

Abstract
Glycans linked to proteins and lipids play key roles in biology; thus, accurate replication of cellular glycans is crucial for maintaining function following cell division. The fact that glycans are not copied from genomic templates suggests that fidelity is provided by the catalytic templates of glycosyltransferases that accurately add sugars to specific locations on growing oligosaccharides. To form new glycosidic bonds, glycosyltransferases bind acceptor substrates and orient a specific hydroxyl group, frequently one of many, for attack of the donor sugar anomeric carbon. Several recent crystal structures of glycosyltransferases with bound acceptor substrates reveal that these enzymes have common core structures that function as scaffolds upon which variable loops are inserted to confer substrate specificity and correctly orient the nucleophilic hydroxyl group. The varied approaches for acceptor binding site assembly suggest an ongoing evolution of these loop regions provides templates for assembly of the diverse glycan structures observed in biology.
Animal cell surfaces are covered with thousands of distinct glycan structures attached to proteins and lipids1. These glycans participate in numerous biological phenomena including modulating cellular communication, mediating immune functions, targeting proteins to specific subcellular organelles, assisting in protein folding, altering recycling of receptors, and protecting proteins from degradation2,3. Cells in diverse tissues, or in different stages of development, have distinct glycan structures on their surfaces, leading to differential functional consequences in those contexts. When a cell divides, these glycan structures are replicated; however, because glycans are not directly synthesized from a genome-encoded template, as proteins and nucleic acids are, the fidelity is dependent on the specificity of the glycosyltransferases (GTs) that synthesize complex glycans by adding one sugar at a time. Most eukaryotic GTs are localized to the lumen of the secretory pathway and modify proteins and/or lipids as they transit to the cell surface or extracellular spaces4. Most GTs catalyze sugar transfer from a donor molecule (nucleotide sugar or lipid-linked sugar) to the hydroxyl group of an acceptor molecule5. For GTs that employ nucleoside diphosphate (NDP) sugar donors, the generalized reaction can be depicted as:
We have learned a great deal about how GTs bind donor nucleotide sugars, but specificities of GTs reside in their ability to selectively modify the correct hydroxyl group on an acceptor containing many equally reactive hydroxyls (for example, complex oligosaccharides). Here we review recent structures of ternary complexes of eukaryotic GTs including donors, donor analogs, and acceptors, providing details of how these enzymes modify the correct hydroxyl group and provide fidelity to accurately replicate the glycan structures on cell surfaces.
The Carbohydrate-Active Enzymes (CAZy) database (http://www.cazy.org) annotates nearly 500,000 GT sequences across all kingdoms of life and organizes them into 106 different families6, and the human genome encodes ~210 GTs4 in 45 CAZy families. The first three-dimensional structure of a GT was that of rabbit glycogen phosphorylase, deposited in 19907. The fact that most eukaryotic GTs in the secretory pathway require disulfide bonding, N-glycosylation, and eukaryotic chaperones for proper folding makes production of sufficient amounts of these enzymes for structural analysis a challenge4,8. This limitation has been a major barrier in the field, but recent availability of eukaryotic expression systems (insect cells; HEK293 cells4) has allowed for elucidation of more structures of eukaryotic enzymes in the past decade. As a result, since 2000 the number of independent GT structures has grown steadily to ~209 from 53 different GT families (56% bacterial, 38% eukaryotic, 4% archaeal, 1% viral; Supplementary Fig. 1; Supplementary Dataset 1).
Although there are many GT families, structural analysis has shown that their catalytic domains fall into a small number of protein folds: GT-A, GT-B, and GT-C (Table 1)5,9. GT-A enzymes contain a single Rossmann-like fold, a domain found in enzymes that bind nucleotides. Most, but not all, GT-A fold enzymes require divalent cations for activity and contain a ‘DxD’ motif that is involved in coordinating the metal and nucleotide sugar. GT-B fold enzymes contain two Rossmann-like folded domains, but do not require divalent cations for activity or contain DxD motifs. GT-C fold enzymes, such as oligosaccharyltransferases, contain multiple transmembrane α-helices and employ lipid-linked sugar donors. For the sake of brevity, we will not discuss GT-C enzymes, CAZy GT1 family enzymes that act on small-molecule metabolites, or the nuclear/cytoplasmic O-GlcNAc transferase, which have all been recently reviewed10–12, but will focus on recent eukaryotic GT-A and GT-B fold structures that synthesize cell surface and secreted glycoproteins.
Table 1 |.
Characteristics of GT-A, GT-B, and GT-C GTs
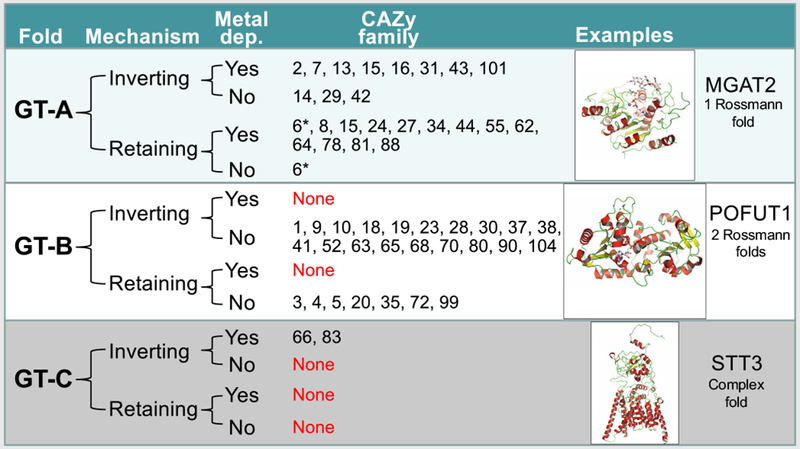 |
The GT structures in the PDB that are listed in Supplementary Dataset 1 were divided by fold type, catalytic mechanism, and metal dependency for sugar donor interaction and listed by CAZy family along with a cartoon protein structure display for a representative member of each fold family (GT-A, MGAT2 (PDB: 5VCM and 5VCS31); GT-B, POFUT1 (PBD: 5KY020); GT-C, STT3 (PDB: 6C2686)). In one case (GT6, asterisk) there is one bacterial family member (BACOVA_0193289) that is a metal-independent retaining enzyme, whereas other eukaryotic GT6 family members (GGTA125 and ABO47) are metal dependent.
Mechanistic strategies
Nucleotide-sugar-dependent GTs use a restricted set of donor molecules to make the diverse collection of glycan structures in cellular systems and generate enzymatic products with distinct anomeric configurations. Each of the GT families can either invert or retain the stereochemistry of the anomeric linkage of the donor sugar during transfer, often to discrete hydroxyl groups on the acceptor structure5,9. For example, β-1,4-galactosyltransferase 1 (B4GALT1) is an inverting enzyme. It transfers galactose (Gal) from UDP-α-Gal to the 4′-hydroxyl of N-acetylglucosamine (GlcNAc) to form a Gal-β-1,4-GlcNAc product (Fig. 1a)13–15. Most inverting enzymes utilize an SN2 single-displacement reaction mechanism (Fig. 1b) in which the acceptor nucleophilic hydroxyl attacks the donor sugar anomeric carbon and displaces the leaving group nucleotide moiety from the opposite face. The result is an inversion of the anomeric configuration of the product5,9. In most instances the enzyme-bound sugar nucleotide donor assumes a ‘folded back’ conformation during sugar transfer, in contrast to the more extended conformation of the donor in solution16–18. Significant conformational change in loop regions of the enzyme may also occur upon binding the sugar donor13. These enzymes employ a catalytic base (typically Glu, Asp, or His), which assists by deprotonating the nucleophile. A few inverting enzymes without an identifiable catalytic base have also recently been identified (POFUT119,20 and FUT121). In both cases an SN1 reaction mechanism was proposed in which the β-phosphate oxygen atom of the GDP-Fuc donor assists in the deprotonation of the acceptor hydroxyl group (Fig. 1c)19–21. An active site water molecule may promote the reaction by providing a proton relay between the hydroxyl and the β-phosphate oxygen20,21.
Fig. 1 |. GT reaction mechanisms.
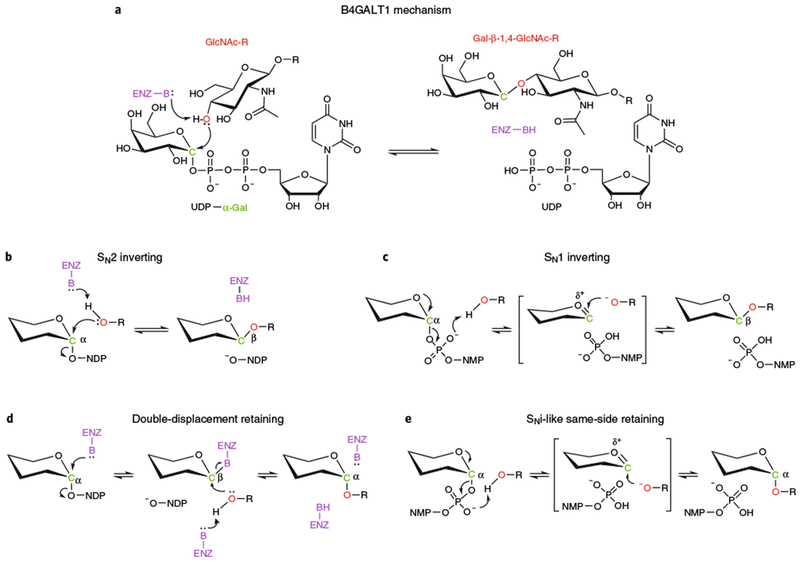
a, B4GALT1 reaction. b, SN2 inverting mechanism. c, SN1 inverting mechanism. d, Double-displacement retaining mechanism. e, SNi-like same-side retaining mechanism. All mechanisms in b-e are drawn using a generic hexose in α-linkage to a nucleoside diphosphate. The β-phosphate is drawn out when it participates in the reaction (panels c and e). Enz-B and Enz-BH (purple) indicate the catalytic base that deprotonates the acceptor nucleophile. NMP and NDP are nucleoside monophosphate and nucleoside diphosphate, respectively. Red O indicates nucleophilic oxygen of acceptor substrate. Green C indicates anomeric carbon of donor sugar. R indicates acceptor substrate. The linkage of anomeric carbon in the hexose is indicated in both substrates and products.
The reaction mechanism for retaining GTs has been debated for many years, although several recent structures have provided significant insights22–25. Similarly to retaining glycosidases that utilize a double-displacement mechanism with a transient covalent enzyme–substrate intermediate5,9, retaining GTs were also originally thought to employ a covalent intermediate (Fig. 1d)5. However, no convincing evidence of a covalent intermediate for a native retaining GT has been obtained, and in most cases there is not an appropriate enzyme nucleophile in proximity to accept the intermediate or a catalytic base to deprotonate the acceptor nucleophile. An alternative mechanism for a same-side SNi-type reaction has recently been proposed in which the acceptor hydroxyl nucleophile is deprotonated by the donor β-phosphate oxygen and attacks the anomeric carbon atom of the donor sugar from the same side as the leaving nucleotide, resulting in retention of anomeric configuration (Fig. 1e). Several structural snapshots of enzyme–donor–acceptor Michaelis complexes for retaining GTs have recently appeared (XXYLT22, GALNT223, GGTA125, and GpgS24). The position of the nucleophilic acceptor hydroxyl group relative to the anomeric carbon atom, together with snapshots showing progression of the reaction, strongly support the SNi-type mechanism.
The contrast of inverting and retaining catalytic mechanisms is illustrated by comparison of two GT-A fold enzymes, B4GALT726 and GGTA24,25 (Fig. 2a,c). Both enzymes employ GT-A folds and metal-dependent interactions with their UDP-Gal donors. B4GALT7, an inverting GT, employs an Asp side chain on a conserved α-helix to deprotonate the Xyl C4 nucleophilic hydroxyl group of the acceptor glycan during its SN2 in-line nucleophilic attack of the donor sugar (Figs. 1 and 2a). By comparison, GGTA1, a retaining GT, maintains a Glu residue at the position equivalent to that of the B4GALT7 catalytic base but shifts the position of the incoming acceptor Gal C3 hydroxyl nucleophile by >2Å away from the respective amino acid side chain, precluding its use as a catalytic base (Fig. 2c). This shift in the position for the acceptor nucleophile has two consequences for the catalytic mechanism. First, the catalytic base function is now provided by the β-phosphate oxygen of the sugar donor. Second, the angle of attack of the nucleophilic hydroxyl on the donor C1 atom is changed such that an in-line, direct displacement reaction is no longer possible (Fig. 2b). In its place, a same-side dissociative SNi-type mechanism has been proposed (Figs. 1 and 2c) to account for the retained anomeric configuration of the product, the altered geometry of nucleophilic attack, and the distinct mechanism for nucleophile deprotonation24,25. Thus, positioning of the acceptor hydroxyl group relative to the donor anomeric carbon is critical in establishing the enzyme catalytic mechanism. These mechanisms have been reviewed in more detail elsewhere25,27–30.
Fig. 2 |. Comparison of representative inverting and retaining catalytic mechanisms and enzyme topologies for substrate interactions.
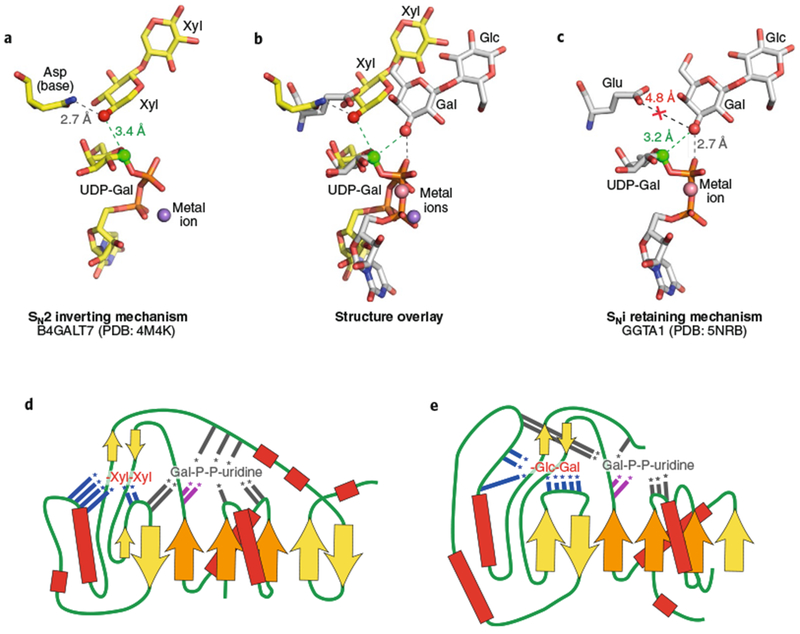
Bound ligands for B4GALT7 (a, PDB: 4M4K26) and GGTA1 (c, PDB: 5NRD25) are displayed along with putative catalytic bases. a, The inverting SN2 catalytic mechanism for B4GALT7 is catalyzed by deprotonation of the Xyl acceptor hydroxyl (red sphere) by the catalytic base (Asp211 in the wild type enzyme mutated to Asn211 in PDB ID 4M4K) and attack of the anomeric carbon (green sphere) in line with the departing nucleotide leaving group. c, GGTA1 has a retaining SNi-like mechanism in which the nucleophile position (red sphere) is shifted relative to the inverting enzymes. The equivalent catalytic base residue (Glu317) is too far away for deprotonation, but the acceptor Gal hydroxyl (red sphere) is positioned adjacent to the β-phosphate oxygen for deprotonation (gray dotted line) and in proximity to attack the anomeric carbon (green sphere). b, Overlay of the two structures based on the Gal C1-phosphate oxygen bond illustrates the differences in geometry of nucleophilic attack on the anomeric carbons. d,e, Donor and acceptor binding sites for B4GALT7 (d) and GGTA1 (e) are displayed as topology diagrams illustrating the positions of residues that interact with donors and acceptors in loops extending from the core β-sheets of the Rossmann fold. β-Sheets are indicated as filled arrows (orange for conserved Rossmann-fold elements and yellow for nonconserved sheets among GT-A fold enzymes) and helices as red boxes connected by loop regions (green lines). Positions of interacting residues are indicated by colored lines with asterisks (gray, interacting with donors; blue, interacting with acceptors; magenta, DxD motif). Acceptors for each enzyme (B4GALT7: Xyl-β-1,4-Xyl and GGTA1: Gal-β-1,4-Glc) are indicated in abbreviated form, but with inverted orientation from standard nomenclature to indicate the proximity of the terminal sugar acting as nucleophile in the glycosyltransfer reaction.
Assembly of active site donor and acceptor modules
The recent structures of a growing number of eukaryotic GTs containing bound donors (or donor analogs) and/or acceptor structures (Supplementary Dataset 1) have also provided insights into how these enzymes can catalyze selective glycan synthesis. These structures demonstrated that GT active sites are assembled as modular components extending from the stable Rossmann-fold scaffolds that bind donor and acceptor molecules with precise geometry and reactivity (Fig. 2d,e). GT-A and GT-B fold enzymes both employ a parallel β-sheet Rossmann-fold structure to anchor loops that bind the nucleotide portion of the donor substrate (for example, the inverting enzyme B4GALT7 complex with UDP-Gal26, Figs. 2d and 3; the retaining enzyme, GGTA1, Figs. 2e and 5). Direct interactions with the nucleotide are surprisingly variable among the collection of GTs31, yet each is capable of providing complementary specificity for the respective nucleotide base and donor sugar (Figs. 2–5). The GT-A fold enzymes that employ divalent cations use a carboxylate residue of the DxD motif (and sometimes His residues26) to coordinate the metal, which interacts with the nucleotide phosphodiester of the donor (for example, MGAT1 interaction with UDP-GlcNAc32, Fig. 3b), whereas metal-independent GT-A fold enzymes generally employ Lys and Arg side chains for phosphodiester interactions (for example, GCNT1 interaction with UDP33,34, Fig. 3b). GT-B fold enzymes employ one of their two Rossmann folds for metal-independent sugar nucleotide interactions using a similar set of Lys and Arg side chains (for example, POFUT1 interaction with GDP-Fuc20), similarly to the metal-independent GT-A fold enzymes (Table 1).
Fig. 3 |. Structural gallery of eukaryotic GT-A fold inverting enzymes.
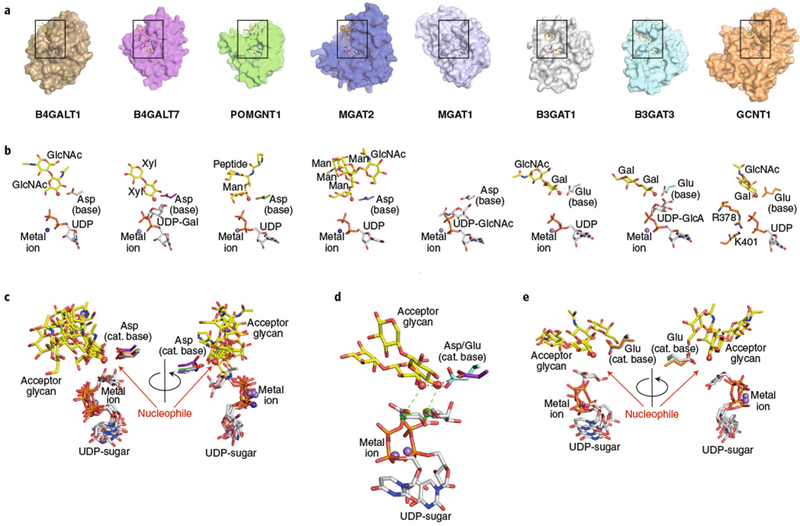
a, Eight eukaryotic GT-A fold inverting enzyme structures were aligned using Coot88 (transparent surface) with bound donor analogs (white sticks), acceptor analogs (yellow sticks), divalent cations (spheres), and catalytic bases (sticks). Structures include B4GALT1 (PDB: 1TW513), B4GALT7 (PDB: 4M4K26), POMGNT1 (PDB: 5GGI44), MGAT2 (overlay of PDB IDs 5VCM and 5VCS31), MGAT1 (PDB: 1FOA32), B3GAT1 (PDB: 1V8445), B3GAT3 (PDB: 1FGG46), and GCNT1 (overlay of PDB IDs 2GAM and 3OTK33,34). Boxes represent the regions of the respective structures shown in b. b, Enlarged views of the bound ligands are displayed in the same orientation and coloring as in a, with individual molecule components labeled. For GCNT1, the Lys and Arg residues that interact with the phosphodiester are indicated (sticks). c, Bound ligand components for enzymes that employ Asp as the catalytic base, shown in two different rotations. e, Bound ligand components for enzymes that employ Glu as the catalytic base, shown in two different rotations. d, Overlay of two representative enzymes (B4GALT7 employing an Asp catalytic base and B3GAT3 employing a Glu catalytic base) illustrating the differences in position for the nucleophiles and sugar donors as a consequence of the differences of length of the catalytic base side chains. Acceptors, donors (C1 as green sphere) metal ions and the nucleophilic hydroxyl (red sphere) are labeled, and the positions of nucleophilic attack (green dotted line) and catalytic base (black dotted line) action are indicated.
Fig. 5 |. Structural gallery of eukaryotic GT-A fold retaining enzymes.
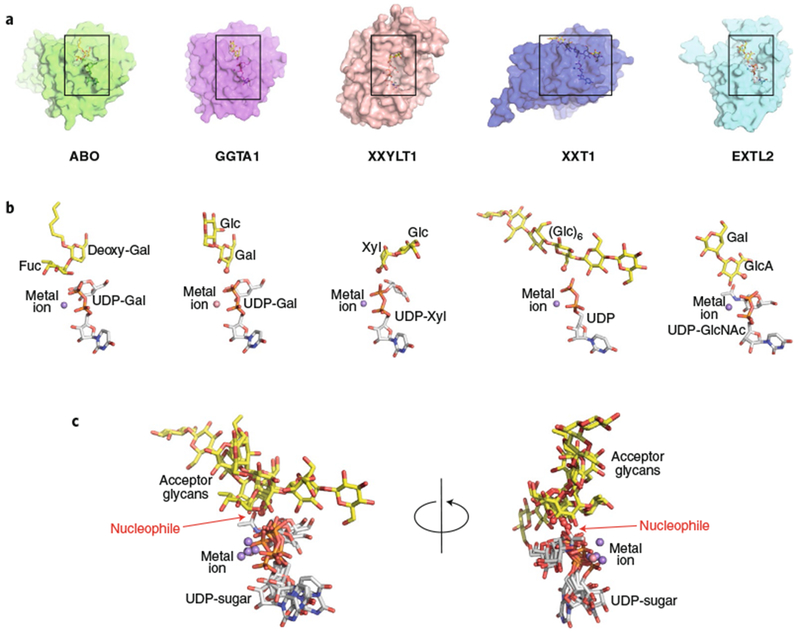
a, Five eukaryotic GT-A fold retaining enzyme structures were aligned using Coot88 (transparent surface) with bound donor analogs (white sticks), acceptor analogs (yellow sticks), and divalent cations (spheres). Structures are ABO (PDB: 2RJ747), GGTA1 (PDB: 5NRD25), XXYLT1 (PDB: 4WNH22), XXT1 (PDB: 6BSW48) and EXTL2 (overlay of PDB IDs 1ON8 and 1OMZ49). Boxes represent the regions of the respective structures shown in b. b, Enlarged views of the bound ligands are displayed in the same orientation and coloring as in a, with donors, acceptors, metal ions, catalytic bases labeled as in Fig. 3. c, Overlay of the aligned ligand components, shown in two different rotations.
Although GT-A and GT-B fold enzymes both use analogous approaches for nucleotide sugar donor interactions, the enzymes differ in their interactions with extended acceptor structures. GT-A fold enzymes employ extensions and insertions into their single Rossmann core fold to assemble acceptor binding site modules31. The diversity in positions and structures of the loop insertions among the GT-A fold enzymes is quite striking (Fig. 2d,e), even among enzymes in the same CAZy family (for example, GT29 NeuAc transferases4,35–38 and GT14 xylosyl and GlcNAc transferases33,39), and unique complementarities for each of the respective acceptors are achieved using these loop extensions (see below).
For GT-B fold enzymes, the two Rossmann folds are linked in tandem with donor (A domain) and acceptor (B domain) binding subsites facing into the cleft between the two domains (Fig. 7)5. The acceptor binding subsites are assembled largely from loops extending from the B domain into the cleft (for example, MGAT540 and FUT121,41), but there are several examples of GTs that employ residues from both domains for acceptor interactions (POFUT120, POFUT242, and POGLUT143). In addition, the geometry of the cleft can vary significantly among the GT-B fold enzymes to accommodate extended, branched glycan structures (for example, MGAT540) and FUT121,41)) or globular protein domain structures (POFUT120, POFUT242, and POGLUT143, Fig. 7).
Fig. 7 |. Domain specific GTs (POFUT1, POFUT2, and POGLUT1) with substrates.
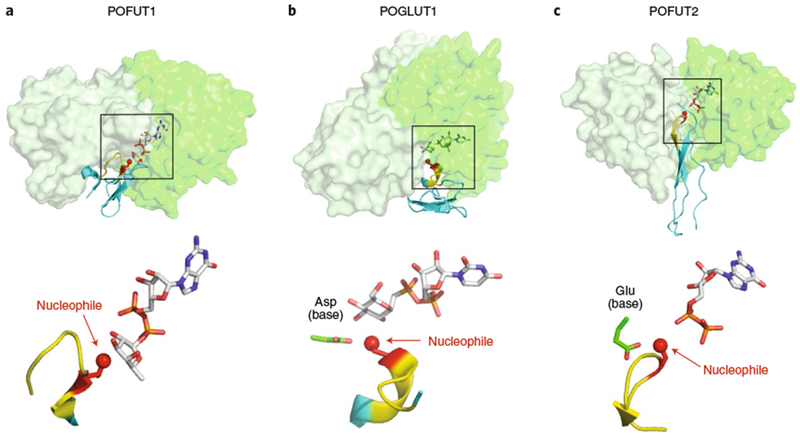
a, POFUT1 (overlay of PDB IDs 5KXH and 5KY320). b, POGLUT1 (overlay of PDB IDs 5L0R and 5L0U43). c, POFUT2 (PDB: 5FOE42). Upper panels: all three GT-B fold enzymes are displayed in transparent surface representations with A domains in light green and B domains in darker green. Folded domains (EGF repeats for POFUT1 and POGLUT2; TSR for POFUT2) are in cyan cartoon with respective consensus sequences in yellow. Acceptor amino acid is shown in red sticks, with the acceptor oxygen as a red sphere. Donor nucleotides, white sticks. Note that the POGLUT1 structure has the donor analog UDP-CH2-Glc. Lower panels: enlarged views of the bound ligands are displayed in the same orientation and coloring as the upper panels, with individual molecule components labeled. Boxes in the upper panels represent the regions of the respective structures shown in the lower panels.
The overall conclusions from these diverse substrate-bound structures are that GTs have a conserved and restricted positioning for the nucleotide sugar in the donor binding subsite based on the constraints of interacting loops that extend from the Rossmann fold (Fig. 2d,e). By contrast, the striking diversity in binding geometries for glycan, peptide, lipid or metabolite acceptors shows a common theme for precise positioning of the nucleophilic hydroxyl group appropriately for catalysis leading to either inversion (SN2- or SN1-type mechanisms) or retention (SNi-type mechanism) in anomeric configuration upon sugar transfer (Figs. 1 and 2a–c)31.
Structures of eukaryotic secretory-pathway GTs containing bound acceptors now total 18 enzymes for glycan-directed GTs and 7 structures for peptide-directed GTs (Supplementary Dataset 1). Glycan-directed eukaryotic enzymes include the metal-dependent inverting GT-A fold enzymes B4GALT113, B4GALT726 (GT7), POMGNT144 (GT13), MGAT231 (GT16), B3GAT145, and B3GAT346 (GT43) (Fig. 3). Metal-independent GT-A fold inverting enzymes include GCNT134 (GT14) (Fig. 3), ST3GAL137, ST6GAL135, and ST8SIA338 (GT29) (Fig. 4). Enzyme–acceptor complexes for metal-dependent retaining GT-A fold enzymes include ABO47 and GGTA125 (GT6), XXYLT122 (GT8), XXT148 (GT34), and EXTL249 (GT64) (Fig. 5). Glycan-directed inverting GT-B enzymes include MGAT540 (GT18) and FUT121,41 (GT37). Polypeptide-directed GT-A fold enzymes include the inverting enzyme XYLT1 (GT14) and the retaining enzymes GALNT2 and GALNT4 (GT27) (Fig. 6). GT-B fold inverting enzymes include POFUT120 (GT65), POFUT242 (GT68), and POGLUT143 (GT90) (Fig. 7), as well as OGT50 (GT41). These structures are described in more detail in the following sections.
Fig. 4 |. Structural gallery of eukaryotic GT29 sialyltransferases as acceptor-bound complexes.
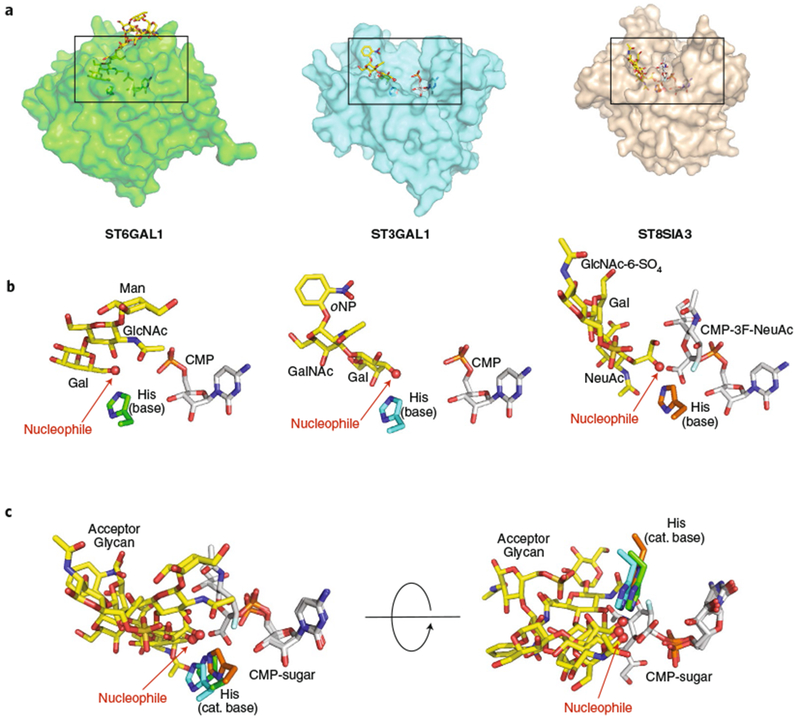
a, Three eukaryotic GT29 sialyltransferase structures (GT-A fold (variant 2)) were aligned using Coot88 (transparent surface) with bound donor analogs (white sticks), acceptor analogs (yellow sticks), and catalytic bases (sticks). Structures include ST6GAL1 (PDB: 4JS235), ST3GAL1 (PDB: 2WNB37), and ST8SIA3 (PDB: 5BO938). Boxes represent the regions of the respective structures shown in b. b, Enlarged views of the bound ligands are displayed in the same orientation and coloring as in a, with individual donors, acceptors, catalytic bases labeled as in Fig. 3. For ST6GAL1, the large Gal2GlcNAc2Man3GlcNAc2-Asn ligand has been simplified to the terminal Gal-β-1,4-GlcNAc-β-1,2-Man acceptor. c, Overlay of the three bound ligand complexes in two different orientations.
Fig. 6 |. Glycosyltransferases recognizing linear peptides.
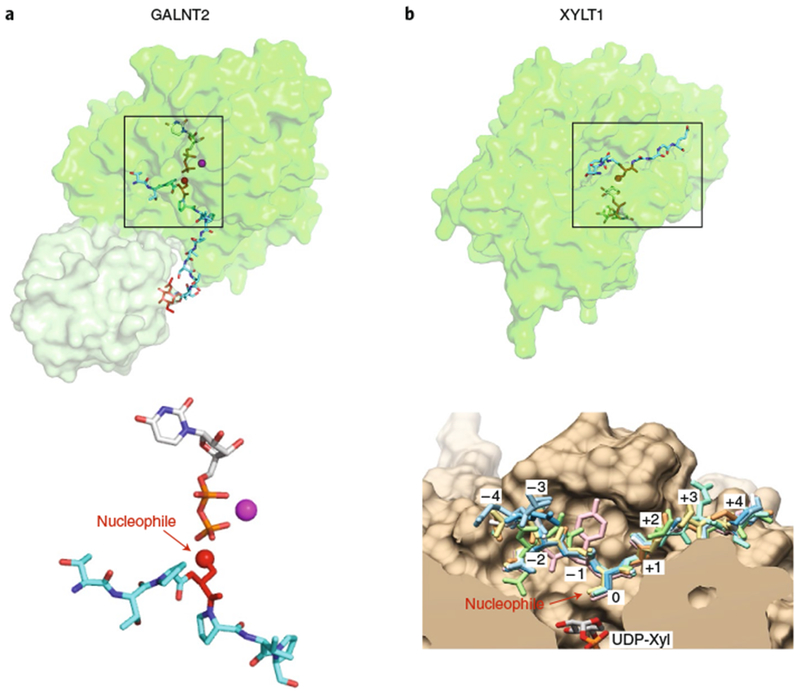
a, GALNT2 (PDB: 5AJP69). Enzyme represented in transparent surface representation, with the catalytic domain in green and lectin domain in light green. Donor nucleotide, white sticks; acceptor peptide, cyan sticks; GalNAc on acceptor peptide, red sticks; manganese, magenta sphere. Lower panel: enlarged view of the bound ligands are displayed in the same orientation and coloring as that shown on top with individual molecule components labeled. Acceptor amino acid is shown in red sticks, with the acceptor oxygen as a red sphere. b, XYLT1 (overlay of PDB IDs 6EJ7 and 6EJ839). The catalytic domain of the enzyme is represented in transparent surface representation in green. Donor nucleotide, white sticks; acceptor peptide, cyan sticks. Lower panel: enlarged cut-through view of the active site in tan with donor UDP-Xyl at the bottom, showing superposition of eight acceptor peptides in sticks (reproduced with permission from ref. 39). Boxes in the upper panels represent the regions of the respective structures shown in the lower panels.
Glycan-modifying glycosyltransferases
Metal-dependent, inverting GT-A fold enzymes.
Several recent glycan-directed enzyme–substrate complexes have been solved for inverting GT-A fold enzymes that provide notable insights into acceptor interactions (Fig. 3). Alignment of six metal-dependent, inverting GT-A fold enzymes containing bound acceptors (B4GALT113, B4GALT726, POMGNT144, MGAT231, B3GAT145 and B3GAT346), as well as MGAT1 as an intact UDP-GlcNAc donor complex and GCNT1 as a representative metal-independent inverting GT-A fold enzyme, revealed conserved catalytic elements (Fig. 3). Each enzyme acts on a distinct glycan acceptor structure and employs a unique collection of acceptor binding interactions assembled from loops and extensions emerging from different positions in the respective core Rossmann folds (Fig. 2d,e). Despite the diversity of interactions, the positioning of the catalytic hydroxyl nucleophile is similar among all of the inverting enzymes in its proximity to the catalytic base and the C1 of the donor sugar residue (Fig. 3). However, two distinct patterns are revealed in the structural alignments. For five of the enzymes, the catalytic base is an Asp residue, and the corresponding position of the nucleophile and C1 of the donor sugar are almost identical (Fig. 3c). By contrast, for B3GAT1, B3GAT3, and GCNT1 the catalytic base is a Glu residue, and the positions for the catalytic base, nucleophile, and C1 of the respective sugar donor are also essentially identical, but in a position that is shifted relative to the enzymes that employ Asp as the catalytic base (Fig. 3e). All eight enzymes position the catalytic bases at the end of a highly conserved α-helix. As a result, the differences in the lengths of the side chains for the catalytic bases result in a coordinated shift in position for both the corresponding nucleophilic hydroxyls and C1 of the sugar donors to accomplish their in-line SN2 catalytic mechanisms (Fig. 3d). Surprisingly, metal-independent GCNT1 and metal-dependent B3GAT1 and B3GAT3 share identical positioning of the catalytic base, nucleophile, and C1 of the sugar donor (or presumed C1 position for GCNT1 and B3GAT1) (Fig. 3e). These data suggest that the identity of the catalytic base, not the metal-dependent or -independent donor sugar interactions, determines the positioning of the donor and nucleophile for the GT-A fold inverting enzymes.
A highlight of these differences in acceptor interactions is the comparison of enzyme–acceptor complexes for MGAT2 (GT16) and POMGNT1 (GT13)31 (Fig. 3). These enzymes are structurally similar in their Rossmann-fold cores: both employ UDP-GlcNAc as donors, and both generate β-1,2-GlcNAc linkages on terminal Man residues using a metal-dependent inverting catalytic mechanism. MGAT2 initiates N-glycan branching in the Golgi during complex type N-glycan synthesis. The structure of its GlcNAcMan3GlcNAc2-Asn acceptor complex reveals substrate interactions through an extended binding site that anchors a terminal GlcNAc-β-1,2-Man-α-1,3-Man ‘recognition arm’ at a distal exosite and positions the Man-α-1,6-Man acceptor arm in the catalytic site31 (Fig. 3). By contrast, POMGNT1 modifies peptide-linked O-Man residues found predominately on α-dystroglycan, a component of the multiprotein dystrophin glycoprotein complex at the cell surface that interacts with the extracellular matrix in many tissues, including muscle51,52. Defects in α-dystroglycan glycosylation can lead to congenital dystroglycanopathies with muscle, eye, and brain pathologies51,53. POMGNT1 recognizes terminal Man residues on dystroglycan, but additional interactions with the peptide backbone also contribute to substrate specificity. Although acceptor substrates for MGAT2 and POMGNT1 are structurally distinct, and the acceptor binding sites are completely different, these two enzymes present their Man acceptor residues and their reactive C2 hydroxyl nucleophiles in essentially identical positions relative to the catalytic base and donor sugars (Fig. 3 and ref. 31). Thus, the extended acceptor binding sites act as templates complementary to their respective acceptor structures to achieve common positioning of the acceptor residue and identical catalytic mechanisms.
The MGAT2 exosite interactions with the GlcNAc-β-1,2-Man-α-1,3-Man ‘recognition arm’ were also found to resemble the bound substrate geometry and interactions for an unrelated enzyme in the N-glycan biosynthetic pathway, Golgi α-mannosidase II (MAN2A1)54. MAN2A1 acts one step before MGAT2 in N-glycan biosynthesis and cleaves two Man residues from its GlcNAcMan5GlcNAc2-Asn substrate to produce the GlcNAcMan3GlcNAc2-Asn acceptor for MGAT2. The two enzymes have completely different protein folds and catalytic mechanisms, yet they both employ convergent exosite architectures for interaction with the same ‘recognition arm’ (GlcNAc-β-1,2-Man-α-1,3-Man) substructure common to both substrates31. This use of convergent exosite interactions by MGAT2 and MAN2A1 for recognition of a common glycan substructure is in contrast to the larger collection of GT-A fold enzymes that use similar core fold elements for sugar donor interactions and highly divergent loop regions to recognize and position a diverse number of acceptor structures for similar mechanisms of sugar transfer (Fig. 3).
Metal-independent, inverting GT-A fold enzymes.
Analogous use of conserved sugar donor interactions and divergent acceptor recognition is also seen for the metal-independent GT29 sialyltrans-ferases4,35–38 (Fig. 4a,b). These enzymes generate distinct NeuAc capping structures on glycoproteins (N- and O-glycans) and glycolipids based upon unique acceptor specificities among the 20 mammalian GT29 isoforms. Terminal sialylated glycan epitopes provide a broad collection of ligand structures involved in developmental, immunological and pathological interactions in mammalian systems55–58. The GT29 sialyltransferases employ a modified GT-A fold structure (GT-A variant 2) comprised of conserved ‘sialylmotif’ element sequences in the core of the Rossmann-fold scaffold to assemble a metal-independent cytidine monophospho-N-acetylneuraminic acid (CMP-NeuAc) binding site using adjoining proximal loop regions (Fig. 4)36,59,60.
GT29 sialyltransferases fall into six sequence subclades that differ in the acceptor monosaccharides they bind and the linkage positions they produce: NeuAc-α-2,3-Gal (two subclades), NeuAc-α-2,6-Gal, NeuAc-α-2,6-GalNAc (two subclades), and NeuAc-α-2,8-NeuAc linkages36. Representative acceptor-bound complexes have been determined for three of the subclades and revealed surprising differences in acceptor interactions among members of this family. Each of the sialyltransferases position the nucleotide or nucleotide sugar donors similarly to the conserved sialylmotif element sequences and employ a conserved His residue as catalytic base. The structure of ST6GAL1, the major N-glycan NeuAc-α-2,6-Gal-synthesizing enzyme60,61, revealed an enzyme-bound Gal2GlcNAc2Man3GlcNAc2-Asn acceptor complex demonstrating interaction of the terminal Gal-β;-1,4-GlcNAc unit in the enzyme active site with the terminal Gal C6 hydroxyl positioned adjacent to the His catalytic base35,36 (Fig. 4a,b). By contrast, ST3GAL1, the major enzyme synthesizing sialylated core 1 O-glycan structures (NeuAc-α-2,3-Gal-β-1,3-GalNAc-Ser/Thr)60,61, was crystallized with a bound Gal-β;-1,3-GalNAc-oNP acceptor37 and revealed a completely different ‘flipped’ geometry for the terminal Gal acceptor residue that positions the C3 hydroxyl adjacent to the His catalytic base as the nucleophile for glycan transfer (Fig. 4a–c)36. By comparison, ST8SIA3, an enzyme involved in generating NeuAc-α-2,8-NeuAc-glycan structures on glycoproteins and glycolipids60,61, was solved as a ternary complex containing a CMP-3F-NeuAc donor analog and a NeuAc-α-2,3-Gal-β;-1,4-GlcNAc-6-SO4 acceptor38. In contrast to the two Gal-specific sialyltransferases, the latter enzyme employed completely different acceptor interactions to position the NeuAc C8 hydroxyl adjacent to the His catalytic base as the nucleophile for transfer (Fig. 4a–c). The recent structure of ST6GALNAC2, an enzyme that synthesizes an alternative sialylated core 1 isomer structure relative to ST3GAL160,61, was determined as a CMP complex, but without a bound acceptor analog4. The positioning of potential loops for acceptor interactions were quite distinct from those of ST6GAL1, ST3GAL1, or ST8SIA3, suggesting that even within this single CAZy family there is considerable diversity in acceptor substrate recognition through distinct binding-site architectures.
Metal-dependent, retaining GT-A fold enzymes.
Substrate interactions for retaining GT-A fold enzymes also share many features with those of GT-A fold inverting enzymes (Fig. 5). Five retaining GT-A fold enzyme–acceptor complexes (ABO47, GGTA125, XXYLT122, XXT148, and EXTL249) demonstrate analogous metal-dependent interactions with the phosphodiester of the nucleotide, mediated by an enzyme-associated DxD motif and a similar overall position for the nucleotide sugar (Fig. 5). Retaining GT-A fold enzymes also assemble their glycan acceptor sites from unique loops emerging from the core Rossmann fold to provide complementary templates for glycan interactions. The enzyme–acceptor complexes displayed in Fig. 5 indicate that the approach of each acceptor nucleophile to the catalytic site is unique, with the exception of the two GT6 enzymes ABO and GGTA1, which employ an analogous approach for their nucleophilic Gal residues. As indicated in the mechanisms summary above (Figs. 1 and 2a–c) these latter enzymes presumably do not use active site amino acids for acceptor deprotonation, but position their respective acceptor nucleophiles adjacent to the donor β-phosphate oxygen for deprotonation and the proposed same-side dissociative SNi-type mechanism.
Inverting GT-B fold enzymes.
Although far fewer structures of eukaryotic glycan-directed GT-B fold enzymes have been determined with bound acceptors, two recent structures have provided insights into glycan acceptor recognition by these enzymes. The first, FUT1, is an inverting enzyme that generates α-1,2-Fuc linkages on the termini of plant cell wall xyloglucan side branches. Xyloglucans are highly abundant cell wall polymers comprised of a β-1,4-glucan backbone decorated with a repeating branching pattern of α-1,6-Xyl, β-1,2-Gal and α-1,2-Fuc substituents. FUT1 structures were determined with bound GDP and octasaccharide acceptor ligands comprised of a Gal-β-1,2-Xyl-α-1,6-modified cellopentaose structure21,41. Unlike the protein-domain-specific fucosyltransferases described below, the octasaccharide acceptor structure assumes a flattened planar arrangement that interacts across the face of the enzyme with only the Gal acceptor residue extending perpendicularly into the active site. The inverting catalytic mechanism for this enzyme is more complex, as there is no apparent catalytic base available for nucleophile deprotonation. As described earlier, an SN1 mechanism (Fig. 1c) has been invoked with a water-mediated proton shuttle for nucleophile deprotonation21.
The structure of a second GT-B inverting enzyme, MGAT5, involved in the branching of complex type mammalian N-glycans, was also recently described. The enzyme extends a β-1,6-GlcNAc branch on GlcNAc2Man3GlcNAc2-Asn glycan biosynthetic intermediates that can then be further extended to form poly-N-acetyllactosamine-containing structures62. MGAT5 expression is upregulated in response to Ras-Raf-Ets activation63,64 and was identified as a cancer-associated GT65,66, contributing to alterations in cell adhesion during metastasis in transformed tissues67. Binding affinity for the UDP-GlcNAc sugar donor is unusually poor for a GT, but the enzyme has higher affinity for glycan acceptor interactions65,66. The MGAT5 structure was solved in complex with a chimeric bisubstrate-type inhibitor (GlcNAc-β-1,2-Man-α-1,6-Man tethered to UDP-GlcNAc), but only the glycan acceptor component could be resolved bound exclusively by the B domain of the GT-B fold40. Further structure determination in the presence of donor analogs will be required before the catalytic mechanism of this latter enzyme can be clarified.
Protein-modifying glycosyltransferases
Glycosyltransferases that directly modify hydroxyls of Ser or Thr in a protein must bind their acceptor protein substrates to orient the respective Ser/Thr hydroxyl nucleophile correctly for sugar transfer (Fig. 1). Two classes of protein-modifying GTs include those that modify linear peptides and those that modify folded, cysteine-rich domains.
GTs that modify linear peptides.
Structures of several eukaryotic enzymes that add sugars to unstructured peptides have been determined, including polypeptide GalNAc-transferases (GALNTs), protein O-xylosyltransferase 1 (XYLT1), O-GlcNAc transferase (OGt), and oligosaccharyltransferase (OST) (Supplementary Dataset 1). Here we will limit our discussion to GALNTs and XYLT1. Twenty GALNTs exist in the human genome, and structures of two of them (GALNT2 and GALNT4) have been solved in complex with acceptor peptides23,68–70. GALNTs are Golgi-localized, membrane bound, retaining enzymes with two lumenally oriented domains: an N-terminal glycosyltransferase domain (metal-dependent GT27 GT-A fold; Supplementary Dataset 1) and a C-terminal lectin domain with a β-trefoil fold held together by a flexible linker27. They transfer GalNAc from UDP-GalNAc to peptides rich in Ser, Thr and Pro, often leading to heavily glycosylated mucin-type regions. Some of the GALNTs prefer unmodified peptides as acceptor substrates, whereas others prefer partially GalNAc-modified peptides. The peptides bind in a hydrophobic groove on the surface of the enzymes, which can accommodate a variety of sequences. For GalNAc-modified peptides, the lectin domain binds to the GalNAc and positions the peptide so that a specific Ser/Thr residue at a particular distance from the GalNAc can be glycosylated (Fig. 6a). For instance, GALNT2 has a preference for adding GalNAc to a Ser/Thr residue about 10 residues N-terminal to the GalNAc-modified residue27,69. Other GALNTs have different specificities, allowing heavy modification of Ser/Thr-rich regions. For instance, GALNT4 preferentially modifies a Ser/Thr residue C-terminal to a previously modified residue70. Interestingly, the flexible linker between the catalytic and lectin domains has been shown to fine-tune acceptor substrate recognition and glycosylation70. The GALNTs are the only known mammalian GTs with a lectin domain that contributes to the specificity of the enzyme. The differing specificities of GALNTs likely explain why a strict consensus sequence for predicting sites of GalNAc modification does not exist, although algorithms for prediction of sites are improving (see http://www.cbs.dtu.dk/services/NetOGlyc/ and http://isoglyp.utep.edu/index.php). In all cases, the enzymes orient the acceptor hydroxyl group on the same side of the anomeric carbon of the sugar as the nucleotide to perform an SNi-like reaction resulting in retention of configuration (Fig. 1e).
Protein O-xylosyltransferases 1 and 2 (XYLT1 and XYLT2, respectively) catalyze transfer of xylose from UDP-Xyl to Ser residues with inversion of configuration, initiating the biosynthesis of glycosaminoglycan chain addition on proteogycans (Heparin Sulfate, Chondroitin Sulfate, Dermatan Sulfate)39. XYLT1 and 2 are anchored to the luminal face of the Golgi complex with an N-terminal glycosyltransferase domain (metal-independent GT14 GT-A fold enzyme similar to GCNT1, Supplementary Dataset 1) linked to a C-terminal Xylo_C domain of unknown function. Transfer is specific for Ser residues generally surrounded by glycines, frequently with acidic residues positioned at −2 and −4 relative to the serine. A structure of ternary complex of XYLT1 with various acceptor peptides and UDP-Xyl revealed the molecular basis for this specificity (Fig. 6b)39. The acceptor peptides bind a groove in the enzyme, placing the hydroxyl of the acceptor Ser residue precisely in position to perform a nucleophilic attack on the anomeric carbon of xylose (SN2 reaction, Fig. 1b). The binding cleft narrows C-terminal to the Ser, explaining the preference for residues with small side chains in the +1 and +2 positions. The binding cleft is larger on the N-terminal side allowing more flexibility, although there is a general positive charge explaining the preference for acidic residues in the −3 and −4 positions.
GTs modifying folded cysteine-rich domains.
Co-crystal structures of domain-specific GTs with bound acceptor domains include protein O-fucosyltransferases 1 and 2 (POFUT1 (GT65) and POFUT2 (GT68)) and protein O-glucosyltransferase 1 (POGLUT1 (GT90)), all of which are inverting GTs with GT-B folds (Supplementary Dataset 1). All three are soluble enzymes localized in the ER that have the unique ability to recognize folded protein domains71–73. POGLUT1 and POFUT1 add O-glucose or O-fucose, respectively, to properly folded epidermal growth factor-like (EGF) repeats containing the appropriate consensus sequences: C1XSX(P/A)C2 for POGLUT1 and C2XXXX(S/T)C3 for POFUT1 (in which C1-3 are conserved cysteines in EGF repeats, and underlined residues are modified)74–77. Although multiple proteins contain EGF repeats with these consensus sequences, the Notch family of receptors appears to be the major biological target for these modifications78. POGLUT1 and POFUT1 are required for Notch receptor function in vivo74–77.
POFUT2 adds O-fucose to properly folded thrombospondin type 1 repeats (TSRs) containing the consensus CXX(S/T)C (in which the C’s are either conserved cysteines 1 and 2, or cysteines 2 and 3, depending on whether the TSR is group 1 or group 2, respectively)76. Nearly 50 proteins contain TSRs with this consensus sequence, including all members of the A disintegrin and metallo-protease with thrombospondin repeats (ADAMTS) family of extracellular proteases78. Deletion of Pofut2 in mice results in an identical embryonic lethal phenotype to that with deletion of Adamts9, suggesting that ADAMTS9 requires addition of O-fucose glycans for proper function79,80.
POFUT1, POFUT2, and POGLUT1 all have a large cleft between the two Rossman-like folds where the folded protein domains bind, covering approximately one third of the surface area of the domains (Fig. 7)20,42,43,81. The cleft in each enzyme shows remarkable surface complementarity for the face of the domain bound in the cleft with multiple hydrogen bonds, especially within the loop containing the consensus sequences (see refs. 20,42,43,81 for details). This orients the domain to place the hydroxyl group of the Ser/Thr acceptor precisely in position to perform nucleophilic attack on the anomeric carbon of the fucose or glucose (SN2 reaction for POGLUT143,81 and POFUT242, SN1 for POFUT119,20, Fig. 1). Interestingly, POFUT2 also has multiple water molecule-mediated hydrogen bonds at the enzyme-substrate interface, presumably allowing flexibility of sequences for different TSRs to bind to the enzyme42. These structures clearly demonstrate why both a consensus sequence and a properly folded domain are needed for modification by these enzymes. All three enzymes have been implicated in quality control pathways for the proper folding of EGF repeats (POFUT1 and POGlUT182) and TSRs (POFUT283) since the glycosylated product stabilizes the folded structural domains76,77,84.
Future directions
The GT structures presently in PDB represent only 53 of the 106 total CAZy families and only 15 of 45 CAZy families with vertebrate members (Supplementary Dataset 1). Since many of these enzyme families have multiple members with distinct acceptor and donor specificities, the present structures clearly represent only a minor fraction of the glycan synthetic capability in biological systems. The rate of new GT structures deposited in PDB has only minimally increased in recent years, but a greater number of recent structures for eukaryotic enzymes have resulted from significant improvements in eukaryotic expression strategies. Hopefully these efforts will expand more effectively in the coming decade and include a greater number of the more challenging GT-C fold enzymes. An increase in more complex enzyme structures determined by cryo-electron microscopy will also be expected in the coming years11,85–87.
The goals of these studies will be to understand not only the structural basis of linkage-specific glycan synthesis, but also broader themes of how the GTs provide catalytic templates for glycan assembly and how faithful replication of glycan structures is accomplished with high fidelity among cells of common lineages. The restricted set of protein folds and the modular nature of the GT active site architectures have allowed independent evolution of specificities within donor, acceptor, and catalytic modules to expand the repertoire of glycan structures in biological systems. A major challenge for the future will be to improve our understanding of how selective pressures of evolution have led to the present collection of enzyme specificities and how novel specificities could evolve or be engineered. These insights will only come from a more comprehensive comparative analysis of GT sequences from varied organism sources paired with enzyme specificity studies and structures determined for GT enzyme–substrate complexes. Challenges for the latter goals will be large-scale production of the full complement of eukaryotic glycoenzymes and the development of glycan, glycopep-tide, and glycolipid libraries for use as substrates and ligands. These efforts will reveal mechanisms that drive evolution of new catalytic templates and enzyme specificities that lead to glycan diversification for cell surface and secreted glycoconjugates. The studies would also provide insights that allow engineering of novel enzyme capabilities for biochemical and biotherapeutic applications that would catalyze glycoscience advances in the future.
Supplementary Material
Acknowledgements
The authors would like to thank M. Tiemeyer and H. Takeuchi for providing useful comments. Original work in the author’s laboratories is supported by NIH grants P01GM107012, P41GM103390, U01GM120408, R01GM130915 (K.W.M.) and GM061126, HD090156, HD096030 (R.S.H.).
Competing interests
K.W.M. acknowledges ownership interest and roles as President and CEO of Glyco Expression Technologies, Inc., a biotechnology spinout commercializing recombinant glycosyltransferases, and may conceivably profit from the results described herein.
Footnotes
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-019-0350-2.
References
- 1.Cummings RD & Pierce JM The challenge and promise of glycomics. Chem. Biol 21, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varki A Biological roles of glycans. Glycobiology 27, 3–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moremen KW, Tiemeyer M & Nairn AV Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol 13, 448–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moremen KW et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol 14, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lairson LL, Henrissat B, Davies GJ & Withers SG Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem 77, 521–555 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM & Henrissat B The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barford D & Johnson LN The allosteric transition of glycogen phosphorylase. Nature 340, 609–616 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Ju T, Aryal RP, Kudelka MR, Wang Y & Cummings RD The Cosmc connection to the Tn antigen in cancer. Cancer Biomark. 14, 63–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rini JM & Esko JD in Essentials of Glycobiology (eds. Varki A, Cummings RD, Esko JD et al. ) 65–75 (Cold Spring Harbor, NY, 2015). [Google Scholar]
- 10.Levine ZG & Walker S The biochemistry of O-GlcNAc transferase: which functions make it essential in mammalian cells? Annu. Rev. Biochem 85, 631–657 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Bai L & Li H Cryo-EM is uncovering the mechanism of eukaryotic protein N-glycosylation. FEBS J. 286, 1638–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujiwara R, Yokoi T & Nakajima M Structure and protein-protein interactions of human UDP-glucuronosyltransferases. Front. Pharmacol 7, 388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramakrishnan B & Qasba PK Crystal structure of lactose synthase reveals a large conformational change in its catalytic component, the beta1,4-galactosyltransferase-I. J. Mol. Biol 310, 205–218 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Qasba PK, Ramakrishnan B & Boeggeman E Structure and function of β-1,4-galactosyltransferase. Curr. Drug Targets 9, 292–309 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramakrishnan B, Balaji PV & Qasba PK Crystal structure of β1,4-galactosyltransferase complex with UDP-Gal reveals an oligosaccharide acceptor binding site. J. Mol. Biol 318, 491–502 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Gagnon SM et al. High resolution structures of the human ABO(H) blood group enzymes in complex with donor analogs reveal that the enzymes utilize multiple donor conformations to bind substrates in a stepwise manner. J. Biol. Chem 290, 27040–27052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner GK, Pesnot T, Palcic MM & Jørgensen R Novel UDP-GalNAc derivative structures provide insight into the donor specificity of human blood group glycosyltransferase. J. Biol. Chem 290, 31162–31172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breton C, Snajdrová L, Jeanneau C, Koca J & Imberty A Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R (2006). [DOI] [PubMed] [Google Scholar]
- 19.Lira-Navarrete E et al. Structural insights into the mechanism of protein O-fucosylation. PLoS One 6, e25365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z et al. Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol 13, 757–763 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Urbanowicz BR et al. Structural, mutagenic and in silico studies of xyloglucan fucosylation in Arabidopsis thaliana suggest a water-mediated mechanism. Plant J 91, 931–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H et al. Notch-modifying xylosyltransferase structures support an SNi-like retaining mechanism. Nat. Chem. Biol 11, 847–854 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lira-Navarrete E et al. Substrate-guided front-face reaction revealed by combined structural snapshots and metadynamics for the polypeptide N-acetylgalactosaminyltransferase 2. Angew. Chem. Int. Edn Engl 53, 8206–8210 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Albesa-Jové D et al. A native ternary complex trapped in a crystal reveals the catalytic mechanism of a retaining glycosyltransferase. Angew. Chem. Int. Edn Engl 54, 9898–9902 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Albesa-Jové D, Sainz-Polo MA, Marina A & Guerin ME Structural snapshots of α-1,3-galactosyltransferase with native substrates: insight into the catalytic mechanism of retaining glycosyltransferases. Angew. Chem. Int. Edn Engl 56, 14853–14857 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Ramakrishnan B & Qasba PK Crystal structure of the catalytic domain of Drosophila beta1,4-galactosyltransferase-7. J. Biol. Chem 285, 15619–15626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurtado-Guerrero R Recent structural and mechanistic insights into protein O-GalNAc glycosylation. Biochem. Soc. Trans 44, 61–67 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ardèvol A, Iglesias-Fernández J, Rojas-Cervellera V & Rovira C The reaction mechanism of retaining glycosyltransferases. Biochem. Soc. Trans 44, 51–60 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Gómez H, Mendoza F, Lluch JM & Masgrau L QM/MM studies reveal how substrate-substrate and enzyme-substrate interactions modulate retaining glycosyltransferases catalysis and mechanism. Adv. Protein Chem. Struct. Biol 100, 225–254 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Schuman B, Evans SV & Fyles TM Geometric attributes of retaining glycosyltransferase enzymes favor an orthogonal mechanism. PLoS One 8, e71077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadirvelraj R et al. Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc. Natl Acad. Sci. USA 115, 4637–4642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon RD et al. X-ray crystal structures of rabbit N-acetylglucosaminyltransferase I (GnT I) in complex with donor substrate analogues. J. Mol. Biol 360, 67–79 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Pak JE et al. X-ray crystal structure of leukocyte type core 2 beta1,6-N-acetylglucosaminyltransferase. Evidence for a convergence of metal ion-independent glycosyltransferase mechanism. J. Biol. Chem 281, 26693–26701 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Pak JE, Satkunarajah M, Seetharaman J & Rini JM Structural and mechanistic characterization of leukocyte-type core 2 β1,6-N-acetylglucosaminyltransferase: a metal-ion-independent GT-A glycosyltransferase. J. Mol. Biol 414, 798–811 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Kuhn B et al. The structure of human α-2,6-sialyltransferase reveals the binding mode of complex glycans. Acta Crystallogr. D Biol. Crystallogr 69, 1826–1838 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Meng L et al. Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem 288, 34680–34698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao FV et al. Structural insight into mammalian sialyltransferases. Nat. Struct. Mol. Biol 16, 1186–1188 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Volkers G et al. Structure of human ST8SiaIII sialyltransferase provides insight into cell-surface polysialylation. Nat. Struct. Mol. Biol 22, 627–635 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Briggs DC & Hohenester E Structural basis for the initiation of glycosaminoglycan biosynthesis by human xylosyltransferase 1. Structure 26, 801–809.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagae M et al. Structure and mechanism of cancer-associated N-acetylglucosaminyltransferase-V. Nat. Commun 9, 3380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rocha J et al. Structure of Arabidopsis thaliana FUT1 reveals a variant of the GT-B class fold and provides insight into xyloglucan fucosylation. Plant Cell 28, 2352–2364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valero-González J et al. A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol 12, 240–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z et al. Structural basis of Notch O-glucosylation and O-xylosylation by mammalian protein-O-glucosyltransferase 1 (POGLUT1). Nat. Commun 8, 185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuwabara N et al. Carbohydrate-binding domain of the POMGnT1 stem region modulates O-mannosylation sites of α-dystroglycan. Proc. Natl Acad. Sci. USA 113, 9280–9285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kakuda S et al. Structural basis for acceptor substrate recognition of a human glucuronyltransferase, GlcAT-P, an enzyme critical in the biosynthesis of the carbohydrate epitope HNK-1. J. Biol. Chem 279, 22693–22703 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Pedersen LC et al. Heparan/chondroitin sulfate biosynthesis. Structure and mechanism of human glucuronyltransferase I. J. Biol. Chem 275, 34580–34585 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Alfaro JA et al. ABO(H) blood group A and B glycosyltransferases recognize substrate via specific conformational changes. J. Biol. Chem 283, 10097–10108 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Culbertson AT, Ehrlich JJ, Choe JY, Honzatko RB & Zabotina OA Structure of xyloglucan xylosyltransferase 1 reveals simple steric rules that define biological patterns of xyloglucan polymers. Proc. Natl Acad. Sci. USA 115, 6064–6069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedersen LC et al. Crystal structure of an alpha 1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J. Biol. Chem 278, 14420–14428 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Lazarus MB et al. Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nat. Chem. Biol 8, 966–968 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barresi R & Campbell KP Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci 119, 199–207 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Yoshida-Moriguchi T & Campbell KP Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 25, 702–713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheikh MO, Halmo SM & Wells L Recent advancements in understanding mammalian O-mannosylation. Glycobiology 27, 806–819 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shah N, Kuntz DA & Rose DR Golgi alpha-mannosidase II cleaves two sugars sequentially in the same catalytic site. Proc. Natl Acad. Sci. USA 105, 9570–9575 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhide GP & Colley KJ Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem. Cell Biol 147, 149–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X & Varki A Advances in the biology and chemistry of sialic acids. ACS Chem. Biol 5, 163–176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen M & Varki A The sialome—far more than the sum of its parts. OMICS 14, 455–464 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Macauley MS, Crocker PR & Paulson JC Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol 14, 653–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Datta AK & Paulson JC The sialyltransferase “sialylmotif” participates in binding the donor substrate CMP-NeuAc. J. Biol. Chem 270, 1497–1500 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Harduin-Lepers A, Mollicone R, Delannoy P & Oriol R The animal sialyltransferases and sialyltransferase-related genes: a phylogenetic approach. Glycobiology 15, 805–817 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Harduin-Lepers A et al. The human sialyltransferase family. Biochimie 83, 727–737 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Dennis JW, Nabi IR & Demetriou M Metabolism, cell surface organization, and disease. Cell 139, 1229–1241 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buckhaults P, Chen L, Fregien N & Pierce M Transcriptional regulation of N-acetylglucosaminyltransferase V by the src oncogene. J. Biol. Chem 272, 19575–19581 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Kang R et al. Transcriptional regulation of the N-acetylglucosaminyltransferase V gene in human bile duct carcinoma cells (HuCC-T1) is mediated by Ets-1. J. Biol. Chem 271, 26706–26712 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Gu J et al. Purification and characterization of UDP-N-acetylglucosamine: alpha-6-d-mannoside beta 1-6N-acetylglucosaminyltransferase (N-acetylglucosaminyltransferase V) from a human lung cancer cell line. J. Biochem 113, 614–619 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Shoreibah MG, Hindsgaul O & Pierce M Purification and characterization of rat kidney UDP-N-acetylglucosamine: α-6-d-mannoside β-1,6-N-acetylglucosaminyltransferase. J. Biol. Chem 267, 2920–2927 (1992). [PubMed] [Google Scholar]
- 67.Boscher C, Dennis JW & Nabi IR Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol 23, 383–392 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Fritz TA, Raman J & Tabak LA Dynamic association between the catalytic and lectin domains of human UDP-GalNAc:polypeptide alpha-N-ace tylgalactosaminyltransferase-2. J. Biol. Chem 281, 8613–8619 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Lira-Navarrete E et al. Dynamic interplay between catalytic and lectin domains of GalNAc-transferases modulates protein O-glycosylation. Nat. Commun 6, 6937 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Las Rivas M et al. The interdomain flexible linker of the polypeptide GalNAc transferases dictates their long-range glycosylation preferences. Nat. Commun 8, 1959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takeuchi H, Kantharia J, Sethi MK, Bakker H & Haltiwanger RS Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. J. Biol. Chem 287, 33934–33944 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luo Y, Nita-Lazar A & Haltiwanger RS Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem 281, 9385–9392 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Wang Y & Spellman MW Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J. Biol. Chem 273, 8112–8118 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Varshney S & Stanley P Multiple roles for O-glycans in Notch signalling. FEBS Lett 592, 3819–3834 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harvey BM & Haltiwanger RS Regulation of Notch function by O-glycosylation. Adv. Exp. Med. Biol 1066, 59–78 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Holdener BC & Haltiwanger RS Protein O-fucosylation: structure and function. Curr. Opin. Struct. Biol 56, 78–86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu H & Takeuchi H Protein O-glucosylation: another essential role of glucose in biology. Curr. Opin. Struct. Biol 56, 64–71 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Schneider M, Al-Shareffi E & Haltiwanger RS Biological functions of fucose in mammals. Glycobiology 27, 601–618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benz BA et al. Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol 416, 111–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Du J et al. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev. Biol 346, 25–38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu H et al. Structural analysis of Notch-regulating Rumi reveals basis for pathogenic mutations. Nat. Chem. Biol 12, 735–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takeuchi H et al. O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J. Biol. Chem 292, 15964–15973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vasudevan D, Takeuchi H, Johar SS, Majerus E & Haltiwanger RS Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol 25, 286–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lira-Navarrete E & Hurtado-Guerrero R A perspective on structural and mechanistic aspects of protein O-fucosylation. Acta Crystallogr. F Struct. Biol. Commun 74, 443–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wild R et al. Structure of the yeast oligosaccharyltransferase complex gives insight into eukaryotic N-glycosylation. Science 359, 545–550 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Bai L, Wang T, Zhao G, Kovach A & Li H The atomic structure of a eukaryotic oligosaccharyltransferase complex. Nature 555, 328–333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Braunger K et al. Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360, 215–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thiyagarajan N et al. Structure of a metal-independent bacterial glycosyltransferase that catalyzes the synthesis of histo-blood group A antigen. Sci. Rep 2, 940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.