

Abstract
Fibrillar amyloid deposits are defining pathological lesions in Alzheimer’s disease brain and are thought to mediate neuronal death. Amyloid is composed primarily of a 39–42 amino acid protein fragment of the amyloid precursor protein (APP), called amyloid β-protein (Aβ). Because deposition of fibrillar amyloid in vitrohas been shown to be highly dependent on Aβ concentration, reducing the proteolytic release of Aβ is an attractive, potentially therapeutic target. Here, the turnover rate of brain Aβ has been determined to define treatment intervals over which a change in steady-state concentration of Aβ could be measured. Mice producing elevated levels of human Aβ were used to determine approximate turnover rates for Aβ and two of its precursors, C99 and APP. Thet½ for brain Aβ was between 1.0 and 2.5 hr, whereas for C99, immature, and fully glycosylated forms of APP695 the approximate t½ values were 3, 3, and 7 hr, respectively. Given the rapid Aβ turnover rate, acute studies were designed using phorbol 12-myristate 13-acetate (PMA), which had been demonstrated previously to reduce Aβ secretion from cells in vitro via induction of protein kinase C (PKC) activity. Six hours after intracortical injection of PMA, Aβ levels were significantly reduced, as measured by both Aβ40- and Aβ42-selective ELISAs, returning to normal by 12 hr. An inactive structural analog of PMA, 4α-PMA, had no effect on brain Aβ levels. Among the secreted N-terminal APP fragments, APPβ levels were significantly reduced by PMA treatment, whereas APPα levels were unchanged, in contrast to most cell culture studies. These results indicate that Aβ is rapidly turned over under normal conditions and support the therapeutic potential of elevating PKC activity for reduction of brain Aβ.
Keywords: protein turnover, amyloid-β protein, amyloid precursor protein, Alzheimer’s disease, phorbol ester, protein kinase C
The behavioral deficits associated with Alzheimer’s disease (AD) result from the loss of neurons and synapses, primarily in the cortex and hippocampus. Amyloid deposits in these regions are an invariant pathological feature of AD and result from the aggregation of a 39–42 amino acid long protein known as amyloid β-protein (Aβ) (Selkoe, 1996). This protein has been shown to be neurotoxic in vitro when present in an aggregated form (Pike et al., 1993). Evidence obtained from the study of familial forms of AD in which mutations exist in either the amyloid precursor or presenilin genes (Hardy, 1997) implicates elevated secretion of the 42 amino acid form as a likely etiological event in disease development. This form is most fibrillogenic in vitro (Jarrett et al., 1993) and is more abundant in amyloid deposits in the AD brain than shorter, less fibrillogenic forms (Iwatsubo et al., 1994; Savage et al., 1995; Yamaguchi et al., 1995). Under normal conditions, however, soluble Aβ ending at residue 40 is more abundant than the 42 residue form (Haass et al., 1992; Seubert et al., 1992; Shoji et al., 1992;Vigo-Pelfrey et al., 1993).
The amyloid precursor protein (APP) is processed to form Aβ and other derivatives by at least three proteases, identified by their cleavage specificities (Selkoe, 1996). α-Secretase generates a secreted N-terminal APP fragment (APPα) and also destroys the Aβ domain. The remaining C-terminal fragment (9 kDa) can be processed further by γ-secretase, leading to the secretion of a 3 kDa protein (P3) and formation of a 6 kDa C-terminal fragment retained by the cells. β-Secretase cleaves APP at the N terminus of Aβ, leading to the secretion of an N-terminal fragment (APPβ) that is 16 residues shorter than APPα and a cell-associated, C-terminal fragment (C99) that can be processed further by γ-secretase to generate the C terminus of the Aβ protein.
To develop an experimental system in which to measure pharmacological effects on APP processing in vivo, we have used a gene-targeted mouse harboring the Swedish familial Alzheimer’s disease (FAD) mutation and a humanized Aβ domain (Reaume et al., 1996). These mice express readily detectable levels of human Aβ and, unlike conventional APP transgenic animals, express endogenous levels of APP under normal developmental and tissue-specific control. In this model, we previously demonstrated increased cleavage at the β-secretase site, a finding well documented in vitro in FAD model systems (Citron et al., 1992; Cai et al., 1993; Citron et al., 1994).
Knowledge of Aβ turnover rate in vivo is necessary to determine treatment intervals over which a change in Aβ levels could be measured. Here, we demonstrate that mechanisms are present in mouse brain that eliminate Aβ, C99, and APP within several hours of their generation. This knowledge was exploited to initiate studies of agents that could lower Aβ levels in the brain. Phorbol 12-myristate 13-acetate (PMA) has been used extensively in vitro to modulate APP processing and, as a result, lower secreted Aβ levels (Buxbaum et al., 1992; Hung et al., 1993). This compound is highly selective for the activation of protein kinase C (PKC) (Newton, 1995) and has been used previously to modulate brain processes in vivo (Cope et al., 1984; Routtenberg et al., 1986; Baranyi et al., 1987; Deitrich et al., 1989). Here, intracortical injections of PMA resulted in short-term reductions in Aβ levels in mouse brain, thus validating in vivo the modulatory effect of PKC activation on Aβ secretion that has been described in cultured cell systems. We have extended these observations by demonstrating specific effects on levels of both Aβ40 and Aβ42. Interestingly, we did not observe increased APPα concentrations in brain coincident with the reduction in levels of Aβ, contrary to effects of PMA seen in cell culture. This highlights the importance of measuring Aβ levels directly, as well as other APP derivatives, when evaluating modulators of APP metabolism in vivo.
MATERIALS AND METHODS
Antibodies. Rabbit polyclonal antibody 1153 was generated against the first 28 amino acids of human Aβ (Savage et al., 1994). Monoclonal antibody 6E10 and biotinylated 6E10 were both purchased from Senetek (Maryland Heights, MO) and recognize an epitope within Aβ1–17. Rabbit polyclonal antibody 97 was directed against the 30 amino acids at the C terminus of APP (Reaume et al., 1996). Aβ40- and Aβ42-selective polyclonal antibodies (affinity-purified; Aβ40, lots 4434804, 4434805, 4434806, and 4434807; Aβ42 lot 4434417) were obtained from Quality Controlled Biochemicals (Hopkinton, MA). Polyclonal antibody 54 was generated against the peptide sequence SEVNL and is specific for Swedish FAD mutant APPβ (Siman et al., 1995). A monoclonal antibody against actin (clone C4) was purchased from Boehringer Mannheim (Indianapolis, IN). Both goat anti-rabbit and goat anti-mouse IgG1 were purchased from Southern Biotechnology Associates (Birmingham, AL).
Infusion of [35S]methionine into gene-targeted mice. Mice at 6 months of age were anesthetized with a mixture of 120 mg/kg ketamine and 12 mg/kg xylazine. [35S]Methionine (New England Nuclear, Boston, MA) at 500 μCi/100 μl (with 0.9% saline diluent) was infused into the femoral vein of each mouse over 30 min. Rate and volume of infusion were chosen to approach steady-state plasma levels of isotope (Garlick and Marshall, 1972). At various time points after the midpoint of the infusion, mice were anesthetized with avertin (1.25% 2,2,2-tribromoethanol and 2.5% 2-methyl-2-butanol), and blood was withdrawn via intraventricular puncture. The mice were then perfused with 15 ml of Ringer’s solution at room temperature over 5 min, and brains were removed, rapidly frozen, and stored at −70°C.
Detection of radiolabeled and steady-state Aβ, C99, and APP. Brain supernatants were processed for immunoprecipitation as described previously (Reaume et al., 1996). Briefly, brains were homogenized in 6 m guanidine and 50 mm Tris, pH 7.5, and centrifuged at 100,000 × g, and supernatants were dialyzed against PBS with protease inhibitors. From three-fourths of the dialysate, C99 and Aβ were immunoprecipitated using the polyclonal antiserum 1153 and Pansorbin (Calbiochem, San Diego, CA). From the remaining one-fourth of the dialysate, APP was immunoprecipitated using polyclonal antiserum 97. Proteins immunoprecipitated with antiserum 1153 were resolved by electrophoresis on 10–20% Tris-tricine polyacrylamide gels (Owl Scientific, Woburn, MA) and transferred to a polyvinylidene difluoride membrane. Proteins immunoprecipitated using antiserum 97 were resolved using Laemmli gels (4–20%, Owl Scientific) and transferred to nitrocellulose. Dried membranes were exposed to phosphorimage screens (Molecular Dynamics, Sunnyvale, CA). Relative intensities of protein bands were determined using ImageQuant software (Molecular Dynamics).
After exposure of the phosphorimage screen to detect radiolabeled proteins corresponding to Aβ, C99, and APP, steady-state levels of these proteins were detected by immunoblotting. Membranes were wetted with transfer buffer and blocked with 5% nonfat dry milk in Tris-buffered saline (TBS). Monoclonal antibody 6E10 and enhanced chemiluminescence were used to detect these APP forms as described previously (Reaume et al., 1996).
Treatment of mice with phorbol ester. Gene-targeted mice from 3–6 months of age were anesthetized with ether. PMA (40 nmol) or 4αPMA (40 nmol) (phorbol esters from Alexis Biochemicals, San Diego, CA) or a corresponding volume of vehicle (2.5 μl, 30% DMSO and 0.9% saline) was injected unilaterally into the parietal cortex, 2.5 mm down from the surface of the head. Six or 12 hr later, animals were anesthetized with avertin, brains were removed, and parietal cortex samples from both hemispheres were Dounce-homogenized together in 2 ml of 0.2% diethylamine (DEA) and 50 mm NaCl. Brain homogenates were centrifuged at 100,000 × g, and recovered supernatants were neutralized to pH 8.0 with 2 mTris-HCl. Extracts were diluted 1:1 with 5% fetal clonal serum (HyClone, Logan, UT) and 1% nonfat dry milk in TBS and analyzed for Aβ concentration using the ELISAs described below.
Aβ40- and 42-specific ELISAs. For the 40-specific ELISA, Fluoronunc plates (Nunc, Naperville, IL) were coated with goat anti-rabbit IgG at 1:300 in 0.1 m sodium bicarbonate. This was followed by an Aβ40-selective polyclonal antibody at 1:300 in 5% fetal clonal serum and 1% nonfat dry milk in TBS. The wells were blocked further in this same solution, without antibody. Tween 20 (0.1%) in TBS was used as the wash solution between each indicated step. Brain DEA extracts containing Aβ were diluted 1:1 in blocking solution and applied to the plates overnight at 4°C. Aβ was detected using biotinylated 6E10 at 1:5000 and avidin–alkaline phosphatase (Cappel/ICN, Costa Mesa, CA) at 1:500. Bound phosphatase was detected using 4-methyl umbelliferyl phosphate (4-MeUP; Sigma, St. Louis, MO) and read at 360/460 nm. Specificity of this ELISA for Aβ1–40 was tested using recombinant C100 (comprising the last 100 amino acid residues of APP; Savage et al., 1994), Aβ1–42, or Aβ1–43 (Bachem, King of Prussia, PA). These proteins were added to standard curves comprising Aβ1–40 (Bachem), and their ability to alter the curve was determined. ELISA signals are reported as femtomoles of Aβ per milligram of total extracted protein based on Aβ standard curves generated in each experiment.
The design of the 42-selective ELISA was the same, except the capture antibody was 6E10 and the detecting antibody was selective for Aβ42. Fluoronunc plates were coated with goat anti-mouse IgG1 as described above. Monoclonal antibody 6E10 was used at 1:1000 in 5% fetal clonal serum and 1% nonfat dry milk, followed by an additional block in 5% nonfat dry milk in TBS. Intermediate washes were as above. Brain DEA extracts were diluted in blocking solution and applied for overnight capture at 4°C. Aβ42 was detected using the 42-specific polyclonal antibody at 1:200, followed by a goat anti-rabbit IgG–alkaline phosphatase conjugate (Southern Biotechnology Associates, Birmingham, AL) at 1:5000. 4-MeUP substrate was used as above. Specificity of this ELISA for Aβ was tested by comparing signals generated with Aβ1–40, Aβ1–43, and recombinant C100 with signals generated using Aβ1–42.
Because C99 and APP could also bind to the 6E10 capture antibody used in the 42-specific ELISA and are present at much higher concentrations than Aβ42 in the mouse brain, we examined DEA extracts for the presence of C99 and APP. Neutralized DEA extracts were adjusted to 1× radioimmunoprecipitation assay buffer (50 mm Tris base, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 2 mm EDTA, and 1 mm benzamidine) and immunoprecipitated with Ab 97 and Pansorbin overnight at 4°C. Some samples were spiked with 5 ng of recombinant C100. Precipitated proteins were electrophoresed on 10–20% Tris-tricine gels or 4–20% Laemmli gels (both from Owl Scientific) to visualize C99 or APP, respectively. Proteins were immunoblotted and detected using 6E10 and ECL as above.
Detection of APPα and APPβ. From the DEA extracts described above, 250 μg aliquots of protein from each sample were methanol-precipitated for 1–2 hr at −20°C. Precipitates were air-dried and resuspended in equal volumes of Laemmli sample buffer. Aliquots of each sample (50 μg) were electrophoresed in triplicate using 4–20% Laemmli gels and transferred to nitrocellulose. Membranes were blocked using 5% nonfat milk powder in TBS and incubated with either 6E10 at 1:2000 or 54 at 1:500 for detection of APPα or APPβ, respectively. Relative band intensity (visualized using ECL as described above) was determined using a Docugel V Scanalytic system (CSP, Inc., Billerica, MA). As a control for protein loading and transfer variabilities between samples, immunoblots were stripped using 100 mm β-mercaptoethanol and 2% SDS at 37°C for 1 hr and reblotted using a monoclonal anti-actin antibody at 1:2000. Band densities representing secreted APP fragments were then normalized to the actin signal densities contained within each respective sample.
RESULTS
Radiolabeling in vivo
We have studied the metabolism of C99, Aβ, and APP in vivo using infusion of [35S]methionine into the femoral vein of the gene-targeted mouse. The turnover of all examined species occurs within 1 d. Identity of these three proteins (Figs. 1,2) is confirmed by molecular weight and reactivity with two Aβ/APP-specific antibodies (1153 and 6E10), one of which is specific for human APP and derivatives (6E10). Radiolabeled proteins immunoprecipitated with 1153 migrated between 3.5 and 6 kDa (Fig. 1A), at 14 kDa (Fig.1A), or between 97 and 200 kDa (Fig.2A) and aligned precisely with proteins immunodetected with 6E10 (Figs. 1D,E,2D). These proteins are, therefore, identified as Aβ, C99, and APP, respectively. All three immunoblotted proteins also co-electrophoresed with APP-related standards (APP, Fig.2E; Aβ and C99, data not shown).
Fig. 1.

Time-dependent changes in specific activity of mouse brain Aβ and C99. [35S]Methionine (500 μCi) was infused into the femoral vein of gene-targeted mice. At indicated hourly time points after midpoint of infusion, mice were perfused with Ringer’s solution, and brain APP fragments were isolated by immunoprecipitation and visualized using electrophoresis and exposure of resolved proteins to phosphorimage screens.A, Representative phosphorimage showing radiolabeled Aβ and C99. Graphs illustrating change in density of C99 (B) or Aβ (C) with time are shown. n = 3 at each time point. D, E, Representative immunoblots confirming equivalent absolute levels of Aβ and C99 during these experiments. Immobilized proteins used to obtain the phosphorimage in A were detected using antibody 6E10. D, Aβ; E, C99 and Aβ (from a longer exposure of D).
Fig. 2.

Change in specific activity of mouse brain APP with time. Mice were treated as reported in the legend to Figure 1 and Materials and Methods. A, Representative phosphorimage showing radiolabeled immature (i) and fully glycosylated (m) APP695. B, C, Time-dependent change in specific activity of immature (B) and fully glycosylated APP695 (C). n = 3 at each time point. D, Representative Western blot showing relatively constant absolute levels of APP, although specific activity was changing. E, Immunoblot confirming the predominant forms of APP synthesized by gene-targeted mouse. The most prominent band in the gene-targeted mouse brain sample (lane 2) co-electrophoreses with immature human APP695 from transgenic rat brain (lane 1) and lowest APP form isolated from human cortex (lane 3). The top band in the full-length APP complex (lane 2) co-electrophoreses with fully glycosylated human APP 695 from transgenic rat (lane 1).
C99 achieved its maximal observed specific activity between 1 and 2 hr after infusion (Fig. 1A). Thet½ estimate based on decay of C99 signal over time was ∼3 hr (Fig. 1B). Peak specific activity of the Aβ peptide was achieved by 4–7 hr after infusion with the signal returning to baseline levels by 16 hr (Fig.1A,C). Because there were no data points on the downward slope of the Aβ decay curve, an approximatet½ for Aβ is best estimated from its rate of formation, because, at steady-state, the rate of protein synthesis equals the rate of turnover. The specific activity of the Aβ protein increased from background at 2 hr after infusion to peak incorporation between 4 and 7 hr (i.e., 2–5 hr later). Therefore, we estimate the t½ to be between 1.0 and 2.5 hr. The coincident decay of C99 and formation of Aβ in vivo supports the precursor–product relationship of these two APP derivatives, respectively (Golde et al., 1992; Cai et al., 1993; Perez et al., 1996). To determine absolute levels of these fragments during the course of the experiment, and also to confirm their identities, immunoblotting was performed using 6E10 (Fig. 1D,E). Absolute levels of Aβ and C99 were relatively constant at each time point, whereas their specific activities were changing. This confirms that the turnover measurements were performed under steady-state conditions. A radiolabeled protein that migrated slightly slower than C99 (Fig. 1A) was not labeled with 6E10 (Fig.1E). This protein, therefore, was not derived from APP and was nonspecifically precipitated.
Full-length APP was immunoprecipitated using the Cterminal-specific Ab 97. Two major proteins were evident on both the phosphorimage (Fig. 2A) and immunoblot with 6E10 (Fig. 2D). On both images, the lowerMr band was sharply focused, and the upper band was more diffuse, as is typically seen with mature, fully glycosylated APPs (e.g., Oltersdorf et al., 1990). Most of the APP made in the brains of our gene-targeted mice (Fig. 2E) is APP695, as expected for rodent brain (Rockenstein et al., 1995). The major proteins immunoprecipitated with Ab 97 and immunodetected with 6E10 electrophoresed precisely with both of the major APP forms extracted from the brain of a transgenic rat overexpressing human APP695. In addition, the lowest Mr band detected in the gene-targeted brains co-electrophoresed with the lowest band isolated from human cortex, which is immature APP695. APP-like proteins (APLPs) are also present in mouse brain (Wasco et al., 1992) and could contribute to the phosphorimage signals. This is unlikely, because proteins seen on the Western blot using 6E10 (which does not recognize APLPs) precisely co-electrophorese with the radiolabeled proteins immunoprecipitated with Ab97.
Immature APP695 attained its peak specific activity by 1 hr (Fig.2B), and the t½ of this material appeared to be ∼3 hr. The specific activity of mature APP695 peaked between 1 and 2 hr and fell to background by 16 hr (Fig.2C). Therefore, the t½was ∼7 hr.
ELISAs detect Aβ40 and Aβ42 in brains of gene-targeted mice
Using sandwich ELISAs selective for Aβ peptides with C termini ending at residue 40 or 42, Aβ was detected in brain extracts. The 40-selective assay (Fig. 3A) was linear to 240 fmol/ml and sensitive to 12 fmol/ml. This assay was >1000-, 5000-, or 10,000-fold more selective for Aβ1–40 compared with Aβ1–43, C100, or Aβ1–42 standards, respectively. Brain Aβ40 levels measured in the homozygous, gene-targeted mouse brain with this ELISA are 120 fmol/mg protein.
Fig. 3.

ELISAs selective for Aβ40 and Aβ42 detect levels of endogenous Aβ, correctly reflecting gene dosage.A, Standard curve of Aβ1–40 generated using ELISA with a polyclonal antibody selective for Aβ40. B,Standard curve of Aβ1–42 using 42-selective ELISA. C,Soluble Aβ extracted from brains of gene-targeted mice having one copy (white bars) or two copies (black bars) of the targeted allele. n = 3 in each group. D, Immunoblot showing that the DEA extraction method does not release detectable levels of C99, an abundant, membrane-spanning form of APP. Lanes 2–4, Extracts immunoprecipitated with antibody 97, which recognizes the C terminus of C99; lane 1, recombinant C100 added into DEA extract before immunoprecipitation; lane 5, 0.5 ng of C100 loaded directly on to the gel.
The 42-selective assay (Fig. 3B) was linear to 480 fmol/ml and sensitive to 7.4 fmol/ml. This assay was 400-, 10,000-, or 16,000-fold more selective for Aβ1–42 compared with Aβ1–43, Aβ1–40, or C100 standards, respectively. Selectivity in the Aβ42 ELISA resided in the detecting antibody, unlike the 40-specific ELISA. Because the DEA extracts contain little membrane-associated C99 (Fig.3D) or APP (data not shown), the nonselective 6E10 capture reagent was not saturated by C99 or full-length APP and was therefore free to capture DEA-extracted Aβ species. Whole-brain Aβ42 levels measured using this ELISA are 16 fmol/mg protein. Both ELISAs also responded appropriately to twofold differences in brain Aβ driven by gene dosage. Extracts from homozygous mice revealed a twofold higher signal in the ELISA compared with heterozygous mice with only one copy of the targeted allele (Fig. 3C), confirming a previous observation made using Western blotting (Reaume et al., 1996).
Phorbol ester reduces cortical Aβ and APPβ levels acutely
Because the Aβ protein is cleared within a few hours of its synthesis in the mouse brain, we examined the acute effect of the phorbol ester PMA on the level of Aβ and other APP derivatives. Highly significant 30–35% reductions in levels of Aβ40 (from 121.4 to 84.1 fmol/mg) and Aβ42 (from 20 to 12.9 fmol/mg) were seen in parietal cortex 6 hr after intracortical injection of 40 nmol of PMA (Fig. 4; p < 0.0001). By 12 hr, the effect on Aβ40 was lost (Fig.5A). Aβ42 levels were not examined at 12 hr. Intracortical injection of 40 nmol of 4αPMA, an analog of PMA unable to activate PKC (Castagna et al., 1982; Kikkawa et al., 1983; Nichols et al., 1987), failed to reduce Aβ40 levels at 40 nmol 6 hr after injection (Fig. 5B). The hippocampus was also examined and was inconsistently affected by PMA injection (data not shown). It is possible that this highly lipophilic compound did not travel a significant distance from the injection site.
Fig. 4.

Effect of PMA on levels of Aβ40 and Aβ42. Aβ proteins were significantly reduced 6 hr after treatment. PMA was injected into the parietal cortex of the gene-targeted mouse. Parietal cortex was removed, and Aβ was extracted using DEA–NaCl buffer, neutralized to pH 8.0, and analyzed by ELISA; vehicle values are inblack, and PMA values are in white. Aβ40 and Aβ42 were reduced by 31 (p < 0.0001) and 35% (p < 0.0001), respectively. Number of animals per group: 40 assay, vehicle, 30; PMA, 39; 42 assay, vehicle, 24; PMA, 33.
Fig. 5.
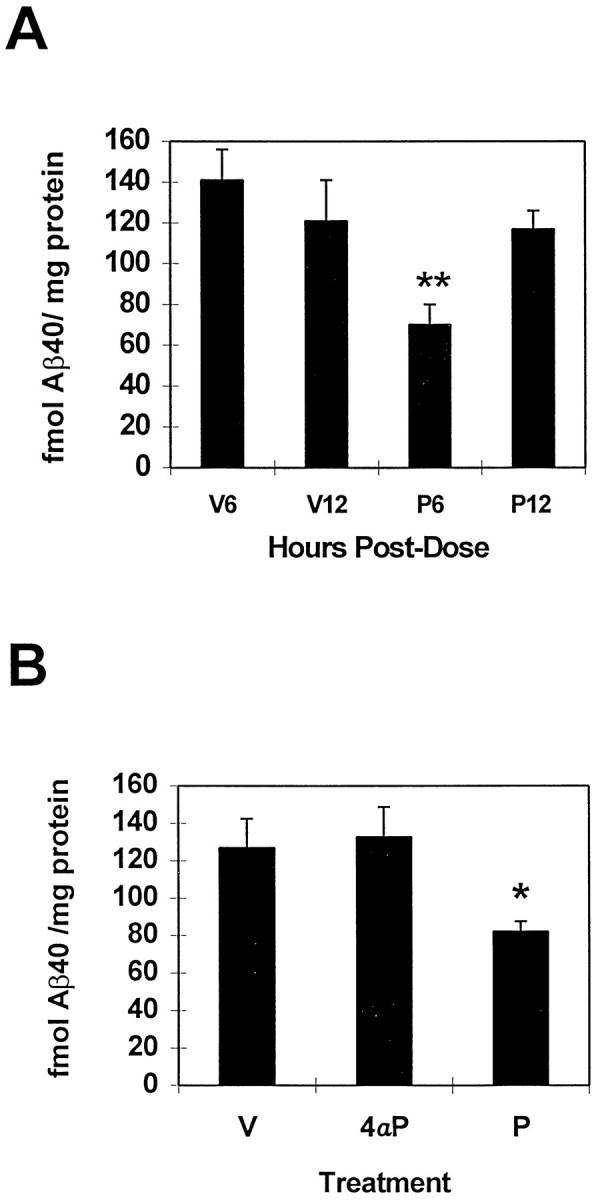
Time-dependent reduction in Aβ levels after PMA treatment. Aβ levels were unaffected by 4αPMA. A,Significant reductions in Aβ40 at 6 hr (P6,p < 0.001) were gone by 12 hr (P12). V, Vehicle; P, PMA. Number of animals per group: V6, 6; V12, 6; P6, 11; P12, 12. B,4αPMA (4αP) did not lower cortical Aβ40 levels after 6 hr compared with the active analog PMA (P, p < 0.03). Number of animals per group: V, 6; 4αP, 7;P, 6.
APPα and APPβ levels were also examined 6 hr after PMA injection from the same extracts used to measure Aβ. In contrast to expectations from cell culture experiments, APPα levels were unchanged by this compound (Fig.6A). APPβ levels, on the other hand, were significantly reduced by 32% in the PMA-treated group (p < 0.02) (Fig. 6B). The degree of difference in levels of the two forms of secreted APP compared with controls was essentially identical whether the values were normalized to the actin band subsequently visualized within each blotted sample.
Fig. 6.

Effect of PMA on levels of APPα and APPβ in parietal cortex 6 hr after treatment. Cortical APPβ levels were significantly reduced by PMA treatment, whereas APPα levels are unchanged. These APP fragments were assayed from the DEA extracts used to measure Aβ. Equivalent amounts of protein were MeOH-precipitated and immunoblotted with either 6E10 (A) to visualize APPα or 54 (B) to visualize APPβ. The reduction of APPβ is significant to p < 0.02. Number of animals per group: vehicle, 19; PMA, 23.
DISCUSSION
We examined the synthesis and turnover of human Aβ present within the gene-targeted mouse brain and used this information to design studies aimed at the reduction of brain Aβ. Under the control of the mouse APP promoter, this model provided appropriate cellular production of human Aβ and allowed the study of Aβ and APP metabolism under physiologically relevant APP concentrations. Aβ, C99, and APP undergo virtually complete clearance within 1 d in this model. The estimated t½ for Aβ is between 1.0 and 2.5 hr and was estimated from the rate of synthesis, because, at steady-state levels of protein, the rate of synthesis equals the rate of turnover. In addition to metabolic turnover of Aβ, rapid transport of this protein out of the brain into either blood or CSF could also contribute to its clearance. Although Aβ transport into brain across the blood–brain barrier has been reported (Zlokovic et al., 1993), transport of Aβ out of brain parenchyma has not yet been demonstrated.
Western blot data show steady-state levels of all APP-related proteins over the course of the experiment. Thet½ values predicted here for immature APP (3 hr), fully glycosylated APP (7 hr), and C99 (3 hr) were based on turnover of those molecules that acquired [S35]methionine during the original pulse and via reuse of label released from rapidly metabolized proteins. Rates of synthesis for C99 and APP could not be used to obtain more precise estimates of t½, because the specific activity of both forms of APP and C99 had already peaked at the earliest time examined. A more precise definition of the half-lives of these proteins based on the rate of synthesis in vivo would require a shorter pulse duration and the measurement of points between time 0 and 1 hr.
We have shown that peripheral infusion of [35S]methionine over 30 min is sufficient to visualize Aβ and its precursors in mouse brain, despite the presence of only one methionine per Aβ molecule, and a background of physiological levels of unlabeled methionine. In studies that examined turnover of total protein in brain using TCA precipitation, there was concern that a stable pool of radiolabeled precursor be maintained to act as a saturating pulse (Garlick and Marshall, 1972; Dunlop et al., 1975; Reith et al., 1978). In these experiments, very long infusion periods or large bolus injections of radiolabel were delivered to saturate the precursor pools. Continuous infusion of [35S]methionine and other radiolabeled amino acids directly into small subregions of the brain has also been used to study the synthesis and transport of substance P (Sperk and Singer, 1982;Torrens et al., 1982; Krause et al., 1984) and other neuropeptides (Kochman et al., 1982; Krause et al., 1982). This report demonstrates whole-brain synthesis and degradation of specific proteins using peripheral infusion of radiolabeled amino acid over a relatively short infusion interval.
The synthesis and turnover of APP and APP fragments have been studied extensively in vitro (Weidemann et al., 1989; Oltersdorf et al., 1990; Haass et al., 1991; Golde et al., 1992; Busciglio et al., 1993; Siman et al., 1993; Perez et al., 1996). Pulse–chase experiments using [35S]methionine showed processing of APP to fully glycosylated, proteolytically cleaved forms within 1 hr in non-neuronal cells (Oltersdorf et al., 1990; Haass et al., 1991; Golde et al., 1992;Busciglio et al., 1993; Siman et al., 1993). In contrast, the half-life of immature APP was 3 hr in the neuronal NT2N cell line (Wertkin et al., 1993), identical to the half-life we estimate in vivo. The gene-targeted mice have the Swedish FAD mutation within the context of mouse APP, unlike the wild-type, human APP present in NT2N cells. When turnover rates of wild-type versus Swedish mutant APP have been compared in vitro, they were found to be identical (Perez et al., 1996). Therefore, the turnover rate estimate of the APP present in the gene-targeted mice likely reflects the in vivo turnover of wild-type APP as well.
The turnover of C-terminal derivatives measured in various cell culture systems has ranged from 1–2 hr to >8 hr (Oltersdorf et al., 1990;Haass et al., 1991; Estus et al., 1992; Busciglio et al., 1993; Siman et al., 1993; Martin et al., 1995). Our estimate of thet½ of C99 in vivo from expression of the Swedish mutant transgene falls within that range. The cellular compartment in which β-secretase activity generates the N terminus of Aβ differs for APPs containing the Swedish FAD mutation compared with wild-type forms (Haass et al., 1995; Thinakaran et al., 1996). A 13.5 kDa fragment (analogous to C99 here) appeared more rapidly in mouse N2a cells transfected with Swedish mutant APP than wild-type APP (Thinakaran et al., 1996). Therefore, the rate of formation of C99 may also be more rapid in our gene-targeted mouse system expressing Swedish mutant APP compared with wild-type APP.
Aβ turnover in vitro has been studied recently (Naidu et al., 1995; Qiu et al., 1996, 1997). The peak specific activity of radiolabeled Aβ secreted from Chinese hamster ovary (CHO) cells transfected to overexpress APP 695 occurred at 6 hr (Naidu et al., 1995), similar to the result in our gene-targeted system. Estimates of Aβ t½ values are also comparable between 1.5 and 3 hr. Similar rates of Aβ secretion have been found using CHO cells expressing either wild-type or Swedish mutant forms of APP, whereas the amount of Aβ secreted was elevated in the presence of the mutation (Perez et al., 1996). We predict that turnover rates of Aβ determined in this study will estimate the rates for Aβ generated from wild-type APP if the compartments in which Aβ is metabolized are the same, and Aβ turnover mechanisms are not saturated.
Specific proteases that metabolize Aβ are beginning to be identified. A serine protease–α2-macroglobulin complex was identified in preparations of pancreatic trypsin or fetal bovine serum that degraded exogenous Aβ, as well as Aβ secreted by cells (Qiu et al., 1996). A nonmatrix metalloprotease and a serine protease secreted by both CHO cells and BV-2 microglial cells have also been reported to catabolize Aβ (Qiu et al., 1997). Microglial cells in brain may, therefore, secrete a protease important for Aβ turnover in vivo. Turnover of Aβ could also occur in an intracellular compartment, because Aβ42 uptake from extracellular medium has been described (Knauer et al., 1992); also, the de novo synthesis of intracellular Aβ has been reported in primary neurons (Tienari et al., 1997), NT2N cells (Wertkin et al., 1993), and 293 or CHO cells transfected with Swedish mutant APP (Martin et al., 1995, Perez et al., 1996). Rates of Aβ synthesis and clearance determine its steady-state level and dysfunction of either process could lead to the elevation of Aβ to concentrations critical for fibril formation.
Using this information concerning the turnover rate of Aβ in mouse brain, we investigated the effects of intracortically injected phorbol esters on brain Aβ metabolism. The development of these gene-targeted mice was critical to our study because Aβ is undetectable in wild-type mouse brain with either our ELISA or immunoblotting methods. A highly significant 30–35% reduction was demonstrated in levels of both Aβ40 and Aβ42 6 hr after administration of PMA. Six hours was chosen as the earliest postdose interval after PMA, allowing 2–6 half-lives for Aβ clearance. This is the first report of the selective effects of PMA on Aβ40 and Aβ42 as measured by ELISA. For the elevation of PKC activity to be considered a therapeutically relevent approach for the treatment of AD, it was necessary to demonstrate reduction in levels of brain Aβ42 with a compound known to impact this pathway. The selective effect of phorbol ester stimulation on Aβ42 was examined previously using an in vitro system, whereas Aβ40 was measured as part of the “total” Aβ that remained (Citron et al., 1996). In our study, cortical levels of Aβ40 returned to baseline by 12 hr, possibly reflecting the half-life of PMA in mouse brain, which was determined to be 9.6 hr (Dietrich et al., 1989). We expect the Aβ42 followed a similar time course of recovery, although this fragment was not measured at 12 hr.
The activity of PMA on APP processing in mouse brain is presumably through its selective PKC-stimulatory function (Newton, 1995;Nishizuka, 1995), because an analog that is unable to activate PKC, 4α-PMA, had no effect on Aβ levels. We were unable to measure changes in PKC translocation after PMA delivery in mouse brain, because the DEA extraction buffer led to the release of membrane-associated PKC (data not shown). Therefore, it was not possible to measure concomitant effects of PMA on Aβ levels and PKC activity.
The role of PKC in modulating APP processing was determined by the discovery that stimulation of PKC either directly (Caporaso et al., 1992) or by activation of muscarinic receptor subtypes linked to this second messenger pathway (Buxbaum et al., 1992; Nitsch et al., 1992;Farber et al., 1995), led to an increase in the secretion of APPα and concomitant reduction in both Aβ (Buxbaum et al., 1993; Hung et al., 1993) and APPβ (Felsenstein et al., 1994; Jacobsen et al., 1994). These results suggested either increased processing of APP via an α-secretase-mediated pathway (by increased secretase activity or a rerouting of full-length APP to compartments involved in α-secretase processing) or decreased activity via a β-secretase-mediated pathway. We chose to study the effect of a phorbol ester in vivo, because PKC itself is ubiquitously expressed, whereas select cell surface receptors linked to PKC have a more focal distribution.
In contrast to Aβ, APPα levels were unchanged 6 hr after PMA delivery, whereas APPβ levels decreased significantly. This correlation of Aβ reduction with APPβ reduction suggests that there is reduced cleavage of APP by β-secretase in the presence of phorbol esters, as suggested previously (Buxbaum et al., 1993; Hung et al., 1993; Felsenstein et al., 1994; Jacobsen et al., 1994). The lack of significant elevation in levels of APPα after phorbol ester treatmentin vivo could be attributed to more efficient turnover of this protein compared with in vitro systems or, alternatively, to a dissociation of systems regulating Aβ/APPβ secretion and APPα secretion in the mouse brain. These processes have been reported to be dissociated in certain in vitro systems (Gabuzda et al., 1993; Dyrks et al., 1994; Fuller et al., 1995). Recently, however, increased APPα secretion was reported in a rat model in which PKC activity was constitutively upregulated after treatment with menthylazoxymethanol in utero (Caputi et al., 1997), supporting the influence of this pathway on APP processingin vivo. Together, these studies emphasize the importance of measuring all APP fragments of interest when testing pharmacological modulators of APP processing in brain. In drug discovery efforts, for example, the degree to which a compound is effective in lowering Aβ levels should not be based exclusively on the measurement of a surrogate marker, such as APPα. In addition, test systems must effectively model both synthesis and clearance of brain Aβ and other APP-processing fragments; cultured cells may only partially represent these processes.
Collectively, our data show that the gene-targeted mouse is a useful model for the study of agents that modulate Aβ levels in brain. Turnover of brain Aβ protein under physiological conditions occurs within several hours. This makes possible the design of further studies to investigate agents that modulate Aβ levels in vivowhether by modulation of PKC activity or by alternative pathways independent of PKC.
Footnotes
We thank Drs. Matthew Miller, Bruce Jones, and Dorothy Flood of Cephalon and Dr. Jim Krause of Washington University for discussions that helped initiate this work, Thomas Emmons for help with preparation of figures, Renee Simmons and Edwin McCabe for their excellent care of the gene-targeted animals, and Drs. Frank Baldino and Jeffry Vaught for their continuing support of this work.
Correspondence should be addressed to Dr. Mary Savage, Cephalon, Inc., 145 Brandywine Parkway, West Chester, PA 19380.
REFERENCES
- 1.Baranyi A, Szente MB, Woody CD. Intracellular injection of phorbol ester increases the excitability of neurons of the motor cortex of awake cats. Brain Res. 1987;424:396–401. doi: 10.1016/0006-8993(87)91486-7. [DOI] [PubMed] [Google Scholar]
- 2.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of β-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer β/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid β/A4 peptide. Proc Natl Acad Sci USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai XD, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 6.Caporaso GL, Gandy SE, Buxbaum JD, Ramabhadran TV, Greengard P. Protein phosphorylation regulates secretion of Alzheimer β/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:3055–3059. doi: 10.1073/pnas.89.7.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caputi A, Barindelli S, Pastorino L, Cimino M, Buxbaum JD, Cattabeni F, Di Luca M. Increased secretion of the amino-terminal fragment of amyloid precursor protein in brains of rats with a constitutive upregulation of protein kinase C. J Neurochem. 1997;68:2523–2529. doi: 10.1046/j.1471-4159.1997.68062523.x. [DOI] [PubMed] [Google Scholar]
- 8.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 9.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 10.Citron M, Vigo-Pelfrey C, Teplow DB, Miller C, Schenk D, Johnston J, Winblad B, Venizelos N, Lannfelt L, Selkoe DJ. Excessive production of amyloid β-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc Natl Acad Sci USA. 1994;91:11993–11997. doi: 10.1073/pnas.91.25.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid β protein are generated from the β-amyloid precursor protein by different protease activities. Proc Natl Acad Sci USA. 1996;93:13170–13175. doi: 10.1073/pnas.93.23.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cope FO, Conrad EA, Staller JM, Boutwell RK. Induction of mouse brain ornithine decarboxylase by 12-O-tetradecanoylphorbol-13-acetate is independent of TPA receptor concentration. Cancer Lett. 1984;23:331–342. doi: 10.1016/0304-3835(84)90101-0. [DOI] [PubMed] [Google Scholar]
- 13.Deitrich RA, Bludeau PA, Baker RC. Investigations of the role of protein kinase C in the acute sedative effects of ethanol. Alcohol Clin Exp Res. 1989;13:737–745. doi: 10.1111/j.1530-0277.1989.tb00413.x. [DOI] [PubMed] [Google Scholar]
- 14.Dunlop DS, van Elden W, Lajtha A. A method for measuring brain protein synthesis rates in young and adult rats. J Neurochem. 1975;24:337–344. doi: 10.1111/j.1471-4159.1975.tb11885.x. [DOI] [PubMed] [Google Scholar]
- 15.Dyrks T, Mönning U, Beyreuther K, Turner J. Amyloid protein precursor secretion and βA4 amyloid generation are not mutually exclusive. FEBS Lett. 1994;349:210–214. doi: 10.1016/0014-5793(94)00671-7. [DOI] [PubMed] [Google Scholar]
- 16.Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu X, Tabira T, Greenberg BD, Younkin SG. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992;255:726–728. doi: 10.1126/science.1738846. [DOI] [PubMed] [Google Scholar]
- 17.Farber SA, Nitsch RM, Schulz JG, Wurtman RJ. Regulated secretion of β-amyloid precursor protein in rat brain. J Neurosci. 1995;15:7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Felsenstein KM, Ingalls KM, Hunihan LW, Roberts SB. Reversal of the Swedish familial Alzheimer’s disease mutant phenotype in cultured cells treated with phorbol 12, 13-dibutyrate. Neurosci Lett. 1994;174:173–176. doi: 10.1016/0304-3940(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 19.Fuller SJ, Storey E, Li QX, Smith AI, Beyreuther K, Masters C. Intracellular production of βA4 amyloid of Alzheimer’s disease: modulation by phosphoramidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry. 1995;34:8091–8098. doi: 10.1021/bi00025a015. [DOI] [PubMed] [Google Scholar]
- 20.Gabuzda D, Busciglio J, Yankner BA. Inhibition of β-amyloid production by activation of protein kinase C. J Neurochem. 1993;61:2326–2329. doi: 10.1111/j.1471-4159.1993.tb07479.x. [DOI] [PubMed] [Google Scholar]
- 21.Garlick PJ, Marshall I. A technique for measuring brain protein synthesis. J Neurochem. 1972;19:577–583. doi: 10.1111/j.1471-4159.1972.tb01375.x. [DOI] [PubMed] [Google Scholar]
- 22.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 23.Haass C, Hung AY, Selkoe DJ. Processing of β-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostazewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 25.Haass C, Capell A, Citron M, Teplow DB, Selkoe DJ. The vacuolar H+-ATPase inhibitor bafilomycin A1 differentially affects proteolytic processing of mutant and wild-type β-amyloid precursor protein. J Biol Chem. 1995;270:6186–6192. doi: 10.1074/jbc.270.11.6186. [DOI] [PubMed] [Google Scholar]
- 26.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 27.Hung AY, Haass C, Nitsch RM, Qui WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid β-protein. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- 28.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42/43 and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42/43. Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 29.Jacobsen JS, Spruyt MA, Brown AM, Sahasrabudhe SR, Blume AJ, Vitek MP, Muenkel HA, Sonnenberg-Reines J. The release of Alzheimer’s disease β amyloid peptide is reduced by phorbol treatment. J Biol Chem. 1994;269:8376–8382. [PubMed] [Google Scholar]
- 30.Jarrett JT, Berger EP, Lansbury PT. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 31.Kikkawa U, Takai Y, Miyake R, Nishizuka Y. Protein kinase C as a possible receptor protein for tumor-promoting phorbol esters. J Biol Chem. 1983;258:11442–11445. [PubMed] [Google Scholar]
- 32.Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/β protein. Proc Natl Acad Sci USA. 1992;89:7437–7441. doi: 10.1073/pnas.89.16.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kochman K, Kerdelhue B, Ostrowska A, Chomike L, Domanski E, Justisz M. Biosynthesis, in vivo, of gonadotropin-releasing hormone in the hypothalamus of normal and ovariectomized female rats. Mol Cell Endocrinol. 1982;25:193–198. doi: 10.1016/0303-7207(82)90052-1. [DOI] [PubMed] [Google Scholar]
- 34.Krause JE, Advis JP, McKelvy JF. In vivo biosynthesis of hypothalamic luteinizing hormone releasing hormone in individual free-running female rats. Endocrinology. 1982;111:344–346. doi: 10.1210/endo-111-1-344. [DOI] [PubMed] [Google Scholar]
- 35.Krause JE, Reiner AJ, Advis JP, McKelvy JF. In vivo biosynthesis of [35S] and [3H]substance P in the striatum of the rat and their axonal transport to the substantia nigra. J Neurosci. 1984;4:775–785. doi: 10.1523/JNEUROSCI.04-03-00775.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin BL, Schrader-Fischer G, Busciglio J, Duke M, Paganetti P, Yankner BA. Intracellular accumulation of β-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J Biol Chem. 1995;270:26727–26730. doi: 10.1074/jbc.270.45.26727. [DOI] [PubMed] [Google Scholar]
- 37.Naidu A, Quon D, Cordell B. β-amyloid peptide produced in vitro is degraded by proteinases released by cultured cells. J Biol Chem. 1995;270:1369–1374. doi: 10.1074/jbc.270.3.1369. [DOI] [PubMed] [Google Scholar]
- 38.Newton A. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 39.Nichols RA, Haycock JW, Wang JKT, Greengard P. Phorbol ester enhancement of neurotransmitter release from rat brain synaptosomes. J Neurochem. 1987;48:615–621. doi: 10.1111/j.1471-4159.1987.tb04137.x. [DOI] [PubMed] [Google Scholar]
- 40.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 41.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid protein derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- 42.Oltersdorf T, Ward P, Henriksson T, Beattie EC, Neve R, Lieberburg I, Fritz LC. The Alzheimer amyloid precursor protein: identification of a stable intermediate in the biosynthetic/degradative pathway. J Biol Chem. 1990;265:4492–4497. [PubMed] [Google Scholar]
- 43.Perez RG, Squazzo SL, Koo EH. Enhanced release of amyloid β-protein from codon 670/671 “Swedish” mutant β-amyloid precursor protein occurs in both secretory and endocytic pathways. J Biol Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- 44.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu WQ, Borth W, Ye Z, Haass C, Teplow DB, Selkoe DJ. Degradation of amyloid β-protein by a serine protease-α2-macroglobulin complex. J Biol Chem. 1996;271:8443–8451. doi: 10.1074/jbc.271.14.8443. [DOI] [PubMed] [Google Scholar]
- 46.Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem. 1997;272:6641–6646. doi: 10.1074/jbc.272.10.6641. [DOI] [PubMed] [Google Scholar]
- 47.Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the β-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Aβ sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- 48.Reith MEA, Schotman P, Gispen WH. Measurements of in vivo rates of protein synthesis in brain, spinal cord, heart and liver of young versus adult rats, intact versus hypophysectomized rats. J Neurochem. 1978;30:587–594. doi: 10.1111/j.1471-4159.1978.tb07812.x. [DOI] [PubMed] [Google Scholar]
- 49.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 50.Routtenberg A, Colley P, Linden D, Lovinger D, Murakami K, Sheu FS. Phorbol ester promotes growth of synaptic plasticity. Brain Res. 1986;378:374–378. doi: 10.1016/0006-8993(86)90940-6. [DOI] [PubMed] [Google Scholar]
- 51.Savage MJ, Iqbal M, Loh T, Trusko S, Scott RW, Siman R. Cathepsin G: localization in human cerebral cortex and generation of amyloidogenic fragments from the β-amyloid precursor protein. Neurosci. 1994;60:607–619. doi: 10.1016/0306-4522(94)90490-1. [DOI] [PubMed] [Google Scholar]
- 52.Savage MJ, Kawooya JK, Pinsker LR, Emmons TL, Mistretta S, Siman R, Greenberg BD. Elevated Aβ levels in Alzheimer’s disease brain are associated with selective accumulation of Aβ42 in parenchymal amyloid plaques and both Aβ40 and Aβ42 in cerebrovascular deposits. Amyloid Int J Exp Clin Invest. 1995;2:234–240. [Google Scholar]
- 53.Selkoe DJ. Amyloid β-protein and the genetics of Alzheimer’s disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 54.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 55.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tinter R, Frangione B, Younkin SG. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 56.Siman R, Mistretta S, Durkin JT, Savage MJ, Loh T, Trusko S, Scott RW. Processing of the β-amyloid precursor: multiple proteases generate and degrade potentially amyloidogenic fragments. J Biol Chem. 1993;268:16602–16609. [PubMed] [Google Scholar]
- 57.Siman R, Durkin JT, Husten EJ, Savage MJ, Murthy S, Mistretta S, Chatterjee S, Dembofsky B, Poorman R, Greenberg BD. Genesis and degradation of Aβ protein by cultured human neuroblastoma cells. In: Iqbal K, Mortimer JA, Winblad B, Wisniewski HM, editors. Research advances in Alzheimer’s disease and related disorders. Wiley; Chichester, England: 1995. pp. 675–684. [Google Scholar]
- 58.Sperk G, Singer EA. In vivo synthesis of substance P in the corpus striatum of the rat and its transport to the substantia nigra. Brain Res. 1982;238:127–135. doi: 10.1016/0006-8993(82)90776-4. [DOI] [PubMed] [Google Scholar]
- 59.Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells: evidence that cleavage at the “β-secretase” site occurs in the Golgi apparatus. J Biol Chem. 1996;271:9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- 60.Tienari PJ, Ida N, Ikonen E, Simons M, Weidemann A, Multhaup G, Masters CL, Dotti CG, Beyreuther K. Intracellular and secreted Alzheimer β-amyloid species are generated by distinct mechanisms in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1997;94:4125–4130. doi: 10.1073/pnas.94.8.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torrens Y, Michelot R, Beaujouan JC, Glowinski J, Bockaert J. In vivo biosynthesis of 35S-substance P from 35S-methionine in the rat striatum and its transport to the substantia nigra. J Neurochem. 1982;38:1728–1734. doi: 10.1111/j.1471-4159.1982.tb06655.x. [DOI] [PubMed] [Google Scholar]
- 62.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of β- amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 63.Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid β protein precursor. Proc Natl Acad Sci USA. 1992;89:10758–10762. doi: 10.1073/pnas.89.22.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weidemann A, König G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 65.Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, Lee VM-Y. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular β-amyloid or A4 peptides. Proc Natl Acad Sci USA. 1993;90:9513–9517. doi: 10.1073/pnas.90.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamaguchi H, Sugihara S, Ishiguro K, Takashima A, Hirai S. Immunohistochemical analysis of COOH-termini of amyloid beta protein (Aβ) using end-specific antisera for Aβ40 and Aβ42 in Alzheimer’s disease and normal aging. Amyloid Int J Exp Clin Invest. 1995;2:7–16. [Google Scholar]
- 67.Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood-brain barrier transport of circulating Alzheimer’s amyloid β. Biochem Biophys Res Commun. 1993;197:1034–1040. doi: 10.1006/bbrc.1993.2582. [DOI] [PubMed] [Google Scholar]