

Abstract
Cellular migration is a tightly regulated process that involves actin cytoskeleton, adaptor proteins, and integrin receptors. Forces are transmitted extracellularly through protein complexes of these molecules, called adhesions. Adhesions anchor the cell to its substrate, allowing it to migrate. In Chinese hamster ovary cells, three classes of adhesion can be identified: nascent adhesions (NAs), focal complexes, and focal adhesions, ranked here ascendingly based on size and stability. To understand the dynamics and mechanosensitive properties of NAs, a biophysical model of these NAs as colocalized clusters of integrins and adaptor proteins is developed. The model is then analyzed to characterize the dependence of NA area on biophysical parameters that regulate the number of integrins and adaptor proteins within NAs through a mechanosensitive coaggregation mechanism. Our results reveal that NA formation is triggered beyond a threshold of adaptor protein, integrin, or extracellular ligand densities, with these three factors listed in descending order of their relative influence on NA area. Further analysis of the model also reveals that an increase in coaggregation or reductions in integrin mobility inside the adhesion potentiate NA formation. By extending the model to consider the mechanosensitivity of the integrin bond, we identify mechanical stress, rather than mechanical load, as a permissive mechanical parameter that allows for noise-dependent and independent NA assembly, despite both parameters producing a bistable switch possessing a hysteresis. Stochastic simulations of the model confirm these results computationally. This study thus provides insight into the mechanical conditions defining NA dynamics.
Significance
This manuscript makes valuable contributions in deciphering the dynamics of nascent adhesions (NAs). It presents a novel mathematical model that determines how mechanical stress applied to these adhesions governs their assembly and disassembly. The model is biophysically relevant; it provides a framework that links NA area to the number of mobile or immobile integrins along with adaptor proteins and considers a multitude of experimental observations defining their mechanosensitive properties. The model is then examined to uncover how four components of this systems interact: NA area, integrin and adaptor proteins coaggregation, number of integrins and adaptor proteins within NAs, and force exerted on NAs. It is the first study that uniquely identifies adhesion stress, rather than its total load, as the mechanical parameter conserved during NA formation.
Introduction
Cellular migration plays a fundamental role in many physiological and pathophysiological systems, including embryonic morphogenesis and continual tissue regeneration (1), immune responses (2), tissue repair (3), and the metastatic properties of cancers (4). Typically, cellular migration occurs in response to an extracellular signal in the form of a chemical or mechanical gradient (5, 6), leading to cellular polarization through nonlinear processes governing biochemical pathways that internally amplify the extracellular gradients to establish a front and rear of the cell (7, 8). Cells attach themselves to their external environment by forming integrin-based adhesion complexes that anchor to the substrate and act as an extension of the cytoskeleton. This results in an active remodeling of the cytoskeleton and causes a net displacement of the cell along the substrate (9, 10).
Adhesions serve as a point of force transmission to the external environment for motile cells. This force transmission is primarily mediated through integrin receptors, which span the cellular membrane and bind to their extracellular ligands (e.g., collagen and fibronectin). They form partially immobilized traction points that a cell can use to move itself or to deform the external environment. The cytoplasmic domains of integrins are linked to the actin cytoskeleton through a variety of adaptor proteins that interact through biochemical signaling pathways (11, 12, 13), creating a biologically regulated mechanical coupling system that links the actin cytoskeleton with the extracellular matrix (ECM). The interactions between these proteins, along with mechanical forces arising from active processes in the cytoskeleton, establish complex spatiotemporal patterns of activity (7, 8). These patterns regulate the dynamics of adhesion assembly and disassembly (14, 15) and cytoskeletal organization (9, 16, 17), producing cellular migration in response to extracellular cues (10).
Adhesions are formed in a stepwise manner, starting as nascent adhesions (NAs), a class of dynamic nanoscale clusters of integrin receptors (referred to hereafter as integrins) that contain on average 20–50 integrins (18, 19). Many mechanisms, such as increases in the long-range lateral mobility of integrins (e.g., reduced corralling by actin cytoskeleton), local changes in their free diffusivity, and modulation of their binding affinity through interactions with the adaptor protein talin, have been found to be relevant in the formation and regulation of integrin clusters (20, 21, 22, 23, 24, 25, 26). Experimentally dissecting the effect of each mechanism on adhesion formation has often been obscured by the fact that all these effects are simultaneously present in physiological conditions (21, 24, 27). Theoretical models of adhesions allow for independent modulation of these mechanisms to study their effects. Existing models of adhesions either have idealized their physical model of an adhesion (10, 28, 29, 30, 31) or have focused on certain phenomenological aspects of the system (30, 32, 33, 34) that are not applicable during the formation of NAs. Others are more biologically realistic with many variables, making it difficult to understand the underlying dynamics (35, 36, 37, 38, 39, 40, 41).
Integrins are intrinsically mechanosensitive, capable of exhibiting a catch-bond behavior (42). In contrast to the more common slip bonds (43), catch bonds exhibit a nonmonotonic profile in their lifetime when plotted as a function of the applied force. This profile suggests that there exists a nonzero optimal applied force that maximizes the bond lifetime, for which the increase in bond lifetime is associated with a conformational change to a long-lived bound state (44, 45). It has been proposed that this phenomenon may reflect the activation of integrins (i.e., the induction of a high-affinity bound state) by mechanical forces (45, 46). Because of their position in the mechanical linkage between the cell and its environment, the mechanosensitivity of integrins is likely to be implicated in adhesion dynamics. This has been investigated thoroughly from a theoretical perspective by considering a finite number of bonds that may rupture because of force and rebinding (28, 29, 34, 47). These studies have reported disassembly of clusters of bonds but have not yet examined how assembly is manifested in the presence of applied force. Thus, determining the effect(s) of force on NA dynamics during assembly and disassembly remains incomplete.
The process of adhesion assembly into a cluster containing tens of molecules starting from a single molecular-complex initial condition (e.g., a single integrin-adaptor protein complex) was previously demonstrated through stochastic simulations of a molecular model that considered many possible state transitions (41). However, mean-field analysis of this molecular model was performed using a simplified one-variable ordinary differential equation (ODE) model, predicting an unstable steady state that blocks the assembly of very small adhesions (41). In theory, it should be possible to jump over this unstable state because of stochastic effects even in one dimension. However, the conditions necessary to produce such a jump remain unexplored. Moreover, other studies that produce similar configurations of steady states also predict a net loss in both the number of bound integrins and adaptor proteins when adhesions are very small (39, 40, 47). On the one hand, this is congruent with the proposition that NAs are thermodynamically unfavorable structures and thus are the product of stochastic fluctuations (30, 48, 49). On the other hand, NAs have been shown to be stable for hundreds of seconds in appropriate mechanical conditions, suggesting that they are, in fact, thermodynamically favorable structures (18, 49). To further investigate this, we develop here a mathematical model that possesses an elevated equilibrium representing NAs that can either 1) allow for NA assembly independently of stochastic effects or 2) be used to understand in more detail the conditions required for noise-induced NA formation.
Because of the limited understanding of NA assembly dynamics, we present in this study this biophysically based mathematical model of NA formation and stabilization, which considers a multitude of experimental observations and provides precise characterization of their underlying behavior in various conditions. We then use the model, which not only considers the integrin bonds but also the adhesion plaque formed by adaptor proteins, to demonstrate how assembly and disassembly are affected by force and their dynamics are manifested under various physiologically relevant contexts.
Methods
Mathematical model of NA formation
We propose a biophysical model of NA formation based on a mechanism we term coaggregation, in which the interaction of integrins in the membrane with adaptor proteins in the cytoplasm leads to the colocalized aggregation of both into discrete structures (see Supporting Materials and Methods for a detailed derivation of the model). This model is constructed to incorporate a number of experimental observations:
O1. NAs have a circular geometry, and they grow isotropically (50). This is in contrast to the growth of larger and typically elongated focal adhesions, which is highly anisotropic (48, 50).
- O2. Adaptor proteins exhibit a wide range of mobilities that vary in a location-dependent manner.
-
(a)Adaptor proteins diffuse rapidly in the cytosol, whereas they diffuse more slowly inside adhesions (51).
- (b)
-
(c)Some adaptor proteins have a longer residency time in adhesions containing a higher density of integrins, suggesting that a reduction in adaptor mobility is due to interactions (binding or tethering) with integrin (54).
-
(a)
O3. Integrins diffuse laterally along the membrane. Their free diffusion is slower within the adhesion, and this effect is likely due to interactions with adaptor proteins on the cytoplasmic side of the membrane (25).
O4. Integrins become immobilized for extended periods of time upon binding to ligand, and this process is favored inside adhesions (55).
O5. The integrin-ligand bond lifetime varies with force (42), exhibiting a biphasic profile with a maximal value at nonzero applied force (∼37 pN).
Furthermore, in the derivation of model equations, we have also made a number of assumptions that are primarily based on experimental observations:
M1. As suggested in O1, the model defines an NA as a two-dimensional region of space on the membrane demarcated by the boundary of a membrane-proximal circular adhesion plaque with area A (see Fig. 1).
- M2. The adhesion plaque is a condensed phase of interacting adaptor proteins tethered to the membrane by integrins. Adaptor proteins are assumed to be incorporated with a fixed density through adsorption. The density, ρ, is determined by the mean volume of all adaptor proteins in the plaque, and the length of the bonds between them.
-
(a)According to O2a, we assume that the cytoplasmic pool of adaptor proteins is well-mixed (see Eq. S6), whereas adaptor proteins in the adhesion plaque exhibit reduced mobility because of interactions with other adaptor proteins in the plaque and integrins in the membrane.
-
(b)The adhesion-plaque dynamics are derived based on the assumption that it can grow or shrink because of the adsorption or loss of adaptor proteins to or from any point in its interior, respectively; this assumption corresponds to the “bulk-on/bulk-off” formalism considered previously (30). The choice of this formalism is motivated by the notion that NAs are not strongly associated with the actin cytoskeleton during their assembly (56, 57), allowing for the adsorption or loss of adaptor proteins through the dorsal face of the adhesion. This assumption would not be appropriate for models dealing with the maturation of NAs, as that process seems to be intrinsically tied to an increased association with organized actin filaments (50, 56, 58) that protect the adhesion from degradation by loss of matter through its dorsal face (30).
-
(c)From O2b and O2c, the multiple mobility states of adaptor proteins in the adhesion are interpreted as being due to their tethering to the membrane by integrin receptors (i.e., in a state with lower mobility if the adaptor protein is tethered to the membrane but in a state with higher mobility if it is only associated with other adaptor proteins). This implies that a nonlinear relation between the adhesion’s disassembly rate and its integrin content exists (see Eq. S7).
-
(a)
- M3. Underneath the adhesion plaque, there are a total of Nin integrins. Lin of these integrins are liganded and immobilized while Min are mobile and diffuse freely (i.e., Nin = Lin + Min; see Fig. 1).
-
(a)Per O3, we use a position-dependent diffusion coefficient to abruptly change the diffusivity of integrins from Dout to Din when they pass from the outside to the inside of the adhesion (Dout > Din; see Eq. S2).
-
(b)Per O4, integrins inside the adhesion bind to their ligands with a rate kbind. Furthermore, we assume that bound integrins are completely immobile. To account for the density of extracellular ligand, we take the binding rate to be proportional to ligand density (i.e., ).
-
(c)Per O5, bound integrins inside the adhesion unbind from their ligands with a rate kunbind(f), where f is the force applied to a single integrin (see Eq. S14 for details on the form of kunbind(f) and assumption M4a for the determination of f). Bound integrins outside the adhesion unbind with a rate kunbind(0), with this situation only occurring when the adhesion plaque is shrinking.
-
(a)
- M4. The total load applied to the adhesion is distributed evenly among all bound integrins. Therefore, when an integrin unbinds, the load is distributed over the (fewer) remaining bound integrins such that each receptor feels an increase in applied force.
-
(a)The force, f, applied to each integrin is computed by assuming that the NA is under fixed stress, , and that each bound integrin bears an equal load (see Eq. S17).
-
(a)
M5. For simplicity, we consider a periodic square lattice of adhesions with an interadhesion spacing of h (see Fig. 1). The flux of matter between the square lattices is at equilibrium and thus the total matter in each square is assumed to be conserved. Model parameters are chosen in such a way that to prevent the adhesion from extending beyond the dimension of a single lattice cell (see Geometric Constraints).
Figure 1.

Schematic of the model describing adhesion formation. (A) Integrins and adaptor proteins coaggregate to form a membrane-proximal adhesion plaque, under which sits a cluster of integrins that exhibit a decreased diffusion coefficient (Din ≤ Dout) and become immobilized upon binding to ligand. Adaptor proteins that are in the proximity of integrins are assumed to be reversibly tethered to the membrane. Untethered adaptor proteins leave the plaque with a rate koff, whereas tethered ones leave at a rate (1 − δ)koff. (B) Geometry of the h × h-square lattice containing the adhesion domain Ω is shown. NAs possess a circular geometry confined to grow in this square lattice cell. The area of the adhesion plaque (A(t)) and the number of integrins it contains (Nin(t)) are dynamic variables that vary because of adsorption and diffusion, respectively. To see this figure in color, go online.
Mathematically, the coaggregation mechanism discussed above is implemented through the interaction of the integrin-dependent tethering mechanism (M2c) and the reduced integrin mobility inside the adhesions (M3a and M3b).
The behavior of the model is investigated through two complementary approaches: 1) by investigating its full spatiotemporal dynamics using stochastic simulations and 2) by characterizing the outcomes of the stochastic simulations using a simplified differential equation model of NAs describing the dynamics near equilibrium. The simplified model is given by the following set of ODEs:
| (1a) |
| (1b) |
| (1c) |
where is a kinetic parameter related to the rate of adsorption of adaptor proteins into the adhesion (see Parameter Estimation for more details), koff is the off-rate of adaptor proteins from the adhesion in the absence of integrins, δ is the fractional reduction in the off-rate of adaptor proteins when tethered to the membrane by integrins, b is the concentration of integrin inside the adhesion that results in the tethering of 50% of adaptor proteins in the adhesion plaque, n is the degree of cooperativity between integrins for the tethering of adaptor proteins, and CI (CP) is the mean density of integrins (adaptor proteins) taken by averaging over the h × h lattice cell. For detailed description and derivation of the model, see Supporting Materials and Methods.
Conditional expectation analysis
We have developed a computational method to infer the conditional mean of one physical variable given the value of another dependent physical variable. This technique is used to assess the relation between NA area, A, and the number of integrins it contains, Nin, by using only the two terms PA and , representing the marginal distributions of these quantities, respectively. Briefly, we assume , where denotes the conditional expectation of the variable x given the value of another variable y, and then use Bayesian methods to quantitatively estimate f(⋅)using the change-of-variable formula given by . See Supporting Materials and Methods for further details.
Model parameters
We have used a complex systems approach to parameter determination, applying different estimation techniques to guarantee that our model produces predictions that are consistent with experimental findings while also exhibiting the emergent behavior of the system under consideration (i.e., NAs). This is accomplished by deriving analytical expressions (e.g., steady states of the model) that can be equated to experimental measurements (18, 25, 42, 51) and then solving the resulting set of nonlinear equations to quantify model parameters. This approach is used to determine the parameters b, h, CI, Kin = Dout/Din, , and Kbind = kbind/kunbind. Furthermore, the previously published data describing the distributions of protein cluster sizes and number of integrins within these protein clusters (18) are digitized and utilized to derive a relation between A and Nin using conditional expectation analysis (described above). The nonzero A-nullcline, obtained by setting Eq. 1a to zero while assuming no cooperativity (i.e., by letting n = 1), is then fitted to this relation using a nonlinear least-squares fitting procedure that yields estimates for the two identifiable parameters
and
Estimating these identifiable parameters is necessary to determine the values of b and Kon (see Supporting Materials and Methods). The parameters δ, Dout, kunbind, and CP are estimated directly from experimental data (25, 42, 55, 59). Finally, the remaining two parameters koff (or ) and ρ are manually tuned to obtain stochastic simulations that are comparable to experimental recordings of adhesion assembly and disassembly (50). Parameter values are used to explain how the analytical results of our model manifest themselves numerically when constrained by experimental data. The qualitative behavior of the model does not depend critically on parameter choices made; this is to be expected because almost all results presented here have been obtained analytically. For more details about parameter estimation, see Supporting Materials and Methods.
Software
Stability analysis for fixed protein density and binding affinity is performed symbolically using Mathematica Version 11.3 (Wolfram Research, Champaign, IL). On the other hand, for fixed adhesion stress, stability analysis is conducted numerically using the continuation software AUTO-07p to obtain the location of bifurcation points (60), The stability of branches obtained are then cross-validated using Mathematica and MATLAB (The MathWorks, Natick, MA). Stochastic simulations and nonlinear least-squares fitting are performed using MATLAB. Parameter estimation is also done using Mathematica. The code used for ODE analysis, numerical continuation, and stochastic simulations can be obtained online (61).
Results
The relation between NA area and its integrin content
To understand NA dynamics, we first aim to establish how their area and integrin content are related. If this relation is linear, then the density of integrins inside the adhesion is constant, and the area of the adhesion can be explained by Nin alone. Such a relation could be used to estimate the effective membrane area of a single integrin and motivates the theoretical assumption of a fixed ratio of integrin/adaptor proteins (32, 33, 34, 36). If this relation is nonlinear, on the other hand, then the area of the adhesion cannot be explained solely by the number of integrins and their physical dimensions. In such a case, it would be inaccurate to use models that assume a fixed ratio of integrin/adaptor proteins.
To investigate this, we have digitized previously published distributions of NA diameter and their integrin content (18). The distribution of NA diameter is first converted to a distribution of NA area A by assuming a circular geometry, followed by applying conditional expectation analysis (see Methods) to quantify the relation between the two random variables A and Nin. This analysis is motivated by the significant differences in skewness between the distributions, suggesting the possibility of a nonlinear relation. Indeed, our results (see Fig. 2 A) reveal that this relation appears to have a saturating “sigmoidal” phase for NAs containing up to ∼190 integrins, followed by a linear phase for larger adhesions. This indicates that the integrin density inside the adhesion, Nin/A, is not fixed in small NAs (see dotted line in Fig. 2 B), and therefore, the size of these NAs cannot be explained by the number of integrins it contains. This sigmoid relation thus must be taken into consideration when formulating the model for NAs.
Figure 2.

(A) Inferred relation between NA area A and its integrin content Nin as determined by conditional expectation analysis. Bayesian inference is used to determine an empirical relation between A and Nin under the assumption that NAs are circular. After digitizing the distributions of NA diameters and their integrin contents published in (18), a relation between them is quantified. A 99.95% confidence interval is plotted in gray, demonstrating that the data generally exhibits a nonlinear relation between the two variables. The maximal a posteriori (MAP) estimate of this relation is also plotted as a dotted line, suggesting that NA area depends sigmoidally on Nin for small adhesions, whereas this dependence is linear for large adhesions (compare to dashed line). The nontrivial A-nullcline of the model, describing the relation between the two variables near equilibrium as defined by Eq. 2 (solid line), is fitted to the MAP estimate in the nonlinear phase and found to be in agreement. (B) The computed integrin density inside the adhesion, Nin/A(Nin), is given. Estimates are computed from the data-derived MAP estimate of A(Nin) (dotted line) and from the model prediction for A(Nin) given by Eq. 2 (solid line). Both curves exhibit a linear increase in integrin density with respect to Nin for Nin ∈ [50, 190]. (C) The probability distribution of integrin densities is shown. Black line: probability density function P(Nin/A) = P(Nin)[d(Nin/A(Nin))/dNin]−1, where A(Nin) is given by Eq. 2; gray line: cumulative density function.
To account for this, we have incorporated assumption M2c into the model. By setting the left-hand side of Eq. 1a to zero, we solve for the nonzero A-nullcline. This is equivalent to assuming that the observed differences in NA area are due to local variations in integrin availability (i.e., differences in CI). For simplicity, we consider the absence of cooperativity between integrins in the tethering of adaptor proteins (n = 1). The resulting expression for the A-nullcline is given by
| (2) |
where A0 = h2[(CP/ρ) − (1/Kon)] is the (possibly negative) area of the adhesion plaque in the absence of coaggregation (i.e., when δ → 0) with and A∞ = h2[(CP/ρ) − (1 − δ)/Kon] is the maximal area of the adhesion plaque (attained when the mean integrin density in the membrane is arbitrarily high; see Geometric Constraints). The shape of the curve described by Eq. 2. is in agreement with the profile of the relation inferred by conditional expectation analysis (solid line in Fig. 2 A), supporting the hypothesis that the aforementioned variation in integrin density (inside NAs) may be explained by the tethering mechanism used to formulate Eq. 1a.
We note that the model suggests that integrin density may grow unboundedly (see solid line in Fig. 2 B), which is due to the treatment of integrins as infinitesimally small point particles. In reality, the physical size of the integrins will eventually have non-negligible effects on their flux as the adhesion becomes increasingly crowded with integrins. Our data analysis demonstrates that adhesions containing integrin densities up to ∼17,000 μm−2 are not strongly influenced by these molecular crowding effects, as can be seen from the saturating relation between A and Nin for Nin < 190. Conversely, above this threshold in density, we have A ∝ Nin, suggesting that the crowding of integrins plays a significant role in determining the size of larger adhesions.
The maximal integrin density we have computed is significantly higher than the mean densities of ∼1000 μm−2 previously reported in Wiseman et al. (62), or the ∼6600 μm−2 computed by Changede et al. (18). Firstly, we would like to note that the most likely integrin density according to our analysis is ∼4940 μm−2 (see black line in Fig. 2 C), which is comparable to the value reported by Changede et al. Secondly, it has been previously suggested that Wiseman et al. (62) estimated the mean density of integrins on the membrane because their diffraction-limited imaging would not allow for proper resolution of NA boundaries (18). As such, we note that the value we have used for the mean density of integrins, CI = 1844 μm−2, is comparable to what was reported by Wiseman et al. (62) (see Parameter Estimation for more details).
Furthermore, our approach quantifies the variability of integrin density inside adhesion. Therefore, although our analysis suggests that this density may go as high as ∼17,000 μm−2, it also predicts that 80% of adhesions have an integrin density that is less than 12,000 μm−2 (see gray line in Fig. 2 C). Changede et al. (18) previously computed a maximal theoretical density of ∼25,000 μm−2 based on an estimated integrin footprint of 40 nm2. Using a hypothetical model (63) built from multiple partial crystal structures of integrin (64, 65, 66, 67), we estimate a rectangular bounding box footprint of 7.6 nm × 4.5 nm ≈ 34 nm2 and 12 nm × 5 nm ≈ 60 nm2 for the closed and open conformations of integrin, respectively. This provides us with corresponding bounds on the maximal possible density of integrins, given by 29,400 and 16,667 μm−2, respectively. Interestingly, our estimate of 17,000 only marginally surpasses the lower bound on the theoretical maximal density (i.e., when all integrins are in the open conformation) and remains well below the upper bound. This suggests that the majority of the integrins within these high-density NAs are in a closed conformation.
In this study, we focus our analysis on the nonlinear regime identified for Nin < 190 because NAs typically have Nin ∈ [20, 50] (19). In what follows, we examine the chemical and mechanical regulation of this sigmoidal relation between NA areas and their integrin content through the biophysical parameters, which control both Nin and Eq. 2.
Stabilization of the adhesion plaque by integrin
One of the equilibria of Eqs. 1a, 1b, and 1c is the unclustered steady state (A, Min, Lin) = (0, 0, 0). The stability of this steady state depends on the values of model parameters. To reduce the complexity of our analysis, we will first consider the model in the absence of force (f = 0). In subsequent sections, we will extend our steady-state analysis to include force and then investigate the time course of the model under applied force.
By analyzing the stability conditions of this steady state, we find that it becomes unstable once model parameters cross a threshold (see Supporting Materials and Methods); we will now demonstrate how this corresponds to the induction of adhesion formation. Given that the existence of the unclustered steady state (0, 0, 0) is independent of the degree of cooperativity n, we first restrict our analysis to the case when n = 1 for simplicity. This will allow us to build intuition about the behavior of the system and determine what happens for n > 1. Under these conditions, the unclustered steady state loses stability through a transcritical bifurcation occurring whenever , , or [Ligand] > [Ligand]‡ (see Eqs. S11–S13). That is, once there are enough integrins, adaptor proteins, or ligands at the interface between the cell and its environment, the unclustered steady state becomes unstable, and stable adhesion plaques form (see Fig. 3). This transition occurs when the system undergoes a transcritical bifurcation (see Fig. 3, A–C), forming a stable nontrivial steady state, given by
| (3) |
| (4) |
where ĈI = (1 + Kbind)KinKonCI, Kbind = kbind/kunbind, Kin = Dout/Din ≥ 1, , ω = (βϕ + 1)Kon, φ = (CP/ρ) − (1 − δ)/Kon, and is defined by Eq. S8. These expressions thus determine analytically how different parameters of the model affect the nontrivial steady state and the threshold (the transcritical bifurcation).
Figure 3.

Effects of coaggregation on NA area. Bifurcation diagrams of equilibrium adhesion area A∗ with respect to (A) CI, (B) CP, and (C) [Ligand] are given, showing branches of stable (solid lines) and unstable (dashed lines) steady states separated by thresholds defined by transcritical bifurcation points (open circles). The bifurcation points occur when CI = in (A), CP = in (B), and [Ligand] = [Ligand]‡ in (C). The stable branches represent the (un)clustered steady states (before) after the threshold. Notice that increasing CI leads to a saturating response, eventually plateauing at A∞ (dotted line in A), whereas increasing CP leads to an unbounded growth in adhesion area (B). Increasing [Ligand] also produces a saturating response, plateauing at (C).
As discussed in the previous section, increasing the number of integrins inside the NA causes an increase in their area. According to the model, this may be accomplished directly through two approaches: 1) increasing the total number of integrins on the membrane through the parameter CI or 2) increasing the likelihood that an integrin is inside the adhesion through the ligand concentration [Ligand] (see how Kbind is defined in M3b) or the reduction in diffusivity Kin. In the limit CI → ∞, the first approach yields adhesions with a finite size A∞ < h2 (see Eq. S10; Fig. 3 A). On the other hand, the limiting case for the second approach is equivalent to all integrins being inside the adhesion, where (according to Eq. 2) the area of the clustered steady state is given by
with = KonCI and
We note that (see Fig. 3 C) and , which implies that ligand-dependent regulation of adhesion area cannot overcome integrin-dependent regulation. Furthermore, a stable adhesion plaque may also grow if more plaque material is introduced into the system. Within the model, this is controlled by the parameter CP, which is linearly proportional to A∞. Therefore, increasing CP leads to unbounded growth of the adhesion area (see Fig. 3 B). Thus, the maximal area of NAs is regulated intracellularly through the parameters CI and CP, whereas the formation of adhesions and modulation of their area may be regulated intracellularly through these same parameters or extracellularly through [Ligand]. This, as a result, establishes a hierarchy in the determination of NA area by these three physiologically regulated densities, ranked according to their relative importance as follows: CP > CI > [Ligand].
Effects of the magnitude and degree of coaggregation
To further understand the dynamics of the model, we will analyze the model under appropriate limits. Thus, we first consider the integrin-independent adhesion-plaque formation when δ → 0. In this case, the model reduces to a fully deterministic version of the “bulk-on/bulk-off” adsorption model (30) with one added component for the conservation of matter. In this limit, we have A∗ → A0, which implies that adhesion area should grow linearly with adaptor concentration (see Fig. 3 B). This demonstrates the basic chemical kinetic control system of the model. Regardless of integrin density, adhesion plaques may form if a large enough pool of adaptor proteins is present (i.e., A0 > 0). In the absence of coaggregation, the cell must control both the formation and area of adhesions through the concentration of adaptor proteins. Interestingly, reductions in adaptor protein concentration have been found to decrease adhesion size (68, 69, 70, 71, 72). On the other hand, if there is coaggregation between integrins and adaptor proteins, the cell can set the maximal area of adhesions, A∞, by varying adaptor protein concentration while controlling their formation with the density of integrins. This is in line with the recent findings that NA size saturates as integrin density (or activation) is increased (see Fig. 2; (18)), suggesting that the coaggregation of the two proteins is a critical component of the model needed to capture experimental observations.
By tracking the location of the threshold (the transcritical bifurcation in Fig. 3) as the values of both CI and CP are varied in a two-parameter bifurcation, we obtain two regimes of behavior separated by a monotonically decreasing boundary at a given value of δ (see Fig. 4 A). This boundary divides the first quadrant into the two regimes of unclustered (below) and clustered (above) steady states, with the former expanding at the expense of the latter as the magnitude of coaggregation δ is decreased. These results suggest that the adaptor protein concentration needed to induce clustering is always lower in the presence of coaggregation (see Fig. 4 A). The amount of reduction in adaptor protein density generated by the inclusion of coaggregation is given by
Figure 4.

The effects of coaggregation and cooperativity on steady-state dynamics of protein clustering. (A) Two-parameter bifurcation of adhesion area A with respect to CI and CP are given, showing the boundary between the regimes of unclustered (below) and clustered (above) steady states for various values of coaggregation δ. The boundary is defined by the transcritical bifurcation points in Fig. 3 when CI and CP are both varied. Increasing δ reduces the adaptor protein density needed to induce aggregation. In the limit δ → 0, integrin density CI has no effect on the aggregation of adaptor proteins. (B) Stable branches of the clustered steady states shown in Fig. 3A for various degrees of cooperativity n are given. By increasing n, the density thresholds for clustering decreases, and the steepness of the stable branch of clustered steady states increases.
Interestingly, in the limit of infinite integrin density, we have
This suggests that even when integrin density is arbitrarily high, the cell can still turn off clustering by reducing CP (or δ) to make ϕ negative.
To study the effects of altering the degree of cooperativity n on the steady-state dynamics of adhesion area, we plot in Fig. 4 B the stable branch of clustered steady states (i.e., the stable branch to the right of the threshold in Fig. 3 A) at various values of n. Our results reveal that increasing n increases the slope of A∗ with respect to CI and that in the limit as n → ∞, we obtain a step-like switching response in which protein clusters form beyond a threshold in integrin density occurring right at the transcritical bifurcation point (see Fig. 4 B). The area of adhesions formed in this case remain roughly the same regardless of integrin density. Finally, we have used the maximal a posteriori estimate of A(Nin) (see Fig. 2) as means of estimating n numerically. This is done by solving for the nonzero A-nullcline with n = 2, 3, 4, where visual comparison with the data for A(Nin) suggests n = 2–3 (data not shown).
The effects of reduced integrin lateral mobility
The model assumes that integrin mobility is significantly reduced inside the NA (see O3 and O4) and that, at equilibrium, this area is in turn dependent on the number of integrins it contains. This makes understanding the dependence of NA dynamics on changes in integrin lateral mobility nontrivial. We can quantitatively understand this by first noting that the area specified by Eq. 2 is a monotonically increasing function of the number of integrins inside the NA, . Therefore, we may simply study the effects of the two parameters, which control changes in integrin lateral mobility (namely, Kin and Kbind), on the variable . From Eq. 4 and Eq. S8, we have
We can thus conclude that depends on integrin mobility only through the term Kin(1 + Kbind) that appears implicitly in ĈI, ω, and β. This means that we cannot distinguish between the effects induced by the two parameters purely based on adhesion area or integrin content. On the other hand, the fraction of bound integrins, given by
depends explicitly on the binding affinity. This means that although both reduced diffusivity (Kin) and ligand binding (Kbind) will result in an increase in the area and integrin content of adhesions, only ligand binding can increase the fraction of bound integrins.
Because of the similarity of the effects of ligand binding and reduced diffusivity, we focus our attention on analyzing more closely the effects of ligand binding, noting that the same analysis can be performed to study the effects of reduced diffusivity. In the absence of binding, we obtain the following unbound steady state
which has the associated critical protein thresholds
| (5) |
Based on Eq. S11, we can conclude that is larger than by a factor of 1 + Kbind. Similarly, from Eq. S12, it can be shown that . Thus another effect of binding (and reduced diffusivity) is that it increases the potency of integrins and adaptor proteins in the formation of adhesions by decreasing the threshold for clustering (i.e., shifting the transcritical bifurcation to the left) and expanding the range of stability of the clustered steady state (see Fig. 5 A).
Figure 5.

Distinct regions in parameter space are distinguished by their dependence on ligand binding. (A) A bifurcation diagram of adhesion area A with respect to [Ligand] is given, showing branches of stable (solid lines) and unstable (dashed lines) steady states separated by thresholds of transcritical bifurcation points (open circles). Decreasing Kon from 0.7 (black lines) to 0.3 (gray lines) shifts the transcritical bifurcation point to the left, allowing for the unclustered stable steady state to be attained at [Ligand] = 0. (B) Two-parameter bifurcation of adhesion area A with respect to [Ligand] and Kon is given, showing the boundary (black line) between the regions of unclustered (region 1) and clustered steady states lying below and above the boundary, respectively. The region of clustered steady states is further divided into three regions: region 2, defined by [Ligand]‡ > 0, requiring the presence of both ligand and integrin to induce clustering; region 3, defined by [Ligand]‡ < 0 with A0 < 0, requiring the presence of integrin, but not necessarily ligand, to form stable adhesion plaques; and the unphysiological region 4, defined by A0 > 0, which exhibits clustering irrespective of integrin or ligand densities.
For the more general scenario when Kbind > 0, we note that the state is a biophysically relevant metastable steady state only if all its coordinates are non-negative. This is equivalent to the experimentally verifiable criterion [Ligand]‡ > 0, which can be used to further divide parameter space into ligand-dependent and independent clustering regions (see Fig. 5). These regions are differentiated by their behavior in the limit as [Ligand] → 0+, where in the former region the stable unclustered steady state is attained (black lines in Fig. 5 A, with Kon = 0.7), whereas in the latter, the stable clustered steady state is attained (gray lines in Fig. 5 A, with Kon = 0.3). This implies that if the interactions between adaptor proteins are strong enough (e.g., if Kon is large enough; see Fig. 5 B), then adhesion plaques will form even in the absence of ligand binding. As discussed in the previous section, it is even possible for these interactions to be strong enough to induce the formation of an adhesion plaque in complete absence of integrins (as suggested by region 4, where A0 > 0). Together, these considerations allow us to identify four regions in parameter space:
-
1)
No Clustering (region 1 in Fig. 5 B): This occurs when [Ligand] < [Ligand]‡, and the model predicts there is no clustering of adaptor proteins.
-
2)
Ligand-Dependent Clustering (region 2 in Fig. 5 B): This region is bounded by [Ligand]‡ ≤ [Ligand] with [Ligand]‡ > 0. Within this region, the model predicts clustering occurs only when [Ligand] is high enough.
-
3)
Ligand-Independent Clustering (region 3 in Fig. 5 B): This region is defined by [Ligand]‡ ≤ [Ligand] with [Ligand]‡ < 0 and A0 < 0. Within this region, the model predicts clustering to occur regardless of the value of [Ligand].
-
4)
Integrin-Independent Clustering (region 4 in Fig. 5 B): This region is defined by A0 > 0. Within this region, the model predicts clustering regardless of the value of [Ligand] and CI. This appears to be unphysiological.
Because clustering is absent in region 1 and unphysiological in region 4, one may conclude that cells have likely tuned the interaction of adaptor proteins and integrins to that in regions 2 or 3, which differ by whether or not adhesions form in the absence of ligand. Whereas some have hypothesized that activation of integrins by adaptor proteins may be sufficient to induce clustering of integrins (73), an observation consistent with the model in region 3, others have shown that a ligand spacing greater than ∼60 nm drastically reduces the spreading of cells (74, 75, 76), an observation more consistent with crossing the transcritical bifurcation point in region 2. These two mutually exclusive hypotheses are considered to determine the two distinct sets of parameter values used to generate the curves in Fig. 5 A (see Parameter Estimation).
Mechanosensitive ligand binding dynamics
Thus far, we have not explicitly considered the effects of force on NA formation, although it is known that adhesions transmit force to the ECM. Mechanical forces can accelerate the dissociation of the integrin-ligand bond (see O5 and M3c in Methods). Moreover, a collection of catch bonds under fixed load will catastrophically fail once the number of bound integrins drops below a threshold (47). Therefore, to effectively transmit force to its environment, an NA must contain a sufficient number of bound integrins. This raises the question of how the dynamics of force generation in NAs are manifested or, more specifically, whether adhesion load (the total force) or stress (force per unit area) is the fixed mechanical parameter that allows for NA assembly from a single integrin-adaptor protein complex (32, 36, 77).
If adhesion load is held constant, then adhesions needs to build up a sufficient integrin content to be able to bear the load. As shown earlier, a large equilibrium integrin content at steady state can be achieved by either increasing Kin or Kbind. However, previous work has found that clusters of catch bonds will disassemble when there are a small number of bound integrins (47). Therefore, it is unclear how adhesions reach their stable equilibrium under fixed load. Alternatively, if stress is held constant, then the fraction of bound integrins must be large enough to bear the variable load that increases as the adhesion grows. The latter requires that Kbind be large enough (as discussed before). The mechanosensitivity of the integrin-ligand bond and the complex spatial dynamics of integrins make resolving this question nontrivial.
Bistable switch in regions 2 and 3 with respect to stress
To explore the effect of mechanical force on dynamics, we expand our equilibrium mean-field model described by Eqs. 1a, 1b, and 1c to include the mechanosensitive binding affinity of integrin, as described by Eqs. S16 and S17 (for further details, see Supporting Materials and Methods). Our initial analysis, as well as previous studies (28, 29, 47), suggest that there is a maximal stress, σc, that NAs can sustain. To further expand on this result, we plot in Fig. 6 the dependence of the steady-state adhesion area A∗ (and in region 3) with respect to stress σ and label the different branches of the curve based on the stability properties of the steady states belonging to each branch (using the subscripts “s” and “u” to label the stable and unstable ones, respectively). The resulting bifurcation diagram shows that the dynamics are governed by a bistable switch possessing a hysteresis, similar to those previously observed in systems of the Rho family of GTPases (8, 59). The switch has a plateaued branch of elevated steady states (black line), labeled , merging with another branch of unstable saddle points beneath it (dashed black line), labeled , at a saddle-node bifurcation when σc ≈ 470 kPa. In region 3, bistability is produced by another lower branch of metastable states formed by (gray line in Fig. 6 B), whereas the branch A = 0 is unstable. In region 2, the unstable branch intersects A = 0 at some value of stress σt (0 ≤ σt ≤ σc), leading to the formation of a transcritical bifurcation that produces a region of monostability for σ < σt (horizontal dashed line in Fig. 6 A) and bistability for σ > σt (horizontal solid line in Fig. 6 A).
Figure 6.

The effects of integrin mechanosensitivity on NA area and integrin content. Bifurcation diagram of adhesion area with respect to stress as determined by (A) region 2, and (B) region 3, showing a bistable switch with two semiplateaued stable branches of steady states (solid lines), one of which is elevated, representing (black), whereas the other is lower, representing (gray), separated by an unstable branch of saddle points (short-dashed black line). and merge at a threshold determined by a saddle-node bifurcation point at σ = σc. Beyond this critical stress σc, all integrins rapidly unbind and adhesions disassemble to (A) the unclustered steady state or (B) the unbound steady state . In region 2 (A), the unstable branch undergoes a transcritical bifurcation at σ = σt (open circles), producing a region of monostability where adhesions may directly assemble to the stable branch. Thick lines: [Ligand] = 8050 μm−2; thin lines: [Ligand] = 2012.5 μm−2.
The folded elevated stable and unstable branches seen in Fig. 6 have been previously reported in studies that considered only the number of liganded bonds (28, 29, 47) or adaptor proteins in the adhesion (41); these studies have reported either a monotonically decreasing elevated stable branch (28, 29) or one that increases significantly with applied force or ECM stiffness (41, 47). In either case, a lower stable/metastable branch (Z = (0, 0, 0) in region 2 with σ > σt and the previously defined in region 3 with σ < σc) has not been previously reported for a model that considered only liganded bonds. Our results thus show that the system possesses bistability between clustered and the unbound or unclustered steady states, indicating that this bistability arises from the consideration of plaque dynamics. We denote the elevated stable and middle unstable branches by S∗ = and U∗ = , respectively. These two steady states correspond to large clusters of integrins bearing a small load and small clusters bearing large loads, respectively. This correspondence may also be used to intuitively understand their stability because the small (unstable) clusters will unbind catastrophically once a single integrin-ligand bond ruptures, whereas the large (stable) clusters contain enough integrins for rebinding effects to outpace unbinding.
NA dynamics as determined by the bistable switch
Near the saddle node of Fig. 6, A and B (i.e., near σ ≈ σc), the basin of attraction of the clustered steady states shrinks significantly until it eventually disappears, along with the steady state, when σ > σc. This means that the number of bound integrins at the steady state S∗ are incapable of supporting the imposed stress, causing integrins within the NA to catastrophically unbind and the NA area to revert to the unbound steady state in region 3 (Fig. 6 A) and to A = 0 in region 2 (Fig. 6 A). This type of behavior corresponds to NA disassembly. On the other hand, in region 3 with σ < σc, stochastic fluctuations in the number of integrins inside the adhesion plaque can cause the system to jump from the unbound steady states to the clustered steady states . In contrast to the previous jump, this type of behavior corresponds to NA assembly. Alternatively, in the monostable regime of region 2 (see dashed horizontal line in Fig. 6 A), there is no barrier to cross for assembly, and NAs may form in a noise-independent manner. Interestingly, in both regions, the model predicts that the stable NA area does not vary significantly for a whole range of stress between [0, σc] because of the plateau nature of the upper stable branch. This suggests that NAs may go through a cycle of adhesion assembly, stability, and disassembly based on the magnitude of stress exerted on them, as previously seen experimentally (18, 50).
We have also investigated the effects of reduced ligand concentration on the mechanosensitivity of adhesions by reducing the value of [Ligand] fourfold (see thin lines in Fig. 6). Here, we observe two notable differences in the model outcomes. Firstly, when ligand concentration is reduced, the area of NAs under zero force (e.g., σ = 0) is reduced, whereas the maximal value it attains remains roughly the same. This suggests that NAs, in these conditions, can exhibit significant reinforcement because of force, increasing in size upon application of force. This reinforcement is similar to what has been previously observed experimentally (78) and in other theoretical models of focal adhesions (36, 48), as well as clusters of catch bonds (47). Secondly, we also find that the critical stress σc decreases when ligand concentration is reduced. This implies that the primary effect of reducing ligand concentration on NAs is to make them more susceptible to forces by 1) making their area more force-dependent and 2) making them more likely to catastrophically disassemble because of force.
Stochastic simulation of the model
It has been previously shown through stochastic simulations that the upper branches of Fig. 6 are metastable equilibria for clusters of catch bonds with a fixed load and that upon crossing the lower branch, the cluster undergoes disassembly in which all bonds rapidly unbind (47).
By running stochastic simulations with σ ≈ σc (see Supporting Materials and Methods for more details on the implementation), the model also produces NA disassembly under fixed stress regardless of the region (see Fig. 7 A for simulations associated with region 3) and for fixed load (data not shown). The stochastic simulation of the model also exhibits NA assembly from a single integrin-adaptor protein complex (see Fig. 7 A). In region 2 with σ < σt, NA assembly reaches S∗ unimpeded by U∗ because of the lack of bistability for 0 ≤ σ < σt, whereas in region 3 (see Figs. 6 B and 7 B), the system must cross the stable manifold of U∗. This manifold forms a threshold (highlighted by the dashed line in Fig. 7) that determines whether the system jumps from 1) the single integrin-adaptor complex to the clustered steady state S∗ (black solid line in Fig. 7 B) during assembly, passing through the unbound metastable state (gray line), or from 2) the clustered steady S∗ (black solid line in Fig. 7 B) to the unbound metastable state (gray line) during disassembly. The mechanical conditions that allow for a stochastic jump to cross the barrier created by the unstable state were not previously determined (41, 47). In this study, we have explored 1) under what conditions noise-driven NA assembly occurs and 2) what role bistability plays in this process.
Figure 7.
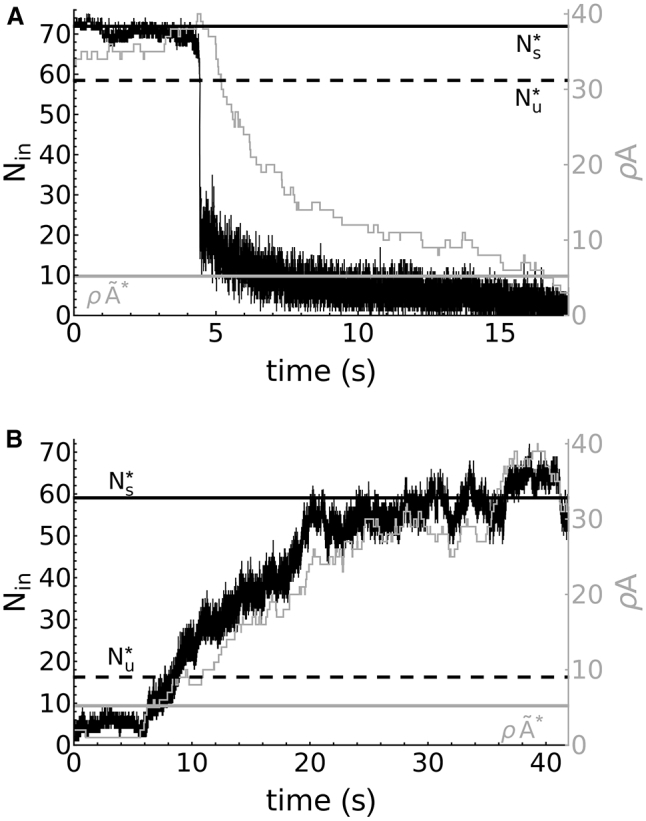
The assembly and disassembly of NAs is a force-dependent process. (A and B) Stochastic simulations of the number of bound integrins (thin black lines) and adaptor proteins (thin gray lines) during assembly and disassembly are shown. (A) Under high stress (σ = 0.92σc) and starting from the equilibrium S∗ (thick black horizontal line), integrins within NAs undergo catastrophic unbinding upon crossing the stable manifold of the saddle U∗ (dotted black horizontal line), leading to disassembly as they diffuse away from the adhesion area. (B) A long-lived NA state is formed, starting from a single integrin-adaptor protein complex, when the applied stress is low (σ = 0.25σc). This process is driven primarily by plaque growth toward the metastable state with (thick gray horizontal line), which, in conjunction with ligand binding, increases Nin, the number of integrins inside the NA, until it crosses the stable manifold of the saddle point U∗. Beyond the stable manifold of U∗, the trajectory flows to the stable steady state S∗. Black horizontal lines are identical to those in (A).
To verify whether the model under fixed adhesion load produces results consistent with those previously seen (28, 29, 34, 47), we also performed stochastic simulations starting from a single integrin-adaptor protein complex, in which adhesion load is held fixed. In this scenario, assembly is only observed for very small fixed loads (less than ∼1% of the critical load), as the force per integrin of small adhesions with a fixed load overwhelmingly favors the unbinding of integrin (data not shown). This is in agreement with the previous finding that fixed loads can only lead to adhesion disassembly (41, 47). Interestingly, with the model presented here, we do observe assembly for fixed stresses arbitrarily close to the critical stress σc, albeit exceedingly rarely. This highlights the unique nature of the model, which identifies fixed stress as a mechanical condition allowing for NA assembly and predicts that cells can occasionally assemble NAs even at very high stresses.
The bistability characterizing this model also plays an important role in reaching the elevated steady state S∗. Near the initial condition of a single integrin-adaptor protein complex, diffusion stochastically adds and removes integrins from underneath the adhesion plaque. However, as can be seen in Fig. 7 A, these stochastic fluctuations do not bring the system close enough to the saddle point U∗ (dotted line) to cross its stable manifold. The metastable state , on the other hand, acts as an attractor for the system, initiating the first phase of a robust growth in the adhesion plaque. In conjunction with ligand binding, this eventually pushes the system past the stable manifold of the saddle U∗, allowing it to reliably reach the elevated stable steady state S∗. As highlighted before, because fixed stress (but not fixed load) is permissive to ligand binding during the growth phase, we may conclude that the mechanical constraint on the system and the bistability with the metastable attractor allows for the system to overcome the kinetic barrier created by the unstable steady state U∗ such that adhesions may form from a single integrin-adaptor protein complex.
Discussion
In this study, we presented a biophysical model of NA formation as a condensed phase of adaptor proteins and integrins that forms along at the interface of the cytoplasm and the cell membrane because of a mechanism we term coaggregation. From this biophysical model, we constructed a three-dimensional minimal mathematical model of adhesion dynamics near equilibrium and a computational framework for realizing discrete simulations of adhesion formation. The nonlinearities included in the mathematical model were adapted from the biophysical framework used to describe NAs. The near-equilibrium dynamics used to analyze the stability of the equilibria showed that adhesions form with an area and integrin content specified by S∗ once integrins, adaptor proteins, or extracellular ligands cross a well-defined density threshold. These thresholds are defined by transcritical bifurcation points at which a stable branch of clustered steady states emerges in each case. We analytically determined the dependence of these thresholds on model parameters as specified by Eqs. S11–S13. Our analysis of these results revealed that the stable branch plateaus at high integrin or ligand density but monotonically increases with respect to adaptor protein density, suggesting that adaptor proteins play a key role in regulating the size of NAs. It also showed that there is a hierarchy in the relative importance of the three protein densities regulating NA area, given by adaptor proteins > integrins > extracellular ligands. The dependence of S∗ on biophysical parameters was then analyzed to demonstrate that the immobilization of integrins upon binding to ligands inside the adhesion area (controlled by Kbind) lowers the density thresholds for adhesion formation.
In contrast to previous models of adhesions (32, 33, 34, 36), we did not assume that the average density of integrins inside the adhesion plaque is a fixed quantity. This assumption is necessary for the model to be consistent with the observation that the integrin density measured in adhesions varies significantly within the same cell (79) and is motivated by the nonlinear relation between NA area and its integrin content (see Fig. 2). Our model accounted for the variable density of integrins by considering their interactions with adaptor proteins forming the adhesion plaque, which spatially delimits the adhesion area. Similar interactions have also been considered in a more biophysically detailed model of adhesion dynamics (38), but the simplicity of the model presented here compared to this previous study allowed for further theoretical analysis of this system. Interestingly, it was previously estimated that the upper limit of Nin/A is ∼25,000 μm−2 using structural considerations (18), but the results of our conditional expectation analysis suggest that the effective limit of integrin density in the adhesion may be as low as ∼17,000 μm−2. Below this threshold, NA area may be a result of the tethering mechanism described by Eq. S7, whereas above this threshold, adhesion area was the result of integrin crowding, which may be indicative of NAs transitioning to more mature classes of adhesions such as focal complexes. To the best of the authors’ knowledge, the tethering mechanism proposed here represents a novel understanding of adhesion stabilization by integrins.
Because of the apparent isotropic growth of NAs (50), we have not considered anisotropic effects of compression and/or stretching on adaptor protein adsorption as has been done in other models (3, 32). This can be justified by the fact that the dynamic nature of adaptor-cytoskeletal interactions leads to slippage under force (80), producing a viscoelastic mechanical response, in which the effects of elasticity is lost on long timescales (e.g., at equilibrium). Furthermore, it was previously argued that the interactions between adaptor proteins are unlikely to be strong enough for the energy of deformation of the adhesion plaque to have a significant effect on the outcome of the adhesion formation process (36). This assumption allowed us to use a simpler model of adaptor protein adsorption (30) and incorporate the effects of force in a manner that can be directly linked to experimental single-molecule observations (42).
The mechanosensitive properties of integrin unbinding were included in the model as a force-dependent bond lifetime (see Eq. S14). Using this to determine integrin binding affinity, we managed to compute the equilibrium area of the adhesion as a function of applied force, revealing a saddle-node bifurcation at a critical value of stress (see Fig. 6 D). Beyond this value of stress, all the integrins in the adhesion rapidly become unbound from their ligands, and tension is no longer transmitted to the extracellular environment. Similar phenomena had been previously observed in other studies for fixed values of adhesion load (28, 29, 47). By considering the effects of integrin content on the area of the adhesion plaque, we were able to use adhesion stress rather than load as a bifurcation parameter and study how its steady-state effects may give rise to a mechanically regulated NA lifecycle. We demonstrated that such a lifecycle is governed by a bistable switch with a saddle-node bifurcation when NA area is plotted against fixed stress. The saddle node acts as a threshold for adhesion disassembly, the last phase of the lifecycle. Increasing the ligand concentration made the upper branch of the bistable switch more plateaued such that increasing or decreasing the stress within a given range has little to no effect on NA area.
Consistent with previous studies, stochastic realizations of our model with high values of stress (or load; data not shown) produced NA disassembly close to the threshold determined by the saddle-node bifurcation, causing the trajectory to jump from the upper to the lower stable branch of the bistable switch. The jump is due either to crossing the threshold or to stochastic effects pushing trajectories beyond the stable manifold of the saddle points in the middle branch, leading to the rapid unbinding of all integrins in the adhesion, followed by slow adhesion-plaque disassembly.
The process of adhesion-plaque assembly, initiated from a single integrin-adaptor complex, was found to have a more diverse set of dynamics than disassembly. First, we discovered that unlike during disassembly, the accumulation of bound integrins inside the adhesion area and the growth of the adhesion plaque occur on a similar timescale during assembly. Second, we identified a region in parameter space (region 2 in Figs. 5 B and 6 A with σ < σt) where assembly occurs independently of stochastic effects. This finding is noteworthy because previous models had reported, in their mean-field analysis, the presence of an unstable steady state that blocks the assembly of very small adhesions (41, 47). The mean-field analyses were performed using a one-variable setting in which the only way to reach the state corresponding to stable adhesions was through stochastic jump effects that were not very well understood. The mean-field analysis performed here was done in a three-variable setting in which it was possible to shift the position of the unstable state to a nonphysical regime, thus minimizing its interference with the flow of trajectories toward the upper stable steady state.
The difference in outcomes between this study and previous ones can also be attributed to the mechanical condition we considered in this study (fixed stress). This may be understood intuitively by the fact that, when stress is fixed, a small adhesion will have a correspondingly small load. However, when load is fixed (28, 29, 34, 47), the integrin bonds break too quickly for the equilibrium with area A∗ to be readily attained. In previous models of clustered bonds, there was no explicit adhesion area, and therefore, adhesion stress could not be defined in these models (28, 29, 34, 47). In this study, we resolved this by considering the adsorption dynamics of the adaptor proteins that form the adhesion plaque (30). Experimentally, NA formation and area were found to be independent of traction force, whereas they disassemble in a force-dependent manner (18). These findings are consistent with our model predictions under fixed stress (but not fixed load), showing that NAs assemble to a roughly constant area for a wide range of stresses and disassemble at high stresses.
Similar to previous studies, the model also showed that there is a regime in parameter space within which the stable manifold of the saddle points acts as a barrier that can be crossed with stochastic effects (see region 3 in Figs. 5 B and 6 B). Within this regime, we demonstrated that fixing adhesion stress (but not load) allows for the assembly of adhesions and that bistability between the elevated steady state S∗ and the metastable state plays a significant role in determining the dynamics of assembly in this region of parameter space. When we fixed the value of load (rather than stress), we found that assembly is exceedingly rare for nonzero values of load (>1% the critical load; data not shown).
As suggested above, the mechanical control of the NA lifecycle, including assembly, stability, and disassembly, may be explained by a bistable switch (possessing a hysteresis) with respect to adhesion stress and that this stress, rather than the total adhesion load, is the mechanical parameter conserved during noise-driven NA assembly. As stress increases, NA area may increase (causing adhesion reinforcement) when extracellular ligand density is low or remain relatively constant when it is high. Once stress becomes large enough, NA disassembly can be initiated either by stochastic effects driving the system beyond a threshold corresponding to the stable manifold of saddle points in the middle branch of the bistable switch or by an excess buildup of force pushing the system beyond the saddle node of this switch.
Increasing the complexity of the model presented here by considering different types of adaptor proteins forming the adhesion plaque to examine their effects on dynamics represents an interesting direction to pursue. This could be also combined with studying the effect of actin branching and bundling on NA dynamics. The latter, although very technically challenging, can provide further insights onto how they may exert force on the adhesion plaque.
Author Contributions
L.M. conceived the biophysical model, analyzed it, wrote the code for simulations, and drafted the manuscript. A.K. substantially revised the manuscript and provided supervision throughout the project.
Acknowledgments
Discussion with Prof. Claire Brown, Department of Physiology, McGill University, greatly benefited the development of this work.
This work was supported by the Fonds Nature et technologies, Gouvernement du Quebec (http://www.frqnt.gouv.qc.ca/en/accueil) team grant to A.K. L.M. was supported by the NSERC-CREATE in Complex Dynamics Graduate Scholarship.
Editor: Vivek Shenoy.
Footnotes
Supporting Material can be found online at https://doi.org/10.1016/j.bpj.2019.08.004.
Supporting Citations
References (81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103) appear in the Supporting Material.
Supporting Material
References
- 1.Friedl P., Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 2.Luster A.D., Alon R., von Andrian U.H. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 3.Li L., He Y., Jiang J. Collective cell migration: implications for wound healing and cancer invasion. Burns Trauma. 2013;1:21–26. doi: 10.4103/2321-3868.113331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark A.G., Vignjevic D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015;36:13–22. doi: 10.1016/j.ceb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Lo C.M., Wang H.B., Wang Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devreotes P., Janetopoulos C. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J. Biol. Chem. 2003;278:20445–20448. doi: 10.1074/jbc.R300010200. [DOI] [PubMed] [Google Scholar]
- 7.Mori Y., Jilkine A., Edelstein-Keshet L. Wave-pinning and cell polarity from a bistable reaction-diffusion system. Biophys. J. 2008;94:3684–3697. doi: 10.1529/biophysj.107.120824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin B., Holmes W.R., Levchenko A. Synthetic spatially graded Rac activation drives cell polarization and movement. Proc. Natl. Acad. Sci. USA. 2012;109:E3668–E3677. doi: 10.1073/pnas.1210295109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas Parsons J., Horwitz A.R., Schwartz M.A. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan C.E., Odde D.J. Traction dynamics of filopodia on compliant substrates. Science. 2008;322:1687–1691. doi: 10.1126/science.1163595. [DOI] [PubMed] [Google Scholar]
- 11.Morse E.M., Brahme N.N., Calderwood D.A. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53:810–820. doi: 10.1021/bi401596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S., Calderwood D.A., Ginsberg M.H. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 2000;113:3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 13.Zaidel-Bar R., Itzkovitz S., Geiger B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiller H.B., Fässler R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 2013;14:509–519. doi: 10.1038/embor.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton E.R., Byron A., Humphries M.J. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015;17:1577–1587. doi: 10.1038/ncb3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiller H.B., Hermann M.R., Fässler R. β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 2013;15:625–636. doi: 10.1038/ncb2747. [DOI] [PubMed] [Google Scholar]
- 17.Smith M.A., Hoffman L.M., Beckerle M.C. LIM proteins in actin cytoskeleton mechanoresponse. Trends Cell Biol. 2014;24:575–583. doi: 10.1016/j.tcb.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Changede R., Xu X., Sheetz M.P. Nascent integrin adhesions form on all matrix rigidities after integrin activation. Dev. Cell. 2015;35:614–621. doi: 10.1016/j.devcel.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Changede R., Cai H., Sheetz M.P. Ligand geometry controls adhesion formation via integrin clustering. bioRxiv. 2018 [Google Scholar]
- 20.Kucik D.F., Dustin M.L., Brown E.J. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest. 1996;97:2139–2144. doi: 10.1172/JCI118651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kucik D.F. Rearrangement of integrins in avidity regulation by leukocytes. Immunol. Res. 2002;26:199–206. doi: 10.1385/IR:26:1-3:199. [DOI] [PubMed] [Google Scholar]
- 22.Nishida N., Xie C., Springer T.A. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–594. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 23.Luo B.H., Carman C.V., Springer T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu T., Wu X., Kucik D.F. Affinity, lateral mobility, and clustering contribute independently to beta 2-integrin-mediated adhesion. Am. J. Physiol. Cell Physiol. 2010;299:C399–C410. doi: 10.1152/ajpcell.00039.2009. [DOI] [PubMed] [Google Scholar]
- 25.Rossier O., Octeau V., Giannone G. Integrins β1 and β3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat. Cell Biol. 2012;14:1057–1067. doi: 10.1038/ncb2588. [DOI] [PubMed] [Google Scholar]
- 26.Welf E.S., Haugh J.M. Signaling pathways that control cell migration: models and analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011;3:231–240. doi: 10.1002/wsbm.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Kooyk Y., van Vliet S.J., Figdor C.G. The actin cytoskeleton regulates LFA-1 ligand binding through avidity rather than affinity changes. J. Biol. Chem. 1999;274:26869–26877. doi: 10.1074/jbc.274.38.26869. [DOI] [PubMed] [Google Scholar]
- 28.Erdmann T., Schwarz U.S. Stochastic dynamics of adhesion clusters under shared constant force and with rebinding. J. Chem. Phys. 2004;121:8997–9017. doi: 10.1063/1.1805496. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz U.S., Erdmann T., Bischofs I.B. Focal adhesions as mechanosensors: the two-spring model. Biosystems. 2006;83:225–232. doi: 10.1016/j.biosystems.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 30.Gov N.S. Modeling the size distribution of focal adhesions. Biophys. J. 2006;91:2844–2847. doi: 10.1529/biophysj.106.088484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cirit M., Krajcovic M., Haugh J.M. Stochastic model of integrin-mediated signaling and adhesion dynamics at the leading edges of migrating cells. PLoS Comput. Biol. 2010;6:e1000688. doi: 10.1371/journal.pcbi.1000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicolas A., Safran S.A. Limitation of cell adhesion by the elasticity of the extracellular matrix. Biophys. J. 2006;91:61–73. doi: 10.1529/biophysj.105.077115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Besser A., Safran S.A. Force-induced adsorption and anisotropic growth of focal adhesions. Biophys. J. 2006;90:3469–3484. doi: 10.1529/biophysj.105.074377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabass B., Schwarz U.S. Modeling cytoskeletal flow over adhesion sites: competition between stochastic bond dynamics and intracellular relaxation. J. Phys. Condens. Matter. 2010;22:194112. doi: 10.1088/0953-8984/22/19/194112. [DOI] [PubMed] [Google Scholar]
- 35.Zhao T., Li Y., Dinner A.R. How focal adhesion size depends on integrin affinity. Langmuir. 2009;25:1540–1546. doi: 10.1021/la8026804. [DOI] [PubMed] [Google Scholar]
- 36.Olberding J.E., Thouless M.D., Garikipati K. The non-equilibrium thermodynamics and kinetics of focal adhesion dynamics. PLoS One. 2010;5:e12043. doi: 10.1371/journal.pone.0012043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welf E.S., Naik U.P., Ogunnaike B.A. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys. J. 2012;103:1379–1389. doi: 10.1016/j.bpj.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Z., Plotnikov S.V., Liu J. Two distinct actin networks mediate traction oscillations to confer focal adhesion mechanosensing. Biophys. J. 2017;112:780–794. doi: 10.1016/j.bpj.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao X., Lin Y., Shenoy V.B. A chemomechanical model of matrix and nuclear rigidity regulation of focal adhesion size. Biophys. J. 2015;109:1807–1817. doi: 10.1016/j.bpj.2015.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao X., Ban E., Shenoy V.B. Multiscale model predicts increasing focal adhesion size with decreasing stiffness in fibrous matrices. Proc. Natl. Acad. Sci. USA. 2017;114:E4549–E4555. doi: 10.1073/pnas.1620486114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walcott S., Kim D.H., Sun S.X. Nucleation and decay initiation are the stiffness-sensitive phases of focal adhesion maturation. Biophys. J. 2011;101:2919–2928. doi: 10.1016/j.bpj.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong F., García A.J., Zhu C. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 2009;185:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 44.Pereverzev Y.V., Prezhdo O.V., Sokurenko E.V. Allosteric role of the large-scale domain opening in biological catch-binding. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2009;79:051913. doi: 10.1103/PhysRevE.79.051913. [DOI] [PubMed] [Google Scholar]
- 45.Li Z., Kong F., Zhu C. A model for cyclic mechanical reinforcement. Sci. Rep. 2016;6:35954. doi: 10.1038/srep35954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J., Springer T.A. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc. Natl. Acad. Sci. USA. 2017;114:4685–4690. doi: 10.1073/pnas.1704171114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novikova E.A., Storm C. Contractile fibers and catch-bond clusters: a biological force sensor? Biophys. J. 2013;105:1336–1345. doi: 10.1016/j.bpj.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicolas A., Geiger B., Safran S.A. Cell mechanosensitivity controls the anisotropy of focal adhesions. Proc. Natl. Acad. Sci. USA. 2004;101:12520–12525. doi: 10.1073/pnas.0403539101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu C.H., Law J.B., Sheetz M.P. Early integrin binding to Arg-Gly-Asp peptide activates actin polymerization and contractile movement that stimulates outward translocation. Proc. Natl. Acad. Sci. USA. 2011;108:20585–20590. doi: 10.1073/pnas.1109485108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi C.K., Vicente-Manzanares M., Horwitz A.R. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolfenson H., Lubelski A., Geiger B. A role for the juxtamembrane cytoplasm in the molecular dynamics of focal adhesions. PLoS One. 2009;4:e4304. doi: 10.1371/journal.pone.0004304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Digman M.A., Brown C.M., Gratton E. Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys. J. 2008;94:2819–2831. doi: 10.1529/biophysj.107.104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chi L.C., Aguilar J.S., Digman M.A. Nanoimaging of focal adhesion dynamics in 3D. PLoS One. 2014;9:e99896. doi: 10.1371/journal.pone.0099896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Dévédec S.E., Geverts B., van de Water B. The residence time of focal adhesion kinase (FAK) and paxillin at focal adhesions in renal epithelial cells is determined by adhesion size, strength and life cycle status. J. Cell Sci. 2012;125:4498–4506. doi: 10.1242/jcs.104273. [DOI] [PubMed] [Google Scholar]
- 55.Tsunoyama T.A., Watanabe Y., Kusumi A. Super-long single-molecule tracking reveals dynamic-anchorage-induced integrin function. Nat. Chem. Biol. 2018;14:497–506. doi: 10.1038/s41589-018-0032-5. [DOI] [PubMed] [Google Scholar]
- 56.Brown C.M., Hebert B., Wiseman P.W. Probing the integrin-actin linkage using high-resolution protein velocity mapping. J. Cell Sci. 2006;119:5204–5214. doi: 10.1242/jcs.03321. [DOI] [PubMed] [Google Scholar]
- 57.Alexandrova A.Y., Arnold K., Verkhovsky A.B. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS One. 2008;3:e3234. doi: 10.1371/journal.pone.0003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupton S.L., Eisenmann K., Waterman-Storer C.M. mDia2 regulates actin and focal adhesion dynamics and organization in the lamella for efficient epithelial cell migration. J. Cell Sci. 2007;120:3475–3487. doi: 10.1242/jcs.006049. [DOI] [PubMed] [Google Scholar]
- 59.Tang K., Boudreau C.G., Khadra A. Paxillin phosphorylation at serine 273 and its effects on Rac, Rho and adhesion dynamics. PLoS Comput. Biol. 2018;14:e1006303. doi: 10.1371/journal.pcbi.1006303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doedel E.J., Fairgrieve T.F., Wang X. Concordia University; 2007. AUT0–07p: continuation and bifurcation software for ordinary differential equations.https://www.macs.hw.ac.uk/∼gabriel/auto07/auto.html [Google Scholar]
- 61.MacKay, L., and A. Khadra. 2019. Data from: dynamics of mechanosensitive nascent adhesion formation. Anmar Khadra Repository 2019. Deposited July 30, 2019. http://www.medicine.mcgill.ca/physio/khadralab/code_BJ1.html. [DOI] [PMC free article] [PubMed]
- 62.Wiseman P.W., Brown C.M., Horwitz A.F. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 2004;117:5521–5534. doi: 10.1242/jcs.01416. [DOI] [PubMed] [Google Scholar]
- 63.Goodsell, D.S. 2019. PDB-101: Molecule of the month: Integrin. 2011. http://pdb101.rcsb.org/motm/134.
- 64.Springer T.A., Zhu J., Xiao T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J. Cell Biol. 2008;182:791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu J., Luo B.H., Springer T.A. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lau T.L., Kim C., Ulmer T.S. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wegener K.L., Partridge A.W., Campbell I.D. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 68.Hamill K.J., Hiroyasu S., Jones J.C.R. Alpha actinin-1 regulates cell-matrix adhesion organization in keratinocytes: consequences for skin cell motility. J. Invest. Dermatol. 2015;135:1043–1052. doi: 10.1038/jid.2014.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim D.H., Wirtz D. Focal adhesion size uniquely predicts cell migration. FASEB J. 2013;27:1351–1361. doi: 10.1096/fj.12-220160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawson C., Lim S.T., Schlaepfer D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Velasco-Velázquez M.A., Salinas-Jazmín N., Mandoki J.J. Reduced paxillin expression contributes to the antimetastatic effect of 4-hydroxycoumarin on B16-F10 melanoma cells. Cancer Cell Int. 2008;8:8. doi: 10.1186/1475-2867-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hagel M., George E.L., Thomas S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 2002;22:901–915. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Changede R., Sheetz M. Integrin and cadherin clusters: a robust way to organize adhesions for cell mechanics. Bioessays. 2017;39:1–12. doi: 10.1002/bies.201600123. Published online December 8, 2016. [DOI] [PubMed] [Google Scholar]
- 74.Arnold M., Cavalcanti-Adam E.A., Spatz J.P. Activation of integrin function by nanopatterned adhesive interfaces. ChemPhysChem. 2004;5:383–388. doi: 10.1002/cphc.200301014. [DOI] [PubMed] [Google Scholar]
- 75.Cavalcanti-Adam E.A., Micoulet A., Spatz J.P. Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly. Eur. J. Cell Biol. 2006;85:219–224. doi: 10.1016/j.ejcb.2005.09.011. Published online October 10, 2005. [DOI] [PubMed] [Google Scholar]
- 76.Schvartzman M., Palma M., Wind S.J. Nanolithographic control of the spatial organization of cellular adhesion receptors at the single-molecule level. Nano Lett. 2011;11:1306–1312. doi: 10.1021/nl104378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saez A., Buguin A., Ladoux B. Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J. 2005;89:L52–L54. doi: 10.1529/biophysj.105.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roca-Cusachs P., del Rio A., Sheetz M.P. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA. 2013;110:E1361–E1370. doi: 10.1073/pnas.1220723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ballestrem C., Hinz B., Wehrle-Haller B. Marching at the front and dragging behind: differential alphaVbeta3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 2001;155:1319–1332. doi: 10.1083/jcb.200107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu K., Ji L., Waterman-Storer C.M. Differential transmission of actin motion within focal adhesions. Science. 2007;315:111–115. doi: 10.1126/science.1135085. [DOI] [PubMed] [Google Scholar]
- 81.Lagarrigue F., Vikas Anekal P., Ginsberg M.H. A RIAM/lamellipodin-talin-integrin complex forms the tip of sticky fingers that guide cell migration. Nat. Commun. 2015;6:8492. doi: 10.1038/ncomms9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cluzel C., Saltel F., Wehrle-Haller B. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J. Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aratyn-Schaus Y., Gardel M.L. Transient frictional slip between integrin and the ECM in focal adhesions under myosin II tension. Curr. Biol. 2010;20:1145–1153. doi: 10.1016/j.cub.2010.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Felsenfeld D.P., Choquet D., Sheetz M.P. Ligand binding regulates the directed movement of β1 integrins on fibroblasts. Nature. 1996;383:438–440. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- 85.Chang A.C., Mekhdjian A.H., Dunn A.R. Single molecule force measurements in living cells reveal a minimally tensioned integrin state. ACS Nano. 2016;10:10745–10752. doi: 10.1021/acsnano.6b03314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Milligen B. Ph., Bons P.D., Sánchez R. On the applicability of Fick’s law to diffusion in inhomogeneous systems. Eur. J. Phys. 2005;26:913–925. [Google Scholar]
- 87.Tupper P.F., Yang X. A paradox of state-dependent diffusion and how to resolve it. Proc. R. Soc. A Math. Phys. Eng. Sci. 2012;468:3864–3881. [Google Scholar]
- 88.Stratonovich R.L. Fokker-planck equation with discontinuous coefficients and conditions at the surface of discontinuity. Sov. Radiophys. 1965;8:500–505. [Google Scholar]
- 89.Pereverzev Y.V., Prezhdo E., Sokurenko E.V. The two-pathway model of the biological catch-bond as a limit of the allosteric model. Biophys. J. 2011;101:2026–2036. doi: 10.1016/j.bpj.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kloeden P.E., Platen E. Springer-Verlag; Berlin Heidelberg: 1992. Numerical Solution of Stochastic Differential Equations. [Google Scholar]
- 91.Vestergaard C.L., Génois M. Temporal gillespie algorithm: fast simulation of contagion processes on time-varying networks. PLoS Comput. Biol. 2015;11:e1004579. doi: 10.1371/journal.pcbi.1004579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kong F., Li Z., Zhu C. Cyclic mechanical reinforcement of integrin-ligand interactions. Mol. Cell. 2013;49:1060–1068. doi: 10.1016/j.molcel.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cavalcanti-Adam E.A., Volberg T., Spatz J.P. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 2007;92:2964–2974. doi: 10.1529/biophysj.106.089730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kingma D.P., Welling M. Auto-encoding variational Bayes. 2013. https://arxiv.org/abs/1312.6114 arXiv, arXiv:1312.6114.
- 95.Rippel O., Adams R.P. High-dimensional probability estimation with deep density models. 2013. https://arxiv.org/abs/1302.5125 arXiv, arXiv:1302.5125.
- 96.Dinh L., Sohl-Dickstein J., Bengio S. Density estimation using Real NVP. 2016. https://arxiv.org/abs/1605.08803 arXiv, arXiv:1605.08803v3.
- 97.Tadavani P.K. University of Waterloo; 2013. Nonlinear dimensionality reduction by manifold unfolding. PhD thesis. [Google Scholar]
- 98.Roberts S., Osborne M., Aigrain S. Gaussian processes for time-series modelling. Philos. Trans. A Math. Phys. Eng. Sci. 2013;371:20110550. doi: 10.1098/rsta.2011.0550. Published online December 31, 2012. [DOI] [PubMed] [Google Scholar]
- 99.Hastie T., Tibshirani R. Generalized additive models for medical research. Stat. Methods Med. Res. 1995;4:187–196. doi: 10.1177/096228029500400302. [DOI] [PubMed] [Google Scholar]
- 100.Rasmussen C.E., Williams C.K.I. MIT Press; Cambridge, MA: 2006. Gaussian Processes for Machine Learning, First Edition; pp. 1–248. [Google Scholar]
- 101.Ozair S., Bengio Y. Deep directed generative autoencoders. https://arxiv.org/abs/1410.0630 arXiv, arXiv:1410.0630v1.
- 102.Perrone D., Favaro P. A clearer picture of total variation blind deconvolution. IEEE Trans. Pattern Anal. Mach. Intell. 2016;38:1041–1055. doi: 10.1109/TPAMI.2015.2477819. [DOI] [PubMed] [Google Scholar]
- 103.Tikhonov A.N., Arsenin V.Y. Winston; Washington D.C.: 1977. Solutions of Ill-Posed Problems. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.