

Abstract
The relationship between bacterial communities and their host is being extensively investigated for the potential to improve the host’s health. Little is known about the interplay between the microbiota of parasites and the health of the infected host. Using nematode co-infection of lambs as a proof-of-concept model, the aim of this study was to characterise the microbiomes of nematodes and that of their host, enabling identification of candidate nematode-specific microbiota member(s) that could be exploited as drug development tools or for targeted therapy. Deep sequencing techniques were used to elucidate the microbiomes of different life stages of two parasitic nematodes of ruminants, Haemonchus contortus and Teladorsagia circumcincta, as well as that of the co-infected ovine hosts, pre- and post infection. Bioinformatic analyses demonstrated significant differences between the composition of the nematode and ovine microbiomes. The two nematode species also differed significantly. The data indicated a shift in the constitution of the larval nematode microbiome after exposure to the ovine microbiome, and in the ovine intestinal microbial community over time as a result of helminth co-infection. Several bacterial species were identified in nematodes that were absent from their surrounding abomasal environment, the most significant of which included Escherichia coli/Shigella. The ability to purposefully infect nematode species with engineered E. coli was demonstrated in vitro, validating the concept of using this bacterium as a nematode-specific drug development tool and/or drug delivery vehicle. To our knowledge, this is the first description of the concept of exploiting a parasite’s microbiome for drug development and treatment purposes.
Subject terms: Microbiome, Infectious diseases
Introduction
Nematode infection is of major concern to human health in middle- and low-income countries, particularly in cases of foodborne disease [1]. In addition, animals infected by pathogenic nematodes are a serious health, welfare and economic burden for countries reliant on agriculture [2]. Effective interventions are therefore necessary to promote human health, protect livestock, and ensure production efficiency. Current standard practices for eradicating helminthic disease focus on the routine and frequent administration of anthelmintics, small-molecule drugs, to infected hosts. However, as with many chemicals, the development of resistance means that these drugs’ effectiveness is reducing [3], and alternative treatments are of paramount importance [4]. Large numbers of new chemical drug classes are unlikely to be synthesised and licensed to combat growing drug resistance in nematodes in the near future, given the large time commitment required for drug research and development [5]. Admittedly, a small number of compounds are at the early stage of investigation for controlling human whipworm infections [6, 7]. Yet, contingency strategies and tools to help expedite drug development are still desirable.
In parasitic disease, attempts have been made to characterise the interplay between helminths and the bacterial populations inhabiting the mammalian gut, elucidating the ways in which the activity of the parasite affects the constituency of the gut microbiota and vice versa [8–10]. These studies have suggested that the co-evolution of these two communities has established a relationship wherein the survival of either population is impacted by the other. Susceptibility and resistance to helminth infection in humans have been linked with certain bacterial taxa, suggesting that there may exist an ideal host microbial profile that guards against such disease [11]. In fact, it has recently been discovered that parasites themselves have a microbiome. The nematode microbiome has become an increasingly popular area of study and has seen considerable advancement over the past 2 years due to 16S rRNA gene-sequencing accessibility: the microbiomes of Caenorhabditis elegans [12], the ruminant parasite Haemonchus contortus [13], the murine parasite Trichuris muris [9], soil- and beetle-associated nematodes [14], the marine nematode Litoditis marina [15] and various other marine nematodes [16] have all been sequenced.
High-throughput technologies are ideally placed to examine the interplay between the microbial communities within nematodes and the microbial communities of the animals they infect. However, while big data have been utilised to expand our understanding of the nematode microbiome, less consideration has been given to how this information might be applied to the therapeutic benefit of parasite-infected organisms. Defining the microbial communities of nematodes and their host opens opportunities for exploiting differences for drug development and/or treatment purposes. Identifying bacterial communities that uniquely colonise the nematode presents an opportunity to investigate their use as oral agents that specifically target the parasite, leaving the host unaffected.
Exploitation of the host microbiota as a means of treating disease in the host is well studied across multiple species —from the use of faecal microbiota transplantation for inducing remission in ulcerative colitis in humans [17] to the treatment of laminitis in horses [18]; however, exploitation of the parasite microbiome as an aid to drug development and treatment has not yet been described. We hypothesised that (i) nematode co-infection of the host would significantly alter the host microbiome over time; (ii) the host microbiome would significantly alter the microbiome of the nematodes and (iii) despite interactions between host and parasite microbiota, key differences between the two would be apparent that would welcome their further investigation as aids to drug development and treatment.
In this study, the microbiomes of the ovine abomasum and intestines were characterised following co-infection of lambs with the pathogenic nematodes H. contortus and Teladorsagia circumcincta. The abomasum is one of four compartments of the ruminant stomach, in which H. contortus and T. circumcincta live [19], and of the four compartments bear the closest resemblance to the anatomy and functionality of the simple stomach of non-ruminants [20]. The microbiomes of both nematodes were also characterised at both the infective larval (L3) and adult stages of their development, marking this as the first report of the T. circumcincta microbiome and the first comparative study where different nematode genera are derived from the same host. The ovine model chosen is appropriate for a proof-of-concept study, and the blood-feeding parasite H. contortus is a good model system for blood-feeding nematodes. This study also offers insights into the effects of parasites on the host, and vice versa. The effects on the host are quantified by monitoring changes in the ovine microbiome over the 28 days of parasitic co-infection. Effects on the parasite are examined by comparing the microbiomes of pre- and post-infection nematode larvae.
Materials and methods
Ovine and parasite samples were collected at various time points over a 28-day infection (Supplementary Fig. 1).
Parasite material—adult nematodes
Four lambs were artificially co-infected per os with 15,000 infective larvae (L3; 5000 H. contortus and 10,000 T. circumcincta). Twenty-eight days post infection (i.e., at the point of culling), adult worms were collected from the abomasa of each lamb [21]. The nematodes were sexed, staged and species-identified using criteria described in the Ministry of Agriculture, Fisheries and Food document [22]. Separate pools of 100 adult male and 100 adult female worms were species-identified, washed twice in sterile phosphate-buffered saline (PBS) to remove surface-adherent bacteria, snap frozen in liquid nitrogen, and transferred to −80 °C storage prior to deoxyribonucleic acid (DNA) extraction. Both worm species were processed separately.
Parasite material—pre-infection and post-infection larvae
To provide an indication of the microbial diversity present within the L3 population that were used to generate the adult material, sub-samples of ~10,000 infective larvae used in the artificial challenge doses were snap-frozen in liquid nitrogen on the day of challenge and stored −80 °C storage prior to DNA extraction. Faecal material containing eggs (both H. contortus and T. circumcincta) from the patent parasite infections were collected from the infected donor lambs at postmortem (d28) and incubated at 22 °C for 14 days. Infective larvae derived from the d28 faeces were extracted, enumerated and identified to species level, snap-frozen in liquid nitrogen and stored at −80 °C in pools of ~ 10,000 larvae. Figure 1 shows the nematode lifecycle, and its association with the ruminant digestive system.
Fig. 1.

The nematode lifecycle and its association with the ruminant digestive system
Ovine faecal and abomasal sample collection
Individual faecal samples were collected per rectum at days 0, 1, 2, 5, 7, 9, 14, 19, 21 and 28 post infection from all donor animals. Faecal samples were transferred to −80 °C storage prior to DNA extraction. Sub-samples of abomasal contents were collected at postmortem from each lamb donor.
Confirmation of bacterial presence within nematodes
To validate the presence of bacteria within ovine nematodes, wax sections from H. contortus adult worms were Gram-stained following standard procedures [23].
Genomic DNA extraction
The adult worms were transferred to 2 -ml Lysing Matrix B tubes (MP Biomedicals) and were re-suspended in 500 μl sterile phosphate-buffered saline (PBS). The larvae were homongenised using a Precellys24 homogeniser (Bertin Technologies) at 6000 rpm for 30 s for three cycles. The DNA extraction was conducted using the DNeasy Blood and Tissue Kit (Qiagen). To homogenate tubes, 500 μl of ATL buffer supplemented with 12 mAU proteinase K (Promega) was added, followed by incubation at 56 °C for 2 h. To pellet the 0.1 -mm glass beads, the Lysing Matrix B tubes were centrifuged at 15,000 × g for 5 min. The supernatant was transferred to a clean 2 -ml microcentrifuge tube, and this step was repeated to ensure no glass beads were transferred to the DNeasy Mini spin columns. The DNeasy Blood and Tissue Kit guidelines for Animal Tissues (Spin-Column Protocol) were followed, eluting the DNA in 100 μl of Buffer AE before DNA quantification using a NanoDrop ND1000 UV–Vis spectrophotometer (NanoDrop Technologies) and the tubes were stored at −80 °C.
Controls
Negative control tubes were included to account for environmental contaminants present throughout the processing of the samples. These consisted of 1 ml of PBS that was exposed to the equipment used during the postmortem, lab environment, DNeasy Blood and Tissue Kits (Qiagen) and Lysing Matrix B tubes (MP Biomedicals) as well as a DNA extraction conducted on the diluent Ultrapure water.
V3–V4 16S rRNA gene sequencing: PCR amplification
Genomic DNA was amplified using 16S rRNA gene amplicon polymerase chain reaction (PCR) primers targeting the hypervariable V3–V4 region of the 16S rRNA gene: V3–V4 forward, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′ and V3–V4 reverse, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3' (Illumina 16S Metagenomic Sequencing Protocol, Illumina, CA, USA). A 35-µl PCR was performed for each sample per the following recipe: 3.5 µl of template DNA, 17.5 µl of KAPA HiFi HotStart ReadyMix (Roche), 0.7 µl of both primers (initial concentration, 10 pmol/µl), 0.1 µg/µl bovine serum albumin fraction V (Sigma), and 8 µl of 10 mM Tris-Cl (Qiagen). Thermal cycling was completed in an Eppendorf Mastercycler per the directions in the ‘Amplicon PCR’ section of the ‘16S Metagenomic Sequencing Library Preparation’ protocol (Illumina). Amplification was confirmed by running 5 µl of PCR product on a 1.5% agarose gel at 70 volts for 80 min, followed by imaging on a Gel Doc EZ System (Bio-Rad). The product was ~450 base pairs (bp) in size.
PCR-positive products were cleaned per the ‘PCR Clean-Up’ section of the Illumina protocol, with the exception that drying times were reduced to half the prescribed duration to account for the additional drying that occurs in a laminar airflow hood. Sequencing libraries were then prepared using the Nextera XT Index Kit (Illumina) and cleaned per the Illumina protocol. Libraries were quantified using a Qubit fluorometer (Invitrogen) using the ‘High Sensitivity’ assay. Sample processing was subsequently completed at Macrogen Inc., Seoul, South Korea. Samples were normalised, pooled and underwent a paired-end 450 bp run on the Illumina MiSeq platform.
Bioinformatics analyses
The quality of the paired-end sequence data was initially visualised using FastQC v0.11.6, and then filtered and trimmed using Trimmomatic v0.36 to ensure a minimum average quality of 25. The remaining high-quality reads were then imported into the R environment v3.4.4 for analysis with the DADA2 package v1.8.0. After further quality filtering, error correction and chimera removal, the raw reads generated by the sequencing process were refined into a table of Amplicon Sequence Variants (ASVs) and their distribution among the samples. It is recommended that ASVs (formerly called ‘Ribosomal Sequence Variants’) are used in place of ‘operational taxonomic units’ (OTU), in part because ASVs give better resolution than OTUs, which are clustered based on similarity [24]. ASVs were then exported back into Linux, and a second stage of chimera removal was carried out using USEARCH v9 in conjunction with the ChimeraSlayer Gold database v6. The remaining ASVs were screened for contamination using the Decontam package in R v1.0.0. The ASVs were classified at genus level using the classify.seqs function in Mothur. Additional species-level classification was performed using SPINGO.
The following statistical analyses were carried out in R: Shannon alpha diversity and Chao1 species richness metrics, and Bray–Curtis distances, for analysis of beta diversity, were calculated using the PhyloSeq package v1.24, and the Vegan package v2.52. Beta-diversity calculations produce distance matrices with as many columns and rows as there are samples; thus, beta diversity is often represented using some form of dimensionality reduction, in this case, using principal co-ordinates analysis (PCoA) with the Ape package v5.1. Hierarchical clustering, an unsupervised method that can reveal key taxa that distinguish their respective environments, was performed with the heat-plot function in the made4 package v1.54. Differential abundance analysis was carried out using Deseq2 v1.2.0, which identifies differentially abundant features between two groups within the data [25]. Tests of means were performed using the Mann–Whitney U test unless otherwise stated, and correlations were calculated using Spearman’s rank-correlation coefficient. Where applicable, false-positive rates were controlled below 5% using the Bonferroni procedure.
The SourceTracker algorithm was implemented to ensure that any differences between pre- and post-infection nematode larvae were not due to the adherence of gut bacteria to the surface of the latter group, following their exposure to the ovine intestinal tract. The 15 larval nematode samples were treated as ‘sink’ samples and compared with five ‘source’ samples to investigate the level of contamination present, if any. SourceTracker v1.0 was implemented in the R environment.
Phylogenetic analyses were carried out by downloading genomic data for well-characterised laboratory and pathogenic bacterial strains from the SILVA database and creating multiple sequence alignments with our own relevant ASVs using the MUSCLE alignment tool, hosted by the European Bioinformatics Institute (EBI). The resulting alignment was then exported to PhyML, where a phylogenetic tree was constructed using the maximum-likelihood method. Lastly, this tree was exported to the iTOL web server for visualisation.
E. coli larval feeding
Eggs of H. contortus MHco3(ISE) were purified and isolated from faecal samples derived from mono-specifically infected donor lambs using a saturated NaCl flotation method. The eggs were washed and re-suspended in water before being added to NGM agar plates supplemented with E. coli OP50-1:GFP (pFPV25.1) and incubated at 22 °C for 48 h to allow hatching of first-stage larvae and subsequent development to second-stage larvae.
Results
Bacterial presence within nematodes
Figure 2a, b shows cross-sectional images of H. contortus gut with Gram-positive bacteria visible throughout.
Fig. 2.
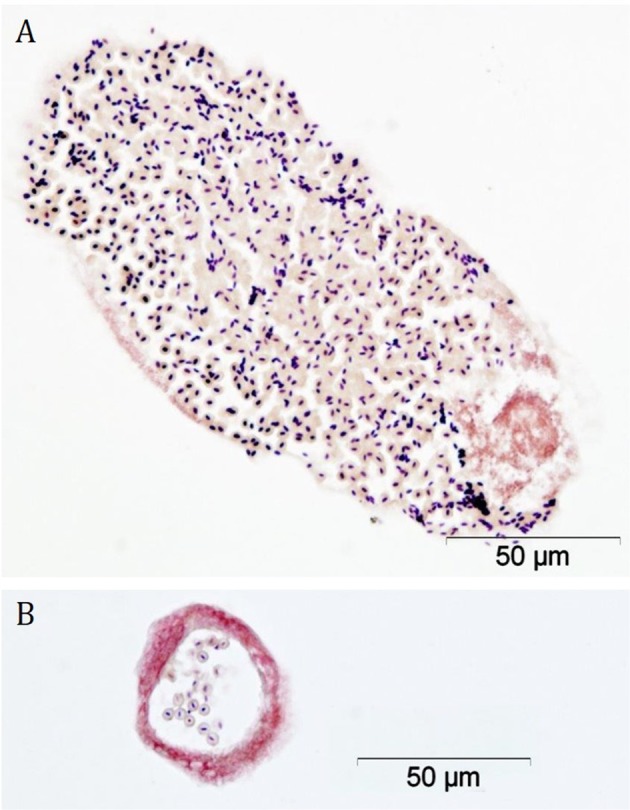
Stained sections through gut of an adult H. contortus. Staining shows the presence of Gram-positive bacteria in cross-sections of the intestinal lumen of an adult H. contortus. Gram-positive organisms stain blue-black, Gram-negative organisms and nuclei stain red (images kindly generated and supplied by Jeanie Finlayson, Moredun Research Institute)
Sample collection and processing
Several samples that proceeded to PCR were not sequenced (Supplementary Fig. 2) because either the amplicon PCR failed to amplify the target gene, or the concentration of the sample fell below the 5 ng/µl threshold for sequencing following the second PCR clean-up, indicating either an imperfect DNA extraction or a low abundance of bacteria in these samples. Neither amplification was evident in the diluent Ultrapure water nor in the PBS exposed to the postmortem laboratory equipment, laboratory environment, Lysing Matrix B tubes and run through the DNA extraction kits; however, control samples proceeded to sequencing regardless, as it is now recognised that sequencing of control samples should be standard practice in microbiome work, especially with low-biomass samples, in which low-level contamination may have a large impact on sample readout [26].
Cohort characteristics
Microbiome analysis was carried out on a total of 5,608,303 error-corrected, non-chimeric ASV reads over the entire data set, with an average read depth of 89,021 reads per sample. This was broken down into a total of 14,351 unique ASVs identified across the four environments studied (Supplementary Fig. 3). Of the four environments sequenced, the larval nematode microbiome was the most distinct, with 84.9% of the total ASVs detected belonging uniquely to the larvae, followed by the faecal microbiome with 73.4% unique ASVs. The mature nematode and abomasal microbiomes were considerably less distinct, with 38.2 and 30% unique ASVs, respectively. Six negative control samples were also sequenced: Ultrapure diluent water, lab environment PBS, postmortem suite PBS and PBS run through two DNA extraction kits and lysing matrix tubes. Considerably fewer error-corrected, non-chimeric ASV reads were generated, with an average of 649. Deeper analysis of these samples showed that there was no crossover between ASVs present in the negative controls and experimental samples (Supplementary Fig. 4). It was therefore concluded that the biological signal from the experimental samples was not influenced by contamination.
General population structure of the ovine and nematode microbiomes
The microbiomes of the four environments studied were initially classified at the phylum level across all individual samples (Fig. 3). Their average, grouped composition was as follows: the abomasum contained 49.5% Firmicutes, 36% Bacteroidetes, 2.9% Fibrobacteres, 1.2% Proteobacteria, 1.1% Actinobacteria, 1% Planctomycetes, 1% Candidatus Saccharibacteria, with the remaining fraction comprising either unclassified or negligible proportions. The lamb faecal microbiome contained 67% Firmicutes, 11% Bacteroidetes, 8.5% Candidatus Saccharibacteria, 3.4% Spirochetes, 2.9% Actinobacteria, 1.2% Verrucamicrobia, with the remaining fraction comprising either unclassified or negligible proportions. The larval nematode microbiome contained 67% Proteobacteria, 18% Bacteroidetes, 8% Actinobacteria, 1.6% Planctomycetes and 1.5% Firmicutes, with the remaining fraction comprising either unclassified or negligible proportions. Finally, the microbiome of the adult nematodes contained 68% Firmicutes, 16% Bacteroidetes, 2.5% Actinobacteria, 2.5% Planctomycetes, 2.2% Candidatus Saccharibacteria, 1.6% Proteobacteria and 1.1% Verrucomicrobia, with the remaining fraction comprising either unclassified or negligible proportions. The four environments are distinguishable even at the phylum level. Nematode larvae have a microbiome dominated by Proteobacteria, a phylum that is not evident in the other environments. The microbiome of the mature nematode more closely resembles the two host sites sampled, suggesting that the host’s environment may influence the microbial populations within the parasite. Despite the resemblance of the adult nematode to the faeces and abomasum of the lambs at this taxonomic level, there are still several phyla that are significantly different in terms of their proportions between these environments.
Fig. 3.

Composition at the phylum level of the ovine microbiome (abomasal lumen contents and faeces) and nematode microbiome (larval and adult nematodes). Each ‘Nematode Larvae’ sample contains ~10,000 pooled larvae, five of which are pre-infection larvae and ten of which are post-infection larvae; each ‘Adult Nematode’ sample contains 100 pooled adult nematodes (five H. contortus (four males, one mixed sex) and seven T. circumcincta (five females, one male, one mixed sex) samples); each ‘Abomasum’ sample is derived from the abomasal washings of one of four lambs; and each ‘Faeces’ sample is derived from one of four lambs across ten time points. Phyla constituting less than 1% of the total phylum distribution were labelled ‘Other’. ‘Nematode Larvae’ were omitted from statistical testing due to their obvious distinctiveness from the other sample groups. The other three samples were compared for proportions of the different phyla identified—initially with a Kruskal–Wallis test, and then a Mann–Whitney U test, making individual comparisons if warranted. Critical values were adjusted using the Bonferroni method
Diversity of the ovine and nematode microbiomes
Alpha diversity, measured using Chao1 species richness showed significant differences between all groups compared, excepting adult nematode and faecal samples, which were similar in terms of species richness (Fig. 4). Larvae were the least diverse group, while the abomasum showed the highest diversity. Beta diversity using Bray–Curtis dissimilarity shows three clusters of samples: lamb faecal samples, nematode larvae and one cluster comprising adult nematodes and lamb abomasa. Hierarchical clustering of the samples based on their composition at ASV level was also performed (Supplementary Fig. 5). This was carried out using the Bray–Curtis distance matrix and the Ward-Linkage method. The Ward-Linkage method revealed the same patterns within the data as those observed in the dimensional reduction of the Bray–Curtis dissimilarity matrix, corroborating these findings. Despite apparent similarities at the phylum level between the adult nematode and ovine faeces, when individual ASVs are compared, the adult nematode bears the closest resemblance to the ovine abomasum, indicating that individual ASVs do not overlap as much as phylum-level annotations between the adult nematode and faeces.
Fig. 4.

Bray–Curtis dissimilarity of the ovine microbiome (abomasal lumen contents and faeces) and nematode microbiome (larval and adult nematodes) correlated with phyla, and Chao1 species richness. a Bray–Curtis dissimilarity of microbiomes studied. For the ‘Abomasum’ samples, each point on the plot is a sample derived from the abomasal washings from one of four lambs, collected 28 days post infection. For the ‘Faeces’ samples, each point on the plot is a sample derived from a stool sample collected from one of four lambs from one of ten time points over a 28-day infection period. For the ‘Nematode Larvae’ samples, each point on the plot is a sample derived from a pooled mixture of ~10,000 larvae, and for the ‘Adult Nematode’ samples, each point on the plot is a sample derived from a pooled mixture of 100 nematodes (five H. contortus (four males, one mixed sex) and seven T. circumcincta (five females, one male, one mixed sex) samples). Ellipses show 80% confidence intervals for their respective groups. The two components of this plot that explained the most variation make up the x- and y-axes. Of the 13 different phyla identified, 10 correlate significantly with one or both of the components of the PCoA based on Spearman’s rank-correlation coefficient. By superimposing this over the PCoA plot, the relationship between these phyla and their environments is visualised. b Horizontal alpha diversity boxplots of microbiomes studied are representative of Chao1 species richness. Significance was determined per the Mann–Whitney U test
Analysis of inter-sex and inter-species differences in the adult nematode microbiome
The nematode microbiomes were probed for variation resulting from differences in sex and species. Alpha and beta diversity between male, female, and mixed-sex pools of adult nematodes were examined (Supplementary Fig. 6). No significant difference was found in terms of alpha diversity based on Chao1 species richness, using the Mann–Whitney U test (P = 0.546). When beta diversity was visualised using a PcOA plot, samples clustered based on the sheep of origin and not based on gender.
The microbiomes of H. contortus and T. circumcincta adult worms were compared at family level (Fig. 5). Due to the novel nature of the microbiomes of both H. contortus and T. circumcincta, 37.6% of ASVs present in H. contortus samples and 34.1% of ASVs present in T. circumcincta samples were not classified to family level. The microbiome of H. contortus comprised the following families: 36.2% Ruminococcaceae, 27.4% Lachnospiraceae, 11.4% Prevotellaceae, 5.7% Acidaminococcaceae, 4.2% Planctomycetaceae, 1.8% Acetobacteraceae, 1.4% Spirochetaceae, 1.2% Veillonellaceae, with the remaining fraction comprising negligible proportions. The microbiome of T. circumcincta comprised the following families: 37% Lachnospiraceae, 26% Ruminococcaceae, 6.5% Prevotellaceae, 3.5% Planctomycetaceae, 3.3% Acidaminococcaceae, 3% Coriobacteriaceae, 2% Bifidobacteriaceae, with the remaining fraction comprising negligible proportions. Veillonellaceae and Acetobacteraceae were present in significantly higher numbers in H. contortus (P = 0.01 and P = 0.005, respectively), while Coriobacteriaceae was significantly more abundant in T. circumcincta (P = 0.005). Significance was determined per the Mann–Whitney U test.
Fig. 5.

Adult nematode microbiome composition at family level of H. contortus and T. circumcincta. The extent to which various bacterial families contribute to the overall make-up of the microbiomes of H. contortus and T. circumcincta. Each column is derived from a pooled mixture of 100 nematodes (five H. contortus (four males, one mixed sex) and seven T. circumcincta (five females, one male, one mixed sex) samples). Nematodes were taken from the ovine abomasum at postmortem, 28 days post infection. Families constituting less than 1% of the total family distribution for a sample were labelled ‘Other’
Alpha diversity in H. contortus was lower than in T. circumcincta (Supplementary Fig. 7). However, the significance of this comparison between the two nematode microbiomes must be considered in the context of sample size (H. contortus n = 5 and T. circumcincta n = 7). Differential abundance analysis using Deseq2 revealed 18 ASVs significantly elevated in one nematode: 5 in H. contortus, and 13 in T. circumcincta (Supplementary Fig. 8). Unlike the Mann–Whitney U test, this method is applied to individual ASVs. Ruminococcaceae/Ruminococcus and Clostridiales dominate the differentially elevated ASVs in T. circumcincta and are absent from the differentially elevated ASVs in H. contortus.
Effect of nematode infection on the faecal microbiome of the host over time
Changes in alpha and beta diversity of the faecal microbiome of infected lambs were examined over several time points between day 0 and day 28 of infection (Fig. 6). Post infection, there is a decrease in species richness within the faecal microbiome, and an increase in dissimilarity over time, compared with the faecal microbiome pre-infection. There is a significant negative Spearman correlation between alpha diversity and time (P = 0.03). Increasing dissimilarity over time is indicated by a strong positive correlation between principal component axis 1 and time. This same principal component, which explains the most variation in the PCoA, also has a statistically significant negative correlation with alpha diversity. This means that the more dissimilar the infected microbiome becomes compared with the pre-infected microbiome, the lower its alpha diversity becomes. Despite the positive correlation between beta diversity and time, when the mean beta diversity of samples at time points 0 and 28 were compared, there was no statistically significant difference (P = 0.89), although visually it appears to decrease slightly (Supplementary Fig. 9).
Fig. 6.

Changes in alpha and beta diversity of the ovine faecal microbiome over time, post infection. Faecal samples were obtained from two-to-four lambs at ten time points over 28 days. All correlation tests used Spearman’s rank-correlation coefficient. a Changes in alpha diversity of the ovine faecal microbiome over time. There is a statistically significant decrease in Chao1 species richness from day 0 to day 28 of infection. b Changes in beta diversity of the ovine faecal microbiome over time. There is a trend in the movement of the lamb faecal microbiome along the x-axis in a positive direction over time, thus becoming more dissimilar to the uninfected lamb microbiome
These diversity metrics inform on changes in the overall relatedness of samples, but give no information about the individual microbes implicated in the faecal microbiome dysbiosis. All ASVs detected were correlated against time using Spearman’s rank-correlation coefficient. There were 39 significant ASVs based on this test, of which 11 showed a positive linear relationship with time and 28 a negative one, post infection (Supplementary Fig. 10). The two most prevalent ASVs associated with time were classified as Bifidobacterium spp. and Sharpea spp., both of which show a negative relationship with time. When blasted against the nr database, these two sequences had 100% identity with Bifidobacterium merycicum, and Sharpea azabuensis. Seven statistically significant ASVs were classified as Ruminococcaceae. Other ASVs, such as the six identified as Candidatus Saccharibacteria, have an ambiguous relationship with time, post infection, as four of these ASVs show positive correlations, and two negative.
Dialister spp. and Clostridium spp. have both been implicated in compromising the human host’s ability to clear nematode infection [11]. Conversely, many other bacterial genera and families are suspected to ‘immunise’ the host against nematode infection (e.g., Subdoligranulum spp., Acinetobacter spp., Paracoccus spp., Gemminger spp., Peptococcaceae, Moraxellaceae, Corynebacteriaceae and Hyphomicrobiaceae). Of these bacteria, we observed only Hyphomicrobiaceae in our data, which was significantly elevated in pre-infection larvae over post-infection larvae (P < 0.05). Moreover, it is known that helminth infection in mice results in increased abundance of the Lactobacillaceae family, leading to the hypothesis that the anti-inflammatory activity of these bacteria may create permissive conditions for nematode survival in the gut [27]. We found similar results with this family in our ovine model, in which a positive correlation with time was observed post-infection (rho = 0.43, P = 0.01).
Effect of the ovine microbiome on the nematode microbiome
In addition to defining the effect of nematode infection on the host, the effect of the host microbiome on the microbial composition of the nematode was also investigated by comparing the microbiomes of larval nematodes pre-infection and post infection. The SourceTracker algorithm failed to detect contamination in the larvae that may have arisen from the ovine intestinal tract (Supplementary Fig. 11).
There is a significant increase in alpha diversity in the pre-infection larvae compared with post-infection larvae as measured by Chao1 species richness (Fig. 7c). The two groups of larvae were also clearly differentiated based on their dissimilarity in the PCoA plot (Fig. 7a), with the clustering by group confirmed statistically by PERMANOVA analysis.
Fig. 7.

a Bray–Curtis dissimilarity between pre-infection and post-infection nematode larvae. b Microbiome composition at family level of pre-infection and post-infection nematode larvae. c Boxplot of Chao1 species richness of pre-infection and post-infection nematode larvae. a Bray–Curtis dissimilarity between pre-infection and post-infection larvae. Each point on the plot is derived from a pooled mixture of ~10,000 larvae (five pre-infection larvae and ten post-infection larvae). Ellipses show 80% confidence intervals for their respective groups. The two groups separate based on the dissimilarity of their microbial composition. Statistical testing was performed by permutational multivariate analysis of variance. b Compositional boxplot of the 19 most prevalent bacterial families. Each column is derived from a pooled mixture of ~10,000 larvae. Significance testing was performed by the Wilcoxon signed-rank test, with critical values adjusted for multiple comparisons using the Bonferroni method. c Boxplot comparing alpha diversity between the two groups as measured by Chao1 species richness. The pre-infection boxplot is derived from five pooled samples of ~10,000 larvae each. The post-infection boxplot is derived from ten pooled samples of ~10,000 larvae each. Statistical testing was performed by the Wilcoxon signed-rank test
The families Planctomycetaceae and Hyphomicrobiaceae are significantly elevated in the pre-infection larvae, while Rhodocyclaceae and Methylobacteriaceae are elevated in post-infection larvae (Fig. 7b). ASVs that were differentially abundant between the two groups were identified using DESeq2. 2037 unique ASVs were identified across all larval nematode samples, of which 97 were elevated in the pre-infection larvae, and 190 in the post-infection larvae. In all cases, this was statistically significant after correcting for multiple testing. A volcano plot depicting this distribution, and a table of all ASVs identified, are also available (Supplementary Figs. 12, 13).
Comparison of the nematode and ovine microbiomes
We investigated the capacity for ovine-adapted bacterial taxa to persist in the nematode microbiome. Firstly, nematode larvae were compared with ovine faecal samples, and adult nematodes were compared with ovine abomasal washings on the basis that these samples originated from a common environment—i.e., the ovine gut and abomasum, respectively. Relatively little convergence was evident between the nematode larvae and ovine faecal samples, with only 227 shared ASVs of a possible 9422 unique ASVs identified across both groups (Supplementary Figs. 14, 15). Conversely, when comparing adult nematodes with ovine abomasal washings, 2494 shared ASVs of a possible 6936 unique ASVs were identified across both groups. Samples clustered definitively based on the host animal of origin.
Next, we reviewed several recent studies that have profiled the ovine microbiome at various sites in the digestive tract according to the abundances of endogenous bacteria present [28, 29]. We then examined our own nematode microbiome data for the presence of bacteria found in sheep in relatively high abundances. Virtually, all taxa present in relatively high abundances in the ovine gut, such as Ruminococcus spp. and Bacteroides spp., were absent from the larvae; however, the Peptostreptococcaceae family was identified in all 32 faecal samples and 14/15 larvae. Abomasum-adapted taxa such as Oscillospira spp., Succinivibrio spp. and Bacteroides spp. were not found in the adult nematodes, but Prevotella spp., one of the most abundant genera in the ovine abomasum, was found in every ovine abomasum and adult nematode sample, along with the abomasally adapted Fibrobacter spp., which was also found in all abomasal samples, and 10/12 nematode samples (data not shown).
Also of interest were potential differences between the adult nematode and the ovine abomasum. The adult nematode and the abomasal lumen content microbiomes were compared using Deseq2. Twelve ASVs were significantly differentially abundant between the nematode microbiome and that of the ovine abomasum (Fig. 8a). The most prevalent differentially abundant ASV was classified as E. coli/Shigella spp. (the taxonomic resolution necessary to distinguish these bacteria is impossible using 16S rRNA gene sequencing analysis [30]). Following this, ASVs classified as E. coli/Shigella were screened for in the data set, resulting in the discovery of four in total. At least one ASVs appeared in every larval sample, and in seven of the twelve adult nematode samples. ASV 75, the most abundant putative E. coli/Shigella ASV, was also present at low levels in some of the lamb faecal samples but all ASVs were absent from the ovine abomasum (Supplementary Fig. 16A). Nematode colonisation by E. coli/Shigella did not appear to be specific for either species of nematode—the two ASVs 75 and 295 combined were found in 4/7 T. circumcincta samples and 3/5 H. contortus samples.
Fig. 8.

a Differentially abundant ASVs between the adult nematode and the ovine abomasum, showing level of fold change between either environment. b Oral ingestion of engineered E. coli by larvae in vitro. a Metabarcoding data for the adult nematodes were derived from 12 pooled samples (five H. contortus (four males, one mixed sex) and seven T. circumcincta (five females, one male, one mixed sex) samples) of 100 nematodes each. Metabarcoding data for the abomasum were derived from the abomasal washings of four lambs. Bacteria are labelled with the most accurate taxonomic classification available for that ASV. Differential abundance was determined with Deseq2. Additional classification to species level with SPINGO is provided. This classification was performed with no confidence cut-offs; thus, it is revealing yet imperfect with respect to the identification of the bacteria. b Eggs of H. contortus MHco3(ISE) were hatched to first-stage larvae and developed to second-stage larvae on NGM agar supplemented with E. coli OP50-1:GFP (pFPV25.1). DIC image left, U.V. Image on right depicting ingestion of GFP labelled OP50 in pharynx and entire length of gut (Mag x250)
Phylogenetic analyses were carried out, comparing the four E. coli/Shigella ASVs found in the data set with other well-characterised and clinically relevant strains to provide evolutionary context (Supplementary Fig. 16B). The bootstrapping values were provided over 1000 iterations. The more distantly related Klebsiella spp. and Salmonella spp. formed the outgroups, as expected; however, the evolutionary distance between E. coli/Shigella genera was limited, as can be seen by the low bootstrapping values at many of the branch points. ASV_295 appears most distantly related to the remaining species, and therefore it is reasonable to suggest that ASV_6240 and E. coli MG1655 form a distinct separate clade, although it is not possible to confirm that evolutionary distance exists between ASV_75, ASV_7656, E. coli 0157:H7 and Shigella spp.
Oral ingestion of engineered E. coli by larvae in vitro
In vitro oral ingestion of engineered E. coli was investigated to assess the potential for exogenous bacteria to reside within the guts of these nematodes, and to locally express heterologous genes. First-stage nematode larvae were grown on a plate seeded with an E. coli strain, genetically modified to express green fluorescent protein (GFP). Fluorescence microscopy showed GFP fluorescence in the pharynx and the entire length of gut, specifically within GFP-expressing, E. coli-fed nematodes. Similar results are observed with T. circumcincta (data not shown).
Discussion
The quality and depth of our sequencing analysis permits a thorough understanding of spatial, kinetic and organism-specific patterns of the microbiomes of helminth-infected hosts. This approach is potentially applicable to parasitic disease at large, including helminthic and ectoparasitic infections, on the condition that differences exist between the host and parasite microbiome. Due to the preferential colonisation of the abomasum by H. contortus and T. circumcincta, it was pertinent to compare these compartments for identification of bacteria that favour nematode cohabitation, and the same rationale was used in the comparison of nematode larvae and ovine faecal samples. The identification of differentially abundant taxa represents valuable knowledge to exploit in future research.
A past study of the H. contortus microbiome with primers targeting both the V3–V4 and V5–V7 regions of the 16S rRNA gene resulted in higher OTU capture using the former primer set, although the latter set contrastingly was capable of detecting the phylum Gemmatimonadetes, albeit in relatively low abundance [13]. The V3–V4 region of the 16S rRNA gene was sequenced for all samples in this study, rather than the V5–V7 because, while targeting the V5–V7 region would be necessary for mapping comprehensively the microbiome of H. contortus by facilitating identification of its less abundant taxa, here our objective was to identify nematode-specific bacteria that are present in relatively high abundance, because these bacteria would be more amenable to concentrating within a nematode, were they administered exogenously. However, there are ways in which less abundant taxa may have important applications for treatment of parasitic disease. For example, there is evidence that bacteria can influence their environment considerably even if their abundance is low [31]. Furthermore, it is known that some bacteria, such as Wolbachia spp., are essential for the development of filarial nematodes, and that antibiotics targeting Wolbachia spp. have filaricidal activity [32]. Thus, the use of antibiotics to target nematode-essential bacteria present either in low or high abundance is a valid treatment strategy. An alternative method could involve feeding the infected host a modified diet that would deprive the bacteria in question of essential nutrients.
The commonalities and differences we observed between the ovine and nematode microbiomes (Figs. 3, 4; Supplementary Fig. 5) are interesting because, in the former case, it presents the possibility that the microbiota of either organism may be influencing that of the other and, in the latter case, it means that the differences between parasite and host could be exploited to the benefit of the infected animal. The abomasum and adult nematode microbiomes are by far the most closely related environments (Fig. 4; Supplementary Fig. 5). This could be considered unsurprising because these environments are in intimate contact with one another; yet, nematode larvae and host faeces, from which the larvae derive, separate into two distinct clusters despite their proximity. We reasoned that there may exist differences between host and parasite amenable to exploitation despite their gross similarity.
H. contortus and T. circumcincta have contrasting lifestyles, the former being a blood feeder and the latter a mucosal grazer [33]. Thus, characterising both species simultaneously in a co-infection model could illuminate the effects of alternate feeding habits on the nematode and ovine microbiome. Analysing different species in isolation across separate studies could complicate the identification of the source of any variation, as inter-study differences in soil composition, animal feed, age and immune status of host and living conditions, for example, could affect the ovine microbiota and therefore the microbiota of the nematode. To our knowledge, this is the first report of a parasite–host microbiome study in ruminant livestock that incorporates a co-infection model. It is also the first characterisation of the microbiome of T. circumcincta.
Co-infection models are important because it is accepted that different parasites co-habiting the same host can affect each other profoundly in ways that would not occur were they infecting the host as lone pathogens [34]. This can result in one parasite creating a permissive environment for the other parasite or, conversely, one parasite negatively affecting the other parasite’s growth. In some cases, parasitic cohabiters can have more influence on their host than on each other [35]. In addition, multiple studies claim that co-infection of humans and livestock with nematodes is common [36, 37], meaning that more microbiome studies of host and parasite should incorporate co-infection models. Admittedly, this study does not examine the parasite–host microbiome interrelationship in a single-infection model. Therefore, the effects of H. contortus or T. circumcincta alone on the ovine microbiome may be different than what is observed here. In response to a critical lack of information regarding the effects of co-infection on co-habiting parasites, a recent study has successfully employed methodology to predict how two nematodes will influence each other in terms of survival, even when they are examined in different host species [34]. Future research would benefit this field by attempting to predict how host co-infection influences the microbiome compared with single-strain infections.
We discovered that the two species of nematode contain microbiomes that are in many ways comparable. This is not unexpected, given the finding that marine nematodes deriving even from different parts of the planet contain similar microbiomes [16]. However, there are statistically significant differences that are worth noting, namely that the families Veillonellaceae and Acetobacteraceae are both elevated in H. contortus, and Coriobacteriaceae is elevated in T. circumcincta (P > 0.01) (Fig. 5). The fact that different species of nematode living in the same host have quantifiable differences in their microbiomes suggests that the contrasting lifestyles between the two species may be directly responsible for significant changes in microbiome constitution.
Microbiomes associated with improved host health are noted for having high levels of microbial diversity. As such, if parasitic nematode infections were to alter the host’s microbiome, they may have more a profound effect on the health of the host than what is currently appreciated. Infection with multiple parasitic species is a natural phenomenon and is underlined as a more crucial determinant of the effects of infection on host health than host-specific and environmental factors [38]; thus, the effects of co-infection on the microbiome could be just as pronounced. We detected an obvious decrease in alpha diversity 21 days post infection. H. contortus and T. circumcincta pre-patent periods are both ~3 weeks [39, 40], suggesting that nematode infection has a lesser impact on the microbiome of the host in the initial stages of the nematode lifecycle, and only begins to have a noticeable effect once the parasites mature and move into the abomasal lumen rather than residing within the tissue. However, the dose administered to the lambs in this study was sub-clinical, which also may explain why the decrease in alpha diversity was not observed until the latter part of the lifecycle. It is possible that the effects on microbiome diversity could become magnified and/or occur earlier if infections were more acute.
Notably, previous work, albeit within goats, showed that H. contortus infection did not result in a shift in abomasal microbiome diversity; however, an effect was seen on the abundances of several bacterial species [41]. Contrastingly, infection of lambs with H. contortus alone was found to increase microbiome diversity in the abomasum [42]. Differences observed may be attributable to inter-species differences and/or inter-study differences. For example, although both studies administered the same dose of H. contortus, the latter study involved pre-treatment of its animals with the anthelmintics ivermectin and levamisole, which may have removed pre-existing infection that otherwise may have affected study outcome. A study of humans, many of whom were infected with multiple nematodes (most commonly Trichuris spp., followed by Ascaris spp., followed by hookworm), concluded that helminth infection resulted in an increase in diversity of the faecal microbiome [37]. It could be the case that the effect of nematode infection on microbiome diversity within the host may be microbiome-specific (i.e., abomasal vs. faecal), and/or species-specific (i.e., ovine vs. caprine vs. human). It is perhaps relevant that Trichuris spp., Ascaris spp. and hookworm are each intestinal helminths, while H. contortus and T. circumcincta are abomasal helminths. It is reasonable to postulate that parasites will have varying impacts on body sites with which they are directly in contact, than if they were persisting remotely. Furthermore, changes that occur as a result of abomasal colonisation may have dramatically different effects on microbial viability and composition in other, downstream in vivo compartments (e.g., the intestines) that would not occur, were the intestines colonised. For example, there is evidence that colonisation with H. contortus decreases the acidity of the ruminant stomach [42], potentially altering microbial growth patterns here and other areas of the gut. Further study is required to fully understand the extent to which parasite lifestyle and host-specific factors come to bear on microbiome diversity.
In addition to a quantifiable decrease in diversity, the quality of the shift is also noteworthy. Bifidobacterium merycicum and Sharpea azabuensis, both of which become reduced over time, would be considered typical constituents of a healthy ruminant microbiome [43, 44]. Similarly, Ruminococcaceae can be considered a dominant ruminant bacterial family [45] and again, all associated ASVs show a negative correlation with time. Unlike the dominant ruminant bacteria which are clearly affected by nematode infection of the host, some other changes in the host microbiome not directly related to parasitic infection are inevitable due to interactions between bacteria. Bacterial species compete for resources in various ecological niches within the host, produce antibiotics, and often rely on syntrophy for their survival [46]. Thus, it is cautioned that the results of microbiome studies must be considered against a potential background of inter-bacteria interactions that may confound precise interpretation of changes observed.
Taxa that have suggested involvement in either maintenance or clearance of human nematode infection, such as Dialister spp. and Lactovum spp. [11], were largely unfound in the ovine microbiome in this study, with the exception of the Hyphomicrobiaceae family, which was elevated in pre-infection nematode larvae over post-infection larvae. Thus, while these bacteria may have an important role to play in human infection, it is improbable that they are fundamental to the establishment or curtailment of nematode colonisation of the ruminant host, and at the very least might only facilitate the establishment or removal of infection. An increase in the level of anti-inflammatory Lactobacillaceae in murine models of other studies [10], and in this ovine study, is suggestive of a symbiotic relationship between bacteria and parasite, wherein Lactobacillaceae thrive in the presence of nematode infection, while nematode infection is sustained by the dampened immune response effected by this altered microbial signature.
The degree of overlap observed in this study between host and parasite microbiomes occupying the same environment within the host provides insight into the origination of the nematode microbiome and is suggestive of the ability of ruminant-adapted taxa to invade a new niche within the host. The data present a strong case for the mature nematode either feeding on or being passively colonised by constituent bacteria of the ovine abomasum. While many taxa associated with the abomasum are absent from the adult nematode microbiome, there is a significant degree of overlap between the two groups at an ASV level, especially by the highly abundant, abomasally adapted genera Prevotella spp. and Fibrobacter spp. All adult nematodes cluster definitively by host organism (Supplementary Fig. 14), suggesting that these common taxa were indeed acquired by the nematode upon reaching the abomasum.
The identification of differentially abundant taxa presents future opportunities for use as research tools, or indeed therapeutic approaches. While invaluable in combatting helminthic disease, anthelmintic drugs have been the victims of their own success. Frequent and routine use of anthelmintic has led to the prevalence of anthelmintic resistance increasing globally, with multiple class anthelmintic resistance being commonplace in H. contortus and T. circumcincta globally [47]. The development of anthelmintic resistance and consumer concerns over chemical residues in the milk and meat products of treated animals [48] are potentially limiting factors in the deployment of these drugs in the future.
Our metabarcoding data suggest that the microbiomes of H. contortus and T. circumcincta are significantly different from their ovine environment most notably with respect to E. coli/Shigella spp. E. coli may be a much more natural coloniser of nematodes than of animals, and there are several pieces of clinical evidence that support this. Firstly, it is known in human subjects that E. coli is not among the most abundant species found in the gastrointestinal tract and that its numbers may in fact be quite low [49]. Moreover, probiotic strains of E. coli, such as E. coli Nissle 1917, are frequently unsuccessful colonisers of the human gut even when administered in relatively high doses [50], and once colonised often do not persist for long in the gut once the dose is stopped [51]. Thus, naturally low levels of E. coli in animals may be sufficient to ensure its selective compartmentalisation in nematodes. Alternatively, it is possible that E. coli is vertically transmitted in nematodes and that migration from the host either does not take place or has a lesser impact than vertical transmission.
This study provides a rationale for the study and use of parasite-specific bacteria in drug development practices. The successful feeding of infective nematodes with a genetically modified bacterium could be exploited in several ways. An example is a bacterial assay formatted to assess the efficacy of anthelminthic drugs. Bacteria have recently been engineered to ‘sense’ molecules that cannot be quantified by non-invasive methods [52]. These bacteria can detect exposure to a drug, and record this exposure using a memory circuit. This could create a platform through which pharmacokinetic studies on anti-parasitic drugs could be easily and non-invasively performed—both on market-approved compounds and drugs still undergoing clinical testing. Alternatively, bacteria could be used as vehicles for drug delivery, which has many advantages beyond conventional chemical medicines, not least of which is the targeted delivery of therapeutics.
E. coli is an ideal candidate for bacteria-mediated drug delivery. It is readily engineered and highly flexible as a drug testing platform and various strains of this species have attracted interest for their probiotic properties [53]. Its preclinical validation in various drug delivery modalities is also a reassuring aspect of this bacterium [52, 54–58]. Thus, the selective colonisation of the nematode microbiome by E. coli/Shigella is encouraging and invites further investigation of bacteria as orally administrable, target-specific agents.
In summary, this study highlights the potential value in exploitation of nematode microbiota in progression of novel treatments for parasitic diseases affecting both animals and humans.
Supplementary information
{kind=link}
Acknowledgements
The authors acknowledge the provision of in vitro larval images with kind permission from Prof Antony Page, University of Glasgow. MT acknowledges relevant support from Science Foundation Ireland (15/CDA/3630 and 12/RC/2273). We also gratefully acknowledge funding from The Scottish Government’s Rural and Environment Science and Analytical Services Division (RESAS). We are grateful to the Bioservices Division, Moredun Research Institute, for expert care and assistance with the animals.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All experimental procedures described here were approved by the Moredun Research Animal Welfare and Ethical Review Body and were conducted under the legislation of a UK Home Office License (reference P95890EC1) in accordance with the Animals (Scientific Procedures) Act of 1986.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Glenn Hogan, Sidney Walker
These authors contributed equally: Mark Tangney, Dave J. Bartley
Contributor Information
Mark Tangney, Phone: +353-21-420-5709, Email: m.tangney@ucc.ie.
Dave J. Bartley, Phone: +44-131-445-6144, Email: dave.bartley@moredun.ac.uk
Supplementary information
The online version of this article (10.1038/s41396-019-0462-4) contains supplementary material, which is available to authorized users.
References
- 1.Torgerson PR, Devleesschauwer B, Praet N, Speybroeck N, Willingham AL, Kasuga F, et al. World health organization estimates of the global and regional disease burden of 11 foodborne parasitic diseases, 2010: a data synthesis. PLoS Med. 2015;12:e1001920. doi: 10.1371/journal.pmed.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenyon F, Hutchings F, Morgan-Davies C, van Dijk J, Bartley DJ. Worm control in livestock: bringing science to the field. Trends Parasitol. 2017;33:669–77. doi: 10.1016/j.pt.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Rose H, Rinaldi L, Bosco A, Mavrot F, de Waal T, Skuce P, et al. Widespread anthelmintic resistance in European farmed ruminants: a systematic review. Vet Rec. 2015;176:546. doi: 10.1136/vr.102982. [DOI] [PubMed] [Google Scholar]
- 4.Peachey LE, Pinchbeck GL, Matthews JB, Burden FA, Behnke JM, Hodgkinson JE. Papaya latex supernatant has a potent effect on the free-living stages of equid cyathostomins in vitro. Vet Parasitol. 2016;228:23–9. doi: 10.1016/j.vetpar.2016.07.036. [DOI] [PubMed] [Google Scholar]
- 5.Hogan G, Tangney M. The Who, What, and Why of Drug Discovery and Development. Trends Pharmacol Sci. 2018;;39:848–52. doi: 10.1016/j.tips.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Partridge FA, Murphy EA, Willis NJ, Bataille CJ, Forman R, Heyer-Chauhan N, et al. Dihydrobenz[e][1,4]oxazepin-2(3H)-ones, a new anthelmintic chemotype immobilising whipworm and reducing infectivity in vivo. PLoS Negl Trop Dis. 2017;11:e0005359. doi: 10.1371/journal.pntd.0005359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Partridge FA, Forman R, Willis NJ, Bataille CJR, Murphy EA, Brown AE, et al. 2,4-Diaminothieno[3,2-d]pyrimidines, a new class of anthelmintic with activity against adult and egg stages of whipworm. PLoS Negl Trop Dis. 2018;12:e0006487. doi: 10.1371/journal.pntd.0006487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaiss MM, Harris NL. Interactions between the intestinal microbiome and helminth parasites. Parasite Immunol. 2016;38:5–11. doi: 10.1111/pim.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White EC, Houlden A, Bancroft AJ, Hayes KS, Goldrick M, Grencis RK, et al. Manipulation of host and parasite microbiotas: Survival strategies during chronic nematode infection. Sci Adv. 2018;4:eaap7399. doi: 10.1126/sciadv.aap7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glendinning L, Nausch N, Free A, W Taylor D, Mutapi F. The microbiota and helminths: sharing the same niche in the human host. Parasitology. 2014;141:1255–71. [DOI] [PubMed]
- 11.Rosa BA, Supali T, Gankpala L, Djuardi Y, Sartono E, Zhou Y, et al. Differential human gut microbiome assemblages during soil-transmitted helminth infections in Indonesia and Liberia. Microbiome. 2018;6:33. doi: 10.1186/s40168-018-0416-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dirksen P, Marsh SA, Braker I, Heitland N, Wagner S, Nakad R, et al. The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol. 2016;14:38. doi: 10.1186/s12915-016-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Ashram S, Suo X. Exploring the microbial community (microflora) associated with ovine Haemonchus contortus (macroflora) field strains. Sci Rep. 2017;7:70. doi: 10.1038/s41598-017-00171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer JM, Baskaran P, Quast C, Susoy V, Rodelsperger C, Glockner FO, et al. Succession and dynamics of Pristionchus nematodes and their microbiome during decomposition of Oryctes borbonicus on La Reunion Island. Environmental Microbiol. 2017;19:1476–89. doi: 10.1111/1462-2920.13697. [DOI] [PubMed] [Google Scholar]
- 15.Derycke S, De Meester N, Rigaux A, Creer S, Bik H, Thomas WK, et al. Coexisting cryptic species of the Litoditis marina complex (Nematoda) show differential resource use and have distinct microbiomes with high intraspecific variability. Mol Ecol. 2016;25:2093–110. doi: 10.1111/mec.13597. [DOI] [PubMed] [Google Scholar]
- 16.Schuelke T, Pereira TJ, Hardy SM, Bik HM. Nematode-associated microbial taxa do not correlate with host phylogeny, geographic region or feeding morphology in marine sediment habitats. Mol Ecol. 2018;27:1930–51. doi: 10.1111/mec.14539. [DOI] [PubMed] [Google Scholar]
- 17.Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389:1218–28. doi: 10.1016/S0140-6736(17)30182-4. [DOI] [PubMed] [Google Scholar]
- 18.Biddle AS. An In Vitro Model of the Horse Gut Microbiome Enables Identification of Lactate-Utilizing Bacteria That Differentially Respond to Starch Induction. PloS One. 2013;8:e77599. doi: 10.1371/journal.pone.0077599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schallig HD. Immunological responses of sheep to Haemonchus contortus. Parasitol. 2000;120:Suppl:S63–72. doi: 10.1017/S003118209900579X. [DOI] [PubMed] [Google Scholar]
- 20.Harfoot CG. Anatomy, physiology and microbiology of the ruminant digestive tract. Prog Lipid Res. 1978;17:1–19. doi: 10.1016/0079-6832(78)90003-4. [DOI] [PubMed] [Google Scholar]
- 21.Patterson DM, Jackson F, Huntley JF, Stevenson LM, Jones DG, Jackson E, et al. Studies on caprine responsiveness to nematodiasis: segregation of male goats into responders and non-responders. Int J Parasitol. 1996;26:187–94. doi: 10.1016/0020-7519(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 22.MAFF. Ministry of Agriculture, Fisheries and Food, Manual of veterinary parasitological laboratory techniques, 3rd edition. Her Majesty’s Stationary Office (HMSO), London, 1986. Reference Book 418.
- 23.Bancroft JD, Gamble M. Theory and practice of histological techniques. London: Churchill Livingstone; 2008. [Google Scholar]
- 24.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2019;27:105–17. doi: 10.1016/j.tim.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Glendinning L, Nausch N, Free A, Taylor DW, Mutapi F. The microbiota and helminths: sharing the same niche in the human host. Parasitology. 2014;141:1255–71. doi: 10.1017/S0031182014000699. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Fan H, Han Y, Zhao J, Zhou Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australasian J Anim Sci. 2017;30:100–10. doi: 10.5713/ajas.16.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng Y, Zeng D, Ni X, Zhu H, Jian P, Zhou Y, et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express. 2017;7:75. doi: 10.1186/s13568-017-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Cai Y, Zhou G, Shi X, Su J, Chen G, et al. Rapid Sanger sequencing of the 16S rRNA gene for identification of some common pathogens. PloS One. 2014;9:e88886. doi: 10.1371/journal.pone.0088886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pester M, Bittner N, Deevong P, Wagner M, Loy AA. ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 2010;4:1591–602. doi: 10.1038/ismej.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slatko BE, Luck AN, Dobson SL, Foster JM. Wolbachia endosymbionts and human disease control. Mol Biochem Parasitol. 2014;195:88–95. doi: 10.1016/j.molbiopara.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Murray J, Smith WD. Ingestion of host immunoglobulin by three non-blood-feeding nematode parasites of ruminants. Res Vet Sci. 1994;57:387–9. doi: 10.1016/0034-5288(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 34.Lello J, McClure SJ, Tyrrell K, Viney ME. Predicting the effects of parasite co-infection across species boundaries. Proc Biol Sci. 2018;285:1–9. [DOI] [PMC free article] [PubMed]
- 35.Murphy L, Pathak AK, Cattadori IM. A co-infection with two gastrointestinal nematodes alters host immune responses and only partially parasite dynamics. Parasite Immunol. 2013;35:421–32. doi: 10.1111/pim.12045. [DOI] [PubMed] [Google Scholar]
- 36.Almeida FA, Bassetto CC, Amarante MRV, Albuquerque ACA, Starling RZC, Amarante A. Helminth infections and hybridization between Haemonchus contortus and Haemonchus placei in sheep from Santana do Livramento, Brazil. J Vet Parasitol. 2018;27:280–8. doi: 10.1590/S1984-296120180044. [DOI] [PubMed] [Google Scholar]
- 37.Lee SC, Tang MS, Lim YA, Choy SH, Kurtz ZD, Cox LM, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. 2014;8:e2880. doi: 10.1371/journal.pntd.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Telfer S, Lambin X, Birtles R, Beldomenico P, Burthe S, Paterson S, et al. Species interactions in a parasite community drive infection risk in a wildlife population. Science. 2010;330:243–6. doi: 10.1126/science.1190333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goossens B, Osaer S, Kora S, Jaitner J, Ndao M, Geerts S. The interaction of Trypanosoma congolense and Haemonchus contortus in Djallonke sheep. Int J Parasitol. 1997;27:1579–84. doi: 10.1016/S0020-7519(97)00094-5. [DOI] [PubMed] [Google Scholar]
- 40.Kenyon F, Sargison ND, Skuce PJ, Jackson F. Sheep helminth parasitic disease in south eastern Scotland arising as a possible consequence of climate change. Veterinary Parasitol. 2009;163:293–7. doi: 10.1016/j.vetpar.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Li RW, Li W, Sun J, Yu P, Baldwin RL, Urban JF. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci Rep. 2016;6:20606. doi: 10.1038/srep20606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Ashram S, Al Nasr I, Abouhajer F, El-Kemary M, Huang G, Dincel G, et al. Microbial community and ovine host response varies with early and late stages of Haemonchus contortus infection. Vet Res Commun. 2017;41:263–77. doi: 10.1007/s11259-017-9698-5. [DOI] [PubMed] [Google Scholar]
- 43.Kamke J, Kittelmann S, Soni P, Li Y, Tavendale M, Ganesh S, et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterised by lactic acid formation and utilisation. Microbiome. 2016;4:56. doi: 10.1186/s40168-016-0201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biavati B, Mattarelli P. Bifidobacterium ruminantium sp. nov. and Bifidobacterium merycicum sp. nov. from the rumens of cattle. Int J Syst Bacteriol. 1991;41:163–8. doi: 10.1099/00207713-41-1-163. [DOI] [PubMed] [Google Scholar]
- 45.Henderson G, Cox F, Ganesh S, Jonker A, Young W, Global Rumen Census C, et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567. doi: 10.1038/srep14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menon R, Ramanan V, Korolev KS. Interactions between species introduce spurious associations in microbiome studies. PLOS Comput Biol. 2018;14:e1005939. doi: 10.1371/journal.pcbi.1005939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaplan RM. Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol. 2004;20:477–81. doi: 10.1016/j.pt.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Fernandes MAM, Gilaverte S, Bianchi MD, da Silva CJA, Molento MB, Reyes FGR, et al. Moxidectin residues in tissues of lambs submitted to three endoparasite control programs. Res Vet Sci. 2017;114:406–11. doi: 10.1016/j.rvsc.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 49.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prilassnig M, Wenisch C, Daxboeck F, Feierl G. Are probiotics detectable in human feces after oral uptake by healthy volunteers? Wien Klin Wochenschr. 2007;119:456–62. doi: 10.1007/s00508-007-0808-1. [DOI] [PubMed] [Google Scholar]
- 51.Joeres-Nguyen-Xuan TH, Boehm SK, Joeres L, Schulze J, Kruis W. Survival of the probiotic Escherichia coli Nissle 1917 (EcN) in the gastrointestinal tract given in combination with oral mesalamine to healthy volunteers. Inflamm Bowel Dis. 2010;16:256–62. doi: 10.1002/ibd.21042. [DOI] [PubMed] [Google Scholar]
- 52.Flores Bueso Y, Lehouritis P, Tangney M. In situ biomolecule production by bacteria; a synthetic biology approach to medicine. J Control Release. 2018;275:217–28. doi: 10.1016/j.jconrel.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 53.Wassenaar TM. Insights from 100 Years of Research with Probiotic E. Coli. European J Microbiol & Immunol. 2016;6:147–61. doi: 10.1556/1886.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy C, Rettedal E, Lehouritis P, Devoy C, Tangney M. Intratumoural production of TNFalpha by bacteria mediates cancer therapy. PLoS One. 2017;12:e0180034. doi: 10.1371/journal.pone.0180034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lehouritis P, Stanton M, McCarthy FO, Jeavons M, Tangney M. Activation of multiple chemotherapeutic prodrugs by the natural enzymolome of tumour-localised probiotic bacteria. J Control Release. 2016;222:9–17. doi: 10.1016/j.jconrel.2015.11.030. [DOI] [PubMed] [Google Scholar]
- 56.Cronin M, Le Boeuf F, Murphy C, Roy DG, Falls T, Bell JC, et al. Bacterial-mediated knockdown of tumor resistance to an oncolytic virus enhances therapy. Mol Ther. 2014;22:1188–97. doi: 10.1038/mt.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byrne WL, Murphy CT, Cronin M, Wirth T, Tangney M. Bacterial-mediated DNA delivery to tumour associated phagocytic cells. J Control Release. 2014;196:384–93. doi: 10.1016/j.jconrel.2014.10.030. [DOI] [PubMed] [Google Scholar]
- 58.Lehouritis P, Hogan G, Tangney M. Designer bacteria as intratumoural enzyme biofactories. Adv Drug Deliv Rev. 2017;118:8–23. doi: 10.1016/j.addr.2017.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.