may have significant implications on the control of EPHB4 activity, which regulates endothelial function and vessel specification. EPHB4 has an important role in vascular development (51, 52). It is expressed most prominently on venous endothelial cells and forms the arterial-venous boundary by interaction with its arterial ligand ephrinB2 (62). Although EPHB4 deletion in mice leads to embryonic lethality because of various cardiovascular defects (63), it also plays a role in postnatal blood vessel remodeling, and regulates permeability of tumor vasculature (50).
Interactions of phosphatases with other Eph receptors have been described also in neurons to be important for axon guidance. The phosphatase GLEPP1 (PTPRO) dephosphorylates and thereby limits the activity of neuronal EPHA2 and EPHB2 (64). The same group also showed that the R3 family of PTPs bind RTKs including some Eph receptors (other than EPHB4) in a mammalian two-hybrid system (65).
In addition to VE-PTP, we identified TIE2 as a novel binding partner of EPHB4 in endothelial cells. Interestingly, EPHB4 reduces tumor vascular permeability via activation of the Ang-1/TIE2 system (50). EPHB4 reverse signaling increases expression of Ang-1 leading to TIE2 activation in tumor endothelial cells, but a possible direct trans-activation was not examined in this report. This however could be shown for the EphB receptor ligand ephrinB1, which appears to be a direct substrate of TIE2 in vitro (66). EphrinB2 was also shown to suppress Ang1-induced proliferation in endothelial cells (67). However, the signaling behind this was based on p120-Ras-GAP, which reduced Tie-2-mediated activation of Ras-MAPK-activities. In agreement with our results (Fig. 7), ephrinB2 did not alter Tie-2 tyrosine phosphorylation. We could demonstrate by various biochemical means that VE-PTP, EPHB4 and TIE2 are present in a novel ternary receptor complex without crosstalk on the receptor level. Instead, preliminary results indicate that both signaling pathways seem to converge downstream of each of the two receptors at the level of MAP kinases, potentially in an antagonistic fashion (not shown). We propose that VE-PTP may play a crucial role in balancing the activity of both transmembrane receptors and their corresponding signaling pathways. Currently, however, it is unknown whether and to what extent the ternary complex of EPHB4, VE-PTP, and TIE2 that we describe here impacts on endothelial cell function. It may be attractive to speculate that this complex could be relevant for the suggested role of EPHB4 in reducing tumor vascular permeability via activation of the Ang-1/TIE2 system (50).
Long regarded as generally nondruggable, tyrosine phosphatases have recently gained considerable interest as therapeutic targets, and this pertains not only to VE-PTP but also to other phosphatases such as SHP2 (68) or LMPTP (69) and others (for recent reviews see (70) and (71)). However, known or suspected off target effects of PTP inhibitors may be a main reason why no clinically approved PTP-targeting drugs are yet available. The VE-PTP inhibitor AKB-9778 has the potential to change this view because it has already shown promising potential in the stabilization of endothelial junctions not only in vitro but also in vivo (16), and in mouse models of retinopathy (14), breast cancer (15), stroke (72), and in an S. aureus sepsis model (73). Interestingly, these vessel stabilizing effects of the VE-PTP inhibitor AKB-9778 were dependent on the presence of TIE2 (16). Most importantly, AKB-9778 was also shown to be beneficial in a Phase 2 clinical study in patients with diabetic macular edema (74), and it is currently also tested in a Phase 2b clinical trial for the treatment of patients suffering from diabetic retinopathy.
Our identification of novel potential substrates of VE-PTP may warrant future research on the application of the VE-PTP inhibitor in other diseases such as pulmonary arterial hypertension (PAH). BMPR2, was among the most enriched (22.7-fold) proteins in our VE-PTP inhibitor-based anti-phospho tyrosine-pulldown approach. Inactivation of BMPR2 has been implicated to contribute to PAH by promoting HMGA1-mediated endothelial-mesenchymal transition to a smooth muscle-like phenotype (75). Further, BMPR2 signaling appears to be modulated by VEGFR3 (FLT4), another tyrosine kinase receptor identified as potential substrate of VE-PTP in our study. Analogous to our observations on the EPHB4/TIE2 interaction, BMPR2 and VEGFR3 have been shown to also physically associate in a complex in which VEGFR3 is required for BMP-mediated receptor stimulation and downstream signaling such as SMAD phosphorylation and ID gene transcription (76). It may be interesting to investigate the impact of VE-PTP on the function of this pair of receptors in the future.
In conclusion, our search for substrates of VE-PTP revealed that proteins relevant for the regulation of endothelial junctions and of cell adhesion are over-represented among the potential targets of VE-PTP, supporting the concept that VE-PTP is an important regulator of vascular integrity. In addition, our results suggest that VE-PTP balances the signaling of several receptor systems, which may open new opportunities for the treatment of diseases of the vascular system.
DATA AVAILABILITY
All mass spectrometry proteomics data have been deposited to the Proteome-Xchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (23) with the data set identifier (PXD011940). Mass-labeled MS/MS spectra can also be inspected using MS-Viewer (University of California, San Francisco) using the keys zkps44natf (label free AKB-9778 experiment) and ikz3vrnybe (SILAC substrate trapping experiment).
Supplementary Material
Acknowledgments
We thank Dr. Astrid Nottebaum for helpful discussions during the preparation of the manuscript and for critical reading. Many thanks to Annalen Büchsenschütz for sample preparation and MS measurements.
Footnotes
* This work was supported by funds from the Deutsche Forschungsgemeinschaft (SFB1348, B1) (D.V.) and from the Max Planck Society and was performed as part of the Deutsche Forschungsgemeinschaft Clusters of Excellence Cells in Motion program.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- PTKs
- protein tyrosine kinases
- bEnd
- brain derived endothelioma
- EPHB4
- ephrin receptor B4
- FDR
- false discovery rate
- GO
- Gene Ontology
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- PTPs
- protein tyrosine phosphatases
- RTKs
- receptor tyrosine kinases
- SILAC
- stable isotope labeling with amino acids in culture
- TIE1/2
- tyrosine kinases with Ig and EGF homology domains
- VEGFR
- vascular endothelial growth factor receptor
- VE-PTP/PTPRB
- vascular endothelial specific protein tyrosine phosphatase
- WB
- Western blot.
REFERENCES
- 1. Tiganis T., and Bennett A. M. (2007) Protein tyrosine phosphatase function: the substrate perspective. Biochem. J. 402, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 3. Yao Z., Darowski K., St-Denis N., Wong V., Offensperger F., Villedieu A., Amin S., Malty R., Aoki H., Guo H., Xu Y., Iorio C., Kotlyar M., Emili A., Jurisica I., Neel B. G., Babu M., Gingras A. C., and Stagljar I. (2017) A Global Analysis of the Receptor Tyrosine Kinase-Protein Phosphatase Interactome. Mol. Cell. 65, 347–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fachinger G., Deutsch U., and Risau W. (1999) Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene 18, 5948–5953 [DOI] [PubMed] [Google Scholar]
- 5. Nawroth R., Poell G., Ranft A., Samulowitz U., Fachinger G., Golding M., Shima D. T., Deutsch U., and Vestweber D. (2002) VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 21, 4885–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baumer S., Keller L., Holtmann A., Funke R., August B., Gamp A., Wolburg H., Wolburg-Buchholz K., Deutsch U., and Vestweber D. (2006) Vascular endothelial cell specific phospho-tyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood 107, 4754–4762 [DOI] [PubMed] [Google Scholar]
- 7. Dominguez M. G., Hughes V. C., Pan L., Simmons M., Daly C., Anderson K., Noguera-Troise I., Murphy A. J., Valenzuela D. M., Davis S., Thurston G., Yancopoulos G. D., and Gale N. W. (2007) Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Winderlich M., Keller L., Cagna G., Broermann A., Kamenyeva O., Kiefer F., Deutsch U., Nottebaum A. F., and Vestweber D. (2009) VE-PTP controls blood vessel development by balancing Tie-2 activity. J. Cell Biol. 185, 657–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mellberg S., Dimberg A., Bahram F., Hayashi M., Rennel E., Ameur A., Westholm J. O., Larsson E., Lindahl P., Cross M. J., and Claesson-Welsh L. (2009) Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. 23, 1490–1502 [DOI] [PubMed] [Google Scholar]
- 10. Nottebaum A. F., Cagna G., Winderlich M., Gamp A. C., Linnepe R., Polaschegg C., Filippova K., Lyck R., Engelhardt B., Kamenyeva O., Bixel M. G., Butz S., and Vestweber D. (2008) VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J. Exp. Med. 205, 2929–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vockel M., and Vestweber D. (2013) How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood 122, 2512–2522 [DOI] [PubMed] [Google Scholar]
- 12. Amarasinghe K. K., Evdokimov A. G., Xu K., Clark C. M., Maier M. B., Srivastava A., Colson A. O., Gerwe G. S., Stake G. E., Howard B. W., Pokross M. E., Gray J. L., and Peters K. G. (2006) Design and synthesis of potent, non-peptidic inhibitors of HPTPbeta. Bioorg. Med. Chem. Lett. 16, 4252–4256 [DOI] [PubMed] [Google Scholar]
- 13. Campochiaro P. A., and Peters K. G. (2016) Targeting Tie2 for Treatment of Diabetic Retinopathy and Diabetic Macular Edema. Curr. Diab. Rep. 16, 126. [DOI] [PubMed] [Google Scholar]
- 14. Shen J., Frye M., Lee B. L., Reinardy J. L., McClung J. M., Ding K., Kojima M., Xia H., Seidel C., Lima e Silva R., Dong A., Hackett S. F., Wang J., Howard B. W., Vestweber D., Kontos C. D., Peters K. G., and Campochiaro P. A. (2014) Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J. Clin. Invest. 124, 4564–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goel S., Gupta N., Walcott B. P., Snuderl M., Kesler C. T., Kirkpatrick N. D., Heishi T., Huang Y., Martin J. D., Ager E., Samuel R., Wang S., Yazbek J., Vakoc B. J., Peterson R. T., Padera T. P., Duda D. G., Fukumura D., and Jain R. K. (2013) Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J. Natl. Cancer. Inst. 105, 1188–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frye M., Dierkes M., Küppers V., Vockel M., Tomm J., Zeuschner D., Rossaint J., Zarbock A., Koh G. Y., Peters K., Nottebaum A. F., and Vestweber D. (2015) Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 212, 2267–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koblizek T. I., Runting A. S., Stacker S. A., Wilks A. F., Risau W., and Deutsch U. (1997) Tie2 receptor expression and phosphorylation in cultured cells and mouse tissues. Eur. J. Biochem. 244, 774–779 [DOI] [PubMed] [Google Scholar]
- 18. Williams R. L., Risau W., Zerwes H. G., Drexler H., Aguzzi A., and Wagner E. F. (1989) Endothelioma cells expressing the polyoma middle T oncogene induce hemangiomas by host cell recruitment. Cell 57, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 19. Röhnelt R. K., Hoch G., Reiss Y., and Engelhardt B. (1997) Immunosurveillance modelled in vitro: naive and memory T cell spontaneously migrate across unstimulated microvascular endothelium. Intern. Immunol. 9, 435–450 [DOI] [PubMed] [Google Scholar]
- 20. Blanchetot C., Chagnon M., Dubé N., Hallé M., and Tremblay M. L. (2005) Substrate-trapping techniques in the identification of cellular PTP targets. Methods 35, 44–53 [DOI] [PubMed] [Google Scholar]
- 21. Ebnet K., Schulz C. U., Meyer-zu Brickwedde M. K., Pendl G. G., and Vestweber D. (2000) Junctional Adhesion Molecule (JAM) interacts with the PDZ domain containing proteins AF-6 and ZO-1. J. Biol. Chem. 275, 27979–27988 [DOI] [PubMed] [Google Scholar]
- 22. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 23. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doncheva N. T., Morris J. H., Gorodkin J., and Jensen L. J. (2019) Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 18, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hulsen T., de Vlieg J., and Alkema W. (2008) BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith A. L., Friedman D. B., Yu H., Carnahan R. H., and Reynolds A. B. (2011) ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PLoS ONE 6, e16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Campochiaro P. A., Sophie R., Tolentino M., Miller D. M., Browning D., Boyer D. S., Heier J. S., Gambino L., Withers B., Brigell M., and Peters K. (2015) Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology 122, 545–554 [DOI] [PubMed] [Google Scholar]
- 28. Braun L. J., Zinnhardt M., Vockel M., Drexler H. C., Peters K., and Vestweber D. (2019) VE-PTP inhibition stabilizes endothelial junctions by activating FGD5. EMBO Rep. 20, e47046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zang G., Christoffersson G., Tian G., Harun-Or-Rashid M., Vågesjö E., Phillipson M., Barg S., Tengholm A., and Welsh M. (2013) Aberrant association between vascular endothelial growth factor receptor-2 and VE-cadherin in response to vascular endothelial growth factor-a in Shb-deficient lung endothelial cells. Cell Signal. 25, 85–92 [DOI] [PubMed] [Google Scholar]
- 30. Flint A. J., Tiganis T., Barford D., and Tonks N. K. (1997) Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 94, 1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beckers C. M., Knezevic N., Valent E. T., Tauseef M., Krishnan R., Rajendran K., Hardin C. C., Aman J., van Bezu J., Sweetnam P., van Hinsbergh V. W., Mehta D., and van Nieuw Amerongen G. P. (2015) ROCK2 primes the endothelium for vascular hyperpermeability responses by raising baseline junctional tension. Vascul. Pharmacol. 70, 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., Cao G., DeLisser H., and Schwartz M. A. (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431 [DOI] [PubMed] [Google Scholar]
- 33. Muller W. A., Weigl S. A., Deng X., and Phillips D. M. (1993) PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 178, 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Privratsky J. R., and Newman P. J. (2014) PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res. 355, 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen X. L., Nam J. O., Jean C., Lawson C., Walsh C. T., Goka E., Lim S. T., Tomar A., Tancioni I., Uryu S., Guan J. L., Acevedo L. M., Weis S. M., Cheresh D. A., and Schlaepfer D. D. (2012) VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timmerman I., Hoogenboezem M., Bennett A. M., Geerts D., Hordijk P. L., and van Buul J. D. (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of β-catenin phosphorylation. Mol. Biol. Cell 23, 4212–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A. F., and Vestweber D. (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by a different tyrosine residue of VE-cadherin. Nat. Immunol. 15, 223–230 [DOI] [PubMed] [Google Scholar]
- 38. Schnoor M., Stradal T. E., and Rottner K. (2018) Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol. 28, 79–98 [DOI] [PubMed] [Google Scholar]
- 39. Schnoor M., Lai F. P., Zarbock A., Kläver R., Polaschegg C., Schulte D., Weich H. A., Oelkers J. M., Rottner K., and Vestweber D. (2011) Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J. Exp. Med. 208, 1721–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kanda S., Naba A., and Miyata Y. (2009) Inhibition of endothelial cell chemotaxis toward FGF-2 by gefitinib associates with downregulation of Fes activity. Int. J. Oncol. 35, 1305–1312 [DOI] [PubMed] [Google Scholar]
- 41. Kanda S., Kanetake H., and Miyata Y. (2007) Downregulation of Fes inhibits VEGF-A-induced chemotaxis and capillary-like morphogenesis by cultured endothelial cells. J. Cell. Mol. Med. 11, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mochizuki Y., Nakamura T., Kanetake H., and Kanda S. (2002) Angiopoietin 2 stimulates migration and tube-like structure formation of murine brain capillary endothelial cells through c-Fes and c-Fyn. J. Cell Sci. 115, 175–183 [DOI] [PubMed] [Google Scholar]
- 43. Laurent C. E., Delfino F. J., Cheng H. Y., and Smithgall T. E. (2004) The human c-Fes tyrosine kinase binds tubulin and microtubules through separate domains and promotes microtubule assembly. Mol. Cell. Biol. 24, 9351–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim L., and Wong T. W. (1995) The cytoplasmic tyrosine kinase FER is associated with the catenin-like substrate pp120 and is activated by growth factors. Mol. Cell. Biol. 15, 4553–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim L., and Wong T. W. (1998) Growth factor-dependent phosphorylation of the actin-binding protein cortactin is mediated by the cytoplasmic tyrosine kinase FER. J. Biol. Chem. 273, 23542–23548 [DOI] [PubMed] [Google Scholar]
- 46. Itoh T., Hasegawa J., Tsujita K., Kanaho Y., and Takenawa T. (2009) The tyrosine kinase Fer is a downstream target of the PLD-PA pathway that regulates cell migration. Sci. Signal 2, ra52. [DOI] [PubMed] [Google Scholar]
- 47. Oh M. A., Choi S., Lee M. J., Choi M. C., Lee S. A., Ko W., Cance W. G., Oh E. S., Buday L., Kim S. H., and Lee J. W. (2009) Specific tyrosine phosphorylation of focal adhesion kinase mediated by Fer tyrosine kinase in suspended hepatocytes. Biochim. Biophys. Acta 1793, 781–791 [DOI] [PubMed] [Google Scholar]
- 48. Kogata N., Masuda M., Kamioka Y., Yamagishi A., Endo A., Okada M., and Mochizuki N. (2003) Identification of Fer tyrosine kinase localized on microtubules as a platelet endothelial cell adhesion molecule-1 phosphorylating kinase in vascular endothelial cells. Mol. Biol. Cell 14, 3553–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wehrle C., Van Slyke P., and Dumont D. J. (2009) Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required for internalization and degradation. Biochem. J. 423, 375–380 [DOI] [PubMed] [Google Scholar]
- 50. Erber R., Eichelsbacher U., Powajbo V., Korn T., Djonov V., Lin J., Hammes H. P., Grobholz R., Ullrich A., and Vajkoczy P. (2006) EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J. 25, 628–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pitulescu M. E., and Adams R. H. (2014) Regulation of signaling interactions and receptor endocytosis in growing blood vessels. Cell Adh. Migr. 8, 366–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pasquale E. B. (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52 [DOI] [PubMed] [Google Scholar]
- 53. Saharinen P., Eklund L., and Alitalo K. (2017) Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Disco. 16, 635–661 [DOI] [PubMed] [Google Scholar]
- 54. Tammela T., Zarkada G., Wallgard E., Murtomaki A., Suchting S., Wirzenius M., Waltari M., Hellstrom M., Schomber T., Peltonen R., Freitas C., Duarte A., Isoniemi H., Laakkonen P., Christofori G., Yla-Herttuala S., Shibuya M., Pytowski B., Eichmann A., Betsholtz C., and Alitalo K. (2008) Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656–660 [DOI] [PubMed] [Google Scholar]
- 55. Karaman S., Leppänen V. M., and Alitalo K. (2018) Vascular endothelial growth factor signaling in development and disease. Development 145, pii: dev151019. [DOI] [PubMed] [Google Scholar]
- 56. Gomez-Puerto M. C., Iyengar P. V., García de Vinuesa A., ten Dijke P., and Sanchez-Duffhues G. (2018) Bone morphogenetic protein receptor signal transduction in human diseases. J. Pathol. 247, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kleinschmidt E. G., and Schlaepfer D. D. (2017) Focal adhesion kinase signaling in enexpected places. Curr. Opin. Cell Biol. 45, 24–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mori M., Murata Y., Kotani T., Kusakari S., Ohnishi H., Saito Y., Okazawa H., Ishizuka T., Mori M., and Matozaki T. (2010) Promotion of cell spreading and migration by vascular endothelial-protein tyrosine phosphatase (VE-PTP) in cooperation with integrins. J. Cell. Physiol. 224, 195–204 [DOI] [PubMed] [Google Scholar]
- 59. Chen H., Duncan I. C., Bozorgchami H., and Lo S. H. (2002) Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc. Natl. Acad. Sci. U.S.A. 99, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stradal T. E., and Scita G. (2006) Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell Biol. 18, 4–10 [DOI] [PubMed] [Google Scholar]
- 61. Choudhary C., and Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 [DOI] [PubMed] [Google Scholar]
- 62. Wang H. U., Chen Z. F., and Anderson D. J. (1998) Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741–753 [DOI] [PubMed] [Google Scholar]
- 63. Gerety S. S., Wang H. U., Chen Z. F., and Anderson D. J. (1999) Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 4, 403–414 [DOI] [PubMed] [Google Scholar]
- 64. Shintani T., Ihara M., Sakuta H., Takahashi H., Watakabe I., and Noda M. (2006) Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat. Neurosci. 9, 761–769 [DOI] [PubMed] [Google Scholar]
- 65. Sakuraba J., Shintani T., Tani S., and Noda M. (2013) Substrate specificity of R3 receptor-like protein-tyrosine phosphatase subfamily toward receptor protein-tyrosine kinases. J. Biol. Chem. 288, 23421–23431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Adams R. H., Wilkinson G. A., Weiss C., Diella F., Gale N. W., Deutsch U., Risau W., and Klein R. (1999) Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 13, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim I., Ryu Y. S., Kwak H. J., Ahn S. Y., Oh J. L., Yancopoulos G. D., Gale N. W., and Koh G. Y. (2002) EphB ligand, ephrinB2, suppresses the VEGF- and angiopoietin 1-induced Ras/mitogen-activated protein kinase pathway in venous endothelial cells. FASEB J. 16, 1126–1128 [DOI] [PubMed] [Google Scholar]
- 68. Chen Y. N., LaMarche M. J., Chan H. M., Fekkes P., Garcia-Fortanet J., Acker M. G., Antonakos B., Chen C. H., Chen Z., Cooke V. G., Dobson J. R., Deng Z., Fei F., Firestone B., Fodor M., Fridrich C., Gao H., Grunenfelder D., Hao H. X., Jacob J., Ho S., Hsiao K., Kang Z. B., Karki R., Kato M., Larrow J., La Bonte L. R., Lenoir F., Liu G., Liu S., Majumdar D., Meyer M. J., Palermo M., Perez L., Pu M., Price E., Quinn C., Shakya S., Shultz M. D., Slisz J., Venkatesan K., Wang P., Warmuth M., Williams S., Yang G., Yuan J., Zhang J. H., Zhu P., Ramsey T., Keen N. J., Sellers W. R., Stams T., and Fortin P. D. (2016) Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152 [DOI] [PubMed] [Google Scholar]
- 69. Stanford S. M., Aleshin A. E., Zhang V., Ardecky R. J., Hedrick M. P., Zou J., Ganji S. R., Bliss M. R., Yamamoto F., Bobkov A. A., Kiselar J., Liu Y., Cadwell G. W., Khare S., Yu J., Barquilla A., Chung T. D. Y., Mustelin T., Schenk S., Bankston L. A., Liddington R. C., Pinkerton A. B., and Bottini N. (2017) Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 13, 624–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stanford S. M., and Bottini N. (2017) Targeting tyrosine phosphatases: Time to end the stigma. Trends Pharmacol. Sci. 38, 524–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Senis Y. A., and Barr A. J. (2018) Targeting receptor-type protein tyrosine phosphatases with biotherapeutics: Is outside-in better than inside-out? Molecules 23, pii: E569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gurnik S., Devraj K., Macas J., Yamaji M., Starke J., Scholz A., Sommer K., Di Tacchio M., Vutukuri R., Beck H., Mittelbronn M., Foerch C., Pfeilschifter W., Liebner S., Peters K. G., Plate K. H., and Reiss Y. (2016) Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 131, 753–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bartz R. R., McCord T., Suliman H., Orr T., Plantadosi C. A., and Kontos C. (2016) Akb-9785, VE-PTP inhibitor, decreases inflammation and alters the angiopoietin/Tie2 system dynamic response in a S. aureus sepsis model. Am. J. Resp. Crit. Care Med. 193, A5751 [Google Scholar]
- 74. Campochiaro P. A., Khanani A., Singer M., Patel S., Boyer D., Dugel P., Kherani S., Withers B., Gambino L., Peters K., Brigell M., and Group T.-S. (2016) Enhanced benefit in diabetic macular edema from AKB-9778 Tie2 activation combined with vascular endothelial growth factor suppression. Ophtalmology 123, 1722–1730 [DOI] [PubMed] [Google Scholar]
- 75. Hopper R. K., Moonen J. R., Diebold I., Cao A., Rhodes C. J., Tojais N. F., Hennigs J. K., Gu M., Wang L., and Rabinovitch M. (2016) In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133, 1783–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hwangbo C., Lee H. W., Kang H., Ju H., Wiley D. S., Papangeli I., Han J., Kim J. D., Dunworth W. P., Hu X., Lee S., El-Hely O., Sofer A., Pak B., Peterson L., Comhair S., Hwang E. M., Park J. Y., Thomas J. L., Bautch V. L., Erzurum S. C., Chun H. J., and Jin S. W. (2017) Modulation of endothelial bone morphogenetic protein receptor type 2 activity by vascular endothelial growth factor receptor 3 in pulmonary arterial hypertension. Circulation 135, 2288–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry proteomics data have been deposited to the Proteome-Xchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (23) with the data set identifier (PXD011940). Mass-labeled MS/MS spectra can also be inspected using MS-Viewer (University of California, San Francisco) using the keys zkps44natf (label free AKB-9778 experiment) and ikz3vrnybe (SILAC substrate trapping experiment).