

Mitochondrial dysfunction promotes the etiology of neurodegenerative syndrome SCAN1.
Abstract
A homozygous mutation of human tyrosyl-DNA phosphodiesterase 1 (TDP1) causes the neurodegenerative syndrome, spinocerebellar ataxia with axonal neuropathy (SCAN1). TDP1 hydrolyzes the phosphodiester bond between DNA 3′-end and a tyrosyl moiety within trapped topoisomerase I (Top1)-DNA covalent complexes (Top1cc). TDP1 is critical for mitochondrial DNA (mtDNA) repair; however, the role of mitochondria remains largely unknown for the etiology of SCAN1. We demonstrate that mitochondria in cells expressing SCAN1-TDP1 (TDP1H493R) are selectively trapped on mtDNA in the regulatory non-coding region and promoter sequences. Trapped TDP1H493R-mtDNA complexes were markedly increased in the presence of the Top1 poison (mito-SN38) when targeted selectively into mitochondria in nanoparticles. TDP1H493R-trapping accumulates mtDNA damage and triggers Drp1-mediated mitochondrial fission, which blocks mitobiogenesis. TDP1H493R prompts PTEN-induced kinase 1–dependent mitophagy to eliminate dysfunctional mitochondria. SCAN1-TDP1 in mitochondria creates a pathological state that allows neurons to turn on mitophagy to rescue fit mitochondria as a mechanism of survival.
INTRODUCTION
Spinocerebellar ataxia with axonal neuropathy (SCAN1) is an autosomal recessive neurodegenerative disorder that is linked with a homozygous point mutation (H493R) in human tyrosyl-DNA phosphodiesterase 1 (TDP1) (1). TDP1 is primarily involved in the repair of DNA strand breaks linked with abortive topoisomerase 1 (Top1) activity (2–8). Cells derived from patients with SCAN1 are hypersensitive to camptothecin that selectively traps Top1-DNA covalent complexes (Top1cc) (5, 9–12). Patients with SCAN1 develop progressive cerebellar ataxia and peripheral neuropathy, which becomes noticeable at late childhood (13 to 15 years), indicating a slow onset of neurodegeneration (1). However, three independently developed TDP1 knockout mouse models revealed no obvious behavioral phenotypes related to patients with SCAN1 (8, 13, 14). Nevertheless, in one of these mouse models, loss of TDP1 resulted in gradual age-related cerebellar atrophy (8). This suggests that the SCAN1 phenotype is a complex phenomenon and has a multifactorial mechanism that has not been elucidated.
TDP1 hydrolyzes the phosphodiester bond between the Top1-tyrosyl moiety and the DNA 3′-end that involves a two-step mechanism: The first nucleophilic attack is initiated by catalytic H263 on the phosphate bond linking the DNA and the tyrosyl-containing peptide. The C-terminal domain of TDP1 harboring H493 residue acts as a general acid and donates a proton to the apical tyrosine-containing peptide-leaving group. This results in the generation of a TDP1-DNA covalent intermediate (step 1). The H493 acts as a general base catalyst and activates a water molecule that subsequently hydrolyzes the phosphoenzyme intermediate (step 2) (3, 4). The SCAN1 point mutation (H493R) retains partial TDP1 activity and results in defective turnover owing to the formation of TDP1H493R-DNA complexes (8, 10, 14); therefore, SCAN1 cells do not provide a true knockout model of TDP1 (14). The nuclear encoded TDP1 is transported to the mitochondria for mitochondrial DNA (mtDNA) repair (15–18), and the ability of TDP1 to resolve 3′-phosphotyrosyl linkages is not limited to the removal of nuclear Top1-DNA adducts (3–5) but is also required for the excision of mitochondrial Top1 (Top1mt)–DNA covalent complexes (Top1mtcc) that are exclusively formed in the mitochondria (18). TDP1 can also hydrolyze a broad spectrum of 3'-DNA blocking lesions that are formed during oxidative DNA damage (4, 9, 15, 19–22), including 3′-phosphoglycolate and 3′-deoxyribose phosphate ends (19, 21). Accordingly, TDP1-deficient cells are defective in repairing mtDNA damage associated with trapped Top1mtcc or 3'-DNA lesions related to oxidative damage and chain-terminating anticancer or antiviral nucleosides (15, 18, 23, 24). Posttranslational modifications by phosphorylation, (ADP)-ribosylation, SUMOylation, arginine methylation, and ubiquitylation are common regulators for the recruitment, turnover, and modulation of enzymatic activity and stability of TDP1 (12, 25–29).
Vertebrate mtDNA constitutes approximately 5% of the total cellular DNA content where several thousand copies of mtDNA are distributed across hundreds of mitochondria as covalently closed circular DNA (16.5 kb) encoding critical components of the mitochondrial adenosine 5′-triphosphate (ATP) production machinery (30, 31). Mitochondrial network dynamics uses the process of fission or fusion as a complex physiological adaptation to coordinate the segregation or unification of two physically distinct mitochondria, which is essential for regulating mitochondrial morphology, biogenesis, and stress response mechanisms (32, 33). In the context of mtDNA copy number maintenance, the mitochondrial network undergoes fusion to complement the mtDNA lesion by transfer of nucleoids (34). In contrast, mitochondrial fission predominates under conditions of nonrepaired mtDNA lesions, which results in mitochondrial dysfunction (35, 36). To preserve mitochondrial homeostasis, increased mitochondrial fission ensues the degradation of dysfunctional mitochondria by autophagy (lysosome-dependent degradation), specifically termed as mitophagy (32, 36–39).
Unlike chromosomal DNA, mtDNA is more vulnerable to oxidative DNA damage, as they are profoundly exposed to reactive oxygen species (ROS) generated from the electron transport chain (40–42). Moreover, the mitochondria are completely dependent on the nucleus for mtDNA repair and integrity (42). Marked elevation of mtDNA damage has been attributed to the progression of several neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, myopathies and diabetes, aging, and age-related disorders (30, 43). Thus, efficient repair of mtDNA in postmitotic cells like neurons is critical for normal brain functioning. However, the role of mitochondria in the pathoetiology of the SCAN1 phenotype has not been elucidated.
This study provides the first evidence for connecting mitochondrial dysfunction with etiology of SCAN1. We show that cells expressing SCAN1-TDP1 are trapped on the mtDNA, which is escalated in the presence of mitochondria-targeted Top1 poison (mito-SN38). We show that trapping TDP1H493R on mtDNA increases the mitochondrial fission rate and abrogates mitobiogenesis. Last, we show that increased mitochondrial degradation may allow identification of dysfunctional daughter mitochondria expressing TDP1H493R and their subsequent removal through mitophagy, which may serve as a mechanism to restore fit mitochondria for cell survival.
RESULTS
Mitochondria harboring SCAN1 mutant TDP1 (H493R) are hypersensitive to mitochondria-targeted Top1 poison
To investigate the role of SCAN1 mutant TDP1 in mitochondria, we stably complemented TDP1 knockout (TDP1−/−) mouse embryonic fibroblasts (MEFs) with lentiviral constructs harboring FLAG-tagged human SCAN1 mutant TDP1H493R. The presence of the exogenous FLAG-TDP1 variants in the mitochondria of TDP1−/− MEFs (TDP1−/−/WT or TDP1−/−/H493R) was confirmed by cell fractionation and Western blotting, and the relative purity of the mitochondrial fraction was further assessed by analysis of mitochondrial cytochrome c oxidase (COX IV) protein (Fig. 1A), which is consistent with previous reports showing the import of exogenous TDP1 into the mitochondria (15–18).
Fig. 1. Mito-SN38 containing nanoparticle poisons Top1mt (Top1mtcc).
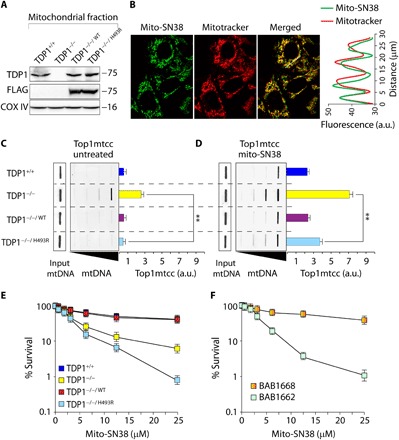
(A) Western blot analysis of mitochondrial lysates extracted from the indicated MEFs. TDP1−/−/WT and TDP1−/−/H493R: TDP1−/− complemented with exogenous FLAG-tagged human wild-type or SCAN1 mutant (H493R) TDP1, respectively. Blots were probed with anti-TDP1, anti-FLAG, and anti–COX IV antibodies. COX IV served as a positive mitochondrial marker. (B) Representative confocal images showing accumulation of mitochondria-targeted cationic nanoparticle containing Top1 poison irinotecan (mito-SN38; intrinsic green fluorescence) inside the mitochondria (labeled with MitoTracker red). MEFs were incubated with mito-SN38 (5 μM for 20 min), and the fluorescence patterns were recorded under live-cell microscopy. Colocalization is shown in the merged image. Quantitation of the pixel intensity of fluorescence along the indicated white line in the merged image (right). The white line was drawn arbitrarily along the region of interest (ROI). a.u., arbitrary units. (C and D) Poisoning of Top1mt (Top1mtcc) in the indicated MEFs expressing TDP1 variants; either untreated (C) or treated with mito-SN38 (5 μM for 3 hours) (D). Top1mtcc was detected by ICE (immunocomplex of enzyme) bioassay. MtDNA at increasing concentrations (0.5, 1, 2, and 4 μg) was immunoblotted with an anti-Top1mt–specific antibody. The mtDNA input was probed with anti-dsDNA (double-stranded DNA) antibody. Densitometry analysis of trapped Top1mtcc band intensity was quantified and expressed as fold increase relative to mtDNA input (error bars represent means ± SEM). Asterisks denote statistically significant differences (**P < 0.01, t test). (E and F) Cell survival curves of indicated MEF variants (E) and a SCAN1 patient–derived lymphoblastoid cell line (BAB1662) and its wild-type counterpart (BAB1668) (F). Mito-SN38–induced cytotoxicity (%) was calculated with respect to the untreated control. Each point corresponds to the mean ± SD of at least three experiments. Error bars represent SDs (n = 3).
The active metabolite of irinotecan (SN38) stabilizes Top1-cleavage complexes (Top1cc). Irinotecan is a widely used anticancer drug (44). We engineered mitochondrial targeting cationic nanoparticles containing irinotecan (mito-SN38) (45) to selectively trap Top1mt on the mtDNA (Top1mtcc). To test the intracellular delivery of mito-SN38 into the mitochondria, we performed live-cell confocal microscopy in cells treated with mito-SN38 (intrinsically green fluorescent) by costaining with the mitochondrion-specific dye MitoTracker red. Colocalization of mito-SN38 with MitoTracker red (Fig. 1B; see merged image) within 30 min of drug exposure confirmed the accumulation of the mito-SN38 nanoparticle exclusively inside the mitochondria. The correlation between the pixel intensities along a line in the two channels (Fig. 1B) substantiated the colocalization. We further validated the activity of mito-SN38 in the mitochondria by directly measuring in vivo trapping of Top1mtcc in isolated mitochondria using ICE (immunocomplex of enzyme) assays (Fig. 1, C and D) (46, 47). Under similar conditions, mito-SN38 failed to trap nuclear Top1 (fig. S1A).
Mitochondrial ICE assays detected covalently trapped Top1mt linked to mtDNA (Top1mtcc) without mito-SN38 in TDP1−/− MEFs (P < 0.01; Fig. 1C), which was markedly increased (~7-fold) after mito-SN38 treatment. TDP1-proficient MEFs (TDP1+/+ or TDP1−/−/WT) show reduced (~3-fold) Top1mtcc compared to TDP1-deficient cells, consistent with the role of TDP1 in excision of trapped Top1mtcc (Fig. 1D) in the mitochondria. Although TDP1−/−/H493R MEFs partially rescued (~1.5-fold) mito-SN38–induced Top1mtcc compared to TDP1−/− (P < 0.01; Fig. 1D), SCAN1-TDP1 was significantly defective in unhooking trapped Top1mtcc in the mitochondria compared to TDP1−/−/WT or TDP1+/+ cells (Fig. 1D).
We further performed survival assays to test the impact of mito-SN38 (Fig. 1E). We noted a substantial increase in mito-SN38–induced cell death in TDP1−/−/H493R MEFs compared to TDP1−/− MEFs (Fig. 1E); however, this effect was not due to increased accumulation of Top1mtcc (Fig. 1D). Under similar conditions, TDP1−/− MEFs complemented with wild-type human TDP1 (TDP1−/−/WT) or TDP1+/+ MEFs rescued the mito-SN38–mediated hypersensitivity (Fig. 1D). Consistent with TDP1−/−/H493R cells, the SCAN1 patient–derived lymphoblastoid cell lines (BAB1662), harboring TDP1 (H493R) mutation, were also hypersensitive to mito-SN38 compared to its wild-type counterpart (BAB1668) (Fig. 1F). Together, these results suggest that defective TDP1 activity is detrimental to the mitochondria challenged with a Top1 poison.
TDP1H493R trapping accumulates mtDNA damage
Because SCAN1 patient–derived lymphoblastoid cells and TDP1−/−/H493R MEFs are hypersensitive to mito-SN38 (Fig. 1, E and F), we tested whether the additional mito-SN38–mediated toxicity was due to trapping of TDP1H493R in the isolated mitochondria using ICE assays. In the absence of mito-SN38, we detected a significant increase (~1.5- to 2-fold) in TDP1H493R-mtDNA complexes (P < 0.1; Fig. 2A), which increased (~4- to 5-fold) after mito-SN38 treatment in TDP1−/−/H493R MEFs compared to TDP1−/− MEFs (P < 0.001; Fig. 2A). Similarly, we also detected mito-SN38–induced (~4- to 5-fold) increase in trapping of TDP1H493R (P < 0.001; Fig. 2A, right) in human SCAN1 cells (BAB1662), confirming that defective SCAN1-TDP1 activity generates TDP1H493R-mtDNA lesions.
Fig. 2. Induction of irreversible mtDNA damage through selective trapping of TDP1H493R.

(A) Detection of trapped TDP1-mtDNA complexes (mtTDP1cc) by ICE bioassays in the indicated cells following no treatment or treated with mito-SN38 (5 μM for 3 hours). MtDNA at increasing concentrations (0.5, 1, 2, and 4 μg) was immunoblotted with an anti-TDP1–specific antibody. The mtDNA input was probed with anti-dsDNA antibody. Densitometry analysis of the trapped mtTDP1cc band intensity was quantified and expressed as fold increase relative to mtDNA input (error bars represent means ± SEM). Asterisks denote statistically significant difference (*P < 0.1 and ***P < 0.001, t test). (B) Catalytically defective SCAN1-TDP1 was hypothesized to be trapped at the Top1mtcc binding sites; this is shown schematically. (C) Detection of TDP1H493R trapping sites on mtDNA by chromatin immunoprecipitation (ChIP) followed by mtDNA-specific quantitative polymerase chain reaction (qPCR) analysis. FLAG-TDP1-DNA adducts were immunoprecipitated with anti-FLAG antibody in the indicated cells after treatment with mito-SN38 treatment (5 μM for 3 hours), and the putative TDP1-binding site was quantified by qPCR. The mtDNA copy numbers of each cell line were concomitantly measured using primers for the ND2 (mitochondrial) and B2M (nuclear) genes. Enrichment of TDP1-bound mtDNA is expressed as percent input, which is then normalized to the mtDNA copy number of the cell line. Data represent means ± SE of independent experiments. Asterisks denote statistically significant differences (***P < 0.001, t test). (D and E) Cells were treated with mito-SN38 for the indicated times. After mito-SN38 removal (R), cells were cultured in drug-free medium for 12 hours (top). Long-range qPCR was used to evaluate mtDNA damage. (D) Induction of mito-SN38–induced mtDNA damage in indicated cell types. Representative images are shown for mtDNA long- and short-fragment PCR after treatment (mito-SN38, 20 μM) and drug removal (R) for the indicated time. Mouse and human mtDNA-specific primers were used. (E) Quantification of mtDNA damage using the ratio of the long-fragment versus short-fragment PCR products. Data represent means ± SE of independent experiments. Asterisks denote statistically significant difference (**P < 0.01 and ***P < 0.001, t test). kbp, kilobase pair; bp, base pair.
To gain further insight into the sites on mtDNA where TDP1H493R becomes trapped, we performed chromatin immunoprecipitation (ChIP) followed by mtDNA-specific quantitative polymerase chain reaction (qPCR) analysis (18, 48). TDP1 is primarily involved in hydrolysis of trapped Top1cc complexes (Fig. 2B); therefore, we tested the enrichment of TDP1H493R in specific mtDNA segments associated with Top1mtcc binding sites (46). Figure 2B shows that specific trapping of TDP1H493R corresponds to the nucleotide residues 15420, 15690, and 16060 as well as the heavy and light strand promoter (HSP/LSP) along the regulatory noncoding region of mtDNA (mtNCR). Under similar conditions, mito-SN38–induced TDP1H493R enrichment within the coding sequences of the mtDNA was not detected (fig. S1B). Therefore, mito-SN38 recruits TDP1 at sites corresponding to Top1mtcc binding sites on mtDNA (46, 48), generating secondary TDP1-mtDNA cross-links in SCAN1 cells (P < 0.001; Fig. 2B).
We next tested whether TDP1H493R trapping instigates mtDNA damage using long-range PCR, a well-established method to evaluate mtDNA damage (15). Top1ccs reverse within minutes after washing out the drug (44); we therefore analyzed the subsequent mtDNA repair by incubating the cells in drug-free medium for an additional 12 hours. Figure 2D shows that mitochondria harboring defective TDP1 (H493R) display rapid depletion of mtDNA (~2-fold) and were significantly deficient in recovery of mtDNA even after 12 hours of repair in drug-free medium (~4-fold) compared to TDP1-deficient (TDP1−/−) or TDP1-proficient MEFs (TDP1−/−/WT or TDP1+/+) (P < 0.001; Fig. 2, D and E). Similarly, SCAN1 patient–derived lymphoblastoid cells show a marked depletion (~3-to 4-fold) and impaired recovery of mtDNA (~6- to 7-fold) even after 12 hours of culture in drug-free medium (P < 0.001; Fig. 2, D and E), suggesting that mito-SN38–induced hypersensitivity in TDP1−/−/H493R cells is primarily attributed to toxic mtDNA lesions linked to TDP1H493R.
Mitochondrial network harboring TDP1H493R undergoes fission upon mito-SN38 treatment
Mitochondrial dynamics is critical in regulating morphology and function (32). Therefore, to investigate the role of TDP1H493R on the dynamics of the mitochondrial network, i.e., the fission and fusion rates, we used a fluorescence recovery after photobleaching (FRAP) technique to analyze the rate of redistribution of mitochondrial matrix–localized mito-YFP (yellow fluorescent protein), ectopically expressed in MEFs expressing TDP1 variants (49). In the absence of mito-SN38, mitochondrial FRAP recovery of TDP1-proficient (TDP1+/+ and TDP1−/−/WT) and TDP1−/− MEFs was fast (65 to 75% in 60 s), indicating that, under steady-state conditions, there is a rapid and large-scale diffusion of mito-YFP due to the existence of a closely associated mitochondrial network (see Fig. 3A and the quantification in Fig. 3B). However, the mitochondrial fluorescence recovery of TDP1−/−/H493R MEFs was slower after 30 s of bleaching and only reached a maximum intensity of 55 to 60% in 60 s (Fig. 3B), suggesting a delay in mitochondrial fusion rate. Mito-SN38 pretreatment significantly blocked the mitochondrial FRAP recovery (∼10 to 15% in 60 s) in TDP1−/−/H493R MEFs (Fig. 3, C and D) compared to TDP1−/− MEFs. Under similar conditions, catalytically active TDP1 (TDP1+/+ or TDP1−/−/WT) rescued (∼20 to 25%) the FRAP recovery (Fig. 3, C and D), suggesting the existence of a dispersed mitochondrial network due to increased mitochondrial fission in SCAN1-TDP1–expressing MEFs.
Fig. 3. TDP1H493R trapping promotes mitochondrial fission.

Mitochondrial network dynamics were analyzed in the indicated cells using live-cell microscopy and photobleaching (FRAP analysis) of cells ectopically expressing the mitochondrial targeted fluorescence protein construct (mito-YFP). Cells were either untreated (A) or pretreated with mito-SN38 (5 μM for 3 hours) (C). A submitochondrial spot indicated by a circle was bleached (BLH) with a 514-nm laser for 30 ms and imaged at regular intervals of 500 ms thereafter. Successive images taken for 60 s after bleaching illustrate the level of return of fluorescence into the bleached areas. (B and D) Quantitative mitochondrial FRAP data (n = 25). Error bars represent means ± SEM. (E and F) Mitochondrial length was measured in the indicated MEFs by staining with MitoTracker red with or without mito-SN38 (5 μM for 3 hours). Nuclei were stained with Hoechst 33342 (blue). The bar graph (n = 25) represents means ± SEM. (G and H) Western blot analysis of Mfn1 (mitofusin 1) and Drp1 (dynamin-related protein 1) in total lysates obtained from the indicated cells after treatment with mito-SN38 (5 μM for 6 hours). Actin served as the loading control. Migration of protein molecular weight markers is indicated on the right. Bar graphs represent the fold change in densitometry analysis of Mfn1 and Drp1 normalized to actin (error bars represent means ± SEM).
Further evidence for mitochondrial fragmentation in SCAN1-TDP1–harboring cells was seen by live-cell microscopy with the mitochondria-specific dye MitoTracker red. The average length of a mitochondrion was substantially shorter in TDP1−/−/H493R MEFs (~2.7 μm), as compared with TDP1−/−/WT MEFs (~4.3 μm) and TDP1−/− MEFs (~3.4 μm; Fig. 3, E and F; untreated). Mito-SN38 treatment markedly induced mitochondrial fragmentation in TDP1−/−/H493R MEFs (~1.2 μm) compared to TDP1−/− (1.7 μm) or TDP1−/−/WT (2.8 μm) MEFs (Fig. 3, E and F; treated) consistent with increased fission rates (Fig. 3, C and D).
To further validate the mitochondrial fragmentation in TDP1−/−/H493R MEFs, we directly measured the expression of the key proteins associated with mitochondrial fission and fusion like Drp1 (dynamin-related protein 1; mitochondrial fission protein) and Mfn1 (mitofusin 1; mitochondrial fusion protein) (32) in cells expressing TDP1 variants in the presence or absence of mito-SN38 (Fig. 3G). We detected a ~2-fold reduction in Mfn1 expression in TDP1−/−/H493R MEFs compared to TDP1−/−/WT MEFs after mito-SN38 treatment (Fig. 3E). In contrast, under similar conditions, the expression of Drp1 was enhanced ~2.5-fold in TDP1−/−/H493R MEFs compared to TDP1−/−/WT MEFs, confirming mitochondrial fragmentation (Fig. 3H), which is in agreement with an increased fission rate (Fig. 3, C and D). Similarly, we also detected a mito-SN38–induced increase (∼2.5-fold) in Drp1 levels in human SCAN1 cells (BAB1662) compared to their wild-type counterpart (BAB1668) (Fig. 3F), confirming that TDP1H493R trapping directs mitochondrial dynamics toward mitochondrial fission.
TDP1H493R-mtDNA lesions impair mitochondrial biogenesis and OXPHOS function
We next tested the impact of TDP1H493R trapping on the integrity of the mitochondrial membrane potential (Δψm) in cells expressing TDP1 variants by tetramethylrhodamine methyl ester (TMRM) staining and flow cytometry analysis. Figure 4A shows that mito-SN38 treatment caused pronounced depolarization of Δψm in TDP1−/−/H493R MEFs, which were rescued by complementation of wild-type human TDP1 (Fig. 4A). SCAN1 cells treated with mito-SN38 showed a marked reduction (∼15-fold) in the mRNA expression of OXPHOS subunits encoded by mtDNA such as ND2, ND5, ND6, CytB, Cox1, Cox2, and ATP8 along the mitochondrial polycistronic heavy- and light-chain transcript (fig. S1C). We also detected a ∼3- to 4-fold depletion of mitochondrial respiratory complex V subunit (nuclear encoded) (fig. S1D) and (∼4-fold) reduction in ATP content in SCAN1 cells (fig. S1E). Collectively these data indicate that irreversible mtDNA damage in TDP1−/−/H493R (Fig. 2D) impaired the assembly of mitochondrial respiratory chain complexes and maintenance of membrane potential (Fig. 4A).
Fig. 4. TDP1H493R trapping promotes mitochondrial dysfunction in SCAN1 cells.
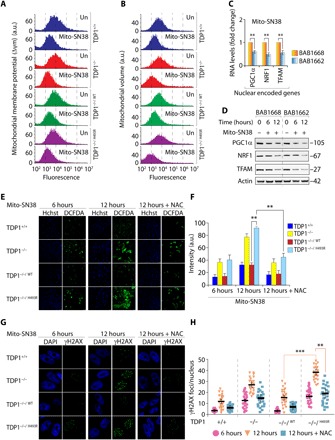
(A) Fluorescence-activated cell sorting (FACS) analysis of mitochondrial membrane potential (Δψm) using TMRM before or after treatment with mito-SN38 (5 μM for 6 hours) in the indicated cells. TMRM fluorescence was plotted against cell numbers (count). Data represent means ± SE of independent experiments. (B) FACS analysis of mitochondrial volume in the indicated cells using MitoTracker green before and after treatment with mito-SN38 (5 μM for 6 hours). Data represent means ± SE of independent experiments. (C) The gene expression profile of nuclear-encoded genes [nuclear respiratory factor 1 (NRF1), peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α), and mitochondrial transcription factor A (TFAM)] for mitochondrial biogenesis by reverse transcription PCR. Indicated cells were either not treated or treated with mito-SN38 (5 μM for 6 hours). Data represent means ± SE of three independent experiments. Asterisks denote statistically significant differences (**P < 0.01, t test). (D) Representative Western blots for nuclear-encoded NRF1, PGC1α, and TFAM in SCAN1 patient–derived lymphoblastoid cell lines (BAB1662) and their wild-type counterpart (BAB1668) before and after treatment with mito-SN38 (5 μM) for the indicated time periods. Actin is shown as the loading control. (E and F) ROS formation was measured by fluorescent dye CM-H2DCFDA in live-cell microscopy after treatment with mito-SN38 (5 μM) or with pretreatment of N-acetyl-l-cysteine (NAC) (10 mM for 2 hours) for the indicated time. The ROS intensity is shown in green, and nuclei were stained with Hoechst 33342 (blue) in the indicated cells. Plots shown on the right represent means ± SDs of at least three experiments. Asterisks denote statistically significant difference (**P < 0.01, t test). (G and H) Representative γH2AX visualization by immunofluorescence microscopy in MEFs expressing TDP1 variants after treatment with mito-SN38 (5 μM) or with pretreatment of NAC (10 mM for 2 hours) for the indicated time. The γH2AX is shown in green, and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scattergrams are shown on the right for at least three experiments; means ± SDs are indicated. Asterisks denote statistically significant differences (**P < 0.01 and ***P < 0.001, t test).
Furthermore, we quantitatively measured mitochondrial mass by staining cells with MitoTracker green using flow cytometry (50). Figure 4B indicates a subtle reduction in the overall mitochondrial volume in TDP1−/−/H493R MEFs without treatment, while mito-SN38 treatment caused steady reduction in mitochondrial volume in TDP1−/−/H493R MEFs, which is in keeping with a marked increase of mitochondrial fission rate as obtained from FRAP experiment (Fig. 4B).
Mitobiogenesis is a coordinated process licensing increased production of nuclear and mitochondria-encoded proteins to increase mitochondrial function or to compensate for removal of the damaged mitochondria (32, 51). This prompted us to test the expression of nuclear genes in SCAN1 cells that are solely responsible for maintaining the mitochondrial homeostasis. We detected ~1.5-fold reduction in the mRNA and the protein levels of PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1-α), NRF1 (nuclear respiratory factor 1), and TFAM (mitochondrial transcription factor A) after mito-SN38 treatment for 6 hours (P < 0.01; Fig. 4, C and D). Prolonged treatment (mito-SN38, 12 hours) resulted in a ~5-fold reduction in protein levels for TFAM, NRF1, and PGC1α in SCAN1 cells (Fig. 4D).
To investigate the mechanistic link between mitochondrial dysfunction and nuclear gene function essential for the mitochondrial biogenesis and OXPHOS, we tested ROS formation and accumulation of DNA damage in the nucleus by γH2AX staining (52). TDP1−/−/H493R MEFs treated with mito-SN38 for 12 hours accumulate (~2.5-fold) increased ROS formation (P < 0.01; Fig. 4E and quantitation in Fig. 4F) and ~3-fold increase in nuclear γH2AX compared to 6-hour treatment (P < 0.001; Fig. 4G and quantitation of γH2AX foci in Fig. 4H). We further confirmed that pretreatment with ROS inhibitor N-acetyl-l-cysteine significantly blocked ROS formation (P < 0.01; Fig. 4, E and F) and nuclear DNA damage in TDP1−/−/H493R MEFs (P < 0.01; Fig. 4, G and H) after treatment with mito-SN38 for 12 hours. Collectively, we provide evidence that selective trapping of TDP1H493R in the mitochondria triggers nuclear DNA damage as a bystander effect through ROS formation.
Mito-SN38 activates autophagy in SCAN1 cells
Accumulation of damaged mitochondria in cells expressing TDP1H493R prompted us to test the mechanism of its removal through autophagy (38, 53, 54). We directly measured steady-state levels of autophagosomes by testing the level of endogenous microtubule-associated protein 1A/1B light chain 3B (LC3-II) lipidation using immunoblotting in cells expressing TDP1 variants after treatment with mito-SN38. Figure 5 (A and B) shows a marked increase (~2-fold) in LC3-II conversion after incubation with sublethal dosage of mito-SN38 in TDP1−/−/H493R MEFs compared to TDP1−/−/WT MEFs, which is consistent with human SCAN1 cells showing marked increase in LC3-II conversion compared to their wild-type counterpart after mito-SN38 treatment (Fig. 5, A and B).
Fig. 5. SCAN1-TDP1 in mitochondria activates autophagy.
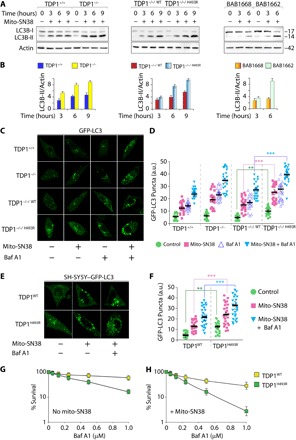
(A) Immunoblotting for LC3-II lipidation with an anti-LC3B–specific antibody with the indicated cell lysates before or after treatment with a sublethal dose of mito-SN38 (2.5 μM) for the indicated time periods. The positions of LC3B-I and LC3B-II are indicated. Actin is shown as the loading control. (B) Plots represent fold increase in LC3B-II band intensity in the indicated cells, normalized to actin (error bars represent means ± SEM). (C) Representative images of GFP-LC3 puncta formation using live-cell confocal microscopy in the indicated cells by ectopically expressing GFP-LC3. The cells were either untreated or treated with mito-SN38 (2.5 μM for 12 hours) or the autophagy inhibitor Baf A1 (200 nM for 4 hours) alone or in combination with Baf A1 (200 nM for 4 hours) + mito-SN38 (2.5 μM for 12 hours). The translocation of GFP-LC3 to phagosome triggers steady-state levels of autophagosomes, as indicated by GFP-LC3 puncta formation. (D) Quantification of GFP-LC3 puncta per cell after the indicated treatment obtained from immunofluorescence confocal microscopy was calculated for 20 to 25 cells (mean ± SEM) from independent experiments. Asterisks denote statistically significant differences (**P < 0.01 and ***P < 0.001, t test). (E) Representative images of differentiated neuronal cells derived from human neuroblastoma cells (SH-SY5Y) stably expressing GFP-LC3 were transfected with lentiviral constructs of TDP1WT or TDP1H493R and were analyzed for GFP-LC3 puncta formation under live-cell confocal microscopy. The cells were either kept untreated or treated with mito-SN38 (2.5 μM for 12 hours) or the autophagy inhibitor Baf A1 (200 nM for 4 hours) alone or in combination with Baf A1 (200 nM for 4 hours) + mito-SN38 (2.5 μM for 12 hours). (F) Quantification of GFP-LC3 puncta per cell after the indicated treatments was obtained from live-cell microscopy calculated for 20 to 25 cells (mean ± SEM) from independent experiments. Asterisks denote statistically significant differences (**P < 0.01 and ***P < 0.001, t test). (G and H) Cell survival of differentiated neuronal cells from SH-SY5Y cells expressing TDP1WT or TDP1H493R. Baf A1–induced cytotoxicity (%) was calculated with respect to the untreated control (G). Combination of mito-SN38 (2.5 μM for 12 hours) + Baf A1 (for the next 24 hours)–induced cytotoxicity (%) was calculated with respect to the untreated control (H). Each point corresponds to the mean ± SD of at least three experiments. Error bars represent SD (n = 3).
To further confirm autophagy activation in cells harboring SCAN1-TDP1, we performed green fluorescent protein (GFP)–LC3 puncta formation assay by using live-cell confocal microscopy (53, 55). Bafilomycin A1 (Baf A1; an autophagy inhibitor) (56) alone increases the number of GFP-LC3 puncta in TDP1−/−/H493R MEFs that were markedly elevated (~2- to 3-fold) when combined with mito-SN38 (P < 0.001; Fig. 5C; see the quantification in Fig. 5D) in contrast to its wild-type counterpart.
To further investigate the role of TDP1H493R trapping in neuronal cells, we differentiated human neuroblastoma cells (SH-SY5Y) to neurons (fig. S2A) (34) and then ectopically expressed TDP1WT or TDP1H493R (fig. S2A). The SH-SY5Y cells were stably selected with GFP-LC3, so we tested GFP-LC3 puncta formation in differentiated SH-SY5Y expressing TDP1 variants under live-cell microscopy. We detected notable increase in the basal levels of GFP-LC3 puncta formation in the differentiated SH-SY5Y cells only by expressing TDP1H493R, which was significantly increased after treatment with the autophagy inhibitor Baf A1 (P < 0.001; Fig. 5E; see the quantification in Fig. 5F) compared to wild-type TDP1 expression. GFP-LC3 puncta were markedly elevated by ~3-fold in the presence of mito-SN38 + Baf A1 in differentiated SH-SY5Y cells expressing TDP1H493R compared to wild-type TDP1 (P < 0.001; Fig. 5E; see the quantification in Fig. 5F).
To test the functional role of autophagy in the survival of the differentiated neuronal cells expressing TDP1H493R, we performed survival assays with the autophagy inhibitor Baf A1 alone or in combination with a sublethal dose of mito-SN38 in the neuronal cells expressing TDP1 variants. We noted that inhibition of autophagy by Baf A1 alone significantly induced cell death in TDP1H493R-expressing neuronal cells (Fig. 5G), which was markedly increased in combination with a sublethal dose of mito-SN38 (Fig. 5G; see mito-SN38 + Baf A1) compared to cells expressing the TDP1 wild-type counterpart, suggesting that autophagy rescues mito-SN38–induced mitochondrial toxicity in SCAN1 cells. Collectively, our data provide evidence that TDP1H493R trapping promotes the steady-state level of autophagosome formation, and upon inhibition of autophagy, this shifts toward apoptosis.
Trapping of TDP1H493R activates PTEN-induced kinase 1–dependent mitophagy
Elimination of damaged mitochondria occurs through a specialized form of autophagy referred to as mitophagy (32, 33, 53). Thus, for the quantitative assessment of mitophagy in cells expressing TDP1 variants, we used a specific traffic light mitophagy reporter (mito-mRFP-EGFP) construct consisting of a pH-stable red fluorescent protein (RFP) fused to an acidic pH-labile GFP with a mitochondria-targeting signal (36). Localization of autophagic mitochondria inside the lysosome leads to formation of red-only fluorescence against yellow fluorescence for nonautophagic mitochondrial counterparts in cells expressing the mito-mRFP-EGFP construct (Fig. 6A). Without mito-SN38 treatment, we detected significant increases in red-only fluorescence in 30 to 35% of mitochondria in TDP1−/−/H493R MEFs compared to TDP1-proficient cells expressing the traffic light construct (P < 0.01; Fig. 6B), which is in keeping with the significant increase in the number of GFP-LC3 puncta without drug treatment in TDP1−/−/H493R MEFs (Fig. 5, C and D) or differentiated neuronal cells expressing TDP1H493R (Fig. 5, E and F) and related to drug-independent trapping of TDP1H493R (Fig. 2A). Mito-SN38 treatment markedly increased mitophagy in TDP1H493R MEFs as revealed by more than 80% red-only fluorescence in the mitochondria (P < 0.001; Fig. 6C) compared to TDP1+/+ or TDP1−/−/WT MEFs. Furthermore, we confirmed that the red-only fluorescent mitochondria in TDP1H493R MEFs correspond to mitochondria engulfed by lysosomes, as shown by colocalization with the lysosomal-specific LysoTracker dye (fig. S2C).
Fig. 6. TDP1H493R trapping in mitochondria activates mitophagy.
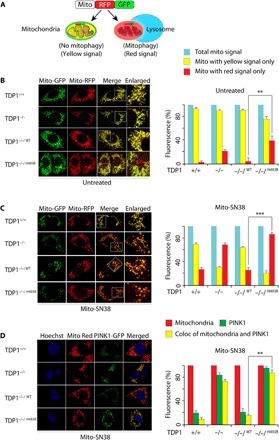
(A) Schematic representation for mitophagy monitoring using the dual fluorescence reporter construct p–mito-mRFP-EGFP (pAT016). Lysosomal delivery of the tandem fusion protein mito-mRFP-EGFP along with entire mitochondria results in pH-dependent quenching of green fluorescence resulting in red-only fluorescence for visual analysis of mitophagic flux. (B and C) Representative confocal live images of indicated MEFs ectopically expressing mito-mRFP-EGFP targeting mitochondria. Cells were kept untreated (B) or treated with mito-SN38 (2.5 μM for 12 hours) (C) and were analyzed under live-cell microscopy. The yellow fluorescence signals denote no mitophagy (merged image); red-only fluorescence signals denote mitophagy or mitochondria inside lysosomes. The enlarged panel shows higher-magnification image. Quantification of the indicated fluorescence obtained from live-cell confocal microscopy was calculated for 20 to 25 cells (calculated value ± SEM) in at least three independent experiments. Asterisks denote statistically significant differences (**P < 0.01 and ***P < 0.001, t test). (D) Representative confocal images of live cells for the indicated MEFs showing accumulation of ectopic PTEN-induced kinase 1 (PINK1)–GFP after mito-SN38 (2.5 μM for 3 hours) treatment. Mitochondria are labeled with MitoTracker red; the colocalization of PINK1-GFP (green) in the mitochondrial network (red) is indicated in the merged image. Quantification of the indicated fluorescence obtained from live-cell confocal microscopy was calculated for 20 to 25 cells (calculated value ± SEM) obtained from independent experiments. Asterisks denote statistically significant differences (**P < 0.01, t test).
The sequential events that promote mitophagy is initiated by mitochondrial depolarization and accumulation of PINK1 (PTEN-induced kinase 1) in the outer mitochondrial membrane of the dysfunctional mitochondria (32, 37). Therefore, we ectopically expressed GFP-PINK1 in cells harboring TDP1 variants and investigated PINK1 accumulation in the mitochondria by costaining the cells with the mitochondrion-specific dye MitoTracker red under live-cell confocal microscopy. Figure 6D shows that 70 to 80% of TDP1−/−/H493R MEFs accumulate significantly higher expression of GFP-PINK1 in the mitochondria (P < 0.01; see merged image Fig. 6D) within 3 hours of mito-SN38 treatment compared to (20 to 25%) TDP1+/+ or TDP1−/−/WT MEFs. Therefore, mitochondrial depolarization in TDP1 defective MEFs (Fig. 4A) activates PINK1-dependent mitophagy. These observations highlight the role of SCAN1-TDP1 trapping in mitochondrial fragmentation, a phenomenon in mitophagy associated with mitochondrial clearance.
DISCUSSION
To our knowledge, the present study provides the first evidence that mitochondria in cells harboring SCAN1-TDP1 (TDP1H493R) are selectively trapped on the mtDNA at Top1mt cleavage sites, are shorter in length, and have increased fission rates. We demonstrate that TDP1H493R trapping prevents mitochondrial transcription, energy production, and mitobiogenesis. SCAN1-TDP1 trapping triggers autophagy that allows identification of dysfunctional mitochondria through PINK1 accumulation and their subsequent removal through mitophagy. Our current work offers further evidence that inhibition of autophagy in the neuronal cells expressing TDP1H493R is detrimental, suggesting that mitophagy is critical for SCAN1.
MtDNA is essential for cell viability, as it encodes subunits of OXPHOS as well as mitochondrial mRNA and tRNA (30). The mitochondrial genome accumulates topological stress from bidirectional transcription and replication, resolved by recruiting the Top1mt to the regulatory noncoding region of mtDNA (mtNCR) to relax mtDNA supercoiling generated during these processes (46, 48, 57). Accordingly, Top1mt knockout mice accumulate increased negative supercoiling in the mtDNA that impairs mtDNA replication (58, 59), promoting mitochondrial dysfunction (52). A number of things enhance the trapping of Top1mtcc in mitochondria including anticancer drugs like marine alkaloids lamellarin D (47), mitochondria-targeted irinotecan (mito-SN38; Fig. 1), and camptothecin treatment of purified mitochondria (46). Top1cc can also be trapped by endogenous DNA breaks, oxidized bases, nicks, and DNA modifications generated by ROS (6, 60).
MtDNA repair is particularly critical for the mitochondrial genome, as the mtNCR region is mostly prone to genome instability (61), because it is the most exposed region, which harbors the primary replication initiation site and the critical promoter elements both for the heavy and light strands (30). Thus, the ability of TDP1 to resolve 3′-phosphotyrosyl linkages is consistent with the role of the enzyme in protecting cells against cytotoxic Top1-DNA lesions generated both in the nucleus and in mitochondria (Fig. 1) (4, 12, 18, 27). We detected significant trapping of TDP1H493R in mitochondria without drug treatment (Fig. 2A), plausibly corresponding to the defective catalytic turnover of SCAN1-TDP1 at endogenous mtDNA lesions (10, 20, 21) or drug-independent Top1mtcc break sites, which were markedly enhanced after mito-SN38 treatment (Fig. 2A). TDP1H493R is selectively trapped at mitochondrial nucleotide residues 15420, 15690, and 16060 along the mtNCR (Fig. 2C) at Top1mtcc binding sites (46, 48). SCAN1-TDP1 displays a rapid depletion of mtDNA (~2-fold) and was deficient in recovery of mtDNA even after 12 hours of repair in drug-free medium (Fig. 2, D and E). Subsequently, TDP1H493R trapping induces transcriptional collapse of OXPHOS genes and is defective in organelle assembly of oxidative phosphorylation complexes in SCAN1 cells, which results in reduction in the steady-state level of ATP. This is in agreement with the recent report showing that TDP1 promotes the integrity of electron transport chain (18). Further evidence for dysregulated oxidative phosphorylation across the mitochondrial inner membrane is revealed by depolarization of the membrane potential in TDP1H493R harboring MEFs after mito-SN38 treatment (Fig. 4D).
The dynamic equilibrium between mitochondrial fusion and fission preserves the mitochondrial network (32–34, 39). Here, our live-cell imaging shows that cells harboring SCAN1-TDP1 have reduced mitochondrial networks because of an increase in mitochondrial fission rate (Fig. 3C), which results in shortening of mitochondrial length (Fig. 3, E and F) together with the quantitative loss of mitochondrial volume (Fig. 4E). This is further supported by increased levels of mitochondrial fission–associated protein Drp1 in SCAN1 cells (Fig. 3F) and is in agreement with the observation that expression of catalytically active TDP1 rescues the mitochondrial fission-fusion dynamics, mitochondrial length (Fig. 2), and respiratory capacity (Fig. 3) in TDP1−/− MEFs.
Fusion and fission dynamically regulate mitochondrial morphology and mitobiogenesis (32, 51), which occurs by division of preexisting organelles in coordination with nuclear encoded proteins and cellular proliferation. In SCAN1 cells, we observed a significant reduction in the transcription (Fig. 4F) and protein level (Fig. 4G) of nuclear encoded mitobiogenesis genes including PGC1α, NRF1, and TFAM after prolonged treatment with mito-SN38, which is associated with nuclear DNA damage mediated through ROS (Fig. 4, H and I). We provide evidence that trapping of TDP1H493R in the mitochondria produces potentially toxic DNA lesions that not only dampens mitochondrial activity but also triggers nuclear DNA damage as a bystander effect through ROS formation (Fig. 4). Maneuvering of fission or fusion improves mitochondrial physiology. Disruption of mitochondrial dynamics is common in severe disorders, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, which show neurodegeneration (32, 33, 38, 39, 42, 43, 54).
Because SCAN1 cells accumulate a fragmented mitochondrial network and disrupted respiratory complexes that fail to manage the cellular energy requirements, they have low levels of ATP. To match the metabolic demand, SCAN1-TDP1–harboring cells increase autophagosomes (Fig. 5) to retain a lower number of fit mitochondria and channel the damaged ones to mitophagy by PINK1 accumulation on the outer mitochondrial membrane of the damaged mitochondria (Fig. 6).
In conclusion, the present study establishes the significance of autophagy to overcome TDP1H493R-induced mitochondrial toxicity in neuronal cells (Fig. 5, E to G). Through evolution, cells that are critically dependent on mitochondria for energy production like neurons have acquired useful mechanisms to sequester and eliminate dysfunctional mitochondria for cell survival. It is worth mentioning that caloric restriction and mammalian target of rapamycin inhibition show life span–extending interventions via increased autophagy and mitophagy (62). This supports the argument that mitophagy in the SCAN1 phenotype may play a crucial role in neuroprotection and plausibly is associated with the late onset of neurological disorders as in patients with SCAN1.
MATERIALS AND METHODS
Drugs, reagents, and antibodies
Puromycin (P8833), Baf A1 (J61835), polybrene (H9268), irinotecan (I1406), all-trans-retinoic acid (R2625), and N-acetyl-l-cysteine (A9165) were all purchased from Sigma-Aldrich (USA). Anti-Top1mt–specific antibody (47, 52) (Top1mt, polyclonal mouse) was a gift from Y. Pommier [Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH), USA]. Rabbit polyclonal antibodies [TDP1 (Ab4166), PGC1α (Ab54481), NRF1 (Ab34682), and mtTFAM (Ab47517)] and mouse monoclonal antibodies [COX IV (Ab14744), DRP1 (Ab56788), Mfn1 (Ab57602), and double-stranded DNA (dsDNA) (Ab27156)] were purchased from Abcam (Cambridge, USA). Mouse monoclonal antibodies [FLAG (M2) (F3165)] and rabbit polyclonal antibodies [FLAG (F7425) and LC3B (L7543)] were purchased from Sigma-Aldrich (St. Louis, MO, USA). Mouse monoclonal γH2AX (05-636) antibody was purchased from Millipore, USA. Rabbit monoclonal GFP (G10362) and anti OXPHOS complex kit (457999) antibodies were purchased from Invitrogen. Mouse monoclonal actin (SC-8432) and secondary antibodies [horseradish peroxidase–conjugated anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG] were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). MitoTracker red (M22426), MitoTracker green (M7514), TMRM (T668), CM-H2DCFDA (C6827), and LysoTracker red DND-99 (L7528) were obtained from Molecular Probes.
Synthesis of mitochondria-targeted nanoparticle containing irinotecan (mito-SN38)
Production of a mitochondria-targeting cationic nanoparticle containing Top1 poison irinotecan (mito-SN38) was described previously (45). Briefly, 6.0 mg of l-α-phosphatidylcholine, 1.0 mg of α-tocopherylsuccinate–triphenylphosphine conjugate, 1.0 mg of irinotecan, and 0.6 mg of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol) 2000] were dissolved in 5.0 ml of dichloromethane-methanol mixture. Solvent was evaporated thoroughly to form a thin uniform lipid film. The lipid film was hydrated with 1 ml of water for 1 hour at 60°C. The nanoparticles formed were passed through Sephadex G-25 followed by extrusion through a 200-nm filter. The nanoparticles (mito-SN38) were stored at 4°C for further use.
Cell culture, plasmids, and generation of stable cell lines
Cells were cultured at 37°C with 5% CO2 in Dulbecco’s minimum essential medium (DMEM) or RPMI containing 10% fetal bovine serum (FBS) (Life Technologies) (12, 15, 27, 28, 63). TDP1+/+ and TDP1−/− primary MEF cells were a gift from C. F. Boerkoel (University of British Columbia, Vancouver, British Columbia, Canada). Lymphoblastoid cell lines homozygous for the TDP1-H493R mutation (BAB1662; SCAN1) and normal individual (BAB1668) were a gift from J. R. Lupski (Departments of Pediatrics and Molecular and Human Genetics, Baylor College of Medicine, Houston, USA). The human embryonic kidney 293T [American Type Culture Collection (ATCC)] and human SH-SY5Y cells (ATCC) were a gift from D. Biswas (Council of Scientific and Industrial Research–Indian Institute of Chemical Biology, Kolkata, India). The cloning of pCMV-Tag2B-FLAG-TDP1 construct was described previously (12, 27, 28). Plasmid MDK124-FLAG-TDP1 was generated by digesting a FLAG-TDP1 fragment from pCMV-Tag2B-FLAG-TDP1 construct using Nhe I and Eco RI restriction sites and cloned into the pMDK124 lentiviral vector. The following point mutation (pMDK124-FLAG-TDP1H493R) was generated using the “QuikChange” protocol (Stratagene, La Jolla, CA, USA). All constructs were confirmed by DNA sequencing. The plasmids were then used for lentivirus generation using a standard protocol and were transduced into TDP1−/− MEFs using lentivirus particles containing FLAG-TDP1WT or FLAG-TDP1H493R in the presence of polybrene (5 μg/ml) (Sigma-Aldrich) and were subjected to puromycin selection (4 μg/ml) for 2 weeks. Individual colonies obtained by this method were further amplified and screened for expression of target proteins using Western blotting against epitope tag–specific antibodies.
The pYFP-Mito construct (15, 49) (protein targeted to the mitochondrial matrix) was a gift from R. J. Youle (National Institutes of Neurological Disorders and Stroke, NIH), the traffic light mitophagy reporter construct pAT016 (p–mito-mRFP-EGFP) was a gift from A. Till (University of Bonn, Germany), the pEGFP-LC3 construct was a gift from A. Saha (Presidency University, India), and the pPINK1 C-GFP construct was a gift from P. Mukherjee (Presidency University, India). All plasmid DNA constructs were transfected with Lipofectamine 2000 (Invitrogen) or FuGENE (Roche) according to the manufacturer’s protocol.
Human SH-SY5Y cells were grown in DMEM containing 10% FBS and 1% penicillin and streptomycin and were stably selected for pEGFP-LC3 construct with puromycin (10 μg/ml). Differentiation of SH-SY5Y was performed (34) using 10 μM all-trans-retinoic acid (Sigma-Aldrich) dissolved in dimethyl sulfoxide for 7 to 10 days with changing retinoic acid–containing media on alternate days. Microscopic images of differentiated SH-SY5Y cells were taken using a Leica TCS SP8 microscope. The lentiviral particles containing FLAG-TDP1WT or FLAG-TDP1H493R were transduced into differentiated SH-SY5Y cells in the presence of polybrene (5 μg/ml) (Sigma-Aldrich) and incubated for 48 hours for ectopic expression of proteins. Expression of the target proteins was checked by Western blotting using epitope tag–specific antibodies. Cells were treated with different concentrations of mito-SN38 and Baf A1, as indicated.
Live-cell confocal microscopy and immunocytochemistry
Live-cell imaging was carried out as described previously (12, 27, 63, 64). Briefly, indicated cells were grown on confocal dishes (Genetix Biotech Asia Pvt. Ltd.). All the plasmid DNA were transfected as above and examined by live-cell confocal microscopy, as indicated. Nuclei were stained with Hoechst 33342 (Sigma-Aldrich). Fluorophores were excited using either separately or in combination with ultraviolet (Diode 405), 488-nm (argon), 561-nm (DPSS 561), or 633-nm (HeNe 633) laser lines using a confocal laser scanning microscope (Leica TCS SP8) with 63×/1.4 numerical aperture oil objective equipped with a heated environmental chamber set to 37°C with an optimal CO2 facility. The percentage of cells displaying the indicated fluorescence was determined using Adobe Photoshop 7.0 from at least 20 to 25 cells expressing individual constructs.
Immunofluorescence staining and confocal microscopy were performed as described previously (12, 27, 28, 63, 64). Briefly, cells were fixed with 4% paraformaldehyde for 20 min at room temperature. Primary antibodies against γH2AX were detected with anti-mouse IgG secondary antibodies labeled with Alexa Fluor 488 (Invitrogen). Cells were mounted in anti-fade solution with 4′,6-diamidino-2-phenylindole (Invitrogen) and examined using a laser scanning confocal microscope (Leica TCS SP8). Images were collected and processed using the Leica LAS X software and sized in Adobe Photoshop 7.0. The γH2AX intensity per nucleus was determined using Adobe Photoshop 7.0 by measuring the fluorescence intensities normalized to the number of cell count.
Measurements of mitochondrial connectivity using FRAP assays
Photobleaching experiments were carried out as described previously (12, 27, 49, 63) to measure mitochondrial connectivity using a confocal laser scanning microscope (Leica TCS SP8). FRAP analyses were carried out with MEFs stably expressing TDP1 variants and ectopically expressing mitochondrial matrix targeted (pYFP-Mito) with or without mito-SN38. Bleaching of YFP-mito was applied at randomly chosen mitochondrial regions using a 514-nm laser for 30 ms and photographed at intervals of 500 ms thereafter. Successive images taken for 60 s after bleaching illustrated the level of recovery of fluorescence into the bleached areas. Relative fluorescence intensities of the bleached region were corrected for background. To show the FRAP curves, the fluorescence signal measured in a region of interest (ROI) was individually normalized to the pre-bleach signal in the ROI according to the following equation: ROI = (It − Ibg)/(Io − Ibg) × 100, where Io is the intensity in the ROI during pre-bleach, It is the intensity in the ROI at time point t, and Ibg is the background signal determined in a region outside of the mitochondria.
Cell extracts and immunoblotting
Preparation of whole cell extracts and immunoblotting were carried out as described previously (12, 27, 63). Briefly, cells were harvested and lysed in a lysis buffer [10 mM tris-HCl (pH 8), 150 mM NaCl, 0.1% SDS, 1% NP-40, and 0.5% Na-deoxycholate supplemented with complete protease inhibitors] (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 1 from Sigma-Aldrich). After thorough mixing and incubation at 4°C for 2 hours, lysates were then centrifuged at 12,000g at 4°C for 20 min. Supernatants were collected and stored in aliquots at −80°C. Immunoblots were carried out following standard procedures, and immunoreactivity was detected using enhanced chemiluminescence reaction (170-5061, Bio-Rad) under ChemiDoc MP System (Bio-Rad).
Preparation of mitochondria
Mitochondria were prepared as described previously (15). Briefly, cell pellets were suspended in 10 mM NaCl, 1.5 mM CaCl2, and 10 mM tris-HCl (pH 7.5) at 25°C for 5 min. Following osmotic shock, cells were homogenized using a glass Dounce homogenizer and mixed with stabilizing buffer [2 M sucrose, 35 mM EDTA, and 50 mM tris-HCl (pH 7.5) at 25°C]. Cell lysates were centrifuged at 750g for 5 min to remove nuclei and cell debris. Mitochondria were spun down from the supernatant at 10,000g for 20 min, washed three times with MT buffer [250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 5 mM dithiothreitol (DTT), and 20 mM Hepes-KOH (pH 7.4) at 25°C], and resuspended in MT buffer. For all experiments, freshly prepared mitochondria were used.
ICE bioassay
Detection of Top1mt, TDP1, and nuclear Top1-cleavage complexes by ICE bioassay was performed as described previously (47). Briefly, cells (5 × 106) expressing TDP1 variants were either untreated or treated with the indicated concentration of drugs. Mitochondria were isolated as described above and lysed by adding lysis buffer [6 M guanidinium thiocyanate, 10 mM tris-HCl (pH 6.5), 20 mM EDTA, 4% Triton X-100, 1% sarosyl, and 1% DTT]. Mitochondrial lysates were mixed with 0.4 ml of 100% ethanol, incubated at −20°C for 5 min, and cleared by centrifugation (12,000g for 10 min). Supernatants were discarded, and pellets were washed two times with 100% ethanol and then dissolved in 0.2 ml of 8 mM NaOH (freshly made) and sonicated for 10 to 20 s at 20% power. For immunodetection, mtDNA at varying concentrations were spotted onto a nitrocellulose membrane (Millipore, USA) using a slot-blot vacuum system (Bio-Rad, USA). For detection of nuclear Top1cc, whole cells were lysed by adding lysis buffer, cleared by centrifugation, and the pellet was dissolved in 8 mM NaOH and slot-blotted as indicated above. Immunoblotting was carried out with anti-Top1mt (47, 52) or anti-TDP1 (15) and anti-nuclear Top1-specific antibodies. Anti-dsDNA was used for loading control. Immunoblots were visualized using enhanced chemiluminescence reactions on a ChemiDoc MP System.
Cell survival assays
Cell survival was carried out as described previously (28, 45, 65). Briefly, TDP1+/+ MEFs, TDP1−/− MEFs, and TDP1−/− MEF cells stably expressing FLAG-TDP1 variants (TDP −/−/WT, TDP −/−/H493R, or TDP +/+/H493R) or human lymphoblastoid cells (BAB1662 or BAB168) or differentiated SH-SY5Y cells ectopically expressing FLAG-TDP1WT or FLAG-TDP1H493R (1 × 103) cells were seeded in 96-well plates (BD Biosciences, USA). After 24 hours, cells were treated with the indicated drugs and kept for a further 48 hours. Cell survival was then assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, M5655). Plates were analyzed on a Molecular Devices Spectra MaxM2 Microplate Reader at 570 nm. The percent inhibition of viability for each concentration of drugs was calculated with respect to the control. Data represent means ± SD for three independent experiments.
Quantification of mtDNA damage
To compare the levels of mtDNA damage and repair in cells expressing TDP1 variants, cells were treated with mito-SN38 (20 μM) for the indicated times or further cultured in drug-free medium to measure repair using mtDNA long-range PCR as described previously (15, 66). We used human and mouse mtDNA sequence–specific primers to amplify a 9- or 10-kbp (kilo–base pair) fragment of mtDNA, respectively. A small 110-bp (for human) or 117-bp (for mouse-specific) mtDNA fragment was also amplified for normalization. PCR reactions were limited to 18 cycles to ensure that the amplification process was still in the exponential phase. The damage index was determined by the ratio LR/SR of long-range PCR product (LR) by the short-range PCR product (SR). The sequences of the primers are listed in table S1.
ChIP and reverse transcription qPCR
ChIP was performed as described previously (18, 48). Briefly, MEF cells expressing TDP1 variants (1 × 107) were treated with mito-SN38 (5 μM) for 6 hours followed by fixation with formaldehyde (1.1% final concentration) for 10 min at room temperature, and reactions were stopped by adding 125 mM glycine for 5 min. Cells were then washed with phosphate-buffered saline (PBS), scraped, and lysed in 0.5 ml of ChIP lysis buffer [50 mM Hepes-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8), 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 1× protease inhibitor] on ice for 30 min. Lysates were sonicated 10 times for 30 s at 30% power to yield fragments of 200 to 300 bp and cleared by centrifugation at 14,000g for 10 min at 4°C. The supernatant (50 μl) was stored at −20°C as input and labeled as “no-IP.” The remainder was diluted fourfold in dilution buffer [50 mM tris-HCl (pH 8), 150 mM NaCl, 2 mM EDTA (pH 8), 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 1× protease inhibitors] and incubated with 10 μl of FLAG antibody on a rotating platform overnight at 4°C. The following day, 150 μl of A/G PLUS-Agarose beads (Santa Cruz Biotechnology) was added and further incubated on a rotating platform for 2 hours at 4°C. The beads were then washed twice with low-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8), and 150 mM NaCl], twice with high-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8), and 500 mM NaCl], followed by one wash with lithium chloride buffer [0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, and 10 mM tris-HCl (pH 8)], and twice with TE buffer. The immunoprecipitated complex was then eluted from the beads in 150 μl of elution buffer (1% SDS and 100 mM NaHCO3). The eluent and the no-IP control samples were then treated with RNase A and proteinase K. The DNA was purified using phenol-chloroform extraction and ethanol precipitation. The pellets were resuspended in 30 μl of distilled water.
For qPCR, the no-IP or input and ChIP samples were diluted 1:10; then, 5 μl was mixed with 2.5 μl of 5 μM forward and reverse primers (table S 1) with 10 μl of 2× SYBR Green PCR mix (Applied Biosystems). The PCR mix was aliquoted into reaction volumes (20 μl) in triplicate and amplified using an ABI 7500 thermocycler (Applied Biosystems). The PCR reactions were carried out with thermocycling conditions of 95°C for 10 min and then 40 cycles at 90°C for 15 s, 50°C for 15 s, and 72°C for 30 s, with signal acquisition at the end of each cycle. Quantification of chromatin enrichment was calculated by percent input normalized to the mtDNA copy number in the input samples. The sequences of the primers are listed in table S1.
Quantification of mitochondrial gene transcription by real-time PCR
The total RNA was extracted from indicated cells (1 × 106) using TRIzol reagent (15596018, Invitrogen) according to the manufacturer’s protocols, which includes deoxyribonuclease (AMPD1; Sigma-Aldrich) treatment for each sample (52). An aliquot of 1 μg of RNA was reverse-transcribed using a reverse transcription kit (4368814, Applied Biosystems). Real-time PCR was performed with SYBR Green PCR master mix (A25742, Applied Biosystems) on the ABI 7500 thermocycler (Applied Biosystems). Reaction mixtures contained 5 μl of 2× SYBR-Green PCR master mix and 2 μl of reverse transcriptase–generated cDNA diluted 10-fold in a final volume of 10 μl containing primers at 25 nM. The thermocycling conditions were 95°C for 5 min, followed by 40 cycles at 95°C for 50 s, 50°C (variable) for 50 s, and 72°C for 60 s. Relative gene expression was expressed as a ratio of the expression level of the gene of interest to that of β-actin RNA, with values in wild-type cells defined as 100%. The sequences of the primers are listed in table S1.
Analysis of mitochondrial length
Mitochondrion length was measured as described previously (49). Briefly, MEFs expressing TDP1 variants were incubated with or without mito-SN38, as indicated. Then, cells were stained with MitoTracker red and were subjected to live-cell confocal microscopy (Leica TCS SP8). Individual mitochondrial length analysis was performed using the line measurement property of Leica LAS X software. An average length of ~20 to 25 mitochondria per cell was measured.
Mitochondrial membrane potential
Determination of Δψm was performed using TMRM (200 nM for 30 min at 37°C) (50, 52), which is a positively charged, colorless dye that enters mitochondria in a membrane potential–dependent manner and emits bright red-orange fluorescence that was analyzed by flow cytometry (BD, FACSAria III).
Determination of mitochondrial mass
Mitochondrial mass was measured by MitoTracker Green FM (Molecular Probes) staining (50, 52). Cells were trypsinized and resuspended in PBS with 200 nM MitoTracker Green FM (30 min at 37°C) in the dark and were analyzed by flow cytometry (BD, FACSAria III).
ATP determination
ATP level in indicated cells was determined using an ATP determination kit (A22066; Molecular Probes) following the manufacturer’s protocol (52).
Intracellular ROS detection by confocal microscopy
For intracellular ROS detection by live-cell confocal (Leica TCS SP8) microscopy, MEFs expressing TDP1 variants were seeded in confocal dishes and treated with or without mito-SN38 for the indicated time periods and then washed twice with PBS (pH 7.4) and 10 μM CM-H2DCFDA in PBS was applied to live cells and kept at 37°C for 5 min (28, 45, 65). The CM-H2DCFDA diffuses into cells where the free nonfluorescent 2′,7′-dichlorodihydrofluorescein in the cytoplasm is oxidized to the green fluorescent moiety, dichlorofluorescein, by intracellular ROS upon excitation with 488-nm argon laser. Nuclei were stained with Hoechst 33342.
Supplementary Material
Acknowledgments
We thank Y. Pommier of the NCI, NIH, USA, for the reagents and help during the study. We are also thankful to C. Austin of the Institute for Cell and Molecular Biosciences, Newcastle University, UK, for critically reading the manuscript. Funding: B.B.D.’s team was supported by a Wellcome Trust/DBT India alliance intermediate fellowship grant (award no. IA/I/13/1/500888), a DST-SERB core research grant (EMR/2017/001652), and the Indian Association for the Cultivation of Science intramural fund. A.G. and S.B. are the recipients of CSIR-NET Senior and Junior Research Fellowship, respectively, in India. A.M. and S.B. thank CSIR-UGC doctoral fellowship and DST-Nanomission [SB/NM/NB-1083/2017 (G)], respectively, for financial support. B.B.D. is a Wellcome Trust/DBT India alliance intermediate fellow. Author contributions: A.G., S.B., S.P.C., A.M., I.R., and S.B. performed the experiments. A.G. and B.B.D. designed and analyzed the data and wrote the manuscript. B.B.D. provided supervision. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9778/DC1
Fig. S1. Mito-SN38 does not trap nuclear-Top1cc but impairs mitochondrial metabolism through SCAN1-TDP1 trapping in the mitochondria.
Fig. S2. Differentiation of SH-SY5Y cells showing expression of FLAG-TDP1 and lysosomal localization of SCAN1 mitochondria showing mitophagy.
Table S1. List of primers used.
REFERENCES AND NOTES
- 1.Takashima H., Boerkoel C. F., John J., Saifi G. M., Salih M. A. M., Armstrong D., Mao Y., Quiocho F. A., Roa B. B., Nakagawa M., Mutation of TDP1, encoding a topoisomerase I–dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 32, 267–272 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Yang S.-W., Burgin A. B. Jr., Huizenga B. N., Robertson C. A., Yao K. C., Nash H. A., A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. U.S.A. 93, 11534–11539 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawale A. S., Povirk L. F., Tyrosyl–DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 46, 520–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y., Huang S.-y. N., Gao R., Das B. B., Murai J., Marchand C., Tyrosyl-DNA-phosphodiesterases (tdp1 and tdp2). DNA Repair 19, 114–129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Khamisy S. F., To live or to die: A matter of processing damaged DNA termini in neurons. EMBO Mol. Med. 3, 78–88 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pommier Y., Sun Y., Huang S.-y. N., Nitiss J. L., Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 17, 703–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katyal S., Lee Y., Nitiss K. C., Downing S. M., Li Y., Shimada M., Zhao J., Russell H. R., Petrini J. H., Nitiss J. L., Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci. 17, 813–821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katyal S., El-Khamisy S. F., Russell H. R., Li Y., Ju L., Caldecott K. W., McKinnon P. J., TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 26, 4720–4731 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Khamisy S. F., Saifi G. M., Weinfeld M., Johansson F., Helleday T., Lupski J. R., Caldecott K. W., Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434, 108–113 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Interthal H., Chen H. J., Kehl-Fie T. E., Zotzmann J., Leppard J. B., Champoux J. J., SCAN1 mutant Tdp1 accumulates the enzyme–DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 24, 2224–2233 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldecott K. W., Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Das B. B., Antony S., Gupta S., Dexheimer T. S., Redon C. E., Garfield S., Shiloh Y., Pommier Y., Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 28, 3667–3680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano R., Interthal H., Huang C., Nakamura T., Deguchi K., Choi K., Bhattacharjee M. B., Arimura K., Umehara F., Izumo S., Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 26, 4732–4743 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins A. J., Subler M. A., Akopiants K., Wiley J. L., Taylor S. M., Rice A. C., Windle J. J., Valerie K., Povirk L. F., In vitro complementation of Tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation. DNA Repair 8, 654–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das B. B., Dexheimer T. S., Maddali K., Pommier Y., Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 107, 19790–19795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai J., Huang S.-y. N., Das B. B., Dexheimer T. S., Takeda S., Pommier Y., Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 287, 12848–12857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fam H. K., Choi K., Fougner L., Lim C. J., Boerkoel C. F., Reactive oxygen species stress increases accumulation of tyrosyl-DNA phsosphodiesterase 1 within mitochondria. Sci. Rep. 8, 4304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang S.-C., Meagher M., Kassouf N., Hafezparast M., McKinnon P. J., Haywood R., El-Khamisy S. F., Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical–induced DNA damage. Sci. Adv. 3, e1602506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inamdar K. V., Pouliot J. J., Zhou T., Lees-Miller S. P., Rasouli-Nia A., Povirk L. F., Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem. 277, 27162–27168 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Interthal H., Chen H. J., Champoux J. J., Human Tdp1 cleaves a broad spectrum of substrates including phosphoamide linkages. J. Biol. Chem. 280, 36518–36528 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T., Lee J. W., Tatavarthi H., Lupski J. R., Valerie K., Povirk L. F., Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 33, 289–297 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassine S. B., Arcangioli B., Tdp1 protects against oxidative DNA damage in non-dividing fission yeast. EMBO J. 28, 632–640 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W., Hu X., Shen Q., Xing D., Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 10, 1704 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang S.-y. N., Murai J., Dalla Rosa I., Dexheimer T. S., Naumova A., Gmeiner W. H., Pommier Y., TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 41, 7793–7803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang S.-C., Carroll J., El-Khamisy S. F., TDP1 serine 81 promotes interaction with DNA ligase IIIα and facilitates cell survival following DNA damage. Cell Cycle 9, 588–595 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Hudson J. J. R., Chiang S.-C., Wells O. S., Rookyard C., El-Khamisy S. F., SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 3, 733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das B. B., Huang S.-y. N., Murai J., Rehman I., Amé J.-C., Sengupta S., Das S. K., Majumdar P., Zhang H., Biard D., PARP1–TDP1 coupling for the repair of topoisomerase I–induced DNA damage. Nucleic Acids Res. 42, 4435–4449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rehman I., Basu S. M., Das S. K., Bhattacharjee S., Ghosh A., Pommier Y., Das B. B., PRMT5-mediated arginine methylation of TDP1 for the repair of topoisomerase I covalent complexes. Nucleic Acids Res. 46, 5601–5617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao C., Beveridge R., Hudson J. J., Parker J. D., Chiang S.-C., Ray S., Ashour M. E., Sudbery I., Dickman M. J., El-Khamisy S. F., UCHL3 regulates topoisomerase-induced chromosomal break repair by controlling TDP1 proteostasis. Cell Rep. 23, 3352–3365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace D. C., A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holt I. J., Mitochondrial DNA replication and repair: All a flap. Trends Biochem. Sci. 34, 358–365 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Chan D. C., Mitochondria: Dynamic organelles in disease, aging, and development. Cell 125, 1241–1252 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Youle R. J., Van Der Bliek A. M., Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., Chan D. C., Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lieber T., Jeedigunta S. P., Palozzi J. M., Lehmann R., Hurd T. R., Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature 570, 380–384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S.-J., Khan M., Quan J., Till A., Subramani S., Siddiqui A., Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLOS Pathog. 9, e1003722 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Youle R. J., Narendra D. P., Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubinsztein D. C., Bento C. F., Deretic V., Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 212, 979–990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chocron E. S., Munkácsy E., Pickering A. M., Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 285–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yakes F. M., Van Houten B., Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A 94, 514–519 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richter C., Park J.-W., Ames B. N., Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. U.S.A. 85, 6465–6467 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheibye-Knudsen M., Fang E. F., Croteau D. L., Wilson D. M. III, Bohr V. A., Protecting the mitochondrial powerhouse. Trends Cell Biol. 25, 158–170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Souza-Pinto N. C., Wilson D. M., Stevnsner T. V., Bohr V. A., Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair 7, 1098–1109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pommier Y., Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 8, 82–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallick A., Kuman M. M., Ghosh A., Das B. B., Basu S., Cerberus nanoparticles: Cotargeting of mitochondrial DNA and mitochondrial topoisomerase I in breast cancer cells. ACS Appl. Nano Mater. 1, 2195–2205 (2018). [Google Scholar]
- 46.Zhang H., Pommier Y., Mitochondrial topoisomerase I sites in the regulatory D-loop region of mitochondrial DNA. Biochemistry 47, 11196–11203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khiati S., Seol Y., Agama K., Dalla Rosa I., Agrawal S., Zhang H., Neuman K., Pommier Y., Poisoning of mitochondrial topoisomerase I by lamellarin D. Mol. Pharmacol. 86, 193–199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalla Rosa I., Huang S.-y. N., Agama K., Khiati S., Zhang H., Pommier Y., Mapping topoisomerase sites in mitochondrial DNA with a poisonous mitochondrial topoisomerase I (Top1mt). J. Biol. Chem. 289, 18595–18602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karbowski M., Norris K. L., Cleland M. M., Jeong S.-Y., Youle R. J., Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658–662 (2006). [DOI] [PubMed] [Google Scholar]
- 50.S. Dingley, K. A. Chapman, M. J. Falk, in Mitochondrial Disorders (Springer, 2012), pp. 231–239. [Google Scholar]
- 51.Ploumi C., Daskalaki I., Tavernarakis N., Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 284, 183–195 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Douarre C., Sourbier C., Dalla Rosa I., Das B. B., Redon C. E., Zhang H., Neckers L., Pommier Y., Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLOS ONE 7, e41094 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galluzzi L., Baehrecke E. H., Ballabio A., Boya P., Bravo-San Pedro J. M., Cecconi F., Choi A. M., Chu C. T., Codogno P., Colombo M. I., Cuervo A. M., Debnath J., Deretic V., Dikic I., Eskelinen E. L., Fimia G. M., Fulda S., Gewirtz D. A., Green D. R., Hansen M., Harper J. W., Jäättelä M., Johansen T., Juhasz G., Kimmelman A. C., Kraft C., Ktistakis N. T., Kumar S., Levine B., Lopez-Otin C., Madeo F., Martens S., Martinez J., Melendez A., Mizushima N., Münz C., Murphy L. O., Penninger J. M., Piacentini M., Reggiori F., Rubinsztein D. C., Ryan K. M., Santambrogio L., Scorrano L., Simon A. K., Simon H. U., Simonsen A., Tavernarakis N., Tooze S. A., Yoshimori T., Yuan J., Yue Z., Zhong Q., Kroemer G., Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boya P., Reggiori F., Codogno P., Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mizushima N., Yoshimori T., Levine B., Methods in mammalian autophagy research. Cell 140, 313–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubinsztein D. C., Cuervo A. M., Ravikumar B., Sarkar S., Korolchuk V. I., Kaushik S., Klionsky D. J., In search of an “autophagomometer”. Autophagy 5, 585–589 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Zhang H., Barceló J. M., Lee B., Kohlhagen G., Zimonjic D. B., Popescu N. C., Pommier Y., Human mitochondrial topoisomerase I. Proc. Natl. Acad. Sci. U.S.A. 98, 10608–10613 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dalla Rosa I., Zhang H., Khiati S., Wu X., Pommier Y., Transcription profiling suggests that mitochondrial topoisomerase IB acts as a topological barrier and regulator of mitochondrial DNA transcription. J. Biol. Chem. 292, 20162–20172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H., Zhang Y.-W., Yasukawa T., Dalla Rosa I., Khiati S., Pommier Y., Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases IIα and IIβ in vertebrate mitochondria. Nucleic Acids Res. 42, 7259–7267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pommier Y., Barcelo J. M., Rao V. A., Sordet O., Jobson A. G., Thibaut L., Miao Z. H., Seiler J. A., Zhang H., Marchand C., Agama K., Nitiss J. L., Redon C., Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 81, 179–229 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothfuss O., Gasser T., Patenge N., Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 38, e24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson S. C., Rabinovitch P. S., Kaeberlein M., mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das S. K., Rehman I., Ghosh A., Sengupta S., Majumdar P., Jana B., Das B. B., Poly (ADP-ribose) polymers regulate DNA topoisomerase I (Top1) nuclear dynamics and camptothecin sensitivity in living cells. Nucleic Acids Res. 44, 8363–8375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kundu B., Das S. K., Paul Chowdhuri S., Pal S., Sarkar D., Ghosh A., Mukherjee A., Bhattacharya D., Das B. B., Talukdar A., Discovery and mechanistic study of tailor-made quinoline derivatives as topoisomerase 1 poison with potent anticancer activity. J. Med. Chem. 62, 3428–3446 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Das S. K., Ghosh A., Paul Chowdhuri S., Halder N., Rehman I., Sengupta S., Sahoo K. C., Rath H., Das B. B., Neutral porphyrin derivative exerts anticancer activity by targeting cellular topoisomerase I (Top1) and promotes apoptotic cell death without stabilizing Top1-DNA cleavage complexes. J. Med. Chem. 61, 804–817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khiati S., Dalla Rosa I., Sourbier C., Ma X., Rao V. A., Neckers L. M., Zhang H., Pommier Y., Mitochondrial topoisomerase I (Top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin. Cancer Res. 20, 4873–4881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax9778/DC1
Fig. S1. Mito-SN38 does not trap nuclear-Top1cc but impairs mitochondrial metabolism through SCAN1-TDP1 trapping in the mitochondria.
Fig. S2. Differentiation of SH-SY5Y cells showing expression of FLAG-TDP1 and lysosomal localization of SCAN1 mitochondria showing mitophagy.
Table S1. List of primers used.