

Abstract
The angiogenin (ANG) gene is mutated frequently in individuals with amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease characterized by the progressive loss of motor neurons. Delivering human ANG to mice that display ALS-like symptoms extends their lifespan and improves motor function. ANG is a secretory vertebrate RNase that enters neuronal cells and cleaves a subset of tRNAs, leading to the inhibition of translation initiation and the assembly of stress granules. Here, using murine neuronal and astrocytic cell lines, we find that ANG triggers the activation of the Nrf2 (nuclear factor erythroid 2-related factor 2) pathway, which provides a critical cellular defense against oxidative stress. This activation, which occurred in astrocytes but not in neurons, promoted the survival of proximal neurons that had oxidative injury. These findings extend the role of ANG as a neuroprotective agent and underscore its potential utility in ALS management.
Keywords: amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), astrocyte, neuroprotection, nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), ribonuclease, angiogenin (ANG), motor neuron, neurodegeneration, oxidative stress
Introduction
Amyotrophic lateral sclerosis (ALS)4 is a progressive, late-onset, and fatal neurodegenerative disease that is characterized by selective motor neuron loss in the spinal cord, brainstem, and motor cortex (1). Approximately 10% of ALS cases are inherited dominantly. The most common genetic determinants of ALS are the expansion of noncoding GGGGCC repeats in C9ORF72 and mutations in the copper/zinc superoxide dismutase 1 (SOD1) locus (2, 3). The search for other gene mutations that segregate with disease in ALS pedigrees has led to loss-of-function mutations in the human angiogenin (ANG) gene (4–6). Indeed, providing ALS-like transgenic mice that overexpress human SOD1G93A with human ANG increases their lifespan and improves their motor function (7).
ANG belongs to the pancreatic-type RNase (ptRNase) superfamily (8). This secretory protein is able to enter cells and catalyze the cleavage of the anticodon loops of mature tRNAs to produce 5′ and 3′ fragments that are designated as tRNA-derived, stress-induced RNAs (tiRNAs) (9). 5′-tiRNAs recruit the translational silencer protein YB-1 and sequester the eukaryotic translation initiation factor 4G/A complex to inhibit translation. Specific 5′-tiRNAs also trigger the assembly of stress granules at sites of ANG localization (10–12).
Translation repression is critical to overcoming oxidative stress, which is a hallmark of neurological disorders (13, 14). Oxidative stress results from an imbalance in the production and detoxification of free radicals from reactive oxygen species (ROS) (15–17). To neutralize ROS toxicity, cells replenish antioxidants by activating Nrf2 (nuclear factor erythroid 2-related factor 2) (18, 19). This transcription factor is usually latent within cells. Under basal conditions, the dimeric multidomain protein Keap1 (Kelch-like ECH-associated protein 1) binds to Nrf2 and promotes its ubiquitination and proteasomal degradation by acting as an adaptor for the Cul3-based E3 ligase. Oxidants react with key sulfhydryl groups of Keap1, which then loses its ability to target Nrf2 for degradation. Consequently, Nrf2 enters the nucleus, where it forms a heterodimer with small Maf proteins. This heterodimer binds to antioxidant-response elements (AREs) to drive the expression of antioxidant enzymes that compensate for the physiological and pathophysiological outcomes of oxidant exposure (20–22). Crossing mice in which the Nrf2 gene is overexpressed selectively in astrocytes with two ALS mouse models leads to double transgenic mice with a significant delay in onset and extended survival compared with the single transgenic ALS mice (23, 24). Moreover, activation of the Nrf2 pathway in astrocytes increases neuronal survival (25).
We recognized that this synergism between astrocytes and neurons resembles aspects of ANG-mediated neuroprotection. ANG is enriched in motor neurons and protects them against various ALS-related insults, such as excitotoxicity, hypoxia, and endoplasmic reticulum stress (7, 27, 28). These relationships provoked us to ask: Does ANG activate Nrf2? Here, we reveal that ANG does indeed activate the astrocytic Nrf2 pathway. Moreover, this activation transmits survival-promoting signals to proximal neurons, protecting them from oxidative stress.
Results
ANG activates the Nrf2 pathway selectively in astrocytes
The Nrf2 pathway mediates the transcriptional induction of a battery of genes that comprise the antioxidant response system. We examined whether ANG activates Nrf2 in cultured cells that were derived from ARE–hPAP transgenic mice. Upon induction, the ARE drives hPAP expression, which increases the level of human placental alkaline phosphatase (hPAP). The phosphatase activity of hPAP was measured as a readout of Nrf2/ARE-dependent promoter activation (29).
tert-Butylhydroquinone (tBHQ) is a known inducer of Nrf2 and was used as positive control in these experiments (30). tBHQ increased hPAP activity to various extents, depending on the cell type. tBHQ treatment led to a 15-fold increase in hPAP activity in astrocytes (Fig. 1A) and only a 7-fold increase in neurons (Fig. 1C). Remarkably, in a mixed culture of astrocytes and neurons, hPAP activity was elevated by 249-fold (Fig. 1E), suggesting that cross-talk between neurons and astrocytes amplifies activation of the Nrf2 pathway.
Figure 1.

ANG activates ARE-dependent promoters selectively in astrocytes. A, C, and E, graphs showing that treatment with tBHQ (40 μm) increased hPAP activity by 15-fold in astrocytes (A), 7-fold in cortical neurons (C), and 249-fold in a mixed culture (E). B, D, and F, graphs showing that treatment with WT ANG (5 μg/ml) increased hPAP activity by 4-fold in astrocytes (B) and 49-fold in a mixed culture (F) but did not change the activity significantly in cortical neurons (D). hPAP activity remained unchanged upon treatment with defective ANG variants (H114R, S28N, and C39W). Significant changes in hPAP activity compared to vehicle controls are marked by asterisks, where * refers to p ≤ 0.05.
WT ANG activated hPAP comparably to tBHQ. Relative to vehicle, WT ANG induced the highest hPAP signal in a mixed culture, induced the second-highest signal in astrocytes, and caused no change in neurons (Fig. 1, B, D, and F). Moreover, the effect of ANG was dose-dependent, because ANG treatment at 5 μg/ml produced greater hPAP activity than at 1 μg/ml. To demonstrate signal specificity, we evaluated the phosphatase activity produced by ALS-associated ANG variants. As described previously (4, 31), H114R ANG is an inactive catalyst, S28N ANG is deficient in nuclear localization, and C39W ANG lacks conformational stability. None of these variants promoted hPAP gene expression (Fig. 1, B, D, and F).
ANG drives the expression of endogenous Nrf2/ARE-dependent genes in astrocytes
Next, we sought to demonstrate the intrinsic activation of ARE-dependent gene expression upon ANG treatment. Nrf2 induces the transcription of an array of antioxidant genes, and we selected three for analysis. NAD(P)H:quinone oxidoreductase 1 (NQO1) is involved in the reduction of quinones to hydroquinones to prevent redox cycling, which often generates free radicals (32). Glutamate-cysteine ligase modifier subunit (GCLM) is part of the rate-limiting enzyme complex for the synthesis of GSH, a free radical scavenger (33). GSH S-transferase α4 (GSTα4) catalyzes the conjugation of reduced GSH to electrophilic substrates, detoxifying endogenous and xenobiotic alkylating agents (34).
Using qPCR, we evaluated the expression of these antioxidant genes following treatment with ANG. In this experiment, tBHQ again served as a positive control. tBHQ treatment significantly up-regulated the expression of NQO1, GCLM, and GSTα4, consistent with the results of the reporter assay, in both neurons and astrocytes (Fig. 2, A and B). Consistent with the hPAP activity data, ANG activated Nrf2-dependent genes in astrocytes but not in neurons. Taken together, data from the reporter assay and on antioxidant gene expression indicate that WT ANG activates Nrf2-dependent gene expression.
Figure 2.

ANG drives the expression of ARE-dependent genes in astrocytes. A and B, graphs of quantitative RT-PCR data demonstrating that treatment with tBHQ (40 μm) increased the expression of antioxidant genes encoding NQO1, GCLM, and GSTα4 in both astrocytes and cortical neurons relative to vehicle. In contrast, WT ANG promoted the expression of these genes in astrocytes (A) but not cortical neurons (B), and higher ANG concentration induced greater gene expression.
ANG-mediated change in gene expression depends on Nrf2
Then we asked whether Nrf2 is required for this ANG-mediated induction of gene expression. In WT astrocytes derived from ARE-hPAP reporter mice, there was a significant increase in hPAP activity with ANG treatment (Fig. 3A). This ANG-mediated increase was attenuated in cultures from ARE-hPAP/Nrf2−/− mice (Fig. 3B). As expected, none of the ANG variants increased hPAP activity in WT or Nrf2-deficient astrocytes. Further, the increase in NQO1 and GSTα4 expression following ANG treatment was reversed almost completely in Nrf2-deficient cultures (Fig. 3B). These results demonstrate that ANG-mediated changes are dependent on Nrf2.
Figure 3.

ANG depends on Nrf2 to induce ARE-dependent gene expression. A and B, in astrocytes, ANG-mediated hPAP activation (A) and induction of the expression of antioxidant genes (B) were comparable with those in Figs. 1A and 2A. These activities were absent in Nrf2-deprived astrocytes (Nrf2−/−). Treating cells with heparin prior to ANG stimulation likewise prevented ANG-induced changes in hPAP activity (A). Significant changes in gene-expression level compared to Nrf2−/− astrocytes are marked by asterisks, where * refers to p ≤ 0.05 and ** refers to p ≤ 0.01.
In astrocytes, ANG is internalized after it binds to syndecan-4, a transmembrane heparan sulfate proteoglycan (35, 36). To compete with the heparan sulfate–binding site within syndecan-4, we applied a saturating amount of heparin, which mimics heparan sulfate and is known to prevent the intracellular accumulation of ANG (37). Pretreatment with heparin did indeed prevent the ANG-mediated increase hPAP activity (Fig. 3A). These data support the hypothesis that the binding of ANG to syndecan-4 is essential for activation of the Nrf2 pathway.
ANG treatment selectively protects against oxidative stress-induced cell death
As described above, Nrf2 is the master regulator of antioxidant responses (18). Small-molecule Nrf2 activators can provide cells with powerful protection from oxidative damage (19). Accordingly, tBHQ treatment protected cells from H2O2-mediated toxicity (Fig. 4, A, C, and E). The degree of protection did, however, vary among cell types; astrocytes were most responsive to tBHQ treatment, followed by a mixed culture, and then neurons.
Figure 4.

ANG protects neurons from oxidative stress via astrocyte communication. A and B, astrocytes; C and D, cortical neurons; E and F, mixed culture. A, C, and E, tBHQ treatment (40 μm) protected cells from H2O2-mediated toxicity. The degree of protection varied between cell types: astrocytes were the most responsive to the treatment, followed by the mixed culture, and then neurons. B, D, and F, treating cells with WT ANG, at two doses, protected only astrocytes and the mixed culture, but not neurons. Three or more biological replicates were performed for each experiment. Significant cell death compared with vehicle controls is marked by asterisks, where * refers to p ≤ 0.05, ** refers to p ≤ 0.01, *** refers to p ≤ 0.001, and **** refers to p ≤ 0.0001.
We note that WT ANG treatment likewise activated the Nrf2/ARE pathway (Figs. 1–3). The robustness of activation appeared to correlate positively with the degree of cellular protection against H2O2-mediated toxicity (Fig. 4). As expected, WT ANG treatment protected astrocytes and a mixed culture less potently than did tBHQ (Fig. 4, B and F). Nonetheless, the protective effect of WT ANG in these cultures was significant. No protection was observed in neuronally enriched cultures (Fig. 4D). Once again, the data suggest that neuron–astrocyte communication is necessary to support neuronal survival against the deleterious consequences of oxidative stress.
Neurons use ANG as a messenger to signal for astrocyte protection
Previous studies have demonstrated that stressed neurons secrete ANG, which is then taken up by astrocytes (36). We replicated these findings, detecting a high level of secreted ANG in conditioned medium collected from neurons exposed to a low nontoxic dose of H2O2. Using a zymogram that provides a highly sensitive assessment of ribonucleolytic activity (38, 39), we calibrated known ANG concentrations and the intensity of bands on a gel. Then we estimated that normal and stressed neuronal conditioned medium contained 0.5 and 4.0 μg of ANG/ml, respectively (Fig. 5A).
Figure 5.
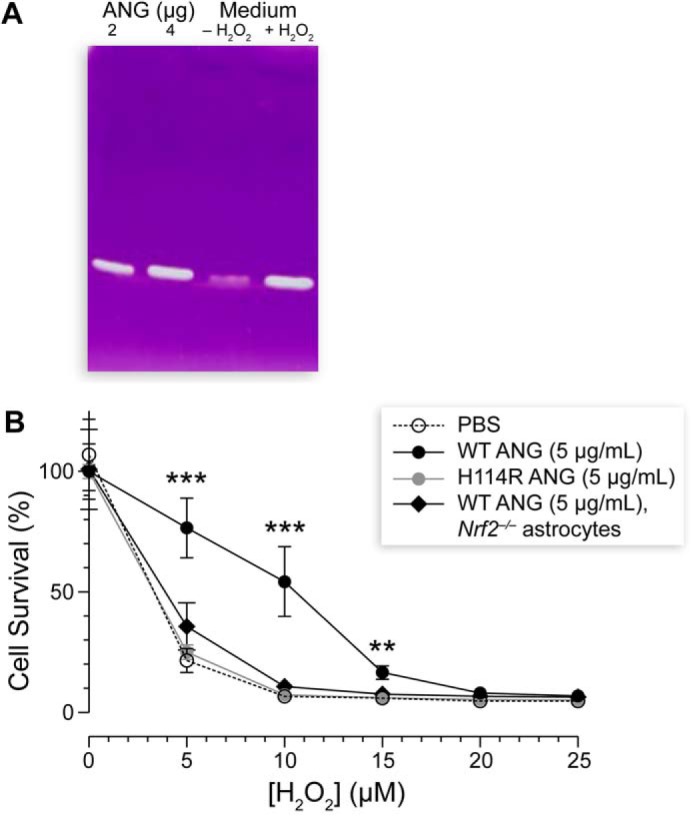
Neurons release ANG to signal their need for protection to astrocytes. A, pre-exposing cortical neurons to H2O2 (1 μm) caused the release of ANG. A zymogram gel of conditioned medium from stressed neurons revealed a high level of active ANG (4.0 μg/ml) compared with that from normal neurons (0.5 μg/ml). B, neurons were protected by conditioned medium collected from astrocytes that were pre-exposed to WT ANG (5 μg/ml). Three or more biological replicates were performed for each experiment. Protection depended on Nrf2 and the ribonucleolytic activity of ANG. Significant cell death compared with Nrf2−/− astrocytes is marked by asterisks, where ** refers to p ≤ 0.01 and *** refers to p ≤ 0.001.
Then we investigated how well stressed neurons are protected when treated with astrocytic conditioned medium that was pre-exposed to WT ANG. First, we treated astrocytes with 5 μg/ml of WT ANG or the inactive H114R variant. After 24 h, we collected the astrocytic conditioned medium and treated neurons with that medium for 24 h. The neurons were then subjected to H2O2-mediated toxicity. Only conditioned medium from astrocytes that were stimulated by WT ANG protected the neurons; conditioned medium from astrocytes exposed to the H114R ANG variant had no effect. Likewise, no change was observed from conditioned medium derived from Nrf2-deficient astrocytes, even upon stimulus with WT ANG (Fig. 5B).
Discussion
Members of the ptRNase superfamily have evolved to be efficient, nonspecific catalysts of RNA degradation (40). Unlike its homologs, ANG has nearly unmeasurable ribonucleolytic activity toward model substrates (41, 42). Moreover, whereas other ptRNases function in the extracellular space, ANG acts within cells (43–46).
Previous studies have shown that ANG cleaves tRNA to mediate neuroprotective activity (10–12). Most ANG mutations that segregate with ALS do not significantly alter the secondary structure or conformational stability of ANG. Instead, they disrupt its ribonucleolytic activity or subcellular distribution (47–50). This observation has stimulated interest in understanding the molecular basis of the role of ANG in neuroprotection.
We find that ANG activates the Nrf2 pathway in astrocytes and that this activation protects neurons from oxidative injury. In this mechanism of action (Fig. 6), stressed neurons secrete high levels of ANG, which then binds to syndecan-4 receptors on astrocytes and enters the astrocytes via endocytosis (51). The binding of ANG to the receptor activates PKCα, which phosphorylates Nrf2, endowing it with the ability to evade the Keap1 inhibitor (52). Phosphorylated Nrf2 translocates to the nucleus and forms heterodimers with Maf. These dimers bind to AREs to stimulate antioxidant gene expression that defends against H2O2-mediated toxicity. A fraction of the ANG in endosomes escapes into the cytosol. Cytosolic ANG is sequestered in granules through the recruitment of 5′-tiRNAs, which are produced by the ANG-mediated cleavage of tRNAs (9). These tiRNAs interact with the translational silencer protein YB-1 and sequester the eukaryotic translation initiation factor 4E/G/A complex to suppress protein translation (10–12). Notably, this mechanism of action (Fig. 6) is consistent with PKCα being activated when ANG binds to the syndecan-4 receptor (35, 36, 53) and with the phosphorylation of Nrf2 by PKCα up-regulating the transcriptional activity of Nrf2 (52, 54).
Figure 6.

Putative neuroprotective pathway of ANG. Upon ANG binding, syndecan-4 activates PKCα to phosphorylate Nrf2. Phosphorylation enables Nrf2 to evade its binding partner, Keap1. Phosphorylated Nrf2 translocates into the nucleus and forms a heterodimer with Maf. The dimer binds to ARE, driving the expression of antioxidant genes to counteract cellular oxidative injury. In addition, ANG participates in stress-induced protein translation repression by generating tiRNAs. ANG executes both actions—activating the Nrf2 pathway and cleaving tRNAs—in a coordinated manner to manifest neuroprotection.
We have demonstrated that ANG activates the Nrf2 pathway. The inactive H114R variant does not, however, trigger the Nrf2 pathway (Fig. 5B). This finding suggests that the ribonucleolytic activity of ANG against tRNAs is essential for Nrf2-dependent protection from H2O2-mediated toxicity. Likewise, a recent study has shown that increasing ribonucleolytic activity of ANG improves astrocyte survival when the cells suffer oxidative stress (39). These two effects of ANG—activating the Nrf2 pathway and cleaving tRNAs—apparently coordinate to manifest antioxidant activity (Fig. 6).
Oxidative stress irritates neurons, leading to the release of ANG, which acts as a “distress” signal that is conveyed to astrocytes. Within astrocytes, catalysis by ANG generates specialized tiRNAs that form G-quadruplexes that arrest protein translation. Delivery of these tiRNAs to neurons is known to be neuroprotective (12). In addition, Nrf2 activation in astrocytes protects proximal neurons (24, 33). This protection is transmitted in ANG-treated astrocytic conditioned medium (Fig. 5B), perhaps by tiRNAs that enter neurons and induce ROS clearance.
ANG elicits manifold effects within astrocytes (55). Like typical ligands that bind to the extracellular face of membrane-bound receptors, ANG activates the intracellular Nrf2 pathway. This activation leads, ultimately, to changes in gene expression. In addition, ANG enters astrocytes and migrates to stress granules to suppress protein translation. ANG, in part, directs the translational machinery to focus on the synthesis of antioxidant enzymes and specifically induces ARE-dependent gene expression through Nrf2. Notably, the mechanism of ANG-mediated neuroprotection is distinct from that of its mechanism for promoting cell proliferation and neovascularization, in which ANG migrates to the nucleolus and promotes rDNA transcription (43, 45, 56, 57).
Nrf2 is the master regulator of the cellular antioxidant system. In this role, Nrf2 senses the presence of oxidants and is responsible for the production of a vast array of antioxidants to counterbalance ROS. Many attempts have been made to develop small-molecule activators of the Nrf2 pathway to combat free radicals. Nevertheless, only one Food and Drug Administration–approved drug (dimethyl fumarate) and one herbal dietary supplement (Protandim) claim to be Nrf2 activators (58, 59). Here, we report that an endogenous protein, ANG, activates the Nrf2 pathway and counteracts the deleterious consequences of ROS. Its activation of Nrf2 underscores the role of ANG as a neuroprotective agent to combat oxidative stress–mediated cellular toxicity and highlights its potential utility in the treatment of ALS.
Experimental procedures
Materials
All chemicals and reagents were from Sigma–Aldrich unless indicated otherwise. Medium and added components trypsin (0.25% w/v), and Dulbecco's PBS were the Gibco brand from Thermo Fisher Scientific.
Conditions
All procedures were performed at ambient temperature (∼22 °C) and pressure (1.0 atm) unless indicated otherwise.
Angiogenins
Human ANG and its variants were produced by heterologous expression in Escherichia coli and were purified as described previously (39, 42, 45).
Animals
ARE-hPAP transgenic mice were created by installing 51 bp of the NQO1 promoter upstream of a reporter gene that encodes hPAP, as described previously (29). Nrf2 knockout mice (Nrf2−/−) were graciously provided by Dr. Kaiman Chan and Dr. Yuet Wai Kan (Howard Hughes Medical Institute, University of California, San Francisco) (60). All mice were on a C57Bl/6 background. All animal procedures were approved by the University of Wisconsin–Madison Institutional Animal Care and Use Committee, and all animal experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Mammalian cell culture
Cultures of primary astrocytes (>98% astrocytes) were prepared from the cortices of 1-day-old mice as described previously (24, 25). Astrocytes were plated at a density of 2 × 104 cells/cm2 in 6- or 96-well collagen-coated plates and maintained in complete medium (CEMEM). CEMEM contained minimum Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 10% (v/v) horse serum, 0.5 mm l-glutamine, 1% (w/v) penicillin (100 IU/ml), and streptomycin (100 μg/ml). Neuronally enriched cultures (>98% neurons) were prepared from embryos on embryonic day 15 or 16 as described previously (29). The neurons were plated at a density of 3 × 104 cells/cm2 in 6- or 96-well poly(d-lysine)–coated plates and maintained in CEMEM for 45 min before the medium was replaced with fresh neurobasal medium (NBM). Every 2–3 days, half of the old NBM was replaced with fresh NBM. A mixed culture (50% neurons and 50% astrocytes) was prepared similarly to neuronal cultures, with the following exceptions. After the cells were plated in CEMEM and incubated for 45 min, the medium was replaced with fresh CEMEM. On day 2, the medium was switched to NBM. Every 2 or 3 days, half of the old NBM was replaced with fresh NBM. The cells were incubated at 37 °C in a tri-gas incubator with 5% O2, 5% CO2, and 90% N2.
hPAP reporter assay
Intracellular hPAP levels were quantified by using a luminescence assay. The cells were grown in 96-well plates and seeded at a density that was appropriate for a particular cell type. Fully differentiated cultures were treated with vehicle (PBS), tBHQ, ANG, or an ANG variant for 24 h. Whole-cell extracts were prepared in the plates by adding lysis buffer (50 mm Tris-HCl, pH 7.5, containing 100 mm NaCl, 5 mm MgCl2, and 1% (w/v) CHAPS), and the extracts were incubated at 65 °C for 30 min to inactivate endogenous alkaline phosphatase activity. Next, an hPAP substrate and its enhancer (CSPD and Emerald, respectively, from Tropix) were added, and hPAP activity was quantified based on the appearance of luminescence, which was recorded with an M1000 plate reader (Tecan).
Cell survival assay
The surviving cells were quantified by using an assay based on the ability of intracellular enzymes to catalyze the reduction of a tetrazolium dye, MTS (61). The cells were grown in 96-well plates and seeded at the density indicated for the various cell types. Fully differentiated cultures were treated with vehicle (PBS), tBHQ, WT ANG, or an ANG variant for 24 h and then with increasing concentrations of H2O2 for an additional 48 h. The cells were then incubated with CellTiter 96 MTS reagent (Promega) for 1–4 h, depending on cell type. Absorbance at 490 nm was recorded with an M1000 plate reader, and the data were analyzed with Prism 5.0 software.
Quantitative RT-PCR
Total cellular RNA was isolated by extraction with TRIzol (Invitrogen), and the RNA samples were then treated with DNase I (Invitrogen) at 37 °C for 15 min. The RNA was purified by phenol-chloroform extraction followed by ethanol precipitation. RNA concentrations and purities were assessed with a NanoVue instrument (GE Healthcare Life Sciences). Purified cellular RNA (∼1 μg) was used in a reverse transcription reaction along with random hexamers from the SuperScript III reverse transcriptase kit (Invitrogen). A 1-μl aliquot of the resultant cDNA solution was used in qPCRs in conjunction with PerfeCTa SYBR Green FastMix reaction mixes (Quanta Biosciences). Amplified cDNAs were evaluated with an ABI Prism 7200 sequence detector (PerkinElmer). The primers used for qPCR were described previously (24) and are listed in Table 1.
Table 1.
Primers used for qPCR
| Target | Forward (5′ → 3′) | Reverse (3′ → 5′) |
|---|---|---|
| Rnase1 | CTGCAAGAACAGGAAGAGCAAC | GAGTGGTCTTGTAGTCACAGTTGG |
| Rnase4 | AACGGTTCCTTCGACAGCAT | GCGTTTGCACTGGACAGAAG |
| Ang1 | TCTGCAGGGTTCAGACATGT | TCTGGGCTATGAGGGGAGAT |
| Rnh1 | CCCAGCTGTAAGCTCAGGAC | CTCTGCTTGGCTCTGAGGAC |
| Gapdh | CTCCCACTCTTCCACCTTCG | CCACCACCCTGTTGCTGTAG |
| Rpl13a | GCTGAAGCCTACCAGAAAGT | TCCGTTTCTCCTCCAGAGT |
| Hprt1 | CTAGTCCTGTGGCCATCTGC | GGGACGCAGCAACTGACATT |
Zymogram assay
Solutions of WT ANG and conditioned medium were diluted 1:1 with 2× Laemmli buffer from Bio-Rad, and the resulting solutions were loaded on to a polyacrylamide gel (15%, w/v) containing poly(cytidylic acid). The loaded gel was subjected to electrophoresis for 1.5 h at 100 V. Subsequent washing, refolding and staining with toluidine blue were performed as described previously (38, 39). Zymograms were quantified by scanning the stained gel with an ImageQuantLAS4000 instrument (GE Healthcare Bio-Sciences) and by using the program ImageJ (26).
Statistical analyses
The data were analyzed by using the program Prism (GraphPad). The values are represented as the means ± S.D. Significance was determined by using an unpaired Student's t test or a one-way analysis of variance followed by post hoc analysis to determine significant paired comparisons based on experimental design.
Author contributions
T. T. H., D. A. J., and J. A. J. conceptualization; T. T. H. data curation; T. T. H., D. A. J., R. T. R., and J. A. J. formal analysis; T. T. H. validation; T. T. H. investigation; T. T. H. methodology; T. T. H. writing-original draft; T. T. H., D. A. J., R. T. R., and J. A. J. writing-review and editing; D. A. J., R. T. R., and J. A. J. supervision; R. T. R. and J. A. J. resources; R. T. R. and J. A. J. funding acquisition; R. T. R. and J. A. J. project administration.
This work was supported by National Institutes of Health Grants R01 CA073808 (to R. T. R.), P50 AG033514 (to J. A. J.), and T32 GM007215 (to T. T. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ALS
- amyotrophic lateral sclerosis
- hPAP
- human placental alkaline phosphatase
- ROS
- reactive oxygen species
- tBHQ
- tert-butylhydroquinone
- tiRNA
- tRNA-derived, stress-induced RNA
- qPCR
- quantitative PCR
- ptRNase
- pancreatic-type RNase
- ARE
- antioxidant-response element
- CEMEM
- complete medium
- NBM
- neurobasal medium.
References
- 1. Taylor J. P., Brown R. H. Jr., and Cleveland D. W. (2016) Decoding ALS: from genes to mechanism. Nature 539, 197–206 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang K., Donnelly C. J., Haeusler A. R., Grima J. C., Machamer J. B., Steinwald P., Daley E. L., Miller S. J., Cunningham K. M., Vidensky S., Gupta S., Thomas M. A., Hong I., Chiu S.-L., Huganir R. L., et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaur S. J., McKeown S. R., and Rashid S. (2016) Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. 577, 109–118 10.1016/j.gene.2015.11.049 [DOI] [PubMed] [Google Scholar]
- 4. Greenway M. J., Andersen P. M., Russ C., Ennis S., Cashman S., Donaghy C., Patterson V., Swingler R., Kieran D., Prehn J., Morrison K. E., Green A., Acharya K. R., Brown R. H. Jr., and Hardiman O. (2006) ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 38, 411–413 10.1038/ng1742 [DOI] [PubMed] [Google Scholar]
- 5. Wu D., Yu W., Kishikawa H., Folkerth R. D., Iafrate A. J., Shen Y., Xin W., Sims K., and Hu G.-F. (2007) Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann. Neurol. 62, 609–617 10.1002/ana.21221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gellera C., Colombrita C., Ticozzi N., Castellotti B., Bragato C., Ratti A., Taroni F., and Silani V. (2008) Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics 9, 33–40 10.1007/s10048-007-0111-3 [DOI] [PubMed] [Google Scholar]
- 7. Kieran D., Sebastia J., Greenway M. J., King M. A., Connaughton D., Concannon C. G., Fenner B., Hardiman O., and Prehn J. H. (2008) Control of motoneuron survival by angiogenin. J. Neurosci. 28, 14056–14061 10.1523/JNEUROSCI.3399-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strydom D. J., Fett J. W., Lobb R. R., Alderman E. M., Bethune J. L., Riordan J. F., and Vallee B. L. (1985) Amino acid sequence of human tumor derived angiogenin. Biochemistry 24, 5486–5494 10.1021/bi00341a031 [DOI] [PubMed] [Google Scholar]
- 9. Lyons S. M., Fay M. M., Akiyama Y., Anderson P. J., and Ivanov P. (2017) RNA biology of angiogenin: current state and perspectives. RNA Biol. 14, 171–178 10.1080/15476286.2016.1272746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamasaki S., Ivanov P., Hu G.-F., and Anderson P. (2009) Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 185, 35–42 10.1083/jcb.200811106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivanov P., Emara M. M., Villen J., Gygi S. P., and Anderson P. (2011) Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 43, 613–623 10.1016/j.molcel.2011.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ivanov P., O'Day E., Emara M. M., Wagner G., Lieberman J., and Anderson P. (2014) G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. U.S.A. 111, 18201–18206 10.1073/pnas.1407361111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim G. H., Kim J. E., Rhie S. J., and Yoon S. (2015) The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340 10.5607/en.2015.24.4.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peters O. M., Ghasemi M., and Brown R. H. Jr. (2015) Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 125, 1767–1779 10.1172/JCI71601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. D'Amico E., Factor-Litvak P., Santella R. M., and Mitsumoto H. (2013) Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 65, 509–527 10.1016/j.freeradbiomed.2013.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., and Telser J. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84 10.1016/j.biocel.2006.07.001 [DOI] [PubMed] [Google Scholar]
- 17. Lei X. G., Zhu J.-H., Cheng W.-H., Bao Y., Ho Y.-S., Reddi A. R., Holmgren A., and Arnér E. S. (2016) Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 96, 307–364 10.1152/physrev.00010.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson D. A., and Johnson J. A. (2015) Nrf2: a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 88, 253–267 10.1016/j.freeradbiomed.2015.07.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanninen K. M., Pomeshchik Y., Leinonen H., Malm T., Koistinaho J., and Levonen A. L. (2015) Applications of the Keap1–Nrf2 system for gene and cell therapy. Free Radic. Biol. Med. 88, 350–361 10.1016/j.freeradbiomed.2015.06.037 [DOI] [PubMed] [Google Scholar]
- 20. Li J., Calkins M. J., Johnson D. A., and Johnson J. A. (2007) Role of Nrf2-dependent ARE-driven antioxidant pathway in neuroprotection. Methods Mol. Biol. 399, 67–78 10.1007/978-1-59745-504-6_6 [DOI] [PubMed] [Google Scholar]
- 21. Johnson J. A., Johnson D. A., Kraft A. D., Calkins M. J., Jakel R. J., Vargas M. R., and Chen P. C. (2008) The Nrf2–ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann. N.Y. Acad. Sci. 1147, 61–69 10.1196/annals.1427.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calkins M. J., Johnson D. A., Townsend J. A., Vargas M. R., Dowell J. A., Williamson T. P., Kraft A. D., Lee J. M., Li J., and Johnson J. A. (2009) The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox. Signal. 11, 497–508 10.1089/ars.2008.2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kraft A. D., Resch J. M., Johnson D. A., and Johnson J. A. (2007) Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 207, 107–117 10.1016/j.expneurol.2007.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vargas M. R., Johnson D. A., Sirkis D. W., Messing A., and Johnson J. A. (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 28, 13574–13581 10.1523/JNEUROSCI.4099-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vargas M. R., and Johnson J. A. (2010) Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics 7, 471–481 10.1016/j.nurt.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rueden C. T., Schindelin J., Hiner M. C., DeZonia B. E., Walter A. E., Arena E. T., and Eliceiri K. W. (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 10.1186/s12859-017-1934-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sebastià J., Kieran D., Breen B., King M. A., Netteland D. F., Joyce D., Fitzpatrick S. F., Taylor C. T., and Prehn J. H. (2009) Angiogenin protects motoneurons against hypoxic injury. Cell Death Differ. 16, 1238–1247 10.1038/cdd.2009.52 [DOI] [PubMed] [Google Scholar]
- 28. Cho G.-W., Kang B. Y., and Kim S. H. (2010) Human angiogenin presents neuroprotective and migration effects in neuroblastoma cells. Mol. Cell Biochem. 340, 133–141 10.1007/s11010-010-0410-0 [DOI] [PubMed] [Google Scholar]
- 29. Johnson D. A., Andrews G. K., Xu W., and Johnson J. A. (2002) Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J. Neurochem. 81, 1233–1241 10.1046/j.1471-4159.2002.00913.x [DOI] [PubMed] [Google Scholar]
- 30. Kraft A. D., Johnson D. A., and Johnson J. A. (2004) Nuclear factor E2–related factor 2–dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 24, 1101–1112 10.1523/JNEUROSCI.3817-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shapiro R., and Vallee B. L. (1989) Site-directed mutagenesis of histidine-13 and histidine-114 of human angiogenin: alanine derivatives inhibit angiogenin-induced angiogenesis. Biochemistry 28, 7401–7408 10.1021/bi00444a038 [DOI] [PubMed] [Google Scholar]
- 32. Guo Y., Zhang Y., Wen D., Duan W., An T., Shi P., Wang J., Li Z., Chen X., and Li C. (2013) The modest impact of transcription factor Nrf2 on the course of disease in an ALS animal model. Lab. Invest. 93, 825–833 10.1038/labinvest.2013.73 [DOI] [PubMed] [Google Scholar]
- 33. Kanno T., Tanaka K., Yanagisawa Y., Yasutake K., Hadano S., Yoshii F., Hirayama N., and Ikeda J. E. (2012) A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2–ARE pathway: therapeutic implications for ALS. Free Radic. Biol. Med. 53, 2028–2042 10.1016/j.freeradbiomed.2012.09.010 [DOI] [PubMed] [Google Scholar]
- 34. Benedusi V., Martorana F., Brambilla L., Maggi A., and Rossi D. (2012) The peroxisome proliferatoractivated receptor γ (PPARγ) controls natural protective mechanisms against lipid peroxidation in amyotrophic lateral sclerosis. J. Biol. Chem. 287, 35899–35911 10.1074/jbc.M112.366419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aparicio-Erriu I. M., and Prehn J. H. (2012) Molecular mechanisms in amyotrophic lateral sclerosis: the role of angiogenin, a secreted RNase. Front. Neurosci. 6, 167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skorupa A., King M. A., Aparicio I. M., Dussmann H., Coughlan K., Breen B., Kieran D., Concannon C. G., Marin P., and Prehn J. H. (2012) Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 32, 5024–5038 10.1523/JNEUROSCI.6366-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeo K. J., Hwang E., Min K.-M., Jee J.-G., Lee C.-K., Hwang K. Y., Jeon Y. H., Chang S.-I., and Cheong H.-K. (2014) The dual binding site of angiogenin and its inhibition mechanism: the crystal structure of the rat angiogenin–heparin complex. Chem. Commun. (Camb.) 50, 12966–12969 10.1039/C4CC05175K [DOI] [PubMed] [Google Scholar]
- 38. Bravo J., Fernández E., Ribó M., de Llorens R., and Cuchillo C. M. (1994) A versatile negative-staining ribonuclease zymogram. Anal. Biochem. 219, 82–86 10.1006/abio.1994.1234 [DOI] [PubMed] [Google Scholar]
- 39. Hoang T. T., Smith T. P., and Raines R. T. (2017) A boronic acid conjugate of angiogenin that shows ROS-responsive neuroprotective activity. Angew. Chem. Int. Ed. Engl. 56, 2619–2622 10.1002/anie.201611446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raines R. T. (1998) Ribonuclease A. Chem. Rev. 98, 1045–1066 10.1021/cr960427h [DOI] [PubMed] [Google Scholar]
- 41. Curran T. P., Shapiro R., and Riordan J. F. (1993) Alteration of the enzymatic specificity of human angiogenin by site-directed mutagenesis. Biochemistry 32, 2307–2313 10.1021/bi00060a023 [DOI] [PubMed] [Google Scholar]
- 42. Leland P. A., Staniszewski K. E., Park C., Kelemen B. R., and Raines R. T. (2002) The ribonucleolytic activity of angiogenin. Biochemistry 41, 1343–1350 10.1021/bi0117899 [DOI] [PubMed] [Google Scholar]
- 43. Moroianu J., and Riordan J. F. (1994) Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc. Natl. Acad. Sci. U.S.A. 91, 1677–1681 10.1073/pnas.91.5.1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kishimoto K., Liu S., Tsuji T., Olson K. A., and Hu G.-F. (2005) Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene 24, 445–456 10.1038/sj.onc.1208223 [DOI] [PubMed] [Google Scholar]
- 45. Hoang T. T., and Raines R. T. (2017) Molecular basis for the autonomous promotion of cell proliferation by angiogenin. Nucleic Acids Res. 45, 818–831 10.1093/nar/gkw1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomas S. P., Hoang T. T., Ressler V. T., and Raines R. T. (2018) Human angiogenin is a potent cytotoxin in the absence of ribonuclease inhibitor. RNA 24, 1018–1027 10.1261/rna.065516.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crabtree B., Thiyagarajan N., Prior S. H., Wilson P., Iyer S., Ferns T., Shapiro R., Brew K., Subramanian V., and Acharya K. R. (2007) Characterization of human angiogenin variants implicated in amyotrophic lateral sclerosis. Biochemistry 46, 11810–11818 10.1021/bi701333h [DOI] [PubMed] [Google Scholar]
- 48. Thiyagarajan N., Ferguson R., Subramanian V., and Acharya K. R. (2012) Structural and molecular insights into the mechanism of action of human angiogenin-ALS variants in neurons. Nat. Commun. 3, 1121 10.1038/ncomms2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Padhi A. K., Jayaram B., and Gomes J. (2013) Prediction of functional loss of human angiogenin mutants associated with ALS by molecular dynamics simulations. Sci. Rep. 3, 1225 10.1038/srep01225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Padhi A. K., Banerjee K., Gomes J., and Banerjee M. (2014) Computational and functional characterization of angiogenin mutations, and correlation with amyotrophic lateral sclerosis. PLoS One 9, e111963 10.1371/journal.pone.0111963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ferguson R., and Subramanian V. (2018) The cellular uptake of angiogenin, an angiogenic and neurotrophic factor is through multiple pathways and largely dynamin independent. PLoS One 13, e0193302 10.1371/journal.pone.0193302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang H. C., Nguyen T., and Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 10.1074/jbc.M206911200 [DOI] [PubMed] [Google Scholar]
- 53. Couchman J. R. (2003) Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell Biol. 4, 926–937 10.1038/nrm1257 [DOI] [PubMed] [Google Scholar]
- 54. Bloom D. A., and Jaiswal A. K. (2003) Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 278, 44675–44682 10.1074/jbc.M307633200 [DOI] [PubMed] [Google Scholar]
- 55. Skorupa A., Urbach S., Vigy O., King M. A., Chaumont-Dubel S., Prehn J. H., and Marin P. (2013) Angiogenin induces modifications in the astrocyte secretome: Relevance to amyotrophic lateral sclerosis. J. Proteomics 91, 274–285 10.1016/j.jprot.2013.07.028 [DOI] [PubMed] [Google Scholar]
- 56. Pizzo E., Sarcinelli C., Sheng J., Fusco S., Formiggini F., Netti P., Yu W., D'Alessio G., and Hu G.-F. (2013) Ribonuclease/angiogenin inhibitor 1 regulates stress-induced subcellular localization of angiogenin to control growth and survival. J. Cell Sci. 126, 4308–4319 10.1242/jcs.134551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sheng J., Yu W., Gao X., Xu Z., and Hu G.-F. (2014) Angiogenin stimulates ribosomal RNA transcription by epigenetic activation of the ribosomal DNA promoter. J. Cell Physiol. 229, 521–529 10.1002/jcp.24477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suneetha A., and Raja Rajeswari K. (2016) Role of dimethyl fumarate in oxidative stress of multiple sclerosis: a review. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 1019, 15–20 10.1016/j.jchromb.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 59. The ALSUntangled Group (2016) ALSUntangled no. 31: Protandim. Amyotroph. Lat. Scler. Frontotemporal Degener. 17, 154–156 10.3109/21678421.2015.1088707 [DOI] [PubMed] [Google Scholar]
- 60. Chan K., Lu R., Chang J. C., and Kan Y. W. (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943–13948 10.1073/pnas.93.24.13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cory A. H., Owen T. C., Barltrop J. A., and Cory J. G. (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 3, 207–212 10.3727/095535491820873191 [DOI] [PubMed] [Google Scholar]