

Abstract
Focal microinjection of tetrodotoxin (TTX), a potent voltage-gated sodium channel blocker, reduces neurological deficits and tissue loss after spinal cord injury (SCI). Significant sparing of white matter (WM) is seen at 8 weeks after injury and is correlated to a reduction in functional deficits. To determine whether TTX exerts an acute effect on WM pathology, Sprague Dawley rats were subjected to a standardized weight-drop contusion at T8 (10 gm × 2.5 cm). TTX (0.15 nmol) or vehicle solution was injected into the injury site 5 or 15 min later. At 4 and 24 hr, ventromedial WM from the injury epicenter was compared by light and electron microscopy and immunohistochemistry. By 4 hr after SCI, axonal counts revealed reduced numbers of axons and significant loss of large (≥5 μm)-diameter axons. TTX treatment significantly reduced the loss of large-diameter axons. In addition, TTX significantly attenuated axoplasmic pathology at both 4 and 24 hr after injury. In particular, the development of extensive periaxonal spaces in the large-diameter axons was reduced with TTX treatment. In contrast, there was no significant effect of TTX on the loss of WM glia after SCI. Thus, the long-term effects of TTX in reducing WM loss after spinal cord injury appear to be caused by the reduction of acute axonal pathology. These results support the hypothesis that TTX-sensitive sodium channels at axonal nodes of Ranvier play a significant role in the secondary injury of WM after SCI.
Keywords: spinal cord injury, TTX, electron microscopy, white matter, glia, microinjection
With the development of experimental models of contusive spinal cord injury (SCI) (Blight, 1996), researchers obtained the means to study changes that take place in the cord after injury. From these studies, it is apparent that injury occurs in two phases. The first phase, “primary injury”, is the mechanical trauma initially sustained. The second phase, termed “secondary injury”, involves a number of trauma-induced physiological and biochemical changes (e.g., ischemia, anoxia, excitotoxicity) that occur in the ensuing hours and days and exacerbate the consequences of the mechanical injury (Tator and Fehlings, 1991;Young, 1993).
The functional deficits produced by SCI are largely caused by the loss of white matter (WM), particularly the long tracts through which descending and ascending communication occurs (Blight and Decrescito, 1986; Noble and Wrathall, 1989; Wrathall et al., 1994). Recovery of hindlimb locomotion after SCI is highly correlated to WM sparing (Noble and Wrathall, 1989; Blight, 1991; Basso et al., 1996). Initially after experimental contusion injury, the WM appears largely intact (Bresnahan, 1978; Blight, 1983; Noble and Wrathall, 1985; Rosenberg and Wrathall, 1997). Over the next 4 hr, pathology increases (Bresnahan, 1978; Anthes et al., 1995; Fehlings and Tator, 1995; Rosenberg and Wrathall, 1997), suggesting that secondary injury mechanisms are involved in the loss of WM.
Calcium appears to play a critical role in WM pathology (Balentine and Greene, 1984; Stys et al., 1991; Waxman et al., 1991, 1994). Injury causes an influx of Ca2+ into the axon that is believed to initiate a series of pathobiological events (Schlaepfer and Zimmerman, 1981; Balentine and Dean, 1982; Povlishock, 1993; Maxwell, 1996). New evidence suggests that Ca2+ influx after injury is mediated, in part, by Na+. In vitro studies with the optic nerve found axonal conductance after an anoxic episode was enhanced if Ca2+ or Na+ was eliminated from the bathing medium or if Na+ influx was reduced through application of the Na+ channel blocker tetrodotoxin (TTX) (Ransom et al., 1990; Waxman et al., 1991, 1992). An in vitro model of WM compression injury also found recovery of compound action potentials was enhanced when Ca2+ was removed from the bathing medium or when the Na+ channel blocker TTX was added in the presence of Ca2+ (Agrawal and Fehlings, 1996). These findings suggest that Na+ influx and the voltage-gated Na+ channels on axons play critical roles in WM injury.
Based on these results, we examined the effect of TTX in vivo, using a standardized model of SCI (Wrathall et al., 1985). Rats received a contusion SCI followed by focal injection of 0.15 nmol of TTX into the injury site. The effects of TTX treatment on acute WM pathology were examined at 4 and 24 hr after SCI using a protocol for evaluating the extent of WM pathology that was developed in our laboratory (Rosenberg and Wrathall, 1997; Rosenberg et al., 1999). The results indicate that acute axonal pathology is significantly reduced with TTX treatment, supporting the hypothesis that axonal Na+ channels contribute to secondary injury of WM after SCI.
MATERIALS AND METHODS
Spinal cord injury. Female, Sprague Dawley rats (225–250 gm) were anesthetized with 4% chloral hydrate (360 mg/kg, i.p.) and SCI produced with a well characterized weight-drop device (Gale et al., 1985; Wrathall et al., 1985; Panjabi and Wrathall, 1988;Raines et al., 1988; Noble and Wrathall, 1989). A laminectomy was performed at T8, exposing a circle of dura ∼2.8 mm in diameter. For the contusion injury, angled Allis clamps were attached at the seventh and ninth spinous processes to stabilize the animal. A plastic impounder with a tip diameter of 2.4 mm was lowered onto the exposed dura. A 10 gm weight was dropped onto the impounder from a height of 2.5 cm. Rats were kept on absorbent bedding with ad libitum access to food and water after surgery. The bladder was manually expressed twice daily. No prophylactic antibiotics were used in this study.
TTX administration. Drug administration was performed as described previously (Teng and Wrathall, 1997). Briefly, TTX (Research Biochemicals, Natick, MA) was dissolved in citrate buffer (1.5 mm, pH 4.8) at a final concentration of 300 μm and sterilized through a 0.22 μm syringe filter (Millipore, Bedford, MA). Drug delivery was via microinjection. Sterotaxically, a 33 gauge needle was inserted directly into the lesion site with the tip of the needle placed in the dorsal funicular region at the midline, 1 mm below the dura. TTX was infused into the cord, starting either at 5 or at 15 min after SCI at a delivery rate of 0.1 μl/min for a total dose of 0.15 nmol. Vehicle (VEH) controls received an equal volume of the citrate buffer alone 5 or 15 min after SCI. In addition, TTX (0.15 nmol) was microinjected into the spinal cord at T8 of an uninjured rat to examine the effects of TTX alone on ventromedial WM.
Tissue collection and processing for electron microscopy.Two sets of rats were used for assessment of acute WM pathology. The first set of rats was injured and treated with either TTX (n = 5) or VEH (n = 6), beginning at 15 min after SCI. At 4 hr after SCI, they were reanesthetized with 4% chloral hydrate and intracardially perfused with saline followed by fixative (2% glutaraldehyde and 2% paraformaldehyde in 0.1m sodium cacodylate, pH 7.4). The second set of rats (TTX,n = 4 or VEH, n = 5) were similarly injured, treated with TTX beginning at 5 min after SCI, and perfused at 24 hr. Five additional sex- and age-matched uninjured rats were perfused to serve as normal controls.
After perfusion, cords were removed from the vertebral column and placed in fresh fixative overnight. A 2 cm segment, centered on the epicenter of the injury, was cut from each cord and embedded in 4% agar. The embedded tissue blocks were mounted on a tissue chopper stage (Sorvall, Newtown, CT) and cut into 250 μm cross sections (four sections = 1 mm of tissue). From each cord, tissue sections from the epicenter and from 1 mm rostral and caudal to the epicenter were post-fixed for 1 hr in 1% osmium–1% potassium ferricyanide, en bloc stained with 1% uranyl, and flat-embedded in Spurr resin (Ted Pella, Redding, CA). One micrometer sections were cut from each tissue block, stained with 1% toluidine blue, and viewed with light microscopy (LM).
Two blocks that appeared representative of each location (epicenter, +1 mm, −1 mm) were trimmed, leaving ∼1 mm2 of ventromedial WM. Ultra-thin (70–90 nm) sections were cut and placed on 200 mesh nickel grids (Ted Pella). Sections were viewed with a JEOL Jem 1200 EX (Tokyo, Japan) transmission electron microscope. Electron micrographs (2000×) were made of four fields of ventromedial WM, as defined by the 200 mesh grids on which the tissue sections were viewed, as described previously (Rosenberg and Wrathall, 1997; Rosenberg et al., 1999).
Assessment of WM pathology. In this model of incomplete contusive SCI, a rim of residual WM remains at the lesion epicenter (Noble and Wrathall, 1985, 1989). The ventromedial region of WM consistently demonstrates some chronically spared tissue, albeit with reduced numbers of axons and abnormal myelination (Noble and Wrathall, 1985; Wrathall et al., 1998). The development of pathology in this region appears to be caused, in part, to secondary injury over the first 24 hr after SCI (Rosenberg and Wrathall, 1997; Rosenberg et al., 1999). Therefore, assessment and quantification of the effect of TTX on WM pathology was performed on electron micrographs (2000×) of the ventromedial WM. Approximately 1600 μm2 of tissue was represented by each micrograph. The axons were counted, the intramyelin diameter was determined, and each axon was assigned a numerical value representative of the pathology that was present. The assigned values are based on three categories of pathology present in WM tissue after injury that include the axoplasm, myelin, and the presence of abnormal periaxonal spaces (Table1). For each rat, the pathology scores given the individual axons are summed and then divided by the total number of axons evaluated to calculate an axonal injury index (AII) for that animal, as previously described (Rosenberg and Wrathall, 1997;Rosenberg et al., 1999).
Table 1.
Calculation of the axonal injury index (AII)a
| Pathology | Points assigned |
|---|---|
| Myelin1-b | 1 |
| Axoplasm1-c | 2 |
| Periaxonal Space1-d | |
| 25–49% | 4 |
| 50–74% | 6 |
| ≥75% | 10 |
| Necrotic Axoplasm1-e | 10 |
The AII for an axon is the sum of points assigned for myelin and axoplasmic pathology and for periaxonal space, as described previously in detail.
Myelin pathology was defined as unwinding of the myelin lamellae that can be either slight or severe unraveling, appearing as whorls of membranous matter, abnormal spaces in between the lamellae, hypermyelination or hypomyelination, and fragmentation of the myelin sheath. One point was assigned to any axon displaying one or more of these pathologies in addition to points assigned for the other categories of pathology.
Axoplasmic pathology was defined as mitochondria swelling, presence of vacuoles/vesiculation, and condensation or loss of neurofilaments and microtubules. Axons with one or more of these pathologies were assigned 2 points, in addition to points assigned for the other categories of pathology.
Periaxonal spaces were defined as spaces between the axolemma and inner lamella of the myelin sheath. These spaces were occasionally seen in axons in uninjured tissue but never constituted >25% of the intramyelin area. Therefore, only spaces >25% of the intramyelin area were scored with assigned points increased to reflect greater degrees of spacing.
Axons devoid of cytoskeletal structures in which the axoplasm was comprised of a flocculent substance were classified as having “necrotic” axoplasm and given the maximum score of 10.
Glial quantification. In plastic 1 μm sections, a study area measuring 0.5 × 0.7 mm of ventromedial WM, bordered by the ventralmost region of the spinal cord and the ventromedial sulcus, was defined on each side of the sulcus for each cross section of spinal cord that was analyzed. All glia within this defined area with clearly visible, intact nuclei were counted at 400× magnification. For each rat, at least two spinal cord cross sections, separated by 250 μm, were evaluated at the lesion epicenter and also at 1 mm rostral and 1 mm caudal to the injury epicenter.
Immunohistochemical identification of glia. To identify glial subtypes, 12 rats were injured and injected with either TTX (n = 6) or VEH (n = 6), beginning at 5 min after injury, as described above. Six additional rats were included as uninjured (normal) controls. At 24 hr after SCI, rats were intracardially perfused with saline followed by buffered 4% paraformaldehyde, pH 7.4. The cords were post-fixed for 1 hr then equilibrated in sucrose (10–20% in PBS) and left overnight in 20% sucrose. The next day cords were frozen, and serial cross sections (10 and 20 μm) were cut with a Jung Frigicut 2800E cryostat (Leica, Germany) and mounted five sections (50 or 100 μm of tissue) per slide. One slide from each millimeter of tissue was stained with luxol blue/hematoxylin and eosin and evaluated by LM to determine the location of the lesion epicenter.
Tissue sections representing the epicenter and 1 mm rostral and 1 mm caudal to the epicenter were examined by immunocytochemistry with antibodies to the astrocyte marker glial fibrillary acid protein (GFAP; Dako, Carpinteria, CA), the microglia/macrophage marker OX-42 (Serotec; Harlan, Westbury, NY), and the antibody CC1 (APC-7; Oncogene Research Products, Cambridge, MA) that recognizes the adenomatous polyposis coli (APC) gene expressed in oligodendrocytes (Bhat et al., 1996; Crowe et al., 1997; Shuman et al., 1997). Sections were also stained with the antibody MYT1 (a gift from Dr. Lynn Hudson, Laboratory of Developmental Neurogenetics, National Institute of Neurological Diseases and Stroke) that recognizes the Myelin Transcription Factor 1 DNA-binding protein (Armstrong et al., 1997) found in nuclei of oliogodendrocytic precursor cells (Kim et al., 1997). Primary antibodies were used at a dilution of 1:1000 for CC1 and OX-42 and 1:100 for GFAP and MYT1.
Endogenous peroxidase was quenched with 0.3% hydrogen peroxide in 0.1m Tris buffer, pH 7.4, for 20 min. Sections were blocked for 1 hr with 3% serum in 0.1 m Tris buffer. The serum was removed and the tissue exposed overnight at 4°C to the primary antibody. The next day, the tissue was washed with Tris buffer and exposed to the secondary antibody for 30–45 min. Labeling was visualized using the ABC peroxidase technique (Vector Laboratories, Burlingame, CA) with 3,3′-diaminobenzidine with 1% nickel chloride or the VIP substrate kit (Vector Laboratories). Triton X-100 (0.1% final concentration) was added to the diluent, and Tris buffer was used in the MYT1 protocol to insure the antibody penetrated the nucleus.
Counts of labeled glial cells were performed within the same ventromedial WM region described for glial nuclei counts. In each rat, two or three sections at the epicenter of the lesion and at 1 mm rostral and 1 mm caudal to the epicenter were examined. A minimum distance of 50 μm separated the sections counted. Only intensely labeled cells with a clearly defined round nucleus were counted.
Statistical analysis. Comparison of the TTX and VEH groups to uninjured controls was performed with a Dunnett one-factor ANOVA with post hoc multiple comparisons using the Tukey test. Pathology in the TTX and VEH groups was compared with the Student’s t test for the tissues analyzed at 4 and at 24 hr after SCI. Because the 4 and 24 hr experiments were conducted at different times, we did not statistically compare 4 and 24 hr data. Statistical evaluation was performed using the Sigmastat program (SPSS, San Rafael, CA). In all cases, statistical significance was established with a p value of <0.05.
RESULTS
Qualitative assessment of pathology
When cross sections of normal spinal cord tissue were examined in 1 μm plastic sections, a clear line of demarcation separating the ventral gray and white matter was seen. The dorsal half of ventral WM, closest to the ventral horns of gray matter, contained mainly small- and medium-diameter axons. The ventral (more peripheral) half contained small-, medium-, and also large-diameter axons (Fig.1A). The axonal profiles were round or slightly oval and surrounded by a compact rim of myelin. Glial cell bodies and processes filled the space in between the axons.
Fig. 1.
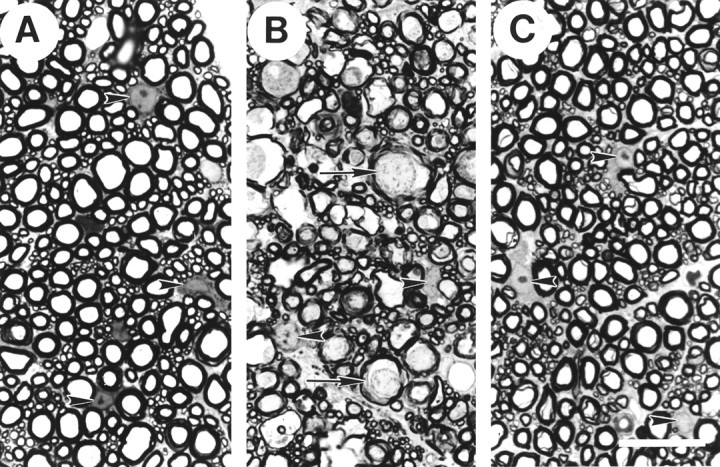
Light microscopy of 1 μm plastic sections of the ventromedial WM in normal thoracic spinal cord (A) and at the injury epicenter 4 hr after SCI and treatment with either VEH (B) or TTX (C). In normal tissue (A), glial nuclei are visible (arrowheads) in between the numerous profiles of myelinated axons of small, medium, and large diameters. In the injured and VEH-treated tissue (B), fewer glial nuclei are evident, and those present appear abnormal (arrowheads). There are numerous abnormally large axonal profiles with periaxonal spaces, darkened axoplasm (arrows), and myelin abnormalities. In the injured and TTX-treated tissue (C), little axonal pathology is evident, although increased spacing between the axons is seen. The glia nuclei (arrowheads) appear similar to those in uninjured controls. Scale bar, 50 μm.
SCI caused dramatic changes to the appearance of the tissue at the lesion epicenter by 4 hr after injury, as previously described in detail (Rosenberg and Wrathall, 1997; Rosenberg et al., 1999). Consistent with our earlier reports, the ventromedial WM in the VEH-treated group (Fig. 1B) demonstrated many swollen axonal profiles, as well as scattered empty-appearing myelin microcysts. The axoplasm, particularly of the larger axons, appeared more electron-dense than in normal tissue. Glial pathology was also evident. There appeared to be fewer glial nuclei, and those present were typically deformed with chromatin clumping and an absence of nucleoli. Examination of tissue sections distal to the epicenter showed that by 4 hr after injury pathology was also evident in ventromedial WM up to 2 mm rostral and caudal to the injury epicenter.
The TTX-treated group showed considerably less pathology in the ventromedial WM at 4 hr after SCI (Fig. 1C). Most of the axons in the TTX-treated tissue retained their normal appearance at the LM level, even at the injury epicenter. Indications of pathology were restricted to slight separations between the myelin sheaths and the axoplasm. The glial nuclei in the TTX-treated group appeared more numerous and generally normal in appearance (Fig.1F). Furthermore, the rostrocaudal extent of ventromedial WM pathology was dramatically attenuated with TTX treatment. Tissue in sections ±1 mm from the epicenter demonstrated no signs of pathology by light microscopy at 4 hr after SCI.
At 24 hr after SCI, the pathology in the VEH-treated group was still considerably more severe than that in the TTX-treated group (Fig.2A). In the VEH-treated group, WM pathology extended upward of 3 mm rostral and caudal of the epicenter. At the epicenter, numerous myelin microcysts were evident. However, many appeared to be collapsed, causing them to appear smaller than those observed at 4 hr after SCI (Fig. 1B). In addition, some of the axons at the epicenter appeared to have collapsed.
Fig. 2.

Light microscopy of 1 μm plastic sections of epicenter ventromedial WM at 24 hr after SCI with VEH (A) or TTX (B) treatment. In the VEH-treated tissue (A), many axons appear collapsed. Areas devoid of axons are present. In the TTX-treated (B) tissue, numerous microcysts (arrows) are seen, some containing small amounts of darkened axoplasm. Relatively normal-appearing axons are visible in between the microcysts, particularly near the pial edge of the ventral sulcus. Scale bar, 50 μm.
The TTX-treated tissue at 24 hr after injury (Fig.2B) showed considerably fewer collapsed myelin microcysts: the myelin microcysts present generally resembled those seen at 4 hr after SCI. There were also fewer collapsed axons. Many of the axons, particularly along the ventral sulcus and pial edge, appeared normal by light microscopy. Furthermore, in the TTX-treated group ventromedial WM pathology often did not extend beyond 2 mm rostral and caudal of the epicenter, further supporting the observation that pathology was attenuated in the TTX-treated compared with the VEH group.
To better evaluate WM pathology present after injury, ventromedial WM at the epicenter was examined with transmission electron microscopy. In normal tissue (Fig. 3A), axons were typically surrounded by a rim of myelin composed of multiple lamellae tightly wrapped around the axon. The axolemma was juxtaposed to the inner lamella of myelin. Numerous mitochondria, neurofilaments, and microtubules were present throughout the axoplasm, giving it a granular appearance. The space between the myelinated axons was filled with glial elements, primarily astrocytic processes.
Fig. 3.
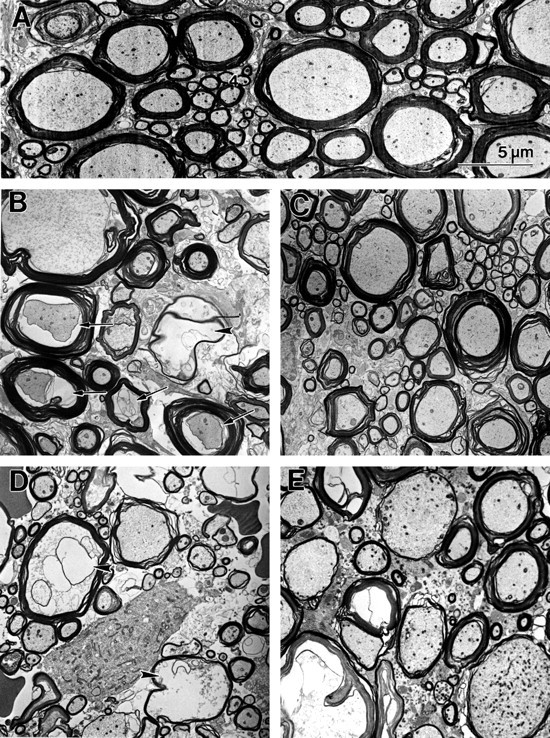
Electron micrographs of ventromedial WM from normal thoracic spinal cord (A) and at 4 (B, C) and 24 (D, E) hr after SCI at the injury epicenter. In normal tissue (A) myelinated axons of various diameters are visible. Axoplasm fills the intramyelin space with longitudinally oriented neurofilaments and microtubules viewed in cross section, giving the axoplasm a slightly granular appearance. Numerous mitochondria are visible. At 4 hr after SCI, axons in tissue from the VEH-treated group (B) demonstrate unwinding of the myelin sheaths, periaxonal spaces (arrows), and swollen mitochondria. Remnants of degenerated axons are also present (arrowhead) within myelin microcysts. In the TTX-treated tissue (C) at 4 hr after SCI, myelin pathology is present, primarily associated with larger diameter axons. Swollen axonal mitochondria can be seen, but little other axoplasmic pathology is evident. At 24 hr after injury, axons remaining in the VEH-treated tissue (D) are primarily of small diameter. Many of the larger axon profiles demonstrate profound axoplasmic pathology (arrowheads). Others appear to have degenerated, as indicated by the presence of empty-appearing myelin microcysts. In the TTX-treated tissue at 24 hr after injury (E), both small- and large-diameter axons with relatively normal looking axoplasm and myelin sheaths can be seen. Other axons demonstrate myelin pathology in the form of thin myelin sheaths or swelling between the lamellae. A few of the larger axons contain axoplasm with abnormally high numbers of organelles, particularly mitochondria.
At 4 hr after SCI in the VEH-treated group (Fig. 3B), axoplasmic, myelin, and glial abnormalities were seen. The myelin sheaths frequently had lucent spaces present between the lamellae. In many axons, axonal mitochondria appeared swollen, the axoplasm appeared dark, and there were often periaxonal spaces between the axolemma and the inner lamella of myelin. Examination at higher magnification revealed that darkened axoplasm was caused by compacted neurofilaments in affected axons (data not shown). In other axons, the axoplasm appeared as a flocculent substance devoid of organelles or cytoskeleton. Most of the pathology observed at 4 hr after SCI was seen in the medium- and large-diameter axons, whereas smaller axons appeared only occasionally affected.
At 4 hr after injury in the TTX-treated group (Fig. 3C), we observed myelin pathology as indicated by unraveling of the myelin sheath. We did not see the widespread axoplasmic pathology observed in the VEH group. Although periaxonal spaces and swollen mitochondria were visible in some axons, their presence appeared restricted to the larger diameter axons. The actual numbers of axons affected by injury in the TTX-treated group appeared reduced compared with the VEH-treated group.
By 24 hr after SCI, small-diameter axons constituted the majority of surviving axons in tissue from the VEH-treated group (Fig.3D). There were considerable amounts of debris from degenerating axons and glia present in the extracellular space. There were also a number of empty myelin sheaths. In contrast, the TTX-treated tissue still contained some large-diameter axons with relatively normal-looking axoplasm and myelin (Fig. 3E). Some of the medium- and large-diameter axons in the TTX-treated tissue had abnormal concentrations of organelles reminiscent of terminal clubs (Kao et al., 1977) or restrictions within axoplasm that impede axonal transport of organelles (Povlishock, 1993). Empty myelin sheaths were also observed in the TTX group, although there appeared to be fewer in comparison to the VEH group. In contrast to the VEH-treated tissue, the glial matrix that surrounds the axons appeared somewhat denser in the TTX-treated tissue (Fig. 3E).
Quantitative assessment of pathology
Axonal counts
Quantitative analyses were based on tissue from four to six rats and at least 1200 axons per group (Table2). In normal tissue, we found on average 112.1 ± 10.2 (SE) total myelinated axons per 1600 μm2 of ventromedial WM (Fig.4C). Axonal numbers in VEH-treated tissue averaged 81.4 ± 12.6 at 4 hr after SCI and 76.6 ± 14.8 at 24 hr after injury. In the TTX-treated tissue, axonal numbers were 103.2 ± 5.6 at 4 hr and 102.4 ± 23.9 at 24 hr after SCI. Although there were higher average numbers of axons in the TTX-treated group compared with VEH treatment, statistical analysis indicated the difference was not significant.
Table 2.
Axonal databases used for quantification of WM pathology
| Uninjured controls | 4 hr | 24 hr | |||
|---|---|---|---|---|---|
| Vehicle | TTX | Vehicle | TTX | ||
| Rats, n= | 5 | 6 | 5 | 5 | 4 |
| Micrographs/rat | 3 | 3 | 3 | 3 | 3 |
| Total axons/group | 1687 | 1422 | 1547 | 1149 | 1229 |
Fig. 4.

A histogram showing the distribution of the various axonal diameters in the ventromedial WM 4 (A) and 24 (B) hr after SCI and treatment with 0.15 nmol of TTX. Computed means and SEs are based on counts per animal for 1600 μm2 total area of tissue. Comparison of axon numbers in normal ventromedial WM and at 4 and 24 hr after SCI showed a nonsignificant trend toward reduction after SCI in the VEH-treated groups and less apparent effect of injury with TTX treatment. B, No change in numbers of small-diameter (≤2.5 μm) axons was observed at either 4 or 24 hr after SCI (Dunnett ANOVA; p = 0.46).C, Medium-diameter (>2.5–4.45 μm) axon numbers were reduced at 4 hr after SCI in the VEH group but not in the TTX group. At 24 hr after SCI, numbers of medium-diameter axons in both the VEH and TTX-treated groups were significantly reduced compared with uninjured controls. D, At 4 hr after injury, a significant reduction of large-diameter (≥4.5 μm) axons was seen in the VEH group compared with uninjured controls but not in the TTX group. There were significantly more large axons in the TTX as compared with the VEH group at 4 hr after injury. At 24 hr after SCI, the number of large axons in both treatment groups had been significantly reduced compared with controls. TTX treatment attenuated the loss of large axons at 24 hr after injury compared with VEH treatment. Bars represent the means ± SE for groups of four to six rats per group, as shown in Table 2. Asterisk indicates significantly different from uninjured controls (0 hr group). p values shown are for the indicated comparisons of the VEH and TTX groups (Student’st test).
Because a preferential effect of injury on axons of large diameters has been previously reported (Blight, 1983; Blight and Decrescito, 1986;Rosenberg and Wrathall, 1997; Rosenberg et al., 1999), axonal loss was evaluated according to axon diameter. We examined the frequency distribution of ventromedial axons in VEH- and TTX-treated animals (Fig. 4A,B) and uninjured controls (data not shown). After SCI there was an overall tendency toward reduction in axon numbers in each size category. The TTX-treated group showed less effect of injury than the VEH-treated group (Fig.4A,B). In normal uninjured animals, ∼50% of the population of ventromedial axons was composed of small axons with intramyelin diameters ≤2.5 μm (Fig.4D). Approximately 25% of the axons were medium-size (2.5–4.45 μm diameter), and the remaining 25% were large-diameter axons (≥4.5 μm) (Fig.4E,F). We compared the numbers of axons in each of these size categories with those in the VEH- and TTX-treated groups at both 4 and 24 hr after SCI (Fig.4D–F).
Both the frequency histograms (Fig.4A,B) and size group data (Fig.4D–F) showed medium- and large-diameter axons at 4 hr after SCI to be particularly responsive to TTX treatment. There were significantly more 3, 4, 5, 6, and 7 μm diameter axons in the TTX group compared with the VEH group (Fig.4A). The number of small-diameter axons was not significantly affected by SCI and/or TTX treatment at either 4 or 24 hr after injury (Fig. 4D). The number of medium-diameter axons tended to be reduced in the VEH-treated group at 4 hr after SCI as compared with uninjured controls, but the effect was not statistically significant (Fig. 4E). However, by 24 hr there was a significant loss of medium axons in both the VEH and TTX-treated groups.
An important finding was the significant loss of large-diameter axons (≥4.5 μm) after SCI (Fig. 4F). By 4 hr after injury, the numbers of large-diameter axons had been reduced by half in the VEH group. At 24 hr after SCI, approximately one-tenth of the normal number of large-diameter axons in the ventromedial area remained in the VEH-treated group. Treatment with TTX significantly reduced the loss of large-diameter axons at both 4 (p = 0.005) and 24 hr (p = 0.04) after injury, doubling the number of large-diameter axons at 24 hr after injury.
TTX effects on white matter pathology
Assessment of overall WM pathology (i.e., axoplasmic, myelin, and periaxonal spacing) with the AII indicated that, whereas both injured groups demonstrated considerable pathology compared with uninjured controls, pathology was greatly reduced with TTX treatment (Fig.5). Statistical comparison confirmed that TTX treatment significantly reduced overall pathology at 4 (p = 0.036) and 24 hr (p= 0.043) after SCI.
Fig. 5.

Assessment of overall axonal pathology with the AII. Both the VEH and TTX groups exhibit considerable WM pathology at 4 and 24 hr after SCI. However, the AII is significantly less in the TTX-treated groups at both 4 hr (p = 0.036) and 24 hr (p = 0.043) after injury. Bars represent the means ± SE for four to six rats per group, as shown in Table 2. Asterisk indicates significant difference between treatment groups (Student’s t test).
Assessment of axoplasmic and myelin pathology independently of one another showed that >50% of the axons in the VEH-treated tissue demonstrated some form of axoplasmic pathology at 4 hr after injury, compared with ∼28% of axons in the TTX-treated tissue (Fig.6A), a significant difference (p = 0.007). The TTX-treated group also demonstrated significantly less axoplasmic pathology at 24 hr after SCI compared with the VEH group (p = 0.045).
Fig. 6.

Assessment of axoplasmic (A) and myelin (B) pathology at 4 and 24 hr after SCI. Both axoplasmic and myelin pathology was significantly greater than in uninjured controls (0 hr group) at 4 and 24 hr after injury (Dunnett ANOVA; p < 0.001). Comparison of the treatment groups showed axoplasmic pathology was significantly reduced with TTX treatment at both 4 (p < 0.004) and 24 hr (p = 0.045) after SCI. Examination of myelin pathology showed significantly more pathology in the VEH group compared with the TTX group at 4 hr (p< 0.001), but by 24 hr both groups demonstrated similar degrees of myelin pathology (p = 0.799). Bars represent the means ± SE for four to six rats per group, as shown in Table2. Asterisk indicates significant difference between treatment groups (Student’s t test).
Approximately 12% of the axons in uninjured control tissue had some unraveling of the myelin sheath (Fig. 6B). Comparison of the uninjured control group to the two treatment groups revealed that myelin pathology was significantly greater in both the VEH and TTX groups at 4 and at 24 hr after injury. The proportion of axons with myelin pathology was significantly less in the TTX-treated compared with the VEH group at 4 hr after SCI (p = 0.004). However, by 24 hr after injury, myelin pathology associated with the remaining axons was similar in the VEH and TTX-treated groups (p = 0.799).
The electron micrographs showed considerably more periaxonal space (Fig. 7A) present in axons in the VEH group than in the TTX-treated group. Quantitative analysis confirmed that the percentage of axons with abnormal periaxonal space (i.e., space >25% of the intramyelin diameter) was significantly reduced with TTX treatment at both 4 and 24 hr after injury compared with VEH controls (Fig. 7B). Furthermore, linear regression analysis showed that periaxonal space was significantly correlated with axon diameter (R2 = 0.38;p = 0.002) in the VEH group, with the larger axons demonstrating greater amounts of periaxonal space than smaller diameter axons (Fig. 7C). Treatment with TTX abolished this relationship (Fig. 7D; R2= 0.091; p = 0.237).
Fig. 7.

Assessment of periaxonal space after SCI.A, An electron micrograph of an axon (arrow) demonstrating 50% periaxonal space.B, Quantification of axons with periaxonal spacing >25% found that at 4 hr after SCI both VEH- and TTX-treated groups demonstrated significantly more periaxonal space than uninjured controls (Dunnett ANOVA followed by post hoc Tukey,p < 0.05). Compared to VEH controls, TTX reduced the proportions of axons with abnormal periaxonal space at 4 hr after injury (p = 0.009). By 24 hr, the proportion of axons with abnormal periaxonal space in the VEH-treated tissue was still significantly higher than in uninjured controls (Dunnett ANOVA followed by post hoc Tukey, p < 0.05) but in the TTX group it was not significantly from uninjured controls. At 24 hr there was a significant difference between the VEH and TTX groups (p = 0.032). Bars represent the means ± SE for four to six rats per group, as shown in Table2. Asterisk indicates significant difference between treatment groups (Student’s t test). C, Linear regression analysis of the percent intramyelin area that was perixaxonal space for axons of different diameters in the VEH-treated group shows that the amount of periaxonal space present at 4 hr after SCI is significantly correlated to axonal size. D, Linear regression analysis of similar data from the TTX group indicates no significant correlation.
In the single rat microinjected with TTX but not injured, quantification of WM pathology found the average number of axons (100) per 1600 μm2 area of tissue was within the range (89–129) observed for uninjured controls. Assessment of axoplasm and myelin pathologies found only 0.3% of the axons demonstrated axoplasmic pathology, and only 2.7% of the axons had myelin pathology in the noninjured TTX-treated tissue. Furthermore, none of the 300 axons we examined contained any abnormal periaxonal spaces, nor did we observe any axons with necrotic axoplasm.
Effects of TTX on WM glia
Glial counts
Examination of 1 μm plastic sections of tissue from the epicenter and 1 mm rostral and caudal to the epicenter at high magnification (400×), allowed identification of intact glial cell nuclei (Fig. 1). Nuclear counts revealed there was no significant effect of TTX treatment on glial numbers at 4 or 24 hr after SCI (Fig.8). Glia counts obtained from tissue from the noninjured, TTX microinjected animal were within the range for uninjured controls. We also observed no morphological changes in these cells to indicate there was glial toxicity, e.g., swelling, chromatin aggregation, as a result of treatment with TTX. Identification of glial subtypes based on morphology alone was difficult after injury. Even at the EM level, distinguishing astrocytes from oligodendrocytes was problematic because fibrils associated with astrocytes (Peters et al., 1991) appeared to be affected by injury and were not always visible. Therefore, we used the molecular markers CC1, GFAP, and OX42 to determine whether TTX treatment had a preferential effect on a specific subtype of glia.
Fig. 8.

Effects of TTX treatment on glial survival in ventromedial WM at 4 and 24 hr after SCI. Quantification of glial nuclei in 1 μm plastic sections showed that, compared with uninjured controls, there were significantly fewer glia in the VEH group at 4 hr after injury, but there was no difference between control and the TTX-treated groups. Comparison of VEH and TTX groups at 4 hr after SCI found glial numbers were higher in the TTX group, but the difference was not statistically significant (Student’s t test;p = 0.053). By 24 hr, both the VEH and TTX group had significantly fewer glia than uninjured controls. There was no statistical difference between treatment groups (Student’st test; p = 0.161).
The antibody CC1, which recognizes oligodendrocytes in the brain and spinal cord (Bhat et al., 1996; Crowe et al., 1997; Shuman et al., 1997; Rosenberg et al., 1999), labeled a population of cells in normal tissue that had oval cell bodies and two or more processes (Fig.9A). CC1-labeled cells were evenly distributed throughout ventral WM. Injury caused the cell bodies of CC1-positive cells to round and lose cellular processes (Fig.9B). GFAP-labeled glia were observed primarily in more ventral regions, close to the pial surface, or aligned along axonal arrays that originated from the ventral horns and passed through the ventral WM. GFAP-labeled astrocytes had cell bodies that were slightly smaller than those identified with CC1 (15–20 vs 25–30 μm, respectively). The cellular processes of these cells extended long distances (Fig. 9C). At 24 hr after SCI, GFAP-positive cells usually retained their physical features, although there appeared to be fewer of them. In addition, we observed that the cell processes, especially the finer ones running through the WM, stained more intensely after injury (Fig. 9D). A small number of cells scattered throughout normal ventral WM labeled positively for the microglia–macrophage marker OX-42. The cell bodies of these cells were smaller (10–15 μm, respectively) than those stained with the antibodies to CC1 or GFAP. OX-42-positive cells in normal tissue tended to have a star-like appearance because of the presence of multiple, branching thin processes (Fig. 9E). The labeled cell bodies appeared enlarged, and the thin processes thickened at 24 hr after SCI (Fig. 9F).
Fig. 9.

Immunohistochemical identification of glia in spinal cord ventromedial WM. A, CC1-positive oligodendrocytes in normal uninjured WM and (B) at 24 hr after SCI. C, GFAP-positive astrocytes in normal WM and (D) at 24 hr after SCI.E, OX-42-positive microglia in normal WM and (F) at 24 hr after injury.
Quantification of the immunohistochemically labeled cells revealed that CC1- and GFAP-positive cells were significantly reduced at 24 hr after SCI compared with uninjured controls (Table3). Treatment with TTX did not significantly enhance survival of either cell type. Numbers of OX-42-positive cells in both the VEH- and TTX-treated groups at 24 hr after SCI were found to be statistically similar to uninjured controls.
Table 3.
Glial subtypes in ventromedial WM at 24 hr after SCI3-a
| Uninjured | Vehicle | TTX | |
|---|---|---|---|
| Hematoxylin (Total nuclei) | 66.0 ± 2.1 | 35.3 ± 4.3* | 41.9 ± 3.6* |
| CC1 (Oligodendrocytes) | 29.8 ± 1.9 | 14.6 ± 0.8* | 17.2 ± 1.3* |
| GFAP (Astrocytes) | 19.7 ± 1.1 | 11.4 ± 1.3* | 12.0 ± 1.1* |
| OX 42 (Microglia/ macrophages) | 10.1 ± 1.1 | 10.2 ± 1.1 | 10.5 ± 1.6 |
| MYT 1 (Precursor cells) | 2.9 ± 1.3 | 6.0 ± 0.6* | 4.9 ± 0.7 |
Data represents mean ± SE for counts from tissue sections at the epicenter and at ±1 mm;n = 6 rats per group.
*Indicates statistically significant difference (p < 0.05) compared to the uninjured control group (Dunnett ANOVA with Tukey post hoc multiple comparison).
By immunohistochemistry, we also identified a small population of cells that were MYT1-positive. The labeling was primarily nuclear with an occasional cell demonstrating some cytoplasmic labeling (data not shown). Quantitative analysis indicated significantly more MYT1-positive nuclei in the VEH-treated tissue compared with uninjured controls (Table 3). In the TTX-treated group, the numbers of MYT1-positive nuclei were not significantly different from that in either the VEH group or uninjured controls.
DISCUSSION
In this study, we found that focal injection of the sodium channel blocker TTX into the injury site after a standardized spinal cord contusion dramatically reduced acute WM pathology. Specifically, TTX significantly attenuated the loss of large (≥5 μm)-diameter axons. The surviving axons in the TTX group demonstrated less axoplasmic pathology in comparison to those in the VEH group. The effectiveness of TTX appears to be in its ability to reduce axonal pathology per se as opposed to reducing injury-induced loss of glia or myelin pathology after SCI.
We previously showed that focal microinjection of TTX (0.15 nmol) into the lesion site significantly reduced WM loss at the injury epicenter chronically at 8 weeks after SCI (Teng and Wrathall, 1997). Morphometry analysis showed a three-fold sparing of WM in the TTX-treated group compared with VEH controls. This led us to hypothesize that TTX treatment was sparing axons. The finding that TTX treatment spared large axons is particularly interesting because large-diameter axons are preferentially lost after SCI (Blight and Decrescito, 1986;Fehlings and Tator, 1995; Rosenberg and Wrathall, 1997; Rosenberg et al., 1999) supports this hypothesis.
Because large axons occupy a greater area than small axons may explain the increase in WM seen with TTX treatment at 8 weeks after injury (Teng and Wrathall, 1997). Reducing WM loss after SCI can potentially produce important functional improvement. Sparing as little as 5–10% of the original axon population has been reported to be sufficient for recovery of locomotion after SCI (Blight, 1983). In the case of TTX treatment, the chronic sparing of WM with TTX treatment was significantly and highly correlated to enhanced chronic hindlimb function after SCI (Teng and Wrathall, 1997). Not known is the extent to which recovery of locomotion depends on the type of axons spared. Sparing large-diameter, fast-conducting axons may be especially important.
Comparison of TTX and VEH-treated groups found WM pathology differed greatly, particularly with respect to the injury-mediated periaxonal spaces that develop afterward. Data suggests periaxonal spaces result from the condensation of axoplasm when neurofilaments collapse on themselves because of the loss of neurofilament sidearms (Pettus and Povlishock, 1996; Okonkwo et al., 1998). We found abnormal periaxonal spaces are present in axons as early as 15 min after SCI (Rosenberg and Wrathall, 1997). Approximately 40% of the large-diameter axons in the ventromedial WM demonstrated periaxonal spaces occupying 75% or more of the intramyelin area at 15 min after injury. By 4 hr after injury, ∼50% of the large axons had been lost, implying a connection between the formation of extensive periaxonal spaces and acute loss of large axons.
Linear regression analysis confirmed a correlation between axon diameter and development of extensive (>50% of the intramyelin area) periaxonal spaces. More importantly, TTX treatment significantly reduced the extensive periaxonal spaces that developed in larger diameter axons and increased the survival of large axons. TTX was found to attenuate the development of periaxonal spaces in optic nerve after anoxia as shown by Waxman et al. (1994). They found that if 1 μm TTX was given 10 min before the onset of a 60 min anoxic episode, the formation of periaxonal spaces were dramatically reduced. Our results also suggest that the formation of periaxonal space are similarly mediated by TTX-sensitive sodium channels.
Although overall WM pathology was significantly reduced with TTX treatment, assessment of the axoplasm, myelin, and periaxonal space indicates that the overall reduction may primarily reflect an attenuation of axoplasmic abnormalities. TTX treatment reduced by half the number of axons with axoplasmic pathology at 4 hr after SCI. We continued to see significantly less axoplasmic pathology in the TTX-treated group compared with VEH controls at 24 hr after injury. TTX treatment had no significant effect on myelin pathology after SCI.Waxman et al. (1994) also reported that TTX treatment did not prevent the anoxic injury-mediated detachment of the oligodendroglial myelin loops at the nodes of Ranvier in their in vitro optic nerve preparation.
Glial loss is observed after SCI (Crowe et al., 1997; Liu et al., 1997;Rosenberg et al., 1999). Sodium channels are present on glia. Astrocytes, in particular, are known to contain voltage-gated Na+ channels (Bowman et al., 1984; Bevan et al., 1987) that may play a role in maintaining ionic homeostasis (Sontheimer et al., 1996). We found that oligodendrocytic loss can be substantially reduced through blockade of AMPA/kainate receptors with the antagonist NBQX (Rosenberg et al., 1999), possibly by attenuating Na+ influx (Seeburg, 1993; Wisden and Seeburg, 1993). However, if glial cell loss after SCI is partly caused by increased intracellular Na+, we would expect to see enhanced glial survival in the current study because Na+ channels are blocked with TTX (Sontheimer et al., 1996; Macfarlane and Sontheimer, 1998). However, compared with VEH controls, we found that neither oligodendrocytes nor astrocytes were protected from injury with TTX treatment. We also have no reason to believe that TTX caused additional stress (toxicity) to glia based on the fact that TTX microinjected into a normal spinal cord produced no glia loss 24 hr later.
A small population of cells in the ventromedial WM did demonstrate intense MYT1 nuclear labeling after SCI. This antibody labels precursor cells present during development (Kim et al., 1997) and in the adult human subventricular zone (Armstrong et al., 1997). In the adult rat, the numbers of such cells are few in the normal spinal cord but appear greatly increased chronically after SCI (Wrathall et al., 1998). In the current study, we found that the numbers of MYT1-positive nuclei were significantly increased in the VEH group at 24 hr after SCI compared with uninjured controls, suggesting that injury stimulated MYT1 expression and/or induced proliferation, or migration, of these cells.
The development of in vitro models of WM injury has given rise to the concept that WM injury results from elevations of intraaxonal Ca2+ brought about through an injury-mediated increase in intraaxonal Na+ (Ransom et al., 1990; Stys et al., 1990, 1993; Waxman et al., 1994; Imaizumi et al., 1997; Stys, 1998). There is strong evidence that under pathological conditions, Na+ enters the axon not only through voltage-gated Na+ channels but also through “leak” channels that do not inactivate (Stys et al., 1993). Consequently, intraaxonal levels of Na+ are elevated, reaching concentrations that could drive the Na+/Ca2+ exchangers located in the axolemma to operate in reverse (Stys et al., 1991, 1992; Waxman et al., 1991). Normally, these exchangers extrude Ca2+from the axon in exchange for Na+. Reverse operation causes Ca2+ to be pumped in and Na+ extruded. The loss of axonal conductivity has also been linked to Na+/H+antiporter dysfunction. Pharmacological blockade of this exchanger prevented the loss of compound action potentials after an anoxic (Imaizumi et al., 1997) or mechanical insult (Agrawal and Fehlings, 1996) to WM in vitro. These findings suggest that WM injury either involves multiple mechanisms or different mechanistic pathways are activated by specific components of injury, i.e., anoxia, mechanical stretch, etc. In either case, the common endpoint is an elevation in intraaxonal Ca2+ (Stys, 1998). In vitro studies of dendrite transection showed that increasing internal Ca2+ levels alone is insufficient to cause cell loss (Rosenberg and Lucas, 1996; Lucas et al., 1997). Reducing external Ca2+ preserved neurofilaments and microtubules but did not prevent severe damage to the smooth endoplasmic reticulum (SER) or mitochondria (Lucas et al., 1990). Reducing external Na+ concentration (normal Ca2+) dramatically decreased cytoskeletal and organelle damage (Lucas et al., 1997). The SER and mitochondria, both important in the regulation of internal Ca2+, appear highly vulnerable to Na+-mediated damage. Preventing elevations in intracellular Na+ after injury may decrease damage to these structures and preserve important means of regulating internal Ca2+.
Mitochondrial Na+/Ca2+ exchangers operate somewhat differently from those in the plasma membrane in that they extrude a single Na+ for a single Ca2+ (Carafoli, 1988a,b). Therefore, it is possible that when internal levels of Na+ are high, the mitochondrial Na+/Ca2+ exchanger transporter may operate in reverse, extruding Ca2+into the cytosolic environment while sequestering Na+. Uptake of Na+ by the mitochondria could then elevate internal concentrations, causing water to enter, which in turn could produce swelling and possible mitochondrial lysis (Trump et al., 1988). Mitochondrial loss would result in a decrease of ATP needed to maintain the Na+/K+-ATPase and Ca2+-ATPase transporters (Carafoli, 1988a,b). This in turn could cause a release of mitochondrial Ca2+into the cytosol and in addition to the loss of ATP could potentially exacerbate axonal injury. It is our hypothesis that the benefits of TTX in SCI stem not only from its ability to block the reverse operation of the exchangers but, also, in its ability to attenuate Na+-mediated destruction of important organelles such as the mitochondria and SER. Thus, we postulate that by preserving the mitochondria we are potentially preserving a vital source of metabolic energy needed to drive cellular functions, particularly mechanisms that my be involved in regulating internal Ca2+.
In summary, the chronic sparing of WM seen with TTX treatment appears to be associated with an acute reduction in axonal pathology. Our results suggest that a significant amount of the acute axonal pathology present after SCI is secondary to the initial mechanical trauma and mediated through TTX-sensitive Na+ channels.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS-35647. We extend a special thanks to Mr. Hai Ning Dai for his assistance in the preparation of ultrathin sections and Ms. Sadia Aden for the preparation of the cryosections.
Correspondence should be addressed to Dr. Jean R. Wrathall, Georgetown University, Department of Cell Biology, 3900 Reservoir Road, Washington, DC 20007.
REFERENCES
- 1.Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anthes DL, Theriault E, Tator CH. Characterization of axonal ultrastructural pathology following experimental spinal cord compression injury. Brain Res. 1995;702:1–16. doi: 10.1016/0006-8993(95)01028-6. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RC, Migneault A, Shegog ML, Kim JG, Hudson LD, Hessler RB. High-grade human brain tumors exhibit increased expression of myelin transcription factor 1 (MYT1), a zinc finger DNA-binding protein. J Neuropathol Exp Neurol. 1997;56:772–781. [PubMed] [Google Scholar]
- 4.Balentine JD, Dean DL., Jr Calcium-induced spongiform and necrotizing myelopathy. Lab Invest. 1982;47:286–295. [PubMed] [Google Scholar]
- 5.Balentine JD, Greene WB. Ultrastructural pathology of nerve fibers in calcium-induced myelopathy. J Neuropathol Exp Neurol. 1984;43:500–510. doi: 10.1097/00005072-198409000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 7.Bevan S, Lindsay RM, Perkins MN, Raff MC. Voltage gated ionic channels in rat cultured astrocytes, reactive astrocytes and an astrocyte-oligodendrocyte progenitor cell. J Physiol (Lond) 1987;82:327–335. [PubMed] [Google Scholar]
- 8.Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 9.Blight AR. Cellular morphology of chronic spinal cord injury in the cat: analysis of myelinated axons by line-sampling. Neuroscience. 1983;10:521–543. doi: 10.1016/0306-4522(83)90150-1. [DOI] [PubMed] [Google Scholar]
- 10.Blight AR. Morphometric analysis of a model of spinal cord injury in guinea pigs, with behavioral evidence of delayed secondary pathology. J Neurol Sci. 1991;103:156–171. doi: 10.1016/0022-510x(91)90159-5. [DOI] [PubMed] [Google Scholar]
- 11.Blight AR. An overview of spinal cord injury models. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. McGraw-Hill; New York: 1996. pp. 1367–1380. [Google Scholar]
- 12.Blight AR, Decrescito V. Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience. 1986;19:321–341. doi: 10.1016/0306-4522(86)90025-4. [DOI] [PubMed] [Google Scholar]
- 13.Bowman CL, Kimelberg HK, Frangakis MV, Berwald-Netter Y, Edwards C. Astrocytes in primary culture have chemically activated sodium channels. J Neurosci. 1984;4:1527–1534. doi: 10.1523/JNEUROSCI.04-06-01527.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bresnahan JC. An electron microscopic analysis of axonal alterations following blunt contusion of the spinal cord of the Rhesus monkey (Macaca mulatta). J Neurol Sci. 1978;37:59–82. doi: 10.1016/0022-510x(78)90228-9. [DOI] [PubMed] [Google Scholar]
- 15.Carafoli E. Calcium homoeostasis in excitable cells. Biochem Soc Trans. 1988a;16:519–520. doi: 10.1042/bst0160519. [DOI] [PubMed] [Google Scholar]
- 16.Carafoli E. The intracellular homeostasis of calcium: an overview. Ann NY Acad Sci. 1988b;551:147–157. doi: 10.1111/j.1749-6632.1988.tb22333.x. [DOI] [PubMed] [Google Scholar]
- 17. Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3 1997. 73 76[Erratum (1997) 3:240]. [DOI] [PubMed] [Google Scholar]
- 18.Fehlings MG, Tator CH. The relationships among the severity of spinal cord injury, residual neurological function, axon counts, and counts of retrogradely labeled neurons after experimental spinal cord injury. Exp Neurol. 1995;132:220–228. doi: 10.1016/0014-4886(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 19.Gale K, Kerasidis H, Wrathall JR. Spinal cord contusion in the rat: behavioral analysis of functional neurologic impairment. Exp Neurol. 1985;88:123–134. doi: 10.1016/0014-4886(85)90118-9. [DOI] [PubMed] [Google Scholar]
- 20.Imaizumi T, Kocsis JD, Waxman SG. Anoxic injury in the rat spinal cord: pharmacological evidence for multiple steps in Ca(2+)-dependent injury of the dorsal columns. J Neurotrauma. 1997;14:299–311. doi: 10.1089/neu.1997.14.299. [DOI] [PubMed] [Google Scholar]
- 21.Kao CC, Chang LW, Bloodworth JM., Jr Electron microscopic observations of the mechanisms of terminal club formation in transected spinal cord axons. J Neuropathol Exp Neurol. 1977;36:140–156. doi: 10.1097/00005072-197701000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Kim JG, Armstrong RC, v Agoston D, Robinsky A, Wiese C, Nagle J, Hudson LD. Myelin transcription factor 1 (Myt1) of the oligodendrocyte lineage, along with a closely related CCHC zinc finger, is expressed in developing neurons in the mammalian central nervous system. J Neurosci Res. 1997;50:272–290. doi: 10.1002/(SICI)1097-4547(19971015)50:2<272::AID-JNR16>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 23.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas JH, Emery DG, Higgins ML, Gross GW. Neuronal survival and dynamics of ultrastructural damage after dendrotomy in low calcium. J Neurotrauma. 1990;7:169–192. doi: 10.1089/neu.1990.7.169. [DOI] [PubMed] [Google Scholar]
- 25.Lucas JH, Emery DG, Rosenberg LJ. Physical injury of neurons: important roles for sodium and chloride ions. The Neuroscientist. 1997;3:89–111. [Google Scholar]
- 26.Macfarlane SN, Sontheimer H. Spinal cord astrocytes display a switch from TTX-sensitive to TTX-resistant sodium currents after injury-induced gliosis in vitro. J Neurophysiol. 1998;79:2222–2226. doi: 10.1152/jn.1998.79.4.2222. [DOI] [PubMed] [Google Scholar]
- 27.Maxwell WL. Histopathological changes at central nodes of Ranvier after stretch-injury. Microsc Res Tech. 1996;34:522–535. doi: 10.1002/(SICI)1097-0029(19960815)34:6<522::AID-JEMT4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 28.Noble LJ, Wrathall JR. Spinal cord contusion in the rat: morphometric analyses of alterations in the spinal cord. Exp Neurol. 1985;88:135–149. doi: 10.1016/0014-4886(85)90119-0. [DOI] [PubMed] [Google Scholar]
- 29.Noble LJ, Wrathall JR. Correlative analyses of lesion development and functional status after graded spinal cord contusive injuries in the rat. Exp Neurol. 1989;103:34–40. doi: 10.1016/0014-4886(89)90182-9. [DOI] [PubMed] [Google Scholar]
- 30.Okonkwo DO, Pettus EH, Moroi J, Povlishock JT. Alteration of the neurofilament sidearm and its relation to neurofilament compaction occurring with traumatic axonal injury. Brain Res. 1998;784:1–6. doi: 10.1016/s0006-8993(97)01075-5. [DOI] [PubMed] [Google Scholar]
- 31.Panjabi MM, Wrathall JR. Biomechanical analysis of experimental spinal cord injury and functional loss. Spine. 1988;13:1365–1370. doi: 10.1097/00007632-198812000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Peters A, Palay SL, HD W. The fine structures of the nervous system, Ed 3. Oxford; New York: 1991. [Google Scholar]
- 33.Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- 34.Povlishock JT. Pathobiology of traumatically induced axonal injury in animals and man. Ann Emerg Med. 1993;22:980–986. doi: 10.1016/s0196-0644(05)82738-6. [DOI] [PubMed] [Google Scholar]
- 35.Raines A, Dretchen KL, Marx K, Wrathall JR. Spinal cord contusion in the rat: somatosensory evoked potentials as a function of graded injury. J Neurotrauma. 1988;5:151–160. doi: 10.1089/neu.1988.5.151. [DOI] [PubMed] [Google Scholar]
- 36.Ransom BR, Stys PK, Waxman SG. The pathophysiology of anoxic injury in central nervous system white matter. Stroke. 1990;21(III):52–57. [PubMed] [Google Scholar]
- 37.Rosenberg LJ, Lucas JH. Reduction of NaCl increases survival of mammalian spinal neurons subjected to dendrite transection injury. Brain Res. 1996;734:349–353. [PubMed] [Google Scholar]
- 38.Rosenberg LJ, Wrathall JR. Quantitative analysis of acute axonal pathology in experimental spinal cord contusion. J Neurotrauma. 1997;14:823–838. doi: 10.1089/neu.1997.14.823. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg LJ, Teng YD, Wrathall JR. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline reduces glial loss and acute white matter pathology after experimental spinal cord contusion. J Neurosci. 1999;19:464–475. doi: 10.1523/JNEUROSCI.19-01-00464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlaepfer WW, Zimmerman UP. Calcium-mediated breakdown of glial filaments and neurofilaments in rat optic nerve and spinal cord. Neurochem Res. 1981;6:243–255. doi: 10.1007/BF00964040. [DOI] [PubMed] [Google Scholar]
- 41.Seeburg PH. The Trends Neurosci/TIPS Lecture. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 42.Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 43.Sontheimer H, Black JA, Waxman SG. Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci. 1996;19:325–331. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 44.Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab. 1998;18:2–25. doi: 10.1097/00004647-199801000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Stys PK, Ransom BR, Waxman SG, Davis PK. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci USA. 1990;87:4212–4216. doi: 10.1073/pnas.87.11.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stys PK, Waxman SG, Ransom BR. Reverse operation of the Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian CNS white matter. Ann NY Acad Sci. 1991;639:328–332. doi: 10.1111/j.1749-6632.1991.tb17321.x. [DOI] [PubMed] [Google Scholar]
- 47.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stys PK, Sontheimer H, Ransom BR, Waxman SG. Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci USA. 1993;90:6976–6980. doi: 10.1073/pnas.90.15.6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms [see comments]. J Neurosurg. 1991;75:15–26. doi: 10.3171/jns.1991.75.1.0015. [DOI] [PubMed] [Google Scholar]
- 50.Teng YD, Wrathall JR. Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long-term functional deficits resulting from experimental spinal cord injury. J Neurosci. 1997;17:4359–4366. doi: 10.1523/JNEUROSCI.17-11-04359.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trump BF, Balentine JD, Berezesky IK. Mechanisms of cellular injury and death. J Neurotrauma. 1988;5:215–218. doi: 10.1089/neu.1988.5.215. [DOI] [PubMed] [Google Scholar]
- 52.Waxman SG, Ransom BR, Stys PK. Non-synaptic mechanisms of Ca(2+)-mediated injury in CNS white matter. Trends Neurosci. 1991;14:461–468. doi: 10.1016/0166-2236(91)90046-w. [DOI] [PubMed] [Google Scholar]
- 53.Waxman SG, Black JA, Stys PK, Ransom BR. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve. Brain Res. 1992;574:105–119. doi: 10.1016/0006-8993(92)90806-k. [DOI] [PubMed] [Google Scholar]
- 54.Waxman SG, Black JA, Ransom BR, Stys PK. Anoxic injury of rat optic nerve: ultrastructural evidence for coupling between Na+ influx and Ca(2+)-mediated injury in myelinated CNS axons. Brain Res. 1994;644:197–204. doi: 10.1016/0006-8993(94)91680-2. [DOI] [PubMed] [Google Scholar]
- 55.Wisden W, Seeburg PH. Mammalian ionotropic glutamate receptors. Curr Opin Neurobiol. 1993;3:291–298. doi: 10.1016/0959-4388(93)90120-n. [DOI] [PubMed] [Google Scholar]
- 56.Wrathall JR, Pettegrew RK, Harvey F. Spinal cord contusion in the rat: production of graded, reproducible, injury groups. Exp Neurol. 1985;88:108–122. doi: 10.1016/0014-4886(85)90117-7. [DOI] [PubMed] [Google Scholar]
- 57.Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wrathall JR, Li W, Hudson LD. Myelin gene expression after experimental contusive spinal cord injury. J Neurosci. 1998;18:8780–8793. doi: 10.1523/JNEUROSCI.18-21-08780.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young W. Secondary injury mechanisms in acute spinal cord injury. J Emerg Med. 1993;11:13–22. [PubMed] [Google Scholar]