

Abstract
Calcium ions play crucial roles in a large variety of cell functions. The recent proposal that changes in the intracellular calcium concentration ([Ca2+]i) in astrocytes underline a reciprocal communication system between neurons and astrocytes encourages the interest in the definition of the various components participating in this novel Ca2+signaling system. We investigate here whether functional voltage-operated calcium channels (Ca2+ VOCs), which are clearly expressed in cultured astrocytes, participate in the regulation of [Ca2+]i also in astrocytes in situ. Depolarization with 40–60 mm K+ was used to analyze the activity of Ca2+ VOCs in Indo-1-loaded astrocytes in acute slices from the visual cortex and the CA1 hippocampal region of developing rats. We demonstrate here that the depolarization-induced [Ca2+]i increases in astrocytes are solely attributed to the activation of metabotropic receptors by neurotransmitters, such as glutamate, released by synaptic terminals on depolarization. In fact, (1) the K+-induced [Ca2+]i increases in astrocyte [Ca2+]i were potently reduced by α-methyl-4-carboxyphenylglycine, a metabotropic glutamate receptor competitive inhibitor; (2) after emptying intracellular Ca2+ stores with cyclopiazonic acid, none of the astrocytes displayed a [Ca2+]iincrease on the depolarizing stimulus; and (3) after inhibiting neurotransmitter secretion in neurons by incubating the slices with tetanus neurotoxin, no [Ca2+]iincrease on K+ stimulation was observed in astrocytes. Finally, patch-clamp whole-cell recordings from hippocampal astrocytes in acute brain slices failed to reveal any voltage-dependent calcium currents. On the basis of these results, the various roles proposed for astrocyte Ca2+ VOCs in the CNS should be reconsidered.
Keywords: calcium channels, glutamate, astrocyte-neuron interactions, confocal microscope, patch-clamp, brain slices, rat
Numerous stimuli such as neurotransmitters, membrane depolarization, or mechanical stress elicit in astrocytes increases in the concentration of intracellular calcium ([Ca2+]i) that are quite variable in terms of amplitude and spatiotemporal organization: [Ca2+]i peaks and prolonged plateaus and oscillations (Cornell-Bell et al., 1990; Glaum et al., 1990; Jensen and Chiu, 1991; Pasti et al., 1995), often followed by propagating [Ca2+]i waves (Cornell-Bell et al., 1990; Charles et al., 1991). This complexity reflects the expression of a variety of molecules that control Ca2+ entry from the extracellular space as well as Ca2+ release from intracellular compartments (Berridge, 1993; Verkhratsky and Kettenmann, 1996). In particular, cultured astrocytes from different brain regions express a variety of ion channels, including both T- and L-type Ca2+ channels. The first evidence for Ca2+ channel activity in cultured astrocytes was obtained by the discovery of Ca2+-dependent action potential firing on intracellular current injections (MacVicar, 1984). The presence of voltage-operated Ca2+ channels (Ca2+ VOCs) was then demonstrated by recording L-type Ca2+ current sensitive to nifedipine (MacVicar and Tse, 1988). The expression of Ca2+channels in cultured astrocytes appears to depend either on the presence of neurons (Corvalan et al., 1990) or on treatments with agents that increased cAMP intracellular levels (MacVicar and Tse, 1988; Barres et al., 1989). Using classical Ca2+imaging techniques, [Ca2+]i elevations attributed to Ca2+ influx through Ca2+ VOCs were observed in astrocytes on a depolarizing stimulus (Jensen and Chiu, 1991; MacVicar et al., 1991). Although the evidence for the expression of Ca2+VOCs in cultured astrocytes is beyond any doubt, results obtained in culture do not necessarily represent faithfully the behavior of astrocytes in vivo. To address this criticism, several authors have studied this problem under conditions that more closely resemble the in vivo situation. Astrocytes acutely isolated from the optic nerve displayed Ca2+ currents typical of T- and L-type Ca2+ channel activity (Barres et al., 1990). Several authors also demonstrated that acutely dissociated astrocytes from the hippocampus (Duffy and MacVicar, 1994) as well as astrocytes from acute brain slices (Porter and McCarthy, 1995; Duffy and MacVicar, 1996), increased their [Ca2+]i on K+stimulation. This response was either inhibited (Duffy and MacVicar, 1996) or significantly reduced (Porter and McCarthy, 1995) by the Ca2+ channel antagonist verapamil.
Studies with sharp microelectrodes or patch-clamp techniques from astrocytes either acutely isolated or from brain slices failed, however, to reveal voltage-dependent Ca2+ currents (Duffy and MacVicar, 1994; Jabs et al., 1994; Kressin et al., 1995;Steinhäuser et al., 1994). These negative results have been interpreted as being attributable to an inadequate control of membrane potential, because these cells extensively communicate through gap junctions, thereby forming an electrotonically coupled network. The visualization of [Ca2+]i changes on a depolarizing stimulus and their inhibition by pharmacological tools remain the only results supporting an active role of Ca2+ VOCs in Ca2+ signaling of astrocytes in situ and, implicitly, the presence of Ca2+ VOCs in these cells. The expression of VOCs in astrocytes represents, therefore, an open question. Noteworthy, subpopulations of astrocytes probably exist in the CNS (McKhann et al., 1997). For example, a recent study performed in acute brain slices demonstrated the presence of Ca2+ currents in the so-called “complex” cells from the mouse hippocampus (Akopian et al., 1996). These cells were neither GFAP-positive nor dye-coupled and were therefore proposed to represent a distinct type of hippocampal glial cells, perhaps immature astrocytes (Kressin et al., 1994; Akopian et al., 1996). Given the potential importance of these channels in astrocyte function under physiological as well as pathological conditions, we addressed this problem in detail. A confocal fluorescence microscope and the Ca2+ indicator Indo-1 were used to follow the [Ca2+]ichanges in neurons and astrocytes from both hippocampal and cortical slices. In addition, the patch-clamp technique was applied to record possible voltage-activated calcium currents from astrocytes in situ as well as from astrocytes after detaching their cell body off from the slice by gentle withdrawal of the patch pipette. Our results indicate that the [Ca2+]iincrease observed in astrocytes in response to a depolarizing stimulus with high K+ is not caused by Ca2+ entry through VOCs but rather to Ca2+ release from intracellular Ca2+ stores after activation of G-protein-linked receptors by glutamate and possibly other neurotransmitters released from depolarized synaptic terminals.
MATERIALS AND METHODS
Slice preparation and dye loading. Transverse brain slices (150–250 μm) from both the visual cortex and the hippocampus were prepared from Wistar rats at postnatal days 5–18 (P5–P18) as described previously (Carmignoto and Vicini, 1992). After evidence that antioxidant agents can protect neurons from degeneration (Rice et al., 1994), the physiological saline for slice cutting was as follows (in mm): 120 NaCl, 3.1 KCl, 1.25 NaH2PO4, 25 NaHCO3, 4 dextrose, 2 MgCl2, 1 CaCl2, 2 Na-pyruvate, 0.5 myoinositol, and 0.1 ascorbic acid, pH 7.4, with 5% CO2 and 95% O2. After cutting, slices were allowed to recover for 10–15 min at 37°C in the physiological saline used for cutting. Dye loading was performed in the same physiological saline used for cutting supplemented with the cell-permeant Indo-1 AM (20 μm; Molecular Probes, Eugene, OR) and 0.02% pluronic acid at 37°C for 40–50 min under continuous influx of the gas mixture (5% CO2 and 95% O2). In several experiments, tetanus neurotoxin (TeNT, 100 μg/ml) was added for the entire time of the Indo-1 loading. Continuous mild stirring was found to be crucial for optimizing the loading of the dye. After this procedure, the loading of neurons was as good as that of astrocytes.
Image acquisition. After incubation with Indo-1 AM, slices were mounted in a chamber that was placed on the stage of a Nikon Diaphot 300 inverted microscope, equipped with a 40× water immersion objective (numerical aperture, 1.1) (Nikon) connected to a real-time confocal microscope (Nikon RCM8000). The 351 nm band of the argon ion laser was used for excitation, and the emitted light, separated into its two components (405 and 485 nm) by a dichroic mirror was collected by two separate photomultipliers. The ratio of the intensity of the light emitted at the two wavelengths (405 and 485 nm) was displayed as a pseudocolor scale. Images were acquired with a frame interval of 2 or 30 sec, and 16 images were averaged for each frame. Recording sessions were performed at room temperature (20–23°C). Slices were continuously perfused (3 ml/min) with physiological saline of the following composition (in mm): 120 NaCl, 3.1 KCl, 1.25 NaH2PO4, 25 NaHCO3, 5 dextrose, 1 MgCl2, and 2 CaCl2, pH 7.4, with 5% CO2 and 95% O2. The 405:485 nm emission ratio (R405:485) in basal conditions was observed to vary little in different cells. Occasionally, a slight decrease was observed in R405:485 basal levels. Indeed, prolonged UV irradiation of Indo-1 can cause overall photobleaching and conversion to a fluorescent, but Ca2+-insensitive, species (Scheenen et al., 1996). In several experiments, we used 100 μm Trolox, a vitamin E analog that inhibits formation of Indo-1 photodegradation products (Scheenen et al., 1996). No substantial differences were, however, observed in our conditions. The stimulation with high-K+ extracellular solution was obtained by isosmotic replacement of Na+ with K+.
Electrophysiological recordings. Standard procedures were used for pipette preparation and patch-clamp recording in the whole-cell configuration (Edwards et al., 1989; Carmignoto and Vicini, 1992). Acute hippocampal slices were continuously perfused (3–5 ml/min) with a bath solution of the following composition (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, to pH 7.4 with KOH. The solutions were continuously bubbled with 5% CO2 and 95% O2. Cells were viewed with an upright Zeiss Axioskop microscope equipped with differential interference contrast, Nomarski optics (UEM; Zeiss, Oberkochen, Germany), and an electrically insulated water immersion 40× objective with a long working distance (2 mm). For the investigation of calcium currents, we used an extracellular solution of the following composition (in mm): 135 tetraethylammonium chloride (TEA), 10 BaCl2, 5 glucose, and 10 HEPES, to pH 7.4 with tetraethyammonium hydroxide (TEAOH). With respect to Ca2+, Ba2+ slows the rate of channel inactivation and is more permeant than Ca2+ through most of Ca2+ channels, thus producing larger currents. The pipette solution contained (in mm): 60N-methyl-d-glucamine (NMDG)-Cl, 60 CsCl, 2 MgCl2, 20 TEA, 5 EGTA, 3.0 Na2-ATP, 0.2 Na2-GTP, and 10 HEPES, to pH 7.2 with TEAOH. In a number of experiments, 120 mm Cs-methanesulfonate replaced NMDGCl and CsCl. Lucifer yellow (LY; 0.1%) was included in the patch pipette. Recordings were performed in current and voltage clamp with a patch-clamp amplifier (EPC 7; List Electronics, Darmstadt, Germany), sampled at 5 or 10 kHz, filtered at 1.5 kHz (eight-pole low-pass Bessel filter; Frequency Devices, Haverhill, MA), and digitized by a Digidata 1200A interface. pCLAMP-6 software (Axon Instruments) was used for acquisition and analysis of data. Linear capacity and leakage currents were measured and subtracted during acquisition except in the experiments in which subtraction of the current traces recorded before and after slice perfusion with 100 μmCd2+ was applied off-line. Series resistance compensation (20–40%) was used to ameliorate the voltage-clamp control. To reduce access resistance, pipette resistance was normally <4 MΩ. In several experiments, after establishing the whole-cell configuration, astrocytes were detached from the slice by carefully withdrawing the patch pipette and applying a mild suction through the pipette. The possible presence of Ca2+ currents in three detached astrocytes was investigated.
Drugs. The excitatory amino acid receptor agents NMDA, α-methyl-4-carboxyphenylglycine (MCPG), 2-amino-5-phosphonopentanoic acid (d-AP-5), and 1-aminocyclo-pentane-1,3-dicarboxylic acid (t-ACPD) were from Tocris (Buckhurst Hill, UK); cyclopiazonic acid (CPA), tetrodotoxin (TTX), Trolox, lucifer yellow (dilithium salt), and verapamil were from Sigma (Milan, Italy). These compounds were dissolved in water, NaOH, or dimethylsulfoxide and diluted in the physiological saline used for recordings. Purified TeNT (Schiavo and Montecucco, 1995) was a gift from C. Montecucco, Department of Experimental Biomedical Sciences, University of Padova.
RESULTS
High-K+-induced [Ca2+]i increase in astrocytes
In a series of previous experiments aimed at clarifying the role of [Ca2+]i oscillations in astrocytes from acute brain slices, we observed that, although the application of a depolarizing stimulus such as 40–60 mm KCl resulted in a rise of the [Ca2+]i in astrocytes, this increase was delayed several seconds with respect to that in neurons. We described previously the experimental approach that allows us to identify astrocytes and neurons in situ on the basis of their morphological, electrophysiological, immunocytological, and pharmacological criteria (Pasti et al., 1997) and demonstrated that the different kinetics of the response to K+ stimulation is a characteristic feature of the astrocyte and can thus be used as a diagnostic tool to distinguish neurons and astrocytes in situ. The sequence of pseudocolor images of Figure1A illustrates the typical behavior of the [Ca2+]i change in neurons and astrocytes from the hippocampal CA1 region on perfusion with 40 mm KCl, as revealed by the fluorescent indicator Indo-1. Whereas pyramidal neurons, cells marked with closed arrowheads in Figure 1Aa, displayed a prompt [Ca2+]i increase on depolarization with high extracellular K+, astrocytes, cells marked with open arrowheads, initially failed to respond but displayed a significant [Ca2+]iincrease several seconds after that of pyramidal neurons (Fig.1A). In two experiments, cells were followed for at least 10 min, but no further delayed responses were observed in astrocytes. The same pattern of responses was observed in electrophysiologically classified astrocytes and neurons loaded with Indo-1 through the patch pipette (Pasti et al., 1997). Given that the kinetics of the astrocyte [Ca2+]iincrease on high K+ stimulation (Fig.1B) is hardly compatible with a participation of Ca2+ VOCs, what might be a plausible explanation for this delayed response? We hypothesized that it could be, to a large extent, attributable to the release of glutamate and/or other neurotransmitters by depolarized synaptic terminals and to the subsequent activation of metabotropic receptors linked to inositol trisphosphate (IP3) production. This event may also account for the biphasic response in neurons (Fig.1A,B). Noteworthy, the pattern of [Ca2+]i changes in neurons and astrocytes on high K+ stimulation described above was observed to be qualitatively similar in slices from rats at P5, P7, P12, and P18.
Fig. 1.

Response in neurons and astrocytes to stimulation with 40 mm KCl. A, Time series of pseudocolor images of the [Ca2+]ichanges occurring in Indo-1-loaded cells from CA1 hippocampal region of a young rat (at P6) after perfusion of the slice with 40 mmKCl. The sequence shows the [Ca2+]iincrease in pyramidal neurons (closed arrows) occurring several seconds before that in astrocytes (open arrows). The R405:485 is displayed as a pseudocolor scale. Sampling rate, 2 sec; scale bar, 10 μm. B, Kinetics of the [Ca2+]i changes in the neurons (continuous lines) and the astrocytes (dotted lines), indicated by the arrows inA, after K+ stimulation, as expressed by the ratio between Indo-1 emission wavelength at 405 and 485 nm.a–h, Images a–h inA.
A first result that gave support to the above-mentioned hypothesis was obtained by investigating the origin of the biphasic response in neurons. The second [Ca2+]i peak that usually occurred in neurons at approximately the same time of the [Ca2+]i increase in astrocytes was found to be caused by the activation of NMDA receptors. It was, in fact, abolished by the application of the NMDA receptor antagonistd-AP-5 (Watkins et al., 1990) (Fig.2, left) and recovered after the second high K+ challenge performed 10 min after the washout of the antagonist (Fig. 2, right). In contrast, the response from astrocytes was unchanged (data not shown). Similar results were obtained in neurons and astrocytes from the visual cortex.
Fig. 2.

Kinetics of the [Ca2+]i change in two representative hippocampal pyramidal neurons to K+ stimulation in the presence of the NMDAR antagonist d-AP-5 (left) and 15 min after the onset of its washout (right).
We previously obtained evidence (Pasti et al., 1997) that the application of 1 mm MCPG, a metabotropic glutamate receptor (mGluR) competitive inhibitor (Watkins and Collingridge, 1994), drastically reduced the [Ca2+]iincrease triggered in astrocytes by synaptically released glutamate as well as that induced by the specific mGluR agonist t-ACPD (10 μm; Palmer et al., 1989). We have found that the [Ca2+]i increase induced by K+ stimulation in the presence of 1 mmMCPG was reduced to 60.5 ± 3.14% (mean ± SE;n = 7; two experiments) with respect to controls.
The delay in the astrocyte response with respect to the response in neurons on a first high K+ challenge was significantly increased (t test, p < 0.01) after a second high K+ stimulation (12.4 ± 0.6 vs 15.6 ± 1.1 sec; mean ± SE; n = 23) performed 10 min after slice perfusion with 10 μm TTX. The amplitude of the response was unchanged. The occurrence of the second [Ca2+]i peak in the response of neurons displayed a parallel delay, whereas the amplitude was unchanged. These results suggest that the entry of Na+ through voltage-dependent Na+channels is involved in the release of glutamate triggered by high K+ stimulation most likely by affecting the kinetics of the depolarization in neurons.
Ca2+ VOCs do not contribute to the K+-induced [Ca2+]iincrease in astrocytes
Altogether, although the results from the experiments described above cannot allow definitive conclusions on the possible expression of functional Ca2+ VOCs in astrocytes, they cast important doubts as to the interpretation of some previous data. The role of Ca2+ VOCs in the [Ca2+]i increase induced in astrocytes by K+ stimulation was, therefore, subjected to more stringent experiments, whose results are reported in Figures3 and 4. If the activation of mGluRs by glutamate fully accounts for the [Ca2+]i increase triggered in astrocytes by K+ stimulation, one would predict that (1) depletion of intracellular Ca2+ stores should inhibit the response of astrocytes to K+stimulation; and (2) inhibition of glutamate release from neurons should block the [Ca2+]i rise induced by high K+ stimulation in astrocytes. The first of such predictions was verified in the experiment presented in Figure 3. Astrocytes were challenged with 60 mm K+after depleting their [Ca2+]i stores with CPA, a potent and selective inhibitor of endoplasmic reticulum Ca2+ ATPase (Mason et al., 1991). As in the representative experiment reported in Figure 4, on slice perfusion with CPA (50 μm), the great majority of astrocytes (14 of 15; three experiments), previously identified by their delayed response to a first challenge with 60 mm KCl, displayed a slow, progressive increase in their [Ca2+]i. Within 20–30 min from the onset of CPA application, a new steady state, slightly higher than the resting [Ca2+]i, was reached. In these conditions, the stimulation with t-ACPD did not induce any significant [Ca2+]i elevations (data not shown), although the second challenge with K+caused in 14 of 15 astrocytes a slow decrease in the [Ca2+]i, as in the example reported in Figure 3, and no [Ca2+]ichange in the remaining one. The behavior of neurons was different. After CPA, their increase in [Ca2+]iwas negligible, as in the case of Figure 3. A clear [Ca2+]i increase during CPA application was observed in only 10% (2 of 20) of the neurons analyzed. In contrast to the behavior of astrocytes, neurons responded to the two episodes of K+ stimulation, the first in the absence and the second in the presence of CPA, with [Ca2+]i increases of similar amplitude.
Fig. 3.

Kinetics of the [Ca2+]i change in two astrocytes (dotted lines) and one pyramidal neuron (continuous lines) on K+ stimulation before and 30 min after slice perfusion with CPA, an inhibitor of the endoplasmic reticulum ATPase. After the depletion of the [Ca2+]i stores by CPA, the response of astrocytes to K+ stimulation was abolished, whereas that of the neuron was unchanged. To reduced photobleaching, from the onset of CPA application until the second episode of K+ stimulation, images were acquired every 30 sec.
Fig. 4.

The response in neurons and astrocytes on K+ stimulation is modified after TeNT treatment.A, Kinetics of the [Ca2+]i changes in two representative neurons (continuous lines) and astrocytes (dotted lines) from the visual cortex of a 10-d-old rat in response to stimulation with 60 mm KCl after slice incubation with TeNT. B, Mean values ± SE of the change in R405:485 representing the amplitude of [Ca2+]i elevations on K+ stimulation in neurons (N) and astrocytes (A) from control slices (42 neurons and 27 astrocytes, 6 experiments) and TeNT-treated slices (63 neurons and 37 astrocytes, 6 experiments). *p < 0.001.
The second prediction was verified in the experiment presented in Figure 4. We reported previously that in slices incubated with TeNT (100 μg/ml), a highly specific blocker of neurotransmitter secretion in neurons (Calabresi et al., 1989; Schiavo et al., 1992), synaptic transmission was blocked, and stimulation of neuronal afferents failed to trigger any significant [Ca2+]iincrease in neurons as well as in astrocytes (Pasti et al., 1997). Preliminary observations also suggested that the [Ca2+]i increase normally occurring in astrocytes on K+ stimulation was grossly reduced by TeNT. Here we confirm and expand those initial observations. Figure4A shows the kinetics of [Ca2+]i changes on high K+ stimulation in four representative cells, i.e., two neurons and two astrocytes, from a slice incubated for 40 min in 100 μg/ml TeNT. In these conditions, the response from these as well as the other astrocytes in six different experiments was virtually abolished (Fig. 4A,B). In two additional experiments, cells from TeNT-treated slices were followed for at least 10 min, but any [Ca2+]iincrease was observed in astrocytes. In neurons the amplitude of the first [Ca2+]i peak after a KCl challenge was unchanged by toxin treatment (Fig. 4B), although the second peak was either greatly reduced or absent (Fig.4A). Figure 4B reports the mean amplitude of the response of neurons (open bars) and astrocytes (stripped bars) to high K+stimulation in the absence and presence of TeNT. Noteworthy, TeNT has no specific effects on astrocyte Ca2+ handling. Indeed, after stimulation of the mGluR with t-ACPD (10 μm), astrocytes from slices incubated with TeNT responded with a typical pattern of [Ca2+]ichanges, including [Ca2+]ioscillations (data not shown).
Last but not least, if the preincubation with TeNT was reduced to 15–20 min, instead of the usual 30–40 min, the stimulation with 60 mm KCl induced a significant, although reduced, [Ca2+]i increase in astrocytes (n = 11; two experiments). Under these conditions, the response was abolished in all astrocytes when K+stimulation was performed in the presence of the mGluR antagonist MCPG (1 mm). On the contrary, MCPG did not affect the response in neurons.
In previous studies on acutely isolated astrocytes (Duffy and MacVicar, 1994) and astrocytes from acute brain slices (Porter and McCarthy, 1995; Duffy and MacVicar, 1996), it has been demonstrated that the Ca2+ VOC antagonist verapamil potently inhibits the [Ca2+]i increase in astrocytes caused by high K+ stimulation. This observation was interpreted as an indication for the expression of Ca2+ VOCs in astrocytes in situ and for their crucial role in mediating the [Ca2+]i increase in these cells on K+ stimulation. Here we have replicated that experiment and confirmed the observation. Verapamil was found to reduce by ∼50% the [Ca2+]i increase induced in astrocytes by 40 mm KCl, as in the typical example reported in Figure 5. However, under these conditions, the response from pyramidal neurons (both the first and second [Ca2+]i peaks) was also similarly inhibited (Fig. 5A). The mean reduction in the amplitude of the [Ca2+]i increase in neurons and astrocytes is reported in Figure 5B. Noteworthy, two subsequent applications of 40 mm KCl performed in the absence of verapamil resulted in comparable responses from both astrocytes and neurons (data not shown). Taken together, the data of Figure 5 indicate that the inhibition by verapamil cannot be taken as evidence for the existence of Ca2+ VOCs in astrocytes, because its effect could be indirect and attributed to a partial inhibition of the glutamate release from neurons.
Fig. 5.

The Ca2+ channel blocker verapamil reduced the response of both neurons and astrocytes.A, Kinetics of the [Ca2+]i change in one representative neuron (continuous line) and one astrocyte (dotted line) from the CA1 hippocampal region on K+ stimulation before and after slice perfusion with 100 μm verapamil. B, Mean relative decrease ± SE in the amplitude of the response to K+ stimulation from neurons (open bars labeled N1 and N2 report the amplitude of the first and second [Ca2+]i peaks respectively;n = 33, 2 experiments) and astrocytes (A, stripped bars; n= 8, 2 experiments) after slice perfusion with 100 μmverapamil.
Whole-cell patch clamping from astrocytes failed to reveal voltage-dependent Ca2+ currents
The possible presence of voltage-dependent Ca2+currents in astrocytes was finally investigated in acute hippocampal brain slices by using intracellular and extracellular solutions designed to reveal even small Ca2+ currents and to abolish K+ and Na+ currents. After establishing the whole-cell configuration, depolarizing current pulses of increasing amplitude were applied to trigger action potential discharges that could reveal the possible neuronal identity of the patched cell. Immediately after this procedure, which did not take >1 min, the perfusion with the solution containing TEA and 10 mm BaCl2 was started, thus minimizing the time necessary for current analysis and limiting the rundown of the Ca2+ currents. The membrane was then hyperpolarized to −110 mV for 0.5 sec from a holding potential of −80 mV and then depolarized to +10 mV for 100 msec by a single step. Successive episodes were separated by 4 sec intervals. After perfusion with the extracellular solution aimed to isolate Ca2+currents, the Na+ current, which was observed in 15 of a total of 18 recorded astrocytes, was rapidly reduced in amplitude and then blocked (Fig.6A). No inward currents remained detectable under these conditions (Fig. 6B). As already reported in astrocytes acutely isolated from mouse hippocampus (Steinhäuser et al., 1994), the astrocyte Na+ current was TTX-sensitive, as it rapidly disappeared after perfusion with 1 μm TTX added to the standard extracellular solution (n = 3). After the disappearance of the Na+ current, in nine astrocytes we compared the current traces before and after perfusion with 100 μm Cd2+, but no differences were detectable and, in no cases did the subtraction of the current traces disclose a Cd2+-sensitive inward current (Fig.6C). In several experiments, LY was included in the patch pipette. All the astrocytes recorded (n = 8) were dye-coupled with at least two other astrocytes. Under the same experimental conditions, the Na+ current recorded from a CA1 pyramidal neurons was progressively inhibited on slice perfusion with TEA and BaCl (10 mm), and a distinct Ca2+ current appeared (Fig. 6D). In a few experiments, by gentle withdrawing the patch pipette, we attained the detachment of the astrocyte cell body from the slice. Visual inspection revealed that some processes were relatively intact. Under these conditions, gap junction communication with other astrocytes was abolished, and the voltage control was presumably much better than in the astrocytes in situ. No Ca2+ currents were, however, detected in three successfully detached astrocytes.
Fig. 6.
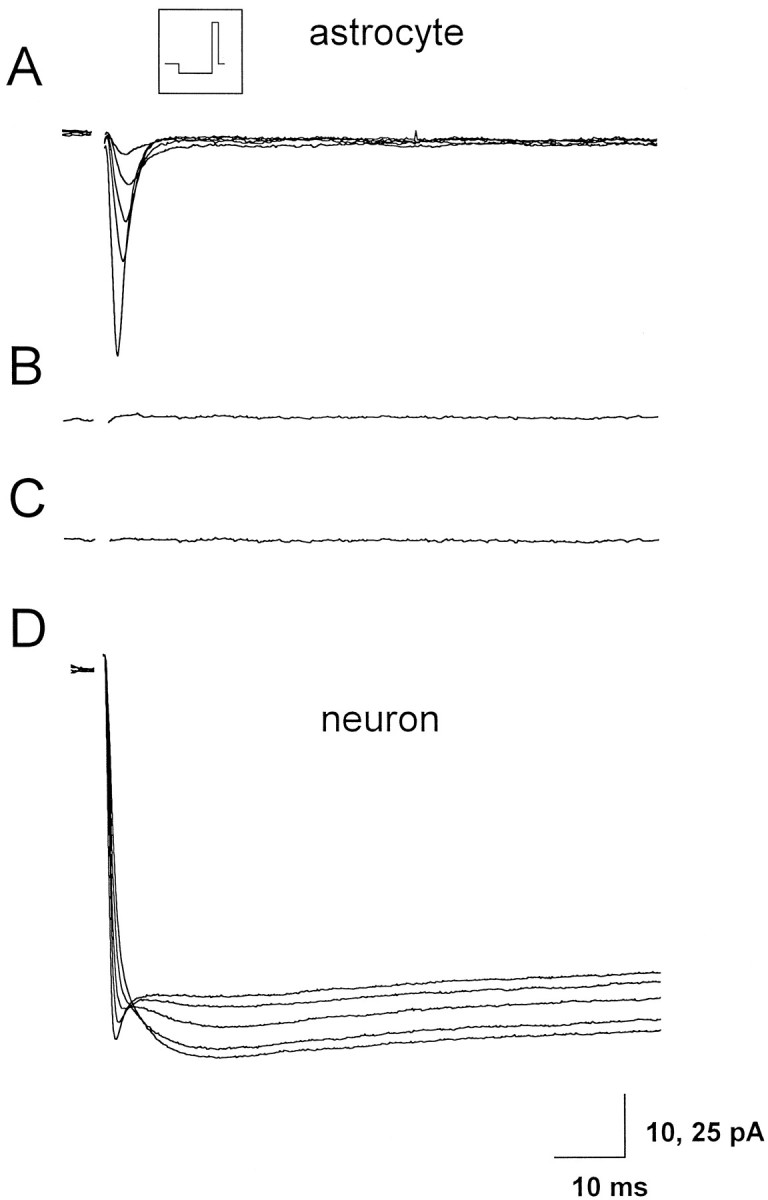
Whole-cell recordings of Ba2+and Na+ currents in hippocampal astrocytes and neurons. A, Progressive reduction of the Na+ current recorded from one astrocyte from the CA1 hippocampal region of a 10-d-old rat on slice perfusion with 10 mm BaCl2 and TEA in substitution of Na+ ions. The inset shows the pulse protocol used; the membrane was hyperpolarized to −110 mV from a holding potential of −80 mV for 0.5 sec and then depolarized to 0 mV for 100 msec. Successive voltage pulses were separated by a 4 sec interval. The capacitive transients have been blanked. LY included in the patch pipette diffused through gap junction from the recorded astrocyte into two other astrocytes (data not shown). B, No inward currents remain detectable after switching to Na+-free solution. C, Trace obtained after subtraction of the current traces recorded before and after 100 μm Cd2+ from the same astrocyte.D, Whole-cell recording from a CA1 pyramidal neuron of a 10-d-old rat with Ba2+ as charge carrier. Note the progressive increase of the Ba2+ current and the parallel reduction of the Na+ current on slice perfusion (which started immediately after establishing the whole-cell configuration) with 10 mm BaCl2 and TEA in substitution of Na+ ions. Stimulation protocol as inA.
DISCUSSION
The main finding of this study is that the neuronal release of neurotransmitters is responsible for the [Ca2+]i increase observed in astrocytes in situ on high K+stimulation. The contribution of Ca2+ VOCs in this event is, therefore, either absent or negligible. This conclusion derives from experiments performed in astrocytes from both rat visual cortex and CA1 hippocampal region at P5–P18; therefore, it does not necessarily hold for astrocytes from other brain regions or at different developmental stages.
The remarkable delay occurring in astrocyte responses on a depolarizing stimulus with 40–60 mm KCl with respect to the prompt [Ca2+]i increase in neurons (Pasti et al., 1997) represents the initial observation that induced us to further investigate the role of Ca2+ VOCs in astrocytes. A plausible hypothesis that could account for the delayed [Ca2+]i increase in astrocytes on high-K+-induced depolarization is that this response is caused by the action of neurotransmitters, such as glutamate and GABA, released by synaptic terminals. In other words, the [Ca2+]i increase in astrocytes could represent a secondary response after activation by glutamate, and probably other neurotransmitters, of metabotropic receptors that trigger IP3-mediated release of Ca2+from intracellular organelles. The significant reduction in the [Ca2+]i increase in astrocytes induced by K+ stimulation by MCPG is in agreement with the above-mentioned hypothesis. It should be noted that MCPG is a relatively weak competitive mGluR antagonist and could not completely prevent the action of glutamate massively released on the depolarizing stimulus. In addition, MCPG cannot block the action of other neurotransmitters, such as GABA, that can be released by neurons on depolarization and trigger [Ca2+]iincrease in astrocytes (Kettenmann et al., 1988; Nilsson et al., 1993).
The observation that the NMDAR antagonist d-AP-5 abolished the second [Ca2+]i peak observed in neurons on K+ stimulation demonstrated that (1) significant amounts of glutamate are released on the depolarizing stimulus; and (2) Ca2+ entry through the NMDA receptor is responsible for the biphasic pattern of the neuronal response. Interestingly, in contrast to the lack of recovery observed in some neurons in the absence of d-AP-5, all neurons recovered [Ca2+]i basal levels, and the time of recovery was also much faster in the presence ofd-AP-5. These results confirm that neurons can hardly face a [Ca2+]i rise outside the physiological range when it derives from excessive stimulation of NMDA receptors.
Two pieces of experimental evidence conclusively demonstrate that the [Ca2+]i increase occurring in astrocytes on a depolarizing stimulus is exclusively a glutamate-mediated response: (1) after emptying [Ca2+]i stores with CPA, no astrocyte displayed a detectable [Ca2+]iincrease on the depolarizing stimulus; and (2) no response to high K+ stimulation was observed in astrocytes from slices incubated in TeNT. As to the first, after store depletion, opposite to expectation if Ca2+ VOCs were expressed in these cells, the depolarizing stimulus failed to cause any [Ca2+]i increase in astrocytes but, rather, induced a progressive [Ca2+]idecrease. This latter observation is consistent with the expression in astrocytes of a Ca2+ release-activated Ca2+ influx (Hoth and Penner, 1992; Fasolato et al., 1994). Accordingly, the [Ca2+]idecrease caused by depolarization likely depends on a decrease in the driving force for Ca2+ influx. As to the second, TeNT is known to be highly neuron-specific and to exert its action on VAMP/synaptobrevin, a protein that plays a crucial role in the neuroexocytosis process (Schiavo et al., 1992; Matteoli et al., 1996). The inhibitory effect of the neurotoxin on neurotransmitter exocytosis was confirmed in each slice by the observations that (1) electrical stimulation of Schaffer collaterals failed to induce [Ca2+]i increases in hippocampal neurons otherwise observed in TeNT-untreated slices; (2) pyramidal neurons still responded to K+-induced depolarization with an early [Ca2+]i increase, but the second [Ca2+]i peak in their response was either greatly reduced or abolished; and (3) [Ca2+]i basal levels were recovered much faster than in neurons from TeNT-untreated slices. The absence of [Ca2+]i changes evoked in astrocytes from TeNT-treated slices conclusively confirms the hypothesis that glutamate released by depolarized synaptic terminals is responsible for the K+-induced [Ca2+]i increase in astrocytes. The observation that astrocytes from TeNT-treated slices displayed typical [Ca2+]i oscillations on slice perfusion with the mGluR agonist t-ACPD demonstrated that astrocytes can respond normally in the presence of TeNT.
Our conclusion is clearly in contrast with several studies supporting a role of Ca2+ VOCs in the K+-induced [Ca2+]iincrease in astrocytes in situ. In particular, in the presence of Ca2+ channel antagonists, the [Ca2+]i increase on high K+ depolarization in both acutely dissociated astrocytes (Duffy and MacVicar, 1994) and astrocytes from hippocampal slices was observed to be, at least partially, inhibited (Porter and McCarthy, 1995; Duffy and MacVicar, 1996). Although we obtained similar results, our interpretation is that Ca2+ VOC antagonists reduce the [Ca2+]iincrease in astrocytes on K+ stimulation by blocking neuronal Ca2+ channels, thus causing a reduction in neurotransmitter release. The observation that verapamil also decreased the amplitude of the response from neurons supports our interpretation. In particular, given that the second [Ca2+]i peak in the neuronal response to K+ stimulation is caused by the release of glutamate and to the consequent activation of the NMDA receptor, the finding that verapamil decreased the amplitude not only of the first but also of the second [Ca2+]i peak suggests that it directly affects the process of neurotransmitter release. Because the release of the neurotransmitter in the mammalian CNS is known to be controlled by various Ca2+channels but not the L-type channel (Dunlap et al., 1995), verapamil most likely interferes, at least at the concentration used, not only with the L-type but also with other Ca2+ channels, such as the N- and P-type, that regulate exocytosis in hippocampal neurons.
The results we obtained are hardly compatible with the presence of functional VOCs in astrocytes in situ. Nevertheless, the possibility cannot be ruled out that astrocytes either express Ca2+ VOCs at very low density or express exclusively rapidly inactivating T-type Ca2+ channels. In both cases, the [Ca2+]i increase resulting from their activation might be too small or too rapid to be detectable, at least by our experimental approach with the confocal microscope.
Despite the various experimental conditions we used to detect even a small Ca2+ current, our patch-clamp study failed to reveal any Ca2+ currents from astrocytes in situ. Negative results were also obtained by recording from three astrocytes after their detachment from the slice. Under these conditions, gap junction communication with other astrocytes was most likely abolished, thereby allowing an adequate voltage control. Although unlikely, the possibility still remains that a low density of VOCs is expressed exclusively in the processes that were lost in the detached astrocytes.
We would like, however, to point out that even if Ca2+ VOCs are expressed in astrocytes, they do not contribute to the dramatic [Ca2+]ichange occurring in these cells on depolarization. This finding, therefore, has implications for the mechanism by which alterations in the extracellular K+ possibly affect the function of astrocytes in vivo. In fact, it has been reported that pathological conditions such as ischemia or spreading depression lead to large elevations of extracellular K+ (60–80 mm). These changes are accompanied by strong reductions in the concentration of extracellular Ca2+ (Nicholson et al., 1978). It has been suggested that the [Ca2+]i increase observed in astrocytes under these conditions is the result of Ca2+ influx through Ca2+ VOCs. The hypothesis was then advanced that astrocytes can efficiently buffer Ca2+ in the extracellular space as much as they can buffer K+. By lowering Ca2+ in the synaptic cleft, astrocytes may reduce Ca2+influx in neurons mediated by Ca2+ VOCs and ionotropic glutamate receptors, thus modifying Ca2+-dependent synaptic transmission and protecting neurons from cell death because of excessive elevations of [Ca2+]i (Duffy and MacVicar, 1994). After the results reported here, the role of astrocyte Ca2+ VOCs in these processes should be reconsidered, at least at the developmental stages and in the brain regions investigated in this study. Noteworthy, we recently demonstrated that the activation of the mGluR in cultured astrocytes as well as in astrocytes from acute brain slices triggers, via prostaglandin formation, a significant Ca2+-dependent release of glutamate (Pasti et al., 1997; Bezzi et al., 1998). Through this glutamate-mediated glutamate release, astrocytes may thus contribute to, rather than protect from, the neuronal death that results from the excitoxic action of glutamate.
The possible contribution of Ca2+ VOCs in depolarization-dependent [Ca2+]ichanges in astrocytes under physiological conditions should also be reconsidered. A rise in the [Ca2+]imay modulate in astrocytes multiple events such as Ca2+-dependent K+ channel activation (Quandt and MacVicar, 1986), nitric oxide production (Murphy et al., 1993), propagating [Ca2+]iwaves (Cornell-Bell et al., 1990; Finkbeiner, 1992) and, as already mentioned, the release of glutamate (Parpura et al., 1994; Jeftinija et al., 1996; Pasti et al., 1997; Bezzi et al., 1998). The source of Ca2+ that permits these changes can be either intracellular, i.e., release from stores, and/or extracellular, i.e., entry through plasma membrane channels and ion exchangers. Although a contribution of the Na+ and Ca2+exchanger cannot be excluded (Goldman et al., 1994), our results suggest that the release of Ca2+ from intracellular Ca2+ stores and the Ca2+release-activated Ca2+ influx are major mechanisms used by astrocytes to achieve rises in their [Ca2+]i that could have functional significance for the triggering of Ca2+-dependent events.
Footnotes
This manuscript was supported by Telethon Grant 845, European Union Programs, Human Capital and Mobility Network Grant CHRXCT940500, Human Frontier Science Program Grant RG520/95, Italian University Ministry, Fidia Research Laboratories, and Biotechnology Program Grant 0BIO4CT960382. We thank Aldebaran Hofer and Daniela Pietrobon for helpful discussion and critical reading of this manuscript and Cesare Montecucco for the generous gift of the purified tetanus toxin.
Correspondence should be addressed to Giorgio Carmignoto, Department of Experimental Biomedical Sciences, University of Padova, Viale G. Colombo 3, 35131 Padova, Italy.
REFERENCES
- 1.Akopian G, Kressin K, Derouiche A, Steinhäuser C. Identified glial cells in the early postnatal mouse hippocampus display different types of calcium currents. Glia. 1996;17:181–194. doi: 10.1002/(SICI)1098-1136(199607)17:3<181::AID-GLIA1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 2.Barres BA, Chun LLY, Corey D. Calcium current in cortical astrocytes: induction by cAMP and neurotransmitters and permissive effect of serum factors. J Neurosci. 1989;9:3169–3175. doi: 10.1523/JNEUROSCI.09-09-03169.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barres BA, Koroshetz WJ, Chun L, Corey D. Ion channel expression by white matter glia: the type-1 astrocyte. Neuron. 1990;5:527–544. doi: 10.1016/0896-6273(90)90091-s. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ. Inositol trisphosphate and calcium signaling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 5.Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Lodi Rizzini B, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 6.Calabresi P, Benedetti M, Mercuri NB, Bernardi G. Selective depression of synaptic transmission by tetanus toxin: a comparative study on hippocampal and neostriatal slices. Neuroscience. 1989;30:663–670. doi: 10.1016/0306-4522(89)90159-0. [DOI] [PubMed] [Google Scholar]
- 7.Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- 8.Charles AC, Merrill JE, Dirksen ER, Sanderson MJ. Intracellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation. Neuron. 1991;6:983–992. doi: 10.1016/0896-6273(91)90238-u. [DOI] [PubMed] [Google Scholar]
- 9.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long range glial signalling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 10.Corvalan V, Cole R, De Vellis J, Hagiwara S. Neuronal modulation of calcium channel activity in cultured rat astrocytes. Proc Natl Acad Sci USA. 1990;87:4345–4348. doi: 10.1073/pnas.87.11.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 12.Duffy S, MacVicar BA. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience. 1994;61:51–61. doi: 10.1016/0306-4522(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 13.Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J Neurosci. 1996;1:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from synaptically connected neurones of the mammalian CNS. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 15.Fasolato C, Innocenti B, Pozzan T. Receptor-activated Ca2+ influx: how many mechanisms for how many channels? Trends Pharmacol Sci. 1994;15:77–83. doi: 10.1016/0165-6147(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 16.Finkbeiner S. Calcium waves in astrocytes: filling the gap. Neuron. 1992;8:1101–1108. doi: 10.1016/0896-6273(92)90131-v. [DOI] [PubMed] [Google Scholar]
- 17.Glaum SR, Holzwarth JA, Miller RJ. Glutamate receptors activate Ca2+ mobilization and Ca2+ influx into astrocytes. Proc Natl Acad Sci USA. 1990;86:3454–3458. doi: 10.1073/pnas.87.9.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldman WF, Yarowsky PJ, Juhaszova M, Krueger BK, Blaustein MP. Sodium/calcium exchange in rat cortical astrocytes. J Neurosci. 1994;14:5834–5843. doi: 10.1523/JNEUROSCI.14-10-05834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoth M, Penner R. Depletion of intracellular stores activates a calcium current in mast cells. Nature. 1992;355:353–355. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 20.Jabs R, Kirchoff F, Kettenmann H, Steinhäuser C. Kainate activates Ca2+-permeable glutamate receptors and blocks voltage-gated K+ currents in glial cells of mouse hippocampus. Pflügers Arch. 1994;426:310–319. doi: 10.1007/BF00374787. [DOI] [PubMed] [Google Scholar]
- 21.Jeftinija SD, Jeftinija FK, Stefanovic G, Liu F. Neuroligands-evoked calcium dependent release of excitatory amino acids from cultured astrocytes. J Neurochem. 1996;66:676–684. doi: 10.1046/j.1471-4159.1996.66020676.x. [DOI] [PubMed] [Google Scholar]
- 22.Jensen AM, Chiu SY. Differential intracellular responses to glutamate in type 1 and type 2 cultured brain astrocytes. J Neurosci. 1991;11:1674–1684. doi: 10.1523/JNEUROSCI.11-06-01674.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kettenmann H, Backus KH, Schachner M. GABA receptor on cultured astrocytes. In: Kimelberg HK, editor. Glial cell receptors. Raven; New York: 1988. pp. 95–106. [Google Scholar]
- 24.Kressin K, Kuprijanova E, Jabs R, Seifert G, Steinhäuser C. Developmental regulation of Na+ and K+ conductances in glial cells of mouse hippocampal brain slices. Glia. 1995;15:173–187. doi: 10.1002/glia.440150210. [DOI] [PubMed] [Google Scholar]
- 25.MacVicar BA. Voltage-dependent calcium channels in glial cells. Science. 1984;226:1345–1347. doi: 10.1126/science.6095454. [DOI] [PubMed] [Google Scholar]
- 26.MacVicar BA, Tse FWY. Norepinephrine and cyclic adenosine 3′:5′-cyclic monophosphate enhance a nifedipine-sensitive calcium current in cultured astrocytes. Glia. 1988;1:359–365. doi: 10.1002/glia.440010602. [DOI] [PubMed] [Google Scholar]
- 27.MacVicar BA, Hochman D, Dealy MJ, Weiss S. Modulation of intracellular Ca2+in cultured astrocytes by influx through voltage-activated Ca2+ channels. Glia. 1991;4:448–455. doi: 10.1002/glia.440040504. [DOI] [PubMed] [Google Scholar]
- 28.McKhann GM, II, D’Ambrosio R, Janigro D. Heterogeneity of astrocyte resting membrane potential and intercellular coupling revealed by whole-cell and gramicidin-perforated patch recording from cultured astrocytes. J Neurosci. 1997;17:6850–6863. doi: 10.1523/JNEUROSCI.17-18-06850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular stores and permeability of the plasma membrane: comparison of the effects of thapsigargin, 2,5-Di-(ter-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J Biol Chem. 1991;266:20856–20862. [PubMed] [Google Scholar]
- 30.Matteoli M, Verderio C, Rossetto O, Iezzi N, Cocco S, Schiavo G, Montecucco C. Synaptic vesicle endocytosis mediates the entry of tetanus toxin into hippocampal neurons. Proc Natl Acad Sci USA. 1996;93:13310–13315. doi: 10.1073/pnas.93.23.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy S, Simmonds ML, Agullo L, Garcia A, Feinstein DL, Galea E, Reis DJ, Minc-Golomb D, Schwartz JP. Synthesis of nitric oxide in CNS glial cells. Trends Neurosci. 1993;16:323–328. doi: 10.1016/0166-2236(93)90109-y. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson C, Bruggencate GT, Stockle H, Steinberg R. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J Neurophysiol. 1978;41:1026–1039. doi: 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- 33.Nilsson M, Eriksson PS, Ronback L, Hansson E. GABA induces transients on astrocytes. Neuroscience. 1993;54:605–614. doi: 10.1016/0306-4522(93)90232-5. [DOI] [PubMed] [Google Scholar]
- 34.Palmer E, Monaghan DT, Cotman CW. Trans-ACPD, a selective agonist of the phosphoinositide-coupled excitatory amino acid receptor. Eur J Pharmacol. 1989;166:585–587. doi: 10.1016/0014-2999(89)90383-x. [DOI] [PubMed] [Google Scholar]
- 35.Parpura V, Basarky TA, Liu F, Jeftinija FK, Jeftinija SD, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 36.Pasti L, Pozzan T, Carmignoto G. Long-lasting changes of calcium oscillations in astrocytes. A new form of glutamate-mediated plasticity. J Biol Chem. 1995;25:15203–15210. doi: 10.1074/jbc.270.25.15203. [DOI] [PubMed] [Google Scholar]
- 37.Pasti L, Volterra A, Pozzan T, Carmignoto G. [Ca2+]i oscillations in astrocytes triggers repetitive glutamate-mediated [Ca2+]i increases in neurons in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porter JA, McCarthy KD. GFAP-positive hippocampal astrocytes in situ respond to glutamatergic neuroligand with increased in [Ca2+]i. Glia. 1995;13:101–112. doi: 10.1002/glia.440130204. [DOI] [PubMed] [Google Scholar]
- 39.Quandt FN, MacVicar BA. Calcium activated potassium channels in cultured astrocytes. Neuroscience. 1986;19:29–41. doi: 10.1016/0306-4522(86)90003-5. [DOI] [PubMed] [Google Scholar]
- 40.Rice ME, Pérez-Pinzón MA, Lee EJK. Ascorbic acid, but not glutathione, is taken up by brain slices and preserves cell morphology. J Neurophysiol. 1994;71:1591–1560. doi: 10.1152/jn.1994.71.4.1591. [DOI] [PubMed] [Google Scholar]
- 41.Scheenen WJJM, Makings LR, Gross LR, Pozzan T, Tsien RY. Photodegradation of Indo-1 and its effects on apparent Ca2+ concentration. Chem Biol. 1996;3:765–774. doi: 10.1016/s1074-5521(96)90253-7. [DOI] [PubMed] [Google Scholar]
- 42.Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: isolation and assay. Methods Enzymol. 1995;248:643–652. doi: 10.1016/0076-6879(95)48041-2. [DOI] [PubMed] [Google Scholar]
- 43.Schiavo G, Benfenati F, Puolain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolitic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 44.Steinhäuser C, Kressin K, Kuprijanova E, Weber M, Seifert G. Properties of voltage-activated sodium and potassium currents in mouse hippocampal glial cells in situ and after acute isolation from tissue slices. Pflügers Arch. 1994;428:610–620. doi: 10.1007/BF00374585. [DOI] [PubMed] [Google Scholar]
- 45.Verkhratsky A, Kettenmann H. Calcium signaling in glial cells. Trends Neurosci. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- 46.Watkins JC, Collingridge GL. Phenylglycine derivatives as antagonists of metabotropic glutamate receptors. Trends Pharmacol Sci. 1994;15:333–342. doi: 10.1016/0165-6147(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 47.Watkins JC, Krogsgaard-Larsen P, Honoré T. Structure-activity relationships in the development of excitatory amino acid receptor agonists and competitive antagonists. Trends Pharmacol Sci. 1990;11:25–33. doi: 10.1016/0165-6147(90)90038-a. [DOI] [PubMed] [Google Scholar]