

Cryo-EM reveals the genome ejection mechanism of bacteriophage P68, a potential phage therapy agent against Staphylococcus aureus.
Abstract
Phages infecting Staphylococcus aureus can be used as therapeutics against antibiotic-resistant bacterial infections. However, there is limited information about the mechanism of genome delivery of phages that infect Gram-positive bacteria. Here, we present the structures of native S. aureus phage P68, genome ejection intermediate, and empty particle. The P68 head contains 72 subunits of inner core protein, 15 of which bind to and alter the structure of adjacent major capsid proteins and thus specify attachment sites for head fibers. Unlike in the previously studied phages, the head fibers of P68 enable its virion to position itself at the cell surface for genome delivery. The unique interaction of one end of P68 DNA with one of the 12 portal protein subunits is disrupted before the genome ejection. The inner core proteins are released together with the DNA and enable the translocation of phage genome across the bacterial membrane into the cytoplasm.
INTRODUCTION
Staphylococcus aureus causes a range of illnesses from minor skin infections to life-threatening diseases such as pneumonia, meningitis, and sepsis (1). Medical expenses caused by S. aureus infections in the United States and European Union have been estimated to exceed $2.5 billion, and S. aureus infections result in tens of thousands of deaths every year (2). Many S. aureus strains, particularly those found in hospitals, carry antibiotic resistance genes (3). In 2017, the World Health Organization listed S. aureus among the most threatening antibiotic-resistant pathogens for which new treatments are urgently needed (4). Mouse models showed that phages could be used to treat infections caused by antibiotic-resistant S. aureus strains (5). All previously described phages from the family Podoviridae that infect S. aureus are members of the genus Rosenblumvirus. P68 and several other related phages are promising phage therapy candidates because of their wide host range (6). They were analyzed for their ability to treat animals infected by methicillin-resistant S. aureus and to prevent healthcare-associated human infections (5). Despite the testing for the use in phage therapy, the mechanism of phage P68 infection of the Gram-positive S. aureus cells has not been described in detail.
Three-dimensional (3D) structures of virions of several Podoviridae phages, including T7, φ29, C1, ε15, P22, K1E, and K1-5, were determined using cryo–electron microscopy (cryo-EM) (7–15). Bacteriophages from the family Podoviridae have icosahedral or prolate heads and short noncontractile tails. The assembly of the heads of some bacteriophages is promoted by scaffolding proteins (14). Minor capsid proteins, attached to the outer surface of the phage head, increase the stability of capsids or, in the case of head fiber proteins, enable the initial attachment of phages to bacteria. The heads of some phages contain inner core proteins that are associated with the portal complex. The inner core proteins are released together with the phage genome and were speculated to play a role in the delivery of the DNA into the cell cytoplasm.
The phage tail is attached to one of the fivefold vertices of the head in which a pentamer of capsid proteins is replaced by a dodecahedral portal complex. The tails of podoviruses are variable in size and protein composition; however, they share common organizational motifs. The tails contain specialized protein subunits for receptor binding, cell wall degradation, and cell membrane penetration. Podoviruses that infect S. aureus require wall teichoic acid for their adsorption to cells (16). Podovirus tails are decorated by 6 or 12 tail fibers. Structures of tail fibers from several podoviruses, including P22, φ29, and K1F, were determined using x-ray crystallography (17–20). Tail fibers form trimers in which the N-terminal part is responsible for attachment to the virion and the C-terminal domain enables receptor binding. Two or more types of proteins form the central tubular part of the podovirus tail. The proximal part of the tail, attached to the portal complex, is usually a dodecamer of lower collar proteins (21). In contrast, a hexamer of tail knob proteins usually forms the distal part of the tail tube (10). The tails of some podoviruses also contain tail spike proteins with hydrolase activities, which enable phages to degrade the bacterial cell wall. Tails of podoviruses contain a trimer of tail needle proteins that is located in the tube formed by the lower collar and tail knob proteins. The tail needle is essential for retaining the genome in the phage head after packaging, and it may have a role in the penetration of the cell wall (22). The genome of phage P68 is 18,227 base pairs of double-stranded DNA (dsDNA) and contains 22 open reading frames (23).
Here, we used a combination of cryo-EM and x-ray crystallography to structurally characterize bacteriophage P68 and the mechanism of regulation of its genome ejection. An asymmetric reconstruction of the P68 virion was determined to a resolution of 4.7 Å. The structures of heads of native virions, genome release intermediates, and empty particles of P68 with imposed icosahedral symmetry were determined to resolutions of 3.3, 6.3, and 3.4 Å, respectively. The same structures with imposed fivefold symmetries were determined to resolutions of 3.8, 22.7, and 4.3 Å, respectively. Portal complexes and tails with 12-fold symmetries in native, genome release, and empty particles of P68 were characterized to resolutions of 3.9, 18.8, and 7.1 Å, respectively. X-ray crystallography enabled the determination of the structure of the C-terminal part of P68 tail fiber to a resolution of 2.0 Å. In combination, the structures enabled us to describe the mechanisms of attachment of the portal complex to the capsid, selectivity of binding of head fibers to hexamers of major capsid proteins adjacent to the tail vertex, and the conformational changes of the portal complex required for the genome release.
RESULTS AND DISCUSSION
P68 capsid structure
The virion of P68 has an icosahedral head with a diameter of 480 Å and a 395 Å–long tail, which is decorated with tail fibers (Fig. 1). The structures of heads of native virions, genome release intermediates, and empty particles of P68 with imposed icosahedral symmetry were determined to resolutions of 3.3, 6.3, and 3.4 Å, respectively (Fig. 1, A to D; figs. S1, A to G, and S2; and tables S1 and S2). The capsid proteins in the P68 head are organized in a T = 4 icosahedral lattice (Fig. 2, A and B). The major capsid protein gp20 has the canonical HK97 fold common to numerous tailed phages and herpesviruses. According to the HK97 convention, the protein can be divided into four domains: the N-terminal arm (residues 1 to 84), extended loop (residues 85 to 124), peripheral domain (residues 125 to 182 and 346 to 388), and axial domain (residues 183 to 277, 341 to 345, and 389 to 408) (Fig. 2C). Unlike in HK97, the P68 major capsid protein also contains an insertion domain (residues 278 to 340). The insertion and peripheral domains form a cleft that binds the extended loop of an adjacent major capsid protein and thus contribute to the capsid’s stability (fig. S3, A to E).
Fig. 1. Virion and genome organization of phage P68.
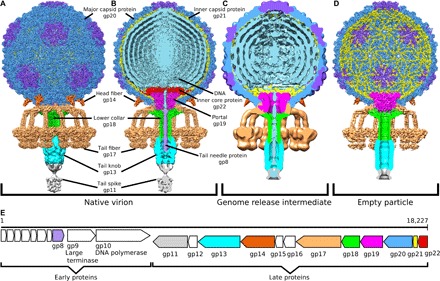
(A and B) Structures of P68 virion, (C) genome release intermediate, and (D) empty particle. The whole P68 virion is shown in (A), whereas particles without the front half are shown in (B) to (D). The structures are colored to distinguish individual types of structural proteins and DNA. (E) Schematic diagram of P68 genome organization, with structural proteins color-coded in accordance with the structure diagrams shown in (A) to (D).
Fig. 2. Capsid structure of P68.
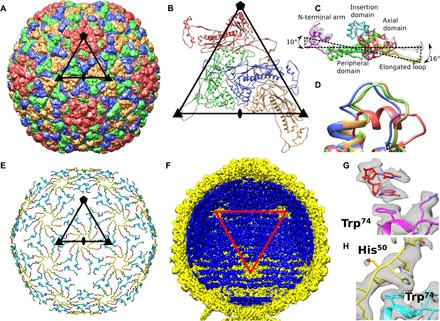
(A) Major capsid proteins of P68 have HK97 fold and form T = 4 icosahedral lattice. Positions of selected icosahedral five-, three-, and twofold symmetry axes are indicated by pentagon, triangles, and oval, respectively. Borders of one icosahedral asymmetric unit are highlighted. (B) Cartoon representation of P68 major capsid proteins in icosahedral asymmetric unit. Positions of icosahedral symmetry axes and borders of icosahedral asymmetric unit are shown. (C) Major capsid proteins from icosahedral asymmetric unit differ in positions of elongated loops and N-terminal domains. Color coding of one of the subunits indicates division of major capsid protein to domains. (D) Residues 253 to 263 from the axial domain of major capsid proteins differ in structure. The residues form an α helix in the subunit that is part of the pentamers, whereas they constitute loops in the other subunits. The color coding of subunits is the same as in (B). (E) The inner capsid protein is organized in a T = 4 icosahedral lattice. Proteins are rainbow-colored from N terminus in blue to C terminus in red. Subunits related by icosahedral threefold axes and quasi-threefold axes of the T = 4 lattice form three-pointed stars in which the C terminus of one subunit is positioned next to the N terminus of another subunit. Borders of a selected icosahedral asymmetric unit are shown. (F) The ordering of the packaged P68 dsDNA genome (shown in blue) is disrupted around the fivefold vertices of the capsid (shown in yellow). Red triangle indicates one face of icosahedron. (G) Stacking interactions of two nucleotides with side chain of Trp74 of major capsid protein located next to fivefold vertex. (H) Side chains of Trp74 of major capsid proteins that form hexamers bind to His50 of inner capsid proteins.
Inner capsid proteins mediate contacts of capsid with genome
The inner face of the P68 capsid is lined by inner capsid proteins gp21, organized with icosahedral T = 4 symmetry (Fig. 2E). Structures of the inner capsid proteins were determined as part of the icosahedral reconstruction of the P68 head to a resolution of 3.3 Å. Except for an α helix formed by residues 14 to 21, the 55-residue-long inner capsid protein “Arstotzka” lacks secondary structure elements. The arrangement of three copies of gp21 (Fig. 2E) is reminiscent of the emblem of the fictional country Arstotzka. The inner capsid proteins related by icosahedral threefold axes and quasi-threefold axes form three-pointed stars (Fig. 2E) and are arranged so that the N terminus of one subunit is located close to the C terminus of another one within the stars (Fig. 2E).
The electron density of P68 dsDNA is resolved inside the regions of the capsid lined by the inner capsid proteins, but it is missing in the proximity of fivefold vertices, where the inner capsid proteins are not structured (Fig. 2, E and F). The electron densities of two nucleotides of single-stranded DNA are stacked against the side chains of Trp74 of the major capsid proteins that form pentamers (Fig. 2G). In contrast, the side chains of Trp74 of major capsid proteins that form hexamers interact with the side chains of His51 of the inner capsid proteins (Fig. 2H) and thus cannot bind phage DNA. Therefore, the inner capsid proteins could enable the packaging of the P68 genome in its head. The location of gp21 in the P68 genome resembles that of the scaffolding protein gp7 of phage phi29 (24). Unlike typical scaffolding proteins, gp21 subunits remain attached to the P68 capsid after maturation. Subunits of gp21 could function in determining the triangulation number of the P68 capsid during assembly, as was shown for the CpmB protein of S. aureus pathogenicity island 1 that is parasitical on staphylococcal phage 80α (25). However, gp21 has no sequence similarity to the gp7 of phi29 or CpmB of S. aureus pathogenicity island 1, and the proteins also differ in their structures. The inner capsid proteins of P68 remain attached to the capsid after genome ejection, indicating that they do not participate in genome delivery (Fig. 1D).
DNA is held inside the P68 head by interaction with one subunit of the portal complex
The structure of the portal complex from the native P68 virion with imposed 12-fold symmetry was determined to a resolution of 3.9 Å. The portal complex of P68 is 80 Å long along its 12-fold axis, with the external diameters of the upper and lower parts of 140 and 100 Å, respectively (Fig. 3, A to C). The structure of the P68 portal protein gp19 could be built except for residues 1 to 6 and 83 to 104 out of 327. According to the convention (26), it can be divided into three domains: the clip (residues 178 to 223), stem (residues 6 to 41, 156 to 177, and 227 to 248), and wing (residues 249 to 327) (Fig. 3, A and B). Unlike the portal proteins of phages SPP1, P22, and T4 but similar to that of Bacillus phage phi29, the portal protein of P68 lacks a crown domain (21, 27–29). The clip domain is composed of helix α4 and antiparallel strands β4 and β5 (Fig. 3B). It forms part of the binding site for the lower collar complex and tail fibers. The stem domain of the portal protein is composed of a “tail fiber hook” (residues 6 to 11) and helices α1, α3, and α5 (Fig. 3B). The tail fiber hook enables the attachment of tail fibers to the portal complex, as discussed in detail below. Helices α1 and α3 of the stem domain form the outer surface of the portal complex that interacts with the capsid. The wing domain, which forms the part of the portal inside the phage head, consists of helices α2 and α6 to α9 and strands β1 to β3. Helix α6 of the wing domain is inserted into the neighboring portal protein subunit and thus stabilizes the dodecamer structure of the portal complex (Fig. 3A).
Fig. 3. Structure of P68 portal complex and its interaction with capsid.
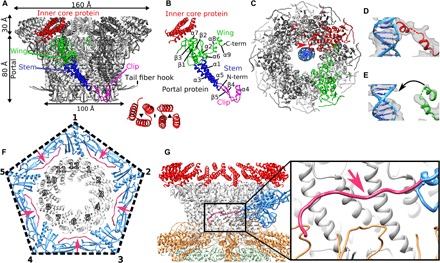
(A) Twelvefold symmetrized structure of portal and inner core complexes of native P68. One of the portal proteins is colored according to domains: clip domain in magenta, wing in green, and stem in blue. Six inner core proteins associated with one portal protein subunit are highlighted in red. The inset shows the symmetry of the arrangement of the six inner core proteins. (B) Division of portal protein into domains. Color coding is the same as in (A). (C) Asymmetric reconstruction of portal complex showing interactions of one of the portal proteins highlighted in red, with DNA shown in blue. The interaction is indicated with a red arrow. One of the portal protein subunits that does not interact with the DNA is highlighted in green. (D) Detail of interaction of helix α9 of portal protein with DNA. Cryo-EM density is shown as gray transparent surface. (E) Structure of portal protein subunit that does not interact with DNA. (F) Interface between portal complex and capsid. Portal proteins are shown in gray, capsid proteins are shown in blue, and N termini of capsid proteins that mediate interactions with the portal are shown in pink and highlighted with pink arrows. (G) Side view of capsid-portal interactions. The single major capsid protein is shown in blue and its N terminus in pink, portal proteins in gray, inner core proteins in red, tail fibers in orange, and lower collar proteins in green. The inset shows detail of interactions between the N-terminal arm of the major capsid protein and stem domains of portal proteins.
Asymmetric reconstruction of the portal complex of native P68 virion, determined to a resolution of 6.6 Å, contains a unique interaction of one of the portal proteins, with the DNA positioned at the center of the portal channel (Fig. 3, C to E). Helix α9 from the wing domain of the unique DNA binding portal subunit binds to a side of the DNA helix (Fig. 3, C and D, and fig. S2D). At a resolution of 6.6 Å, it is not possible to identify residues of the portal protein subunit interacting with the phage DNA. We speculate that the interactions of P68 genome with the portal protein and those of the tail needle protein with the plug domain of the tail knob protein, as discussed below, hold the phage DNA inside the phage head. A similar asymmetric interaction of one of the portal protein subunits with the end of the phage genome has been previously observed for phage phi812 from the family Myoviridae (30).
Interface between capsid and portal complex
Asymmetric reconstruction of the entire P68 virion at a resolution of 4.7 Å enabled characterization of the interface between the capsid and portal complex (Fig. 3, F and G). The binding interface is formed by the outer surface of the wing and stem domains of the portal proteins and N terminus and P domain of the major capsid protein. The interface is in agreement with the previous prediction made on the basis of docking of portal complexes into capsid structures of other phages (27). Residues 1 to 42 from the N-terminal arm of the major capsid proteins adjacent to the portal are not structured. Residues 43 to 59 of the major capsid proteins wrap around the stem domains of portal proteins (Fig. 3F and fig. S4, A and B). Capsid proteins of all tailed phages have HK97 folds that include N-terminal arms. However, there is variability in the structures of the N-terminal arms among phage capsid proteins. Therefore, more high-resolution structures of portal complex–capsid interfaces are needed to determine whether other tailed phages use a mechanism similar to that of P68 for incorporating portal complexes into their capsids.
In the asymmetric reconstruction of the P68 virion, the portal complex exhibits 12-fold symmetry. This is in agreement with the previous studies of phage P22, for which it was speculated that its portal complex can exist in two conformations: an asymmetric one that binds the large terminase to enable genome packaging and a symmetric one present in the mature virion (31).
Inner core proteins interact with the capsid and determine the attachment sites of head fibers
The surface of the portal complex facing toward the center of the P68 head is covered by 72 subunits of the inner core protein gp22 (Figs. 1B and 3, A and B). The structures of the inner core proteins were determined as part of the reconstruction of the portal complex from the native P68 virion with imposed 12-fold symmetry at a resolution of 3.9 Å. Only residues 91 to 114, which form an α helix, are resolved from each 147-residue-long inner core protein. The six inner core proteins associated with each portal protein form two three-helix bundles related to a quasi-twofold rotational axis (Fig. 3A). However, the asymmetric reconstruction of the P68 virion at a resolution of 4.7 Å shows that the inner core proteins positioned closest to the capsid have additional structured residues that interact with axial domains of the adjacent major capsid proteins and, by modifying their conformations, enable the attachment of head fibers to the capsid, as discussed below. Inner core proteins detach from the portal complex during the phage DNA release (Fig. 1C) and contain predicted pore-lining helices (fig. S3, I and J), indicating that they may enable the transport of the phage DNA across the bacterial cytoplasmic membrane.
Head fibers can position P68 particles for genome delivery at cell surface
The P68 head is decorated with five trimers of head fibers gp14, which are attached to the hexamers of major capsid proteins adjacent to the tail vertex (Figs. 1A and 4, A to C). Structures of head fibers are resolved in the asymmetric reconstruction of the P68 virion, which was determined to an overall resolution of 4.7 Å; however, the distant parts of the head fibers are less well resolved (Figs. 1A and 4A). Because of the mismatch of the 5-fold symmetry of the head and 12-fold symmetry of the tail, only three head fibers are stabilized by interaction with the tail fibers (Fig. 4A). P68 head fibers can be divided into the N-terminal capsid-binding domain (residues 1 to 55), α-helical stalk (residues 56 to 339), and receptor binding domain (residues 340 to 481), which is positioned at the level of the receptor binding domains of tail fibers (Fig. 4A). A cryo-EM map of the fivefold symmetrized head of the native P68 virion at a resolution of 3.8 Å enabled the building of the poly-alanine structure of 55 residues of the capsid-binding domain of the head fiber, which is composed of three β sheets and an α helix (Fig. 4, B and C). Residues of the β sheets mediate the attachment of the head fiber to a hexamer of major capsid proteins (Fig. 4C). The α helices form a coiled coil that enables trimerization of the head fibers (Fig. 4, B and C). The selectivity of binding of the head fibers to the hexamers of major capsid proteins adjacent to the tail vertex is determined by interactions of the inner core proteins with the inner face of the capsid (Fig. 4, D and E). Fifteen of the 72 inner core proteins present in the P68 head form structured “arms” that reach the axial domains of the closest hexamers of major capsid proteins (Fig. 4E). The interaction with the inner core proteins forces the side chains of Phe259, from the axial domains of the major capsid proteins, to adopt a threefold symmetrical alternating “in and out” conformation (Fig. 4, F and G, and fig. S3G). In contrast, in hexamers of the major capsid proteins that do not interact with the inner core proteins, two Phe259 side chains point toward the center of the head and four side chains point away from the particle center (Fig. 4, F and H, fig. S3H). In summary, the binding of the inner core proteins causes a change of symmetry of the six Phe259 side chains from twofold to threefold and thus defines the attachment sites for the head fiber trimers.
Fig. 4. Head of P68 is decorated with five head fibers attached to hexamers of major capsid proteins located next to tail vertex.
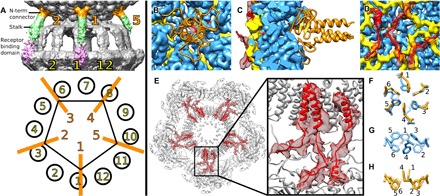
(A) P68 head is decorated with five head fibers that extend toward tail fibers. The head fibers can be divided into the N-terminal connector shown in orange, stalk in green, and receptor binding domain in pink. Because of the mismatch of the 5-fold symmetry of the head and 12-fold symmetry of the tail, only fibers 1, 2, and 4 are stabilized by interactions with tail fibers. (B to D) N-terminal connector domains of head fibers (shown in cartoon representation in orange) are attached to hexamers of major capsid proteins (shown as blue density). Cryo-EM density of inner capsid proteins is shown in yellow, and arms of inner core proteins, which interact with major capsid proteins, are shown in cartoon representation in red. Cryo-EM density of inner core proteins is shown as semitransparent red surface. External view of P68 head (B), section through capsid (C), and internal view of capsid (D). (E) Section through P68 head perpendicular to tail axis at level of inner core complex. Inner core proteins that interact with major capsid proteins are highlighted in red. The electron density of the inner core proteins is shown as a red semitransparent surface. (F to H) Details of organization of Phe259 side chains around quasi-sixfold axis of hexamer of major capsid proteins. In hexamers that interact with inner core proteins, side chains (in blue) are organized with threefold symmetry in alternating up and down conformations (F and G). In hexamers of capsid proteins that do not interact with inner core proteins, Phe259 side chains are organized with twofold symmetry, with two side chains pointing into capsid and four out (F and H).
Residues 340 to 477 of the head fiber are homologous to the receptor binding proteins of lactococcal phages TP901-1, P2, and bIL170 (table S3) (32–34). The putative receptor binding domains of P68 head fibers are positioned next to the receptor binding domains of tail fibers (Fig. 4A). Therefore, the binding of head fibers to receptors can position P68 with its tail orthogonal to the cell surface for genome delivery. This is supported by the broader host range of phage P68 in comparison to the closely related phage 44AHJD, which lacks the gene for the head fiber (23, 35). In contrast, the head fibers of previously structurally characterized phages point in all directions and are thought to function in the reversible attachment of phages to cells in random orientations.
Lower collar complex and tail needle of P68 tail
The structure of the 12-fold symmetrized tail of the P68 virion was determined to a resolution of 3.9 Å. The lower collar complex is attached to the portal complex and forms the central part of the P68 tail (Figs. 1, A and B, and 5A). The dodecamer of lower collar proteins (gp18) has the shape of a mushroom with a head diameter of 162 Å and a total length of 146 Å (Fig. 5, B to D). It contains an axial channel that is continuous with that of the portal complex (Figs. 1B and 5E). The structure of the lower collar protein could be built except for residues 1 and 154 to 186 out of 251. It can be divided into three parts: the curly domain (residues 3 to 116 and 222 to 251), tube domain (residues 116 to 154 and 184 to 222), and knob connector loop (residues 154 to 184) (Fig. 5D). The curly domain, formed by six α helices, mediates the attachment of the lower collar complex to the portal complex and tail fibers. The tube domain is composed of two antiparallel β strands (Fig. 5, C and D). Twelve tube domains form a β barrel with 24 β strands, which is 108 Å long (Fig. 5C). The knob connector loops enable the attachment of the tail knob complex, with sixfold symmetry, to the lower collar complex.
Fig. 5. Structure of P68 tail.
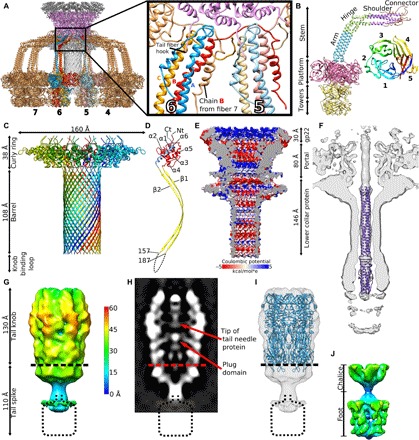
(A) Tail fibers of P68 form skirt around tail tube. Tail fibers are shown in gold; however, individual subunits of two tail fibers are distinguished in red, blue, and orange. Portal proteins are shown in magenta, inner core proteins in dark gray, and lower collar proteins in light gray. The inset shows details of interactions of tail fibers with each other and their attachment to the portal complex. (B) Structure of P68 tail fiber trimer in cartoon representation and its division into domains. The structure of the platform and tower domains was solved by x-ray crystallography to a resolution of 2.0 Å and fitted into the cryo-EM map of the native P68 tail. The inset shows a cartoon representation of the platform domain of the P68 tail fiber rainbow-colored from N terminus in blue to C terminus in red. (C) Structure of lower collar complex with individual subunits distinguished by rainbow coloring. (D) Division of lower collar protein into domains. The curly ring domain is shown in red and blue, the barrel domain is shown in yellow, and the dashed line indicates the knob-binding loop with unknown structure. (E) Surface charge distribution in inner core, portal, and lower collar complex of native P68 particle. (F) Fit of structure of tail needle of phage P22 into 12-fold symmetrized reconstruction of native P68 tail. (G) Sixfold symmetrized reconstruction of P68 tail knob and tail spike complexes. The surface of the cryo-EM map is radially colored on the basis of the distance from the sixfold axis of the complex. (H) Distribution of electron density in central section of tail knob and tail spike complexes. (I) Fit of structure of tail knob of phage C1 into P68 reconstruction. (J) Structure of tail spike with imposed fivefold symmetry shows its chalice and foot domains.
Portal and lower collar complexes of P68 form a channel with a total length of 270 Å (Fig. 5E). The inner diameter of the channel varies from 30 to 55 Å. The surface charge distribution inside the channel is mostly negative, but it is interrupted by neutral and positively charged layers (Fig. 5E). The mostly negative charge of the inner surfaces of the complexes enables ejection of the phage genome.
The central channel of the lower collar complex and the proximal half of that of the tail knob are filled by the tail needle trimer (Fig. 5F). Asymmetric reconstruction of the P68 particle at a resolution of 4.7 Å did not enable building of the atomic structure of the tail needle trimer. However, the structure of the tail needle protein of Salmonella enterica phage P22 fits the tail needle density of P68 with a correlation coefficient of 0.8 (Fig. 5F). On the basis of the high content of α-helical secondary structure elements, we speculate that the tail needle of P68 is made of gp8. It has been shown that the tail needle of bacteriophage P22 is essential for stabilizing packaged DNA inside the virion (22), and there is evidence that it enables DNA delivery by interacting with bacterial membranes.
Tail knob and tail spike
The tail of P68 continues beyond the lower collar protein by tail knob gp13 and tail spike gp11 (Fig. 1, A and B). The two complexes were reconstructed to resolutions of 11 and 7 Å, respectively, indicating that they are more flexible than the parts of the tail near the phage head (Fig. 5, G to J). This flexibility may be required to allow the putative cell wall–degrading enzymes located in the tail spike to cleave a pore in the bacterial cell wall to enable genome delivery.
The previously determined crystal structure of the tail knob of Streptococcus phage C1 fits into the reconstruction of the corresponding part of the P68 tail with a correlation coefficient of 0.65 (Fig. 5I) (table S3) (10). The length of the P68 tail knob is 130 Å along the tail axis. It has an outer diameter of 80 Å and an inner tube diameter of 40 Å. The channel is continuous with that of the lower collar protein (Fig. 1B). One-third of tail knob proximal to the head of native P68 contains a tubular density that probably belongs to the trimer of tail needle protein (Figs. 1B and 5, F and H), which is covalently linked to the end of P68 DNA (23).
An asymmetric reconstruction of the tail spike provides evidence that it has fivefold symmetry (fig. S5, F to H). Fivefold symmetrized, localized reconstruction of the tail spike shows that it is 110 Å long and has a maximum diameter of 70 Å (Fig. 5J). It can be divided into the chalice, which mediates attachment to the tail knob, and the distal lysis domain (Fig. 5J). Sequence comparisons indicate that the tail spike of P68 is homologous to the PlyCb lysin from phage C1 (table S3) (36). However, the structure of PlyCb does not fit into the reconstruction of the P68 tail spike (37). Other proteins with peptidoglycan degradation activities such as the amidase from S. aureus, peptidases from Staphylococcus saprophyticus, and endolysin from staphylococcal phage K are homologous to the last 130 amino acids of the P68 tail spike protein (table S3). This indicates that the tail spike proteins of P68 degrade the bacterial cell wall to enable access of the phage to the cytoplasmic membrane.
Tail fibers
The tail fibers of P68 form a skirt-like structure around the tail (Figs. 1, A and B, and 5A). Each tail fiber is a trimer of 647-residue-long gp17 subunits (Fig. 5B). The tail fiber can be divided into the N-terminal stem domain (residues 1 to 145), platform (residues 151 to 445), and C-terminal tower (residues 446 to 647) (Fig. 5B). Cryo-EM reconstruction of the 12-fold symmetrized tail of P68 virion at a resolution of 3.9 Å enabled the building of the structure of the stem domain, which can be further subdivided into a connector (residues 1 to 45), shoulder (residues 46 to 80), hinge (residues 81 to 115), and arm (residues 116 to 145) (Fig. 5B). Because of the asymmetric shape of the tail fiber, the three constituent subunits (A, B, and C) differ in structure from each other (Fig. 5B). Functionally important differences are found in the connector regions that mediate the attachment of the tail fiber to the portal and lower collar complexes (Fig. 5A). The connector domain of subunit A and shoulder from subunit C form a noose-like structure that encircles the N-terminal tail fiber hook of the portal protein (Fig. 5A). The connector of subunit B binds to the shoulder domains of subunits A and B from the tail fiber positioned counterclockwise when looking at the tail from the direction of the head (Fig. 5A). The first structured residue of the connector of subunit C (Thr24) is located between the clamp domain of subunit B from the tail fiber positioned clockwise and the shoulder domain of subunit C and the clamp domain of subunit A positioned counterclockwise (Fig. 5A). Thus, the N terminus of C subunit mediates interactions between tail fibers that are one position removed from each other.
The shoulder domain of the tail fiber is straight until the hinge domain, which introduces a turn of 110° (Fig. 5B). The hinge of subunit C is formed by two α helices connected by a short loop, which allows the chain to bend and pass under subunits A and B (Fig. 5B). After the hinge, the three subunits form a straight coiled-coil arm (Fig. 5B). The cryo-EM reconstruction of the P68 tail is complemented by the crystal structure of the tail fiber protein determined to a resolution of 2.0 Å (table S4). Although the full-length tail fiber protein was used for crystallization, only the platform and tower domains (residues 139 to 647) were resolved (table S4). The combination of cryo-EM and x-ray results allowed construction of the complete tail fiber.
The platform domain of the P68 tail fiber has a five-bladed β-propeller fold (Fig. 5B). Each of the blades contains four antiparallel β strands. The domain is cyclically enclosed, since the first N-terminal β strand of the domain is part of the same blade as the last three C-terminal β strands (Fig. 5B). It has been shown that the platform domains of various phages, including phi11, PRD1, and PhiKZ, contain receptor binding sites (38–40). The platform of the P68 tail fiber is similar in structure to that of staphylococcal phage phi11 from the family Siphoviridae, with an RMSD (root mean square deviation) of the corresponding Cα atoms of 1.10 Å. The sequence identity of the two proteins is 24%. There are differences in the receptor binding sites within the platform domains of the two phages that may reflect their different receptor requirements (fig. S5, A to D). Whereas P68 binds to wall teichoic acid glycosylated with β-O-N-acetyl-glucosamine, phi11 can attach to both β-O-N-acetyl-glucosamine and α-O-N-acetyl-glucosamine (16).
The tower domain of the P68 tail fiber is composed of two subdomains (residues 454 to 555 and 556 to 645), which are structurally similar to each other (Fig. 5B). Each subdomain is formed of a four-stranded antiparallel β sheet connected by loops and two short helices (Fig. 5B). The β sheets are positioned close to the threefold axis of the fiber, whereas the loops and helices are exposed at the surface. The subdomains are homologous to putative major teichoic acid biosynthesis protein C, muramidases, and receptor binding fibers of R-type pyocin (table S3). Thus, the tower region may be involved in binding to the cell wall or peptidoglycan digestion. Compared to the tail fiber of phage phi11, the platform and tower domains of P68 exhibit domain swapping within the trimer of the tail fiber (fig. S5E) (38).
The 12-fold symmetrized cryo-EM structure of native P68 tail at a resolution of 3.9 Å shows interactions of residues 229 to 271 of platform domain of subunit B with residues 349 to 386 of chain A and residues 180 to 268 of chain B of platform domains from the neighboring tail fiber (Fig. 5A). The interface has a buried surface area of 1100 Å2, and it is likely that it stabilizes the “skirt” structure of P68 tail fibers. The flexibility of the hinge region of tail fibers was proposed to facilitate receptor binding in other phages (38). In contrast, in P68, the structure of tail fibers appears to be rigid.
Changes in P68 particles associated with genome release
The genome release of P68 is connected to the following major conformational rearrangements of its neck region: (i) Helix α9 from the wing domain of the DNA binding portal protein subunit (Fig. 3, C to E) detaches from the side of the DNA, and the portal complex assumes 12-fold symmetry (fig. S4, C and D). (ii) The 12-fold symmetrized reconstruction of the portal complex of the empty particle, which was determined to a resolution of 3.9 Å, contains no density for residues 257 to 267 and 307 to 327 from the wing domain of the portal protein (fig. S4, C and D). The previously determined structure of isolated portal complex of Bacillus phage phi29 also lacked resolved electron density for the corresponding residues from the wing domain. It is therefore possible that these residues only become structured in the presence of DNA in the portal channel. (iii) The inner core proteins are ejected from the phage head (Fig. 1, B to D). Changes in the structure of the wing domains of portal proteins associated with the decreased pressure inside the phage head after partial DNA ejection may trigger release of the inner core proteins.
Phages have to deliver their genomes across the cell wall and cytoplasmic membrane to infect Gram-positive bacterial cells. It has been shown that, after cleavage of the peptidoglycan cell wall, phages from the family Podoviridae, including phi29, release proteins that form pores in the cytoplasmic membrane. The pore-forming proteins attach to the end of the tail tube and enable delivery of the phage DNA into the cytoplasm (41). Unlike virions of phage phi29, particles of P68 do not bind to liposomes at acidic pH (fig. S1, I to P) (41); instead, they aggregate with each other through their tails (fig. S1, I to P). However, liposomes in the mixture with P68 become distorted (fig. S1, I to P). It is possible that the ejected inner core proteins, which contain predicted pore-lining helices (fig. S3, I and J), interfered with the liposome integrity.
The heads of P68 particles in the process of genome release contain shells of packaged DNA that are spaced 26 to 30 Å apart, whereas in the full virions, the DNA spacing is 20 Å (Fig. 1, B and C, and fig. S3F). The resolved structure of the layers of dsDNA in the P68 genome release intermediate indicates that all the particles released similar amounts of DNA and provides evidence of a gradual relaxation of the DNA pressure during the genome release.
Only 2% of P68 genome release intermediates and empty particles retained their tail knobs and tail spikes in vitro (fig. S1, A to G). However, the tail knobs and tail spikes of the complete empty particles contain central channels (Fig. 1D), indicating that the complexes may remain attached to P68 virions during genome release in vivo.
Attachment of P68 virions with S. aureus cell wall
Electron micrographs of P68 virions with fragments of S. aureus cell wall show that most of the P68 virions bind with the axes of their tails at an acute angle to the surface of the cell wall (fig. S1H). This orientation of the tail is not suitable for the delivery of the phage DNA into a cell. The initial binding of P68 virion to the cell wall may be mediated by the head fibers; however, individual head fibers cannot be distinguished in the electron micrographs. The predicted receptor binding sites of tail fibers are located at the sides of the platform domains rather than at the tips of the tower domains (Fig. 5B and fig. S5, A to D). Therefore, the receptor binding sites are located at the sides of the skirt formed by the tail fibers of P68 (Figs. 1A and 5, A and B). The tail knob and tail spike form a 20-nm-long protrusion at the center of the tail fiber skirt (Fig. 1, A and B). Therefore, the P68 virion has to locally degrade the cell wall, by the hydrolase activity associated with its tail spike, to produce a cavity into which the tip of the tail knob can be inserted so that the phage can bind with its tail axis perpendicular to the cell wall.
Mechanism of P68 genome delivery
The structures of the P68 virion, its genome release intermediate, and empty particle in combination with the observation of the attachment of P68 virions to the S. aureus cell wall enabled us to propose the mechanism of P68 genome delivery (Fig. 6). P68 virion binds to the S. aureus cell by either head or tail fibers. At the moment of the initial binding, the tail axis of the phage is not perpendicular to the cell surface (Fig. 6 and fig. S1H). After the attachment, tail spike proteins degrade the cell wall, which allows the phage to position itself with its tail axis perpendicular to the cell surface (Fig. 6). The unique attachment of P68 head fibers exclusively to the hexamers of capsid proteins located next to the tail vertex enables the phage to use its head fibers for orienting itself with the tail axis perpendicular to the cell surface. Further cell wall digestion enables the tip of the P68 tail to reach the S. aureus cytoplasmic membrane. The signal triggering P68 genome release is unknown; however, it may be the binding of the tail spike to a receptor in the membrane, exposure of the tail spike to the hydrophobic environment of the membrane, or a sensing of the transmembrane potential. Subsequently, conformational changes of the P68 portal and disruption of the unique interaction of one of the portal protein subunits with the end of the phage genome enable ejection of the tail needle protein, inner core proteins, and DNA through the tail channel. The tail needle and inner core proteins may form a pore in the bacterial membrane for delivering phage DNA into the bacterial cytoplasm (Fig. 6).
Fig. 6. Mechanism of P68 genome delivery into S. aureus cell.
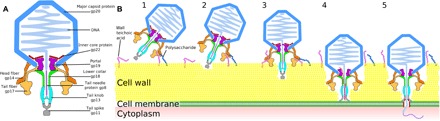
(A) Diagram of P68 virion. (B) Mechanism of P68 genome delivery. P68 virion attaches to cell surface by head or tail fibers (1). This attachment allows enzymes from tail spike to cleave bacterial cell wall (2). This degradation of S. aureus cell wall enables P68 to bind with its tail axis perpendicular to cell surface (3). Further cell wall digestion allows tip of P68 tail to reach cytoplasmic membrane, which triggers release of inner core proteins and DNA (4). Inner core proteins form channel in membrane for ejection of phage DNA into bacterial cytoplasm (5).
The quasi-equivalent structure of the T = 4 icosahedral capsid includes conformational differences in the major capsid proteins from the icosahedral asymmetric unit (Fig. 2, B and C). The N-terminal arms and extended loops are in one plane in the major capsid proteins that connect two hexamers (Fig. 2C). In contrast, the same domains are bent 16° in the major capsid proteins that form pentamers and 8° in subunits that connect hexamers to pentamers (Fig. 2C). Additional differences among the capsid proteins are in the structures of residues 253 to 263 from the axial domain, which fold into α helices in subunits that form pentamers and loops in subunits that belong to hexamers (Fig. 2D).
MATERIALS AND METHODS
P68 growth and purification
Bacteriophage P68 was purchased from the Félix d’Hérelle Reference Center for Bacterial Viruses (Université Laval, Québec, Canada). The propagation strain of S. aureus RN4220 ΔtarM (16) was provided by A. Peschel from the Department of Infection Biology, University of Tübingen. Phage P68 was propagated on S. aureus RN4220 ΔtarM grown at 37°C in meat peptone broth [13 g of nutrient broth CM1 (Oxoid), 3 g of yeast extract powder L21 (Oxoid), and 5 g of peptone L37 (Oxoid) were dissolved in distilled water to a final volume of 1000 ml, and the pH was adjusted to 7.4 using 10 M NaOH].
Phage lysate from 300 ml of bacterial culture was centrifuged at 5000g for 30 min at 4°C. The resulting supernatant was filtered through a 0.45-μm polyether-sulfone syringe filter (Techno Plastic Products, Switzerland) to remove bacterial debris. Phages were pelleted by centrifugation at 64,000g for 2.5 hours at 4°C. The resulting pellets were resuspended in 350 μl of phage buffer [50 mM tris (pH 8.0), 10 mM CaCl2, and 10 mM NaCl] by mild shaking overnight at 5°C. The resulting solution was mixed with an equal amount of chloroform by gently inverting the tube 10×. The mixture was centrifuged at 3000g for 10 min at room temperature. The aqueous fraction from the chloroform mixture was overlaid onto a preformed CsCl step density gradient (1 ml of each 1.45 g/ml, 1.50 g/ml, and 1.70 g/ml of CsCl in phage buffer) and centrifuged at 194,000g for 4 hours at 12°C using an SW55Ti rotor (Beckman Coulter). Phage particles forming a visible band were collected with an 0.8-mm gauge needle and syringe. Cesium chloride was removed from the phage-containing fraction by dialysis against a 5000× excess of phage buffer at 4°C overnight using Visking dialysis tubing type 8/32″, 0.05 mm thick (part no. 1780.1, Carl Roth, Germany).
Liposome preparation
Liposomes were prepared as described previously, with minor modifications (41). The phospholipids (Avanti Polar Lipids) were resuspended in chloroform and mixed in a molar ratio that mimicked the composition of the cell membrane of S. aureus (41): phosphatidyl-ethanolamine, phosphatidyl-dl-glycerol, phosphatidyl-choline, and cholesterol in a molar ratio of 5:6:2:9. The chloroform-liposome mixture was dried for 4 hours in a 200-ml evaporation flask in a rotatory vacuum evaporator at 37°C and 20 rpm to create a homogenous lipid film. The lipid film was resuspended in phage buffer [50 mM tris (pH 8.0), 10 mM CaCl2, and 10 mM NaCl], flash-frozen and thawed five times, and sonicated for 120 s using an ultrasonic cleaner (Sonica). Subsequently, the solution was passed 20 times through a membrane with 400-nm pores in the LiposoFast Basic Extruder (Avestin).
Phage-liposome interaction
The genome ejection of P68 was induced by exposing it to pH 4.2, as described previously for phage phi29 (41). P68 at a concentration of 2 mg/ml in phage buffer [50 mM tris (pH 8.0), 10 mM CaCl2, and 10 mM NaCl] was mixed with liposomes at a concentration of 5 mM in phage buffer in a volume ratio of 1:10. The pH of the mixture was changed to acidic by three rounds of concentrating and diluting with 0.1 M sodium acetate, 300 mM ammonium sulfate (pH 4.2) buffer using 100-kDa Amicon ultra centrifugal filters. The phage was incubated in the acidic buffer for 1 hour. The mixture was applied to holey carbon–coated copper grids (R2/2, mesh 200; Quantifoil), blotted, and flash-frozen in liquid ethane.
Identification of structural proteins of phage P68
The purified P68 virus was resuspended in Laemmli buffer and boiled for 3 min, and the proteins were separated by tricine gradient gel electrophoresis. All protein bands were cut from the gel and used for mass spectrometry (MS) analysis. In addition, P68 at a concentration of 2 mg/ml in phage buffer containing 1% SDS was boiled for 3 min and analyzed by MS. After destaining and washing, the proteins were subjected to trypsin digestion (sequencing grade, Promega). MALDI (matrix-assisted laser desorption/ionization)–MS and MS/MS analyses were performed on an Ultraextreme mass spectrometer (Bruker Daltonics, Bremen, Germany). Data processing and analysis were performed with the software FlexAnalysis 3.4 and MS BioTools (Bruker Daltonics). Mascot software (Matrix Science, London, UK) was used for sequence searches in exported MS/MS spectra against the National Center for Biotechnology Information database and a local database supplied with the expected protein sequences. The mass tolerance of peptides and MS/MS fragments for MS/MS ion searches were 50 parts per million and 0.5 Da, respectively. The oxidation of methionine and propionyl-amidation of cysteine as optional modifications and one enzyme miss-cleavage were set for all searches. Peptides with a statistically significant peptide score (P < 0.05) were considered.
Cryo-EM sample preparation, data acquisition, and initial data processing
Solution containing the phage (3.8 μl) at a concentration of 2 mg/ml was pipetted onto holey carbon–coated copper grids (R2/2, mesh 200; Quantifoil), blotted, and vitrified by plunging into liquid ethane using FEI Vitrobot Mark IV. The vitrified sample was transferred to an FEI Titan Krios electron microscope operated under cryogenic conditions and at an acceleration voltage of 300 kV. The illuminating beam was aligned for parallel illumination in NanoProbe mode, and coma-free alignments were performed to remove residual beam tilt. Imaging was done under low-dose conditions with a total dose of 21 e−/Å2 s−1. Data were collected with underfocus values ranging from −1.0 to −3.0 μm. Data acquisition was performed at a nominal magnification of ×75,000, resulting in a calibrated pixel size of 1.063 Å. Micrographs were acquired using a Falcon II direct electron detector operated in a movie mode. One-second exposure was fractionated into seven frames and saved as separate files. Automated data acquisition was done using the acquisition software EPU (FEI). The micrographs were collected in two separate sessions (1405 and 1486 micrographs) with the same microscope with identical settings. Micrographs from both sessions were merged and processed together.
The seven-frame movies were aligned globally and locally (5 × 5 patch) using the software MotionCor2 (42), and the defocus values were estimated from aligned micrographs using the program CtfFind4 (43). Power spectra of Fourier transforms of micrographs were visually inspected, and micrographs with distorted or missing Thon rings were discarded.
Icosahedral reconstruction of P68 capsid
P68 heads were manually boxed using e2boxer.py from the software package EMAN 2.1 (44). Images of 37,218 particles (600 × 600 pixels) were extracted from the micrographs and background-normalized using RELION 2.1 (45). Multiple rounds of 2D classification of particles were performed using RELION (45). Only particles belonging to high-resolution class averages were used for subsequent reconstruction (36,853 particles). To generate the initial model, we used the random de novo model method as implemented in EMAN2 (44). Particles were divided into two independent sets. From each half, nine subsets of 4× binned images were generated. Each set contained 1000 particles. The particles were assigned random orientations and iteratively reconstructed into 3D volumes using the software package jspr (46). Independent initial models were chosen from both half-datasets by selecting the two models most similar to each other. Afterward, the initial models were unbinned 4× to match the size of the original images and low-pass–filtered to a resolution of 40 Å. The refinement was performed using the program RELION 2.1 (45). After initial auto refinement, 3D classification was performed without the alignment step. Particles belonging to the best class (21,625 particles) were selected for further RELION auto refinement with imposed icosahedral symmetry and maximum allowed deviations from previous orientations of 10°. The resulting map was threshold-masked, divided by the modulation transfer function, and B-factor sharpened during the post-processing in RELION (45).
Capsids of genome release intermediates and empty particles were determined using the same reconstruction strategy. After 2D classification, 4040 and 10,259 images were available for the reconstructions of capsids of genome release intermediate and empty particles, respectively. After 3D classification, the numbers of particles were 2332 and 8580, respectively.
Asymmetric reconstruction of native P68 virion
For the asymmetric reconstruction of the whole virion, the particles were extracted from the original micrographs using a box size of 1200 × 1200 pixels. The images were 2× binned in Fourier space using xmipp_transform_downsample (47), resulting in a pixel size of 2.126 Å. Initial 2D classification with a circular mask with a diameter of 1000 Å led to the selection of 34,717 particles for further processing. The structure of phage P22 (EMD-1222) scaled to 73% of its original size was used as the initial model for 3D reconstruction (48). Asymmetric refinement, using the auto refine procedure, was performed using the software package RELION. This was followed by 3D classification, which used orientations and center offset values from the previous round of refinement. The class with the best-resolved details contained 33,612 particles, which were used for further 3D refinement. A mask that covered the whole virion was generated using the programs UCSF Chimera, Segger, and relion_mask_create (4-pixel expansion and 5-pixel soft edges) (45, 49, 50). The mask was used in the final rounds of refinement with local angular searches using the RELION auto refine procedure (45).
Reconstruction of capsid with fivefold symmetry
Images of capsids of native virions (33,612 particles) from the 3D classification of the asymmetric reconstruction were used as the initial dataset for fivefold symmetrized reconstruction. Refinement with imposed C5 symmetry was followed by 3D classification without alignment. After 3D classification, 28,826 particles were retained for further reconstructions. These particles were re-extracted in boxes of 600 × 600 pixels. A mask that included the capsid was created using the programs UCSF Chimera, Segger, and relion_mask_create.py (4-pixel expansion and 5-pixel soft edges) (45, 49, 50). Masked 3D refinement with local searches around the previously determined orientations was performed using the RELION auto refine procedure (45). Identical strategies were used for the reconstructions of fivefold symmetrized capsids of genome release intermediates and empty particles.
Localized reconstruction of P68 portal and tail
Reconstruction of the P68 head with imposed fivefold symmetry provided Euler angles describing particle orientations with a phage tail along the positive direction of the coordinate z axis. The distance of the center of the tail from the center of the capsid was determined on the basis of the fivefold symmetrized map of the P68 virion using the program UCSF Chimera (49). The script localized_reconstruction.py (51) was used to extract subparticles centered on the tail (512 × 512 pixels). 2D classification of the tails of native P68 virions resulted in the selection of 35,490 images. An initial model of the tail was calculated using relion_reconstruct.py by applying the Euler angles determined in the C5 reconstruction of the capsid (45). The model of the tail with C5 symmetry was low-pass–filtered to 40 Å and used as an initial reference for RELION auto refinement with imposed C12 symmetry. Segments of the reconstruction corresponding to the portal and tail were segmented using the programs UCSF Chimera and Segger and combined to generate a mask covering the portal and tail with the program relion_mask_create.py (4-pixel expansion and 5-pixel soft edges) (45, 49, 50). 3D classification with the applied mask, omitted alignment, and imposed C12 symmetry was used to select 21,702 particles. The particles were subjected to auto refinement and post-processing in RELION (45). The poly-alanine structure was manually built into the resolved parts of the P68 tail. The Protein Data Bank (PDB) was used to prepare an improved mask covering the portal complex, N-terminal parts of tail fibers, and lower collar protein, using the program e2pdb2mrc.py from the EMAN2 package and relion_mask_create.py (4-pixel expansion and 5-pixel soft edges) (44, 45). This mask was used in one more round of auto refinement, in which orientation and offset searches were limited to ±9° and ±3 pixels, respectively. The refinement strategy is summarized in fig. S6.
The reconstruction strategies of tails of P68 particles in the process of genome release and empty particles were identical to those of the full particle. For the genome release intermediates, the initial number of particles was 4040. After 3D classification, 1506 particles were selected for the final 3D auto refinement. The initial set of empty particles contained 10,288 images. After 2D classification, 6123 particles were selected. 3D classification resulted in the selection of 3246 particles.
2D classifications of the particles in the process of genome release and empty particles identified subsets of particles with intact tail knobs and tail spikes. 3D classifications of these subsets with imposed C12 symmetry yielded 98 and 112 particles for the genome release intermediate and empty particles, respectively.
Localized reconstruction of P68 tail knob and tail spike
Side views of native P68 virions were selected for determining the structure of the tail knob and tail spike (21,016 particles). Boxed particles (1440 × 1440 pixels) were 2× binned using xmipp_transform_downsample (47). Projection images of a properly oriented reconstruction of the whole P68 virion, lacking the tail knob and tail spike, were subtracted from the particle images using the program relion_project.py (45).
Parts of the images containing the tail knob were boxed (128 × 128 pixels) from the difference images using the program localized_reconstruction.py (51). After 2D classification, 8589 particles were selected for reconstruction. An initial model of the tail knob was calculated using the particle orientations from the asymmetric reconstruction of the whole virion. 3D auto refinement in RELION was used to calculate the structure of the complex with imposed C6 symmetry. The resulting map was used to generate a mask (extended by 4 pixels and a soft edge of 5 pixels) using the program relion_mask_create.py (45). The final structure of the complexes was determined in 3D auto refinement with the use of the mask in the RELION package (45).
Parts of the images containing the tail spike were extracted from the same set of difference images as the tail knobs (128 × 128 pixels). After 2D classification, 6121 particles were selected for reconstruction. The initial model was calculated using the particle orientations from the asymmetric virion reconstruction. The asymmetric reconstruction indicated that the tail spike complex has fivefold symmetry; therefore, it was enforced during the subsequent rounds of structure refinement. A mask was generated from the tail spike map using the program relion_mask_create (extended by 4 pixels and a soft edge of 5 pixels) (45). The final rounds of reconstruction in RELION were performed with the use of the mask (45). The final map was threshold masked, MTF corrected, and B-factor sharpened using the program relion_postprocess. The refinement strategy is summarized in fig. S6.
Asymmetric reconstruction of portal
Asymmetric reconstruction of the portal complex of native P68 was determined using symmetry expansion from the C12 symmetrized localized reconstruction of the portal (21,702 particles). Initially, the particle orientations from the previous C12 reconstruction were symmetry expanded to C1 using the program relion_particle_symmetry_expand.py (45). Mask covering the portal complex was generated from the C12 symmetrized structure. Focused 3D classification with 12 classes using the symmetry-expanded orientations was performed in RELION (45). The alignment step was omitted to prevent overfitting. Regularization factors in the range of 20 to 50 were tested to establish the optimal value to detect asymmetric features in the portal. The best reconstruction as judged by map interpretability was obtained with the regularization factor 33. The class from the 3D classification showing an asymmetric interaction between one of the portal protein subunits and dsDNA was chosen for further refinement. Duplicate particle images (C12 symmetry–related images of the same particle) were discarded from the reconstruction, resulting in 7568 unique particles. Final refinement was performed using RELION auto refine, with the application of a mask, but without the orientation search (45). The refinement strategy is summarized in fig. S6.
Cryo-EM structure determination and refinement
PDB structures were built manually using the program COOT (52). Maps of P68 capsids with imposed icosahedral symmetries were first rotated to the standard 222 orientation (one of the fivefold symmetry axes of the particle in the xy plane is rotated 31.717° from the y axis). The maps were cropped and normalized, the center of the particle was moved to the center of the box, and the map was converted to crystallographic space group P23. The map of the portal and tail with C12 symmetry was cropped, normalized, and interpolated to a crystallographic P6 space group. The high-symmetry space groups were used to reduce the computational resources required for refinement of the structures.
Initially, the structures were built as poly-alanine chains. Noting the positions of residues with large side chains and aligning them to sequences of P68 structural proteins identified individual proteins in the reconstructions. Final models were iteratively refined in real space using the program PHENIX real_space_refine.py and, in reciprocal space in the program REFMAC5, corrected manually in COOT (52–54).
Preparation of S. aureus cell walls
S. aureus strain RN4220 ΔtarM was grown in a meat peptone broth until the optical density at 600 nm (OD600) of 0.8. Cell culture (20 ml) was centrifuged at 8000g for 10 min. Resulting pellet was resuspended in a phage buffer to an OD600 of 40. Afterward, 1 ml of the cell suspension was mixed with 1 ml of Lysing Matrix B (MP Biomedicals) containing 0.1-mm silica spheres in a 2-ml FastPrep tube (MP Biomedicals). The 2-ml FastPrep tube was placed in the Microtube Homogenizer FastPrep-24 Classic Instrument (MP Biomedicals). The cells were homogenized for 5 min. The resulting lysate was centrifuged at 300g for 1 hour. The resulting supernatant was carefully pipetted to a 1.5-ml tube. The supernatant was centrifuged at 20,000g for 2 hours. The peptidoglycan appeared as a small pellet at the bottom of the tube. Last, the pellet was resuspended in 50 μl of the phage buffer.
Purification and crystallization of P68 tail fiber
The gene encoding the tail fiber of P68 in plasmid pMUH17 was a gift from U. Bläsi from Max F. Perutz laboratories in Vienna (35). The tail fiber protein was purified as described previously (35), with minor changes. The His-tagged tail fiber protein was expressed in Escherichia coli strain BL21 (DE3). After overnight cultivation in LB medium at 37°C and 200 rpm, the preculture was diluted 100× into 1000 ml of fresh LB medium. The culture was grown at 37°C with 200 rpm shaking until an OD600 of 0.7 was reached. The expression was induced by the addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM. After 4 hours of protein expression at 37°C and 200 rpm shaking, the culture was centrifuged at 4000g for 30 min. The cell pellet was resuspended in 50 ml of lysis buffer [300 mM NaCl, 40 mM imidazole, 50 mM NaH2PO4 (pH 8.0), 1 mM phenylmethylsulfonyl fluoride (PMSF), and lysozyme (1 mg/ml)], incubated for 1 hour at 4°C, and homogenized by sonication using a Qsonica Q700 sonicator (2-s sonication, 2-s pause, 4-min sonication, 50 W). The cell lysate was centrifuged at 20,000g for 40 min at 4°C and loaded onto a HisTrap column (GE Healthcare) equilibrated with lysis buffer. Most of the impurities were eluted with wash buffer [70 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4 (pH 8.0), and 1 mM PMSF]. His-tagged tail fiber protein was eluted from the column using elution buffer [500 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4 (pH 8.0), and 1 mM PMSF]. The solution containing tail fiber protein was dialyzed against 25 mM tris-HCl (pH 7.5), 150 mM NaCl buffer, followed by separation in a HiLoad 16/600 Superdex 200 pg size exclusion column (GE Healthcare). The tail fiber protein eluted in the void volume of the column. The protein at a concentration of 5 mg/ml in 25 mM tris-HCl (pH 7.5), 150 mM NaCl buffer was used for crystallization screening. Crystals were obtained in a CrystalEX 96-Well conical bottom plate (Hampton Research) by the sitting-drop vapor diffusion method by mixing 0.5 μl of protein solution with 0.5 μl of 200 mM MgCl2, 100 mM Hepes (pH 7.0), and 20% (w/v) PEG 6000 (polyethylene glycol, molecular weight 6000). Hexagonal crystals with dimensions up to 200 μm formed within 30 days.
X-ray data collection, structure determination, and refinement
Crystals of tail fiber protein in mother liquor solution were vitrified by plunging into liquid nitrogen. Diffraction data were collected at the SOLEIL synchrotron on beamline PROXIMA-1 equipped with a Pilatus 6M detector. The data were integrated and scaled using the package XDS (55). The model for molecular replacement was prepared by converting the tail fiber of phage phi11 (PDB 5EFV) to poly-alanine and rebuilding it according to the threefold averaged cryo-EM map of the P68 tail fiber (56). The quality of the cryo-EM map was sufficient for building the poly-alanine chain of residues 150 to 570, except for several loops in poorly resolved regions of the map. Molecular replacement was performed using the program Phaser (57). The trimers of the tail fiber proteins sit on threefold axes of the R3 space group. Because of the crystal packing, the stem domain has to be bent nonphysiologically within the arm region. It is likely that the bending of the arm region differs among subunits within the crystal, which results in a local disorder. Therefore, the x-ray electron density map does not contain resolved features corresponding to the arm domain of the tail fiber protein. The structure was subjected to several rounds of automated building using the programs Phenix_auto_build and ARP/wARP combined with manual corrections in COOT and refinement in REFMAC5 (52, 54, 58).
Supplementary Material
Acknowledgments
We acknowledge the CEITEC Core Facilities Cryo-Electron Microscopy and Tomography, Proteomics, and Biomolecular Interactions, supported by the CIISB project LM2015043 funded by the MEYS of the Czech Republic. X-ray data were collected at synchrotron SOLEIL beamline PROXIMA-1. Funding: This research was carried out under the project CEITEC 2020 (LQ1601), with financial support from the MEYS of the Czech Republic under National Sustainability Program II. This work was supported by IT4I project (CZ.1.05/1.1.00/02.0070) and funded by the European Regional Development Fund and the national budget of the Czech Republic via the RDI-OP, as well as the MEYS via the Grant (LM2011033). The research leading to these results has received funding from the Ministry of Health of the Czech Republic grant 16-29916A to R.P., Czech Science Foundation grants 15-21631Y and 18-17810S, and EMBO installation grant 3041 to P.P. Author contributions: D.H. and P.P. designed research. D.H., D.S., K.S., and T.F. performed research. D.H., K.S., T.F., R.P., and P.P. analyzed data. D.H., T.F., and P.P. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Cryo-EM electron density maps of the P68 reconstructions were deposited with the Electron Microscopy Data Bank (EMDB): EMD-4438 icosahedral reconstruction of a capsid of the native phage P68; EMD-4436 5-fold symmetrized reconstruction of the native phage P68; EMD-4437 asymmetric reconstruction of the native phage P68; EMD-4440 reconstruction of head fiber of the native phage P68; EMD-4435 12-fold symmetrized reconstruction of a portal and tail of the native phage P68; EMD-4449 6-fold symmetrized reconstruction of a tail knob of the native phage P68; EMD-4450 5-fold symmetrized reconstruction of a tail spike of the native phage P68; EMD-4451 icosahedral reconstruction of a capsid of a genome release intermediate of P68; EMD-4453 5-fold symmetrized reconstruction of a capsid of the genome release intermediate of P68; EMD-4454 12-fold symmetrized reconstruction of a portal and tail of a genome release intermediate of P68; EMD-4455 12-fold symmetrized reconstruction of a portal and tail with knob of the genome release intermediate of P68; EMD-4442 icosahedral reconstruction of a capsid of the empty particle of P68; EMD-4456 5-fold symmetrized reconstruction of the empty particle of P68; EMD-4457 12-fold symmetrized reconstruction of a portal and tail of the empty particle of P68; EMD-4458 12-fold symmetrized reconstruction of a portal and tail with knob of the empty particle of P68; and EMD-4459 map of the whole native bacteriophage P68 combined from localized reconstructions. The corresponding coordinates were deposited in the PDB under the following accession numbers: 6IAT, icosahedral capsid of a native phage; 6IAW, head fiber; 6IAC, portal and tail of native P68; 6IB1, icosahedral capsid of empty particle; and 6Q3G, structure of the whole bacteriophage P68. The atomic coordinates of the tail fiber protein together with the structure factors were deposited in the PDB under the code 6IAB. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw7414/DC1
Fig. S1. Purified sample of P68 contains native virions, particles in process of genome release, and empty particles; attachment of P68 virions to S. aureus cell walls; and interactions of P68 virions with liposomes.
Fig. S2. Resolution and interpretability of cryo-EM reconstructions.
Fig. S3. Details of P68 head.
Fig. S4. Incorporation of P68 portal complex into capsid and changes in the structure of P68 portal complex upon genome release.
Fig. S5. Structures of P68 tail fiber and tail spike.
Fig. S6. Schemes of cryo-EM reconstruction strategies.
Table S1. Cryo-EM structure quality indicators.
Table S2. List of P68 proteins.
Table S3. HHpred searches for homologs of P68 tail fiber, head fiber, tail knob, and tail spike.
Table S4. X-ray structure quality indicators.
REFERENCES AND NOTES
- 1.Chhibber S., Kaur T., Sandeep K., Co-therapy using lytic bacteriophage and linezolid: Effective treatment in eliminating methicillin resistant Staphylococcus aureus (MRSA) from diabetic foot infections. PLOS ONE 8, e56022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Centre for Disease Prevention and Control/European Medicines Agency, “The bacterial challenge: Time to react” (ECDC/EMEA Joint Technical Report, European Centre for Disease Prevention and Control/European Medicines Agency, 2009).
- 3.Kurlenda J., Grinholc M., Alternative therapies in Staphylococcus aureus diseases. Acta Biochim. Pol. 59, 171–184 (2012). [PubMed] [Google Scholar]
- 4.World Health Organization, Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics (World Health Organization, 2017).
- 5.Takemura-Uchiyama I., Uchiyama J., Osanai M., Morimoto N., Asagiri T., Ujihara T., Daibata M., Sugiura T., Matsuzaki S., Experimental phage therapy against lethal lung-derived septicemia caused by Staphylococcus aureus in mice. Microbes Infect. 16, 512–517 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Aleshkin A. V., Ershova O. N., Volozhantsev N. V., Svetoch E. A., Popova A. V., Rubalskii E. O., Borzilov A. I., Aleshkin V. A., Afanas’ev S. S., Karaulov A. V., Galimzyanov K. M., Rubalsky O. V., Bochkareva S. S., Phagebiotics in treatment and prophylaxis of healthcare-associated infections. Bacteriophage 6, e1251379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leiman P. G., Battisti A. J., Bowman V. D., Stummeyer K., Mühlenhoff M., Gerardy-Schahn R., Scholl D., Molineux I. J., The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 371, 836–849 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Hu B., Margolin W., Molineux I. J., Liu J., The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 339, 576–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang Y., Morais M. C., Battisti A. J., Grimes S., Jardine P. J., Anderson D. L., Rossmann M. G., Structural changes of bacteriophage phi29 upon DNA packaging and release. EMBO J. 25, 5229–5239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aksyuk A. A., Bowman V. D., Kaufmann B., Fields C., Klose T., Holdaway H. A., Fischetti V. A., Rossmann M. G., Structural investigations of a Podoviridae streptococcus phage C1, implications for the mechanism of viral entry. Proc. Natl. Acad. Sci. U.S.A. 109, 14001–14006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang W., Baker M. L., Jakana J., Weigele P. R., King J., Chiu W., Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature 451, 1130–1134 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Baker M. L., Hryc C. F., Zhang Q., Wu W., Jakana J., Haase-Pettingell C., Afonine P. V., Adams P. D., King J. A., Jiang W., Chiu W., Validated near-atomic resolution structure of bacteriophage epsilon15 derived from cryo-EM and modeling. Proc. Natl. Acad. Sci. U.S.A. 110, 12301–12306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W., Chang J., Jakana J., Weigele P., King J., Chiu W., Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature 439, 612–616 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D.-H., Baker M. L., Hryc C. F., DiMaio F., Jakana J., Wu W., Dougherty M., Haase-Pettingell C., Schmid M. F., Jiang W., Baker D., King J. A., Chiu W., Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U.S.A. 108, 1355–1360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lander G. C., Khayat R., Li R., Prevelige P. E., Potter C. S., Carragher B., Johnson J. E., The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure 17, 789–799 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X., Gerlach D., du X., Larsen J., Stegger M., Kühner P., Peschel A., Xia G., Winstel V., An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of Podoviridae. Sci. Rep. 5, 17219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinbacher S., Baxa U., Miller S., Weintraub A., Seckler R., Huber R., Crystal structure of phage P22 tailspike protein complexed with Salmonella sp. O-antigen receptors. Proc. Natl. Acad. Sci. U.S.A. 93, 10584–10588 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinbacher S., Seckler R., Miller S., Steipe B., Huber R., Reinemer P., Crystal structure of P22 tailspike protein: Interdigitated subunits in a thermostable trimer. Science 265, 383–386 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Xiang Y., Leiman P. G., Li L., Grimes S., Anderson D. L., Rossmann M. G., Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell 34, 375–386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stummeyer K., Dickmanns A., Muhlenhoff M., Gerardy-Schahn R., Ficner R., Crystal structure of the polysialic acid–degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 12, 90–96 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Olia A. S., Prevelige P. E. Jr., Johnson J. E., Cingolani G., Three-dimensional structure of a viral genome-delivery portal vertex. Nat. Struct. Mol. Biol. 18, 597–603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhardwaj A., Sankhala R. S., Olia A. S., Brooke D., Casjens S. R., Taylor D. J., Prevelige P. E. Jr., Cingolani G., Structural plasticity of the protein plug that traps newly packaged genomes in Podoviridae Virions. J. Biol. Chem. 291, 215–226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vybiral D., Takáč M., Loessner M., Witte A., von Ahsen U., Bläsi U., Complete nucleotide sequence and molecular characterization of two lytic Staphylococcus aureus phages: 44AHJD and P68. FEMS Microbiol. Lett. 219, 275–283 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Morais M. C., Kanamaru S., Badasso M. O., Koti J. S., Owen B. A. L., McMurray C. T., Anderson D. L., Rossmann M. G., Bacteriophage φ29 scaffolding protein gp7 before and after prohead assembly. Nat. Struct. Biol. 10, 572–576 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Dearborn A. D., Wall E. A., Kizziah J. L., Klenow L., Parker L. K., Manning K. A., Spilman M. S., Spear J. M., Christie G. E., Dokland T., Competing scaffolding proteins determine capsid size during mobilization of Staphylococcus aureus pathogenicity islands. eLife 6, e30822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wikoff W. R., Liljas L., Duda R. L., Tsuruta H., Hendrix R. W., Johnson J. E., Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289, 2129–2133 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Sun L., Zhang X., Gao S., Rao P. A., Padilla-Sanchez V., Chen Z., Sun S., Xiang Y., Subramaniam S., Rao V. B., Rossmann M. G., Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 6, 7548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lebedev A. A., Krause M. H., Isidro A. L., Vagin A. A., Orlova E. V., Turner J., Dodson E. J., Tavares P., Antson A. A., Structural framework for DNA translocation via the viral portal protein. EMBO J. 26, 1984–1994 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson A. A., Tao Y., Leiman P. G., Badasso M. O., He Y., Jardine P. J., Olson N. H., Morais M. C., Grimes S., Anderson D. L., Baker T. S., Rossmann M. G., Structure of the bacteriophage φ29 DNA packaging motor. Nature 408, 745–750 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nováček J., Šiborová M., Benešík M., Pantůček R., Doškař J., Plevka P., Structure and genome release of Twort-like Myoviridae phage with a double-layered baseplate. Proc. Natl. Acad. Sci. U.S.A. 113, 9351–9356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lokareddy R. K., Sankhala R. S., Roy A., Afonine P. V., Motwani T., Teschke C. M., Parent K. N., Cingolani G., Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat. Commun. 8, 14310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tremblay D. M., Tegoni M., Spinelli S., Campanacci V., Blangy S., Huyghe C., Desmyter A., Labrie S., Moineau S., Cambillau C., Receptor-binding protein of Lactococcus lactis phages: Identification and characterization of the saccharide receptor-binding site. J. Bacteriol. 188, 2400–2410 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricagno S., Campanacci V., Blangy S., Spinelli S., Tremblay D., Moineau S., Tegoni M., Cambillau C., Crystal structure of the receptor-binding protein head domain from Lactococcus lactis phage bIL170. J. Virol. 80, 9331–9335 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desmyter A., Farenc C., Mahony J., Spinelli S., Bebeacua C., Blangy S., Veesler D., van Sinderen D., Cambillau C., Viral infection modulation and neutralization by camelid nanobodies. Proc. Natl. Acad. Sci. U.S.A. 110, E1371–E1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takáč M., Bläsi U., Phage P68 virion-associated protein 17 displays activity against clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 49, 2934–2940 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson D., Schuch R., Chahales P., Zhu S., Fischetti V. A., PlyC: A multimeric bacteriophage lysin. Proc. Natl. Acad. Sci. U.S.A. 103, 10765–10770 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGowan S., Buckle A. M., Mitchell M. S., Hoopes J. T., Gallagher D. T., Heselpoth R. D., Shen Y., Reboul C. F., Law R. H. P., Fischetti V. A., Whisstock J. C., Nelson D. C., X-ray crystal structure of the streptococcal specific phage lysin PlyC. Proc. Natl. Acad. Sci. U.S.A. 109, 12752–12757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koç C., Xia G., Kühner P., Spinelli S., Roussel A., Cambillau C., Stehle T., Structure of the host-recognition device of Staphylococcus aureus phage ϕ11. Sci. Rep. 6, 27581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu L., Benson S. D., Butcher S. J., Bamford D. H., Burnett R. M., The receptor binding protein P2 of PRD1, a virus targeting antibiotic-resistant bacteria, has a novel fold suggesting multiple functions. Structure 11, 309–322 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Sycheva L. V., Shneider M. M., Sykilinda N. N., Ivanova M. A., Miroshnikov K. A., Leiman P. G., Crystal structure and location of gp131 in the bacteriophage phiKZ virion. Virology 434, 257–264 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Xu J., Gui M., Wang D., Xiang Y., The bacteriophage ϕ29 tail possesses a pore-forming loop for cell membrane penetration. Nature 534, 544–547 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Scheres S. H., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo F., Jiang W., Single particle cryo-electron microscopy and 3-D reconstruction of viruses. Methods Mol. Biol. 1117, 401–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de la Rosa-Trevín J. M., Otón J., Marabini R., Zaldívar A., Vargas J., Carazo J. M., Sorzano C. O., Xmipp 3.0: An improved software suite for image processing in electron microscopy. J. Struct. Biol. 184, 321–328 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Chang J., Weigele P., King J., Chiu W., Jiang W., Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure 14, 1073–1082 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Pintilie G. D., Zhang J., Goddard T. D., Chiu W., Gossard D. C., Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170, 427–438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ilca S. L., Kotecha A., Sun X., Poranen M. M., Stuart D. I., Huiskonen J. T., Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 6, 8843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabsch W., Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleywegt G. J., Read R. J., Not your average density. Structure 5, 1557–1569 (1997). [DOI] [PubMed] [Google Scholar]
- 57.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langer G., Cohen S. X., Lamzin V. S., Perrakis A., Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaaw7414/DC1
Fig. S1. Purified sample of P68 contains native virions, particles in process of genome release, and empty particles; attachment of P68 virions to S. aureus cell walls; and interactions of P68 virions with liposomes.
Fig. S2. Resolution and interpretability of cryo-EM reconstructions.
Fig. S3. Details of P68 head.
Fig. S4. Incorporation of P68 portal complex into capsid and changes in the structure of P68 portal complex upon genome release.
Fig. S5. Structures of P68 tail fiber and tail spike.
Fig. S6. Schemes of cryo-EM reconstruction strategies.
Table S1. Cryo-EM structure quality indicators.
Table S2. List of P68 proteins.
Table S3. HHpred searches for homologs of P68 tail fiber, head fiber, tail knob, and tail spike.
Table S4. X-ray structure quality indicators.