

Abstract
In auditory fear conditioning, pairing of a neutral acoustic conditioned stimulus (CS) with an aversive unconditioned stimulus (US) results in an enhancement of neural responses to the CS in the amygdala and auditory cortex. It is not clear, however, whether cortical plasticity governs neural changes in the amygdala or vice versa, or whether learning in these two structures is determined by independent processes. We examined this issue by recording single-cell activity in the auditory cortex (areas Te1, Te1v, and Te3) of freely behaving, amygdalectomized rats using a movable bundle of microwires. Amygdala damage did not affect short-latency (0–50 msec) tone responses, nor did it interfere with conditioning-induced increases of these onset responses. In contrast, lesions of the amygdala interfered with the development of late (500–1500 msec) conditioned tone responses that were not present before conditioning. Furthermore, whereas onset conditioned responses in the control group remained elevated after 30 extinction trials (presentation of CS alone), onset responses in lesioned animals returned to their preconditioning firing level after approximately 10 extinction trials. These results suggest that the amygdala enables the development of long-latency (US anticipatory) responses and prevents the extinction of short-latency onset responses to threatening stimuli. The findings further suggest that auditory cortex cells may participate differently in explicit and implicit memory networks.
Keywords: emotion, cognition, declarative memory, implicit memory, amygdala, auditory cortex, fear
The firing properties of cells are modified in various brain areas during learning (Olds et al., 1972). For example, after the association of an acoustic conditioned stimulus (CS) with an electric shock unconditioned stimulus (US) in a standard fear conditioning paradigm, spike discharges change in a number of brain regions, including areas of the auditory cortex, hippocampal formation, and amygdala (Diamond and Weinberger, 1984, 1986; Weinberger et al., 1984; Laroche et al., 1989; Quirk et al., 1995, 1997). Although damage to the amygdala prevents the expression of behavioral fear responses conditioned to the CS, damage to the auditory cortex or hippocampus does not (Selden et al., 1991; Phillips and LeDoux, 1992;Romanski and LeDoux, 1992; Armony et al., 1997a). These findings might imply that redundant, behaviorally irrelevant traces are established in areas such as the auditory cortex and hippocampus. It is also possible, however, that experiences are encoded and stored in parallel by complementary memory systems (Eichenbaum et al., 1994,1996; Squire and Knowlton, 1995). Plasticity occurring in a particular brain area thus might be uninvolved in one aspect of learning but crucial to another.
The complementary nature of memory systems is nicely illustrated by the effects of different brain lesions in humans on fear conditioning. Patients with amygdala lesions fail to exhibit conditioned fear responses to a CS [but remember that the CS and US were systematically related (Bechara et al., 1995; LaBar et al., 1995)], whereas patients with damage to the hippocampus and related cortical areas exhibit conditioned responses to the CS but have no memory of having experienced the CS and US together (Bechara et al., 1995). The amygdala is thus required for establishing a linkage between the CS and response control pathways, whereas explicit or declarative knowledge about stimulus relations is acquired through hippocampal–cortical circuits that function more or less independently of the amygdala.
Although the amygdala is not required for the establishment of declarative memories about emotional situations, it may influence mnemonic processing in declarative memory circuits. For example, amygdala damage in humans eliminates the ability of emotional arousal to enhance declarative memory performance (Babinsky et al., 1993;Cahill et al., 1995). The amygdala can interact with hippocampal–cortical circuits by way of direct synaptic connections, as well as through various indirect pathways (Amaral et al., 1992;LeDoux, 1996). Nevertheless, little is known about the manner in which the amygdala might influence plasticity in cortical areas.
Auditory cortex, like other areas of sensory cortex, is reciprocally connected with the hippocampus (by way of parahippocampal circuits) and is a putative component of the declarative memory circuit for explicit auditory memory (Amaral, 1987; Squire and Zola, 1996). The auditory cortex is also reciprocally connected with the amygdala (Amaral et al., 1992; Romanski and LeDoux, 1993) and is thus a potential site of interaction between declarative and emotional memory networks. In the present study we have begun to explore these interactions by examining the effects of amygdala lesions on the plasticity of cells in the auditory cortex during fear conditioning.
MATERIALS AND METHODS
Animals and surgery
Male Sprague Dawley rats (300–350 gm) were pretreated with atropine (0.24 mg/kg), anesthetized with nembutal (50 mg/kg, i.p.), and placed in a Kopf stereotaxic frame with blunt ear bars. Supplemental doses of anesthetic were administered throughout surgery as needed, and body temperature was regulated by a gel heating pad.
Bilateral amygdala ablation was achieved by placing two electrolytic lesions on each side, with the following coordinates (in millimeters) relative to bregma (LaBar and LeDoux, 1996): (1) anterioposterior (AP) = −2.3, mediolateral (ML) = ±4.8, dorsoventral (DV) = −8.4; (2) AP = −2.8, ML = ±5.2, DV = −8.5. Lesions were made by passing anodal constant current (1 mA, 15 sec) through a monopolar, stainless steel electrode (200 μm diameter; Kopf NE-300) with an exposed tip of 500 μm. The skull holes were then covered with sterile bone wax. The intent was to destroy much of the lateral and basal nuclei, but no effort was made to exclude damage to other regions of amygdala.
Electrode implant procedures were identical to those described in Quirk et al. (1997). A movable electrode bundle of 20 insulated nichrome microwires (25 μm diameter each; adapted from Kubie, 1984) was implanted in the auditory cortex (5.0 mm AP, 5.1 mm ML, 6.0 mm DV, relative to bregma) (Paxinos and Watson, 1986). The wires were insulated except for the cut tip (impedance ∼2–3 MΩ at 1 kHz) and contained in a stainless steel tube (0.45 mm diameter). Each wire was attached to one pin of two 10-pin connectors (Augat) embedded in a triangular platform made of dental acrylic. The platform could be advanced by turning three screws (1 turn = 460 μm). Placement accuracy of the electrode bundle was improved by passing the tube and wires through an additional, larger guide tube (0.81 mm diameter), implanted just above the cortex. The tips of the electrode bundle were aimed to rest just dorsal to auditory association cortex (areas Te3/Te1v) (Zilles and Wree, 1985). The diameter of the bundle at the recording sites was ∼0.5 mm. At the end of surgery, the incision was closed with suture around the acrylic block, a topical antibiotic was applied, and the analgesic Buprenorphine (0.02 mg/kg, i.m.) was administered. Rats were allowed 5 d to recover before electrophysiological recordings began. Control animals were treated in the same way, except that no lesions were made. All aspects of the behavioral training and unit recording were identical for both groups. Data for the control group were reported in Quirk et al. (1997). The present study was run concurrently with the latter.
Unit recording
After recovery, the rat was placed in a test box (25 × 30 × 35 cm) with an electrifiable grid floor (Coulbourn Instruments), enclosed in a sound attenuating chamber (Industrial Acoustics, Bronx, NY). The electrode was connected to a headstage containing field-effect transistors (FETs) (#U401; Marshall Electronics, El Monte, CA) operating as source-followers with unitary gain. Signals from the headstage were sent to multichannel amplifiers (AM Systems, Everett, WA) through a slip-ring commutator (Plastics One, Roanoke, VA). The signal was amplified (10,000× gain), passively filtered (300–5000 Hz), digitized at 16 kHz (DataWave Technologies, Longmont, CO), and displayed on a computer monitor and a digital oscilloscope. Signals exceeding a selected voltage threshold were captured in 2 msec epochs and stored on disk for later analysis. Signal waveforms corresponding to single cells were sorted off-line using the DataWave software on the basis of waveform parameters (e.g., peak voltage, spike height, time to peak, etc.). Only those spike clusters, corresponding to a single cell, that could be separated from background activity throughout all phases of the experiment were defined as coming from single units (Quirk et al., 1995, 1997). This procedure ensures that only individual cells are analyzed across an entire session (for examples of typical waveforms, see Quirk et al., 1996). Typically, neural activity could be recorded simultaneously from several wires, and one to three individual cells could be isolated from each active wire.
The electrode was advanced in 50–100 μm steps until discriminable single units were found in several wires. Units were tested for tone responsivity by delivering 50 msec tone pips (80 ± 5 dB, open field) of various frequencies, as well as white noise, delivered through a speaker mounted in the ceiling of the sound-attenuating chamber. Auditory signals were generated with a Digital Signal Processing board controlled by a DataWave Discovery clocked sequence (MOZART) running on the same computer as the data acquisition software.
Conditioning protocol
We used the same conditioning protocol as in previous studies of the amygdala and auditory cortex (Quirk et al., 1995, 1997) to facilitate comparisons between amygdala and auditory cortex recordings as well as to allow comparisons of cortical recordings between amygdala-lesioned and control animals. Briefly, the conditioned stimulus (CS) was a 2 sec pure tone (5 kHz, 80 dB) and the unconditioned stimulus (US) was a mild electric footshock (0.5 mA, 0.5 sec) delivered through the grid floor of the test box.
The experiment consisted of three phases. Duringsensitization, 10 CSs and 10 USs were explicitly unpaired. This was immediately followed by 20 conditioning trials in which the CS coterminated with the US (that is, the tone and shock were always paired). After the conditioning phase, the rat was placed in its home cage for 1 hr, after which it was returned to the test chamber for the extinction phase, during which 30 tone-alone trials were delivered (i.e., no US was presented during the extinction phase). In all phases, the intertrial interval was 1–3 min.
Before the sensitization and extinction phases, behavioral responses to the CS were determined in a different context, using a 20 sec tone, calibrated to the same intensity and frequency as the CS during conditioning (Bruel and Kjaer). Cumulative seconds of freezing during the CS were counted with a digital timer and used as a measure of conditioned fear (Blanchard and Blanchard, 1969; Bolles and Fanselow, 1980). Freezing was defined as absence of all movement, except those associated with breathing. The entire experiment took ∼4 hr. To compare cell responses before (i.e., in naive animals) and after conditioning, each rat could be conditioned only once. This was a limiting factor in the number of cells that could be obtained from each animal.
Data analysis
Data were analyzed, after off-line spike discrimination, using STRANGER (Biographics, Winston-Salem, NC). Statistical analysis was performed using commercial software (Excel and Statistica).
Onset tone responses. Onset tone responses (i.e., unit responses locked to the onset of the stimulus) were determined, before conditioning, by comparing the average firing rate in the first 50 msec of the tone with the firing rate 500 msec before tone onset, during the 10 sensitization trials. Significant onset tone responses were determined using a z test (p < 0.05). Onset tone responses after conditioning were determined in a similar manner, using the firing rates from the first 10 extinction trials.
Long-term conditioned increases in the onset responses were investigated in a similar manner by comparing onset tone responses (with the average pretone firing rate subtracted) before conditioning (10 sensitization trials) with those after conditioning (first 10 extinction trials), for the first 50 msec after tone onset. Because we were interested in whether conditioning had an effect on the signaling of the CS, we restricted the analysis of onset conditioning to those cells that showed a positive tone response after the conditioning phase (i.e., their firing rate in the first 50 msec of the tone was higher than their rate before the tone).
Late tone responses. We investigated the effects of conditioning on responses occurring after tone onset by analyzing firing rates during the entire tone in 100 msec bins before (10 sensitization trials) and after (first 10 extinction trials) conditioning. For each bin, the number of tone-responsive cells was calculated using z scores (2 SD above pretone firing rate). Only cells that were significantly tone responsive in at least two adjacent 100 msec bins were included. This analysis revealed a “region of interest,” between 500 and 1500 msec, in which the majority of late tone responses occurred (Quirk et al., 1997). Thus, this region was further analyzed with Student’s paired t tests (p < 0.05, two-tailed) to determine tone responsiveness, comparing the rate in the 500–1500 msec interval with the pretone rate. (It should be pointed out that because this latter analysis combines the rates of 10 consecutive bins, it will yield a larger number of tone-responsive cells than that seen for any individual bin.) Late tone responses could not be measured during conditioning trials because inputs to the amplifiers were grounded shortly before shock presentation to avoid shock artifact contamination of the unit recordings.
Histology
At the end of the experiment, a small lesion was made by passing current (8 μA, 10 sec) through one of the recording wires. Animals were perfused transcardially with 10% buffered formalin. The brains were removed and stored in a formalin–sucrose solution with 2% nitroferrocyanide. The brains were frozen and cut on a sliding microtome (40 μm sections), and mounted sections were stained for nissl bodies and examined with a light microscope. The site of the Prussian blue reaction (caused by the reaction of iron deposits from the lesion with the nitroferrocyanide) was used to locate the region of the recorded cells. Cases in which the amygdala lesions were incomplete or thalamocortical fibers running through the striatum appeared to be disrupted by the lesion electrode tracks were discarded. Photomicrographs of the lesions were obtained with a CatsEye digital camera (Sony) and acquired on a Macintosh PowerPC 8100 with commercial software (Adobe Photoshop).
RESULTS
Histology
Figure 1 illustrates a typical amygdala lesion. Lesions were centered primarily on the lateral and basal nuclei and in some cases extended medially into the central nucleus. Some animals also had some damage to the amygdalostriatal zone. Those cases exhibiting severe damage of the caudate-putamen, near the electrode track, were discarded. Although caudate-putamen lesions do not interfere with the conditioning of fear behavior (LeDoux et al., 1985; Iwata et al., 1986), auditory thalamocortical fibers pass through the caudate, and normal auditory transmission to the cortex could be affected by damage to this region.
Fig. 1.
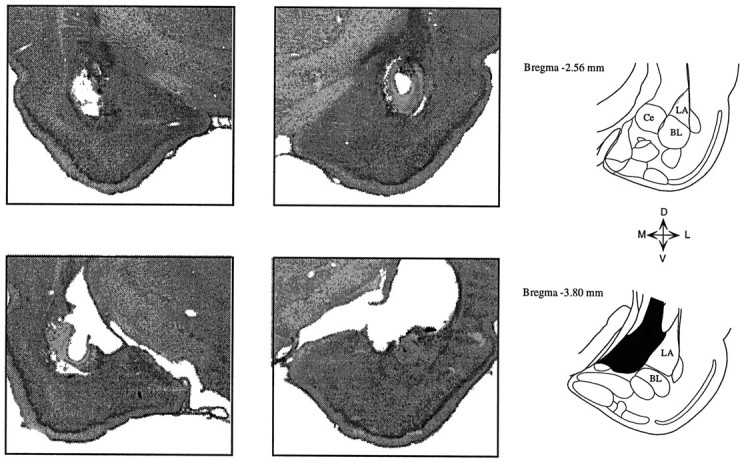
Photomicrographs of coronal sections showing a typical bilateral amygdala lesion. Lesions were centered at the lateral and basolateral nuclei, and in some cases damage extended into the central nucleus. Damage to the caudate nucleus above the amygdala was variable from case to case. Ce, Central nucleus;LA, lateral nucleus; BL, basolateral nucleus.
Recording electrode placements were located in auditory cortex (mainly in association areas Te1v and Te3). Figure2 illustrates the sites of the individual electrode bundles for all the lesion and control animals used in this study, showing a clear overlap of recording locations between the two groups.
Fig. 2.

Drawing of a coronal section, at ∼5 mm posterior to bregma, showing the electrode placements for control (open circles) and lesioned (filled circles) animals. Each circle corresponds to one electrode placement (each rat was implanted only once). Most placements were located in areas Te1v/Te3, which are part of the auditory association cortex in the rat. Te1, Te1v, Te3, Temporal cortex, areas 1–3. Adapted from Swanson (1992).
Behavior
The effects of CS presentation on fear behavior in the animals from which successful recordings were made was assessed by measuring the amount of time accounted for by freezing behavior during a 20 sec exposure to the CS outside of the conditioning context before and after conditioning. Before the sensitization phase, no rats in either of the groups (control and amygdala lesion) showed any substantial freezing to the CS (0.07 ± 0.07 sec and 0.13 ± 0.13 sec, respectively). After conditioning, control animals exhibited a significant increase in freezing to the CS [8.7 ± 2.0 sec; t(12)= 4.45; p < 0.001), whereas rats with amygdala lesion showed no increase in their freezing to the CS, even after 20 pairings (mean freezing time after conditioning = 0.50 ± 0.40 sec;t < 1). These results, shown in Figure3, are consistent with a large body of literature demonstrating a crucial role for the amygdala in the acquisition and expression of conditioned fear responses (for review, see Davis, 1992; Kapp et al., 1992; LeDoux, 1995, 1996; Gallagher and Chiba, 1996; Maren and Fanselow, 1996).
Fig. 3.

Freezing responses to the 20 sec tone conditioned stimulus (CS) before and after conditioning for control (n = 14) and lesion (n = 11) groups. After conditioning, control rats exhibited a significant increase in freezing to the CS (p < 0.001), whereas amygdala-lesioned animals showed no change.
Unit activity
A total of 19 lesioned animals completed the full conditioning protocol. Eight of those were later discarded because of incomplete amygdala lesions or significant damage in the caudate-putamen (see Materials and Methods), or because no individual cells could be isolated satisfactorily throughout the entire experiment. Thirty-two cells were isolated, held, and analyzed from the remaining 11 rats. The average number of cells recorded per animal was similar to the control cases (40 cells in 14 rats). Furthermore, the average spontaneous firing rate of cells before conditioning—collected for 500 msec before each tone trial—in control and lesioned animals was similar: 3.9 ± 0.7 and 5.5 ± 1.1 Hz, respectively. Overall, conditioning did not have a significant effect on the spontaneous firing of cells in either group: the average spontaneous rates after conditioning were 3.1 ± 0.6 and 5.6 ± 1.1 Hz for the control and lesioned animals, respectively. That is, amygdala lesions did not have an effect on the number cells that could be discriminated, the quality of the recordings, or the basic properties of the cells. The number of cells studied was comparable to other conditioning studies of the auditory cortex (Cruikshank and Weinberger, 1996).
Onset tone responses before and after conditioning
Before conditioning, cells in auditory cortex in both control and lesioned animals responded to the tone at short latencies, within the first 50 msec after tone onset. A typical example of an auditory cortex neuron showing onset tone responses in a rat with amygdala damage is shown in Figure 4. Overall, 8/32 (25%) of cells exhibited significant excitatory onset responses in the lesioned animals compared with 14/40 (35%) cells in the control group. These percentages are statistically indistinguishable (χ2= 0.84; p = 0.36).
Fig. 4.

Poststimulus time histograms showing the tone response of a cell in an amygdala-lesioned rat. This cell exhibited a short-latency (<50 msec) onset response before conditioning. As a result of conditioning, the magnitude of the onset response increased relative to the sensitization phase (Pre-Conditioning). Each histogram represents 10 trials; bin width is 50 msec.
After conditioning, 12 cells (30%) in the control group exhibited an increase in their firing rate in the first 50 msec after tone onset (see Materials and Methods). A similar proportion of neurons (12 cells; 37%) showed conditioning-induced onset increases in the amygdalectomized rats (χ2 = 0.45; p = 0.50). Two (5%) and three (9%) of these cells in the control and lesion groups, respectively, did not respond to the tone during the sensitization phase but developed onset tone responses as a result of conditioning. An example of conditioned increases in a cell from a lesioned animal is shown in Figure 4. Thus, amygdala lesions affected neither short-latency (0–50 msec) tone responses nor the likelihood that conditioning would induce onset plasticity in those responses.
Conditioning-induced late tone responses
In addition to the short-latency onset plasticity described above, conditioning also produced increases in tone responses that started around 500 msec after tone onset and lasted for ∼1 sec (Quirk et al., 1997). We refer to these as late conditioned tone responses. An example of such late conditioned increase in an intact animal is shown in Figure 5. Before conditioning, this cell did not respond to the tone. Conditioning did not have an effect on the onset response of the cell to the tone. After CS–US pairing, however, a significant late response to the tone developed, starting at ∼700 msec after tone onset.
Fig. 5.

Example of a cell in a control rat showing long-latency conditioned tone responses. Before conditioning, the cell did not respond to the tone. After conditioning, however, the cell developed a significant tone response, starting at ∼500 msec after tone onset and achieving its peak at 1500 msec after tone onset, which was the time of footshock onset during conditioning (indicated by thearrow). Shock was not delivered during these tests, because they occurred during extinction trials. Each histogram represents 10 trials; bin size is 100 msec. No cells showing these late conditioned responses were found in lesioned animals (see Results).
The development of late tone responses as a result of conditioning was a general phenomenon in cells of control animals. Figure6 (top) shows the percentage of tone-responsive neurons in intact rats throughout the tone, with a 100 msec bin resolution, before and after conditioning. Before conditioning, most tone responses were concentrated at the onset and offset of the tone, with little signaling during the rest of the stimulus. After conditioning, however, we observed a substantial increase in the number of cells exhibiting late tone responses in two or more bins (see Materials and Methods). As in the example shown in Figure 5, these late responses started at ∼500 msec after tone onset and peaked at 1500 msec, which was the latency of the shock onset during the conditioning phase of the experiment. In contrast, as shown in Figure 6 (bottom), no evidence for late conditioned tone-responsive cells was seen in lesioned animals.
Fig. 6.
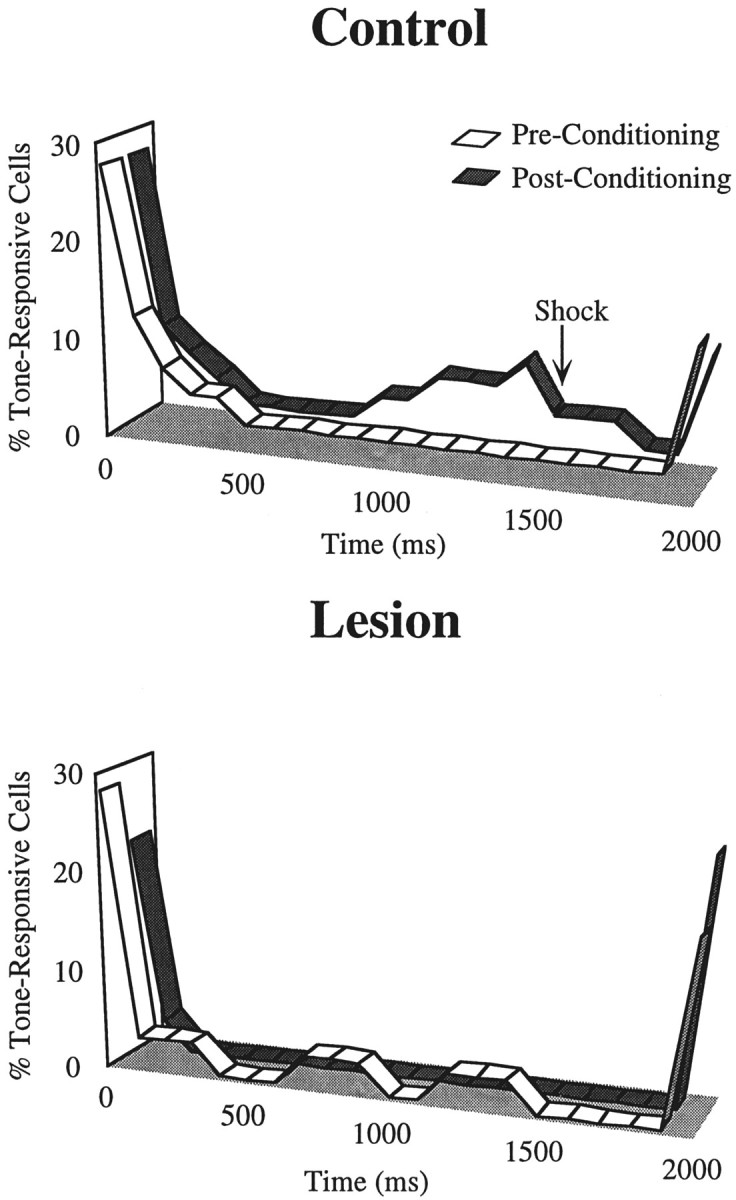
Ribbon plots showing the percentage of cells that were significantly tone responsive (see Materials and Methods) before (white) and after (gray) conditioning for the control and lesion groups. Although there were prominent onset and offset responses before and after conditioning in both groups, conditioning resulted in an increase in the number of cells in the control group showing late responses to the tone CS, starting at ∼900 msec after tone onset and reaching a peak at 1500 msec (time of shock onset during conditioning). This conditioning-induced late response was absent in lesioned animals. The duration of the tone was 2 sec; bin width is 100 msec.
To make a statistical comparison of the number of neurons showing conditioning-induced late tone responses in control and lesion groups, we defined a region of interest between 500 msec (end of all onset responses) and 1500 msec (onset of footshock). The number of tone-responsive cells in each group in this region was calculated using two-tailed, paired t tests (p < 0.05) (see Materials and Methods). After conditioning, 11 of 40 cells (28%) in control animals showed late tone responses. (A color-coded raster plot of the response of these cells throughout training is shown in Fig. 7A.) In contrast, only 2 of 32 cells (6%) in lesioned animals exhibited tone responses in the 500–1500 msec interval. A χ2 test revealed that the difference in these late conditioning-induced tone-responsive cells between control and lesion groups was significant (χ2 = 5.43; p < 0.02).
Fig. 7.

A, Color-coded raster plot showing the average firing rate of cells in the control group that exhibited significant late tone responses (500–1500 msec after tone onset; see Results) after conditioning (n = 11). Thetop of the figure shows the sensitization trials (preconditioning) and the bottom shows extinction trials (the conditioning phase is not shown). Each row is one trial, and the bin size is 100 msec. Firing rates were averaged across cells and smoothed with a nearest-neighbor average. Note that there was no indication of late responses before conditioning, and that the long-latency conditioned response is maximal (red) just before the time of the shock onset during conditioning (indicated by the arrow), similar to the example shown in Figure 5.B, Color-coded raster plot showing the average firing rate of cells that develop short-latency (<50 msec) onset conditioned increases, for the control (n = 12) and lesion (n = 12) groups. Each row is one trial, showing the first 500 msec after tone onset; bin size is 10 msec. The top half shows the 10 sensitization and 20 conditioning trials, and the bottom half shows the 30 extinction trials. Animals were returned to their home cages for 1 hr after the end of conditioning and before the beginning of extinction. Note that in the control group the magnitude of onset responses after 30 extinction trials remained elevated relative to the response before conditioning (sensitization). In contrast, cells in lesioned animals returned to their preconditioning levels after 10 extinction trials.
In summary, conditioning resulted in a significant increase in the number of cells displaying a late (500–1500 msec) tone response in intact animals, but no such changes were observed in cells recorded in animals with amygdala lesions. Unlike onset conditioned increases (see below), late conditioning-induced tone diminished with extinction trials in the control animals.
Extinction of onset conditioned responses
One hour after the end of the conditioning phase, the animals were brought back to the test chamber and exposed to 30 extinction trials (tone alone). As reported previously (Quirk et al., 1997), auditory cortex cells in intact animals retained the conditioning-induced increases of their tone responses after 30 extinction trials (in contrast, extinction occurred in lateral amygdala cells under the same conditions) (Quirk et al., 1997). An example of an auditory cell from a control animal exhibiting extinction-resistant onset conditioning in the present study is shown in Figure8A. In contrast, cells recorded in the auditory cortex of amygdalectomized rats returned to their preconditioning level during extinction, as shown in Figure8B. Figure 7B shows a color-coded average raster plot of all the cells that exhibited an increase in their onset response (0–50 msec) during conditioning (see above) as a function of the trials in the different training phases (sensitization, conditioning, and extinction). Before conditioning, a slightly smaller number (but not statistically significant; see above) of cells in the lesion group responded to the tone onset (hence the lower firing rate in this group seen in Fig. 7B, when compared with controls), but immediately after conditioning, both groups showed a similar number and strength of onset tone responses. Cells in lesioned and control animals, however, exhibited very different response patterns during extinction. Whereas in the control group animals the conditioned onset response in auditory cortex was still higher than the preconditioning (sensitization) response after 30 extinction trials, responses of auditory cortex cells in amygdala-lesioned rats extinguished their conditioned onset increases after approximately 10 extinction trials. This effect was confirmed statistically by comparing the responses at the beginning (average of trials 1–10) and end (average of trials 20–30) of extinction. Cells in control animals showed no extinction in their onset responses (paired t test; p > 0.1), whereas cells in the lesioned rats significantly decreased their onset response to the tone as a function of extinction trials (p < 0.005), as shown in Figure9.
Fig. 8.

Poststimulus time histograms showing the effects of extinction on short-latency (<50 msec) onset conditioning in cells from control (left) and lesioned (right) animals. The cell from the control animal developed onset conditioned increases after conditioning (extinction trials 1–10) that lasted through the entire extinction phase (30 trials). In contrast, the cell from the lesioned rat developed a short-lasting onset conditioned increase and returned to its preconditioning response level at the end of the extinction phase.
Fig. 9.
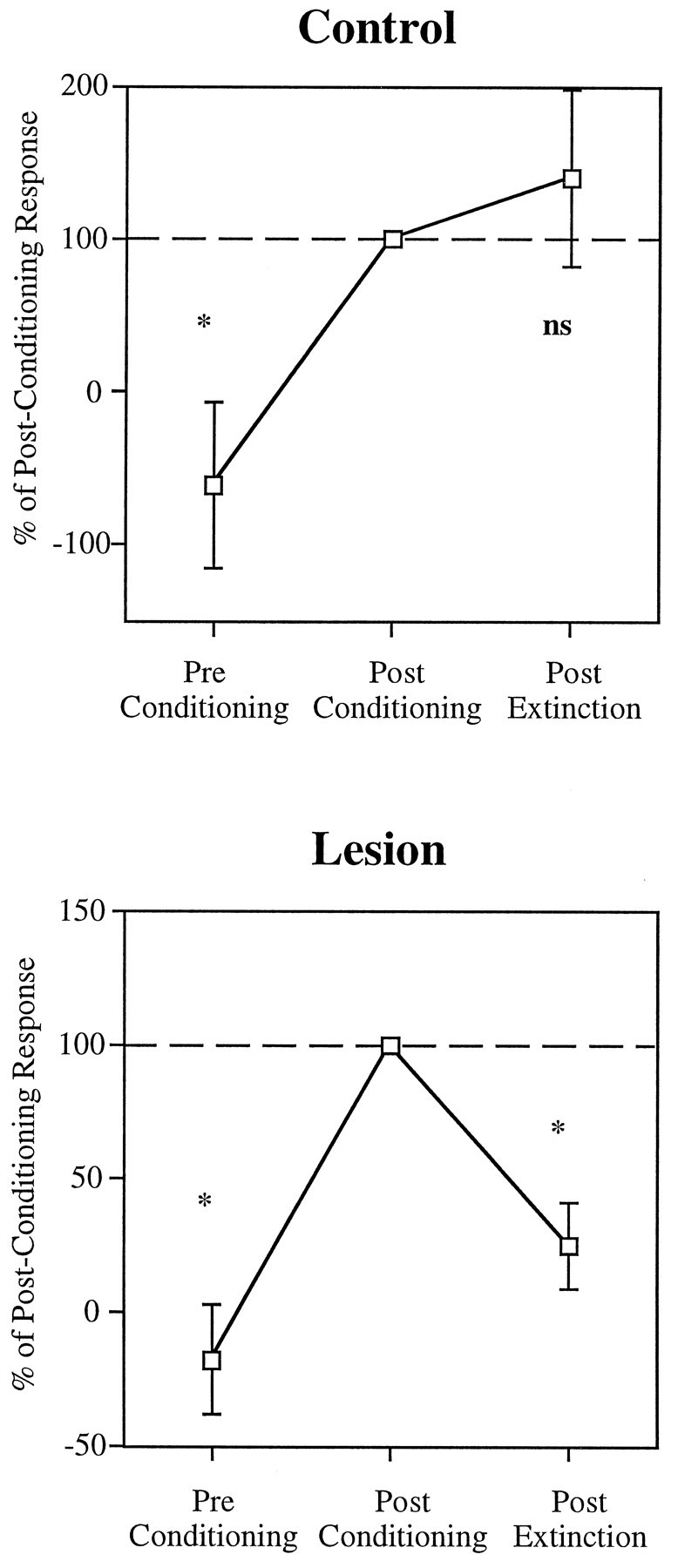
Average onset tone responses (0–50 msec) of cells that conditioned, from the control and lesion groups, plotted for the 10 sensitization trials (Pre Conditioning), the first 10 trials of extinction (Post Conditioning), and the last 10 trials of extinction (trials 20–30; Post Extinction), relative to the postconditioning tone response. In both groups, tone responses before conditioning were significantly smaller than after conditioning (p < 0.01). In the control group, the responses at the end of extinction did not differ from the postconditioning values (p> 0.1), whereas in the lesion group extinction trials resulted in a significant reduction of the tone responses (p < 0.005).
DISCUSSION
We recorded from individual neurons in auditory cortex in rats with bilateral amygdala lesions during fear conditioning and compared the results with those obtained in intact animals (Quirk et al., 1997).
Short-latency (onset) responses
Amygdala lesions did not affect short-latency (0–50 msec) tone responses in auditory cortex that exist before conditioning. Moreover, there was no effect of the lesions on the conditioning-induced modification of these short-latency responses. These results are consistent with the idea that short-latency conditioned tone responses in auditory cortex are driven by direct inputs from the thalamus (Quirk et al., 1996) and thus do not need to rely on an intact amygdala. Although onset conditioning develops in the auditory thalamus (Disterhoft and Stewart, 1976; Gabriel et al., 1976; Ryugo and Weinberger, 1978; Edeline et al., 1990), it has not been established whether thalamic cells that project to the amygdala are plastic, nor is it known whether such plasticity requires an intact amygdala. [A recent study by Poremba and Gabriel (1997) showed that amygdala lesions interfere with thalamic plasticity—although not in the auditory thalamus—in an avoidance conditioning task in rabbits.]
Late conditioned responses
The majority of analyses of plasticity in auditory cortex during conditioning have focused on short-latency (<50msec) responses (for review, see Weinberger, 1995). However, we have observed recently that several cells (11 of 40; 28%) in auditory cortex in control animals showed a late conditioned response to the CS, which lasted from ∼500 to 1500 msec (Quirk et al., 1997). As illustrated in Figure7A, most cells exhibiting this late conditioned response did not show any response to the CS before conditioning. Thus, conditioning resulted in a recruitment of cells that started to respond to the late part of the CS. Interestingly, this conditioned neural response peaked at 1500 msec, which was the latency of the US during the conditioning phase. We have hypothesized that this response may reflect some attentional or anticipatory mechanism (Armony et al., 1997b; Quirk et al., 1997). No evidence of this late response was observed in cells recorded in animals with amygdala lesions, suggesting that this “anticipatory” response may be amygdala-dependent (see below). This amygdala dependency could reflect direct projections to the cortex from the amygdala, feedback to the cortex from various amygdala-triggered responses, including nonspecific arousal responses within the brain (described in more detail below), or somatic, autonomic, or endocrine responses expressed peripherally and returning to the brain (LeDoux, 1996).
Extinction of onset conditioned increases
Extinction trials (presentation of the CS alone) result in a reduction of behavioral conditioned responses. In previous studies involving a conditioning protocol identical to the one used in the present experiment, we observed that cells in the lateral nucleus of the amygdala, the sensory gateway to the amygdala, lost their onset conditioned increases (i.e., they returned to their preconditioning levels of tone-elicited spike trains) after 30 extinction trials (Quirk et al., 1995). In contrast, cells in the auditory cortex in intact animals did not show any significant extinction in their short-latency responses to the CS (Quirk et al., 1997), consistent with the idea that the cortex, specifically sensory areas, may be involved in the storage of some aspect of fear memories.
Interestingly, although amygdala lesions did not interfere with the development of onset conditioning in auditory cortex, they did have an effect on the rate of extinction of such increases. Conditioned cells returned to their preconditioning level of responses after ∼10 extinction trials, as shown in Figure 7B. That is, although the lateral nucleus of the amygdala itself does not seem to be a site of extinction-resistant storage of the CS–US pairing information, the amygdala nonetheless appears to be necessary for the consolidation or maintenance of extinction-resistant responses in auditory cortex. Whether other amygdala subnuclei downstream from the lateral nucleus, such as basal or central nuclei, maintain or extinguish their conditioned responses after presentations of the unreinforced CS is unknown and is an important question for future research, especially because these downstream areas are the source of fibers that project back to the cortex (Amaral et al., 1992) and to other areas that allow the amygdala to influence the cortex indirectly (for a summary, seeLeDoux, 1996).
Role of the amygdala in fear processing
A large body of evidence lends support for a critical role of the amygdala in controlling conditioned fear responses, through its projections to brainstem structures (Davis, 1992; Kapp et al., 1992;LeDoux, 1995, 1996). Little is known, however, about the role of the amygdala in modulating cortical areas during fear conditioning. Nuclei of the amygdala have extensive projections back to cortical areas, and these could play a role in modulating cortical processing of emotional stimuli (Amaral et al., 1992; LeDoux, 1996). A number of hypotheses have been proposed suggesting that the amygdala may modulate mnemonic and attentional processing in cortical areas (Kapp et al., 1992; Rolls, 1992; Gallagher and Holland, 1994; McGaugh et al., 1995; Packard et al., 1995; Weinberger, 1995; Cahill and McGaugh, 1996; Armony et al., 1997b). For example, stimulation of the central nucleus produces neocortical arousal, which is mediated by cholinergic projections from the basal forebrain in rabbit (Kapp et al., 1994), and stimulation of the nucleus basalis produces facilitation of neuronal responses to tones in auditory cortex in awake rats (Edeline et al., 1994). Furthermore, pairing of a tone with iontophoretic application of ACh induces frequency-specific changes in auditory cortex neurons, similar to those observed during fear conditioning (Metherate and Weinberger, 1990). Findings such as these have led Weinberger and colleagues to propose a conceptual model in which cortical plasticity in fear conditioning requires concurrent auditory input from the thalamus and amygdala-elicited diffuse cholinergic input from the basal forebrain (for review, see Weinberger, 1995). According to this hypothesis, a lesion of the amygdala would completely abolish the development of cortical plasticity. The present results do not lend support to that model, because we found that amygdala lesions failed to affect the induction of short-latency (0–50 msec) fear-conditioned cortical plasticity in auditory cortex. Nonetheless, amygdala lesions did block the development of late (500–1500 msec) tone responses after conditioning. Thus, the model of Weinberger and coworkers might be more apt for this component of the conditioned response.
We have recently developed a computational model of emotion–cognition interactions grounded on the anatomy and physiology of the fear conditioning circuit (Armony et al., 1997b). The model is consistent with the results reported here. According to the model, auditory cortex cortical plasticity induced by direct thalamocortical projections, through Hebbian learning, should be independent of the amygdala. Auditory thalamic projections to the amygdala are also thought to be responsible for the plasticity in the amygdala, both in the model (Armony et al., 1995) and in experimental recordings (Quirk et al., 1995). In turn, the amygdala may influence later cortical responses, by way of either direct or indirect projections. Indirect sources include the various nonspecific arousal or modulatory systems in the brain, as well as feedback from peripherally expressed responses (for a summary, see LeDoux, 1996). These late, amygdala-dependent responses may redirect attentional resources to the CS, possibly in anticipation of, and in preparation for, the expected arrival of the US. This hypothesis is still fairly speculative, but it does integrate much of the data described above.
Conclusion
The results presented in this study are consistent with the notion of complementary memory systems that form neural traces in parallel during emotional arousal. Through amygdala-based plasticity, an association is formed between the CS and the aversive US such that the CS gains access to evolutionarily programmed defense responses mediated by amygdala outputs. Plasticity also occurs in cortical areas and makes possible the establishment of explicit or declarative memories about the situation through interactions with the medial temporal lobe memory system (McClelland et al., 1995; Squire and Knowlton, 1995; Eichenbaum et al., 1996). At the same time, activation of the amygdala during the emotional event modulates the strength of explicit, declarative memories represented in various cortical memory circuits (McGaugh et al., 1995, 1996; Cahill and McGaugh, 1996).
Our findings begin to make explicit the manner in which the amygdala might influence cortically based memories. Specifically, the relatively early (within 50 msec) representation is processed in the auditory cortex independent of the amygdala. This plasticity involves, for the most part, the enhancement of preexisting responses to the CS.
Late responses to the CS (500–1500 msec), responses that did not exist before training, anticipate the arrival of the US and are dependent on the amygdala. We hypothesize that the early and late auditory cortex conditioned responses are part of the explicit or declarative representation of the experience processed in conjunction with the medial temporal lobe memory system and that the amygdala modulates the late but not the early component of this representation of the experience. This hypothesis is guiding additional studies in our attempt to understand how complementary memory systems interact during emotional learning.
Footnotes
This work was supported by Public Health Service Grants MH38774, MH46516, and MH00956 to J.E.L. We are grateful to Mian Hou for help with histology.
Correspondence should be addressed to Joseph E. LeDoux, Center for Neural Science, New York University, 4 Washington Place, Room 809, New York, NY 10003-6621.
Dr. Armony’s present address: Department of Experimental Psychology, University of Oxford, South Parks Road, Oxford OX1 3UD, UK.
Dr. Quirk’s present address: Department of Physiology, Ponce School of Medicine, P.O. Box 7004, Ponce, Puerto Rico 00732-7004.
REFERENCES
- 1.Amaral DG. In: Memory: anatomical organization of candidate brain regions. Handbook of physiology. Section 1: The nervous system. Plum F, editor. American Physiological Society; Bethesda, MD: 1987. pp. 211–294. [Google Scholar]
- 2.Amaral DG, Price JL, Pitkanen A, Carmichael ST. Anatomical organization of the primate amygdaloid complex. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 1–66. [Google Scholar]
- 3.Armony JL, Servan-Schreiber D, Cohen JD, LeDoux JE. An anatomically constrained neural network model of fear conditioning. Behav Neurosci. 1995;109:246–257. doi: 10.1037//0735-7044.109.2.246. [DOI] [PubMed] [Google Scholar]
- 4.Armony JL, Servan-Schreiber D, Romanski LM, Cohen JD, LeDoux JE. Stimulus generalization of fear responses: effects of auditory cortex lesions in a computational model and in rats. Cereb Cortex. 1997a;7:157–165. doi: 10.1093/cercor/7.2.157. [DOI] [PubMed] [Google Scholar]
- 5.Armony JL, Servan-Schreiber D, Cohen JD, LeDoux JE. Computational modeling of emotion: explorations through the anatomy and physiology of fear conditioning. Trends Cognit Sci. 1997b;1:28–34. doi: 10.1016/S1364-6613(97)01007-3. [DOI] [PubMed] [Google Scholar]
- 6.Babinsky R, Calabrese P, Durwen HF, Markowitsch HJ, Brechtelsbauer D, Heuser L, Gehlen W. The possible contribution of the amygdala to memory. Behav Neurol. 1993;6:167–170. doi: 10.3233/BEN-1993-6310. [DOI] [PubMed] [Google Scholar]
- 7.Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–1118. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- 9.Bolles RC, Fanselow MS. A perceptual-defensive-recuperative model of fear and pain. Behav Brain Sci. 1980;3:291–323. [Google Scholar]
- 10.Cahill L, McGaugh JL. Modulation of memory storage. Curr Opin Neurobiol. 1996;6:237–242. doi: 10.1016/s0959-4388(96)80078-x. [DOI] [PubMed] [Google Scholar]
- 11.Cahill L, Babinsky R, Markowitsch HJ, McGaugh JL. The amygdala and emotional memory. Nature. 1995;377:295–296. doi: 10.1038/377295a0. [DOI] [PubMed] [Google Scholar]
- 12.Cruikshank SJ, Weinberger NM. Receptive-field plasticity in the adult auditory cortex induced by Hebbian covariance. J Neurosci. 1996;16:861–875. doi: 10.1523/JNEUROSCI.16-02-00861.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis M. The role of the amygdala in conditioned fear. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 255–306. [Google Scholar]
- 14.Diamond DM, Weinberger NM. Physiological plasticity of single neurons in auditory cortex of the cat during acquisition of the pupillary conditioned response: II. Secondary field (AII). Behav Neurosci. 1984;98:189–210. [PubMed] [Google Scholar]
- 15.Diamond DM, Weinberger NM. Classical conditioning rapidly induces specific changes in frequency receptive fields of single neurons in secondary and ventral ectosylvian auditory cortical fields. Brain Res. 1986;372:357–360. doi: 10.1016/0006-8993(86)91144-3. [DOI] [PubMed] [Google Scholar]
- 16.Disterhoft J, Stuart D. Trial sequence of changed unit activity in auditory system of alert rat during conditioned response acquisition and extinction. J Neurophysiol. 1976;39:266–281. doi: 10.1152/jn.1976.39.2.266. [DOI] [PubMed] [Google Scholar]
- 17.Edeline J-M, Neuenschwander-El Massioui N, Dutrieux G. Discriminative long-term retention of rapidly induced multiunit changes in the hippocampus, medial geniculate and auditory cortex. Behav Brain Res. 1990;39:145–155. doi: 10.1016/0166-4328(90)90101-j. [DOI] [PubMed] [Google Scholar]
- 18.Edeline J-M, Hars B, Maho C, Hennevin E. Transient and prolonged facilitation of tone-evoked responses induced by basal forebrain stimulations in the rat auditory cortex. Exp Brain Res. 1994;97:373–386. doi: 10.1007/BF00241531. [DOI] [PubMed] [Google Scholar]
- 19.Eichenbaum H, Otto T, Cohen NJ. Two functional components of the hippocampal memory system. Behav Brain Sci. 1994;17:449–518. [Google Scholar]
- 20.Eichenbaum H, Schoenbaum G, Young B, Bunsey M. Functional organization of the hippocampal memory system. Proc Natl Acad Sci USA. 1996;93:13500–13507. doi: 10.1073/pnas.93.24.13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabriel M, Slatwick SE, Miller JD. Multiple unit activity of the rabbit medial geniculate nucleus in conditioning, extinction, and reversal. Physiol Psychol. 1976;4:124–134. [Google Scholar]
- 22.Gallagher M, Chiba AA. The amygdala and emotion. Curr Opin Neurobiol. 1996;6:221–227. doi: 10.1016/s0959-4388(96)80076-6. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher M, Holland PC. The amygdala complex: multiple roles in associative learning and attention. Proc Natl Acad Sci USA. 1994;91:11771–11776. doi: 10.1073/pnas.91.25.11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwata J, LeDoux JE, Meeley MP, Arneric S, Reis DJ. Intrinsic neurons in the amygdaloid field projected to by the medial geniculate body mediate emotional responses conditioned to acoustic stimuli. Brain Res. 1986;383:195–214. doi: 10.1016/0006-8993(86)90020-x. [DOI] [PubMed] [Google Scholar]
- 25.Kapp BS, Whalen PJ, Supple WF, Pascoe JP. Amygdaloid contributions to conditioned arousal and sensory information processing. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 229–254. [Google Scholar]
- 26.Kapp BS, Supple WF, Whalen PJ. Effects of electrical stimulation of the amygdaloid central nucleus on neocortical arousal in the rabbit. Behav Neurosci. 1994;108:81–93. doi: 10.1037//0735-7044.108.1.81. [DOI] [PubMed] [Google Scholar]
- 27.Kubie JL. A driveable bundle of microwires for collecting single-unit data from freely-moving rats. Physiol Behav. 1984;32:115–118. doi: 10.1016/0031-9384(84)90080-5. [DOI] [PubMed] [Google Scholar]
- 28.LaBar KS, LeDoux JE. Partial disruption of fear conditioning in rats with unilateral amygdala damage: correspondence with unilateral temporal lobectomy in humans. Behav Neurosci. 1996;110:991–997. doi: 10.1037//0735-7044.110.5.991. [DOI] [PubMed] [Google Scholar]
- 29.LaBar KS, LeDoux JE, Spencer DD, Phelps EA. Impaired fear conditioning following unilateral temporal lobectomy in humans. J Neurosci. 1995;15:6846–6855. doi: 10.1523/JNEUROSCI.15-10-06846.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laroche S, Doyere V, Bloch V. Linear relation between the magnitude of long-term potentiation in the dentate gyrus and associative learning in the rat. A demonstration using commissural inhibition and local infusion of an N-methyl-d-aspartate receptor antagonist. Neuroscience. 1989;28:375–386. doi: 10.1016/0306-4522(89)90184-x. [DOI] [PubMed] [Google Scholar]
- 31.LeDoux JE. Emotion: clues from the brain. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- 32.LeDoux J. The emotional brain. Simon & Schuster; New York: 1996. [Google Scholar]
- 33.LeDoux JE, Ruggiero DA, Reis DJ. Projections to the subcortical forebrain from anatomically defined regions of the medial geniculate body in the rat. J Comp Neurol. 1985;242:182–213. doi: 10.1002/cne.902420204. [DOI] [PubMed] [Google Scholar]
- 34.Maren S, Fanselow MS. The amygdala and fear conditioning: has the nut been cracked? Neuron. 1996;16:237–240. doi: 10.1016/s0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- 35.McClelland JL, McNaughton BL, O’Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev. 1995;102:419–457. doi: 10.1037/0033-295X.102.3.419. [DOI] [PubMed] [Google Scholar]
- 36.McGaugh JL, Cahill L, Parent MB, Mesches MH, Coleman-Mesches K, Salinas JA. Involvement of the amygdala in the regulation of memory storage. In: McGaugh JL, Bermudez-Rattoni F, Prado-Alcala RA, editors. Plasticity in the central nervous system: learning and memory. Lawrence Erlbaum; Hillside, NJ: 1995. pp. 17–40. [Google Scholar]
- 37.McGaugh JL, Cahill L, Roozendal B. Involvement of the amygdala in memory storage: interaction with other brain systems. Proc Natl Acad Sci USA. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metherate R, Weinberger NM. Cholinergic modulation of responses to single tones produces tone-specific receptive field alterations in cat auditory cortex. Synapse. 1990;6:133–145. doi: 10.1002/syn.890060204. [DOI] [PubMed] [Google Scholar]
- 39.Olds J, Disterhoft J, Segal M, Kornbluth C, Hirsh R. Learning centers of rat brain mapped by measuring latencies of conditioned unit responses. J Neurophysiol. 1972;35:202–219. doi: 10.1152/jn.1972.35.2.202. [DOI] [PubMed] [Google Scholar]
- 40.Packard MG, Williams CL, Cahill L, McGaugh JL. The anatomy of a memory modulatory system: from periphery to brain. In: Spear NE, Spear LP, Woodruff ML, editors. Neurobehavioral plasticity: learning, development, and response to brain insults. Lawrence Erlbaum; Hillside, NJ: 1995. pp. 149–150. [Google Scholar]
- 41.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 42.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 43.Poremba A, Gabriel M. Amygdalar lesions block discriminative avoidance learning and cingulothalamic training-induced neuronal plasticity in rabbits. J Neurosci. 1997;17:5237–5244. doi: 10.1523/JNEUROSCI.17-13-05237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quirk GJ, Repa JC, LeDoux JE. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron. 1995;15:1029–1039. doi: 10.1016/0896-6273(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 45.Quirk GJ, Armony JL, Repa JC, Li XF, LeDoux JE. Emotional memory: a search for sites of plasticity. In: Stillman B, editor. Cold Spring Harbor Symposia on Quantitative Biology, Vol 61. Cold Spring Harbor; New York: 1996. pp. 247–257. [PubMed] [Google Scholar]
- 46.Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 47.Rolls ET. Neurophysiology and functions of the primate amygdala. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 143–165. [Google Scholar]
- 48.Romanski LM, LeDoux JE. Equipotentiality of thalamo-amygdala and thalamo-cortico-amygdala projections as auditory conditioned stimulus pathways. J Neurosci. 1992;12:4501–4509. doi: 10.1523/JNEUROSCI.12-11-04501.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romanski LM, LeDoux JE. Information cascade from primary auditory cortex to the amygdala: corticocortical and corticoamygdaloid projections of temporal cortex in the rat. Cereb Cortex. 1993;3:515–532. doi: 10.1093/cercor/3.6.515. [DOI] [PubMed] [Google Scholar]
- 50.Ryugo DK, Weinberger NM. Differential plasticity of morphologically distinct neuron populations in the medial geniculate body of the cat during classical conditioning. Behav Biol. 1978;22:275–301. [Google Scholar]
- 51.Selden NRW, Everitt BJ, Jarrard LE, Robbins TW. Complementary roles for the amygdala and hippocampus in aversive conditioning to explicit and contextual cues. Neuroscience. 1991;42:335–350. doi: 10.1016/0306-4522(91)90379-3. [DOI] [PubMed] [Google Scholar]
- 52.Squire LR, Knowlton BJ. Learning about categories in the absence of memory. Proc Natl Acad Sci USA. 1995;92:12470–12474. doi: 10.1073/pnas.92.26.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Squire LR, Zola SM. Structure and function of declarative and nondeclarative memory systems. Proc Natl Acad Sci USA. 1996;93:13515–13522. doi: 10.1073/pnas.93.24.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swanson LW. Brain maps: structure of the rat brain. Academic; New York: 1992. [Google Scholar]
- 55.Weinberger NM. Retuning the brain by fear conditioning. In: Gazzaniga MS, editor. The cognitive neurosciences. MIT; Cambridge, MA: 1995. pp. 1071–1090. [Google Scholar]
- 56.Weinberger NM, Hopkins W, Diamond DM. Physiological plasticity of single neurons in auditory cortex of the cat during acquisition of the pupillary conditioned response: I. Primary field (AI). Behav Neurosci. 1984;98:171–188. doi: 10.1037//0735-7044.98.2.171. [DOI] [PubMed] [Google Scholar]
- 57.Zilles K, Wree A. Cortex: Areal and laminar structure. In: Paxinos G, editor. The rat nervous system. Academic; New York: 1985. pp. 375–416. [Google Scholar]