

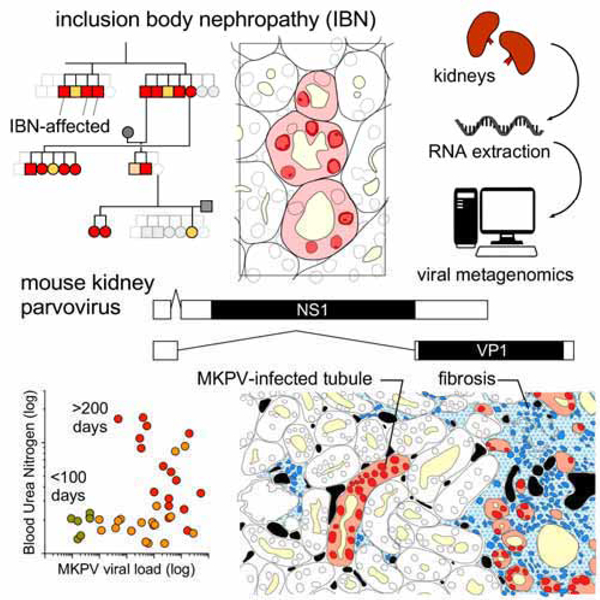
Summary
The occurrence of a spontaneous nephropathy with intranuclear inclusions in laboratory mice has puzzled pathologists for over four decades. While the condition is more severe in immunodeficient animals suggesting an infectious cause, its etiology has remained elusive. Using metagenomics, we identify the causative agent as an atypical virus, mouse kidney parvovirus (MKPV), belonging to a divergent genus of Parvoviridae. MKPV was identified in animal facilities in Australia and North America, is transmitted via fecal-oral or urinary-oral route and controlled by the adaptive immune system. Detailed analysis of the clinical course and histopathological features demonstrated a stepwise progression of pathology ranging from sporadic tubular inclusions to tubular degeneration and interstitial fibrosis culminating in renal failure. In summary, we identify a widely-distributed pathogen in laboratory mice and establish MKPV-induced nephropathy as a new tool for elucidating mechanisms of tubulointerstitial fibrosis that shares molecular features with chronic kidney disease in humans.
Graphical Abstract
In Brief
A kindly parvovirus found in multiple laboratory mouse colonies causes spontaneous nephropathy and represents a new tool for studying chronic kidney disease
Introduction
Chronic kidney disease (CKD), defined as kidney dysfunction of at least three months duration (Webster et al., 2016), affects 3–18% of adults globally (Glassock et al., 2017). CKD arises from a range of conditions that alter the structure and function of the kidneys, including diabetes mellitus and hypertension. Irrespective of the contributing injury, kidney failure in CKD occurs as a result of irreversible fibrosis (‘scarring’) of the kidney parenchyma (Duffield, 2014). Viral infections may also lead to kidney fibrosis, particularly in transplantation, where immunosuppression can result in reactivation of latent viruses within the donor allograft. Of note, polyomavirus-associated nephropathy has emerged as a significant cause of morbidity in transplant patients (Ramos et al., 2009). The most common infectious agent in this regard is BK virus, which propagates within the tubular epithelial cells of the donor kidney to provoke fibrosis, compromise renal function and promote graft loss.
In aged mice, chronic nephropathy occurs naturally with moderate to high incidence in the 129S4/SvJae and B6C3F1 strains, and in Swiss stocks (Brayton, 2007). However, this condition is not used as a disease model because its occurrence and progression are difficult to predict. The development of effective interventions for renal fibrosis has been hindered by a paucity of suitable small animal models of CKD (Nanthakumar et al., 2015). Existing models of tubulointerstitial nephropathy and fibrosis commonly involve the administration of antibodies or nephrotoxic compounds, or surgical interventions such as sub-total (5/6) nephrectomy in rats and unilateral ureteral obstruction (UUO) in mice (Yang et al., 2010). The acute injury induced in these models can either lead to death within weeks or affects only a single kidney and is compensated by the other (Becker and Hewitson, 2013). Thus, most murine models of CKD fail to recapitulate the chronic, insidious nature of fibrotic disease and the failure to resolve that typify human CKD.
The occurrence of intranuclear inclusions of undetermined etiology in mouse renal tubular epithelium has been anecdotally reported by veterinary pathologists for over 40 years (Barthold et al, 2016). These inclusions, associated with chromatin margination and karyomegaly, have been reported in both immunocompetent and immunodeficient mice in the USA and Australia (Baze et al., 2006; McInnes et al., 2015). Known mouse pathogens were not detected in affected mice and electron microscopy failed to demonstrate viral particles in the inclusions. Therefore, the etiology of this condition remained undetermined.
Here, we identify a widely-distributed and atypical viral pathogen that drives the inclusion body nephropathy (IBN) in mice. We have created a detailed, longitudinal map of the kidney disease driven by this virus. Based on our observations, we propose that this infection system represents a novel murine model of chronic tubular injury and interstitial fibrosis that holds relevance for our understanding of virus-associated nephropathy and CKD in humans.
Results
Occurrence of IBN in mice and identification of the causative pathogen
In 2010, staff at the Centenary Institute Animal Facility (Sydney, Australia) noticed unexpected deaths in middle-aged mice within disparate immunodeficient breeding colonies. Further investigation identified the likely cause of death as kidney failure; both kidneys in moribund mice appeared shrunken, pale and pitted upon necropsy, and highly fibrotic by histology (Fig. 1A and B). All other tissues from diseased mice appeared morphologically and histologically normal (data not shown). Retrospective analysis of the necropsy records revealed a high incidence of kidney disease through generations of B6.Cg Rag1−/− and B6.Cg Prkdcscid/scid mice with the age of death or euthanasia usually occurring between 200 and 300 days (Fig. 1C and D). Mice older than 200 days from affected lines exhibited elevated serum creatinine and blood urea nitrogen, and were anemic, consistent with end-stage kidney disease (Fig. 1, E and F, and Fig. S1, A and B).
Figure 1. Renal disease in immunodeficient mice.

(A,B) Representative macroscopic images (A) and Milligan’s trichrome staining (B) of normal (left) and disease-affected (right) kidneys. Bar = 500 µm. (C) Necropsy-confirmed renal disease incidence (red) in immunodeficient B6.Cg Prkdcscid/scid generations at the Centenary Institute. (D) Age of death for necropsy-confirmed renal disease in 3 separate colonies of immunodeficient mice housed at the Centenary Institute from 2010–2017. Colony 1: B6.Cg Prkdcscid/scid, B6.Cg Rag1−/− and Prkdcscid/scid Rag1−/− double-knockout mice; Colony 2: Des T cell receptor (TCR) transgenic (Tg) mice crossed to B6.Cg Rag1−/−; Colony 3: Cxcr6gfp/gfp Rag1−/− mice. Male (black boxes) and female (white circles) mice; n = 38–46 as shown. (E,F) Analysis of serum biochemistry (E) or hemoglobin and haematocrit (F) in mouse strains affected by renal disease (Cxcr6gfp/gfp Rag1−/−; <200 days, black, E (n = 20), F (n = 8); >200 days, red, E (n = 10), F (n = 8)) and in unaffected controls (Rag1−/− and C57BL/6 from Australian BioResources (ABR), white, E (n = 10), F (n = 3)). Each dot represents an individual mouse. BUN, blood urea nitrogen. **P = 0.04, ***P = 0.002, ****P < 0.0001 (one-way ANOVA, Holm-Sidak’s multiple comparison). See also Fig. S1.
Histopathological assessment of affected kidneys identified tubular degeneration and necrosis, with epithelial cells displaying enlarged nuclei and chromatin margination associated with the formation of numerous large, amphophilic, intranuclear inclusions (Fig. 2A and Fig. S1C). Tubular loss, interstitial fibrosis and medullary papillary necrosis were also observed (Fig. S1D). Intranuclear inclusion bodies are a classic hallmark of certain viral infections (Liptak et al., 2006). Electron microscopy of the inclusion bodies proved inconclusive, with no discernable viral particles or crystalline or paracrystalline features (Fig. 2B). However, staining of affected tissue with acridine orange, which emits red fluorescence when bound to single-stranded (ss) nucleic acids, suggested that at least some affected cells contained ssDNA or ssRNA (Fig. 2C), potentially reflecting viral presence.
Figure 2. Identification of MKPV.

(A) Left: Histopathology of a disease-affected kidney stained with hematoxylin & eosin. Arrows, chromatin clearing and nuclear inclusion bodies within affected tubular cells. Bar = 40 µm. Right: Schematic demarking epithelial tubules, lumina (pale yellow), diseased epithelial cells (pink) and nuclei with inclusions (red). (B) TEM of a single tubular epithelial cell nucleus from a diseased kidney, depicting the electron-lucent nuclear inclusion (ib) and electron-dense marginated chromatin (mc). (C) Confocal microscopy of a diseased kidney stained with acridine orange and DAPI. DAPI fluorescence (blue) and acridine orange fluorescence identifies either double-strand (ds) DNA (green) or single-strand (ss) DNA/RNA (red). Arrows indicate ssDNA/RNA in a diseased nucleus. Bar = 20 µm. (D) Workflow for RNA sequencing of diseased kidneys. (E) Identification of viral mRNA sequences from diseased mice. Left and middle: Abundance and sequence lengths of unannotated mRNA sequences (left) and predicted amino acid lengths of the most abundant mRNAs from the more moribund mouse (middle). Right: Homologous viral sequences in a second diseased mouse. (F) mRNA reads from the two diseased mice (black line and blue histogram) mapped to the complete viral genome confirmed by PCR from kidney DNA and Sanger sequencing. Black and red arrows indicate major splice donor and acceptor sites, respectively (see Fig. S2). Below: Schematic of the mouse kidney parvovirus sequence. Red, inverted tandem repeats (ITRs); White boxes; non-structural (NS1; rep) and capsid (VP1; cap) protein coding regions.
Since all murine colonies at the Centenary Institute and its vendors are free of known specified infectious agents, it was likely that any causative agent would be hitherto uncharacterized. We therefore took a metagenomic approach. RNA was extracted from diseased kidneys and sequenced (Fig. 2D). Sequencing of RNA was chosen to accommodate the possibility that the disease was driven by an RNA virus. Sequences were pre-screened in silico for plant, bacterial, fungal and mouse origins prior to assembly, which left ~55,000 unassigned sequences from each mouse, many of which represented unannotated mouse genes, endogenous retroviruses and low-abundance DNA contaminants (Fig. 2E).
Examination of the longest and most abundant sequences isolated from a moribund mouse identified two coding sequences with homology to parvoviral non-structural (NS1) and structural (VP1) proteins (Fig. 2E). No other genes resembling replication-competent viruses were found. A BLAST nucleotide search of RNA-seq data from a second mouse identified four homologous parvoviral sequences (Fig. 2E). Manual alignment of six sequences revealed a complete, 4442 nucleotide (nt) viral genome, with fidelity confirmed by Sanger sequencing of PCR products generated from diseased kidney DNA. Possession of the complete DNA sequence enabled us to measure RNA coverage across the viral genome within each sample (Fig. 2F). This revealed that expression of NS1 and VP1 probably occurred by alternative splicing in both animals (Fig. S2), consistent with productive infection within the kidneys of these mice. We termed this virus mouse kidney parvovirus (MKPV).
Detection and transmission of MKPV
We designed diagnostic PCR primers specific for MKPV and tested the serum and urine of immunodeficient mice for the presence of viral DNA. Using 25 cycles of PCR, viral amplicons were detected in mouse colonies in which kidney disease had been observed but were absent in disease-free lines (Fig. 3A). Viral DNA was also detected within the feces of affected mice, although we observed no pathological changes within the gastrointestinal tract (Fig. S3A).
Figure 3. Transmission of MKPV and kidney disease in immunodeficient mice.

(A) PCR amplification (25 cycles) of MKPV DNA (rep gene) from urine and serum of diseased colonies (Cxcr6gfp/gfp Rag1−/−, Des TCR Tg Rag1−/−, red) but not from unaffected colonies (black). (B) Detection of viral rep DNA (25 PCR cycles) in serum of imported (from ABR) Rag1−/− mice (white box) after co-housing with infected Cxcr6gfp/gfp Rag1−/− mice. (C) Top: Weight changes in individual Rag1−/− mice co-housed with the infected Cxcr6gfp/gfp Rag1−/− mice (colored, n = 3) and in control Rag1−/− littermates (monochrome, n = 3). Co-housed Rag1−/− mice became MKPV PCR+ (urine) by day 74 of co-housing (purple line). Bottom: Weights (mean ± s.d.) during co-housing **P = 0.0013 (unpaired t-test). Representative of 3 independent experiments. (D) Histopathology of unaffected (top) and diseased (bottom) kidneys from NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice from the Memorial Sloan Kettering Cancer Center (MSKCC), depicting tubular epithelial cell nuclei with chromatin margination (black arrows), small eosinophilic inclusions (arrowhead) and tubular necrosis (asterisk). Bar = 40 µm. (E) PCR amplification of MKPV rep DNA from kidneys and urine of diseased NSG mice, but not unaffected mice, from MSKCC. Healthy C57BL/6 and diseased Cxcr6gfp/gfp Rag1−/− mice served as negative and positive controls, respectively. (F) MKPV in situ hybridisation signals (RNAscope®) in kidney sections from NSG mice with moderate (middle) and severe (left) inclusion body nephropathy, at 40X (bar = 500 µm) and 600X (bar = 40 µm) magnification. MKPV nucleic acids were distributed throughout the nucleus and cytoplasm of affected tubular cells. MKPV signals were undetectable in unaffected NSG mice (rightmost). (G-I) Development and use of anti-MKPV serum. (G) C57BL/6 were co-housed with MKPV-infected Cxcr6gfp/gfp Rag1−/− mice and sera collected and pooled from the C57BL/6 mice after 8 weeks, and from C57BL/6 controls. (H) Western blots of recombinant GST-tagged VP1. (I) Immunostaining of diseased (top) and unaffected (bottom) kidney sections. Green, antiserum staining; Red, DAPI staining of nuclei; Blue, autofluorescence. Bar = 30 µm. (J) The most abundant proteins (>0.005% of total spectra) detected by MS/MS in extracts from a normal (left) and a diseased (right) kidney. Grey, proteins identified in both samples; black, proteins unique to each group; red, MKPV NS1 and VP1 – detected only in diseased kidney. See also Fig. S3 and S4.
To test for horizontal transmissibility, MKPV PCR+ Cxcr6gfp/gfp Rag1−/− mice were co-housed with Rag1−/− mice, obtained from a virus-free vendor. After 50–80 days, viral DNA was detectable in serum and urine of co-housed animals (Fig. 3B), which subsequently lost weight and succumbed to kidney disease (Fig. 3C). Control mice remained PCR− and disease-free (Fig. 3B,C). Embryonic rederivation of MKPV+ Cxcr6gfp/gfp Rag1−/− mice removed MKPV from the line (Fig. S3B) and rederived mice remained free of kidney disease (not shown). When virus-bearing mice were co-housed with immunocompetent mice, we detected viral DNA in feces but not serum or urine by PCR (not shown). However, MKPV was detected in urine of three Swiss Sentinel mice that received bedding from Cxcr6gfp/gfp Rag1−/− mice (Fig. S3C and data not shown). Collectively, these data suggest that MKPV is transmitted via a fecal-oral or urinary-oral route and its dissemination to and/or persistence within the kidney is controlled by the adaptive immune system.
The mice at Centenary Institute from which we identified MKPV had histological features identical to those associated with morbidity and mortality in NSG mice at Memorial Sloan Kettering Cancer Center (MSKCC) (Santagostino et al., 2017). We tested whether MKPV was present in NSG mice from MSKCC by PCR (Fig. 3D,E). MKPV was readily detected within NSG mice with IBN but was undetectable in unaffected mice (Fig. 3E). Urine pooled from disease-affected NSG mice was also MKPV PCR+ (Fig. 3E). These results indicate a close correlation between MKPV presence and inclusion body nephropathy.
In situ detection of MKPV in inclusion body nephropathy
As shown in Fig. 3F and Fig. S3D, in situ hybridization localized MKPV sequences to inclusion body-affected tubular epithelial cells. Moreover, the abundance of MKPV sequences detected by in situ hybridization correlated with histological disease severity. In situ hybridization did not detect MKPV nucleic acids in unaffected mice (Fig. 3F).
To assess the distribution of viral proteins within affected kidneys, we used antisera derived from immunocompetent C57BL/6 mice co-housed with MKPV+ Cxcr6gfp/gfp Rag1−/− mice in the Centenary Institute (Fig. 3G). Sera from co-housed, but not control, mice reacted with recombinant MKPV VP1 (Fig. 3H) and predominantly stained the enlarged nuclei of disease-affected, but not unaffected, mice (Fig. 3I and Fig. S4, A and B). Identical results were observed using sera from immunocompetent Swiss mice co-housed with disease-affected NSG mice in MSKCC (Fig. S4, C and D). Two thirds of antiserum-reactive tubules expressed the collecting duct tubular epithelial cell marker and cellular stress response protein Cytokeratin-19 (Krt19) (Djudjaj et al., 2016) (Fig. S4E).
Finally, we sought to demonstrate the presence of MKPV in affected kidneys through mass spectrometry (MS/MS). Both MKPV NS1 and VP1 peptides were detected by MS/MS in an MKPV+ Cxcr6gfp/gfp Rag1−/− mouse but not in an uninfected Rag1−/− control (Fig. 3J), indicating a high abundance of MKPV protein consistent with productive infection. Collectively, we concluded that MKPV is the causative agent of IBN in immunodeficient mice.
Distribution of MKPV in Australian and US facilities and correlation with disease severity
Since some of the histopathological features of IBN have also been reported in immunosufficient mouse strains (Baze et al., 2006), we determined whether MKPV was broadly responsible for this pathology. Veterinary pathologists at the MSKCC Laboratory of Comparative Pathology (LCP) had noted IBN in immunosufficient mice, immunodeficient NSG and athymic nude mice. We performed PCR for MKPV on archived paraffin sections from 34 affected mice and a further six negative controls. Results are summarized in Table 1 and Table S1. While 15/17 (88%) IBN-affected NSG mice were PCR+, MKPV was never detected in unaffected mice. We identified MKPV in a sentinel wild-type mouse and 9/10 nude mice with mild IBN. Moreover, MKPV+ immunodeficient mice were present in multiple separate animal facilities serviced by LCP dating back to 2007. We further associated MKPV to IBN in two other separate sites, one 2006 USA case and five Australian cases from 2010, all of which exhibited severe nephropathy (Fig. S5, A and B, and Table S1).
Table 1. Pathological assessment and MKPV status in mice with histopathologically-confirmed inclusion body nephropathy at the MSKCC Laboratory of Comparative Pathology.
Immunocompetent mice comprise a Swiss Webster (Sentinel) mouse and two mice of an unknown strain presumed to be immunocompetent based on intact lymphoid architecture by histopathology. IBN, inclusion body nephropathy. NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ. TRP, B6.Cg-Rag1tm1MomTyrp1B-wTg (Tcra, Tcrb)9Rest/M. Nude, Crl:NU(NCr)-Foxn1nu. TCR, T cell receptor. Tg, transgenic. n = 34 mice. Letters designate individual facilities.
| Strain/stock | Kidney Pathology | Cause of Death | Immune status | Mice MKPV+ (PCR)1 | Dates of Collection | Facilities MPKV+ (PCR)2 | ||
|---|---|---|---|---|---|---|---|---|
| T cells | B cells | Innate immunity | ||||||
| Immuno-competent strains/stocks | Inclusions and moderate lymphoplasmacytic infiltrate (3/3), with moderate tubular degeneration and mild fibrosis(1/3) | other | present | present | normal | 1/3 | 2007–2008 | 1/2 (B,C) |
| NSG | inclusions, moderate to marked tubular degeneration, mild to moderate interstitial fibrosis, papillary necrosis | IBN, other | absent | absent | defective | 16/18 | 2007–2017 | 3/3 (A,B,D) |
| TRP [Rag1−/−] | inclusions, moderate to marked tubular degeneration, mild to moderate interstitial fibrosis | IBN | TCR Tg T cells only | absent | normal | 3/3 | 2015 | 1 (D) |
| Nude | inclusions, minimal to mild tubular degeneration, mild lymphoplasmacytic interstitial infiltrate | other | absent | present | normal | 9/10 | 2009 | 1 (A) |
Number of mice PCR positive for MKPV / number of mice histologically positive for IBN.
Number of facilities PCR positive for MKPV / number of facilities histologically positive for IBN. See also Table S1
To better understand the differences in disease severity between immunosufficient and nude mice compared to severely immunocompromised NSG, Rag1−/− and Prkdcscid/scid mice (Table 1), we used quantitative PCR (qPCR) to precisely determine the number of MKPV copies in samples (Fig. S5C). As with the diagnostic 25 cycles PCR, some samples did not sufficiently amplify. For those that did, the abundance of MKPV DNA was an order of magnitude lower in wild-type and nude mice compared to more immunocompromised mice (Fig. S5D). These data demonstrate an association between MKPV load and the severity of pathology, presumably determined by the level of immunological control.
Evolutionary history of MKPV
Our metagenomics approach had enabled the assembly of a complete parvovirus sequence; remarkably including the 5’ and 3’ inverted tandem repeats (ITRs) that form the ‘bubble’ stem-loop structures typical of parvoviruses (Fig. 4A). Parvoviral ITRs are not normally transcribed, so our identification of the ITRs may have been due to contamination of the extracted kidney RNA with traces of MKPV DNA.
Figure 4. Phylogenetic and sequence analysis of mouse kidney parvovirus (MKPV).

(A) Predicted secondary structures of the 5’ (top) and 3’ (bottom) ITRs of MKPV. Arrows indicate ‘bubbles’. (B) NS1 amino acid comparison between MKPV and selected parvoviruses of varying sequence similarity. (C) Phylogenetic analysis of the Parvovirinae family based on conserved areas of NS1. Bootstrap support values >70% are shown for key nodes. Branch lengths are scaled according to the number of amino acid substitutions per site, and the tree is rooted to show distinction between Parvovirinae and Densovirinae. (D) Scatterplot depicting capsid vs. total genome lengths for Parvoviridae family members (color-coded).
Analysis of coding potential indicated that MKPV was highly divergent from the known mouse parvoviruses. We performed a phylogenetic analysis using a conserved region of Parvoviridae NS1 protein. Strikingly, MKPV exhibited a close evolutionary relationship with viruses isolated from feces of pigs and wild rats, and was most closely related to parvoviruses isolated from Old World fruit bats and common vampire bats in Ghana and Brazil, respectively (Souza et al., 2017) (Fig. 4B). Hence, MKPV is a member of what we propose is a divergent and currently unclassified Parvovirinae genus (Fig. 4C). The capsid protein sequences were too divergent for an equivalent evolutionary analysis of the VP1 polypeptide. However, analysis of capsid length suggested that MKPV and its nearest known relatives were ~25% smaller than ‘typical’ Parvovirinae viruses, and almost as small as some Desnovirinae viruses (Fig. 4D).
Course of MKPV-induced kidney disease
We then further investigated the pathology and disease progression in MKPV-infected mice. Moribund mice exhibited extensive microscopic and macroscopic fibrotic changes in their kidneys (Fig. 5A and Fig. S6, A and B) and had significantly reduced renal mass after 200 days of age (Fig. 5B). Fibrosis appeared to expand from perivascular locations (Fig. S6C) and was intimately associated with renal epithelial cells bearing nuclear inclusions (Fig. 5C and Fig. S6D).
Figure 5. Kinetics of MKPV infection and disease progression.

(A) Sirius Red staining of kidneys from uninfected Rag1−/− mice (top, n = 3) and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice (bottom, n = 5). Bar = 400 µm. Right: Quantification. *P = 0.0345 by unpaired t-test of means (bars). (B) Kidney weights from uninfected wild-type and Rag1−/− mice (monochrome, n = 10) and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice (colored < 200 days: n = 4, > 200 days: n = 14), stratified according to age. Left (paler symbols) and right (darker symbols) kidneys are identified. ****P < 0.0001 (unpaired t-test). (C) Left & middle: Confocal microscopy of an MKPV-infected Cxcr6gfp/gfp Rag1−/− kidney stained with Sirius Red and DAPI. ECM, extracellular matrix. Right: DAPI stain (inversed) of indicated region in middle panel. Dotted lines indicate nuclei associated with ECM, likely belonging to myofibroblasts (red), and epithelial cell nuclei bearing the pathological features of IBN (blue). Bar = 200 µm (left), 30 µm (middle) or 15 µm (right). (D–G) Chronology of (D) viremia (25 cycle PCR, n = 100–127) and (E) blood urea nitrogen (BUN) levels in the MKPV-infected Cxcr6gfp/gfp Rag1−/− colony (colored, n = 84) and uninfected wild-type and Rag1−/− controls (black, n = 39). (F,G) Quantitative PCR (qPCR) for MKPV DNA in the same infected cohort (n = 32). Green, <100d; orange, 100–200d; red, >200d; brown, culled sick. Viral load in viral genome copies (vgc) per µL urine/serum. (F) Urine vs serum MKPV load. (G) Serum MKPV load vs BUN. (H–J) qPCR for MKPV DNA in organs (vgc/20ng extracted DNA) and fluids (vgc/µL) from another cohort of infected Cxcr6gfp/gfp Rag1−/− mice, pre-viremia (H,J; 0–60d, n = 6) and post-viremia (I,J; after 60 days of age, n = 6 in 61–100 and 101–150 d, n = 5 in other age groups). See also Fig. S6.
We tracked viremia by 25-cycle PCR in a cohort of Cxcr6gfp/gfp Rag1−/− mice, one of the disease-affected lines (Fig. 1D and Fig. 3A). Mice were universally MKPV PCR+ in the serum and urine after ~100 days of life (Fig. 5D), suggesting the virus is highly infectious. From 200 days of age, blood urea nitrogen (BUN) of infected animals became progressively elevated (Fig. 5E), consistent with severe kidney dysfunction. Between 150 and 250 days of age, proteinuria fell dramatically below the usual levels of normal mice (Fig. S6E), coinciding with weight loss in MKPV-infected animals (Fig. S6F). Using qPCR, we confirmed a strong concordance between virus levels in urine and serum (Fig. 5F) and between urine viral load and BUN with age (Fig. 5G). Interestingly, serum viremia dropped in moribund mice, possibly reflecting depletion of virus-targeted cells (Fig. 5G).
Prior to 61 days, when mice appeared normal and virus could not be found in blood by PCR, MKPV was detected in the kidneys but not other organs (Fig. 5H). Between 61 and 100 days, when some mice became viremic (Fig. 5D), viral levels were about 20 times higher in the kidneys (Fig. 5I). MKPV continued to propagate unabated within both kidneys as mice aged (Fig. 5J), and we estimated that each kidney of moribund mice contained 1012–1014 MKPV genome copies.
Finally, at necropsy, the kidneys of affected mice appeared shrunken and pale (Fig. 1A), suggesting atrophy and fibrosis. Collectively, these data demonstrate that MKPV-infected mice experienced kidney dysfunction for 4–5 months before succumbing to the disease, indicative of CKD.
Pathogenesis of MKPV-induced kidney disease
We were next interested in identifying the molecular and cellular mechanisms underlying the fibrotic disease in MKPV-infected mice, using the RNA sequencing and mass spectrometry data we collected to identify and detect MKPV, respectively.
The two most highly upregulated genes in disease-affected kidneys were Havcr1 (encoding kidney injury molecule-1) and Lcn2 (encoding lipocalin 2) (Fig. 6A), both established kidney injury biomarkers (Han et al., 2002; Viau et al., 2010). Consistent with the histological data, we observed an upregulation of genes coding for TGFβ and collagen (Tgfb1, Col1a1, Col3a1) and their regulators (Fosl2, Wnt4, Serpine1), as well as classical myofibroblasts markers (Acta2, Vim) (Fig. 6A). We also found increased expression of Ctgf (encoding connective tissue growth factor), a mediator of tissue remodeling and fibrosis (Lipson et al., 2012). A number of immune system genes were also upregulated, most notably the macrophage-associated Adgre1 (F4/80), Itgam (CD11b), Lyz2 (lysozyme 2), Cd163 and Mrc1 (mannose receptor; CD206). Consistent with this, we detected an expansion of macrophages in MKPV-infected kidneys, with upregulated cell surface CD11b, CD86 and CD206 indicating activation (Fig. 6B). Furthermore, transcription of complement and complement-related genes (C1qa, C1qb, C1qc, C3, C3ar1, C4b, Cfi, Cfp, Cfh), as well as fibrinogen-associated genes, was also upregulated in MKPV-infected kidneys (Fig. S7A).
Figure 6. Molecular and cellular changes with MKPV infection.

(A) Comparison of gene expression in healthy kidneys (n = 2 replicates) vs. MKPV-infected kidneys (n = 2) by volcano plot. Selected genes implicated in TGFβ signalling and fibrosis are marked in black (centre) and blue (left). Genes implicated in immune system processes (GO: 0002376) are marked in red (right) or dark grey (middle). (B) Top: Flow cytometry dotplots identifying CD88+ MHC-II+ macrophages isolated from kidneys of uninfected Rag1−/− (left) and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice (right). Bottom: cell surface expression of markers on these macrophages; black, controls; red, MKPV-infected; grey, fluorescence-minus-one controls. (C,D,E) tSNE plots of flow cytometry data from live (DAPI−) cells isolated from uninfected Rag1−/− and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice. (C) Schematic defining populations in D & E. (E) Relative expression of markers. (F) Flow cytometry dotplots of myofibroblast conversion and epithelial cell loss. Left: CD45+ leukocytes and FAP+ fibroblasts isolated from normal kidneys. Top right: CD24 (heat-stable antigen) and CD29 (β1 integrin) expression by FAP+ fibroblasts from unaffected and IBN kidneys. Red gate: CD24+ CD29lo myofibroblasts. Bottom: EpCAM (epithelial cell adhesion molecule) and CD29 expression by CD45− FAP− cells. Black & red gates identify EpCAMhi CD29+ epithelial cells. Representative of 3 independent experiments. (G) Venn diagrams depicting the numbers of genes highly (>log2 1.5-fold) up- (top) or down-regulated (bottom) in unilateral ureteral obstruction (UUO) (Arvaniti et al., 2016) and MKPV IBN. Colours denote genes unique to UUO (red), MKPV (blue) or shared (purple). (H,I) Urinary EGF (H) and LTBP2 (I) levels in uninfected wild-type and Rag1−/− mice (black; n = 17 in H, n = 10 in l) and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice (red; n = 66 in H, n = 48 in I). ***P = 0.0004 (unpaired t-test). See also Fig. S7 and Table S2.
Whole kidney mass spectrometry identified ~2500 proteins, of which 13% were observed only in the normal kidneys and 22% only in the diseased (Fig. S7B). Moreover, many proteins detected in both groups were differentially regulated between the two conditions (Fig. S7C), including downregulation of proteins normally expressed by kidney tubular epithelial cells (Pcca, Cdh16, Sardh) and upregulation of fibrinogen complex proteins (Fga, Fgb, Fgg), consistent with the RNA sequencing data. Candidate proteins with the largest statistically significant differences are listed in Fig. S7D–F and Table S2. Many of the proteins detected only in normal kidneys were members of the solute carrier family (e.g. Slc6a18 Slc12a3, Slc22a18), likely reflecting the loss of kidney epithelial cells. In contrast, many proteins upregulated in MKPV infection are highly expressed by macrophages (e.g. Lyz2, Ctss, Lgals3, Chil3). Keratins were also upregulated, including Krt18 and Krt19, consistent with a response to tubular injury (Djudjaj et al., 2016) and with our RNA sequencing and immunohistochemistry data (Fig. S4E and data not shown).
We also interrogated the cellular composition of kidneys from uninfected Rag1−/− and MKPV-infected Cxcr6gfp/gfp Rag1−/− mice by flow cytometry. Cells were stained for lineage-defining markers EpCAM (CD326; epithelial cells), CD45 (leukocytes), CD31 (endothelial cells) and fibroblast activation protein (FAP) for fibroblasts. Flow cytometry data were then assessed using nonlinear dimensionality reduction via t-stochastic neighbor embedding (tSNE), which enables unbiased visualization of high-dimensional similarities between cells and populations in a two-dimensional map (Becher et al., 2014). Analysis of the tSNE plots revealed clustering of major cellular populations as well as red blood cells (CD45− CD24hi CD29lo) and ‘non-cellular’ events, which lacked signals in all channels (Fig. 6C–E). GFP+ innate lymphoid cells (Roediger et al., 2013) were also detectable and independently clustered within the Cxcr6gfp/gfp Rag1−/− mice (Fig. 6C,E). Segregation of uninfected and MKPV-infected samples revealed a marked reduction in epithelial cells with MKPV infection, accompanied by a relative enrichment of CD45+ and FAP+ cells and an emergence of a FAP+ CD24hi population resembling myofibroblasts (Schack et al., 2016) which was completely absent in uninfected mice (Fig. 6D,E). Conventional gating of FAP+ fibroblasts and EpCAM+ epithelial cells confirmed these findings (Fig. 6F and data not shown). We conclude that MKPV drives a loss in epithelial cells, expansion of activated macrophages and development of myofibroblasts within the kidney.
These data suggested that MKPV infection may be useful for investigating tubulointerstitial fibrosis. Presently, the most widely-used model of tubulointerstitial fibrosis is unilateral ureteral obstruction (UUO), in which surgical obstruction of the ureter results in hemodynamic changes within the kidney followed by tubular injury and cell death (Chevalier et al., 2009). We compared the transcriptomic changes within MKPV-infected kidneys to those previously observed in UUO (Arvaniti et al., 2016). With MKPV IBN, 657 genes were highly upregulated (>log2 1.5-fold), 67% of which were shared with UUO (Fig. 6G). Similarly, of the 36 genes highly downregulated in MKPV IBN, 69% were also downregulated in UUO (Fig. 6G). Many of the aforementioned transcriptional changes related to kidney injury; TGFβ signaling, collagen production and macrophage expansion were shared between MKPV IBN and UUO (Fig. S7G). Interestingly, two of the uniquely-upregulated genes in MKPV IBN were Clec9a and Itgae, encoding the scavenger receptor Clec9A and integrin ĮE (CD103), respectively (Fig. S7H). Clec9A and CD103 are expressed by a subset of DCs, cDC1, that has previously been implicated in renal fibrosis and CKD in humans (Kassianos et al., 2013). Further investigation revealed an increase in cDC1 in old, Cxcr6gfp/gfp Rag1−/− mice with MKPV IBN compared to young Cxcr6gfp/gfp Rag1−/− and uninfected Rag1−/− mice (Fig. S7I). We conclude that MKPV infection exhibits largely overlapping but also unique molecular changes compared with the traditional UUO model of mouse kidney fibrosis.
Identification of biomarkers of kidney fibrosis and CKD
It is established that the severity of interstitial fibrosis is a major determinant of renal dysfunction (Nath, 1992). However, assessing fibrosis in a clinical setting remains a significant challenge. Many studies of human CKD have focused on the identification of serum and urine biomarkers (Ingelfinger and Alexander, 2013; Ju et al., 2015).
By enriching our RNA-seq data for secreted gene products, we identified a number of putative biomarkers for fibrotic changes within the kidney (Fig. S7J). We focused on epidermal growth factor (EGF; decreased) and latent TGFβ-binding protein 2 (LTBP2; increased), since these have been identified as biomarkers of kidney fibrosis in humans (Haase et al., 2014; Ju et al., 2015). Consistent with the transcriptomics data, urinary EGF was significantly decreased in MKPV-infected mice (Fig. 6H), while LTBP2 was detectable in one third of MKPV-affected mice and undetectable in normal mice, with the exception of one aged animal (Fig. 6I). Collectively, these data indicate that chronic MKPV infection biochemically resembles human CKD.
Discussion
We have identified a murine Parvovirinae pathogen (MKPV) that causes kidney fibrosis and renal failure. MKPV has a distant evolutionary relationship with known mouse parvoviruses, infects renal tubular epithelial cells and induces overt pathology including kidney fibrosis. Dissecting the pathogenesis of MKPV-induced kidney damage may provide new avenues for exploring cellular and molecular pathways in kidney disease, which is particularly relevant in the transplant setting.
Parvoviruses are small, diverse viruses that infect many species (Brown, 2010). In humans, the best-known is parvovirus B19, which causes erythema infectiosum (“fifth disease”) in children and arthritis in adults. In mice, there were four known, closely-related parvoviruses, mouse parvoviruses: MPV-1, -2, and -3 and MVM. These viruses generally do not cause clinical signs, even in immunocompromised mice. In contrast, MKPV is highly divergent from MPV and MVM, exhibits remarkable tropism for renal epithelial cells, and naturally causes significant morbidity and mortality in immunodeficient animals. MKPV appears to belong to an as-yet unclassified Parvovirinae branch that includes viruses isolated from birds, reptiles and mammals (Souza et al., 2017). The tubular tropism of MKPV may prove useful for targeting kidney epithelial cells in vivo using the viral capsid in the future.
Our findings would be strengthened by inoculation experiments using pure MKPV, which we are presently unable to propagate in vitro. Nevertheless, our data fulfil most criteria proposed by Fredericks and Relman to establish a causative relationship between a virus and disease (Fredricks and Relman, 1996). First, MKPV was present in all mice with inclusion body nephritis but not in healthy controls. MKPV was preferentially detected in kidneys but not other organs, which also did not show overt pathology. Second, MKPV viremia universally preceded disease and virus copy numbers correlated with disease severity: numbers increased by 100 million-fold in the kidneys over the course of disease. Third, parvoviruses are known to assemble within the nuclei of infected cells (Cotmore and Tattersall, 2013), thus explaining the occurrence of inclusion bodies within the renal epithelium. Finally, in situ hybridization revealed abundant MKPV sequences in affected but not normal tubular epithelial cells. These data were corroborated by RNA sequencing and mass spectrometry, which revealed alternative splicing of MKPV transcripts and the presence of NS1 and VP1 peptides in diseased kidneys, indicating production and substantial MKPV expansion within the diseased tissue. Thus, the arguments for MKPV being the driver of IBN are compelling.
Although we do not know how and when MKPV was introduced into our colony, the histopathological features of MKPV-infection, i.e. inclusion body nephropathy, have been described previously in immunocompetent and immunocompromised mice in several facilities in Australia and the USA (Baze et al., 2006; McInnes et al., 2015; Santagostino et al., 2017). A viral etiology was surmised, but not proven (Besselsen et al., 2008). Following our discovery of MKPV, assessment of formalin-fixed paraffin-embedded (FFPE) tissues found evidence that MKPV has persisted in multiple colonies for over 10 years. PCR for MKPV showed a strong correlation with histopathology, with the few false negatives possibly due to lower viral copy numbers in immune-sufficient mice and the known limitations associated with nucleic acid detection in FFPE tissues (Okello et al., 2010). Our findings suggest widespread distribution of MKPV in laboratory mouse colonies.
Identifying MKPV as the causal agent of renal pathology in NSG mice holds significance for the biomedical and veterinary research communities. Although the relatively slow kinetics of MKPV infection limit our capacity to detect the virus in early life, the 100% incidence of viremia post 100 days of age indicates that infection is likely established by the time of weaning (3–4 weeks) and that MKPV is highly penetrant in immunodeficient mice. Certainly, the long-term persistence of MKPV within several immunodeficient lines indicates this virus, once established, is not readily removed from host facilities without re-derivation. This strongly suggests that the detection in a single immunodeficient mouse will imply likely MKPV-infection across the entire colony. The adverse effects of MKPV on renal function and lifespan can result in unexpected morbidity and mortality, thus compromising interpretation of research results. We have also demonstrated a milder, subclinical form of IBN in an immunocompetent mouse and in athymic nude mice which have limited immune defects, and similar findings have been described by other authors (Baze et al., 2006). Even mouse viruses that are clinically silent can have significant immunomodulatory effects that influence experimental outcomes (Treuting et al., 2012). For these reasons, future inclusion of MKPV in routine health monitoring of laboratory mice would seem warranted.
MKPV-induced nephropathy shares clinico-pathological similarities with polyomavirus-associated nephropathy (PVAN), which has emerged as a significant cause of morbidity in kidney transplant patients (Ramos et al., 2009). The paucity in understanding mechanisms regulating the host response to kidney infection in immunocompromised individuals results in limited treatment options for PVAN, and treatment efficacy is dictated by how early PVAN is detected. Despite the differences in causative agent between MKPV IBN and PVAN (i.e. parvovirus vs polyomavirus), the affected cell type and accompanying pathology are nearly identical between the two conditions. Hence it is expected the host response would utilize many of the same pathways. We thus propose that MKPV-induced nephropathy in mice represents a new tool for understanding viral nephropathy in immunosuppressed and transplant settings with potential to elucidate mechanisms of viral reactivation and virus-induced tubular damage.
Finally, we have detailed the cellular changes during MKPV IBN. These revealed a marked drop in the proportion of EpCAM+ epithelial cells and emergence of a CD24hi CD29lo FAPhi myofibroblast population. Although much of the reduction in EpCAM+ cells almost certainly reflects direct cell lysis from MKPV, the accompanying fibrotic changes also likely contribute to epithelial cell loss through physical stresses imposed on the remaining renal tubules, or epithelial-to-mesenchymal changes, or both (Zhou and Liu, 2016). Based on these results, we suggest that the decreased EGF that characterizes CKD in both human (Ju et al., 2015) and mice may simply reflect an absolute and irreversible loss of the tubular cells that naturally produce and secrete EGF. Further, we lay the groundwork for a flow cytometry-based assessment of fibroblastic tissue, which should facilitate further investigation and quantitation of the cellular mechanisms of CKD and their response to therapeutic agents.
In summary, the discovery of MKPV resolves the long-standing mystery regarding the etiology of IBN in mice. MKPV is the first virus of its genus to be associated with pathology. Our data open questions on the distribution of MKPV and related viruses in both animals and humans. Finally, we suggest that MKPV-induced nephropathy holds promise as a disease model enabling better understanding of fibrotic processes and pathogenesis of CKD.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ben Roediger (b.roediger@centenary.org.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice maintenance at Centenary Institute
All mice used in this study were on C57BL/6 background, with the exception of Des T cell receptor transgenic (Tg) mice (Schonrich et al., 1991), which were maintained on B10.BR. All mice, purchased or maintained in-house at the Centenary Animal Facility, were backcrossed a minimum of 8–10 generations and any transgenic strains were maintained in homozygosity. Young (8–10 weeks) female C57BL/6 and Rag1−/− (deficient of the adaptive immune system, notably B and T cells) mice were purchased from the Animal Resources Centre, Perth, WA, Australia and the Australian BioResources, Moss Vale, NSW, Australia, primarily for co-housing experiments. A randomised examination into a variety of strains at a spectrum of ages (young and aged mice) was used to determine the presence and prevalence of MKPV within the mouse lines, in the form of serum and urine testing for virus and autopsy and necropsy reports. The transgenic strains studied included Cxcr6gfp/gfp mice (Unutmaz et al., 2000), Des (Schonrich et al., 1991), OT-I (Hogquist et al., 1994), P14 TCR Tg mice (Pircher et al., 1989), Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI) mice (Sakaue-Sawano et al., 2008), mT/mG mice (Muzumdar et al., 2007) and Ubiquitin-eGFP mice (Schaefer et al., 2001), and they were either immunocompetent or crossed and maintained on an immunodeficient Rag1−/− background. Prkdcscid/scid mice were also studied, whereby the immunoglobulin transgenes were segregated (Cook et al., 2003). Of the many strains previously examined, the viral sequencing and PCR-confirmed Cxcr6gfp/gfpRag1−/− strain was selected for further longitudinal and cross-sectional analyses into the infectivity and persistence of MKPV. Both male and female subjects were used (n = 138), since with the exception of the obvious difference in body and kidney weights, the progression of the disease, the viral load burden and any biomarkers examined were comparable between sexes, deeming sex as a negligible factor in this viral study.
Mice were primarily placed in cages of 2–6 mice, with minimal amount of singly housed males. All mice were held in individual ventilated cages (IVC, Tecniplast, Buguggiate, VA, Italy) with 1/4 inch Bed o’Cobs corn-cob bedding (Tecniplast, Buguggiate, VA, Italy) and environment enrichment in the form of autoclaved cardboard boxes and Kleenex tissues for nesting. Mice were provided with standard chow containing necessary protein, vitamin and minerals (Specialty Feeds, Glen Forrest, WA, Australia) and water (pH 2.5–3.0) ad libitum. The facility operated in a 12 h light/dark cycle, with the temperature maintained between 22–26°C and a relative humidity of 30–70%. Rodent health monitoring program showed absence of the following viral agents: Mouse Hepatitis Virus (MHV), Mouse Parvovirus (MPV), Minute Virus of Mice (MVM), Mouse Rotavirus (EDIM), Pneumonia Virus of Mice (PVM), Sendai virus, Theilers Murine Encephalomyelitis Virus (TMEV), Reovirus 3 (Reo-3), Lymphocytic Choriomeningitis Virus (LCMV), Mouse adenovirus (MAdV) type 1 & 2, Ectromelia Virus, Hantavirus, Murine Cytomegalovirus (MCMV), Polyomavirus. Mouse Norovirus (MNV) was present in some but not all breeding rooms, and present in all experimental rooms. Mice were also free of Mycoplasma pulmonis, Citrobacter rodentium, Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus spp, Pasteurella pneumotropica, CAR bacillus, Clostridium piliforme, Corynebacterium kutscheri and Klebsiella pneumoniae, endoparasites, and ectoparasites. Helicobacter spp was present in some but not all breeding rooms. Breeding, ageing and experiments were carried out with and conformed to the approval of the Animal Ethics Committee, University of Sydney (2009–2013) or the Animal Ethics Committee, Royal Prince Alfred Hospital (2012–2018).
Mice maintenance at MSKCC/WCM
Mice in all facilities were housed in solid-bottom polysulfone shoebox cages, maintained in an individually ventilated caging system (Thoren Caging Systems, Hazelton, PA, USA) and autoclaved aspen chip bedding (PWI Industries Canada, Quebec, Canada). Cages were changed weekly in a HEPA-filtered vertical airflow clean air cage changing station (NU619–400, Nuaire, Plymouth, MN, USA). Purina 5053 feed (PMI, Indianapolis, IN, USA), irradiated and placed in the cage changing station, and acidified water (pH 2.5 to 2.8) was provided ad libitum. The holding rooms were ventilated with filtered 100% outside air at 15 air changes hourly. Room conditions included 72±2°F (22±1°C) and relative humidity at 30% to 70%, with a 12:12-h light:dark cycle. Experimental history of each colony is provided in Table S1. Colony D consisted of retired NSG breeders was used for a parasitology study, as described in Santagostino, et al. 2017. In all facilities except facility E (a quarantine facility) a rodent health monitoring program showed absence of the following agents: MHV, Sendai virus, MVM, MPV1/2, TMEV, PVM, Reo-3, EDIM, Ectromelia virus, LCMV, MAdV, K-Virus, Polyomavirus, MCMV, Mouse Thymic Virus, Hantavirus, Mycoplasma pulmonis, Citrobacter rodentium, Clostridium piliforme, Corynebacterioum kutscheri, CAR bacillus., Salmonella sp., Klebsiella pneumoniae, Streptococcus pneumoniae, Streptobacillus moniliformis, Encephalitozoon cuniculi, Myobia musculi, Myocoptes musculinus, Radfordia affinis, Aspiculuris tetraptera, Syphacia obvelata, Giardia muris. Specifically for this study, PCR of urine samples from NSG mice (2 samples pooled from a total of 34 mice) and serology on co-housed Swiss Webster mice (n = 15), for MPV1, MPV2, and MVM, was further performed by the Charles River Research Animal Diagnostic Services, Wilmington, MA, USA. Mouse care and all experimental procedures were approved by the Memorial Sloan Kettering Cancer Center (MSKCC) and Weill Cornell Medicine (WCM) Institutional Animal Care and Use Committee and maintained in accordance to the National Academy of Sciences’ Guide for the Care and Use of Laboratory Animals in AAALAC International-accredited facilities.
Mice Health Screening at Cerberus Sciences
Mice presented to Cerberus Sciences for routine health screening in 2010 underwent complete necropsies and histopathological examination by a board-certified veterinary pathologist as previously described (McInnes et al., 2015). Several of these mice, C3.CgPrkdcscid/scid, demonstrated severe nephropathy with intranuclear inclusion bodies. The health monitoring program showed an absence of the following agents: Mouse Hepatitis Virus (MHV), Mouse Parvovirus (MPV), Minute Virus of Mice (MVM), Mouse Rotavirus (EDIM), Pneumonia Virus of Mice (PVM), Sendai virus, Theilers Murine Encephalomyelitis Virus (TMEV), Reovirus 3 (Reo-3), Lymphocytic Choriomeningitis Virus (LCMV), Mouse adenovirus (MAdV) type 1 & 2, Ectromelia Virus, Hantavirus, Murine Cytomegalovirus (MCMV), Polyomavirus, Mouse Norovirus (MNV), Mycoplasma pulmonis, Citrobacter rodentium, Helicobacter spp., Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus spp., Pasteurella pneumotropica, CAR bacillus, Clostridium piliforme, Corynebacterium kutscheri and Klebsiella pneumoniae, endoparasites and ectoparasites. Numerous methodologies were involved in the health assessment of the mice presented. Directly related to this study, serology and complete necropsy were conducted. For serology analysis, serum samples were obtained from cardiac puncture and left to clot for 30 min before centrifuging for 5 min at 604.8 g. Serum was then assessed primarily by enzyme-linked immunosorbent assay (ELISA) for various range of viruses, and as confirmation, indirect fluorescence antibody test (IFAT) was additionally performed. At complete necropsy, beyond the standard macroscopic observations, kidney samples were fixed in formalin, embedded in paraffin wax, sectioned at 3–4 µm and stained with hematoxylin and eosin and assessed microscopically for abnormalities. All handling and procedures were conducted in accordance with the Australian code for the care and use of animals for scientific purposes. No experimental procedures were undertaken at Cerberus Sciences and results reported were as a consequence of routine monitoring submissions. Cerberus Sciences operates under a Scientific Procedures Premises Licence (SPPL), licence designation SPPL413, issued by the Department of Economic Development, Jobs, Transport and Resources (Agriculture Victoria) and has unrestricted access to the Wildlife and Small Institutions Animal Ethics Committee. Cerberus Sciences is also an Associate Member of the International Council for Laboratory Animal Science and holds ISO 9001:2015 Certification issued by Total Quality Certification Services International.
METHODS DETAILS
Necropsy data and histopathology assessment
Necropsy data were obtained from both the Centenary Institute (CI) and the MSKCC/WCM. In CI, gross necropsies were performed by M.W., M.H., B.R. and Q.L. and Centenary Institute Animal Facility staff. In particular, M.W. initially identified the kidney disease in the Centenary Institute Animal Colony, and together with M.H, tracked the kidney disease within in the Centenary Institute Animal Colony over several years. Necropsy records between 2009 and 2017 were recorded in two customized Filemaker Pro databases (Filemaker Inc, Santa Clara, TX, USA) and were searched with the terms such as “renal/kidney disease” and “small, pitted kidneys”. These records were examined to track the prevalence of the virus across strains, as well as within strain in order to determine transmission throughout generations, as represented in the pedigree (see Fig. 1C). The pedigree was manually generated by B.R. using the software Canvas Draw (ACD Systems, Victoria, Canada). Macroscopic images of the necropsy were taken using a Sony CyberShot through the eyepiece of a Leica M80 digital microscope (‘digiscoping’, Leica Microsystems, Wetzlar, Germany). In MSKCC/WCM, complete necropsies and subsequent histopathological examinations were performed by board-certified veterinary pathologists (S.M., S.F.S), which involved macroscopic organ inspection and dissection, histological examination on various tissues and potential serum chemistry of cage-mates (Santagostino et al., 2017). Necropsy reports from 2004 to 2017 were stored in two laboratory information systems (Filemaker Pro 14, Filemaker, Inc, Santa Clara, TX, USA and VisuaLab, Aurora Systems, Inc, Plano, TX, USA) and were searched for the term “inclusions” and “inclusion bodies” for histopathological findings in kidneys. The formalin-fixed paraffin-embedded (PPFE) kidney blocks of these findings, as well as non-inclusion bodies blocks, were then selected for testing the presence of MKPV by PCR and qPCR, The investigator performing PCR and qPCR was blinded by the histological results during the entire testing. Overall for this study, the pathology in kidney samples was assessed by a MD pathologist specializing in renal disease (J.P.D.) and by a mouse pathology service (Cerberus Sciences), and by three board-certified veterinary pathologists (S.M., S.F.S., L.R.).
Conventional histology
To visualize the micro-structural impact of MKPV on various organs, in particular the kidneys, each organ was fixed in 4 mL of 10% neutral buffered formalin (Sigma, St Louis, MO, USA) for 24–48 hours at room temperature, then replaced with 1x phosphate buffered saline (1xPBS; Mediatech Inc, Manassas, VA, USA) to minimize over-fixation and stored at 4°C until paraffin-embedding. Fixed organs were delivered to Veterinary Pathology Diagnostic Services and the Histopathology Laboratory, University of Sydney, for processing, paraffin-embedding, sectioning (at either 2 µm or 5 µm) and staining with hematoxylin and eosin for general diagnosis on any abnormalities in the structures of the organs or Milligan’s trichrome to demark fibrotic structures. To determine collagen levels in fibrotic kidneys, Sirius Red staining was performed, whereby 5 µm formalin-fixed paraffin-embedded kidney sections were deparaffinized with histolene and rehydrated in an ethanol gradient from 100%, 90%, 75% to 50%, each with 2 min immersion. The slides were then washed in water and incubated in 0.1% Picro-Sirius Red (Sigma, St Louis, MO, USA) at room temperature for 60 min. Sections were then washed twice in containers with acidified water containing 83% glacial acetic acid (Sigma, St Louis, MO, USA), then dehydrated in 100% ethanol and histolene twice, and mounted with 2 drops of Eukitt mounting media (Sigma, St Louis, MO, USA) before applying coverslips. In total, kidneys from 3 individual uninfected mice and 5 MKPV-infected mice were randomly selected and examined, pooled from two separate staining experiments. Imaging was not performed in a blinded fashion. No data were excluded in this analysis. Images of the stained slides were captured with a Leica DM6000B microscope (Wetzlar, Germany) using the Power Mosaic program. Some tiled images were subsequently merged using Adobe Photoshop CS6 (San Jose, CA, USA) under the Automate option, specifically the Photomerge with interactive layout feature. Pathology cartoons were manually generated by B.R. using the software Canvas Draw (ACD Systems, Victoria, Canada).
To examine the presence and localisation of dsDNA and ssDNA/RNA in the infected kidneys, acridine orange/DAPI combination was used and the sections were prepared as per the Sirius red staining. 0.01% (w/v) acridine orange (Sigma, St Louis, MO, USA) was added to the section such that the entire slide was covered for a total of 2 min. It was then washed gently in running water and 60 µL of 5 µg/mL DAPI (4,6-diamidino-2-phenylindole dihydrochloride; Molecular Probes, Invitrogen, Eugene, OR, USA) was added for 5 min before washing once more in running water. 2 kidney sections were stained per experiment and a minimum of 2 independent staining experiments were conducted. The region that displayed all 3 signals, namely DAPI (450 nm), green fluorescence (520 nm) from acridine orange binding to dsDNA and red fluorescence (650 nm) when bound to ssDNA/RNA, was selected as representation of results. The images were acquired on a Leica TCS SP5 confocal microscope system (Leica, Wetzlar, Germany) using the tile-scanning mode and processed with ImageJ software (National Institutes of Health, Bethesda, MD, USA) to visualise single channel images and to form merged-channels image.
Micro-CT scan
To visualize the structural damage occasioned by MKPV to the kidneys without sectioning, which could have potentially introduced cutting artefact, randomly-selected uninfected and MKPV-infected kidneys were subjected to micro-computed tomography (Micro-CT scan), where each fixed kidney was placed in 20 mL of Lugol’s Solution (Sigma, St Louis, MO, USA) on a rocker for 48 h at room temperature. For the purpose of comparable results, kidneys were then placed into the same 2 mL screw-cap tube for consistent exposure and detection during scanning with a Xradia MicroXCT-400 system (Carl Zeiss XRM, Pleasanton, CA, USA). Images were acquired using an unfiltered source of 50 kV and 10 W. Reconstructed image stacks from each scanned kidney were visualized and analyzed using 3D image processing software Avizo (Version 9.0, FEI Visualization Sciences Group, Bordeaux, France). The technician was blinded to the status of the kidneys until all images and analyses were completed. With the purpose of providing a representative image, the experiment was only conducted once with one kidney per group.
Electron microscopy
To image nuclear inclusion bodies, freshly isolated kidneys from MKPV-infected mice were fixed in a mixture of 2% paraformaldehyde (Thermo Scientific, Rockford, IL, USA) and 2.5% glutaraldehyde (ProSciTech, Kirwan, QLD, Australia) overnight prior to microdissection into ~1 mm3 pieces and secondary fixation with 1% osmium tetroxide (ProSciTech, Kirwan, QLD, Australia) for 10 min. Samples were then dehydrated in an alcohol series, resin-infiltrated and embedded, ultramicrotome sectioned, stained with 2% w/v uranyl acetate (ProSciTech, Kirwan, QLD, Australia) and Reynolds Lead Citrate (ProSciTech, Kirwan, QLD, Australia), and imaged in a JEOL JEM-1400 Transmission Electron Microscope (Peabody, MA, USA) operated at 100 kV. The experiment was conducted twice. The technician was not blinded in order to locate and image the region of interest, i.e. the nuclear inclusion bodies.
RNA extraction
In preparation that the disease might be driven by an RNA virus, RNA was extracted from four kidneys from 2 healthy, wild-type (C57BL/6) mice and 2 disease-affected Cxcr6gfp/gfp Rag1−/− mice for RNA sequencing (n = 2 replicates). The extraction was performed using an RNeasy® Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions with slight modifications. Briefly, each kidney was snap frozen in liquid nitrogen immediately following organ harvest and then ground manually using a sterile mortar and pestle in liquid nitrogen. The sample was ground to a fine powder and extreme care was taken to ensure it did not thaw. Post-grinding, the fine suspension (tissue + liquid nitrogen) was immediately transferred to liquid nitrogen-cooled, appropriately sized, pre-weighed tubes. The ground tissue from each individual kidney was transferred into multiple tubes and weighed quickly. Lysis buffer was immediately added at this step and homogenization carried out using a 20G needle and syringe. Lysate was passed through the syringe at least 5–10 times to obtain a homogenous lysate. Post-lysis, ethanol was added to the sample, which was then mixed and applied to the RNeasy® column. Total RNA bound to the membrane at this stage. On-column DNase digestion was carried out followed by multiple washes before the RNA was finally eluted. RNA quality was analyzed employing Nanodrop ND-1000 (Thermo Scientific, Rockford, IL, USA) and Bioanalyzer (Agilent Technologies, Waldbronn, Germany).
RNA sequencing/viral metagenomics
After RNA extraction, RNA libraries were prepared using Illumina’s Ribo-zero Gold protocol to remove ribosomal RNA (rRNA) from the total RNA (see Fig. 2D) with their Ribo-Zero Gold rRNA Removal Kit Human/Mouse/Rat (Illumina, San Diego, CA, USA). This began with washing the magnetic beads twice with RNAse-free water (vortexing and onto the magnetic column to remove water) and then resuspended in 65 µL of the resuspension solution. To hybridise the probes in the removal solution to any rRNA present, the sample was added to a cocktail containing the removal solution and reaction buffer, with the final volume of 40 µL made up with RNase-free water. The sample was then heated at 68°C for 10 min, followed by a brief centrifugation and incubation at room temperature for a further 5 min. To remove the rRNA, the entire 40 µL was then pipetted up and down immediately to the 65 µL magnetic beads previously prepared and vortexed for 10 seconds before 5 min room temperature incubation. It was heated at 68°C for another 5 min before immediately placing onto the magnetic stand. ~80 µL of the supernatant now cleared of rRNA was removed to a 1.5 mL tube, placed on ice in preparation for cleaning up residual salt from the buffers used prior. An extra 100 µL of RNAse-free water was added to the sample, followed by 18 µL of 3 M sodium acetate and 2 µL of 10 mg/mL glycogen and vortexed. 600 µL of 100% ethanol was then added, vortexed and stored at −20°C for 1 h for RNA precipitation to occur. RNA was centrifuged down at 10,000 g for 30 min at 4°C, with the supernatant removed and washed twice with 200 µL of 70% ethanol and spinning at 10,000 g for 5 min at 4°C. Tube was air-dried for a minimum of 5 min before resuspending in RNase-free water. Stranded total RNA samples were then sequenced in a 100 bp paired end run on an Illumina HiSeq 2000 platform.
The primary bioinformatics analysis involved quality control (QC) checks and demultiplexing using a QC pipeline developed in-house at the Australian Genomics Research Facility (AGRF). The data were processed through a RNA-seq expression analysis workflow, which included alignment, transcript assembly, quantification and normalization. Differential gene expression analysis was also performed.
The per base sequence quality for all four samples was excellent, with >95% bases above Q30 across all samples. The raw sequence reads were screened for Illumina adapters, cross-species contamination and low quality bases. The low quality bases, and any possible adapter sequence, were trimmed using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)(Krueger, 2012) and Cutadapt (Martin, 2011). A minimum quality score of 20 was used, with a minimum length requirement of 50 bp.
The Mus musculus mm10 genome was used to create a BWA (Burrows-Wheeler Aligner) index (Li and Durbin, 2010). The index was used as a reference within the application Deconseq (Schmieder and Edwards, 2011) to remove non-viral Mus musculus sequences as well as bacterial, plant and fungal sequences, thereby enriching for unidentified viral sequence reads. The sequence read viral/non-viral classification was determined by applying identity threshold ranges of 70%, 80% and 90% to Percentage Identity and Coverage cut-offs. The identity threshold ranges allowed for the retaining of viral sequence reads and endogenous retroviral sequences that were initially considered as potential causative agents of disease. Each range resulted in a bin of viral/endogenous viral sequence reads that were used for the assemblies. Each bin of reads for each sample was assembled separately with the assemblers IDBA (Peng et al., 2012), Trinity (Grabherr et al., 2011) and a Velvet/Oases (Schulz et al., 2012; Zerbino and Birney, 2008) pipeline. These assemblers were chosen due to the unknown quantity, coverage and identity of the viral reads, with each offering distinct assembly advantages. Briefly, IDBA offered an iterative approach in assembling sequences from short read data with highly variable sequencing depth; Trinity offered efficient and robust reconstruction of sequences; and the Velvet/Oases pipeline produces fragmented but accurate reconstructions of viral RNA sequences. IDBA-Tran was used with default settings to perform the assembly of the viral sequences. IDBA iteratively assembled the sequences using a kmer step of 20.
The list of candidate sequences (~55,000 per mouse) were sorted according to abundance and length and manually assessed by BLASTp searches of the predicted amino acid sequences. Gene identity (mouse, junk/pseudogene, bacterial, endogenous retrovirus or viral) was assigned based on whether the predicted amino acid sequence was coherent and free of stop codons (i.e. non-junk) and the homology to known proteins.
MKPV was initially identified from two highly-abundant sequences within the 90% to Percentage Identity IDBA-assembled sequences, which, when assembled, identified a 4286 nt contig of MKPV. A further 68 nt of MKPV, representing the final 3’ ITR, was subsequently identified in the Velvet/Oases build through a text search for the final 20 nt of the 4286 nt contig. Final assembly of the 4442 nt MKPV genome was achieved from Sanger sequencing of genomic PCR products, which identified an additional 88 nt of intronic DNA between the 5’ ITR and the NS1 translational start site.
The read coverage for assembled parvovirus segments was determined by aligning the trimmed raw sequences to the assembled parvovirus contigs using BWA.
Digital gene expression – raw count
The RNA sequence reads that passed Quality Control procedures were aligned against the Mus musculus genome (Build version mm10) using Tophat Aligner (Version 2.0.14) (Trapnell et al., 2009) to identify exon-exon splice junctions. The resulting BAM files were used in the downstream analysis. The counts of reads mapping to each known gene were summarized at gene level using the featureCounts v1.4.6-p2 utility of the subread package (http://subread.sourceforge.net/) (Liao et al., 2013).
Reference guided transcript analysis
The transcripts were assembled from RNA-seq alignment using Stringtie tool (v1.0.4) (Pertea et al., 2015). The alignment BAM files from the Tophat alignment and the reference annotation based assembly option (RABT) were used to create known and potentially novel transcripts.
Differential gene expression
The differential gene expression analysis was performed using Bioconductor (V3.2) (Gentleman et al., 2004) and the limma package (https://bioconductor.org/packages/release/bioc/html/limma.html) (Ritchie et al., 2015). The raw gene count data from the tophat alignment stage was used as input to determine the list of differentially expressed genes.
Phylogenetic analysis
To understand the phylogenetic relationship MKPV has with other viruses, a Blastx analysis of the full length MKPV NS1 gene was initially conducted to find viruses closely related to our mouse parvovirus. The top five hits – accession numbers KP925531, KU563733, KX272741, JX885610, NC_032097 – were all unclassified members of the family Parvovirdae of single-strand DNA viruses. A previously described reference data set of 133 virus variants representing each classified species of the Parvoviridae (Cotmore et al., 2014) was compiled from GenBank. Then, sequences were trimmed and translated to contain only the NS1 gene, and aligned using MAFFT version 7 employing the E-INS-I algorithm (Katoh and Standley, 2013). After removing all ambiguously aligned regions using trimAl (Capella-Gutierrez et al., 2009), a final NS1 sequence alignment of length 398 amino acids (n = 139) was determined (see Fig. 4B). A phylogenetic tree based on this alignment was then inferred using the maximum likelihood (ML) procedure implemented in PhyML 3.0 package (Guindon and Gascuel, 2003), employing the LG amino acid substitution model with a discrete gamma distribution with four rate categories (Γ4), and SPR branch-swapping. Bootstrap support values for individual nodes were generated using 100 bootstrap replicates. A bootstrap support value of >70% for a particular node was determined to be of reasonable good confidence and the values of these nodes were specified in Fig. 4C. To enhance clarity, monophyletic groups corresponding to different parvovirus genera were collapsed and the branch lengths were scaled based on the number of amino acid substitutions per site.
DNA extraction
To extract DNA in preparation for detecting viral presence using PCR and measuring viral load with qPCR, DNA extraction was carried out using a QIAamp® DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions with slight modifications. 5 sections of 5 µm-thick from formalin-fixed, paraffin-embedded blocks were collected in 2 mL tubes. After xylene was added to dissolve the paraffin, tubes were placed on the shaker set at the lowest setting for 20–30 min. Tubes were centrifuged at full speed (18,000 g) for 10 min at room temperature, before carefully removing the xylene (Sigma, St Louis, MO, USA), such that tissues tentatively adhered to the side of the tubes. 100% ethanol was added to remove residual xylene and shaken for 10–15 min, then centrifuged at full speed for 6 min. This was repeated, before incubating the tubes with lids open at 56°C to evaporate the ethanol. Tissue pellet was resuspended in tissue lysis buffer and then proceeded as per frozen organs. Frozen organs were crushed using mortar and pestle in liquid nitrogen to prevent thawing. No more than 25 mg of powdered organ was transferred to autoclaved 1.5 mL tubes. Tissue lysis buffer and proteinase K were added and subjected to 56°C incubation for 2–3 h with regular vortexing for efficient lysis. Additional lysis buffer was added and incubated at 70°C for 10 min. After lysis, 100% ethanol (Sigma, St Louis, MO, USA) was added and the mixture was transferred to the spin column and centrifuged at full speed (18,000 g) for 1 min. Filtrate was decanted and centrifuged at full speed for 1 min. Two wash buffers were added and centrifuged at full speed before incubating the QIAamp membrane in the spin column with buffer AE for a minimum of 5 min, then centrifuged at full speed for 1 min to elute DNA. The subsequent DNA quality and concentration was measured with Nanodrop ND-1000 (Thermo Scientific, Rockford, IL, USA).
PCR Reaction
In this study, various experiments utilised the conventional PCR reaction to detect the presence of the MKPV DNA (NS1, rep gene). In the screening of various strains in Fig. 3A for the presence of MKPV, both male and female were used. These selected strains were all on Rag1−/− background, since the renal pathology was mostly observed in the immunodeficient colonies. Urine and serum were collected, where serum was obtained after coagulation and centrifugation at 4000 g for 10 min at 22°C. A replicate of two were performed to affirm this screening. In the cross-sectional study of Cxcr6gfp/gfp Rag1−/− mice to track the progression of disease, serum (n = 127) and urine (n = 100) were routinely collected and screened. Both male and female were used and randomly selected, as mentioned in the Experimental Model section. For other regular screening, such as the sentinel mice, urine was mostly collected due to the non-invasive nature of the collection process. DNA from formalin-fixed paraffin-embedded kidneys samples from MSKCC, Cerberus and John Hopkins were screened at the Centenary Institute and the investigator was blinded to any information to the samples except the mouse ID until after the results were completed. These samples, as with the urine, were generally screened once unless further affirmation was required or if quantitation of viral load was needed, then qPCR proceeded.
As specified by the experimental conditions above, 1 µL of serum or urine or 100 ng DNA was added to PCR cocktail comprising Phire II hot start Mastermix (Thermo Scientific, Vilnius, Lithuania), 0.5 µM PCR Forward Primer (5’-TACATGGCCAAAGATCCACA) and 0.5 µM PCR Reverse Primer (5’GTGGCAGTCACCCAGCTAAT). The cycling conditions were: initial denaturation at 98°C for 30 s, follow ed by 25 cycles of denaturation at 98°C for 5 s, annealing at 60°C for 5 s, and extension at 72°C for 15 s, and eventually concluded with a final extension at 72°C for 5 min. 7 µL of the completed PCR product was then loaded onto 1.5% agarose (Vivantis, Selangor Darul Ehsan, Malaysia) gels prepared in 1X TAE buffer (Invitrogen, Grand Island, NY, USA) and 1:10,000 dilution of GelRed (Biotium, Fremont, CA, USA). Electrophoresis was conducted at 110 A for 60 min before visualizing with SynGene G:BOX (Cambridge, UK) using their GeneSnap v7.05 software.
Quantitative PCR
Viral copy number in serum, urine, kidneys and other solid organs was measured using quantitative PCR (qPCR) by interpolation against a standard curve consisting of serial dilutions of plasmids of known copy number. For the construction of standard curves, amplicons of viral DNA were cloned into pGEM-T Easy vectors (Promega, Madison, WI, USA) according to the manufacturers’ instructions. Clones containing the desired insert were selected for plasmid extraction using the PureLink Quick Plasmid Miniprep kit (Invitrogen, Löhne, Germany). The DNA sequence of the insert was confirmed using standard Sanger sequencing. After determining the size, concentration and mass of DNA per plasmid, a serial dilution was performed to obtain a standard curve with copy number ranging from 4 x 101 to 4 x 108. qPCR reactions were performed in duplicates per sample in 20 µL volumes containing 2 µL of DNA samples (20 ng) or serum/urine at 1:10 dilution, 1 × iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and 0.3 µM of the qPCR forward and reverse primers: qPCR Forward: 5’-GCAACACAACTGTAAATCGG; qPCR Reverse: 5’-TGGATAATCGTGCCTAAACC. All qPCR reactions were performed using the CFX96 Real Time PCR Machine (Bio-Rad, Hercules, CA, USA). Cycling conditions were: 95°C for 3 min, followed by 40 cycles at 95°C for 10 s, the an nealing temperature of 48°C for 30 s, and extension at 72°C for 30 s. The fluorescence output signal was measured during the extension step. Viral copy number in each sample was directly obtained from the qPCR output generated by the CFX Manager software (BioRad, Hercules, CA, USA) and re-plotted using GraphPad Prism v.7 (La Jolla, CA, USA). A limit of detection at 40 viral genome copies (vgc) was determined and any values below that were considered either too low to detect or negative. Each qPCR plate/experiment had a water blank control and a known negative control, whether wild-type urine, serum or 20 ng of DNA from frozen or FFPE kidney.
To quantify viral load, existing serum and urine samples from the cross-sectional Cxcr6gfp/gfp Rag1−/− study were subjected to qPCR in technical duplicates as specified above. Samples with a matching serum and urine collected from the same mouse and day (n = 32) were selected, collated and then plotted (Fig. 5F). Similarly, sera which had blood urea nitrogen (BUN) measured at the Centenary Institute were selected, collated and then plotted (n = 45, Fig. 5G). To assess kidneys were the original infected organ of MKPV that led to subsequent dissemination, qPCR was conducted and favoured due to the higher sensitivity than conventional PCR. Left and right kidneys, liver, lungs, small intestine, large intestine, bladder, urine and serum in two age groups <61 days old and 61–100 days old (n = 6 in both) Cxcr6gfp/gfp Rag1−/− mice were assessed (Fig. 5H,I). All remaining stratified age groups (days 101–150, 151–200, >201 and >201 culled sick) had left and right kidneys assessed only (n = 5–6 per group). To quantify the viral load in FFPE DNA samples from four facilities in MSKCC, Cerberus, John Hopkins and the Centenary Institute as control (n = 49) as a result of the standard PCR screening, technical duplicates were performed and results were analysed as described above (Fig. S5C).
Chromogenic RNA in situ hybridization
Since mRNA production is indicative of an active replication of the virus, chromogenic RNA in situ hybridization (ISH) was performed to affirm active production of the MKPV in the kidney epithelial tubules. Target probes were designed to target a 978-base sequence of MKPV mRNA, specifically nucleotides 2444–3421 of the MKPV genome corresponding to the peak mRNA expression based on the RNA sequencing data (Fig. 2F) using RNAscope 2.5 LS Probe (Advanced Cell Diagnostics [ACD], Newark, CA, USA), covering both VP1 and NS1 regions. 5 —m formalin-fixed paraffin-embedded kidney sections of the severely and moderately affected, as well as unaffected NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (status determined by histological assessment prior) were ISH stained on a Leica Bond RX automated stainer (Leica Biosystems, Buffalo Grove, IL, USA), using the RNAscope 2.5 LS Reagent Kit-Red (ACD, Newark, CA, USA) and the Bond Polymer Refine Red Detection (Leica Biosystems, Buffalo Grove, IL, USA). For positive and negative control stains performed on the same specimens, positive (mouse PPIB, ACD, Newark, CA, USA) and negative (dapB, ACD, Newark, CA, USA) control probes were substituted for the target probe, and adequate staining of controls was confirmed (data not shown). The chromogen indicating RNA hybridization signal was fast red and the counterstain was hematoxylin. The ISH experiment was replicated twice with identical results. Once the stain was completed by the automated system, each slide was examined by board-certified veterinary pathologists (S.M. and S.F.S). Image analysis of the nuclear sizes (µm2) of MKPV RNAscope+ tubular epithelial cells (n = 22) compared to RNAscope− nuclei (n = 239) was performed in ImageJ (National Institutes of Health, Bethesda MD, USA) The graphs and unpaired t-test statistical analysis were performed in Prism 7 software (GraphPad Software, La Jolla, CA, USA).
Chromogenic Immunohistochemistry
Chromogenic Immunohistochemistry was performed at MSKCC, completely independent from the fluorescence immunohistochemistry, to detect viral presence in kidney sections. This began by heating the slides at 58–60°C for 30 min, followed by placing in xylene to remove paraffin and then in graded alcohols and water to rehydrate. They were then subjected to 1% hydrogen peroxide (in 1xPBS) for 15 min to block endogenous peroxidase activity, with 3 subsequent washes in 1xPBS. Antigens were retrieved by steaming in 10 mM citrate buffer at pH 6 for 30 min, followed by a 20 min cooling period. Slides were then washed in distilled water for 5 min before being transferred to 1xPBS. They were subsequently incubated in blocking buffer (10% normal horse serum (MP Biomedicals, Santa Ana, CA, USA, 2% BSA in 1xPBS) for 30 min in a humid chamber, then aspirated and incubated with co-housed or non-co-housed Swiss Webster serum at 1:1000 dilution in 2% BSA (1x PBS) overnight at 4°C in the humid chamber. To further detail on the co-housed serum used as the primary antibody above, it was derived from Swiss Webster (SW:Tac) mice co-housed with NSG mice affected by IBN in MSKCC. Serum samples from 3 co-housed SW mice were pooled and used for staining. These 3 mice were co-housed with NSG for a period ranging from 3 to 13 weeks. Serum from one non-co-housed SW mouse was used as negative control. On the next day, the slides were washed three times in 1xPBS, followed by incubation in the secondary antibody biotin-conjugated horse anti-mouse IgG (Vector Labs, Burlingame, CA, USA) at 1:500 dilution in 1% BSA (1xPBS) for 1 h in the humid chamber. Slides were washed three times in 1xPBS before adding avidin-biotin complete elite (part of the Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA, USA) at 1:25 dilution in 1xPBS for 30 min in the humid chamber. Following another three washes in 1xPBS, the substrate 3,3’-diaminobenzidine (DAB; Sigma, St Louis, MO, USA) was added and incubated until an acceptable color intensity was reached. They were immediately washed in water and subsequently counterstained with hematoxylin (Fisher Scientific, Hampton, NH, USA) and then washed in running water. Sections were dehydrated in three changes of 95% alcohol, 100% alcohol and cleared in three changes of xylene before coverslips were applied. A minimum of 2 experiments were conducted, where one of which involved an optimization of both co-housed and non-co-housed sera using dilutions of 1:50 to 1:1000 on the kidneys sections. Slides were examined by board-certified veterinary pathologists (S.M. and S.F.S).
Fluorescence Immunohistochemistry
Fluorescence immunohistochemistry was performed at the Centenary Institute, completely independent from the chromogenic immunohistochemistry, to visualise the viral presence in the disease-affected kidneys. Formalin-fixed paraffin-embedded kidney blocks were initially sectioned at 5 —m onto slides before incubating at 60°C for 1 h. Slides were submerged in histolene (Fronine, Thermo Fisher Scientific, Riverstone, NSW, Australia) for 25 min at room temperature to remove paraffin, followed by rehydrating in 100% ethanol for 2 min and 1 min each in 90%, 75% and 50% ethanol before transferring into 1xPBS. The slides immediately proceeded to antigen retrieval, whereby the slides were fully immersed in 10 mM sodium citrate buffer (pH 6) within a glass container and heated in the microwave at 100% power for 3 min, followed by a lower power at 20% for another 15 min. The glass container with the slides was then cooled in a water bath at room temperature for 30 min before transferring the slides to 1x PBS. After demarcating the kidney sections with a PAP pen, each section on the slide was incubated in 120 —L of blocking buffer (2.5% FBS [Hyclone, GE Life Sciences, Mordialloc, VIC, Australia], 2 mM EDTA, 3% rat serum [Sigma, St Louis, MO, USA], 3% rabbit serum [Applied Biological Products Management, Aldinga Beach, SA, Australia] in 1xPBS) for 1 h at room temperature. The blocking buffer was then drained off and 120 —L of the diluent (2.5% FBS, 2mM EDTA in 1x PBS) containing 3% of co-housed or non-co-housed mouse serum and anti-cytokeratin 19 at 1:60 dilution (Abcam, Cambridge, UK) was added to each kidney section for 1 h at room temperature. The slides were washed three times in 1xPBS at 10 min intervals before incubating in 120 —L diluent for 40 min at room temperature in the dark, containing the secondary antibody Alexa Fluor594 conjugated rabbit anti-mouse IgG (Invitrogen, Eugene, OR, USA) at 1:500 dilution (antiserum staining only) or Alexa Fluor488 conjugated donkey anti-mouse IgG (Invitrogen, Eugene, OR, USA) at 1:200 dilution together with Alexa Fluor594 conjugated goat anti-rabbit IgG (Invitrogen, Eugene, OR, USA) at 1:200 dilution (co-staining for cytokeratin 19). The slides were further washed twice in 1xPBS at 10 min intervals and then overnight at 4°C before adding 60 —L of DAPI (4,6-diamidino2–2-phenylindole dihydrochloride; Molecular Probes, Invitrogen, Eugene, OR, USA) at 5 —g/mL for 5 min at room temperature. Excess DAPI was drained off and the slides were mounted in 70 mM DABCO (1,4-diazobizyclo(2,2,2)octane, Sigma, St Louis, MO, USA) in PBS/glycerol (1:10) before applying coverslip. The kidney sections were then imaged on a Leica TCS SP5 confocal microscope (Leica, Wetzlar, Germany).
Image analysis was performed using ImageJ (National Institutes of Health Bethesda, MD USA), which included the generation of multi-channel images and quantification of antisera staining signals. In Fig. S7A, anti-sera staining signals were analyzed against normal and enlarged nuclei (n = 817–2073 nuclei analyzed). The staining was performed in a minimum of 3 replicates and a representative image was shown in this study. In Fig. S7E, the percentage of antiserum+ nuclei observed in cytokeratin 19 (Krt19)− and Krt19+ was determined from 10 fields of view (n = 129 cells) from two independent experiments. For antisera and immunohistochemical staining, kidneys from 100–120 day old mice were used, since nuclear inclusions were reliably indefinable on all tissue sections at this age.
Recombinant GST-VP1 protein preparation
DNA encoding residues 1–496 of VP1 was synthesized (Genscript, Piscataway, NJ, USA) and integrated into the glutathione S-transferase (GST) fusion expression vector pGEX-4T-1 (GE Healthcare, Chicago, IL, USA). The resulting open reading frame translates to a GST-VP1 fusion construct. The protein was overexpressed in BL21 (DE3) cells (Invitrogen, Carlsbad, CA, USA) grown in Luria Broth at 37°C until OD600 reached 0.6. The temperature was subsequently lowered to 20°C and protein expression was induced by the addition of 0.2 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG; Astral Scientific, Taren Point, NSW, Australia). The cells were harvested after 12 h by centrifugation (8,000 rpm, 10 min, 4°C), resuspended in buffer A (20 mM Tris pH 8, 300 mM NaCl) and stored at −20°C until further use.
To purify the GST-VP1 fusion protein, the cells were thawed and lysed in a high-pressure homogeniser (10 kPsi, Emulsiflex-C3; Avestin Inc, Ottawa, ON, Canada). Cellular debris was cleared by centrifugation (20,000 rpm, 45 min, 4°C) and the supernatant containing the protein was immobilized on GST-affinity resin (Glutathione Sepharose 4B, GE Healthcare, Chicago, IL, USA), pre-equilibrated with Buffer A, by incubation under stirring for 1 h. The protein-bound resin was loaded onto a Poly-prep gravity flow column (Bio-Rad, Hercules, CA, USA) and washed with 30 column volumes of Buffer A. The protein was subsequently eluted with 5 column volumes of 50 mM Tris pH 8, 100 mM NaCl, 25 mM glutathione (Sigma, St Louis, MO, USA), concentrated to approximately 10 mg/mL and stored at −80°C until further use. Recombinant GST-VP1 was validated using MALDI-TOF mass spectrometry.
Western blot of GST-tagged VP1 with antiserum
Western blotting was performed to confirm the presence of anti-MKPV antibodies, specifically anti-VP1, in the serum of co-housed C57BL/6 mice. To denature and reduce the recombinant glutathione S-transferase (GST) tagged VP1 protein in preparation of performing SDS polyacrylamide gel electrophoresis (PAGE), the recombinant GST-VP1 protein was added to a buffer containing 1x lithium dodecyl sulfate (LDS) sample buffer (NuPAGE, Invitrogen, Carlsbad, CA, USA) and 1x reducing agent containing dithiothreitol (NuPAGE, Invitrogen, Carlsbad, CA, USA) in MilliQ water. This mixture was heated at 95°C for 25 min before cooling down on ice for 5 min. 2 —g of GST-VP1 protein was loaded to each allocated well in a 1.0mm, 15 well 4–12% Bis-Tris protein gel (NuPAGE, Invitrogen, Carlsbad, CA, USA) and 3.5 —L of the PageRuler™Plus pre-stained ladder (Thermo Scientific, Vilnius, Lithuania) was loaded for referencing protein sizes between 10 to 250 kDa. The gel was subsequently run in 1x MES buffer (NuPAGE, Invitrogen, Carlsbad, CA, USA) within a mini gel tank (Invitrogen, Carlsbad, CA, USA) at 160 V for 50 min. Protein transfer onto a methanol-activated polyvinylidene difluoride (PVDF) membrane (Immobilon, Millipore, Billerica, MA, USA) used a Mini Blot Module (Invitrogen, Carlsbad, CA, USA) with cold 1x transfer buffer (NuPAGE, Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s instructions. The wet transfer was conducted in the mini gel tank containing deionised water at 30 V for 1 h. To confirm the presence of protein in the PVDF membrane, the membrane was incubated in 0.1% w/v Ponceau S Red (Sigma, St Louis, MO, USA) at room temperature for 5 min. The blot was excised in preparation for incubating in different sera, followed by 2 washes of 1xPBST (0.05% Tween-20, Sigma, St Louis, MO, USA, in 1xPBS) at 2 min each for de-staining. The destained excised blot was then placed into a 15 mL tube (Corning, NY, USA) containing 2.5 mL of 5% skim milk (Fonterra, Mount Waverly, VIC, AUS in 1xPBS) and rolled for 1.5 h at room temperature for blocking. Once the blocking buffer was decanted, co-housed or non-co-housed control serum was added at 1:100 dilution in 2.5 mL of 5% skim milk to the respective blot for overnight 4°C roller incubation. After decanting the diluted serum, the blot was washed 5 times for 5–10 min in 1xPBST on the rocker at room temperature. The blot was then placed into a 15 mL tube containing 2.5 mL of goat anti-mouse IgG (H+L) HRP (Invitrogen, Rockford, IL, USA) at 1:10000 dilution in 5% skim milk for 1.5 h on the roller at room temperature in the dark. The secondary antibody was subsequently aspirated and washed 5 times 5–10 min in 1xPBST on the rocker at room temperature. Following the addition of the chemiluminescent HRP substrate (Immobilon, Millipore, Billerica, MA, USA) solution, the blot was imaged with ChemiDoc Imaging System (Bio-Rad, Hercules, CA, USA) using the application chemi high sensitivity with signal accumulation mode for detecting chemiluminescent signals and using the application white for imaging the ladders. The two images were then merged using the Image Lab image acquisition and analysis software (Bio-Rad, Hercules, CA, USA) for simultaneous display. The experiment was repeated for a minimum of three times and one was selected as representation.
Tissue Lysate Preparation for Mass Spectrometry
To prepare kidney tissue lysates, snap frozen kidneys (one kidney per mouse) were homogenized in 500 —L RIPA buffer (1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl, 0.1% SDS, 0.1% Triton X-100, in MilliQ water) with 1x Halt™ Protease Inhibitor Single-Use Cocktail (PIC, Thermo Scientific, Rockford, IL, USA). The residual volume remaining on the homogenizer was washed in another 500 —L RIPA buffer containing PIC, before pooling both into a single 1.5 mL tube. Tissue lysate was kept on ice for 30 min before centrifuging at 18,000 g for 10 min at 4°C. Without disturbing the pellet, the supernatant was carefully aliquoted into a new 1.5 mL tube. Protein levels were subsequently measured with the Pierce™ BCA protein assay kit (Thermo Scientific, Rockford, IL, USA), whereupon 10 —L of the diluted lysate (1:5 dilution in 1xPBS) was mixed with 200 —L of working reagent (Reagent A:B, 1:50), followed by 30 min incubation at 37°C. Absorbance was measured at 562 nm with a POLARstar Omega Spectrophotometer (BMG LABTECH, Offenburg, Germany) and the tissue lysate concentration was interpolated from the standard curve. For mass spectrometry, 100 —g of tissue lysate was transferred to a fresh Eppendorf tube, reduced and alkylated with 1 M DTT (Dithiothreitol) and TBP (tributylphosphene), then quenched with 1 M acrylamide monomer. Trypsin was then added at 50 ng/—g of total protein and allowed to digest overnight at 37°C. The digests were assayed via Qubit peptide analysis (Thermo Scientific, Rockford, IL, USA) so that 1 —g could be accurately loaded into the mass cytometry instrument per run. Runs were performed in technical triplicate for infected and control.
Mass Spectrometry (LC/MS/MS)
Using an Acquity M-class nanoLC system (Waters, Milford, MA, USA), 5 —L of the sample (1 —g) was loaded at 15 —L/min for 3 min onto a nanoEase Symmetry C18 trapping column (180 —m x 20 mm) before being washed onto a PicoFrit column (75 —m ID x 250 mm; New Objective, Woburn, MA, USA) packed with Magic C18AQ resin (Michrom Bioresources, Auburn, CA, USA). Peptides were eluted from the column and into the source of a Q Exactive™ Plus mass spectrometer (Thermo Scientific, Rockford, IL, USA) using the following program: 5–30% MS buffer B (98% acetonitrile + 0.2% formic acid) over 90 min, 30–80% MS buffer B over 3 min, 80% MS buffer B for 2 min, 80–5% for 3 min. The eluting peptides were ionized at 2000V. A Data Dependent MS/MS (dd-MS2) experiment was performed, with a survey scan of 350–1500 Da performed at 70,000 resolution for peptides of charge state 2+ or higher with an AGC target of 3e6 and maximum Injection Time of 50 ms. The Top 12 peptides were selectively fragmented in the HCD cell using an isolation window of 1.4 m/z, an AGC target of 1e5 and maximum injection time of 100 ms. Fragments were scanned in the Orbitrap analyzer (Thermo Scientific, Rockford, IL, USA) at 17,500 resolution and the product ion fragment masses measured over a mass range of 120–2000 Da. The mass of the precursor peptide was then excluded for 30 s.
Mass Spectrometry Analysis
The MS/MS data files were searched using the Mascot search engine (www.matrixscience.com) against the Mouse Proteome database (SWISS-PROT) “spiked” with the MKPV NS1 and VP1 proteins, using the following parameter settings: Variable mods: oxidation of methionine. Peptide mass tolerance: ± 50ppm. MS/MS mass tolerance: ± 0.6Da. Peptide charge state: 1+, 2+ and 3+. Data format: Mascot Generic (MGF). Monoisotopic peak selection. Enzyme: Trypsin. Number of allowed missed cleavages: 1. MS/MS data were compiled and exported using Scaffold Q+ V4 (Proteome Software Inc, Portland, OR, USA).
MKPV secondary structure
Secondary structures of the 5’ and 3’ ITRs of MKPV were generated using the Mfold web server for nucleic acid folding and hybridization prediction (Zuker, 2003).
Tissue processing and flow cytometry
Kidneys were cut into small pieces and incubated in collagenase (1 mg/mL; Sigma, St Louis, MO, USA) in 5 mL DMEM with 10% fetal bovine serum (FBS; Hyclone, GE Life Sciences, Mordialloc, VIC, Australia) at 37°C for 40 min. Samples were then filtered through a 70 µm cell strainer (Falcon, Corning, NY, USA) into a 50 mL tube (Falcon Corning, NY, USA) with a further 25 mL DMEM (Gibco Life Technologies, Grand Island, NY, USA) with 10% FBS to wash cells through. Cells were then pelleted and the supernatant decanted. For leukocyte isolation, cells were resuspended in 4 mL flow running buffer (2.5% FBS, 2 mM EDTA, and 0.02% sodium azide, Sigma, St Louis, MO, USA in 1x PBS) and further filtered using 50 µm nylon filter (Sefar, Meadowbrook, QLD, Australia) into 15 mL tube (Corning, Corning, NY, USA). Each tube was subsequently underlaid with 3 mL Lympholyte M (Cedarlane, Burlington, ON, Canada) and centrifuged at 1500 g for 30 min at 22°C (breaks off). The leukocytes were then removed at the interface and washed twice with flow running buffer. For fibroblasts preparation, cells were then resuspended in 15 mL of 1xPBS prior to the addition of 9 mL ‘isotonic’ Percoll (1:9 10X PBS:Percoll, Uppsala, Sweden) and centrifuged at 931 g for 8 min (brakes on). After carefully decanting, cells were resuspended in 20 mL of DMEM with 10% FBS and spun at 524 g for 5 min at 4°C. Red blood cells (RBC) were then lysed in 2 mL RBC lysis buffer (ACK buffer, prepared in-house, 145 mM NH4Cl, 0.1 mM EDTA and 12 mM NaHCO3 in MilliQ water) for 1 min at room temperature. Cells were then washed and antibody-stained in running buffer for 1 h at 4°C. Immediately prior to flow cytometry, cell suspensions were resuspended in running buffer containing 0.5 µg/mL DAPI (4,6-diamidino-2-phenylindole dihydrochloride; Molecular Probes, Invitrogen, Eugene, OR, USA) for exclusion of dead cells. For quantification of cells, 20 µL of Flow-Count fluorospheres (Beckman Coulter, Galway, Ireland) counting beads equating to 20,000 beads were additionally added. The antibodies used in this study were: B220 FITC (clone RA3–6B2; BD) CD11b PE, APC-Cy7 (clone M1/70; BD), CD11c PE (clone HL3; BD), CD14 PE (Sa2–8; eBioscience), CD16/32 Purified (clone 2.4G2; BD); CD24 PerCP-Cy5.5 (clone M1/69; eBioscience), CD29 PE (clone HMβ1–1; eBioscience), CD45 BUV395 (clone 30-F11; BD), CD86 PE (clone GL1; eBioscience), CD88 APC (clone 20/70; BioLegend), CD103 PE (clone 2E7; BD), CD206 PE (clone C069C2; BioLegend), Ly6C FITC (clone HK1.4; BioLegend), Ly6G PerCP-Cy5.5 (clone 1A8; BioLegend), MHC-II Pacific Blue (clone M5/114.15.2; BioLegend). To detect fibroblast activation protein (FAP), we used a FAP-selective inhibitor Compound 4613B conjugated to the infra-red fluorophore IRDye800CW (M.G., B.R., J. H. L., W.Wu., W.W.B. and J.H., manuscript in preparation, synthesis described below). Cells were analyzed on a custom 10-laser LSR II (BD Biosciences, Franklin Lakes, NJ, USA) equipped with an infra-red laser for detection of IRDye800CW.
Flow cytometry data were analyzed with FlowJo software (TreeStar, Ashland, OR, USA). All analyses were pre-gated on time to remove any fluidic issues, followed by forward scatter and side scatter singlets to remove doublets and then DAPI− live cells. To phenotype kidney macrophages as in Figure 6B, CD45+ leukocytes were gated upon, followed by excluding Ly6G+ neutrophils and Ly6C+ monocytes. CD88+MHC-II+ macrophages were subsequently gated and the expression levels in CD11b, CD14, CD86 and CD206 were compared between the uninfected and MKPV. 2 replicates of the same experiment were performed. To quantitate kidney macrophages (Figure S7I), cDC1 and cDC2, n = 4 mice with separated left and right kidneys were analysed in 3 groups – young Rag1−/− (<100 days old), young Cxcr6gfp/gfp Rag1−/− (<100 days old) and old Cxcr6gfp/gfp Rag1−/− (> 200 days old). After gating upon CD45+ leukocytes, CD88+ MHC-II+ macrophages, CD88− MHC-II+ CD103+ CD24hi CD11blo cDC1 and CD88− MHC-II+ CD103− CD24lo CD11b+ cDC2 were identified and quantified. For identifying fibroblasts and epithelial cells in the kidneys as in Fig. 6F, three replicates from the uninfected and MKPV-infected were performed. CD45− FAPhi identified CD24hiCD29lo and CD24hiCD29lo fibroblasts, whereas CD45− FAP−identified EpCAMhiCD29hi epithelial cells. To investigate the expressions of various markers in a wide range of cellular populations between uninfected and MKPV-infected (Fig. 6C–E), colored t-SNE plots were generated in R using a custom-built script (Ashhurst, 2017). Clusters of populations were identified based on the expression of markers, as per described above. Experiment shown in Figure 6 is representative of 3 independent experiments.
Synthesis of Compound 4613B
The FAP-selective inhibitor Compound 4613B was initially generated from N-Boc-D-Ala-OH in 40 mL anhydrous DMF, whereby 10 mmol of the compound was added to 10.5 mmol of L-boroPro-pn.HCl, 10.5 mmol of HATU and 23 mmol of DIEA and stirred at room temperature for 2 h before condensing in vacuo. The residue was then dissolved in ethyl acetate before washing thrice in 40 mL KHSO4, thrice in 40 mL aq. NaHCO3, once in 30 mL brine and dried over anhydrous MgSO4 to form N-Boc-D-Ala-L-boroPro-pn. This was then added to 30 mL of 4 N HCl in dioxane before stirring at room temperature for 2 h and condensing in vacuo. Residue was evaporated with 3 times of 30 mL dichloromethane, and then added into 1 mmol 6-(Nƍ-Boc-hydrazino)-nicotinic acid (in 4 mL anhydrous DMF), 10.5 mmol of HATU and 2.3 mmol of DIEA. The mixture was stirred at room temperature for 2 h before condensing in vacuo, which was then dissolved in 50 mL dichloromethane and washed thrice in 10mL aq. NaHCO3, 10 mL brine and dried over anhydrous MgSO4. The intermediate compound was dissolved in 5 mL dry dichlormethane and cooled to - 78°C, then dropwise added 5.0mL of BCl3 in 1 M dichloromethane and stirred at −78°C for 1 h before concentrating in vacuo. The residue was partitioned in 5 mL ether and 5 mL water, where the aqueous layer was selected, washed twice with 5 mL ether and concentrated in vacuo. This was purified by semi-preparative RP-HPLC to yield a compound where 0.3 mmol was conjugated to 0.1 mmol IRDye 800CW NHS Ester in 10 mL of phosphate buffer at pH 7.8. The mixture was stirred at room temperature for 3 h and subsequently purified by semi-preparative RP-HPLC to yield the green powder IRDye 800CW-conjugated FAP-selective inhibitor known as Compound 4613B.
Blood assessment
Red blood cell (RBC), hemoglobin and hematocrit measurements were performed using a Sysmex XS-1000i Automated Hematology Analyzer (Toa Medical Electronics, Kobe, Japan). This involved a prior 1:20 dilution of mouse blood collected in Mini Collect EDTA coated tubes (Greiner bio-one, Kremsmünster, Austria) with 1x PBS before subjecting 80 µL for analysis. Each of the MKPV-infected mice (n = 16) was selected to represent a particular age in order to cover the age range. The uninfected Rag1−/− (n = 3) were selected to match the age of the infected that would have widespread MKPV infection within the body (>150 days old). At analysis, the MKPV-infected mice were stratified to under and over 200 days of age and they were compared with the uninfected using one-way ANOVA, Holm-Sidak’s multiple comparison on Prism 7 software (GraphPad Software, La Jolla, CA, USA).
Serum Biochemistry
Serum biochemistry was performed by the Laboratory of Comparative Pathology at the MSKCC using a Beckman Coulter AU680 clinical chemistry analyzer (Beckman Coulter, Brea, CA, USA), with the technicians blinded to any information regarding to the samples except the mouse identification number. Samples were selected to cover the age range of the infected Cxcr6gfp/gfp Rag1−/− colony (up to 350 days old; n = 30) and the respective uninfected wild-type control (n = 10). 130 µL of each mouse serum sample was subjected to the same analysis, encompassing: Blood Urea Nitrogen (BUN; mg/dL), Creatinine (mg/dL), Alkaline Phosphatase (ALP; U/L), Alanine Transaminase (ALT; U/L), Aspartate Aminotransferase (AST; U/L), Creatine Kinase (CK; U/L), Total Protein (g/dL), Albumin (g/dL), Tryglycerides (mg/dL), Cholesterol (mg/dL), Glucose (mg/dL), Potassium (K; mEq/L), Chloride (Cl, mEq/L), Total Carbon Dioxide (TCO2; mEq/L), Sodium (Na; mEq/L), Calcium (mg/dL) and Phosphorus (mg/dL). Globulin (g/dL) was calculated based on the albumin and total protein levels. Data were generated into an Excel format and used for further analyses in the Prism 7 software (GraphPad Software, La Jolla, CA, USA), where the infected strain was stratified into 2 age groups, under and over 200 days of age. The 2 groups, along with the uninfected wild-type, had each of their serum biochemistry examined and compared using one-way ANOVA, Holm-Sidak’s multiple comparison.
Urinary protein measurement
To determine the effect of MKPV in proteinuria, it was measured using the Bradford protein assay. 5 µL of undiluted urine (n = 17 uninfected and n = 101 MKPV-infected) or bovine serum albumin (BSA; Sigma, St Louis, MO) standards was added to each well of a 96-well flat bottom plate (Falcon, Corning, NY, USA), followed by mixing with 250 µL Bradford Reagent (BioRad, Hercules, CA, USA) and a 5 min incubation at room temperature. Absorbance levels were measured at 595 nm using a POLARstar Omega Spectrophotometer (BMG LABTECH, Offenburg, Germany). Protein concentrations were interpolated from the BSA standard curve. The data were pooled from 3 independent experiments. Each sample was run as a single technical replicate. Both male and female samples were collected. Samples of each group were selected to provide a comprehensive age range.
Blood urea nitrogen measurement
In this study, blood urea nitrogen (BUN), the biomarker associated with kidney function, was measured in two ways. In Fig. 1E and Fig. S1A, BUN was measured at MSKCC as part of the serum chemistry analysis using a Beckman Coulter AU680 clinical chemistry analyzer (Beckman Coulter, Brea, CA, USA), as described earlier in the Serum Biochemistry section. For BUN measurements shown in Fig. 5E, sera from 84 MKPV-infected and 39 uninfected wild-type and Rag1−/− control mice had their BUN measured using the Blood Urea Nitrogen (BUN) Colorimetric Detection Kit (Invitrogen, Frederick, MD, USA). Serum samples from a broad age range of both groups were selected. 25 µL of the diluted serum (at a minimum of 1:10) or the standard was added to each well of a 96-well flat bottom plate (Falcon). This was followed by 37.5 µL of Color Reagent A and subsequently 37.5 µL of Color Reagent B. Sample absorbance at 450 nm was measured after 30 min incubation at room temperature using a POLARstar Omega Spectrophotometer (BMG LABTECH, Offenburg, Germany). BUN concentration (mg/dL) was then calculated from the absorbance level of each well and the standard curve. Each sample was assessed once and the data were pooled from 3 independent experiments. Data were plotted in Prism 7 (GraphPad Software, La Jolla, CA, USA) against age (days).
Epidermal growth factor measurement
Urinary Epidermal Growth Factor (EGF) concentrations were measured using the Mouse EGF DuoSet® ELISA kit (R&D Systems, Northeast, Minneapolis, MN, USA) and conducted as per the manufacturer’s instructions. Subsequent to overnight coating of a 96-well plate (Corning Costar, Corning, NY, USA) with the capture antibody at 200 ng/mL, the plate was washed twice with 0.05% Tween® 20 (Sigma, St Louis, MO, USA) in 1x PBS. The plate was then blocked with 1% BSA (Sigma, St Louis, MO, USA) in 1x PBS for 1 h and washed twice. A 100 µL sample (or standard) was added for a 2 h incubation at room temperature, followed by 2 washes. The following urine dilutions were achieved with the diluent (1% BSA in 1x PBS), as determined by the age group and the mouse strain. For all wild-type (C57BL/6 and Rag1−/−, n = 17) samples and disease affected Cxcr6gfp/gfp Rag1−/− (n = 66) mice at 1–130 days old, 1:12800, 1:25600, 1:51200 were used. For the diseased 131–190 days old mice, 1:6400, 1:12800, 1:25600 were used. Lastly, any diseased mice greater than 191 days old had 1:800, 1:1600, 1:3200 dilutions. Detection antibody with the final concentration of 200 ng/mL was then added for another 2 h at room temperature and washed twice. Streptavidin-HRP (provided in the kit) at 1:200 dilution in the diluent was further incubated for 20 min at room temperature in the dark, and twice washed. 1-Step™ Ultra TMB-ELISA Substrate Solution (Thermo Scientific, Rockford, IL, USA) was incubated at room temperature for 20 min, followed by adding 2 N sulfuric acid to stop the reaction. Absorbance was measured by a POLARstar Omega Spectrophotometer (BMG LABTECH, Offenburg, Germany) at 450 nm and 570 nm to account for optical correction of the plate. Following optical correction, absorbance of each sample and the standard curve were used to calculate the concentration of urinary EGF. As mentioned prior, each sample was conducted in triplicates at 3 different dilutions, however, only the two closest dilutions within the range of the standard curve were accepted as duplicates for the EGF measurement. Data were collated from a minimum of 3 independent experiments.
Latent TGFβ binding protein 2 measurement
Urinary Latent TGFβ binding protein 2 (LTBP2) levels were measured using the Mouse LTBP2 Sandwich ELISA Kit (LifeSpan Biosciences Inc, Seattle, WA, USA), as part of the investigation into biomarkers for kidney fibrosis. Any urine collection over 100 µL from uninfected wild-type (n = 10) and Cxcr6gfp/gfp Rag1−/− (n = 48) mice were considered into LTBP2 measurement. Even distribution of age range in the samples was also a collection criterion. 100 µL of mouse urine samples at neat or 1:2 dilution (with the diluent provided) or standard were added to the capture antibody pre-coated plate for 2 h at 37°C, followed by aspirating the liquid. Detection Reagent A at 1:100 was added for another 1 h incubation at 37°C. After aspiration, the wells were washed thrice with the 1x wash buffer provided. Detection reagent B at 1:100 was then added for another 1 h incubation at 37°C, followed by aspirating and washing 5 times. TMB substrate from the stock solution was directly added to each well and incubated for 30 min at 37°C, followed by the stop solution provided. The absorbance levels were then measured using a POLARstar Omega Spectrophotometer (BMG LABTECH, Offenburg, Germany) at the wavelength of 450 nm. Concentrations of LTBP2 were calculated from the absorbance values and the standard curve. Samples were limited to one replicate due to the limited volume of urine available per collection/timepoint. Data were pooled from two independent experiments.
Co-housing
Cxcr6gfp/gfp Rag1−/− female mice confirmed positive for the virus by serum and urine PCR (typically >100 days of age) were co-housed with Rag1−/− female mice (8–10 weeks old) from Australian BioResources (Moss Vale, NSW, Australia) to demonstrate that the virus could be horizontally transmitted. Each co-housing experiment typically consisted of 2–3 infected mice with 3 uninfected Rag1−/−, as well as a separate cage of 3 uninfected Rag1−/− littermates as negative control. All mice were initially screened to confirm their MKPV status and then periodically examined at a 2-week interval to detect any positivity for the virus from urine and serum collection using the standard PCR described earlier. Co-housed mice were declared MKPV positive when the faint amplicons could be detected for at least 2 consecutive time points, to be conservative. The increasing intensity in the amplicon appearance on the PCR gel as the virus progressed was a further confirmation of an active MKPV infection. All mice were additionally weighed twice weekly to monitor body weight loss, as another means to track and confirm the progression of the disease. Mice were subsequently sacrificed at the point where weight loss exceeded 10% of the initial weight or appeared sufficiently ill from body posture, whichever occurred first. A total of 3 independent co-housing experiments/replicates were conducted in this study. In order to prevent cross contamination, control mice were always weighed first, necessitating that the technician was not blind to the study.
Embryonic re-derivation
To demonstrate a resolution in removing MKPV from a mouse colony, transplantation of Cxcr6gfp/gfp Rag1−/− embryos from MKPV+ mothers into disease-free pseudo-pregnant Rag1−/− dams was surgically performed by the Australian BioResources (Moss Vale, NSW, Australia). Rederived mice were imported back to the Centenary Animal Facility and the strain was subsequently screened for viral positivity by PCR on the urine and serum collected as described earlier. To confirm the virus had been eliminated, minimum of 3 older mice (>200 days old) were randomly selected from the clean strain for screening, since at this age, in the virus-infected strain, MKPV would have been prevalent in the body. Mice were also kept alive past at least 250 days to compare against the infected strain which have either lost weight or succumbed to the disease. Embryonic re-derivation was conducted once only and the subsequent screening of the re-derived was conducted in n = 2 or 3 mice and in a minimum of 2 independent experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details and tests used, the number and representation of n and any other forms of quantification present are specified in the respective figure legends and results section. All statistical analyses in this study were performed using the Prism 7 software (GraphPad Software, La Jolla, CA, USA). For two-group comparison, unpaired Student’s t-test and non-parametric unpaired Mann Whitney test were used. For multiple groups comparison, one way ANOVA (Holm-Sidak’s multiple comparison) was used. Unless otherwise specified, statistical significance was defined by the P-value greater than 0.05 (* p>0.05).
Supplementary Material
(A) Expanded analysis of serum biochemistry in mouse strains affected (Cxcr6gfp/gfp Rag1−/−; black and red) and unaffected (white, n = 10) by renal disease, as shown in Fig. 1E. Each dot represents an individual mouse. Control mice comprise disease-free Rag1−/− and C57BL/6 mice. Cxcr6gfp/gfp Rag1−/− mice were stratified according to age (<200 days, black, n = 20; >200 days, red, n = 10). ALP, alkaline phosphatase. ALT, alanine transaminase. AST, aspartate aminotransferase. BUN, blood urea nitrogen. CO2, carbon dioxide. CK, creatine kinase. (B) Expanded evaluation of hemoglobin and red blood cells in Cxcr6gfp/gfp Rag1−/− mice (black and red) and control Rag1−/− mice (white, n = 3), as shown in Fig. 1F. Each dot represents an individual mouse. Cxcr6gfp/gfp Rag1−/− mice were stratified according to age (<200 days, black, n = 8; >200 days, red, n = 8). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (one-way ANOVA, Holm-Sidak’s multiple comparison). RBC, red blood cells. (C,D) Representative microscopic images of a disease-affected kidney from a moribund Cxcr6gfp/gfp Rag1−/− mouse, stained with hematoxylin and eosin. (C) High magnification image depicting tubular epithelial cell nuclei with chromatin margination and amphophilic intranuclear inclusions (black arrows) and, less frequently, eosinophilic intranuclear inclusions (white arrow head). Bar = 40 µm. (D) Lower magnification demonstrating renal papillary necrosis (black arrow) in the same mouse. Bar = 500 µm.
(A) Top: MKPV mRNA reads from diseased kidneys mapped to the final assembled viral genome for the two disease-affected mice (black line and blue histogram) detailed in Fig. 2. Black and red arrows indicate major splice donor and acceptor sites, respectively. Lollipops indicate consensus polyadenylation signals (AATAAA). (B) Integrative Genomics Viewer (IGV) snapshots of MKPV mRNA reads mapped to the final assembled viral genome. Individual reads spanning presumptive introns are indicated by grey blocks (reads) connected by pale blue lines (introns). Superimposed black and red lines indicate the locations of consensus-conforming splice donor and acceptor sites, respectively. (C) Putative major transcripts encoding (in black) NS1 (non-structural protein 1), hypothetical protein NS2 (non-structural protein 2) and VP1 (viral protein 1), based on sequence analysis, observed splicing, reads map and location of polyadenylation signals.
(A) Gastrointestinal tract (GI) in an MKPV-infected mouse. Hematoxylin and eosin staining of indicated regions of the GI tract from a moribund mouse with kidney disease. No pathological changes were detected besides the kidney pathology. Bar = 200 µm. (B) Clearance of MKPV through embryonic rederivation, Non-detection of viral DNA by conventional PCR (25 cycles) in the serum (top) and urine (bottom) of 277 days old Cxcr6gfp/gfp Rag1−/− mice (blue, n = 3 littermates) following rederivation through embryo transfer. Resultant Cxcr6gfp/gfp Rag1−/−mice did not develop IBN (not shown). Serum and urine from a non-rederived Cxcr6gfp/gfp Rag1−/− mouse (red) is included as a positive control. Fluids from disease-free Rag1−/− and C57BL/6 colonies served as negative controls. (C) PCR detection of MKPV in the urine of an outbred Swiss mouse. Detection of viral DNA by conventional PCR (25 cycles) in the urine (white box) of an outbred sentinel Swiss mouse. This mouse was a sentinel for a rack that housed MKPV-infected Cxcr6gfp/gfp Rag1−/− mice. MKPV transmission to the Swiss mouse is presumed to have occurred as a result of bedding transfer. (D) Enlargement of MKPV-infected nuclei. Left and center-left: High (600X) magnification image of RNAscope® for MKPV nucleic acids in a kidney section from a NSG mouse with moderate IBN (from Fig 3F). Viral nucleic acids were highly localized to tubular epithelial cells with inclusion bodies. Bar = 40 µm. Center-right: Schematic, demarking epithelial tubules and their lumens (yellow), disease-affected epithelial cells (pink) and nuclei containing inclusions (red). Right: Nuclear area (µm2) in MKPV RNAscope+ tubular epithelial cells (red, n = 22) compared to RNAscope− nuclei (black, n = 239). ****P < 0.0001 (unpaired t-test).
(A) Immunostaining of kidney sections affected (top) and unaffected (bottom) by inclusion body nephropathy, using pooled sera from co-housed mice as described in Fig. 3G. Antiserum staining shown in red; DAPI staining shown in green. Left scale bar = 100 µm, Right scale bar = 50 µm (B) Quantification of antiserum signals within individual nuclei as shown in (A) and in Fig. 3I. Dots indicate individual nuclei (n = 817–2073 as indicated). Graphs depict the total antiserum signal (co-housed serum, left; control serum, right) within each nucleus (arbitrary units) vs nuclear area (µm2). Only co-housed serum applied to disease-affected kidneys produced signals above 1 unit, predominantly in enlarged nuclei (>50 µm2). (C) Development of anti-MKPV serum in outbred Swiss Webster (SW:Tac) mice at the MSKCC. SW mice (n = 3) were co-housed with NSG mice exhibiting inclusion body nephropathy, and the SW sera were collected and pooled after 3–13 weeks of co-housing. Control serum was obtained from a non-co-housed SW mouse. (D) Immunostaining of kidney sections from MSKCC NSG mice affected (top) and unaffected (bottom) by inclusion body nephropathy, using the sera developed in C. Only serum from co-housed SW mice used on disease-affected NSG kidneys produced strong staining, particularly in enlarged nuclei typical of cells with inclusion bodies. Bar = 50 µm. (E) Immunostaining of a disease-affected tubule for MKPV, using serum from a C57BL/6 mouse co-housed with MKPV+ Cxcr6gfp/gfp Rag1−/− mice, counterstained for cytokeratin 19 and DAPI. MKPV staining shown in green or white as indicated; cytokeratin 19 staining in red; DAPI also in red (rightmost panel). Far right: Percentage of antiserum+ nuclei observed in Krt19− (37%) and Krt19+ (63%) cells (n = 129 cells from 10 fields of view from two independent experiments). Scale bars = 50 µm.
(A,B) Detection of MKPV in facilities outside of Centenary Institute and Memorial Sloan Kettering Cancer Center. (A) Left: Histopathology of a disease-affected kidney from a Balb/c Rag1−/− mouse necropsied in 2006 at Johns Hopkins University School of Medicine, Department of Molecular and Comparative Pathobiology. Right: Detection of MKPV rep DNA by PCR (25 cycles) in formalin-fixed paraffin-embedded kidney tissue from the necropsy. Fixed kidney tissue from Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice (Centenary Institute) were included as negative and positive controls, respectively. (B) Left: Histopathology of a disease-affected kidney from a C3.Cg-Prkdcscid mouse necropsied in 2010 at Cerberus Sciences Laboratory (South Australia). Right: Detection of MKPV rep DNA by 25 cycles PCR in formalin-fixed paraffin-embedded kidney tissues from 5 sentinel Prkdcscid mice diagnosed with nephropathy by Cerberus Sciences Laboratory that were housed in an Australian (non-Centenary Institute) animal facility. Fixed kidney tissue from Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice (Centenary Institute) were included as negative and positive controls, respectively. (C,D) Relative MKPV abundance in different strains of disease-affected mice. (C) Quantitation of viral DNA by qPCR from formalin-fixed paraffin-embedded kidney tissues from the indicated facilities (n = 49 mice from seven facilities). Kidney samples from unaffected control colonies are included for comparison as negative controls (n = 7 mice from four facilities). MSKCC, Memorial Sloan Kettering Cancer Center Laboratory of Comparative Pathology; Cerberus, Cerberus Sciences Laboratory; Centenary, Centenary Institute; JHU, Johns Hopkins University School of Medicine Department of Molecular and Comparative Pathobiology; NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; RAG, Cxcr6gfp/gfp Rag1−/− (Centenary) or Balb/c Rag1−/− (JH) or B6.Cg-Rag1tm1MomTyrp1B-wTg (Tcra,Tcrb)9Rest/M (MSKCC); SCID, C3.Cg-Prkdcscid. ***P = 0.007, unpaired Mann Whitney U test. (D) Comparison of viral burden in MKPV+ (>105 vgc/20ng) samples, stratified according to severity of nephropathy (See Table 1). Mild, immune-competent and nude mice. Severe, NSG, RAG and SCID. ****P < 0.0001, unpaired Mann Whitney U test.
(A–D) Fibrotic changes in MKPV-infected mice. (A) Iodine-enhanced micro-computed tomography reconstructions of normal (left) and disease-affected (right) kidneys. (B) Kidney section from a disease-affected Cxcr6−/− Rag1−/− mouse, stained with Milligan’s trichrome. Bar = 100 µm. (C) Annotated schematic of kidney section in (B). Interstitial fibrosis shown in pale blue. glom. glomerulus. bv, blood vessel. (D) Kidney section from a nephropathy-affected NSG mouse, stained with Masson’s trichrome. White arrows indicate disease-affected tubules. Blue staining indicates collagen (interstitial fibrosis). Bar = 100 µm. (E,F) Urine protein levels in MKPV-infected mice. (E) Chronology of urinary protein changes in Cxcr6gfp/gfp Rag1−/− mice with age (red circles). Protein concentration was determined from untimed urine samples and do not indicate fractional protein excretion. Mice from uninfected C57BL/6 wild-type and Rag1−/− colonies have been included for comparison (black circles). (F) Body weight versus proteinuria, stratified according to sex.
(A) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot. Genes encoding complement and complement-associated proteins are marked in black. Fibrinogen-associated genes are marked in magenta. (B–F) Proteomics of unaffected and inclusion body nephropathy-affected kidneys. (B) Venn diagram depicting the numbers of unique proteins that were detected by MS/MS in normal (n = 3 technical replicates) and IBN-affected (n = 3 technical replicates) kidneys. Colors indicate the numbers of proteins that were only detected in normal kidneys (white), unique to IBN (black) or shared (grey). (C) The most abundant proteins (>0.003% of total spectra) detected by MS/MS in both disease-affected (x-axis) versus normal (y-axis) kidneys. Each dot represents an individual protein. (D) Top 50 most abundant proteins that were only detected in normal kidney. (E) Top 50 differentially detected proteins that were either decreased (left) or increased (right) in diseased kidneys relative to normal. Fold-change is indicated on the x-axis. (F) Top 50 most abundant proteins that were only detected in disease-affected kidneys. MKPV NS1 is shown in red. (G–I) Comparison of transcriptional changes in UUO and MKPV, and changes in macrophage and dendritic cell numbers with MKPV-infection. (G,H) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot, restricted to highly differentially-expressed genes (>log2 1.5-fold) and stratified according to whether expression changes were similarly observed in UUO (G) or unique to MKPV (H). Clec9a and Itgae, encoding the scavenger receptor Clec9A and integrin ĮE (CD103), respectively, are marked in red. (I) Left: Absolute numbers of macrophages (CD45+ CD88+ MHC-II+), cDC1 (CD45+ CD88− MHC-II+ CD103+ CD24hi CD11blo) and cDC2 (CD45+ CD88− MHC-II+ CD103− CD24lo CD11b+) isolated from kidneys of young Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice, and old, disease-affected Cxcr6gfp/gfp Rag1−/− mice. Right: cDC1/cDC2 ratio in indicated mice. Left (pale grey triangles and orange circles) and right (dark grey triangles and red circles) kidneys were assessed individually. n = 4 mice per group. ***P = 0.002 (one-way ANOVA, Holm-Sidak’s multiple comparison). (J) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot. Data have been pre-filtered to enrich for genes encoding secreted products (GO:0005615; extracellular space). Egf and Ltbp2, encoding epidermal growth factor (EGF) and latent TGFβ-binding protein 2 (LTBP2), respectively, are marked in red.
Pathological assessment of individual mice identified with IBN(n = 42) and 6 control animals at MSKCC together with their MKPV status as assessed by PCR of paraffin-embedded sections.
Differentially expressed proteins identified by MS/MS in normal and IBN-affected kidneys (P < 0.05, Fischer’s exact test). Proteins that were only detected in normal kidneys or were unique to IBN have been ordered according to abundance (% of total spectra). Proteins that were observed in both samples have been further subdivided into those that were increased and reduced in affected mice relative to controls. The top 100 differentially-expressed proteins for each group are shown except for the proteins only observed in normal mice, for which only 55 were statistically significant.
Highlights.
Inclusion body nephropathy is caused by mouse kidney parvovirus (MKPV)
MKPV is widely distributed in animal facilities in Australia and North America
MKPV is highly divergent from other mouse parvoviruses
Uncontrolled MKPV infection in immunocompromised mice results in renal failure
Acknowledgments
We thank M. Rizk, J. Qin, A. Cooray, C. Yee, C. M. McGuffog, I. Stevanovski, L. Shaw, R. Kwan and K. Bremner for animal husbandry & genotyping; S. J. Cook, M. Jiao, B. Crossett, J. Chen, A. Tatian, N. Keilar, A. Lay, M. Xiang, D. J. Guo, F. Kao, A. Aspland, S. Allen, K. Jahn, & T. M. Ashhurst for technical assistance; C. Hagan, A. C. Ucero, Ö. Uluçkan, W. B. Baze, P. M. Treuting, D. Imai, S. Miller, C. Goldsmith, P. McKenzie, G. Lyons, D. Vanichkina, D. Tscharke, G. Zheng, F. Sierro, M. Palendira, L. Gordon, R. Jain, R. Barugahare, P. Obeidy, E. Machala, R. Bagnall, C. Stuart, N. Lipman & R.J. Ricart Arbona for useful discussions; and L. Cavanagh & N. Littlejohn for administrative assistance. We also acknowledge technical microscopic assistance of ACMM at University of Sydney, particularly I. Kaplin, N. Gokoolparsadh and M. Foley. We also thank R. Stevenson, M. White and the staff at Cerberus Sciences for their assistance. Supported by the Australian National Health and Medical Research Council (W.W., B.R., P.B., C.J.J., M.D.G., E.C.H., J.J.-L.W, L.L), the Cancer Institute NSW (B.R., J.J.-L.W) and National Cancer Institute Cancer Center Support Grant P30 CA008748 (S.F.S., S.M.). M.J. is supported by National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK107309. E.C.H. is funded by an Australian Research Council Australian Laureate Fellowship (FL170100022).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
B.R. and W.W. are co-inventors on international patent application PCT/AU2018/050505 submitted by the Centenary Institute that is related to the detection and use of MKPV in research and commercial applications.
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing data have been deposited at NCBI’s Gene Expression Omnibus, accessible through GEO Series accession number *6(. The GenBank accession numbers for the MKPV sequences determined in this study are MH670587 (Centenary Institute; complete sequence including ITRs) and MH670588 (MSKCC; near-complete sequence lacking ITRs). The raw and analyzed mass spectrometry proteomics data are accessible at the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the dataset identifier PXD010540.
References
- Arvaniti E, Moulos P, Vakrakou A, Chatziantoniou C, Chadjichristos C, Kavvadas P, Charonis A, and Politis PK (2016). Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci Rep 6, 26235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashhurst TM (2017). tSNEplots v1.1.0 (DOI: http://doi.org/10.5281/zenodo.851736 , repository: http://doi.org/10.5281/zenodo.851736https://github.com/sydneycytometry/tSNEplots, repository: https://github.com/sydneycytometry/tSNEplots : Github).
- Baze WB, Steinbach TJ, Fleetwood ML, Blanchard TW, Barnhart KF, and McArthur MJ (2006). Karyomegaly and intranuclear inclusions in the renal tubules of sentinel ICR mice (mus musculus). Comp Med 56, 435–438. [PubMed] [Google Scholar]
- Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW, Low D, Ruedl C, Riccardi-Castagnoli P, Poidinger M, et al. (2014). High-dimensional analysis of the murine myeloid cell system. Nat Immunol 15, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Becker GJ, and Hewitson TD (2013). Animal models of chronic kidney disease: useful but not perfect. Nephrol Dial Transplant 28, 2432–2438. [DOI] [PubMed] [Google Scholar]
- Besselsen DG, Franklin CL, Livingston RS, and Riley LK (2008). Lurking in the shadows: emerging rodent infectious diseases. ILAR J 49, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayton C (2007). Chapter 25 - Spontaneous Diseases in Commonly Used Mouse Strains A2 - Fox, James G In The Mouse in Biomedical Research (Second Edition), Davisson MT, Quimby FW, Barthold SW, Newcomer CE, and Smith AL, eds. (Burlington: Academic Press; ), pp. 623–717. [Google Scholar]
- Brown KE (2010). The expanding range of parvoviruses which infect humans. Rev Med Virol 20, 231–244. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, and Gabaldon T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier RL, Forbes MS, and Thornhill BA (2009). Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75, 1145–1152. [DOI] [PubMed] [Google Scholar]
- Cook AJL, Oganesian L, Harumal P, Basten A, Brink R, and Jolly CJ (2003). Reduced Switching in SCID B Cells Is Associated with Altered Somatic Mutation of Recombined S Regions. J Immunol 171, 6556–6564. [DOI] [PubMed] [Google Scholar]
- Cotmore SF, Agbandje-McKenna M, Chiorini JA, Mukha DV, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D, et al. (2014). The family Parvoviridae. Arch Virol 159, 1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore SF, and Tattersall P (2013). Parvovirus diversity and DNA damage responses. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djudjaj S, Papasotiriou M, Bulow RD, Wagnerova A, Lindenmeyer MT, Cohen CD, Strnad P, Goumenos DS, Floege J, and Boor P (2016). Keratins are novel markers of renal epithelial cell injury. Kidney Int 89, 792–808. [DOI] [PubMed] [Google Scholar]
- Duffield JS (2014). Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124, 2299–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredricks DN, and Relman DA (1996). Sequence-based identification of microbial pathogens: a reconsideration of Koch’s postulates. Clin Microbiol Rev 9, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassock RJ, Warnock DG, and Delanaye P (2017). The global burden of chronic kidney disease: estimates, variability and pitfalls. Nat Rev Nephrol 13, 104–114. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, and Gascuel O (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Haase M, Bellomo R, Albert C, Vanpoucke G, Thomas G, Laroy W, Verleysen K, Kropf S, Kuppe H, Hetzer R, et al. (2014). The identification of three novel biomarkers of major adverse kidney events. Biomark Med 8, 1207–1217. [DOI] [PubMed] [Google Scholar]
- Han WK, Bailly V, Abichandani R, Thadhani R, and Bonventre JV (2002). Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62, 237–244. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Ingelfinger JR, and Alexander SI (2013). One step closer to “Rejectostix”. N Engl J Med 369, 84–85. [DOI] [PubMed] [Google Scholar]
- Ju W, Nair V, Smith S, Zhu L, Shedden K, Song PX, Mariani LH, Eichinger FH, Berthier CC, Randolph A , et al. (2015). Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 7, 316ra193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassianos AJ, Wang X, Sampangi S, Muczynski K, Healy H, and Wilkinson R (2013). Increased tubulointerstitial recruitment of human CD141(hi) CLEC9A(+) and CD1c(+) myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. Am J Physiol Renal Physiol 305, F1391–1401. [DOI] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F (2012). Trim Galore!
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KE, Wong C, Teng Y, and Spong S (2012). CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 5, S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak P, Kemeny E, and Ivanyi B (2006). Primer: histopathology of polyomavirus-associated nephropathy in renal allografts. Nat Clin Pract Nephrol 2, 631–636. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17. [Google Scholar]
- McInnes E, Bennett M, O’Hara M, Rasmussen L, Fung P, Nicholls P, Slaven M, and Stevenson R (2015). Intranuclear Inclusions in Renal Tubular Epithelium in Immunodeficient Mice Stain with Antibodies for Bovine Papillomavirus Type 1 L1 Protein. Vet Sci 2, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nanthakumar CB, Hatley RJ, Lemma S, Gauldie J, Marshall RP, and Macdonald SJ (2015). Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov 14, 693–720. [DOI] [PubMed] [Google Scholar]
- Nath KA (1992). Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis 20, 1–17. [DOI] [PubMed] [Google Scholar]
- Okello JB, Zurek J, Devault AM, Kuch M, Okwi AL, Sewankambo NK, Bimenya GS, Poinar D, and Poinar HN (2010). Comparison of methods in the recovery of nucleic acids from archival formalin-fixed paraffin-embedded autopsy tissues. Anal Biochem 400, 110–117. [DOI] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM, and Chin FY (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, and Zinkernagel RM (1989). Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561. [DOI] [PubMed] [Google Scholar]
- Ramos E, Drachenberg CB, Wali R, and Hirsch HH (2009). The decade of polyomavirus BK-associated nephropathy: state of affairs. Transplantation 87, 621–630. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, et al. (2013). Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol 14, 564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Santagostino SF, Arbona RJR, Nashat MA, White JR, and Monette S (2017). Pathology of Aging in NOD scid gamma Female Mice. Vet Pathol 54, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schack LM, Buettner M, Wirth A, Neunaber C, Krettek C, Hoffmann A, and Noack S (2016). Expression of CD24 in Human Bone Marrow-Derived Mesenchymal Stromal Cells Is Regulated by TGFbeta3 and Induces a Myofibroblast-Like Genotype. Stem Cells Int 2016, 1319578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Schaefer ML, Kappler JW, Marrack P, and Kedl RM (2001). Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell Immunol 214, 110–122. [DOI] [PubMed] [Google Scholar]
- Schmieder R, and Edwards R (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6, e17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonrich G, Kalinke U, Momburg F, Malissen M, Schmitt-Verhulst AM, Malissen B, Hammerling GJ, and Arnold B (1991). Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extrathymic tolerance induction. Cell 65, 293–304. [DOI] [PubMed] [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, and Birney E (2012). Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza WM, Romeiro MF, Fumagalli MJ, Modha S, de Araujo J, Queiroz LH, Durigon EL, Figueiredo LT, Murcia PR, and Gifford RJ (2017). Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J Gen Virol 98, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuting PM, Clifford CB, Sellers RS, and Brayton CF (2012). Of mice and microflora: considerations for genetically engineered mice. Vet Pathol 49, 44–63. [DOI] [PubMed] [Google Scholar]
- Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, and Littman DR (2000). The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol 165, 3284–3292. [DOI] [PubMed] [Google Scholar]
- Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, Pillebout E, Berger T, Mak TW, Knebelmann B, et al. (2010). Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest 120, 4065–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster AC, Nagler EV, Morton RL, and Masson P (2016). Chronic Kidney Disease. Lancet. [DOI] [PubMed]
- Yang HC, Zuo Y, and Fogo AB (2010). Models of chronic kidney disease. Drug Discov Today Dis Models 7, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, and Birney E (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, and Liu Y (2016). Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat Rev Nephrol 12, 68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Expanded analysis of serum biochemistry in mouse strains affected (Cxcr6gfp/gfp Rag1−/−; black and red) and unaffected (white, n = 10) by renal disease, as shown in Fig. 1E. Each dot represents an individual mouse. Control mice comprise disease-free Rag1−/− and C57BL/6 mice. Cxcr6gfp/gfp Rag1−/− mice were stratified according to age (<200 days, black, n = 20; >200 days, red, n = 10). ALP, alkaline phosphatase. ALT, alanine transaminase. AST, aspartate aminotransferase. BUN, blood urea nitrogen. CO2, carbon dioxide. CK, creatine kinase. (B) Expanded evaluation of hemoglobin and red blood cells in Cxcr6gfp/gfp Rag1−/− mice (black and red) and control Rag1−/− mice (white, n = 3), as shown in Fig. 1F. Each dot represents an individual mouse. Cxcr6gfp/gfp Rag1−/− mice were stratified according to age (<200 days, black, n = 8; >200 days, red, n = 8). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (one-way ANOVA, Holm-Sidak’s multiple comparison). RBC, red blood cells. (C,D) Representative microscopic images of a disease-affected kidney from a moribund Cxcr6gfp/gfp Rag1−/− mouse, stained with hematoxylin and eosin. (C) High magnification image depicting tubular epithelial cell nuclei with chromatin margination and amphophilic intranuclear inclusions (black arrows) and, less frequently, eosinophilic intranuclear inclusions (white arrow head). Bar = 40 µm. (D) Lower magnification demonstrating renal papillary necrosis (black arrow) in the same mouse. Bar = 500 µm.
(A) Top: MKPV mRNA reads from diseased kidneys mapped to the final assembled viral genome for the two disease-affected mice (black line and blue histogram) detailed in Fig. 2. Black and red arrows indicate major splice donor and acceptor sites, respectively. Lollipops indicate consensus polyadenylation signals (AATAAA). (B) Integrative Genomics Viewer (IGV) snapshots of MKPV mRNA reads mapped to the final assembled viral genome. Individual reads spanning presumptive introns are indicated by grey blocks (reads) connected by pale blue lines (introns). Superimposed black and red lines indicate the locations of consensus-conforming splice donor and acceptor sites, respectively. (C) Putative major transcripts encoding (in black) NS1 (non-structural protein 1), hypothetical protein NS2 (non-structural protein 2) and VP1 (viral protein 1), based on sequence analysis, observed splicing, reads map and location of polyadenylation signals.
(A) Gastrointestinal tract (GI) in an MKPV-infected mouse. Hematoxylin and eosin staining of indicated regions of the GI tract from a moribund mouse with kidney disease. No pathological changes were detected besides the kidney pathology. Bar = 200 µm. (B) Clearance of MKPV through embryonic rederivation, Non-detection of viral DNA by conventional PCR (25 cycles) in the serum (top) and urine (bottom) of 277 days old Cxcr6gfp/gfp Rag1−/− mice (blue, n = 3 littermates) following rederivation through embryo transfer. Resultant Cxcr6gfp/gfp Rag1−/−mice did not develop IBN (not shown). Serum and urine from a non-rederived Cxcr6gfp/gfp Rag1−/− mouse (red) is included as a positive control. Fluids from disease-free Rag1−/− and C57BL/6 colonies served as negative controls. (C) PCR detection of MKPV in the urine of an outbred Swiss mouse. Detection of viral DNA by conventional PCR (25 cycles) in the urine (white box) of an outbred sentinel Swiss mouse. This mouse was a sentinel for a rack that housed MKPV-infected Cxcr6gfp/gfp Rag1−/− mice. MKPV transmission to the Swiss mouse is presumed to have occurred as a result of bedding transfer. (D) Enlargement of MKPV-infected nuclei. Left and center-left: High (600X) magnification image of RNAscope® for MKPV nucleic acids in a kidney section from a NSG mouse with moderate IBN (from Fig 3F). Viral nucleic acids were highly localized to tubular epithelial cells with inclusion bodies. Bar = 40 µm. Center-right: Schematic, demarking epithelial tubules and their lumens (yellow), disease-affected epithelial cells (pink) and nuclei containing inclusions (red). Right: Nuclear area (µm2) in MKPV RNAscope+ tubular epithelial cells (red, n = 22) compared to RNAscope− nuclei (black, n = 239). ****P < 0.0001 (unpaired t-test).
(A) Immunostaining of kidney sections affected (top) and unaffected (bottom) by inclusion body nephropathy, using pooled sera from co-housed mice as described in Fig. 3G. Antiserum staining shown in red; DAPI staining shown in green. Left scale bar = 100 µm, Right scale bar = 50 µm (B) Quantification of antiserum signals within individual nuclei as shown in (A) and in Fig. 3I. Dots indicate individual nuclei (n = 817–2073 as indicated). Graphs depict the total antiserum signal (co-housed serum, left; control serum, right) within each nucleus (arbitrary units) vs nuclear area (µm2). Only co-housed serum applied to disease-affected kidneys produced signals above 1 unit, predominantly in enlarged nuclei (>50 µm2). (C) Development of anti-MKPV serum in outbred Swiss Webster (SW:Tac) mice at the MSKCC. SW mice (n = 3) were co-housed with NSG mice exhibiting inclusion body nephropathy, and the SW sera were collected and pooled after 3–13 weeks of co-housing. Control serum was obtained from a non-co-housed SW mouse. (D) Immunostaining of kidney sections from MSKCC NSG mice affected (top) and unaffected (bottom) by inclusion body nephropathy, using the sera developed in C. Only serum from co-housed SW mice used on disease-affected NSG kidneys produced strong staining, particularly in enlarged nuclei typical of cells with inclusion bodies. Bar = 50 µm. (E) Immunostaining of a disease-affected tubule for MKPV, using serum from a C57BL/6 mouse co-housed with MKPV+ Cxcr6gfp/gfp Rag1−/− mice, counterstained for cytokeratin 19 and DAPI. MKPV staining shown in green or white as indicated; cytokeratin 19 staining in red; DAPI also in red (rightmost panel). Far right: Percentage of antiserum+ nuclei observed in Krt19− (37%) and Krt19+ (63%) cells (n = 129 cells from 10 fields of view from two independent experiments). Scale bars = 50 µm.
(A,B) Detection of MKPV in facilities outside of Centenary Institute and Memorial Sloan Kettering Cancer Center. (A) Left: Histopathology of a disease-affected kidney from a Balb/c Rag1−/− mouse necropsied in 2006 at Johns Hopkins University School of Medicine, Department of Molecular and Comparative Pathobiology. Right: Detection of MKPV rep DNA by PCR (25 cycles) in formalin-fixed paraffin-embedded kidney tissue from the necropsy. Fixed kidney tissue from Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice (Centenary Institute) were included as negative and positive controls, respectively. (B) Left: Histopathology of a disease-affected kidney from a C3.Cg-Prkdcscid mouse necropsied in 2010 at Cerberus Sciences Laboratory (South Australia). Right: Detection of MKPV rep DNA by 25 cycles PCR in formalin-fixed paraffin-embedded kidney tissues from 5 sentinel Prkdcscid mice diagnosed with nephropathy by Cerberus Sciences Laboratory that were housed in an Australian (non-Centenary Institute) animal facility. Fixed kidney tissue from Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice (Centenary Institute) were included as negative and positive controls, respectively. (C,D) Relative MKPV abundance in different strains of disease-affected mice. (C) Quantitation of viral DNA by qPCR from formalin-fixed paraffin-embedded kidney tissues from the indicated facilities (n = 49 mice from seven facilities). Kidney samples from unaffected control colonies are included for comparison as negative controls (n = 7 mice from four facilities). MSKCC, Memorial Sloan Kettering Cancer Center Laboratory of Comparative Pathology; Cerberus, Cerberus Sciences Laboratory; Centenary, Centenary Institute; JHU, Johns Hopkins University School of Medicine Department of Molecular and Comparative Pathobiology; NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; RAG, Cxcr6gfp/gfp Rag1−/− (Centenary) or Balb/c Rag1−/− (JH) or B6.Cg-Rag1tm1MomTyrp1B-wTg (Tcra,Tcrb)9Rest/M (MSKCC); SCID, C3.Cg-Prkdcscid. ***P = 0.007, unpaired Mann Whitney U test. (D) Comparison of viral burden in MKPV+ (>105 vgc/20ng) samples, stratified according to severity of nephropathy (See Table 1). Mild, immune-competent and nude mice. Severe, NSG, RAG and SCID. ****P < 0.0001, unpaired Mann Whitney U test.
(A–D) Fibrotic changes in MKPV-infected mice. (A) Iodine-enhanced micro-computed tomography reconstructions of normal (left) and disease-affected (right) kidneys. (B) Kidney section from a disease-affected Cxcr6−/− Rag1−/− mouse, stained with Milligan’s trichrome. Bar = 100 µm. (C) Annotated schematic of kidney section in (B). Interstitial fibrosis shown in pale blue. glom. glomerulus. bv, blood vessel. (D) Kidney section from a nephropathy-affected NSG mouse, stained with Masson’s trichrome. White arrows indicate disease-affected tubules. Blue staining indicates collagen (interstitial fibrosis). Bar = 100 µm. (E,F) Urine protein levels in MKPV-infected mice. (E) Chronology of urinary protein changes in Cxcr6gfp/gfp Rag1−/− mice with age (red circles). Protein concentration was determined from untimed urine samples and do not indicate fractional protein excretion. Mice from uninfected C57BL/6 wild-type and Rag1−/− colonies have been included for comparison (black circles). (F) Body weight versus proteinuria, stratified according to sex.
(A) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot. Genes encoding complement and complement-associated proteins are marked in black. Fibrinogen-associated genes are marked in magenta. (B–F) Proteomics of unaffected and inclusion body nephropathy-affected kidneys. (B) Venn diagram depicting the numbers of unique proteins that were detected by MS/MS in normal (n = 3 technical replicates) and IBN-affected (n = 3 technical replicates) kidneys. Colors indicate the numbers of proteins that were only detected in normal kidneys (white), unique to IBN (black) or shared (grey). (C) The most abundant proteins (>0.003% of total spectra) detected by MS/MS in both disease-affected (x-axis) versus normal (y-axis) kidneys. Each dot represents an individual protein. (D) Top 50 most abundant proteins that were only detected in normal kidney. (E) Top 50 differentially detected proteins that were either decreased (left) or increased (right) in diseased kidneys relative to normal. Fold-change is indicated on the x-axis. (F) Top 50 most abundant proteins that were only detected in disease-affected kidneys. MKPV NS1 is shown in red. (G–I) Comparison of transcriptional changes in UUO and MKPV, and changes in macrophage and dendritic cell numbers with MKPV-infection. (G,H) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot, restricted to highly differentially-expressed genes (>log2 1.5-fold) and stratified according to whether expression changes were similarly observed in UUO (G) or unique to MKPV (H). Clec9a and Itgae, encoding the scavenger receptor Clec9A and integrin ĮE (CD103), respectively, are marked in red. (I) Left: Absolute numbers of macrophages (CD45+ CD88+ MHC-II+), cDC1 (CD45+ CD88− MHC-II+ CD103+ CD24hi CD11blo) and cDC2 (CD45+ CD88− MHC-II+ CD103− CD24lo CD11b+) isolated from kidneys of young Rag1−/− and Cxcr6gfp/gfp Rag1−/− mice, and old, disease-affected Cxcr6gfp/gfp Rag1−/− mice. Right: cDC1/cDC2 ratio in indicated mice. Left (pale grey triangles and orange circles) and right (dark grey triangles and red circles) kidneys were assessed individually. n = 4 mice per group. ***P = 0.002 (one-way ANOVA, Holm-Sidak’s multiple comparison). (J) Comparison of gene expression in wild-type kidneys (n = 2 replicates) versus that in MKPV-infected kidneys (n = 2 replicates) by volcano plot. Data have been pre-filtered to enrich for genes encoding secreted products (GO:0005615; extracellular space). Egf and Ltbp2, encoding epidermal growth factor (EGF) and latent TGFβ-binding protein 2 (LTBP2), respectively, are marked in red.
Pathological assessment of individual mice identified with IBN(n = 42) and 6 control animals at MSKCC together with their MKPV status as assessed by PCR of paraffin-embedded sections.
Differentially expressed proteins identified by MS/MS in normal and IBN-affected kidneys (P < 0.05, Fischer’s exact test). Proteins that were only detected in normal kidneys or were unique to IBN have been ordered according to abundance (% of total spectra). Proteins that were observed in both samples have been further subdivided into those that were increased and reduced in affected mice relative to controls. The top 100 differentially-expressed proteins for each group are shown except for the proteins only observed in normal mice, for which only 55 were statistically significant.