

Abstract
RNA structures play a pivotal role in many biological processes and the progression of human disease, making them an attractive target for therapeutic development. Often RNA structures operate through the formation of complexes with RNA-binding proteins, however, much like protein-protein interactions, RNA-protein interactions span large surface areas and often lack traditional druggable properties, making it challenging to target them with small molecules. Peptides provide much greater surface areas and therefore greater potential for forming specific and high affinity interactions with RNA. In this chapter, we discuss our approach for engineering peptides that bind to structured RNAs by highlighting methods and design strategies from previous successful projects aimed at inhibiting the HIV Tat-TAR interaction and the biogenesis of oncogenic microRNAs.
Section 1: Introduction
RNA molecules play crucial roles in regulating healthy and diseased cellular processes (e.g. transcription, splicing, mRNA transport, translation, etc) and are therefore tightly regulated transcriptionally and post-transcriptionally, often through their interactions with other RNAs and with RNA binding proteins. RNAs and RNA-protein interactions regulate viral replication or the expression of proto-oncogenes, and are mis-regulated in many infectious and chronic diseases (Cooper, Wan, & Dreyfuss, 2009; Esteller, 2011) making these RNA structures and RNA-protein surfaces an untapped source of potential drug targets (Burnett, & Rossi, 2012; Ling, Fabbri, & Calin, 2013). However, RNA-protein interactions are much more challenging to target with small molecules than traditional enzymatic active sites (Warner, Hajdin, & Weeks, 2018). These interactions span large surface areas and often lack structural complexity (Jones, Daley, Luscombe, Berman, & Thornton, 2001; Lunde, Moore, & Varani, 2007). Therefore, it is more challenging, though not impossible (Afshar et al, 1999; Bower et al, 2003; Murchie et al, 2004; Davis et al, 2004; Howe et al, 2015; Palacino et al, 2015; Ratni et al, 2016), to discover small molecules that compete with much larger proteins and stabilize the often dynamic single stranded regions of the RNA. Intermediate molecular weight (1.5–2 kDa) peptides can provide much greater surface area and therefore have greater potential to form high affinity and specific complexes (Puglisi, Chen, Blanchard, & Frankel, 1995; Battiste et al, 1996). Thus, our group has used peptides to discover RNA-binding ligands for probing structural and mechanistic aspects of RNA-protein interactions and investigating possible new RNA inhibitors.
Here, we discuss our approach to engineering peptides that bind RNAs by highlighting methods and design strategies (Fig. 1). In Section 2, we describe limitations of targeting RNA with linear peptides (Leulliot, & Varani, 2001) and how conformationally constrained peptide mimetics address some of these issues (Robinson, 2008). Section 3 discusses the design process for building mimetics from protein structure and sequence, based on our successful targeting of the interaction between viral trans-activator of transcription (Tat) and trans-activating response element (TAR) (Athanassiou et al., 2004; Leeper, Athanassiou, Dias, Robinson, & Varani, 2005). Section 4 describes the use of positional scanning libraries to discover a high affinity peptide capable of binding human immunodeficiency virus TAR RNA (Athanassiou et al, 2007; Davidson et al., 2009). Section 5 is dedicated to explaining how structure-based optimization can lead to the discovery of peptides with low pM affinity and exquisite specificity (Davidson, Patora-Komisarska, Robinson, & Varani, 2011; Shortridge et al, 2018). In Section 6, we evaluate how β-turn mimetics can be adapted to target other pharmaceutically relevant RNA stem-loop structures by two cases as examples (Moehle et al, 2007; Shortridge et al, 2017).
Figure 1.

Schematic depicting the three design stages described in Sections 3–5 that led to the discovery of very potent RNA-targeting macrocyclic peptides. From left to right: Bovine immunodeficiency virus trans-activation response (BIV-TAR) is a structured cis-regulatory element in the BIV 5’ UTR mRNA that is recognized by BIV Tat to stimulate viral transcription (PDB ID: 1MNB; Puglisi, Chen, Blanchard, & Frankel, 1995); Section 3 overviews the process of using peptidomimetics to target the BIV-TAR—Tat interaction by grafting the wildtype Tat sequence onto a β-hairpin scaffold. This strategy led to the discovery of peptide BIV-2 and its structure determination by NMR (PDB ID: 2A9X; Leeper, Athanassiou, Dias, Robinson, & Varani, 2005); Section 4 describes how a large positional scanning peptide library was organized to generate structure activity relationship (SAR), from which a BIV-Tat mimetic was adapted to target human immunodeficiency virus TAR (HIV-TAR) (PDB ID: 2KDQ; Davidson et al., 2009); Section 5 describes the structure-based optimization process and peptide design strategies that incorporated non-canonical amino acid substitution (e.g. backbone extension, side-chain truncation) to achieve low picomolar binding, JB-181 (PDB ID: 6D2U; Shortridge et al, 2018).
Section 2: Targeting RNA with Macrocyclic β-Hairpin Peptide Mimetics
2.1. Challenges in targeting RNA with Peptides
Engineering peptides to bind RNA requires an understanding of several fundamental challenges inherent to targeting RNA. First, RNA structures are not abundant in the PDB. Crystallization can be difficult and sometimes impossible because of RNA’s intrinsic flexibility (Leulliot, & Varani, 2001; Williamson, 2000). Our group uses NMR spectroscopy to circumnavigate this issue as it provides characterization of RNA and peptide conformation, even when structures are flexible (Varani, Aboula-ela, & Allain, 1996). Structure determination of peptide-RNA complexes produce atomic resolution accuracy and provides critical insight regarding key peptide-RNA interactions, yet only becomes accessible once a potent enough peptide is identified. Furthermore, overlapped NMR spectra reduce resolution, making unambiguous chemical shift assignment impossible for weakly bound peptides discovered early on in the design process. Second, binding interfaces are sometimes so large that it can be difficult to target them with peptides, particularly for larger multimeric RNA-protein complexes. Third, for any RNA binding ligand, it is difficult to differentiate using only biochemical assays (Kd) how stable the RNA-ligand complex actually is. Even when a molecule is bound, conformational flexibility might remain, or the molecule might bind in multiple locations on the RNA, boosting affinity through avidity and giving the illusion of potency. It is often the case that the resulting affinity results from non-specific interactions from electrostatic interactions between the phosphate backbone or electronegative major groove with basic side chains of peptides, or charged small molecules. Maintaining a single RNA-ligand conformation is also inherently energetically unfavorable because of the entropic penalty associated with rigidification of flexible RNA molecules by induced fit (Leulliot, & Varani, 2001; Williamson, 2000). The penalty is magnified when RNA is targeted with linear peptides with poor conformational stability, since short peptide fragments lack the tertiary interactions that buttress protein binding sites in folded proteins. This is the reason we prefer to use highly structured peptide mimics with stable secondary structure in our targeting efforts.
2.2. Macrocyclic β-Hairpin Scaffolds as an Effective Strategy for Targeting RNA
Structure-based peptidomimetics is a peptide design strategy where inspection of a protein-bound structure provides a rational to design a minimal peptide that mimics the activity of the full protein (Pelay-Gimeno, Glas, Koch, & Grossmann, 2015; Mason, 2010; Avan, Hall, & Katritzky, 2014). The protein structure provides a starting point for the design of structurally rigid peptides that recapitulate intermolecular interactions within a minimal framework and inhibit biological activity (Werder, Hauser, Abele, & Seebach, 1999; Rosenström et al, 2006; Whitby et al, 2011; Muppidi et al, 2012). It is crucial for the mimetic to match both sequence and structure, and this is done most effectively when using secondary structure motifs that stabilize a peptide fold through local intramolecular interactions while allowing incorporation of the desired sequence into small peptide fragments (Fujii et al, 2003; Pelay-Gimeno, Glas, Koch, & Grossmann, 2015). The two most common protein secondary structures are β-hairpins and α-helices, whose backbones act as scaffolds upon which energetically favorably sidechains can be pre-presented for binding in position precisely defined by intramolecular hydrogen bonding and packing interactions. β-hairpins are of particular interest for targeting RNA as many RNA-binding proteins (RBP’s) exploit β-sheet structures (Cléry, Blatter, & Allain, 2008; Sibanda, & Thornton, 1985).
Our group has targeted RNA using macrocyclic β-hairpin peptidomimetic chemistry. Head-to-tail cyclization rigidifies the structure (Pelay-Gimeno, Glas, Koch, & Grossmann, 2015), and cyclized peptides are advantageous from a pharmacological standpoint as well because they are exonuclease resistant (Robinson, 2000) and have increased cellular uptake when arginine-rich (Lättig-Tünnemann et al, 2011). Their structure is comprised of two anti-parallel β-strands stabilized by two β-hairpin favoring turn inducers located at the tip of both strands. β-hairpins strongly favor Type I’ (φi+1 = 60, ψi+1 = 30, φi+2 = 90, ψi+2 = 0) and Type II’ (φi+1 = 60, ψi+1 = −120, φi+2 = −80, ψi+2 = 0) turn inducing residues (Madan, Seo, & Lee, 2014). Most of our published work uses 14-mer β-hairpins with a Type II’ D-Pro/L-Pro turn inducer as it readily favors β-hairpin folding (Fig. 2A)(Nair, Vijayan, Venkatachalapathi, & Balaram, 1979; Bean, Kopple, & Peishoff, 1992). The D-Pro/L-Pro turn developed in the Robinson group acts as a template for grafting mimicked sequences onto the β-hairpin scaffold (Robinson, 2008), favoring formation of stable β-hairpins. The second turn varies in sequence depending on the protein being mimicked with the only requirement that it favors Type I’ or Type II’ (Robinson, 2008).
Figure 2.

(A) Generic 2D structure of a 12-mer β-hairpin macrocycle with a D-Pro/L-Pro template. (B) Representation of a 12-mer β-hairpin macrocycle with sidechains grouped by orientation relative to the backbone plane. Positions 2, 4, 9, 11 point ‘up’ (green); while positions 1, 3, 5, 8, 10, 12 point ‘down’ (blue). (C) β-hairpin scaffolds match the shape and size of the RNA major groove and mold the RNA structure. From left to right: (PDB ID: 1ANR; Aboul-ela, Karn, & Varani, 1996), (PDB ID: 6D2U; Shortridge et al, 2018).
If properly designed, cyclic β-hairpins will possess a rigid backbone structure, stabilized by multiple NH to CO backbone hydrogen bonds. A rigid β-hairpin provides a reliable scaffold for mimicking a protein and makes amino acid substitution less likely to affect the β-hairpin character. A 14-mer cyclic beta-hairpin favors a 2:2 sidechain orientation (Sibanda, Blundell, & Thornton, 1989) presenting 6 residues in one direction (for the purpose of this chapter, positions: 1, 3, 5, 8, 10, 12) and 4 residues in the opposite direction (positions: 2, 4, 9, 11) (Fig. 2B). Considering this positioning is essential during optimization and highlighted in Section 4. This class of cyclic β-hairpins also has size and shape to match major groove RNA features found at apical loop and/or bulge junctions (Fig. 2C), allowing sidechains to make stabilizing contacts with RNA bases and the phosphate backbone. The combination of major groove fit and optimizable intermolecular interactions gives the β-hairpin a higher probability of inducing RNA conformation changes that favor formation of both canonical and non-canonical base-pairs, even when these are not present in the unbound state of the RNA (Bardaro et al, 2009, Borkar et al, 2016). Formation of these base-pairs is influential in developing high affinity RNA-targeted peptides.
A detailed description of synthetic methods used to prepare these peptides can be found in previous publications (Athanassiou et al, 2004; Athanassiou et al, 2007). Briefly, cyclic β-hairpin peptides can be generated following solid-state peptide synthesis followed by a standard cyclization procedure, using Fmoc chemistry on acid sensitive 2-chlorotrityl resin in order to retain protecting groups during resin cleavage and HoBt/HBTU head-to-tail cyclization. HPLC purification is necessary after cyclization and before side-chain deprotection in order to separate cyclized product from unreacted linear peptide. It should be noted that due to the highly constrained nature of β-hairpins, some sequences have unexpected poor yields, likely due to unfavorable reaction geometries. In addition, the cyclization reaction is sensitive to sequences that produce flexible backbones (e.g. incorporation of glycine, backbone kinks) which may inhibit the cyclization reaction and/or require longer reaction times.
Section 3: Peptidomimetic Targeting the BIV-TAR–Tat Interaction
Mimicking the TAR–Tat interaction of the Bovine Immunodeficiency Virus (BIV) provides an illustration of how to mimic an RBP by grafting amino acids onto a cyclic β-hairpin peptidomimetic scaffold (Athanassiou et al., 2004; Leeper, Athanassiou, Dias, Robinson, & Varani, 2005), and in turn shows the feasibility of using β-hairpin scaffolds to target RNA.
3.1. Design Rational
Regulatory RNAs in viral RNA genomes provide attractive targets for discovery of new antiviral drugs because these elements are essential for viral replication, structured, and highly conserved between isolates (Gallego, & Varani, 2001; Wilson, & Li, 2000; Kirk et al, 1998). A well-studied example is provided by the Tat protein, which plays a critical role lentiviral life cycle and contributes significantly to HIV pathogenesis by increasing transcription of the integrated pro-virus by recognition of TAR RNA, a structured cis-regulatory element in viral 5’ UTR mRNA (Gonda, Luther, Fong, & Tobin, 1994; Jones, & Peterlin, 1994; Wei, Garber, Fang, Fischer, & Jones, 1998). The TAR-Tat interaction is essential for viral replication and driven by TAR recognition of the highly basic Tat Arginine Rich Motif (ARM), which, in the BIV protein (though not in the human counterpart) forms a β-hairpin that binds to BIV TAR within the major groove at the apical loop-stem junction (Ye, Kumar, & Patel, 1995; Puglisi, Chen, Blanchard, & Frankel, 1995). Single nucleotide or amino acid substitutions within either the protein or the RNA severely affect transcriptional levels and hence viral replication (Fischer, Huber, Boelens, Mattaj, & Luhrmann, 1995). BIV and HIV-TAR have similar structure and function, but at the time structural information was limited to the BIV TAR–Tat complex, to which we directed our mimicking efforts.
The starting point for this project was the structure of a Tat 14-mer linear peptide (residues 68-RPRGTRGKGRRIRR-81) bound to the BIV TAR stem-loop (residues 4–31) (Fig. 3A) (Puglisi, Chen, Blanchard, & Frankel, 1995). In the structure, TAR grasps the Tat peptide in a glove-like manner within a widened major groove pocket, sandwiching it between the apical loop and thumb-like bulge (Fig. 3B). The 14-mer peptide bound to the RNA forms a β-hairpin that is kinked due to flexible Gly-71, pushing Thr-72 to bulge upwards towards G22 within the BIV-TAR apical loop. The β-hairpin is favored by a Gly-Lys-Gly γ-turn facing downward and away from the apical loop. Significant contact points include: hydrophobic contacts between the methyl group of Thr-72 and the ribose ring of G22; phosphate H-bonding and π-stacking interactions between Arg-70/G14 and Arg-77/C8 and G9; a triple base pair induced by Arg-73 positioned between G9 and U10 and stabilized by hydrophobic packing of Ile-79 neighboring U11 (Fig. 3C). Together, sandwiching of three arginines (Arg-77, Arg-73, Arg-70) between the neighboring bulge nucleotides (C8, G9, U10, G11) are critical elements for BIV-TAR recognition forming arginine sandwich motifs (ASM) (Puglisi, Chen, Blanchard, & Frankel, 1995).
Figure 3.

(A) Secondary structure of BIV-TAR. (B) BIV Tat arginine-rich motif (cyan, 68–81) binds to within the BIV-TAR (grey) major groove at the stem-bulge and stem-apical loop interface (PDB ID: 1MNB; Puglisi, Chen, Blanchard, & Frankel, 1995). (C) Arg-77, Arg-73, and Arg-70 are critical recognition elements for BIV-TAR binding by sandwiching between nucleotides C8, G9, U10, G11 forming arginine sandwich motifs (ASM). (D) BIV-0 cyclization was designed by eliminating R68, P69, and R81 and grafting a D-Pro/L-Pro template (Athanassiou et al., 2004).
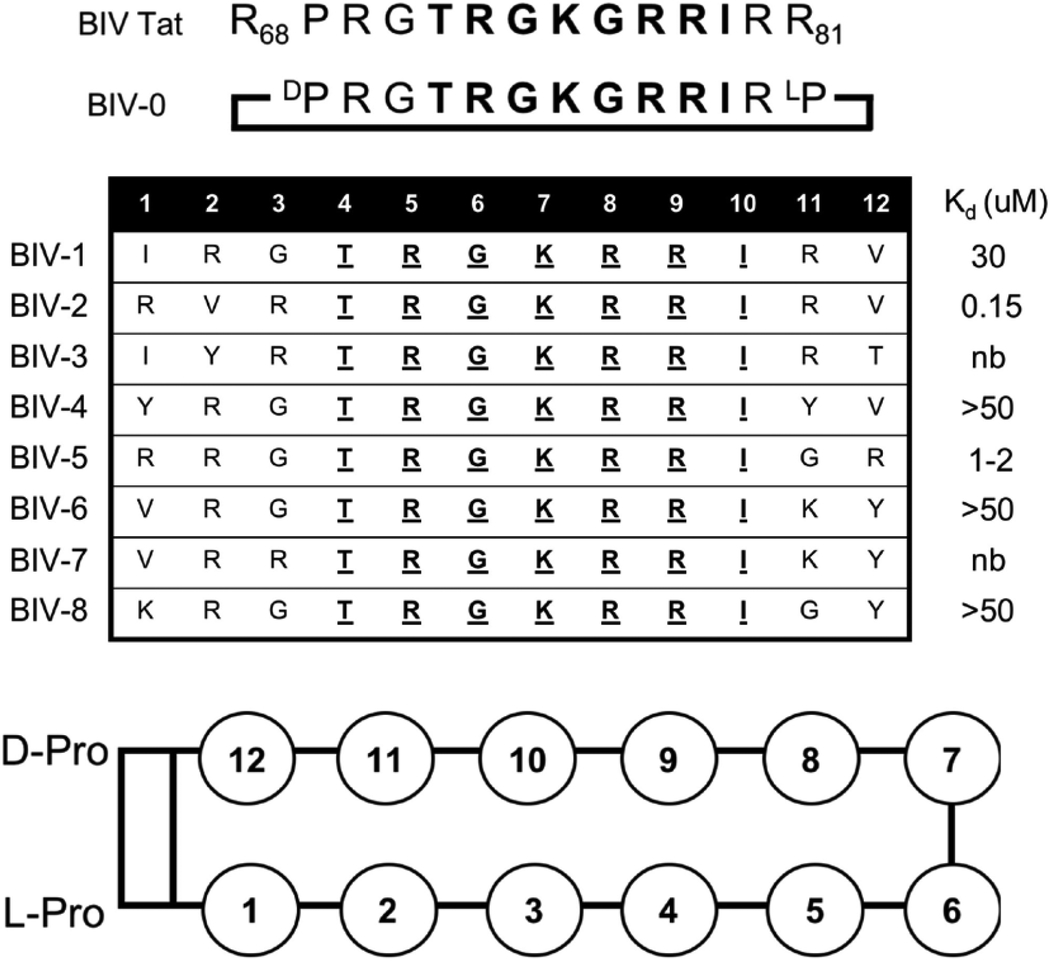
Since BIV Tat forms a β-hairpin when bound to BIV TAR, design of the first mimetic (BIV-0) was straightforward. Mutagenesis data shows that residues critical for binding (Arg-70, Gly-71, Thr-72, Arg-73, Gly-74, Arg-77, and Ile-79) are all located close to the reverse turn formed by residues Gly-74, Lys-75, and Gly-76 (Chen, & Frankel, 1995). Therefore, the peptide was cyclized by replacing Arg-68, Pro-69, and Arg-81 with the D-Pro/L-Pro Type II’ turn template (Fig. 3D). However, this mutagenesis and sequence-based approach to graft the wildtype Tat peptide fragment onto a beta-hairpin scaffold was unsuccessful, since BIV-0, containing an almost identical sequence to wild-type Tat showed very poor, probably non-specific binding (Kd ~ 40uM) (Athanassiou et al., 2004).
This result prompted us to re-consider the importance of peptide stability for β-hairpin design. When we investigated by NMR the structure of BIV-0, we observed essentially a random coil conformation (Athanassiou et al., 2004). In retrospect, a regular 2:2 β-hairpin conformation (Sibanda, Blundell, & Thornton, 1989) requires a particular number of residues on each strand for ideal hydrogen bonding (Griffiths-Jones, Maynard, & Searle, 1999). Turn chemistry may have also destabilized the peptide since the rigid turn-inducing template D-Pro/L-Pro near Gly-71 can induce an irregular backbone conformation. We therefore redesigned the peptide mimic by using 12-residue peptides to promote 2:2 β-hairpin formation and mounting the sequence on a D-Pro/L-Pro template (Favre, Moehle, Jiang, Pfeiffer, & Robinson, 1999; Jiang, Moehle, Dhanapal, Obrecht, & Robinson, 2000). Eight peptides were synthesized (BIV-1–BIV-8, Table 1) with residues Thr-4, Arg-5, Gly-6, Lys-7, Arg-8, Arg-9, and Ile-10 kept constant, corresponding to residues Thr-72, Arg-73, Gly-74, Lys-75, Arg-77, Arg-78, and Ile-79 in BIV Tat, which make critical interactions with BIV-TAR (Athanassiou et al., 2004). The sequence near the apical loop was trimmed by changing positions 1–3, 11, and 12 (positions 68–70, 80, and 81 in wildtype Tat) to residues with charged and aromatic sidechains. Since β-hairpins prefer type I’ and type II’ chemistry, Gly-76 from the γ-turn was also removed to favor a Type II’ turn of Gly-74 and Lys-75. Although all peptides are basic and bind with low uM affinity, only of the eight mimetics (BIV-2) bound with nM affinity (Kd = 150nM) (Athanassiou et al., 2004). BIV-2 is arginine-rich and lacks a mid-strand glycine between turns, further supporting our hypothesis that formation of a stable and regular β-hairpin is essential for peptides to bind RNA.
Table 1.
Sequences of the BIV-Tat ARM (68–81), peptide mimic BIV-0, and β-hairpin mimetics BIV-1 – BIV-8. Reported Kds were measured by electromobility shift assays (EMSA) (nb = no binding) (Athanassiou et al, 2004). Position numbering based on the schematic representation shown below.
|
Next, we investigated which BIV-2 structural features made it bind BIV-TAR strongly by evaluating each peptide structure by NMR. BIV-0, for example had random coil characteristics, but NMR data for BIV-2 showed it had a single set of peaks (Athanassiou et al., 2004). Structures were determined for BIV-2 and BIV-3, and both adopted β-hairpin structures, whereas the other mimetics adopted multiple conformations, yet BIV-2 had a lower RMSD compared to BIV-3, suggesting BIV-2 was more rigid and that peptide stability contributed to binding (Athanassiou et al., 2004).
3.2. Structure and SAR Development
The structure of HIV-TAR bound to BIV-2 was determined using NMR (Leeper, Athanassiou, Dias, Robinson, & Varani, 2005). Inspection of the BIV-TAR–BIV-2 complex reveals that BIV-2 is bound in the same major groove pocket as BIV Tat, but in a completely different orientation, being upside down relative to what was expected from the targeted structure with the D-Pro/L-Pro template positioned downward into the major groove and the Gly-6/Lys-7 turn upwards into the apical loop (Fig. 4A). Yet, despite the flipped orientation, many sidechain interactions with the RNA are similar to BIV Tat. Arg-1, Arg-3, and Arg-5 are sandwiched between bases forming ASMs mimicking BIV Tat sidechain positioning for Arg-70, Arg-73, and Arg-77, respectively. Intercalation occurs between C8, G9, U10, G11, and G14 through a combination of cation-π interactions and hydrogen bonding with purine N7’s and guanosine O2’s (Fig. 4B). Arg-8 is also buried relative to the β-hairpin, yet positioned near flexible apical loop nucleotides and only making transient contacts with multiple apical loop nucleotides. Mimicry is nearly exact between the two structures for Ile-10 and Ile-79, both are positioned deep within the base stack. Ile-10 stabilizes the U10-A13-U24 base triple through van der Waal interactions with A13 and U24 and is a major driving force for TAR recognition (Fig. 4C, D). Solvent exposed Arg-9 and Arg-11 make no hydrogen bonds or cation-π interactions, yet contacts with the phosphate backbone of A21 and G22 favor the apical loop to fold over the peptide (Fig. 4E). Val-2 and Thr-4 point away from the RNA and into the solvent, making little contact with the RNA.
Figure 4.

(A) BIV-2 (green) bound to BIV-TAR (grey) upside down relative to wildtype BIV-Tat. (PDB ID: 2A9X; Leeper, Athanassiou, Dias, Robinson, & Varani, 2005) (B) ASM formed by Arg-1, Arg-3 and Arg-5 intercalating between C8, G9, U10, and G11. (C&D) The base triple induced by BIV-2 mimics that induced by BIV Tat (cyan) (PDB ID: 1MNB; Puglisi, Chen, Blanchard, & Frankel, 1995); Arg-5, Arg-3, Ile-10 mimic Arg-70, Arg-73, Ile-79, respectively. (E) Solvent exposed Arg-9 and Arg-11 make electrostatic interactions with backbone phosphates at A21 and G22.
In summary, the structure revealed that, once a stable β-hairpin conformation is achieved, residue changes that increase or decrease affinity with the RNA are likely to arise from changes in side-chain to RNA interactions. We identified key stabilizing interactions, which contributed to future efforts to characterize the different binding potentials for each position (see Section 4).
Section 4: Discovery of Peptide Mimetics Targeting HIV-TAR
The long-term focus of targeting BIV-TAR project discussed in Section 3 was to discover peptide inhibitors targeting Human Immunodeficiency Virus (HIV) TAR, located at positions 17–45 within the 5’-UTR of the HIV genome (Fig. 5A). Its trinucleotide bulge (U23, C24, U25) nucleotides are functionally analogous to BIV TAR U10, G11, and U12 in that they are necessary for Tat recognition and have been shown to reduce replication rates in mutational studies (Keen, Gait, & Karn, 1996; Dingwall et al, 1990; Roy, Delling, & Rosen, 1990). We used the SAR developed for the BIV-TAR–BIV-2 complex to generate a large positional scanning peptide library, synthesized both to optimize binding affinity, but also to discover new HIV-TAR specific peptides (Athanassiou et al, 2007). Our aim of discovering a peptide which bound to HIV-TAR strongly enough for structure determination was successful (Davidson et al., 2009), which was used for developing an HIV-TAR specific SAR utilized in future rounds of optimization.
Figure 5.

(A) Secondary structure of HIV-TAR. (B) Cartoon depicting SAR for BIV-2 binding to BIV-TAR. Orange dotted lines indicate hydrogen bonding and/or π-stacking interactions, grey dotted indicate electrostatic interactions (Athanassiou et al, 2007).
4.1. Discovery of low nM peptide ligands for HIV-TAR Using a Large Positional Scanning Peptide Library
The structure and initial SAR for the BIV-2 peptide led to the following summary of its requirement for high affinity binding:
Positions 1, 3, 5, 8 are buried against the RNA and exhibit ASM sandwiching with C8, G9, G11, U10, and G14. Arginines are likely favored at these positions because of their multiple hydrogen bond acceptors and charged protonation state. Hydrogen bonding was observed between purine N7’s and guanosine O2’s, along with cation-π and π-π interactions with various bases. Arg-5 contributes significantly to triple base-pair formation based on its location in the binding pocket.
Positions 6 and 7 are non-template turn inducing residues required to be Type I’ and Type II’ turn inducers, since these turns favor β-hairpin formation.
Positions 2 and 4 are solvent exposed and make negligible interactions with the RNA and minor contacts with neighboring side-chains.
Positions 9 and 11 are solvent exposed but make interactions with A21 and G22 that support the RNA conformation through induced fit
Positions 10 and 12 are deeply buried into the RNA interface and form hydrophobic interactions. Ile-10 contributes significantly to triple base-pair formation by packing against A13 and U24.
These interactions are represented on a schematic of a β-hairpin (Fig. 5B) and provide a design rational for the positional scanning peptide library (L-1 to L-86) that led to the discovery of HIV-TAR binding peptides L-22, L-50 and L-51 (Athanassiou et al, 2007). Since optimization was carried out for both BIV and HIV-TAR, patterns discussed are explained as observations for both RNAs unless otherwise specified.
4.1.1. Ala scanning
In a pre-panel screen, each position except the D-Pro/L-Pro template was changed to alanine to identify which residues contributed to binding. Remarkably, every alanine substitution reduced binding significantly, with the exception of solvent exposed Val-2 and Thr-4 where Kds only increased 2-fold relative to BIV-2 (Athanassiou et al, 2007). Changing the buried Ile-10 and Val-12 to alanine also led to low uM binding, likely because of a reduced number of hydrophobic interactions near the base triple. Decreased binding affinity for each residue was also proposed to be correlated to the loss of cross-strand packing with arginine methylene chains. This observation invited us to consider peptide intramolecular stability through side-chain to side-chain interactions when making design decisions.
4.1.2. ASM-forming residues: Arg-1, Arg-3, Arg-5, Arg-8
Arg-1, Arg-3, Arg-5 essentially mimic BIV Tat Arg-77, Arg-73 and Arg-70, while Arg-8 interacts with both A21 associated base and phosphate in the apical loop. Arginines at positions 1,3,5, and 8 were substituted with both charged (Lys, L-ornithine Orn); polar (Asn, Gln, L-citrulline Cit); and aromatic (Tyr, Trp) residues. Arginines are less likely to make non-specific phosphate salt bridges than lysine and its analogue ornithine (Rohs et al, 2010). Therefore, when affinity is retained by mutating an arginine to lysine, the interaction is potentially non-specific. Positions Arg-1, Arg-3, Arg-5 all show no binding when replaced with Lys, but substitution of position 8 to lysine and ornithine only increased its Kd to 2.5uM suggesting some degree of non-specific electrostatic backbone interaction (Athanassiou et al, 2007).
4.1.3. Non-template Beta-Hairpin Turn Inducers: Gly-6, Lys-7
The D-Pro/L-Pro template and Gly-6, Lys-7 form Type II’ turns. Their identity is important (Chen, Lin, Lee, Jan, & Chan, 2001); BIV-0 bound poorly likely due to conformational instability from dis-favorable turn chemistry (Athanassiou et al, 2004). This hypothesis was tested with L-22 and L-51 by switching Gly-6 and Lys-7 to residues favoring a Type I’ turn. This resulted in a significant increase in affinity and selectivity for HIV-TAR over BIV-TAR, and equally important, produced cleaner NMR data for structure determination even when compared to peptides with higher binding affinities (Athanassiou et al, 2007). Lys-7 was also changed to polar, aromatic, and charged residues to improve binding, yet lysine appeared to be the preferred amino acid here. How turn sequences can be used to modulate specificity to other RNAs is an ongoing subject of investigation.
4.1.4. Solvent exposed Val-2, Thr-4
Solvent exposed residues were expected to make negligible contributions to binding. Changing Val-2 and Thr-4 to hydrophobic (Thr, Leu); polar (Asn, Gln); and aromatic (Tyr, Trp) produced mixes results. Val-2 to Thr and Thr-4 to Gln, Val, Tyr increased binding by 2–3-fold (as ordered by largest to smallest increase in affinity) (Athanassiou et al, 2007). Increase in affinity was speculated to be due to changes in peptide conformational stability or lower solvation free energy, but careful analysis of these unbound peptides would be needed to confirm this conjecture.
4.1.5. Apical loop binding residues Arg-9, Arg-11
Residues that might make contacts with the apical loop because of their location on the peptide structure are difficult to optimize. Structures tend to converge poorly in these regions due to the inherent apical loop flexibly and non-specific interactions with the phosphate backbone might drive any energetic contribution. Arg-9 and Arg-11 were substituted, similarly to Arg-1, Arg-3, Arg-5, and Arg-8, by changing arginine to lysine. For BIV-TAR, lysine and ornithine substitutions retained binding (Kd = 150nM) (Athanassiou et al, 2007). When the same peptides were assayed against HIV-TAR, changes at position 9 retained binding, yet changes at position 11 led to 2–3-fold loss in binding affinity suggesting a greater and unexpected dependence on arginine at position 11.
4.1.6. Major Groove Buried Hydrophobic Residues: Ile-10 and Val-12
Inspection of the BIV-2 bound structure shows both of these hydrophobic residues are buried deep within the major groove and contribute a significant faction of the buried interfacial surface. There residues were altered to optimize hydrophobic packing, and for Ile-10, increased stabilization for the triple base pair. Both were substituted to hydrophobic (Leu, cyclohexylalanine); aromatic (Phe, Tyr); and polar (Asn, Gln). Cyclohexylalanine was used as a control due to its hydrophobicity, yet sterically unfavorable size resulting in low uM affinity at each position. All hydrophobic changes retained or increased Kd 2–3-fold, likely due to their increased size leading to a looser fit of the peptide in the major groove (Athanassiou et al, 2007). Val-12 to Ile-12 increased binding affinity (Kd = 100nM), however, and was incorporated in conjunction with changes at other positions (e.g. in L-22). Surprisingly, Ile-10 could be changed to different hydrophobic residues for BIV-TAR without significant changes in binding, yet for HIV-TAR substitutions to other residue led to loss of binding, revealing that isoleucine is optimal at this position for HIV-TAR recognition.
4.1.7. Combining changes at Multiple Positions
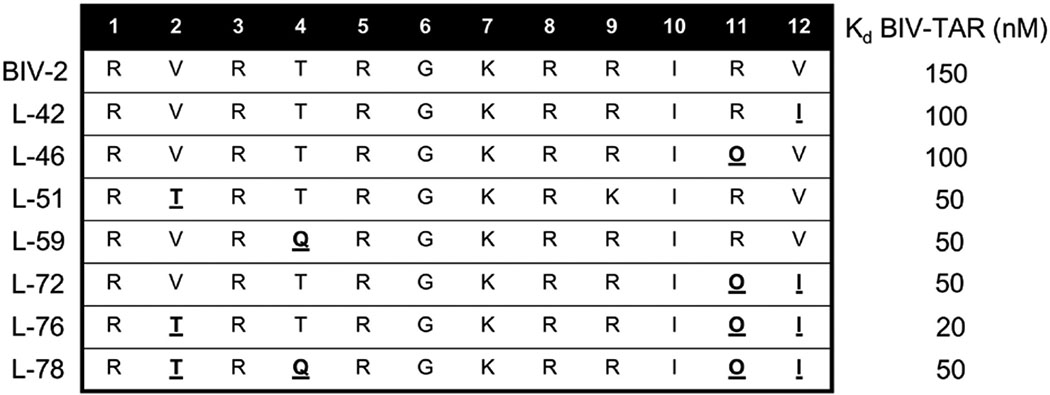
Residue changes that resulted in increased binding affinity were introduced in combination aiming for additive effects. Table 2 provides examples of peptides with several single residue changes (L-42, L-46, L-51, L-59) with improved binding affinity against BIV-TAR. L-72 was designed to incorporate both modifications at position 11 (Arg-to-Orn, Kd = 100nM) and 12 (Val-to-Ile, Kd = 100nM), which together increased binding 2-fold (Kd = 50nM). L-76 incorporated Thr at position 2 for L-72, decreasing the Kd further (Kd = 20nM) (Athanassiou et al, 2007). Not all changes were additive, however, since L-78 incorporated a glutamate into position 4 and reduced binding affinity 2.5-fold (Kd = 50nM). L-76 and L-78 together demonstrates incorporation of multiple positional changes for additivities effects requires careful step-wise design as well, since the effects are not simply additive.
Table 2.
Combination of sequences and Kds of peptides from the L-1 to L-86 peptide where single residue changes improved binding affinity relative to BIV-2 (L-42, L-46, L-51, L-59). Single residue changes were incorporated together as peptides with multiple position changes (L-72, L-76, L-78) (Athanassiou et al, 2007).
|
4.2. HIV-TAR–L-22 Structure and SAR
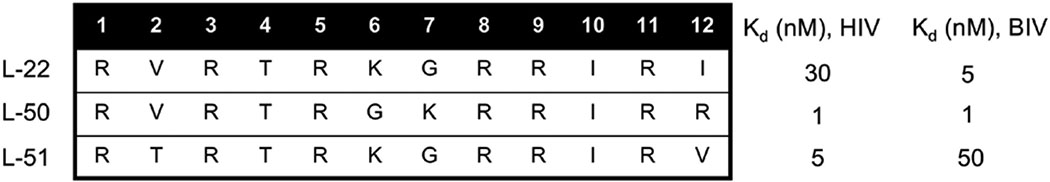
Peptides L-22, L-50 and L-51 were discovered as the highest-affinity peptides targeting HIV-TAR (Davidson et al., 2009) (Table 3). L-51 also demonstrated 10-fold specificity, with binding affinities of 5 nM for HIV-TAR and 50 nM for BIV-TAR, but competition assays showed that L-22 inhibited a 37-mer Tat-derived linear peptide better, since its IC50 was only 2–3-fold above its Kd (Davidson et al., 2009). This promising inhibition data, combined with cleaner NMR data compared to L-50 and L-51, prompted us to determine the structure of HIV-TAR bound to L-22, even if L-50 binds most strongly and L-51 is specific. It is commonly the case that the most potent or highest affinity ligand is chosen as the optimal lead, but we strongly believe that the optimal starting structure is that which provides for structure determination, because of more facile optimization. Not only because of the obvious advantage of the availability of a structure, but also because the quality of the NMR data indicates that the peptide binds in a unique rigid orientation, which is not the case for L-50 and L-51.
Table 3.
Sequences and Kds for peptides with highest-affinity targeting HIV-TAR from the L-1 to L-86 library (Davidson et al., 2009).
|
The structure of the HIV-TAR–L-22 complex was determined using a very high-quality NMR dataset (Davidson et al., 2009). Positioning of L-22 is similar to BIV-2 on BIV TAR, with the D-Pro/L-Pro template facing down into the major groove and a base triple forming between U23, A27, and U38 (Fig. 6A). However, L-22 bound is buried even deeper into the major groove than BIV-2 into BIV TAR, sequestering ~50% of the peptide surface from solvent, including most of its hydrophobic side chains. L-22 SAR with HIV-TAR is as follows: residues 1, 3, 5, 8 make hydrogen bond and/or stacking interactions with the RNA; 10 and 12 make hydrophobic interactions; positions 9 and 11 bind through transient electrostatic interactions near the backbone of G34 and A35 (Fig. 6B). Arg-1 and Arg-3 stabilized the stem-bulge interface, with Arg-1 contacting the RNA backbone and making specific hydrogen bonds with G21 N7 and A22 N7 and Arg-3 with A22 (Fig. 6C). Both side chains are proximal to the phosphate backbone making electrostatic interactions as well. As for the BIV Tat and BIV-2 structures, Arg-5 and Ile-10 stabilize the HIV-TAR base triple, with Arg-5 stacking over the three nucleotides. Very high-quality NMR data provided reliable identification of hydrogen bonding between the Arg-5 guanidium group and G28 N7 and O2 (Fig. 6D). Ile-10 is packed deep into the groove and makes hydrophobic interactions with the H5 and H6 side of U23 (Fig. 6E). A hypothesis for why the KG turn was preferred over the GK turn in L-50 was a newly formed electrostatic interaction between Lys-6 and the phosphate backbone of G28. This long-distance interaction with the RNA was considered in future design and optimized in the later generation peptide JB-181 (Section 5.2).
Figure 6.

(A) L-22 (magenta) bound HIV-TAR (grey). (PDB ID: 2KDQ; Davidson et al, 2009) (B) SAR for L-22 binding to HIV-TAR. Orange dotted lines indicate hydrogen bonding and/or π-stacking interactions, green dotted lines indicate hydrophobic interactions, grey dotted indicate electrostatic interactions. (C) Hydrogen bonds between Arg-1 with G21 N7 and Arg-3 with A22 N7 stabilize the stem-bulge interface. (D) Hydrogen bonds between R5 and U23 and G28 induce the U23-A27-U38 base triple. (E) Ile-10 contributes to base triple stabilization by hydrophobic interactions with the H5 and H6 side of U23.
Section 5: Structure-Based Optimization of L-22 Binding Affinity and Specificity
Further improvements in affinity and specificity were achieved by generating reliable SAR and the structure of the complex as a basis for design hypothesis. In this section, we will dissect these strategies by discussing both a blind alley (Davidson, Patora-Komisarska, Robinson, & Varani, 2011) and success (Shortridge, et al., 2018).
5.1. KP-Z-41: Extending Cyclic β-Hairpin Peptides to 15, 16, and 18-mers
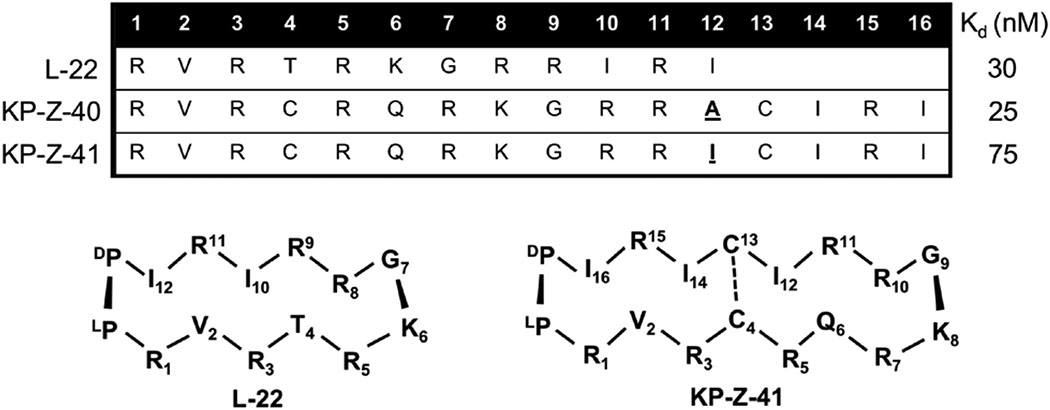
Synthesis of the KP-Z peptide series sought to improve specificity and affinity by extending the peptide to fully engage the apical loop. Inspection of the 14-mer L-22 bound to HIV-TAR shows that several apical loop nucleotides make no interactions with it. Peptide libraries based on the L-22 sequence were designed to take advantage of these available interaction sites by extending the peptides to 15, 16 and 18 amino acids (Davidson, Patora-Komisarska, Robinson, & Varani, 2011). Essential structural requirements for specific recognition of HIV-TAR were observed based on the structure of KP-Z-41 bound to TAR, which revealed an unexpected position of the peptide distinct from our original design.
We synthesized a medium sized cyclic β-hairpin peptide library (~25–50) of 15, 16, and 18 residues in length (Davidson, Patora-Komisarska, Robinson, & Varani, 2011). The L-22 sequence was conserved in these peptides, yet new amino acids were mainly based on our previously established understanding of buried and solvent exposed residues and model-based predictions of new possible contacts in the apical loop by extension of the peptide. Cross-strand disulfide linkages were also introduced to test whether increased rigidity from additional covalent linkage would enhance binding. 15 and 16 amino acid β-hairpins showed poor binding in all cases, perhaps not unexpectedly since these peptides should be unstably folded, whereas 18 amino acid β-hairpins fold similarly to the 14-mers, following the n+4 periodicity for 2:2 beta-hairpins (Griffiths-Jones, Maynard, & Searle, 1999). Binding affinity was enhanced for some 18-mer β-hairpins, improving binding relative to L-22, likely due to an additional arginine positioned on the major groove side of the peptide (positions 1, 3, 5, 7). KP-Z-40 displayed the highest binding affinity for HIV-TAR with a Kd in the pM range without excess tRNA present, and a 25 nM Kd even in the presence of 10,000-fold excess of tRNA (Davidson, Patora-Komisarska, Robinson, & Varani, 2011). However, quality of NMR data for bound KP-Z-40 was poor. We hypothesized that Ala-12 may be occupying the triple base pair pocket normally occupied by Ile-10. KP-Z-41 was therefore designed to have this position changed to an isoleucine and we observed an unexpected 3-fold drop in affinity, yet spectral quality improved allowing for structure determination to be executed (Davidson, Patora-Komisarska, Robinson, & Varani, 2011).
The structure for KP-Z-41 bound to HIV-TAR shows that the peptide is positioned completely opposite to our expectations and farther down into the major groove with the D-Pro/L-Pro template sticking out of the complex (Fig. 7). This sliding might be due to the requirement to form arginine-mediated interaction at or near the UGU bulge. Combined with the upside down BIV-2 mimicry of BIV Tat, this result exemplifies how deviations from a scaffold that might seem conservative, can induce drastic changes in binding position and orientation.
Figure 7.

Overlaid HIV-TAR-bound structures (grey) for L-22 (magenta) and KP-Z-41 (pale green) (PDB ID: 2KX5, Davidson, Patora-Komisarska, Robinson, & Varani, 2011). HIV-TAR backbone phosphates were used as anchor points for the superposition.
5.2. JB-181: Non-Canonical Amino Acids
Peptides L-22, L-50 and L-51 bound to RNA potently, with low nM Kds, but discriminated effectively only against structurally unrelated RNAs, such as tRNA present in all binding assays to reduce non-specific binding (Davidson, A., 2009). When comparing closely-related stem-loop structures such as HIV-TAR mutants or BIV-TAR, binding affinity only decreased by a few to at most 10-folds (Davidson, A., 2009). A new generation of peptides was rationally designed by inspection of the structure of L-22. Since the KP-Z series indicated that extension of the peptide to make additional specific contacts with the apical loop would not be straightforward or possible, new peptides were designed with the goal of allowing deeper penetration into the major groove by incorporating truncated, non-canonical side chains. Using this approach, JB-181 was discovered, a cyclic β-hairpin mimetic of Tat with low picomolar affinity for HIV-TAR and remarkable specificity.
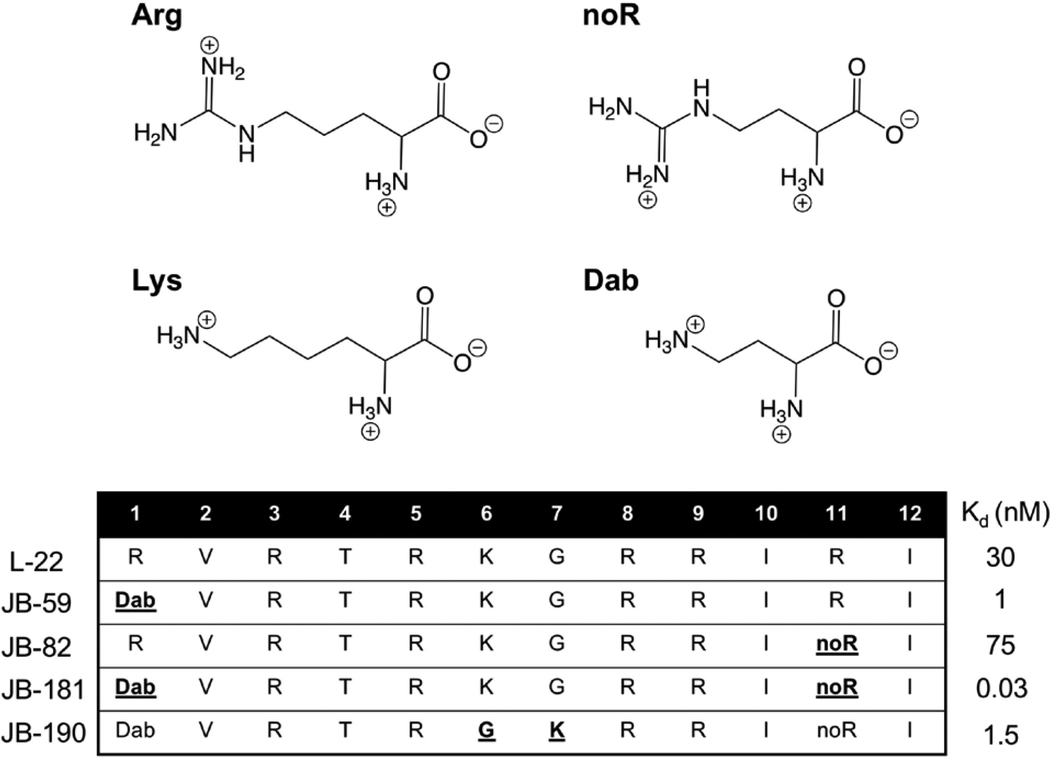
A new library of ~200 peptides was synthesized based on the L-22 sequence, with single residue changes comprising natural L-amino acids, mixing chirality and non-canonical side chains (Table 5, top panel) (Shortridge et al, 2018). The inclusion of L-2,4-diaminobutryric (Dab) at position 1 (JB-59), and L-2-amino-4-guanidinobutyric acid (noR) (JB-82) at position 11 led to the greatest improvement in binding. When both changes were combined into a single peptide, JB-181, it produced pM binding (Kd = 28±4 pM, as measured by a fluorescence assay) and NMR data of unprecedented quality (Table 5) (Shortridge et al, 2018). Although the original β-hairpin mimetics were designed using the BIV structure as a model (Puglisi, Chen, Blanchard, & Frankel, 1995; Athanassiou et al, 2004), binding of JB-181 to BIV-TAR occurs at low nM affinity (Kd = 4 nM) and low uM binding affinity is observed towards the non-coding RNA 7SK-SL4 (Shortridge et al, 2018) that also binds to HIV-Tat, demonstrating that JB-181 has 102–106 fold discrimination against closely related structures.
Table 5.
(top) Most effective non-canonical amino acids used in the JB peptide series. (bottom) Sequences and Kds for peptides with the highest HIV-TAR affinities from the JB library (Shortridge et al, 2018).
|
The quality of the NMR data revealed new interactions responsible for the large increase in binding affinity. JB-181 binds in the same major groove pocket and orientation as the previous Tat-mimetics (Fig. 8A) (Shortridge et al, 2018). The backbone ψ and φ angles for JB-181 and L-22 were nearly identical with the exception of Lys-6, yet their positioning in the TAR binding pocket are different (Fig. 8B). The introduction of truncated side chains (Dab-1, noR-11) essentially allows the D-Pro/L-Pro template and neighboring residues to fit deeper into the major groove pocket. Inspection of the structure shows that substitution to an amine at positioning one maintains a salt bridge with the RNA backbone (2.4Å from O1P of G21 and 3.6Å from O1P of A22) (Fig. 8C), similar to Arg-1, but truncation of the side chain pulls the peptide closer to the major groove by about 2.4Å and allows the KG turn to pitch upwards with respect to the double helical axis. NoR-11 reduces affinity more than 2-fold relative to L-22 (JB-81, Kd = 75 pM), yet we detected more intermolecular NOEs from position noR-11 to A35, which now occupies the syn conformation allowing formation of a more favorable cation-π interaction with the new sidechain (Fig. 8D). Changes at positions 1 and 11 affected other parts of the structure as well. Deeper burial of the peptide favors improved Arg-3 and Arg-5 sandwiching between bases, increasing polar contacts and closer packing of Ile-10 against the base triple. Ile-10 is stabilized by many new NMR observables never observed by us in >20 years of investigation of multiple complexes of TAR such as a 2’-OH in the base triple protected from exchange with solvent. One of the most notable changes is a new hydrogen bond formed between the amine of Lys-6 and the O4 of U25 (Fig. 8E). This novel interaction is supported by mutational analysis such as swapping of the turn from KG to GK in JB-190 (Table 5), or mutating U25 to C25, which both decrease binding from low pM to low nM (Shortridge et al, 2018). Altogether, the remarkable structural and binding properties of JB-181 demonstrate that high affinity and specific RNA recognition of relatively simple RNA secondary structures, hairpin loops, can be achieved using a structure guided approach and macrocyclic β-hairpin scaffolds.
Figure 8.

(A) JB-181 (wheat) bound to HIV-TAR (grey) (PDB ID: 6D2U; Shortridge et al, 2018). (B) Overlaid of HIV-TAR-bound structures (grey) for L-22 (magenta) (PDB ID: 2KDQ; Davidson, 2009) and JB-181 (wheat). HIV-TAR backbone phosphates were used as anchor points for the superposition. (C) Dab-1 forms salt bridges with the G21 and A22 phosphates. (D) noR-11 makes a cation-π interaction with A35 favoring a syn conformation for the A35 base. (E) Change in peptide position favors formation of a novel hydrogen bond between Lys-6 and U25 O4.
Section 6: Targeting other RNAs Using Macrocyclic β-Hairpin Peptides
In this section, we show how the same class of peptides can be used to target RNAs other than TAR, namely the Rev-RRE (Rev Response Element) interaction in HIV (Moehle et al, 2007) and pre-microRNA-21 (pre-miR-21) the precursor to the potent oncomir miRNA-21 (miR-21) (Shortridge et al, 2017). These additional studies demonstrate the versatility of cyclic β-hairpin scaffolds for targeting RNA.
6.1. Targeting the Rev-RRE Interaction
The virally encoded Rev protein plays a critical role in viral replication by regulating the transport of unspliced and partially spliced viral RNA from the nucleus to the cytoplasm of infected cells (Emerman, & Peden, 1989; Malim et al, 1989). Rev functions by binding through an α-helical ARM to stem loop IIb (Fig. 9A) of the RRE a structured ~300 mRNA segment located within the Env coding region of HIV-1 (Mann et al, 1994). Following this high affinity binding event, approximately 10 additional Rev molecules oligomerize through protein–protein and protein–RNA interactions and coat the entire RRE (Mann et al, 1994). Because the Rev-RRE interaction is analogous to the TAR-Tat interaction in that it involves a high-affinity RNA binding protein that binds by a major groove recognizing ARM. We elected to target it with our BIV-TAR mimetics even if this interaction is distinct from the TAR-Tat interactions in that Rev binds the RRE major groove in an α-helical conformation (Fig. 9B) (Battiste et al, 1996). Similar to previous studies where β-hairpin scaffolds were used to mimic an α-helices and block HDM2 binding to p53 (Fasan et al, 2004), we reasoned our mimetics could be adapted to position sidechains in similar orientations as in the Rev α-helix. Indeed, the β-hairpins can be modeled to show that arginines can be positioned on the peptide to moderately mimic the HIV Rev α-helical ARM (Fig. 9C).
Figure 9.

(A) Secondary structure of stem loop IIb of the Rev Response Element (RRE). (*) indicated next to residues changed for increased T7 transcription efficiency. (B) α-helical Rev ARM (33–55, light blue) bound to the RRE major groove (light cyan) (PDB ID: 1ETF; Battiste et al, 1996). (C) A standard 2:2 β-hairpin mimetic (residues 1–12 with a D-Pro/L-Pro template) (green) superimposed onto the Rev residues critical for RRE binding demonstrating how β-hairpin scaffolds can be adapted to mimic similar α-helix sidechain positioning.
We screened the small peptide library develop for BIV TAR (Table 1) for binding to the RRE. BIV-5, which had only a low uM affinity for HIV-TAR (Kd = 1−2uM) bound to the RRE much more strongly (Kd = 0.3uM) (Moehle et al, 2007). Interestingly, BIV-7 which showed no measurable affinity for TAR, bound with ~1uM affinity as well, further suggesting that specificity can be found in arginine rich peptides if the basic side chains are presented on a rigid scaffold like a β-hairpin. BIV-5 showed effective inhibition potential in a competition-based assay as well, by displacing the Rev peptide with IC50 similar to its Kd (Moehle et al, 2007). Similar optimization as for BIV Tat-TAR was carried out using a medium-size library, which led to the discovery of R-27, a low nM RRE binding peptide (Kd = 2nM) that was 50-fold more selective for RRE compared to HIV-TAR (Kd = 100nM) (Moehle et al, 2007). This project demonstrated in a proof of principle manner that molecular interactions observed on the Rev α-helix could be successfully transplanted onto a β-hairpin scaffold by rational design, leading to rapid discovery of cyclic peptides that are more potent than the Rev ARM in binding to the RRE high affinity site.
6.2. Pre-microRNA 21
Lastly, we used this chemistry to develop peptides targeting the potent oncogene miR-21. MicroRNAs (miRNA) are short non-coding RNAs that silence gene translation through sequence specific binding in most cases at complementary 3’ UTR loci inducing mRNA degradation or translational suppression (Carthew, & Sontheimer, 2009). Their dysregulation is also often observed in human disease (Esteller, 2011; Ling, Fabbri, & Calin, 2013). The biogenesis of miRNAs begins with expression of a transcript sometimes thousands of nucleotides long called the pri-microRNA (pri-miRNA). The primary transcript undergoes two exonucleolytic cleavage reactions, first in the nucleus by Drosha-DGCR8 to generate the pre-miRNA of 55–75 nucleotides, and second in the cytoplasm by Dicer-TRBP to form the functional mature miRNA of 21–24 nucleotides (Carthew, & Sontheimer, 2009). We focused on miR-21 which is upregulated in many human cancers (Ribas, & Lupold, 2010; Kjaer-Frifeldt et al, 2010; Yan et al, 2008; Asangani et al, 2008; Meng et al, 2007) and sought to discover a peptide inhibitor of miR-21 maturation by screening our cyclic peptide libraries to find an initial scaffold to subsequently construct pre-miR-21 specific cyclic β-hairpins.
Targeting pre-miR-21 was unlike previous projects because of the lack of structural information and because pre-miR-21, our target, has a large apical loop, that NMR data indicate is largely unstructured and flexible (Fig. 10A). The Dicer cleavage sites are located above the A29 bulge and at the i+1 cross-strand phosphate backbone, between G45 and C46. Therefore, we focused our screen on this apical section of the structure to avoid discovering peptides that bind to other sections of this RNA. Thus, we truncated the full length pre-miR-21 sequence by removing the bottom half of the helix and used this RNA in both NMR experiments and EMSAs when screening our peptide library (Shortridge et al, 2017). Although EMSAs are cumbersome, they allow direct visualization of complexes and distinguish multimeric and non-specific complexes. We prioritized monomeric hits from screening rather than just affinity because of much easier follow-up to optimize affinity and specificity.
Figure 10.

(A) Secondary structure of pre-miRNA-21 (pre-miR-21; numbers based on miRBASE v21 (Kozomara, & Griffiths-Jones, 2014), the Dicer cleavage sites are indicated by red arrows to generate the ~22 nucleotide mature miR-21 (blue). (B) Structure of pre-miR-21-L50 complex with nucleotides that make intermolecular NOEs highlighted in green (PDB ID: 5UZZ; Shortridge et al, 2017). (C) Both Ile-10 and Arg-12 contact the RNA near one of the Dicer cleavage sites.
From the initial screening, L-50 was identified to bind to pre-miR-21 with nanomolar binding affinity even in the presence excess tRNA (Kd = 200nM) (Shortridge et al, 2017), the only peptide to bind with sub-uM affinity. Follow-up studies revealed that L-50 does not bind the A29 deletion mutant, suggesting L-50 bound near the Dicer cleavage site and it out-competed streptomycin (Kd = 100nM) (Bose et al, 2012). These results collectively provided enough evidence to warrant structure determination.
Despite poor definition at this early stage of discovery, the structure revealed critical sites of contacts and contributed an initial SAR. Inspection of the lowest energy structure reveals that L-50 binds in the opposite orientation compared to TAR binding predecessors, in that the D-Pro/L-Pro template is facing upward into the apical loop (Fig. 10B), and charged residues appear to make non-specific contacts with the phosphate backbone (Shortridge et al, 2017). Interestingly, L-50 is the only peptide in our screening library with an arginine at position 12, located near the phosphate backbone of G28, suggesting Arg-12 is necessary for pre-miR-21 binding; both Ile-10 and Arg-12 contact the RNA near the Dicer cleavage site (Fig. 10C). When we tested for activity in an in vitro dicer assay using in-house purified Dicer-TRBP, we observed anomalous Dicer products; cellular assays showed modest but specific inhibition of miR-21 production in HEK293 cell (Shortridge et al, 2017). These preliminary results motivated efforts currently ongoing to optimize L-50 for better pre-miR selectivity, and to incorporate new turn chemistry that deviates from the D-Pro-L-Pro template. Current unpublished results have identified peptides with new turn chemistry and sequences that bind much more strongly to pre-miR-21 compared to TAR, inhibit Dicer processing in vitro, and reduce miR-21 levels in cells with greatly improved potency compared to the original hit.
Section 7: Summary
Peptides provide an appealing solution to inhibiting large intermolecular interfaces, such as protein-protein or protein-RNA interactions, that are less amenable to small molecule inhibitors than traditional enzyme targets. While less pharmaceutically attractive than small molecules, they represent a growing area of pharmacology (Pelay-Gimeno, Glas, Koch, & Grossmann, 2015; Fosgerau, & Hoffmann, 2015; Teixido, & Giralt, 2017) that can sometimes lead to bioactive molecules and even, in favorable cases, oral bioavailability (Renukuntla, Dutt, Patel, Boddu, & Mitra, 2013). Past efforts to use peptides to target RNA focused on peptoids (Hamy et al, 1997) or other backbone modification (Niu, Jones, Wu, Varani, & Cai, 2011) invariably led to highly flexible linear structure from which specific binding activity was very difficult to generate, or very short fragments whose binding activity was too weak to observe any biochemical or cellular effects. We show here that cyclic β-hairpin mimics can yield very potent (pM Kd) binding with strong specificity, and that these can be rapidly identified from a combination of conventional library-generated SAR and structure-based design. Success can be found either by mimicking the known interfaces of RNA-binding protein, or by an agnostic process of library screening to identify imperfect hits that can then be rapidly optimized, due to the relative ease of peptide chemistry compared to small molecules. Conformationally restrained peptides are particularly important for RNA, which often have flexible structure. For this reason, peptide folding and binding must be established. Critical information on the characteristics of the intermolecular interface that transcend and extend binding constants can be obtained by NMR. Namely, whether the complex is formed at a unique rigid binding site on the RNA, as opposed to sliding on the structure as observed, for example, with aminoglycosides like streptomycin (Bose et al, 2012; Maiti, Nauwelaerts, & Herdewijn, 2012; Shortridge, & Varani, 2015). Inspection of the structure of the complex, which can be generated by NMR even for initial relatively low affinity hits if the peptide is rigid, reveals how the peptide is positioned and identifies the location of key sidechains identified through library-based SAR.
While peptides do not generally match small molecules with regards to in vivo application, they are nevertheless more conductive to optimization of pharmacological properties than oligonucleotide analogues (Juliano, 2016; Wang et al, 2015; Stein et al, 2011). Combined with in vitro evolution, our methods rapidly generate leads that can be optimized to generate peptides with improved pharmacological potential through the introduction of side chain and backbone modifications, leading even, in favorable cases, to oral bio-availability (Witt et al, 2001; Adessi, Soto; 2002; Chatterjee, Gilon, Hoffman, & Kessler, 2008; Boehm et al, 2017). We consider this family of peptides to be very useful alternatives to small molecules in discovering RNA targeting bioactive structures and provide herein protocols for their discovery, characterization and optimization of their binding activity to RNA.
Table 4.
Sequences and Kds for peptides with highest-affinity to HIV-TAR from the KP-Z library (Davidson, Patora-Komisarska, Robinson, & Varani, 2011).
|
Section 8: References
- 1.Aboul-ela F, Karn J, Varani G (1996). Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res., 24(20), 3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adessi C, & Soto C (2002). Converting a peptide into a drug: strategies to improve stability and bioavailability. Current medicinal chemistry, 9(9), 963–978. [DOI] [PubMed] [Google Scholar]
- 3.Afshar M, Prescott CD, Varani G. (1999). Structure-based and combinatorial search for new RNA-binding drugs. Curr Opin Biotechnol., 10(1), 59–63. [DOI] [PubMed] [Google Scholar]
- 4.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H (2008). MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene, 27, 2128–2136. [DOI] [PubMed] [Google Scholar]
- 5.Athanassiou Z, Dias R, Moehle K, Dobson N, Varani G, & Robinson J (2004). Structural Mimicry of Retroviral Tat Proteins by Constrained Beta-Hairpin Peptidomimetics: Ligands with High Affinity and Selectivity for Viral TAR RNA Regulatory Elements. JACS Articles, 126, 6906–6913. [DOI] [PubMed] [Google Scholar]
- 6.Athanassiou Z, Patora |K, Dias RL, Moehle K, Robinson JA, & Varani G (2007). Structure-Guided Peptidomimetic Design Leads to Nanomolar Beta-Hairpin Inhibitors of the Tat-TAR Interaction of Bovine Immunodeficiency Virus. Biochemistry, 46, 741–751. [DOI] [PubMed] [Google Scholar]
- 7.Avan I, Hall CD, Katritzky AR (2014). Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev, 43, 3575–3594. [DOI] [PubMed] [Google Scholar]
- 8.Bardaro MF Jr, Shajani Z, Patora-Komisarska K, Robinson JA, & Varani G (2009). How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic acids research, 37(5), 1529–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR (1996). Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science, 273, 1547–1551. [DOI] [PubMed] [Google Scholar]
- 10.Bean JW, Kopple KD, Peishoff CE (1992). Conformational analysis of cyclic hexapeptides containing the D-Pro-L-Pro sequence to fix β-turn positions. J. Am. Chem. Soc, 114, 5328–5334. [Google Scholar]
- 11.Boehm M, Beaumont K, Jones R, Kalgutkar AS, Zhang L, Atkinson K, … & Holder BR (2017). Discovery of Potent and Orally Bioavailable Macrocyclic Peptide–Peptoid Hybrid CXCR7 Modulators. Journal of Medicinal Chemistry, 60(23), 9653–9663. [DOI] [PubMed] [Google Scholar]
- 12.Borkar AN, Bardaro MF, Camilloni C, Aprile FA, Varani G, & Vendruscolo M (2016). Structure of a low-population binding intermediate in protein-RNA recognition. Proceedings of the National Academy of Sciences, 113(26), 7171–7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bose D, Jayaraj G, Suryawanshi H, Agarwala P, Pore SK, Banerjee R, and Maiti S (2012). The tuberculosis drug streptomycin as a potential cancer therapeutic: inhibition of miR-21 function by directly targeting its precursor. Angew. Chem., Int. Ed, 51, 1019–1023. [DOI] [PubMed] [Google Scholar]
- 14.Bower J, Drysdale M, Hebdon R, Jordan A, Lentzen G, Matassova N, Murchie A, Powles J, Roughley S. (2003). Structure-based design of agents targeting the bacterial ribosome. Bioorg Med Chem Lett., 13(15), 2455–2458. [DOI] [PubMed] [Google Scholar]
- 15.Burnett JC, Rossi JJ. (2012). RNA-based therapeutics: current progress and future prospects. Chem Biol., 19(1), 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carthew RW & Sontheimer EJ (2009). Origins and Mechanisms of miRNAs and siRNAs. Cell, 136, 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee J, Gilon C, Hoffman A, & Kessler H (2008). N-methylation of peptides: a new perspective in medicinal chemistry. Accounts of chemical research, 41(10), 1331–1342. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, & Frankel AD (1995). A peptide interaction in the major groove of RNA resembles protein interactions in the minor groove of DNA. Proc. Natl. Acad. Sci. U.S.A., 92, 5077–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen P, Lin C, Lee C, Jan H, & Chan SI (2001). Effects of turn residues in directing the formation of the beta-sheet and in the stability of the beta-sheet. Protein Science, 10, 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cléry A, Blatter M, Allain FH (2008). RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol., 18(3), 290–298. [DOI] [PubMed] [Google Scholar]
- 21.Cooper TA, Wan L, & Dreyfuss G (2009). RNA and Disease. Cell, 136, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, & Varani G (2009). Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U.S.A, 106(29). 11931–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidson A, Patora-Komisarska K, Robinson JA, & Varani G (2011). Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Research, 39(1), 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis B, Afshar M, Varani G, Murchie AI, Karn J, Lentzen G, … & Aboulela F (2004). Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic “hot spots”. J Mol Biol. 2004, 336(2), 343–356. [DOI] [PubMed] [Google Scholar]
- 25.Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, & Skinner MA (1990). HIV-1 tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 9(12), 4145–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emerman M, & Peden K (1989). The rev Gene Product of the Human Immunodeficiency Virus Affects Envelope-Specific RNA Localization. Cell, 57, 1155–1165. [DOI] [PubMed] [Google Scholar]
- 27.Esteller M (2011). Non-coding RNAs in human disease. Nat. Rev. Genet, 12, 861–874. [DOI] [PubMed] [Google Scholar]
- 28.Fasan R, Dias RLA, Moehle K, Zerbe O, Vrijbloed JW, Obrecht D, & Robinson JA (2004). Using a Beta-Hairpin To Mimic an Alpha-Helix: Cyclic Peptidomimetic Inhibitors of the p53–HDM2 Protein–Protein Interaction**. Angew. Chem. Int. Ed, 43, 2109–2112. [DOI] [PubMed] [Google Scholar]
- 29.Favre M, Moehle K, Jiang L, Pfeiffer B, & Robinson JA (1999). Structural Mimicry of Canonical Conformations in Antibody Hypervariable Loops Using Cyclic Peptides Containing a Heterochiral Diproline Template. J. Am. Chem. Soc, 121, 2679–2685. [Google Scholar]
- 30.Fischer U, Huber J, Boelens WC, Mattaj IW, & Luhrmann R (1995). The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell, 82, 475–483. [DOI] [PubMed] [Google Scholar]
- 31.Fosgerau K, & Hoffmann T (2015). Peptide therapeutics: current status and future directions. Drug Discovery Today, 20(1), 122–128. [DOI] [PubMed] [Google Scholar]
- 32.Fujii N, Oishi S, Hiramatsu K, Araki T, Ueda S, Tamamura H, … & Broach JA (2003). Molecular-size reduction of a potent CXCR4-chemokine antagonist using orthogonal combination of conformation-and sequence-based libraries. Angewandte Chemie International Edition, sequence 42(28), 3251–3253. [DOI] [PubMed] [Google Scholar]
- 33.Gallego J, & Varani G (2001). Targeting RNA with Small-Molecule Drugs: Therapeutic Promise and Chemical Challenges. Acc. Chem. Res, 34, 836–843. [DOI] [PubMed] [Google Scholar]
- 34.Gonda MA, Luther DG, Fong SE, & Tobin GJ (1994). Bovine immunodeficiency virus: molecular biology and virus-host interactions. Virus Res., 32, 155–181. [DOI] [PubMed] [Google Scholar]
- 35.Griffiths-Jones SR, Maynard AJ, Searle MS (1999). Dissecting the stability of a beta-hairpin peptide that folds in water: NMR and molecular dynamics analysis of the beta-turn and beta-strand contributions to folding. J Mol Biol., 292(5), 1051–1069. [DOI] [PubMed] [Google Scholar]
- 36.Hamy F, Felder ER, Heizmann G, Lazdins J, Aboul-ela F, Varani G, Karn J, Klimkait T (1997). An inhibitor of the Tat TAR RNA interaction that effectively suppresses HIV-1 replication. Proc Natl Acad Sci U S A., 94, 3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, … & Roemer T (2015). Selective small-molecule inhibition of an RNA structural element. Nature, 526(7575), 672–677. [DOI] [PubMed] [Google Scholar]
- 38.Jiang L, Moehle K; Dhanapal B, Obrecht D, & Robinson JA (2000). Combinatorial Biomimetic Chemistry: Parallel Synthesis of a Small Library of β Hairpin Mimetics Based on Loop III from Human Platelet Derived Growth Factor B. HelV. Chim. Acta, 83, 3097–3112. [Google Scholar]
- 39.Jones KA, & Peterlin BM (1994). Bovine immunodeficiency virus: molecular biology and virus-host interactions. Annu. Rev. Biochem, 63, 717–743. [DOI] [PubMed] [Google Scholar]
- 40.Jones S, Daley DTA, Luscombe NM, Berman HM, & Thornton JM (2001). Protein-RNA interactions: a structural analysis. Nucleic Acids Research, 29(4), 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juliano RL (2016). The delivery of therapeutic oligonucleotides. Nucleic acids research, 44(14), 6518–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keen NJ, Gait MJ, & Karn J (1996). Human immunodeficiency virus type-1 Tat is an integral component of the activated transcription-elongation complex. Proc Natl Acad Sci. U S A. 93, 2505–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirk SR, Luedtke NW, & Tor Y (2000). Neomycin−Acridine Conjugate: A Potent Inhibitor of Rev-RRE Binding. J. Am. Chem. Soc, 122, 980–981. [Google Scholar]
- 44.Kjaer-Frifeldt S, Hansen TF, Nielsen BS, Joergensen S, Lindebjerg J, Soerensen FB, dePont Christensen R, Jakobsen A, Danish Colorectal Cancer Group. (2012). The prognostic importance of miR-21 in stage II colon cancer: a population-based study. Br. J. Cancer, 107, 1169–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozomara A, and Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res., 42, D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leeper T, Athanassiou Z, Dias R, Robinson JA, & Varani G (2005). TAR RNA Recognition by a Cyclic Peptidomimetic of Tat Protein. Biochemistry, 44, 12362–12372. [DOI] [PubMed] [Google Scholar]
- 47.Leulliot N, Varani G (2001). Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry, 40(27), 7947–7956. [DOI] [PubMed] [Google Scholar]
- 48.Ling H, Fabbri M, Calin GA (2013). MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov., 12, 847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lunde BM, Moore C, & Varani G (2007). RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol., 8(6), 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lättig-Tünnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC (2011). Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat Commun., 2, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madan B, Seo SY, Lee SG (2014). Structural and sequence features of two residue turns in beta-hairpins. Proteins., 82(9), 1721–33. [DOI] [PubMed] [Google Scholar]
- 52.Maiti M, Nauwelaerts K, & Herdewijn P (2012). Pre-microRNA binding aminoglycosides and antitumor drugs as inhibitors of Dicer catalyzed microRNA processing. Bioorganic & medicinal chemistry letters, 22(4), 1709–1711. [DOI] [PubMed] [Google Scholar]
- 53.Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR (1989). The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature, 338, 254–257. [DOI] [PubMed] [Google Scholar]
- 54.Mann DA, Mikaelian I, Zemmel RW, Green SM, Lowe AD, Kimura T, Singh M, Butler G, Gait M, Karn J (1994). Co-operative Rev Binding to Stem I of the Rev-response Element Modulates Human Immunodeficiency Virus Type-1 Late Gene Expression. J. Mol. Biol, 241, 193–207. [DOI] [PubMed] [Google Scholar]
- 55.Mason JM (2010). Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med Chem., 2(12), 1813–1822. [DOI] [PubMed] [Google Scholar]
- 56.Mei H-Y, Cui M, Heldsinger A, Lemrow SM, Loo JA, Sannes-Lowery KA, Sharmeen L, & Czarnik AW (1998). Inhibitors of protein-RNA complexation that target the RNA: specific recognition of human immunodeficiency virus type 1 TAR RNA by small organic molecules. Biochemistry, 37, 14204–14212. [DOI] [PubMed] [Google Scholar]
- 57.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology, 133, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moehle K, Athanassiou Z, Patora K, Davidson A, Varani G, & Robinson JA (2007). Design of Beta-Hairpin Peptidomimetics That Inhibit Binding of Alpha-Helical HIV-1 Rev Protein to the Rev Response Element RNA. Angew. Chem. Int. Ed, 46, 9101–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, & Lin Q (2012). Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. Journal of the American Chemical Society, 134(36), 14734–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murchie AI, Davis B, Isel C, Afshar M, Drysdale MJ, Bower J, … & Karn J. (2004). Structure-based drug design targeting an inactive RNA conformation: exploiting the flexibility of HIV-1 TAR RNA. J Mol Biol., 336(3), 625–638. [DOI] [PubMed] [Google Scholar]
- 61.Nair CM, Vijayan M, Venkatachalapathi YV, Balaram P (1979). X-ray crystal structure of pivaloyl-D-Pro-L-Pro-L-Ala-N-methylamide; Observation of a consecutive β-turn conformation. J. Chem. Soc., Chem. Commun, 1183–1184. [Google Scholar]
- 62.Niu Y, Jones A, Wu H, Varani G, & Cai J (2011). Organic & Biomolecular Chemistry, 9, 6604–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, … & Beibel M (2015). SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nature chemical biology, 11(7), 511. [DOI] [PubMed] [Google Scholar]
- 64.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN (2015). Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew Chem Int Ed Engl. 2015, 54(31), 8896–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puglisi JD, Chen L, Blanchard S, & Frankel AD (1995). Solution structure of a bovine immunodeficiency virus Tat-TAR peptide-RNA complex. Science, 270, 1200–1203. [DOI] [PubMed] [Google Scholar]
- 66.Renukuntla J, Dutt A, Patel A, Boddu SHS, & Mitra AK (2013). Approaches for enhancing oral bioavailability of peptides and proteins. International Journal of Pharmaceutics, 447(1–2), 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ribas J, Lupold SE (2010). The transcriptional regulation of miR-21, its multiple transcripts, and their implication in prostate cancer. Cell Cycle, 9, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ratni H, Karp GM, Weetall M, Naryshkin NA, Paushkin SV, Chen KS, … & Zhang X (2016). Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. Journal of medicinal chemistry, 59(13), 6086–6100. [DOI] [PubMed] [Google Scholar]
- 69.Robinson JA (2000). The design, synthesis and conformation of some new β-hairpin mimetics: Novel reagents for drug and vaccine discovery. SynLett, 429–441. [Google Scholar]
- 70.Robinson JA (2008). Beta-hairpin peptidomimetics: design, structures and biological activities. Acc Chem Res., 41(10),1278–1288. [DOI] [PubMed] [Google Scholar]
- 71.Rohs R, Jin X, West SM, Joshi R, Honig B, & Mann RS (2010). Origins of Specificity in Protein-DNA Recognition. Annu. Rev. Biochem 79, 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenström U, Sköld C, Lindeberg G, Botros M, Nyberg F, Karlén A, & Hallberg A (2006). Design, synthesis, and incorporation of a β-turn mimetic in angiotensin II forming novel pseudopeptides with affinity for AT1 and AT2 receptors. Journal of medicinal chemistry, 49(20), 6133–6137. [DOI] [PubMed] [Google Scholar]
- 73.Roy S, Delling U, & Rosen CA (1990). A bulge structure in HIV-I TAR RNA is required for Tat binding and Tat- mediated trans-activation. Genes & Development, 4, 1365–1373. [DOI] [PubMed] [Google Scholar]
- 74.Shortridge MD, Walker MJ, Pavelitz T, Chen Y, Yang W, & Varani G (2017). A Macrocyclic Peptide Ligand Binds the Oncogenic MicroRNA-21 Precursor and Suppresses Dicer Processing. ACS Chem. Biol, 12, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shortridge MD, Wille PT, Jones AN, Davidson A, Bogdanovic J, Arts E, … & Varani G (2018). An ultra-high affinity ligand of HIV-1 TAR reveals the RNA structure recognized by P-TEFb. Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sibanda BL, Blundell TL, & Thornton JMJ (1989). Conformation of β-hairpins in protein structures: A systematic classification with applications to modelling by homology, electron density fitting and protein engineering. Mol. Biol, 206, 759–777. [DOI] [PubMed] [Google Scholar]
- 77.Sibanda BL, Thornton JM (1985). β-Hairpin families in globular proteins. Nature, 316, 170–74. [DOI] [PubMed] [Google Scholar]
- 78.Stein CA, & Goel S (2011). Therapeutic oligonucleotides: the road not taken. Clinical Cancer Research, 17(20), 6369–6372. [DOI] [PubMed] [Google Scholar]
- 79.Teixido M, & Giralt E (2017). Jumping Hurdles: Peptides Able To Overcome Biological Barriers. Acc Chem Res., 50(8), 1847–1854. [DOI] [PubMed] [Google Scholar]
- 80.Varani G, Aboula-ela F, Allain FH (1996). NMR Investigation of RNA Structure. Progress in Nuclear Magnetic Resonance Spectroscopy, 29, 51–127. [Google Scholar]
- 81.Wang J, Lon HK, Lee SL, Burckart GJ, & Pisetsky DS (2015). Oligonucleotide-Based Drug Development: Considerations for Clinical Pharmacology and Immunogenicity. Therapeutic Innovation & Regulatory Science, 49(6), 861–868. [DOI] [PubMed] [Google Scholar]
- 82.Warner KD, Hajdin CE, Weeks KM (2018). Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov., 8, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wei P, Garber ME, Fang S-M, Fischer WH, & Jones KA (1998). A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell, 92, 451–462. [DOI] [PubMed] [Google Scholar]
- 84.Werder M, Hauser H, Abele S, & Seebach D (1999). β-Peptides as Inhibitors of Small-Intestinal Cholesterol and Fat Absorption. Helvetica chimica acta, 82(10), 1774–1783. [Google Scholar]
- 85.Whitby LR, Ando Y, Setola V, Vogt PK, Roth BL, & Boger DL (2011). Design, synthesis, and validation of a β-turn mimetic library targeting protein–protein and peptide–receptor interactions. Journal of the American Chemical Society, 133(26), 10184–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williamson JR (2000). Induced fit in RNA–protein recognition. Nature Structural and Molecular Biology, 7(10), 834. [DOI] [PubMed] [Google Scholar]
- 87.Wilson WD, & Li K (2000). Targeting RNA with small molecules. Curr. Med. Chem, 7, 73–98. [DOI] [PubMed] [Google Scholar]
- 88.Witt KA, Gillespie TJ, Huber JD, Egleton RD, & Davis TP (2001). Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides, 22(12), 2329–2343. [DOI] [PubMed] [Google Scholar]
- 89.Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, Zeng YX, Shao JY (2008). MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA, 14, 2348–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ye X, Kumar RA, & Patel DJ (1995). Molecular recognition in the bovine immunodeficiency virus Tat peptide-TAR RNA complex. Chem. Biol, 2, 827–840. [DOI] [PubMed] [Google Scholar]