

SUMMARY
Munc13 proteins play several roles in regulating shortterm synaptic plasticity. However, the underlying molecular mechanisms remain largely unclear. Here we report that C. elegans UNC-13L, a Munc13-1 ortholog, has three domains that inhibit synaptic vesicle (SV) exocytosis. These include the X (sequence between C2A and C1), C1, and C2B domains. Deleting all three inhibitory domains produces a hyperactive UNC-13 (sUNC-13) that exhibits dramatically increased neurotransmitter release, Ca2+ sensitivity of release, and release probability. The vesicular pool in unc-13 mutants rescued by sUNC-13 exhibits a faster synaptic recovery and replenishment rate, demonstrating an important role of sUNC-13 in regulating synaptic plasticity. Analysis of double mutants suggests that sUNC-13 enhances tonic release by increasing the open probability of UNC-64/syntaxin-1A, whereas its effects on evoked release appear to be mediated by additional functions, presumably by further regulating the activity of the assembled soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) complex.
Graphical Abstract
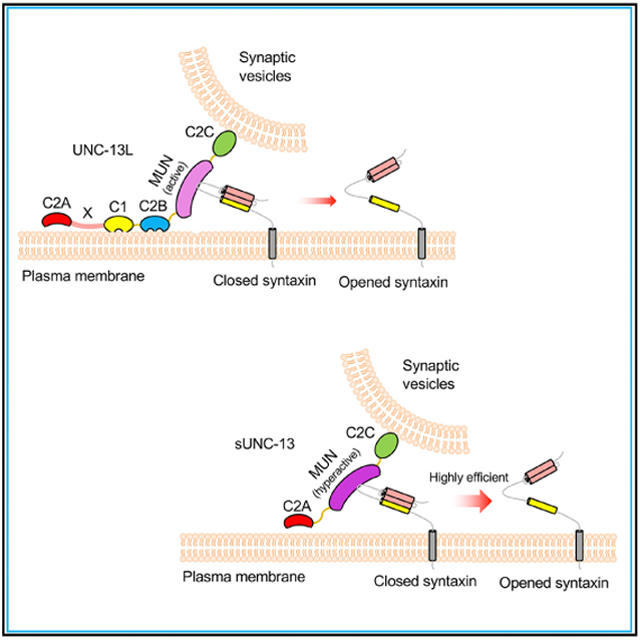
In Brief
Li et al. identify three domains in UNC-13L that inhibit neurotransmitter release. Removal of the three inhibitory domains produces a hyperactive UNC-13 that dramatically increases Ca2+ sensitivity and release probability of vesicle exocytosis by opening syntaxin in a highly efficient manner.
INTRODUCTION
Communication between neurons is determined by the release of neurotransmitters from synaptic vesicles (SVs) at presynaptic nerve terminals. SV release is a complicated and tightly regulated process in which SVs undergo several steps to become fusion competent, including docking and priming (Sudhof, 2004; Südhof and Rizo, 2011). Depolarization of the presynaptic plasma membrane leads to opening of voltage-gated Ca2+ channels, resulting in Ca2+ influx, which, in turn, triggers SV fusion (Borst and Sakmann, 1996). Synaptic efficacy is regulated by many aspects of SV exocytosis, with the release probability (Pvr) being one of the most essential parameters regulating synaptic strength, which is thought to underlie learning and memory formation in the nervous system (Malenka, 1994; Fioravante and Regehr, 2011). The release probability is mainly determined by the fusogenicity of SVs in the readily releasable vesicle pool (RRP). A large number of factors are involved in the regulation of the fusogenicity of SVs, such as the function of Ca2+ channels, the status of the fusion machinery, and the coupling of SVs to Ca2+ entry (Borst and Sakmann, 1999; Meinrenken et al., 2003; Catterall and Few, 2008).
At single synapses, release probability is assessed at both entire synapse and single SV levels (Gerber et al., 2008; Körber and Kuner, 2016). The Pvr of the entire synapse is determined by the ratio of release evoked by an action potential to the one that occurs when the entire RRP is released. The Pvr of individual SVs is typically determined by the frequency of spontaneous SV fusion. In this study, unless specifically designated, release probability represents the Pvr of both the entire synapse and single SVs. In the past two decades, a number of synaptic proteins have been identified for their essential roles in regulating SV release (Südhof and Rizo, 2011). It is believed that the soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) complex, consisting of synaptobrevin/vesicle-associated membrane protein (VAMP) on SVs and syntaxin and SNAP-25 on the plasma membrane, comprises the minimal machinery for SV fusion (Weber et al., 1998). Modulation of the SNARE complex is tightly linked to SV release and release probability (Weber et al., 2010). Several proteins, such as complexin, synaptotagmin, and tomosyn, regulate SV release through binding interactions with the SNARE core complex. Intriguingly, these proteins have been reported to regulate the release probability of entire synapses and single SVs differentially. For example, a dramatically increase in spontaneous release has been reported in synaptotagmin-1 or complexin-1 knockout neurons, resulting in an increased Pvr of single SVs, whereas the Pvr of entire synapses is largely decreased in these neurons (Huntwork and Littleton, 2007; Maximov et al., 2009; Shin et al., 2009; Xu et al.,2009; Martin et al., 2011). In tom-1 mutants, the Pvr of entire synapses is increased, whereas the Pvr of single SVs remains unchanged (Gracheva et al., 2006; McEwen et al., 2006; Chen et al., 2011; Liu et al., 2018). These observations indicate that the release probability of entire synapse and single SVs are regulated by distinct synaptic mechanisms.
As one of the SNARE-binding proteins, Munc13-1 and its homologs are involved in SV docking, priming, and post-priming Ca2+-triggered exocytosis (Aravamudan et al., 1999; Augustin et al., 1999; Richmond et al., 1999; Guan et al., 2008; Chen et al., 2013; Camacho et al., 2017). Munc13 proteins possess multiple functional domains that have different binding partners (Brose et al., 1995; Koch et al., 2000). Of the Munc13 proteins, Munc13-1 and Munc13-2 have been implicated in the regulation of synaptic plasticity (Basu et al., 2007; Chen et al., 2013). However, despite the work done on the Munc13 proteins, the molecular mechanisms by which they regulate synaptic plasticity remain largely unclear. In the present study, we investigated the functional roles of the individual domains in UNC-13L, a Munc13-1 homolog in C. elegans. We identified three different domains (X, C1, and C2B) that inhibit UNC-13-mediated synaptic transmission. Removing all three domains in UNC-13L produced a super-UNC-13 that greatly altered many aspects of synaptic transmission, including increased Ca2+ sensitivity and release probability, decreased synaptic depression, and faster synaptic recovery. We further demonstrated that the super-UNC-13 enhanced the release probability by activating UNC-64/syntaxin-1A, presumably by opening syntaxin in a highly efficient manner, as well as regulating the opened syntaxin activity during SNARE assembly. Our study therefore provides significant molecular insight into the role of UNC-13 in regulating synaptic transmission and plasticity.
RESULTS
Three UNC-13L Domains Inhibit Tonic Release
Unlike the action potential-evoked release found in mammalian synapses, the C. elegans neuromuscular junction (NMJ) exhibits graded synaptic transmission, with endogenous neural activity continuously driving SV fusion. This type of SV release is therefore termed tonic release (Hobson et al., 2011). Despite the difference in definition, tonic release at the C. elegans NMJ requires similar regulatory mechanisms as for spontaneous release, which occurs in the mouse CNS (Augustin et al., 1999; Richmond et al., 1999; Maximov et al., 2009; Martin et al., 2011). Moreover, we recently found that tonic release can still occur in the absence of extracellular Ca2+ at GABAergic and cholinergic synapses (Liu et al., 2018). This therefore provides a means to study tonic release under low endogenous activity.
We examined the functions of individual UNC-13L domains in tonic release. Both miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs, respectively) were recorded from unc-13 mutants rescued by full-length UNC-13L or truncated forms lacking specific domains (Figure 1A). Tonic release was completely eliminated in the unc-13 null mutants (s69) in 0 mM Ca2+, and this defect was fully rescued by transgenes expressing an UNC-13L cDNA (Figures 1B and 1C). Interestingly, we found that UNC-13L mutants lacking single N-terminal domains (i.e., ΔX ΔC1, and ΔC2B mutants) had a significantly increased mIPSC frequency (Figures 1B and 1C), indicating that these domains inhibit tonic release. The mIPSC amplitude was unchanged in all three deletion mutants (Figure 1D), suggesting that postsynaptic receptor abundance and function were unaffected.
Figure 1. The X, C1, and C2B Domains in UNC-13L Inhibit Tonic Release.

Miniature EPSCs were recorded from the body wall muscle of adult worms in a 0 mM Ca2+ solution.
(A) Cartoon depicting the domain structure of UNC-13L.
(B) Representative mIPSC traces from the indicated genotypes.
(C and D) Quantification of the frequency (C) and amplitude (D) of the mIPSCs from the same genotypes as in (B).
(E) Representative mEPSC traces from the indicated genotypes.
(F and G) Average of the frequency (F) and amplitude (G) of the mEPSCs from the same genotypes as in (E).
(H) Sequence alignment of the C1 and C2B domains between worm unc-13 and rat Munc13-1. Identical residues are highlighted (blue). The histidine (H) that is essential for DAG binding in the C1 domain and the five aspartates (D1-D5) that bind Ca2+ in the C2B domain are indicated with stars.
(I) Representative mIPSC traces from the indicated genotypes.
(J and K) Quantification of the frequency (J) and amplitude (K) of the mIPSCs from the same genotypes as in (I).
Data are presented as box-and-whisker plots, with both the median (line) and mean (cross) indicated. ###p < 0.001 compared with the wild type; *p < 0.05, **p < 0.01, ***p < 0.001 compared with UNC-13L rescue; n.s., non-significant compared with UNC-13L rescue; one-way ANOVA test for the data in (D) and (G); oneway ANOVA following Kruskal-Wallis test for the data in (C), (F), (J), and (K). The number of worms analyzed for each genotype is indicated under each box.
In general, deleting these domains had smaller effects on mEPSCs than those observed for mIPSCs. Deleting the X or the C1 domain failed to cause a detectable change (Figures 1E and 1F), whereas deleting the C2B domain led to a significant increase in mEPSC frequency (Figure 1F), indicating stronger inhibition of the C2B domain on tonic release. This led us to ask whether the weaker phenotype in mEPSCs resulted from a functional defect of the postsynaptic receptors at cholinergic synapses under Ca2+-free conditions. To test this, we performed acetylcholine (ACh) puff experiments in the presence or absence of extracellular Ca2+ in wild-type animals. A short pulsed application of ACh (0.5 M, 100 ms) onto the body wall muscle elicited a large current in a 1 mM Ca2+ bath solution, whereas the current was reduced by 50% in a 0 mM Ca2+ solution (Figures S1A and S1B). In contrast, puffing γ-aminobutyric acid (GABA; 0.5 M, 100 ms) onto the body wall muscle in 0 mM Ca2+ produced a current with a similar size as that obtained in 1 mM Ca2+ (Figures S1C and S1D). These results demonstrate that the channel conductance of the postsynaptic receptors is decreased at cholinergic synapses in the absence of Ca2+, which may cause a greater variation in the mEPSCs recorded under the same condition. Therefore, changes in mEPSC frequency may be difficult to see in some cases.
Mutations in the C1 and C2B Domains Mimic the Inhibitory Functions
Despite our mEPSC results, our mIPSC findings clearly showed that the X, C1, and C2B domains inhibit tonic release. Prior studies identified key residues in the C1 and C2B domains that affect synaptic transmission (Lou et al., 2008; Shin et al., 2010; Michelassi et al., 2017). A point mutation in the Munc13-1 C1 domain (H567K) abolishes diacylglycerol (DAG)/phorbol ester binding but leads to increased spontaneous release in the calyx of Held, suggesting increased Munc13-1 function (Basu et al., 2007). Sequence alignment showed that the histidine residue is deeply conserved in other Munc13 isoforms as well as in the two unc-13 isoforms in C. elegans (H696 in UNC-13L; Figure 1H; Hu et al., 2013). We found that the unc-13 mutants rescued by UNC-13L(H696K) exhibited a significantly higher mIPSC frequency that was comparable with that following UNC-13LΔC1 rescue in 0 mM Ca2+ (Figures 1I and 1J), indicating that the H696K mutation in UNC-13L functionally mimics the C1 deletion.
The C2B domain is characterized by Ca2+ and phospholipid binding properties (Shin et al., 2010). It has been reported that the Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis (Shin et al., 2010). Further studies in C. elegans have demonstrated that deleting C2B in UNC-13L significantly increases stimulus-evoked EPSCs (evoked EPSCs) (Michelassi et al., 2017). The C2B domain possesses five conserved aspartates, two of which are located in loop 1, with the other three being located in loop 3 (Figure 1H). These residues are essential for changing the structure of the C2B domain after binding to Ca2+ has occurred (Shin et al., 2010). Prior studies demonstrated that replacing the calcium-binding aspartate residues in loop 3 of C2B with asparagines (D3,4N) mimics the Ca2+-liganded state, whereas the corresponding glutamate substitutions (D3,4E) mimic the Ca2+-free state. Analysis of these mutants has revealed that calcium binding to C2B promotes membrane binding, increasing the evoked EPSC amplitude (Michelassi et al., 2017). Here we examined whether these mutations alter tonic release in 0 mM Ca2+. As shown in Figures 1I and 1J, a significant increase in mIPSC frequency was observed in unc-13 mutants rescued by UNC-13L(D3,4N), with the D3,4E mutations causing a slight but not significant decrease in mIPSC frequency. Mutating all five aspartates to asparagines (D1-5N) did not produce a further increase in mIPSC frequency. Moreover, the mIPSC frequency in the UNC-13L(D3,4N) rescue closely resembled that in the UNC-13LΔC2B rescue (Figures 1C and 1J). Collectively, these results indicate that unliganded C2B inhibits tonic release and that this inhibition is relieved by calcium binding.
A conserved calmodulin (CaM) binding motif was found in the X domain (Figure S2A). Calmodulin binding to Munc13 is required for short-term synaptic plasticity at the calyx of Held (Lipstein et al., 2013). We next tested whether calmodulin binding to UNC-13L accounted for the X domain’s inhibitory function. Co-immunoprecipitation results indicate that calmodulin binds to the N-terminal domain of UNC-13L and that this binding was disrupted by a point mutation in the calmodulin domain (W593R) (Figures S2A and S2B). The mIPSC rate exhibited by wild-type UNC-13L rescue was indistinguishable from that observed in unc-13 mutants rescued by UNC-13L(ΔCaM) or UNC-13L(W593R) (Figures S2C-S2E). These results indicate that calmodulin binding is not required for the inhibitory function of the X domain.
The Probability of Tonic Release Is Greatly Increased by Simultaneously Removing All Three Inhibitory Domains in UNC-13L
We next wondered whether the inhibitory effect of the X, C1, and C2B domains on tonic mEPSCs could be seen more easily by deleting all three domains simultaneously. To test this, a truncated UNC-13L lacking these domains (ΔXΔC1ΔC2B) was constructed (Figure 2A). As shown in Figures 2B and 2D, a 20-fold increase in mEPSC frequency was observed in unc-13 mutants rescued by UNC-13L(ΔXΔC1ΔC2B). Similarly, triple deletion of the X, C1, and C2B domains quadrupled the mIPSC frequency (Figures 2C and 2F). The cumulative probabilities for the interevent intervals of both mEPSCs and mIPSCs were strongly shifted when these three domains were lacking (Figures 2H and 2I). These results demonstrate strong inhibition of tonic release by the three domains. No difference in the amplitude of the tonic release was observed between UNC-13L and the triple deletion, suggesting that the highly increased tonic release frequency arises from a presynaptic mechanism. It is worth noting that the UNC-13L(ΔXΔC1ΔC2B) mEPSC and mIPSC rates in 0 mM Ca2+ exceeded those of all previously described mutants in which tonic release is inhibited. Our results therefore indicate that simultaneously deleting all three inhibitory domains leads to a super-UNC-13 function (hereafter termed sUNC-13).
Figure 2. UNC-13L Lacking the XC1C2B Domains Increases Tonic Release.

(A) Domain structure of full-length UNC-13L and UNC-13LΔXΔC1ΔC2B (i.e., sUNC-13).
(B and C) Representative mEPSC (B) and mIPSC (C) traces recorded from unc-13 mutants rescued by UNC-13L or UNC-13LΔXΔC1ΔC2B in 0 mM Ca2+.
(D–G) Boxplots of mEPSC/mIPSC frequency and amplitude from the indicated genotypes.
(H and I) Cumulative probability distributions of the interevent intervals of the mEPSCs (H) and mIPSCs (I) in UNC-13L and sUNC-13 rescue.
(J and K) Quantification of the mEPSC (J) and mIPSC (K) frequencies at various Ca2+ levels (0, 0.05, 0.1, and 1 mM) from unc-13 mutants rescued by UNC-13L (blue) and sUNC-13 (red).
(L and M) Quantification of the decay of the averaged mEPSCs (L) and mIPSCs (M).
Data in (J) and (K) are presented as mean ± SEM, and all other data are shown as box-and-whisker plots with the median (line) and mean (cross) indicated. ***p < 0.001 compared with UNC-13L rescue; Mann-Whitney test for data in (D) and (G); Student’s t test for data in (E) and (F); one-way ANOVA test for (L) and (M). The number of worms analyzed for each genotype is indicated under each box.
The finding that mEPSCs and mIPSCs were increased in 0 mM Ca2+ by sUNC-13 indicates an enhancement of the probability of tonic release. To confirm this, we analyzed the Ca2+ dependence of tonic release by recording both mEPSCs and mIPSCs in various Ca2+ concentrations. As depicted in Figures 2J and 2K, raising the extracellular Ca2+ concentration from 0 mM to 0.05 mM, 0.1 mM, and 1 mM significantly enhanced the mEPSC and mIPSC frequencies in wild-type animals, suggestive of a strong Ca2+ dependence of tonic release. The mEPSC and mIPSC frequencies in sUNC-13 rescue animals were significantly higher than those in wild-type animals at low Ca2+ levels (0.05 mM and 0.1 mM), demonstrating an increase in the probability of tonic release. Moreover, the mEPSC and mIPSC frequencies in sUNC-13 rescue animals at both 0.05 mM and 0.1 mM Ca2+ were indistinguishable from those in 0 mM Ca2+, indicating strongly increased Ca2+ sensitivity. The decay of the mEPSCs and mIPSCs in sUNC-13 worms were also comparable with those in wild-type and UNC-13L rescue animals, indicating that the increased release probability does not lead to a change in the kinetics of single SV fusion (Figures 2L and 2M). It should be noted that the mEPSCs and mIPSCs in 1 mM Ca2+ were comparable between sUNC-13 rescue and UNC-13L rescue (Figures 2J and 2K). In fact, despite the obvious inhibitory effect of the X, C1, and C2B domains on tonic release in 0 mM Ca2+, we did not observe changes in tonic release rescued by UNC-13L lacking single inhibitory domains or carrying point mutations (histidine-to-lysine mutation [HK] and aspartate-to-asparagine mutation [DN]) in 1 mM Ca2+ (Figure S3). These results demonstrate that sUNC-13 increases the release probability of single SVs in low Ca2+ by enhancing the Ca2+ sensitivity of SV exocytosis.
Stimulus-Evoked Release, but Not Priming, Is Markedly Increased by sUNC-13
We next examined the functional role of sUNC-13 in evoked neurotransmitter release by recording stimulus-evoked EPSCs from the body wall muscle. Compared with UNC-13L, evoked EPSCs mediated by sUNC-13 were dramatically increased, with a 3-fold increase in amplitude and a 10-fold increase in charge transfer (Figures 3A-3C). Kinetic analysis of the evoked EPSCs mediated by sUNC-13 revealed a significant increase in rise time (20%–80%) and a large increase in decay compared with those rescued by UNC-13L. These results demonstrate that evoked neurotransmitter release is strongly inhibited by the X, C1, and C2B domains. Increased evoked EPSCs have also been observed in unc-13 mutants rescued by UNC-13L with single domain deletions (Figure S4; Michelassi et al., 2017). However, the increases in the amplitude, charge transfer, and decay caused by the single domain deletions were far smaller than those observed in sUNC-13 rescue. The decay of the mEPSCs rescued by sUNC-13 was indistinguishable from that of mEPSCs rescued by UNC-13L (Figures 3H and 3I), suggesting that the slower decay of sUNC-13-mediated evoked EPSCs was not due to a change in the decay of the mEPSCs.
Figure 3. sUNC-13 Increases Evoked Neurotransmitter Release without Changing the RRP.

(A) Example traces of stimulus-evoked EPSCs recorded in 1 mM Ca2+ from wild-type (black), UNC-13L rescue (blue), and sUNC-13 rescue (red) animals.
(B–F) Quantification of the evoked EPSC amplitude (B), charge transfer (C), 20%–80% rise time (D), decay (E), and delay (F) from the same genotype as in (A).
(G) Evoked EPSCs recorded from various Ca2+ levels (0.25, 0.5, and 1 mM) from UNC-13L (blue) and sUNC-13 (red) rescue animals.
(H) Example traces of averaged mEPSCs from the indicated genotypes.
(I) Quantification of the decay τ of the mEPSCs.
(J) Hypertonic sucrose-evoked current recorded from wild-type (black) and unc-13 mutants rescued by UNC-13L (blue) or sNC-13 rescue (red).
(K) Averaged charge transfer from the sucrose-evoked currents in (H).
(L) Quantification of the probability of synaptic vesicle release (Pvr) from the indicated genotypes.
(M) Top: representative confocal z stack images for UNC-13L and UNC-10/RIM (left) and sUNC-13 and UNC-10/RIM (right). Scale bar, 5 μm. Bottom: line scans along the dorsal nerve cord.
(N) Quantification of the fluorescence intensity and punctum size of UNC-13L::mApple and sUNC-13::mApple.
Data in (G) are presented as mean ± SEM, and all other data are shown as box-and-whisker plots with median (line) and mean (cross) indicated. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the wild type; **p < 0.01, ***p < 0.001 compared with UNC-13L rescue; Student’s t test for data in (G), one-way ANOVA following Kruskal-Wallis test for data in (B) and (C), and one-way ANOVA for all other data. The number of worms analyzed for each genotype is indicated under each box.
To assess the Ca2+ dependence of evoked release, we assessed the evoked EPSCs mediated by UNC-13L or sUNC-13 in 0.25, 0.5, and 1 mM Ca2+. Our results revealed that sUNC-13 rescue produced larger evoked EPSCs than UNC-13L rescue at all Ca2+ concentrations tested (Figure 3G). The increase in evoked charge transfer mediated by sUNC-13 was even greater at low Ca2+ concentrations (20-fold in 0.25 mM Ca2+ and 10-fold in 1 mM Ca2+). These results, together with our observations in relation to tonic release, demonstrate that sUNC-13 dramatically enhances the Ca2+ sensitivity of SV exocytosis.
To examine whether the increase in evoked EPSCs following sUNC-13 rescue was caused by an increase in SV priming, we estimated the size of the RRP by measuring the synaptic charge evoked by hypertonic sucrose (1 M,2 s; Rosenmund and Stevens, 1996). Using this assay, SV priming was nearly eliminated in unc-13 mutants and restored by expressing full-length UNC-13L in all neurons (Figure 3J), consistent with previous reports (McEwen et al., 2006; Hu et al., 2013; Zhou et al., 2013). However, the sucrose-evoked currents rescued by sUNC-13 were comparable with those rescued by UNC-13L (Figure 3K). Thus, sUNC-13 did not alter SV priming, implying that the dramatically increased tonic and evoked release reflect sUNC-13 stimulation of a post-priming aspect of SV exocytosis. Consequently, the Pvr of the entire synapses, calculated by the ratio of the charge transfer of evoked EPSCs to the averaged charge transfer in sucrose responses (i.e., the RRP), was greatly increased by sUNC-13 (Figure 3L).
To determine whether deleting the three inhibitory domains in UNC-13L alters its synaptic localization, we examined the colocalization of sUNC-13 with UNC-10/RIM, an active zone protein. Our results revealed that UNC-13L (tagged with mApple) displayed good colocalization with UNC-10/RIM (tagged with GFP) (Figure 3M, left). A high degree of colocalization was also observed between sUNC-13 (tagged with mApple) and UNC- 10/RIM (Figure 3M, right), indicating that the inhibitory domains in UNC-13L are not involved in the regulation of protein localization.
The increased tonic and evoked release in sUNC-13 could arise from an increased abundance of sUNC-13 proteins, allowing sUNC-13 to trigger more SV release. To examine the protein levels of UNC-13L and sUNC-13, we quantified the tagged mApple fluorescence intensity and the punctum size. As shown in Figure 3N, both the expression levels and the punctum size of UNC-13L and sUNC-13 were comparable. Thus, our results demonstrate that sUNC-13 increases the release probability of entire synapses and single SVs by enhancing the Ca2+ sensitivity of SV exocytosis without increasing protein abundance.
sUNC-13 Weakens Synaptic Depression and Accelerates Synaptic Recovery
The highly increased release probability induced by sUNC-13 indicates a change in synaptic plasticity. The cholinergic synapses at the worm NMJ exhibit synaptic depression when receiving a train stimulus (Liu et al., 2009). The Pvr of the entire synapse in sUNC-13 rescue animals reached 0.5, which means that half of the total SVs in the primed vesicular pool were released in response to one stimulus, presumably resulting in stronger synaptic depression. To test this, a line expressing ChIEF, a variant of channelrhodopsin (Watanabe et al., 2013), was crossed into the UNC-13L and sUNC-13 rescue lines. Application of a 1-Hz or 5-Hz train light stimulus led to synaptic depression in UNC-13L rescue worms. Unexpectedly, sUNC-13 worms exhibited markedly decreased depression in response to both the 1-Hz and 5-Hz stimulus (Figures 4A and 4D). The ratios of the second, third, and fourth EPSCs to the first EPSC were significantly higher in sUNC-13 rescue worms (Figures 4B and 4E). The depression was slowed 3-fold by sUNC-13 compared with UNC-13L, as determined by the time constant of mono-exponential fits used to describe the normalized amplitude decay (Figures 4C and 4F). Synaptic depression can be caused by both presynaptic vesicular pool depletion and postsynaptic receptor desensitization (Regehr, 2012). Given that the UNC-13 constructs were only expressed in presynaptic neurons, and the mEPSC amplitude was unaltered in the sUNC-13 rescue animals (Figures 2E and 2G), our results indicate that sUNC-13 weakens synaptic depression via a presynaptic mechanism.
Figure 4. sUNC-13 Weakens Synaptic Depression and Accelerates Synaptic Recovery.

Synaptic depression and recovery were investigated by applying a train (1 Hz and 5 Hz) or a paired light stimulus onto the ventral nerve cord of UNC-13L (blue) and sUNC-13 (red) rescue worms with expression of ChIEF in their cholinergic motor neurons.
(A) Example traces of 1-Hz light train stimulus-evoked EPSCs from UNC-13L and sUNC-13 rescue animals.
(B) Quantification of synaptic depression by normalizing the EPSC amplitude (EPSCi) to the first EPSC amplitude (EPSC1) (n = 19 for UNC-13L rescue and n = 16 for sUNC-13 rescue).
(C) Averaged depression τ of the normalized EPSC amplitude in (B).
(D) Example traces of 5-Hz light train stimulus-evoked EPSCs from the same genotypes as in (A).
(E) Quantification of synaptic depression by normalizing the EPSCi to EPSC1 (n = 13 for UNC-13L rescue and n = 9 for sUNC-13 rescue).
(F) Averaged depression τ of the normalized EPSC amplitude in (E).
(G and I) Averaged cumulative EPSC amplitudes during 1-Hz (G) and 5-Hz (I) trains.
(H and J) Quantification of the replenishment rates in (G) and (I), respectively.
(K) Evoked EPSCs triggered by a paired light stimulus with various intervals ranging from 50 ms to 5 s.
(L) Averaged synaptic recovery, calculated by the ratio of EPSC2 to EPSC1, in UNC-13L and sUNC-13 rescue animals (UNC-13L rescue, 50 ms n = 6, 100 ms n = 6, 200 ms n = 10, 500 ms n = 7, 1 s n = 11, 5 s n = 7; sUNC-13 rescue, 50 ms n = 7, 100 ms n = 9, 200 ms n = 7, 500 ms n = 6, 1 s n = 7, 5 s n = 6).
Data in (B), (E), (G), (I), and (L) are presented as mean ± SEM, and all other data are shown as box-and-whisker plots with both median (line) and mean (cross) indicated. **p < 0.01, ***p < 0.001 compared with UNC-13L rescue; Student’s t test for data in (B), (E), and (L); Mann-Whitney test for data in (C), (H), and (J). The number of worms analyzed for each genotype is indicated under each box.
Although the ratio of synaptic depression for sUNC-13 eventually fell to the same level as that for UNC-13L, sUNC-13-mediated EPSCs in the steady state during the train stimulus were 2-to 3-fold larger than those mediated by UNC-13L. Considering the normal size of the RRP, there must have been a faster replenishment rate of the vesicular pool to compensate for the faster depletion of the vesicular pool in the synaptic terminals expressing sUNC-13. The replenishment rate was described by the slope of the cumulative EPSCs and calculated by a line fit through the linear section (Figures 4G and 4I). We found that the replenishment rates were significantly faster in the sUNC-13 rescue than those in the UNC-13L rescue under a 1-Hz or 5-Hz stimulus (Figures 4H and 4J). Thus, our results indicate that sUNC-13 enhances the replenishment rate of the RRP to overcome the fast depletion that occurs during the train stimulus, allowing a continuous high release probability and producing decreased synaptic depression.
To determine whether sUNC-13 is involved in RRP dynamics, we next measured synaptic recovery in response to a paired stimulus with various intervals ranging from 50 ms to 5 s (Figure 4K). The recovery rate of the vesicular pool was calculated by the ratio of the two paired EPSCs (EPSC2/EPSC1). We found that synaptic recovery was significantly accelerated by sUNC-13 compared with UNC-13L (Figure 4L). It took 200 ms to reach 75% recovery of the EPSC in the sUNC-13 rescue, whereas it required 2 s to reach a similar level of recovery in the UNC-13L rescue. Synaptic recovery was also assessed in 0.5 mM Ca2+, under which the initial release probability is supposed to be lower. Indeed, the first EPSCs recorded in 0.5 mM Ca2+ in sUNC-13 and UNC-13L rescues were significantly smaller than those in 1 mM Ca2+ (data not shown). Similar to the results obtained in 1 mM Ca2+, synaptic recovery was faster in the sUNC-13 rescue in 0.5 mM Ca2+ (Figure S5). Moreover, synaptic recovery exhibited an increased trend when release probability was decreased (in 0.5 mM Ca2+) in the UNC-13L rescue (Figure S5C), but we did not observe an obvious change in sUNC-13 rescue (Figure S5D), probably because the initial release probability was not decreased enough in this case. We further compared synaptic depression between UNC-13L and sUNC-13 in 0.5 mM Ca2+. Interestingly, the EPSC train mediated by sUNC-13 exhibited increased depression in the late stage (1 Hz and 5 Hz) (Figures S5E and S5F), corresponding to the increased Pvr of the entire synapse. The unchanged (1 Hz) or decreased (5 Hz) depression at the early stage may arise from a compensation of faster replenishment. Together, these results demonstrate that sUNC-13 accelerates the replenishment rate of the RRP, consistent with our cumulative EPSC results.
The C2A Domain and the Linker Domains Are Essential for sUNC-13 Function
The above results all demonstrate that sUNC-13 is hyperactive in mediating SV release. To understand how this hyperactivity is achieved, we investigated the domain functions in sUNC-13. The N-terminal C2A domain appears to be important because our prior studies have found that the C2A domain in UNC-13L is required for tonic and evoked neurotransmitter release (Liu et al., 2019). Apart from the C2A domain, several linker domains are retained in sUNC-13 (Figure 5A). These linkers separate the X, C1, and C2B domains in UNC-13L but form a large linker between the C2A and Munc13 homology (MUN) domains in sUNC- 13. To determine the functional importance of the C2A domain and the linker domain in sUNC-13, we examined SV release in unc-13 mutants rescued by sUNC-13 lacking the C2A domain (sUNC-13ΔC2A) or the linker domain (sUNC-13Δlinker) (Figure 5A). We observed a large reduction in mEPSC frequency in 0 mM Ca2+ (Figures 5B and 5C) and a pronounced decrease in evoked EPSCs (including their amplitude, charge transfer, and decay) in both sUNC-13ΔC2A and sUNC-13Δlinker rescue animals (Figures 5E and 5F). These results confirmed that both the C2A and linker domains play essential roles in the activity of sUNC-13. However, the sUNC-13Δlinker and sUNC-13ΔC2A still restored the RRP to a wild-type level, indicating that they are not involved in SV priming (Figures 5G and 5H). Consequently, the release probability of both the entire synapse and single SVs were largely decreased by removal of the C2A or linker domain from sUNC-13 (Figure 5I). Moreover, deleting C2A or the linker resulted in stronger synaptic depression compared with that of sUNC-13 (Figures 5J and 5K). The decreased SV release caused by deletion of the C2A or linker domain in sUNC-13 is unlikely to be caused by changes in protein abundance because the expression levels in sUNC-13ΔC2A and sUNC-13Δlinker were indistinguishable from that in sUNC-13 (Figures 5L and 5M). Together, these results demonstrate that the C2A domain and the linker are both important for hyperactivity of sUNC-13.
Figure 5. The C2A and Linker Domains Are Essential for sUNC-13 Function.

(A) Cartoon depicting the domain structure of sUNC-13, sUNC-13Δlinker, and sUNC-13ΔC2A.
(B–D) Representative traces and boxplot of the frequency and amplitude of mEPSCs and mIPSCs recorded from sUNC-13 rescue, Δlinker rescue, and ΔC2A rescue animals in 0 mM Ca2+.
(E and F) Representative traces and summary of the amplitude, charge transfer, and decay of the evoked EPSCs recorded from sUNC-13 rescue and Δlinker rescue and ΔC2A rescue in 1 mM Ca2+.
(G–I) Example traces of hypertonic sucrose-evoked currents (G), averaged charge transfer of sucrose currents (H), and Pvr (I) from the indicated genotypes.
(J) 1-Hz light train stimulus-triggered EPSCs from sUNC-13 rescue, Δlinker rescue, and ΔC2A rescue animals.
(K) Synaptic depression, analyzed by the normalization of the amplitude of the EPSCs to the amplitude of the first EPSC.
(L) Representative confocal z stack images for sUNC-13Δlinker and sUNC-13ΔC2A (tagged with mApple) and their colocalization with UNC-10/RIM (tagged with GFP). Scale bar, 5 μm. Bottom: line scans along the dorsal nerve cord.
(M) Quantification of the fluorescence intensity of sUNC-13Δlinker::mApple and sUNC-13ΔC2A::mApple.
Data in (J) are presented as mean ± SEM, and all other data are shown as box-and-whisker plots with median (line) and mean (cross) indicated. **p < 0.01, ***p < 0.001 compared with sUNC-13 rescue; one-way ANOVA test for data in (D), (H), and (M); one-way ANOVA following Kruskal-Wallis test for data in (C), (F), (I), and (K). The number of worms analyzed for each genotype is indicated under each box.
Similar to the results for mEPSCs, the mIPSC frequency was also largely decreased in 0 mM Ca2+ when the C2A domain was removed in sUNC-13 (Figure 5D). However, deleting the linker in sUNC-13 did not cause a change in mIPSC frequency in 0 mM Ca2+ (Figure 5D), indicating that the hyperactivity of sUNC-13 in GABAergic synapses does not rely on the linker, although the linker is required for sUNC-13 at cholinergic synapses (Figure 5C). This is probably due to a lower threshold of SV fusion in GABAergic synapses (Liu et al., 2018).
Given that the linker between C2A and MUN consists of three small independent linkers (linker 1, linker 2, and linker 3; Figure S6A), we next asked whether these three linkers are all required for sUNC-13 activity. To test this, each linker was removed from sUNC-13 (termed Δlinker1, Δlinker2, and Δlinker3; Figure S6A), leading to a large reduction in mEPSC and mIPSC frequency in 0 mM Ca2+ (Figures S6B-S6G) and a pronounced decrease in the amplitude and charge transfer of the evoked EPSCs (Figures S6H-S6J). These results indicate that a proper length of linker between C2A and MUN is essential for sUNC-13 activity.
sUNC-13 Enhances the Release Probability of Single SVs by Opening Syntaxin-1A
How does sUNC-13 enhance release probability? Of all functional domains in UNC-13/Munc13, the MUN domain has been shown to be of central importance. It has been reported that the MUN domain binds syntaxin and catalyzes its opening, promoting SNARE complex assembly and membrane fusion (Madison et al., 2005; Guan et al., 2008; Yang et al., 2015). Thus, it is likely that the high release probability mediated by sUNC-13 arises from the increased activity of syntaxin. If this is the case, then the synaptic transmission mediated by sUNC-13 should be abolished in the absence of syntaxin. We tested this by introducing a mutation (unc-64(e246)) that reduces syntaxin-1A function in sUNC-13 rescue animals. The mEPSCs and mIPSCs recorded in 0 mM Ca2+ were nearly eliminated in unc-64 mutants (Figures 6A-6F), consistent with our previous findings (Liu et al., 2018). Moreover, sUNC-13-mediated tonic release was completely arrested in sUNC-13;unc-64 double mutants in 0 mM Ca2+ (Figure S7A-S7F) as well as in 0.1bmM Ca2+ (Figures S7C and S7D). These results clearly demonstrate that the tonic release mediated by sUNC-13 requires UNC-64/syntaxin-1A.
Figure 6. sUNC-13 Increases the Release Probability of Single SVs by Opening UNC-64/Syntaxin.

(A and B) Representative traces of the mEPSCs (A) and mIPSCs (B) recorded from the indicated genotypes in 0 mM Ca2+.
(C–F) Quantification of the frequencies and amplitudes of the mEPSCs and mIPSCs from the indicated genotypes in (A) and (B).
(G) Representative traces of the mEPSCs recorded from the indicated genotypes in 0.1 mM and 1 mM Ca2+.
(H–L) Averaged mEPSC frequencies and amplitudes from the same genotypes as in (G).
Data are shown as box-and-whisker plots with median (line) and mean (cross) indicated. *p < 0.05, **p < 0.01, ***p < 0.001 compared with sUNC-13 rescue; ###p < 0.001 compared with UNC-64 rescue in unc-64 mutants; one-way ANOVA test for data in (C), (D), (F), and (H); one-way ANOVA following Kruskal-Wallis test for data in (E) and (J)–(L). The number of worms analyzed for each genotype is indicated under each box.
The elimination of tonic release in sUNC-13;unc-64 double mutants could simply be due to the fact that UNC-64 is required for all tonic release, no matter how it is triggered. If this were true, then it would not be surprising that we did not observe tonic release in sUNC-13;unc-64 double mutants. We therefore tested this possibility using the cpx-1 mutant, in which tonic release is also highly increased in low Ca2+ (Wragg et al., 2013; Liu et al., 2018). Strikingly, we found that tonic release persists in cpx-1;unc-64 double mutants. Particularly in 0.1 mM Ca2+, the mEPSCs and mIPSCs occurred at much higher frequencies in cpx-1;unc-64 double mutants than in sUNC-13;unc-64 double mutants (Figure S7). These results support the idea that sUNC-13 increases tonic release by specifically activating UNC-64/syntaxin-1A.
When in the open conformation, syntaxin is competent to form SNARE complexes. Our results therefore support the hypothesis that sUNC-13 enhances release probability by opening syntaxin in a highly efficient manner. If this is correct, then we would expect to see an increase in the release probability when syntaxin is in an opened conformation. Prior studies have identified two point mutations (L165/166E) in mouse syntaxin that shift its default conformation from closed to open (Dulubova et al., 1999). These two residues are highly conserved in C. elegans UNC-64 (Richmond et al., 2001; McEwen et al., 2006; Hammarlund et al., 2007). We therefore examined whether the release probability was increased by an opened UNC-64(L169/170E) (hereafter termed UNC-64(open)). The impairment in tonic release in unc-64 mutants was restored by neuronal expression of wild-type UNC-64 (Figures 6A-6F). By comparison, expressing UNC-64(open) in unc-64 mutants led to a significant increase in tonic release in 0 mM Ca2+ (increased ratio: mEPSC, 10-fold; mIPSC, 3-fold). These findings clearly demonstrate that open syntaxin facilitates the release probability of single SVs.
We next asked whether the release probability was further increased in the presence of both sUNC-13 and UNC-64(open). Our results showed that the mEPSCs and mIPSCs in sUNC-13;UNC-64(open) animals were similar to those in UNC-64(open) rescue animals in 0 mM Ca2+ (Figures 6A-6F). These results indicate that sUNC-13 is unable to increase the release probability of single SVs when syntaxin is already open. Our findings therefore support a model where sUNC-13 enhances the release probability of single SVs by opening syntaxin more efficiently, perhaps by stabilizing the open conformation. It should be noted that the mEPSC frequency rescued by UNC-64(open) was significantly lower than that rescued by sUNC-13 (Figures 6C and 6E). This could result from some unknown effects of UNC-64(open) in the absence of Ca2+. It has been reported that closed syntaxin has a positive role in stabilizing syntaxin-1 and Munc18-1 and that syntaxin-1 and Munc18-1 levels decrease when syntaxin becomes open (Gerber et al., 2008). To investigate their Ca2+ sensitivity, we recorded mEPSCs in UNC-64(open) animals at 0.1 mM and 1 mM Ca2+. Our results showed that, in the presence of Ca2+, UNC-64(open) restored the mEPSCs to a similar level as that observed in sUNC-13 rescue animals (Figures 6G-6L). Furthermore, no additional increase in tonic release was observed in sUNC-13; UNC-64(open) animals in either 0.1 mM or 1 mM Ca2+. These data support the idea that sUNC-13 enhances the release probability of single SVs by opening UNC-64/syntaxin-1A in a highly efficient manner.
The above findings raise the question of whether sUNC-13 is still required when syntaxin is in an open conformation. To address this, we tested whether UNC-64(open) can bypass UNC-13 and restore tonic release in unc-13 mutants. We observed poor rescue of the tonic release when UNC-64(open) was expressed in unc-13 null mutants. The mEPSCs and mIPSCs in 1 mM Ca2+ were only slightly increased in UNC-64(open);unc-13 double mutants compared with unc-13 mutants (Table S1). This suggests that sUNC-13 is still required to trigger a high probability of SV fusion when syntaxin is open. These results are consistent with previous reports that showed that the SNARE complex cannot be correctly assembled when Munc13 is deleted (McEwen et al., 2006; Hammarlund et al., 2007; Lai et al., 2017).
sUNC-13 Enhances the Release Probability of Entire Synapses via an Additional Mechanism
We next examined whether opening UNC-64/syntaxin-1A with high efficiency could account for the role of sUNC-13 in enhancing the release probability of entire synapses. The evoked EPSCs in sUNC-13;unc-64 double mutants were completely eliminated, indicating that sUNC-13 regulates evoked neurotransmitter release by activating UNC-64/syntaxin-1A (Figures 7A-7C), similar to our observations in relation to tonic release. Neuronal expression of UNC-64 in unc-64 mutants restored the deficits in evoked EPSCs and the RRP, and expression of UNC-64(open) in unc-64 mutants led to a significant increase in the amplitude, charge transfer, and decay of evoked EPSCs (Figures 7A-7D) but not the RRP (Figures 7E and 7F). Consequently, the Pvr was significantly increased 3-fold by UNC-64(open) compared with UNC-64 (Figure 7G), demonstrating that open syntaxin enhances the release probability of entire synapses. However, both the charge transfer and Pvr of evoked EPSCs mediated by UNC-64(open) were much smaller than those mediated by sUNC-13. These results suggest that sUNC-13 may have additional functions in triggering evoked neurotransmitter release.
Figure 7. sUNC-13 Increases the Release Probability of Entire Synapses beyond Opening UNC-64/Syntaxin.

(A) Example traces of stimulus-evoked EPSCs from the indicated genotypes.
(B–D) Quantification of the amplitude (B), charge transfer (C), and decay (D) of the evoked EPSCs in (A).
(E and F) Representative traces of sucrose-evoked current (E) and averaged charge transfer (F) from the indicated genotypes.
(G) Quantification of the release probability of entire synapses (Pvr). Data are shown as box-and-whisker plots with median (line) and mean (cross) indicated. *p < 0.05, ***p < 0.001 compared with sUNC-13 rescue; ##p < 0.01, ###p < 0.001 compared with UNC-64 rescue in unc-64 mutants; one-way ANOVA test for data in (B), (C), and (G); one-way ANOVA following Kruskal-Wallis test for data in (D and (F). The number of worms analyzed for each genotype is indicated under each box.
We tested this idea by assessing the evoked EPSCs in sUNC-13;UNC-64(open) double rescue animals. The charge transfer and decay of evoked EPSCs in sUNC-13;UNC-64(open) animals were significantly increased compared with those in UNC-64(open) animals but were close to those observed in sUNC-13 animals (Figures 7C and 7D). This indicates that sUNC-13 could further increase evoked release despite the fact that syntaxin-1A was open. On the other hand, UNC-64(open) did not cause an additional increase in evoked EPSCs triggered by sUNC-13. In addition, the effect of UNC-64(open) on evoked neurotransmitter release requires the presence of UNC-13 because the evoked EPSCs were poorly restored in UNC-64(open);unc-13 double mutants (Table S1). The size of the RRP was normal in sUNC-13;UNC-64(open) animals, leading to a Pvr of the entire synapses that was close to that in sUNC-13 animals (Figure 7G). These results demonstrate that the function of sUNC-13 in evoked release is not limited to opening UNC-64/syntaxin-1A. Instead, it could function beyond that, presumably by regulating the activity of the opened syntaxin during SNARE assembly, thereby enhancing the probability of evoked neurotransmitter release.
DISCUSSION
In this study, we identified a more active form of UNC-13 (sUNC-13) that lacks three inhibitory domains (X, C1, and C2B). sUNC-13 leads to dramatically increased SV exocytosis because of enhanced Ca2+ sensitivity, increased release probability, decreased synaptic depression, and accelerated RRP refilling and synaptic recovery. Below we discuss the importance of our findings.
Facilitatory and Inhibitory Regulation of UNC-13 in Neurotransmitter Release
The multiple domains in Munc13-1 and their distinct binding partners suggest that Munc13-1 may have various regulatory roles in synaptic transmission and plasticity. Prior studies have shown that SV priming and Ca2+-triggered SV exocytosis are reduced by removal of the Munc13-1 C2A domain, suggestive of a facilitatory role of C2A in SV release (Zhou et al., 2013; Camacho et al., 2017; Liu et al., 2019). A recent study reported that the C2B domain in UNC-13L inhibits neurotransmitter release at the C. elegans NMJ (Michelassi et al., 2017). Notably, the function of the C2B domain appears to be conserved between UNC-13L and mouse Munc13-1 because replacing the UNC-13L C2B domain with Munc13-1 C2B fully restores the synaptic inhibition caused by C2B deletion. Moreover, the results we obtained using UNC-13L(H696K) resemble those observed with the same mutation in the Munc13-1 C1 domain (H567K), indicating that the function of the C1 domain is also conserved. These findings clearly demonstrate that UNC-13L/ Munc13-1 has two opposite regulatory functions in synaptic transmission and that the underlying mechanisms appear to be conserved between worm and mouse.
Although it is still unclear how UNC-13L/Munc13-1 promotes SV fusion, recent work on its crystal structure has suggested that Munc13-1 may act as a bridge between the plasma membrane and SVs (Liu et al., 2016; Xu et al., 2017). In this model, domains flanking the MUN domain bind to the plasma membrane and vesicular membrane, respectively, exposing the MUN domain. This enables it to catalyze SNARE complex assembly and promote membrane fusion. In light of this model, facilitatory and inhibitory regulation of UNC-13L/Munc13-1 in synaptic transmission likely results from different functions of the MUN domain (e.g., position, conformation, or activity) that subsequently affect the status of the SNARE complex. Thus, our studies, together with previous reports, demonstrate that UNC-13L/Munc13-1 can exert both facilitatory and inhibitory effects on SV exocytosis (Zhou et al., 2013; Camacho et al., 2017; Michelassi et al., 2017; Liu et al., 2019) and that the synaptic transmission mediated by UNC-13L reflects an interaction between facilitation and inhibition. These opposing functions of UNC-13L suggest that it could be one of the major substrates that regulates the heterogeneity of synaptic plasticity at synaptic terminals.
sUNC-13 Increases the Release Probability of Entire Synapses and Single SVs
Many factors are involved in the regulation of release probability, with the status or activity of the SNARE complex playing an important role. We have shown that sUNC-13 and UNC-64(open) enhance the release probability of entire synapse and single SVs, implying a common mechanism that increases the fusogenicity of SVs by sUNC-13 and open syntaxin. It has been suggested that UNC-13L/Munc13-1 regulates SV fusion by opening closed syntaxin via its MUN domain, which physically binds to syntaxin (Guan et al., 2008). Moreover, we found that sUNC-13 greatly enhances the Ca2+ sensitivity of evoked and tonic neurotransmitter release. Thus, our results support a model whereby sUNC-13 increases the open probability of syntaxin, thereby enhancing the Ca2+ sensitivity and release probability of SVs. This appears to be true because the mEPSCs mediated by sUNC-13 and UNC-64(open) were comparable in their frequencies in the presence of Ca2+ (0.1 mM and 1 mM Ca2+). Moreover, no additional increase was found in sUNC-13;UNC-64(open) animals (Figures 6G-6L). Our results therefore demonstrate that sUNC-13 enhances the release probability of single SVs by opening syntaxin in a highly efficient manner.
However, the mechanism for tonic release does not appear to account for the role of sUNC-13 in evoked neurotransmitter release. The sUNC-13-mediated evoked charge transfer and Pvr of entire synapses are much higher than those mediated by UNC-64(open) (Figures 7C and 7G). In addition, the evoked release (including amplitude, charge transfer, and decay) and Pvr in sUNC-13;UNC-64(open) animals were nearly comparable with those in sUNC-13 animals. This indicates that sUNC-13 may function beyond enhancing the open probability of syntaxin in evoked neurotransmitter release. However, the sUNC-13-mediated evoked release was completely abolished in sUNC-13;unc-64 double mutants, suggesting that sUNC-13 specifically targets syntaxin in regulating evoked release. Thus, our results indicate that sUNC-13 could further regulate the activity of the opened syntaxin during SNARE assembly to increase the evoked release to a much higher level. It is also possible that the highly increased open probability of syntaxin produces more opened syntaxin, enhancing the average number of assembled SNARE complexes at the release site.
sUNC-13 Regulates the Dynamics of the Priming Pool
A higher initial release probability often leads to stronger synaptic depression and slower synaptic recovery because of faster depletion of the vesicular pool (Regehr, 2012). However, we found that sUNC-13 enhanced the release probability but decreased synaptic depression and accelerated synaptic recovery (Figure 4). These unexpected findings raise the possibility that sUNC-13 has a role in RRP replenishment, presumably by accelerating the replenishment rate, thereby compensating for fast depletion. Indeed, our analysis of the cumulative EPSCs during a train stimulus showed that the replenishment rate was signifcantly increased by sUNC-13. These results demonstrate that the X, C1, and C2B domains inhibit the refilling process in the SV cycle, although it is unclear how each of these three domains contributes to this inhibition.
How are RRP dynamics regulated by sUNC-13? Many factors have been implicated in the regulation of RRP dynamics, among which increases in presynaptic calcium have been shown to be important in accelerating synaptic recovery from depression (Dittman and Regehr, 1998; Stevens and Wesseling, 1998; Xu-Friedman and Regehr, 2004; Sakaba, 2008), presumably by accelerating replenishment of the RRP. It is therefore possible that sUNC-13 influences Ca2+ entry at nerve terminals. This is likely because previous studies have reported that the Munc13-1 C2B domain binds to voltage-gated Ca2+ channels and regulates the effectiveness of Ca2+ influx (Calloway et al., 2015), raising the possibility that the worm C2B domain is also involved in regulation of presynaptic Ca2+ dynamics. The effect on Ca2+ entry could be even stronger in sUNC-13 rescue animals because of the much higher release probability. In addition, the largely enhanced Ca2+ sensitivity because of sUNC-13 could also contribute to faster synaptic recovery. It is also possible that the RRP contains different sub-pools (Neher, 2015) and that the sub-pools in sUNC-13 animals have different characteristics, somehow leading to slow depression and faster recovery. Overall, despite the normal size of the RRP in sUNC-13 animals, sUNC-13 regulates the dynamics of the vesicular pool, and this process appears to be activity dependent.
Function of the Linker Domains in Regulating UNC-13 Activity
Our results indicate that the linker is playing an important role in regulating the activity of the MUN domain, presumably by placing it in the most favorable position and conformation to open syntaxin and catalyze SNARE complex assembly. So far it is still unclear how UNC-13L/Munc13-1 establishes the bridge between SVs and the plasma membrane. One hypothesis is that mobility of the MUNC2C domain promotes the “capture” of a vesicle. The linker between the C2A and MUN domains could make MUNC2C more flexible (increasing its mobility), whereas this fragment becomes more rigid when the linker is removed, decreasing the probability that it will capture vesicles. It is also possible that this domain has other unknown functions.
STAR ★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All new strains and plasmids created in this study will be provided on request with no restrictions. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhitao Hu (z.hu1@uq.edu.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strain maintenance and genetic manipulation were performed as previously described (Brenner, 1974). Animals were cultivated at room temperature on nematode growth medium (NGM) agar plates seeded with OP50 bacteria. On the day before experiments, L4 larval stage animals were transferred to fresh plates seeded with OP50 bacteria for all the electrophysiological recordings and imaging experiments.
METHOD DETAILS
Constructs, transgenes and germline transformation
All the UNC-13 fragments including full-length or mutated cDNAs were amplified by PCR and inserted into the MCSII of the JB6 vector between the KpnI and NotI sites. The snb-1 promoter (3kb) or the unc-129 promoter (2.6kb) was inserted into the MCSI between the SphI and BamHI sites. For fusion constructs, mCherry or mApple was inserted between NotI and MluI. Transgenic strains were isolated by microinjection of various plasmids using either Pmyo-2::NLS-GFP (KP#1106) or Pmyo-2::NLS-mCherry (KP#1480) as the co-injection marker. Integrated transgenes were obtained by UV irradiation of strains carrying extrachromosomal arrays. All integrated transgenes were outcrossed at least seven times.
Fluorescence imaging
Animals were immobilized on 2% agarose pads with 30 mM levamisole. Fluorescence imaging was performed at the Queensland Brain Institute’s Advanced Microscopy Facility using a spinning-disk confocal system (3i Yokogawa W1 SDC) controlled by Slide-book 6.0 software. Animals were imaged with an Olympus 100× 1.4 NA Plan-Apochromat objective. Z series of optical sections were acquired at 0.11 μm steps. Images were deconvolved with Huygens Professional version 16.10 (Scientific Volume Imaging, the Netherlands) and then processed to yield maximum intensity projections using ImageJ 1.51n (Wayne Rasband, National Institutes of Health) (Schneider et al., 2012).
Electrophysiology
Electrophysiology was conducted on dissected C. elegans as previously described (Hu et al., 2012). Worms were superfused in an extracellular solution containing 127 mM NaCl, 5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 20 mM glucose, 1 mM CaCl2, and 4 mM MgCl2, bubbled with 5% CO2,95% O2 at 22°C. The 1 mM CaCl2 was replaced by 1 mM MgCl2 to record miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs) in 0mM of Ca2+. Whole-cell recordings were carried out at −60mV for all EPSCs, including mEPSCs, evoked EPSCs, and sucrose-evoked responses. Stimulus-evoked EPSCs were stimulated by placing a borosilicate pipette (5–10 μm) near the ventral nerve cord (one muscle distance from the recording pipette) and applying a 0.4 ms, 85 μA square pulse using a stimulus current generator (WPI). A pulsed application of sucrose (1M, 2 s), ACh (0.5M, 100ms), or GABA (0.5M, 100ms) was applied onto the ventral nerve cord using a Picospritzer III (Parker). The holding potential was switched to 0mV to record mIPSCs. The internal solution contained 105 mM CH3O3SCs, 10 mM CsCl, 15 mM CsF, 4mM MgCl2, 5mM EGTA, 0.25mM CaCl2, 10mM HEPES, and 4mM Na2ATP, adjusted to pH 7.2 using CsOH. To estimate the readily releasable pool size, a pipette containing 0.5M sucrose solution was placed at the end of the patched muscle cell, and a 20 psi, 5 s pressure pulse was applied by Picospritzer to create a rapid jump in osmolarity at the neuromuscular junction (< 200 ms latency on average). The integrated charge transfer was computed as a function of time throughout the sucrose delivery, and the charge accumulation was corrected for the baseline holding current and spontaneous fusion events prior to sucrose application.
Retinal feeding
NGM plates (35 mm) were seeded with 250 μL of OP50 bacteria and 4 μL of 100 mM all-trans retinal (Sigma). Seeded retinal plates were kept in the dark at 4°C and were used within 7 days. ChIEF transgenic worms were transferred from regular plates to retinal plates at their L4 stage and then grown for an additional 16 h in a dark box before electrophysiological experiments.
Light stimulation
An LED light source (M470L3-C1, Thorlabs) was controlled by TTL signals from a HEKA EPC-10 double amplifier. Blue light (470 nm) through a GFP filter set was used to excite ChIEF. A train of light stimuli (1Hz or 5Hz) or a paired light stimulus was applied onto the ventral nerve cord to evoke postsynaptic currents. The duration of the light pulse was set at 3ms.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data acquisition and analysis
All electrophysiological data were obtained using a HEKA EPC10 double amplifier (HEKA Elektronik) filtered at 2 kHz, and analyzed with open-source scripts developed by Eugene Mosharov (http://sulzerlab.org/Quanta_Analysis_8_20.ipf) in Igor Pro 7 (Wavemetrics). To analyze mEPSCs and mIPSCs, a 4pA peak threshold was preset, under which release events are distinguished clearly from background noise. The analyzed results were re-checked by eye to ensure that the release events were accurately selected.
Statistical analysis
When the data followed a normal distribution, a Student’s t test or one-way ANOVA was used to evaluate the statistical significance. In other cases a Mann-Whitney test or one-way ANOVA following Kruskal-Wallis test was used. Data are presented as box-and-whisker plots showing the median and interquartile range, or the mean ± SEM. A summary of all electrophysiological data is provided in Table S1, with the results presented as mean ± SEM.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets/code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center (CGC) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CaCl2 | Fluka Analytical | Cat#21114 |
| MgCl2 | Fluka Analytical | Cat#63020 |
| NaCl | Sigma-Aldrich | Cat#S9888 |
| NaHCO3 | Sigma-Aldrich | Cat#S6014 |
| Sucrose | Sigma | Cat#S9378 |
| KCl | Sigma | Cat#P5405 |
| NaH2PO4 | Sigma-Aldrich | Cat#S0751 |
| Glucose | Sigma | Cat#G8270 |
| CsCl | Sigma | Cat#C3309 |
| CsF | Aldrich | Cat#289345 |
| EGTA | Sigma | Cat#E3889 |
| HEPES | Sigma | Cat#H4034 |
| Na2ATP | Sigma-Aldrich | Cat#2383 |
| CsOH | Aldrich | Cat#C8518 |
| Acetylcholine chloride | Sigma | Cat#A6625 |
| γ-Aminobutyric acid | Sigma | Cat#A2129 |
| Agarose | Sigma | Lot #SLBR6299V |
| All trans-Retinal | Sigma-Aldrich | CAS #116-31-4 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: N2 | Caenorhabditis Genetics Center | N2 |
| C. elegans: unc-13(s69) I | Caenorhabditis Genetics Center | BC168 |
| C. elegans: unc-64(e246) III | Caenorhabditis Genetics Center | CB246 |
| C. elegans: tom-1 (nu468) I | Dybbs et al., 2005 | KP3293 |
| C. elegans: tom-1 (nu468) I; unc-64(e246) III | This paper | ZTH585 |
| C. elegans: cpx-1(ok1552) I | Caenorhabditis Genetics Center | RB1367 |
| C. elegans: nuEx1515 [Psnb-1::UNC-13L];unc-13(s69) I | Hu et al., 2013 | KP6893 |
| C. elegans: cpx-1(ok1552) I; unc-64(e246) III | This paper | ZTH390 |
| C. elegans: hztEx41 [Psnb-1::UNC-13L(ΔX)]; unc-13(s69) I | This paper | ZTH318 |
| C. elegans: tauEx441 [Psnb-1::UNC-13L(ΔC1)]; unc-13(s69) I | Michelassi et al., 2017 | JSD1067 |
| C. elegans: tauSi3 [Psnb-1::UNC-13L(ΔC2B)]; unc-13(s69) I | Michelassi et al., 2017 | JSD0805 |
| C. elegans: hztEx19 [Psnb-1::UNC-13L(H696K)]; unc-13(s69) I | This paper | ZTH9 |
| C. elegans: tauEx313 [Psnb-1::UNC-13L(D3,4N)]; unc-13(s69) I | Michelassi et al., 2017 | JSD0835 |
| C. elegans: tauEx321 [Psnb-1::UNC-13L(D3,4E)]; unc-13(s69) I | Michelassi et al., 2017 | JSD0849 |
| C. elegans: hztEx50 [Psnb-1::UNC-13L(D1-5N)]; unc-13(s69) I | This paper | ZTH555 |
| C. elegans: hztEx51 [Psnb-1::UNC-13L(ΔCaM)]; unc-13(s69) I | This paper | ZTH497 |
| C. elegans: hztEx52 [Psnb-1::UNC-13L(W593R)]; unc-13(s69) I | This paper | ZTH446 |
| C. elegans: hztEx57 [Psnb-1::sUNC-13]; unc-13(s69) I | This paper | ZTH484 |
| C. elegans: hztEx58 [Psnb-1::sUNC-13(Δlinker)]; unc-13(s69) I | This paper | ZTH480 |
| C. elegans: hztEx109 [Psnb-1::sUNC-13(ΔC2A)]; unc-13(s69) I | This paper | ZTH696 |
| C. elegans: hztEx92 [Psnb-1::sUNC-13(Δlinker1)]; unc-13(s69) I | This paper | ZTH523 |
| C. elegans: hztEx93 [Psnb-1::sUNC-13(Δlinker2)]; unc-13(s69) I | This paper | ZTH519 |
| C. elegans: hztEx94 [Psnb-1::sUNC-13(Δlinker3)J; unc-13(s69) | This paper | ZTH536 |
| C. elegans: hztEx59 [Punc-129::UNC-13L::mCherry]; nuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH108 |
| C. elegans: hztEx60 [Punc-129::sUNC-13::mApple]; nuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH584 |
| C. elegans: hztEx61 [Punc-129::sUNC-13(Δlinker)::mApple]; nuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH583 |
| C. elegans: hztEx66 [Punc-129::sUNC-13(ΔC2A)::mApple]; nuIs165 [Punc-129::UNC-10::GFP] | This paper | ZTH702 |
| C. elegans: nuEx1515 [Psnb-1::UNC-13L]; unc-13(s69) I; oxSi91 [Punc-17::ChIEF::mCherry] | This paper | ZTH504 |
| C. elegans: hztEx57 [Psnb-1::sUNC-13]; unc-13(s69) I; oxSi91 [Punc-17::ChIEF::mCherry] | This paper | ZTH525 |
| C. elegans: hztEx58 [Psnb-1::sUNC-13(Δlinker)]; unc-13(s69) I; oxSi91 [Punc-17::ChIEF::mCherry] | This paper | ZTH506 |
| C. elegans: hztEx62 [Psnb-1::UNC-64]; unc-64 (e246) III | This paper | ZTH418 |
| C. elegans: hztEx63 [Psnb-1::UNC-64(open)::SL2::mApple]; unc-64 (e246) III | This paper | ZTH513 |
| C. elegans: hztEx57 [Psnb-1::sUNC-13];unc-13(s69) I; unc-64(e246) III | This paper | ZTH511 |
| C. elegans: hztEx57 [Psnb-1::sUNC-13]; unc-13(s69) I; hztEx63 [Psnb-1::UNC-64(open)]; unc-64(e246) III | This paper | ZTH556 |
| C. elegans: hztEx63 [Psnb-1::UNC-64(open)::SL2::mApple]; unc-13(s69) I; unc-64(e246) III | This paper | ZTH684 |
| C. elegans: hztEx67 [Psnb-1::UNC-64::SL2::mApple]; unc-13(s69) I; unc-64(e246) III | This paper | ZTH703 |
| Oligonucleotides | ||
| UNC-13L(C2A) forward primer for fusion: acatggtaccatggat gacgttggagattacaatgatg | IDT | N/A |
| UNC-13L(C2A) reverse primer for fusion: gcacagtacttacatc aaaaggaagctcaaatcgaac | IDT | N/A |
| UNC-13L(R) forward primer for fusion: ttttgatgtaagtactgtgc ttgacgggaacg | IDT | N/A |
| UNC-13L(R) reverse primer for fusion: cggcggccgcctatgttc gattgatgttttgacttgatgc | IDT | N/A |
| Recombinant DNA | ||
| Psnb-1::UNC-13L | This paper | Phzt10 |
| Psnb-1::UNC-13L (ΔX) | This paper | Phzt41 |
| Psnb-1::UNC-13L(ΔC1) | Michelassi et al., 2017 | JP975 |
| Psnb-1::UNC-13L(ΔC2B) | Michelassi et al., 2017 | JP679 |
| Psnb-1::UNC-13L(H696K) | This paper | Phzt19 |
| Psnb-1::UNC-13L(D3,4N) | Michelassi et al., 2017 | JP772 |
| Psnb-1::UNC-13L(D3,4E)] | Michelassi et al., 2017 | JP786 |
| Psnb-1::UNC-13L(D1x-5N) | This paper | Phzt417 |
| Psnb-1::UNC-13L(ΔCaM) | This paper | Phzt381 |
| Psnb-1::UNC-13L(W593R) | This paper | Phzt355 |
| Psnb-1::sUNC-13 | This paper | Phzt448 |
| Psnb-1::sUNC-13(Δlinker) | This paper | Phzt420 |
| Psnb-1::sUNC-13(ΔC2A) | This paper | Phzt501 |
| Psnb-1::sUNC-13(Δlinker1) | This paper | Phzt526 |
| Psnb-1::sUNC-13(Δlinker2) | This paper | Phzt503 |
| Psnb-1::sUNC-13(Δlinker3) | This paper | Phzt575 |
| Punc-129::UNC-13L::mCherry | This paper | A011 |
| Punc-129::sUNC-13::mApple | This paper | Phzt653 |
| Punc-129::sUNC-13(Δlinker)::mApple | This paper | Phzt648 |
| Punc-129::sUNC-13(ΔC2A)::mApple | This paper | Phzt1018 |
| Psnb-1::UNC-64 | This paper | Phzt238 |
| Psnb-1::UNC-64(open)::SL2::mApple | This paper | Phzt458 |
| Psnb-1::UNC-64::SL2::mApple | This paper | Phzt459 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | N/A |
| PatchMaster | HEKA Elektronik | V2×73.2 |
| Igor Pro | Wavemetrics | Version 7 |
| SigmaPlot | Systat Software Inc. | Version 13.0 |
| Other | ||
| Microscope Cover Glass | Fisher Scientific | Lot#060214-9 |
| Glass Capillaries | World Precision Instruments, Inc. | Lot#2009330 |
Highlights.
UNC-13L exhibits both facilitatory and inhibitory regulation in synaptic transmission
Removing all inhibitory domains in UNC-13L produces a hyperactive UNC-13
Synaptic recovery is accelerated in sUNC-13 transgenic animals
sUNC-13 increases release probability by opening syntaxin in a highly efficient manner
ACKNOWLEDGMENTS
We thank the C. elegans Genetics Stock Center for strains and reagents. We thank Dr. Jeremy Dittman for providing the following strains: JSD1067, JSD1062, JSD0835, and JSD0849. We thank Rowan Tweedale for critically reading the manuscript. This work was supported by an Australia Research Council Discovery Project grant (DP160100849 to Z.H.); a National Health and Medical Research Council Project grant (GNT1122351 to Z.H.); a National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator grant (24980 to Z.H.); a UQ Foundation Research Excellence Award (to Z.H.); and a National Institutes of Health (NIH) research grant (GM54728 to J.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.018.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, and Broadie K (1999). Drosophila UNC-13 is essential for synaptic transmission. Nat. Neurosci 2, 965–971. [DOI] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, and Brose N (1999). Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. [DOI] [PubMed] [Google Scholar]
- Basu J, Betz A, Brose N, and Rosenmund C (2007). Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J. Neurosci 27, 1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, and Sakmann B (1996). Calcium influx and transmitter release in a fast CNS synapse. Nature 383, 431–434. [DOI] [PubMed] [Google Scholar]
- Borst JG, and Sakmann B (1999). Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos. Trans. R. Soc. Lond. B Biol. Sci 354, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Hofmann K, Hata Y, and Südhof TC (1995). Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem 270, 25273–25280. [DOI] [PubMed] [Google Scholar]
- Calloway N, Gouzer G, Xue M, and Ryan TA (2015). The active-zone protein Munc13 controls the use-dependence of presynaptic voltage-gated calcium channels. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho M, Basu J, Trimbuch T, Chang S, Pulido-Lozano C, Chang SS, Duluvova I, Abo-Rady M, Rizo J, and Rosenmund C (2017). Heterodimerization of Munc 13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat. Commun 8, 15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, and Few AP (2008). Calcium channel regulation and presynaptic plasticity. Neuron 50, 882–901. [DOI] [PubMed] [Google Scholar]
- Chen K, Richlitzki A, Featherstone DE, Schwärzel M, and Richmond JE (2011).Tomosyn-dependent regulation of synaptic transmission is required for a late phase of associative odor memory. Proc. Natl. Acad. Sci. USA 108, 18482–18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Cooper B, Kalla S, Varoqueaux F, and Young SM Jr. (2013). The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci 33, 8336–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, and Regehr WG (1998). Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J. Neurosci 18, 6147–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, and Rizo J (1999). A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 18, 4372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybbs M, Ngai J, and Kaplan JM (2005). Using microarrays to facilitate positional cloning: identification of tomosyn as an inhibitor of neurosecretion. PLoS Genet. 1,6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, and Regehr WG (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol 21, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, deWit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, et al. (2008). Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321, 1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, and Richmond JE (2006). Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol. 4, e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Dai H, and Rizo J (2008). Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry 47, 1474–1481. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, and Jorgensen EM (2007). Open syntaxin docks synaptic vesicles. PLoS Biol. 5, e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, and Jorgensen EM (2011). Complexin maintains vesicles in the primed state in C. elegans. Curr. Biol 21, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Hom S, Kudze T, Tong XJ, Choi S, Aramuni G, Zhang W, and Kaplan JM (2012). Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science 337, 980–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Tong XJ, and Kaplan JM (2013). UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. eLife 2, e00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, and Littleton JT (2007). A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat. Neurosci 10, 1235–1237. [DOI] [PubMed] [Google Scholar]
- Koch H, Hofmann K, and Brose N (2000). Definition of Munc13-homologydomains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem. J 340, 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber C, and Kuner T (2016). Molecular Machines Regulating the Release Probability of Synaptic Vesicles at the Active Zone. Front. Synaptic Neurosci 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, et al. (2017). Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 05, 591–607.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, and Brose N (2013). Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13-1 signaling. Neuron 70, 82–96. [DOI] [PubMed] [Google Scholar]
- Liu Q, Hollopeter G, and Jorgensen EM (2009). Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc. Natl. Acad. Sci. USA 106, 10823–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, and Rizo J (2016). Functional synergy between the Munc13 C-terminal C1 and C2 domains. eLife 5, e13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li L, Wang W, Gong J, Yang X, and Hu Z (2018). Spontaneous Vesicle Fusion Is Differentially Regulated at Cholinergic and GABAergic Synapses. Cell Rep. 22, 2334–2345. [DOI] [PubMed] [Google Scholar]
- Liu H, Li L, Nedelcu D, Hall Q, Zhou L, Wang W, Yu Y, Kaplan JM, and Hu Z (2019). Heterodimerization of UNC-13/RIM regulates synaptic vesicle release probability but not priming in C. elegans. eLife 8, e40585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, and Schneggenburger R (2008). Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J. Neurosci 28, 8257–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, and Kaplan JM (2005). UNC-13 interaction with syntaxin is required for synaptic transmission. Curr. Biol 15, 2236–2242. [DOI] [PubMed] [Google Scholar]
- Malenka RC (1994). Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78, 535–538. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, and Dittman JS (2011). Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr. Biol 21, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, and Südhof TC (2009). Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323, 516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, and Kaplan JM (2006). Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron 51, 303–315. [DOI] [PubMed] [Google Scholar]
- Meinrenken CJ, Borst JG, and Sakmann B (2003). Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at the rat calyx of Held. J. Physiol 547, 665–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelassi F, Liu H, Hu Z, and Dittman JS (2017). A C1-C2 Module in Munc13 Inhibits Calcium-Dependent Neurotransmitter Release. Neuron 95, 577–590.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (2015). Merits and Limitations of Vesicle Pool Models in View of Heterogeneous Populations of Synaptic Vesicles. Neuron 87, 1131–1142. [DOI] [PubMed] [Google Scholar]
- Regehr WG (2012). Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol 4, a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, and Jorgensen EM (1999). UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci 2, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, and Jorgensen EM (2001). An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412,338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, and Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207. [DOI] [PubMed] [Google Scholar]
- Sakaba T (2008). Two Ca(2+)-dependent steps controlling synaptic vesicle fusion and replenishment at the cerebellar basket cell terminal. Neuron 57, 406–419. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Xu J, Rizo J, and Südhof TC (2009). Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. USA 106, 16469–16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J, et al. (2010). Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol 17, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, and Wesseling JF (1998). Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron 21, 415–424. [DOI] [PubMed] [Google Scholar]
- Sudhof TC (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci 27, 509–547. [DOI] [PubMed] [Google Scholar]
- Südhof TC, and Rizo J (2011). Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol 3, a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Liu Q, Davis MW, Hollopeter G, Thomas N, Jorgensen NB, and Jorgensen EM (2013). Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife 2, e00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, and Rothman JE (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772. [DOI] [PubMed] [Google Scholar]
- Weber JP, Reim K, and Sørensen JB (2010). Opposing functions of two sub-domains of the SNARE-complex in neurotransmission. EMBO J. 29, 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, and Dittman JS (2013). Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, and Südhof TC (2009). Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat. Neurosci 12, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Camacho M, Xu Y, Esser V, Liu X, Trimbuch T, Pan YZ, Ma C, Tomchick DR, Rosenmund C, and Rizo J (2017). Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13-1 C1C2BMUN. eLife 6, e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, and Regehr WG (2004). Structural contributions to short-term synaptic plasticity. Physiol. Rev 84, 69–85. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T, and Ma C (2015). Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol 22, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Stawicki TM, Goncharov A, and Jin Y (2013). Position of UNC-13 in the active zone regulates synaptic vesicle release probability and release kinetics. eLife 2, e01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.