

Abstract
The current state, tools, and applications of personalized medicine with special emphasis on inflammatory skin diseases like psoriasis and atopic dermatitis are discussed. Inflammatory pathways are outlined as well as potential targets for monoclonal antibodies and small‐molecule inhibitors.
Keywords: Atopic dermatitis, endotypes, immunology, inflammatory skin diseases, personalized medicine, precision medicine, psoriasis, targeted therapy
Introduction – why?
One size does not fit all! Or does it? For treatment of many – if not most – inflammatory skin conditions, the dermatologists’ first choice over the last 50+ years has been a topical glucocorticoid 1, most often yielding astonishing anti‐inflammatory effects: rapid relief of itch and ease of rash, bringing the inflamed skin back to a ‘near‐normal’ state within a few days (Box 1).
Box 1. Size matters.
One size fits all, the paradigm of traditional medicine.
One size does not fit all, a mantra of personalized medicine, the goal of which is to provide ‘The right dose of the right drug for the right indication for the right patient at the right time.’ This is another mantra of personalized medicine, a much publicized quote ascribed to former FDA Genomics associate director, Felix Frueh, when he captured the essence of personalized medicine at the Annual FDA Science Forum in 2005 2. A variant of the above principles can be found in the ‘5R framework’ for improving research and development productivity in the pharma industry, with focus on ‘right target, right tissue, right safety, right patients, and right commercial potential’ 3.
But if such an efficient, universal, and inexpensive treatment is already available, where, then, is the unmet need for personalized medicine and targeted therapy? One may even argue that treatment with glucocorticoids is targeted therapy! Because glucocorticoids specifically bind to their molecular target, the cytosolic glucocorticoid receptor, and thereby induce downstream anti‐inflammatory effects. These effects are brought about via several mechanisms: non‐genomic direct activation of anti‐inflammatory proteins, DNA‐dependent (genomic) induction of anti‐inflammatory proteins, and protein interference (via transcription factors, such as NF‐κB) causing repression of inflammatory proteins 4, 5. Now, as glucocorticoid receptor activation produces pleiotropic (multiple and diverse) effects, and because the receptor is universally expressed – albeit to a varying degree – in most cell types, this accounts both for the high anti‐inflammatory efficacy, the broad mode of action, and for the adverse effects associated with – in particular: long‐term – glucocorticoid treatment. One such major adverse effect is skin atrophy, possibly mediated by the glucocorticoid receptor chaperone FKBP51 6, but also systemic side‐effects are observed, such as suppression of the hypothalamus‐pituitary‐adrenal (HPA) axis, due to percutaneous glucocorticoid absorption 7. Moreover, if large areas of the skin are covered with lesions, topical treatment is not a feasible solution. Therefore, and because of extensive disease heterogeneity – not all patients (especially, those with severe disease) respond to glucocorticoids, and all patients differ with respect to their genetic makeup – there is still a need for better, and more targeted therapy. In particular, the two most common inflammatory skin diseases, atopic dermatitis (AD) and psoriasis (PSO), have both a complex pathogenesis including several pathophysiological mechanisms 8, and a multitude of clinical manifestations 9, 10, which make them exemplary diseases for a personalized medicine strategy calling for improved stratification, development of targeted treatment, and prevention 11, 12.
Often, the term ‘personalized medicine’ is used synonymously and sometimes confused with precision/stratified/individualized/tailored/P4 medicine, targeted therapy, and pharmacogenomics. Here, I will mainly use ‘personalized medicine’, though, for clarity, the conceptual nuances of this and its related terms are summarized in Box 2.
Box 2. WHAT? The different flavors of personalized medicine.
Numbers in parentheses correspond to count of Google hits as per February 19th 2019
Both American ‘‐ized’ and British ‘‐ised’ spellings have been included.
Personalized medicine
(5.2M) is an approach to both preventive care (e.g. identifying genetic risk factors to guide behavioral changes and preventive treatment, such as statins for hypercholesterolemia) and to drug therapy (e.g. early and accurate diagnostic tests that can guide targeted treatment and diminish side‐effects) based on the individual's genetic (and other relevant) information.
The term ‘personalized medicine’ – albeit with a slightly different, ethical connotation – can be found already in a 1971 article by W.M. Gibson, who envisages the family practitioner's role as a scientist‐physician who ‘Within a few years will likely have available to him a computer programmed for medicine providing him with a great store of knowledge literally at his fingertips’ 13. Thus, in the early years, personalized medicine focused on the ethical dimensions of patient‐centered practice 14. But actually, the foundation for personalized medicine can be traced all the way back to Hippocrates (460–370 BCE), who famously said ‘It's far more important to know what person the disease has than what disease the person has’, thus introducing the patient‐centric concept 15. Interestingly, today, such patient‐centricity is widely embraced by the pharma industry, which is increasingly engaging in a dialog with patients during the drug development process 16.
Due to concern that ‘personalized medicine’ can be misinterpreted as implying that a unique treatment can be designed for each individual, the National Research Council preferred the term ‘precision medicine’ in their 2011 report Toward Precision Medicine 17.
Precision medicine
(5.5M) is defined as ‘tailoring of medical treatment to the individual characteristics of each patient’. 17 But this does not mean that drugs are being developed uniquely for a patient, rather, it means that individual patients can be classified into subpopulations that differ in their response to a specific treatment. Thus, the focus is on identifying which treatments will work for which patients based on their individual genetic – and epigenetic – characteristics (for example, treatment of breast cancer patients with herceptin will only work for patients that overexpress HER2). An issue with the term precision, however, is that interpreted technically, it is a measure of statistical variability, and as such, it can be argued that medicine is not precise 18.
Targeted therapy
(3.6M) is often used synonymously with molecularly targeted therapy, molecular medicine, and biologic therapy, mainly to distinguish it from traditional chemotherapy in the context of cancer treatment. However, targeted therapy is neither limited to cancer, nor to biologics, as today, both small molecules and monoclonal antibodies are used in the targeted treatment of a wide variety of diseases, including asthma, atopic dermatitis, and psoriasis. The target concept is an old one and marks the beginning of modern pharmacology; it was developed by Paul Ehrlich around 1900, when he was studying antibodies and envisioned a hypothetical drug that would reach and kill its target (microbe) without harming the host; the magic bullet (German: Zauberkugel) 19. Indeed, today Ehrlich's vision has become a reality, where numerous highly specific monoclonal antibody‐based therapies are being applied or are in clinical development.
Pharmacogenomics
(2.9M) refers to the study of how genes affect an individual's response to drugs. The term is a combination of pharmacology and genomics, with the aim of developing safe and effective treatments. When it is applied to the study of drug metabolism, it is largely termed pharmacogenetics, while pharmacogenomics is a broader term encompassing all genes that may impact drug response 20. A typical use includes identification of fast and slow metabolizers due to single nucleotide polymorphisms (SNPs) in the CYP450 system, where the former will achieve suboptimal drug levels, while the latter will have increased risk of adverse drug reactions, and in worst case, death 21.
Individualized medicine
(357K) is the term preferred by Eric Topol (founder and director of the Scripps Translational Science Institute), mainly because it relates both to the medicine and the medical information – including both omics and digital technology – that is particularized to an individual, and because it is supposedly less ambiguous compared to the terms personalized and precision medicine 22. Note, however, that individualized medicine can also be understood as ‘truly’ individualized, such as a cancer vaccine based on the patient's particular tumor. In this respect, individualized medicine lies in one end of the therapeutic continuum, empirical medicine is at the other end, while the field of stratified medicine lies in between 23.
Stratified medicine
(112K) aims at matching a therapy with a specific patient population – who will have a therapeutically meaningful benefit of the treatment – by use of clinical biomarkers, which are, therefore, of utmost importance (e.g. as companion diagnostics, such as the FDA‐approved HercepTest that quantifies HER2, identifying patients who are likely to benefit from Herceptin), because they link the patient subpopulation with the therapy 23.
P4 medicine
(40K) stands for predictive, preventive, personalized, and participatory medicine. The term was coined by Leroy Hood (a pioneer of systems biology and co‐founder of the Institute for Systems Biology in Seattle) with special emphasis on the participatory part. The idea is that the digital revolution and rise of the Internet will empower consumers, who by their use of social media, mobile healthcare apps and wearables 24 generate the big data needed for systems medicine 25. Thus, Hood envisaged the emergence of a whole new healthcare system based on systems biology, big data, and networked consumers, who focus on both disease and wellness care, moving toward a holistic view on biological complexity.
Tailored medicine
(15K) emphasizes the move from the ‘one size fits all’ paradigm of traditional drug development and usage, to personalized medicine, where stratification of patient populations allows identification of responder subpopulations. One ethical issue with such an approach is that most participants in clinical trials in the US are white from higher socioeconomic levels, while ethnic minorities, who make up 40% of the population, are underrepresented. This disparity is problematic because certain diseases are more prevalent among ethnic minorities, who have a different genetic makeup and thus are likely to differ both in pathophysiology and response to treatment 26.
From Disease Understanding to Biomarkers, Endotypes, and Targeted Treatment
Theory
In theory, the logic is simple: If we can understand a disease, then we can also treat it. In particular, if we gain sufficient knowledge of its underlying molecular pathophysiology, then we can identify disease‐driving pathways and target relevant proteins. Or, better yet, it may be possible to take preventive measures even before the disease has manifested. Today, preventive medicine is made possible with the advent of new omics technologies, in particular, next‐generation sequencing (NGS) that enables determination of an individual's entire DNA sequence (six billion base‐pairs in a human diploid genome) in less than a day.1 For the 6000+ human (mostly rare) diseases caused by a single gene mutation, including the more than 600 known monogenic dermatoses 27, a correct molecular (genetic) diagnosis is crucial, both in terms of counseling and preventive measures [e.g. statin treatment of familial hypercholesterolemia) and in terms of avoiding ineffective and often stressful, even deadly treatments (such as cancer chemotherapy for multidrug resistant tumors 28]. In the best‐case scenario, it may even guide treatment. A striking example of such a case was recently reported for a seven‐year‐old boy suffering from a life‐threatening skin disease, junctional epidermolysis bullosa. After genetic analysis revealed the cause to be a splice‐site mutation in the LAMB3 gene, the patient was treated successfully with transgenic keratinocyte stem cells, which resulted in regeneration of the entire epidermis 29.
Practice
In practice, most disorders are not as simple as that; they are polygenic, complex, and multifactorial, meaning that multiple genetic, epigenetic, lifestyle, and environmental factors play a role in the clinical manifestation of the disease. Such diseases include diabetes, cancer, and hypertension, as well as many inflammatory conditions, including asthma, inflammatory bowel disease (IBD), psoriasis, and atopic dermatitis. In these cases, a genomic ‘DNA fingerprint’ will give a static picture of the genetic susceptibility of an individual,2 but will not fully capture the dynamic nature of cells or diseases.
To this end, one needs to identify other relevant and robust biomarkers that reflect the various clinical phenotypes, and which eventually can form the basis for stratification of endotypes.3
Biomarkers for personalized medicine can be classified as diagnostic, prognostic, or predictive.
Diagnostic biomarkers
Ideally, diagnostic biomarkers can detect diseases before they become symptomatic. Examples include early detection of prostate cancer by evaluation of serum prostate‐specific antigen (PSA) [albeit with relatively low sensitivity and specificity 35 and detection of other cancers by measuring circulating tumor cells in liquid biopsies 36]. But diagnostic biomarkers are more than just binary indicators of the absence or presence of disease. If they reflect the molecular pathology of the disease, then they may be able to precisely define and stratify its endotypes, and thus guide selection of the most effective targeted therapy. This also points to a need for improved molecular disease taxonomy 11, 37, because currently, for most diseases – including inflammatory skin diseases – endotypes are not incorporated in the WHO's latest revision of International Classification of Diseases, ICD‐11 38.
Prognostic biomarkers
Prognostic biomarkers can, in principle, project the disease trajectory, i.e. indicate the likelihood of progression, remission, and future clinical events.4 In oncology, classical clinicopathologic biomarkers are tumor size, number of tumor‐positive lymph nodes, and distant metastases, which are used for staging and prognosis indication. In clinical trials, prognostic biomarkers are used to enrich for populations that are more likely to progress, as this increases statistical power and thus, reduces cost of drug development, and also guides decisions regarding the aggressiveness of the treatment 40.
Predictive biomarkers
Predictive biomarkers are most important for guiding personalized medicine, because they have the potential to identify individuals that are more or less likely to respond to a given treatment. In clinical trials, predictive biomarkers are used to stratify the study population into biomarker positive (likely responders) and negative (non‐responders) patients, with the hope of meeting the clinical primary endpoint in the biomarker positive group 40.5 Examples of predictive biomarkers include polymorphisms in the cytochrome P450 superfamily [responsible for the hepatic – as well as extrahepatic – metabolism of most drugs, and thus, of immense importance for their pharmacokinetics 42] and variants of the human leukocyte antigen B (HLA‐B, associated with several hypersensitivity reactions, including toxic epidermal necrolysis); many more such gene variant–drug relationships can be found in PharmGKB (The Pharmacogenomics Knowledge Base, http://www.pharmgkb.org), a public, knowledge‐sharing resource that captures, curates, and integrates pharmacogenomics data, and which currently contains around 21 000 variant annotations and 132 PK/PD focused drug‐pathway diagrams 43. As targeted therapies, e.g. those based on monoclonal antibodies, are expensive and display variable response rates both in AD and PSO, it is important to identify and validate biomarkers for prediction of treatment outcome, as recently reviewed by Ovejero‐Benito et al. 44.
Biomarker combinations
Because heterogeneous treatment responses can be due to a combination of factors, including disease complexity (multiple endotypes), genetic, epigenetic, and environmental effects, a single biomarker has only limited ability to capture all these aspects into a prediction of a patient's response to a given drug. Therefore, patient stratification may rely on the identification of multiple biomarkers, entailing multivariate statistical analysis and machine learning for finding the optimal linear and non‐linear biomarker combinations 45, i.e. those with highest sensitivity and specificity (maximizing the AUC of the ROC analysis) for a given classification. For example, we recently identified and validated a diagnostic miRNA classifier based on a linear combination of three miRNAs (miR‐155, miR‐203, and miR‐205) that could discriminate cutaneous T‐cell lymphoma (CTCL) from benign inflammatory skin diseases with 95% classification accuracy 46, 47. One advantage of using a biomarker panel as opposed to a single biomarker is that individual differences in the baseline levels of the biomarkers can be accounted for, in particular if the biomarkers of interest are normalized to a set of reference biomarkers. Other recent examples include a plasma protein (MMP‐2, sTNF‐R2, TSLP) panel for identification of ischemic stroke 48, a cell surface protein (CD25, CD64, CD69) panel for flow cytometric detection of sepsis 49, and a serum nuclear magnetic resonance (NMR) metabolomics‐derived biomarker (alanine, pyruvate, glycine, sarcosine) panel for early detection and grading of prostate cancer 50. For atopic dermatitis, Thijs et al. applied a panel of 147 serum biomarkers to stratify 193 AD patients into four main clusters, which may represent endotypes 51, although their analysis suggests that AD is very heterogeneous and may even reflect a disease spectrum rather than distinct endotypes.
Note, however, that the above examples only consider combinations of biomarkers of the same biological type or layer, such as genomic (DNA), transcriptomic (RNA), proteomic (proteins), or metabolomic (amino acids) markers. With the explosive development, both in omics technologies (summarized in Box 3) and in bioinformatics and computational tools, the natural next step is to move out of and across (between) the individual layers, integrating the various orthogonal (independent) biologic approaches in an integrative ‘multi‐omics’, systems biology strategy, also referred to as integromics 52, 53, 54. Conceptually, the layers in such an integrative approach can be compared to Google Maps (maps.google.com), which render a multilayered visualization of both spatial (locations, streets, landmarks) and real‐time temporal (traffic) data 22. And this is exactly the ambition of integromics6 on a human scale; to be able to visualize the various layers (mapping the genomic, epigenomic, transcriptomic, proteomic, metabolomic, etc. landscapes) of biologic pathways, as well as to be able to predict the dynamic effects of perturbations, such as targeting central molecules in the pathways, creating ‘roadblocks’ that will stop or diverge the traffic (information, signaling) flow in the system, to remain in the Google Maps analogy.
Box 3. Omics technologies for integrative, personalized medicine.
Genomics
Next‐generation sequencing (NGS) is key to generating the vast amounts of DNA data for whole‐genome sequencing (WGS) and whole‐exome sequencing (WES) projects 55. Because the exome only comprises 1.5% (40 Mb) of our genome, it can be sequenced at a deeper coverage (>100× compared to 30×), faster (hours compared to days), and at lower price compared to WGS. However, it appears that most (80%) of the loci involved in complex diseases are located in the 98.5% noncoding – but important regulatory – regions of the genome 22. Therefore, and because the price of WGS continues to drop, it is today cost‐efficient to perform WGS for single‐nucleotide polymorphism (SNP) analysis, genotyping, pharmacogenomics and genome‐wide association studies (GWAS). If cost is a concern, then microarray‐ or bead‐based SNP analysis can be considered, albeit with considerably lower coverage than WGS.
A powerful tool to reveal the cellular complexity of in particular tumors 56, but also of individual genomic variation (mosaicism), is single‐cell sequencing, which is gaining momentum as the methodologies for whole‐genome amplification (WGA) and sequencing mature 57. This technique (as well as scRNA‐seq, see below) can also be used to profile T‐ and B‐cell receptor repertoires at the single‐cell level, thus enabling a full picture of the immune landscape and its dynamics 58.
Transcriptomics
Since the late 1990s, global gene expression analysis has been performed by use of microarrays 59. Today, due to improvements in next‐generation sequencing (NGS) technology (longer and more reads) and a concomitant drop in price,7 RNA sequencing (RNA‐seq) is the preferred method for transcriptomic profiling 60. A major advantage of quantifying gene expression is that it reflects the dynamics of the cellular system under investigation. This is also a major caveat, because what is measured is a snapshot of the transcriptome, which tends to vary extensively with time and space. Thus, when comparing transcriptomic profiles of biologic samples it is of utmost importance that the experimental conditions are as standardized as possible (a notion that also applies to proteomics, and, in particular, metabolomics); i.e. the specimens should be comparable, both with respect to location (more on this later, with special reference to skin biopsies), and timing, including sampling time and time from sampling to freezing and RNA extraction. Optimally, a time course experiment (multiple samples taken at different time points) should be performed to investigate the expression profiles’ temporal dependence 60, 61, which is also of importance for the selection of biomarkers, as some may display transient expression, while others are more stable, and therefore more robust in a clinical setting. Just as for DNA sequencing, single‐cell RNA‐sequencing (scRNA‐seq) is now opening a window to the cellular phenotype, as it allows for unprecedented detail analysis of cellular heterogeneity and development 62, 63. Finally, novel in situ sequencing techniques such as fluorescent in situ sequencing of RNA (FISSEQ) 64 and STARmap 65 allow for determination of the actual, 3‐dimensional location of gene expression in cells and tissues 66.
Epigenomics
At least three types of epigenetic systems co‐exist: DNA methylation, histone modification, and noncoding RNA (ncRNA, including miRNA, lncRNA, snoRNA, and many more).
DNA methylation is typically assessed by bisulfite treatment of the DNA – converting non‐methylated C's to U's, while methylated C's are protected from this conversion – followed by either microarray analysis or WGS (which captures all 29 million CpGs in the human genome, albeit at 10 times the cost of methylation arrays) enabling epigenome‐wide association studies (EWAS; 67). The interpretation of such studies, however, can be difficult, in particular if the starting material contains a mixture of different cell types, each with their own, highly cell‐type specific epigenome. Thus, it is necessary to perform cell‐type specific deconvolution of the signal in order to identify relevant epigenetic changes rather than just a shift in proportion of cell types 66, 68.8
For studying the ‘histone code’, genome‐wide histone modification assays apply chromatin immunoprecipitation (ChIP) and histone modification‐specific antibodies (to precipitate the DNA–histone complexes), followed by NGS (ChIP‐seq) to identify the bound DNA fragments. This has resulted in mapping of several human epigenomes 69 with promise for identification of epigenetic biomarkers 70 and with implications for epigenetic drugs, such as histone deacetylase (HDAC) inhibitors 70, 71.
Numerous microRNAs have already been identified – by microarrays, qRT‐PCR, and small RNA‐seq – as potential diagnostic, prognostic, and predictive biomarkers in cancer 72, 73, 74, 75, 76, 77, diabetes 78, and many other diseases, including inflammatory skin conditions like psoriasis 79, 80 and atopic dermatitis 81, 82. What remains to be seen is the potential of an emerging class of ncRNA, namely the long ncRNA (lncRNA, of which around 16 000 human variants have been found so far), in precision medicine, cancer 83 and inflammatory diseases 84.
Proteomics
Studying the proteome–by various mass spectroscopy methods – is important because gene expression levels are only approximations of the corresponding protein levels 85. Firstly, because not all mRNA is translated into protein – sometimes because miRNAs bind to the 3′UTR of their target genes, thus blocking translation 85, 86, and secondly, because post‐translational modifications, such as phosphorylation/dephosphorylation, are important determinants of protein function, which is why phospho‐proteomics is a relevant measure of protein function and dynamics of cellular signaling 87.
Metabolomics
The analysis – either by NMR or GC–MS – of the complete set of small‐molecule intermediates, including lipids (the lipidome, a subset of the metabolome) in a biological sample, provides a sensitive snapshot of its physiology, and can thus guide discovery of biomarkers 53, 87, 88. Application examples include ‘breathomics’, breath‐based metabolomics, where quantification of volatile organic compounds has diagnostic potential 89, urine metabolite‐based diagnosis of urinary tract symptoms 90, as well as assessment of glucocorticoid‐induced changes of the lipid profile of human skin 91. In particular, when combined with other – orthogonal – omics technologies, one can obtain mechanistic insight, e.g. on metabolic and inflammatory pathways 53.
Glycomics
The study of glycans (polysaccharides) includes analysis of glycosylated proteins (glycoproteins) and lipids (glycolipids), mainly by MS or HPLC. Since most human proteins are glycosylated, and glycans play important roles in many cellular processes, including cell adhesion, trafficking, and inflammation, individual variations in glycosylation patterns may serve as biomarkers for disease risk and response to therapy 92, 93. For example, heterogeneity in N‐glycosylation of immunoglobulin G (IgG) can modulate its inflammatory effect, with implications for regulation of the immune system 94.
Phenomics
A detailed description of the phenome, i.e. an account of the phenotypic traits of an organism, is crucial for building the translational bridge from genome‐scale biology to disease understanding, i.e. for establishing the genotype–phenotype relationship 95. In practice, it entails deep phenotyping of individuals, including collection of multidimensional clinical data (e.g. biochemical tests, pathology reports, physical examination, family history, demographics, and imaging), and importantly, a precise, comprehensive, and standardized description (metadata) of such data. This makes the data accessible and searchable and facilitates its integration with omics data for translation into disease endotypes and eventually, personalized medicine 96. To aid in connecting genomics and phenomics, a formal ontology (standardized vocabulary and annotation of phenotypes and relations to diseases) has been proposed by The Human Phenotype Ontology (HPO) project 97, which today links more than 13 000 phenotypic terms and over 156 000 disease annotations. Additionally, phenomics can be applied for construction of large‐scale disease trajectories based on information on comorbidities pulled from real‐world data (RWD) 98, such as observational data from disease registries and electronic health records (EHR). One such study used the Danish National Patient Registry (covering the whole population of Denmark, 6.2 million patients followed over 15 years) for generation of disease trajectories that can prove useful for predicting (and ultimately, preventing) disease progression of individual patients 99.
Microbiomics
A growing research field, initiated by the human microbiome project 100, and with potential for personalized medicine is the study (by NGS) of our microbiome, which is the sum of microorganisms (bacteria, archaea, fungi, and viruses) in and on our body (skin, mouth, nose, lung, gut, and vagina). In particular, the gut microbiome has been extensively researched and shown to play an important role in nutrition, metabolism, immune function, and numerous diseases, including inflammatory bowel disease (IBD), type II diabetes, cardiovascular disease, asthma, atopy 101, 102, and autism 103. The microbiome is also implied in drug interactions – studied by pharmacomicrobiomics 104, 105 – and e.g. digoxin has been shown to be metabolized and inactivated by specific gut bacteria 106. In the context of chronic, inflammatory skin diseases, dysbiosis of the skin microbiome has been associated with both PSO 107 and AD 108, 109. This opens possibilities for targeted, preventive intervention, such as administration of prebiotics (non‐digestible food components, like fibers) and probiotics (live microorganisms, such as Lactobacillus strains). Notably, the microbiome is dynamic; it undergoes temporal (e.g. circadian) and spatial fluctuations, both in composition and metabolic activity 110. The question of composition is addressed by targeted 16S rDNA taxonomic profiling and by – more comprehensive – metagenomics shotgun strategies (whole‐metagenome sequencing). But to capture the true dynamics of the microbiome, a full functional analysis must include both metatranscriptomics and metabolomics. The former addresses the question of which genes are expressed (collectively by the microbiome at a given time and condition), while the latter provides important information on which metabolites (both microbiota‐ and host‐derived) are present and interplay at the host–microbiome interface 111.
Exposomics
Genetic factors alone explain only a fraction of what we consider genetic diseases, including cancer 112. The remainder, perhaps more than 90%, can be attributed to environmental factors, also known as the exposome. The term exposome was coined by CP Wild in 2005, who broadly defines it as ‘every exposure to which an individual is subjected from conception to death’ 113. It encompasses three domains: internal, specific external, and general external. The internal exposome consists of endogenous factors, including circulating metabolites, hormones, lipids, oxidative stress, and our microbiome 114. The specific external factors include radiation, infections, contaminants, pollutants, diet, medicine, tobacco, and alcohol, while the general external factors encompass socioeconomic status, education, stress, environment (urban/rural), and climate, among others. Thus, due to the diversity of the exposome, and because it is in constant flux, the challenge is to decide what (which biomarkers of exposure are available, if any) and when to measure 113. One approach has been to apply metabolomics on consecutive saliva samples, assessing the ‘saliva exposome’, as it is easy to collect and measure, and can be used to monitor individual health trajectories 115. Biomarkers of exposure also enable exposome‐wide association studies (EWAS)9 116, 117, which have promise in the near future. Why? Because the digital revolution has opened for disruptive technologies, such as continuous, cloud‐based tracking of big data [such as the Internet of Things, IoT, with a plethora of physical devices that connect, collect, and exchange data for IoT‐enabled health care 118], generated by wearable, environmental monitors and biosensors coupled to our smartphones – a realization of the ‘quantified self’ 119.
Integromics
Also known as integrated/integrative omics, combine two or more omics layers in order to identify relevant overlaps between these. For example, a five‐layer approach may include genomics, epigenomics (three sublayers: DNA methylation, histone code, miRNA), transcriptomics, proteomics, and metabolomics, which coupled to phenotype (phenomics) data appears as an ‘obvious’ integrative omics approach, and one that we are currently exploring. However, so far, most published studies are limited to three layers, namely genomics–transcriptomics–proteomics, which will capture post‐transcriptional regulatory mechanisms, whenever there is discrepancy between gene and protein expression 52, but which do not take advantage of the orthogonal information that e.g. metabolomics adds to the (almost) full picture 52, 53. A major concern about integromics analysis and sharing of such big medical data is the difficult question regarding privacy and security 120, which needs to be solved before a massive open online medical (MOOM) repository can become a reality 22.
Next, let us see how the above considerations apply to personalized medicine in inflammatory skin diseases, with special emphasis on atopic dermatitis and psoriasis.10
Personalized medicine in inflammatory skin diseases
See Box 4.
Box 4. Basic characteristics of PSO and AD.
| PSO | AD | |
|---|---|---|
|
ICD‐10 CM codes |
L40 Psoriasis; L40.0 Psoriasis vulgaris, plaque PSO (90%) L40.1 Generalized pustular PSO (GPP, rare) L40.4 Guttate PSO (2%); L40.8 Other |
L20.9 Atopic dermatitis, unspecified L20.8 Other atopic dermatitis |
| Epidemiology & Comorbidity |
Affects 1–8% of the adult population 121, amounting to at least 130 million people worldwide. Two peaks in age of onset: 20–30 years and 50–60 years. PSO is a systemic condition with several serious comorbidities, including psoriatic arthritis (20–30%), inflammatory bowel disease, metabolic syndrome, and cardiovascular diseases 122. |
Affects 10–25% of all children and 2–10% of the adult population 123, corresponding to at least 320 million people worldwide,11 and with wide regional variation 125. 85–95% of all cases begin before the age of 5 years 126 Prevalence has more than doubled within the last 50 years 127, which suggests environmental effects,12 including lifestyle changes – such as ‘Westernization’ and the hygiene hypothesis 128. AD is associated with other atopic diseases, including asthma (50% risk), food allergy (30% risk), and allergic rhinitis/hay fever (up to 75% risk), which underlines its systemic nature 127. |
| Disease burden | Overall, measured by disability‐adjusted life years (DALYs, excluding mortality; i.e. years of healthy life lost due to disease/disability), skin diseases are the fourth leading cause of disability worldwide 129. Due to the chronic and pruritic nature of both PSO and AD, they negatively impact quality of life (QoL) of most patients (and their families) and impose a major socioeconomic burden 130. | |
| Etiology | Unknown, but high heritability and numerous susceptibility loci suggest complex, polygenic predisposition combined with environmental triggering factors, autoantigens, and systemic inflammation 131. | Unknown, but high heritability and several susceptibility loci suggest a complex genetic disease including epidermal barrier dysfunction, immune dysregulation, and environmental triggers 132. |
| Risk factors and triggers | Family history (genetics, HLA‐Cw6), psychogenic stress, skin injury (Koebner phenomenon), streptococcal infections, medications, smoking, obesity 131. | Family history, FLG mutations, cold dry climate, irritants (detergents, wool), infections (S. aureus), allergens (house dust mites, pollen), cats 133, food allergens 132. |
| Pathogenesis |
IL‐23/Th17 axis key driver 134 For details, see Fig. 1. |
Th2 axis (IL‐4/IL‐13/IL‐5/IL‐31) dominating 134 For details, see Fig. 2. |
| Genetics |
Concordance rate, monozygotic twins: 33% Concordance rate, dizygotic twins: 17% Heritability: 60–75% 135 |
Concordance rate, monozygotic twins: 44–86% Concordance rate among dizygotic twins: 10–23% Heritability: 69–86% 136 |
| GWAS |
HLA‐Cw6: strongest known risk allele, OR 4.32 126 Nine PSO susceptibility regions, PSORS1‐9, containing mostly immune‐related genes +60 PSO susceptibility regions 137 |
FLG: strongest known risk factor 138, more than 40 LOF mutations described 139 OR 1.61–1.92 140 31 susceptibility loci, most related to innate immune system 141 OR 0.90–1.14 (except for FLG) |
| Transcriptomics |
+2600 DEG between lesional PSO and healthy skin 143 ~1800 DEG between lesional and non‐lesional skin 144 |
+1300 DEG between lesional AD and healthy skin 145 ~ 600 DEG between lesional and non‐lesional skin 146 |
| Potential biomarkers |
IL‐19 blood levels correlate with disease activity 147 IL‐2, IL‐5, IL‐10, IL‐12, IL‐22, GM‐CSF serum levels correlate with treatment effect 148 Skin transcriptome response to etanercept 149, ixekizumab 150, brodalumab 151, guselkumab 152, risankizumab vs ustekinumab 153 |
FLG stratifies for early‐onset persistent AD 154 IgE blood levels stratify for intrinsic/extrinsic AD 155 TARC (CCL17) in serum correlates with disease activity 155 IL‐31 levels associated with itch 155 IL‐33 serum levels correlate with disease severity 156 Skin transcriptome response to UVB 157, cyclosporin A 158, dupilumab 159, apremilast 160, fezakinumab (IL‐22) 161. |
| Top‐20 targets13 | CARD14 TYK2 IL12B TRAF3IP2 JAK2 PDE4A ITGB2 TNF IL17RA IL17A VDR ERAP1 IL23R TNFAIP3 NOD2 JAK1 JAK3 IL23A CD2 NR3C1 |
IL13 FLG IL4R RXRA SPINK5 PPIA JAK2 FKBP1A CD2 NR3C1 VDR HRH1 CYSLTR1 JAK1 PLA2G7 IGHE RXRB PDE4B RARG RXRG |
| Current treatment guidelines |
Topical coal tar: antipruritic, combined with UVB 162 Topical corticosteroids: anti‐inflammatory Topical vitamin D analogues: calcipotriol (often in combination with betamethasone dipropionate) inhibits epidermal hyperproliferation, induces differentiation, anti‐inflammatory Topical salicylic acid: keratolytic effect Oral: methotrexate, cyclosporin A, acitretin (for severe PSO), apremilast (PDE4 inhibitor), fumaric acid esters Biologics: etanercept, infliximab, adalimumab (TNF‐α); ustekinumab (IL‐12/IL‐23); secukinumab, ixekizumab (IL‐17A); brodalumab (IL‐17RA); guselkumab, tildrakizumab (IL‐23) |
Emollients: for moisturizing the skin (lipid‐rich) Antiseptics: bleach (sodium hypochlorite 0.0005%) bath 163 Topical corticosteroids: anti‐inflammatory, relieve itch, e.g. hydrocortisone, betamethasone valerate, clobetasol Topical calcineurin inhibitors: tacrolimus (Protopic) or pimecrolimus (Elidel) Oral calcineurin inhibitor: cyclosporin A (severe AD) Antibiotic creams: to fight skin infections, e.g. fucidin/fucicort Biologics (monoclonal antibodies), injectable: targeted therapy, e.g. dupilumab (anti‐IL‐4R) |
Psoriasis
Psoriasis typically presents as thick, erythematous, scaly plaques due to hyperproliferation of keratinocytes. Therefore, it was originally considered an epidermal, keratinocyte‐specific disorder, and it was not until the mid‐1980s, when first, immunosuppression by cyclosporine 164 and later, bone marrow transplantation 165 resulted in remarkable clearance of psoriatic plaques that a major paradigm shift occurred, and psoriasis appeared as a Th1 cell driven, systemic disease 131. Another paradigm shift was precipitated by the discovery of a new T‐cell subset of IL‐23‐regulated IL‐17‐producing Th17 cells in the experimental autoimmune encephalomyelitis (EAE) mouse model 166. This, together with the findings of increased levels of Th17 cells 167 and of IL‐23, the ‘master’ regulator of Th17 development, in psoriatic lesions 168, identified psoriasis as a mixed Th1/Th17 disease.
Today, the central role of the IL‐23/Th17 inflammatory pathway in the immunopathogenesis of PSO (summarized in Fig. 1) is firmly established and has paved the way for development of novel targeted therapies that disrupt IL‐23/IL‐17 signaling (Fig. 1A) 12, 169. And the results are impressive: For moderate‐to‐severe plaque psoriasis, PASI 7514 was obtained for 75–91% of patients treated for 12 weeks with the IL‐17A antagonists ixekizumab or secukinumab, PASI 90 was reached for 54–73% 172, 173, while 78% and 53% of patients treated with the IL‐17RA inhibitor brodalumab achieved PASI 90 and PASI 100 (complete clearance), respectively, after 52 weeks 174. Brodalumab blocks signaling by the five IL‐17 dimers (IL‐17A/F/C/E/AF) through the IL‐17RA subunit (Fig. 3A). This causes inhibition of the downstream pleiotropic effects of IL‐17RA and probably explains the potentially higher clinical efficacy obtainable by receptor blockade compared to neutralization of a single ligand 175, 176, 177. In line with the efficacy of blocking downstream cytokine signaling are the impressive Phase II data on the TYK2 inhibitor BMS‐986165, where PASI75 was obtained for 75% of patients at week 12 178.
Figure 1.
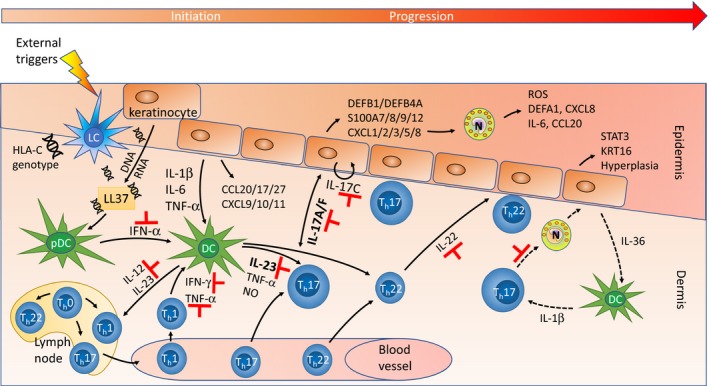
Pathways in the pathogenesis of PSO. Environmental triggers (e.g. drugs, infections, physical and psychological trauma) cause predisposed individuals to develop an autoimmune reaction, although the exact initiation mechanism is still poorly understood. One explanatory model 258 suggests that the autoantigen is LL37 (cathelicidin antimicrobial peptide, encoded by CAMP), which complexes with DNA and RNA released from stressed keratinocytes. This induces plasmacytoid dendritic cells (pDCs) to produce IFN‐α, which activates dermal dendritic cells (DCs). These cells migrate to skin‐draining lymph nodes, where they secrete IL‐12 and IL‐23, hereby stimulating naïve T‐cells to differentiate into Th1, Th17, and Th22 cells. The Th cells are attracted into the dermis by chemokines (CCL20, CCL17, CCL27, CXCL9/10/11) released by keratinocytes. Th1 cells produce IFN‐γ and TNF‐α, while Th17 cells release IL‐22 and IL‐17 family cytokines. The latter (IL‐17A/F) trigger epidermal keratinocytes to a feed‐forward inflammatory response 169, inducing numerous psoriasis‐associated genes [defensins, S100 proteins, chemokines; keratinocytes also produce IL‐17 cytokines, shown is a putative, autocrine IL‐17C loop 175] and stimulating keratinocyte proliferation. The released chemokines CXCL1/2/3/5/8 recruit neutrophils (N), which generate ROS (reactive oxygen species), α‐defensin (DEFA1), CXCL8, CCL20, and IL‐6. IL‐23 (released by activated DCs) stimulates differentiation and expansion of Th22 cells, which secrete IL‐22 that induces STAT3 and KRT16 expression. This causes further epidermal hyperplasia and eventually formation of the psoriatic plaque. To the right (punctuated arrows) is shown the IL‐36/IL‐1 pathway prevalent in pustular psoriasis, which is characterized by accumulation of neutrophils; here, IL‐17 activated neutrophils trigger increased IL‐36 activity, which stimulates DC's to produce IL‐1β reinforcing the Th17 axis 179. Indicated with ⊣ are targets of approved and emerging drugs, most of which are monoclonal antibodies (see Table 2). Figure modified, mainly from van de Kerkhof & Nestle in 131, but also from Noda et al. 134, and Conrad & Gilliet 179.
Figure 3.
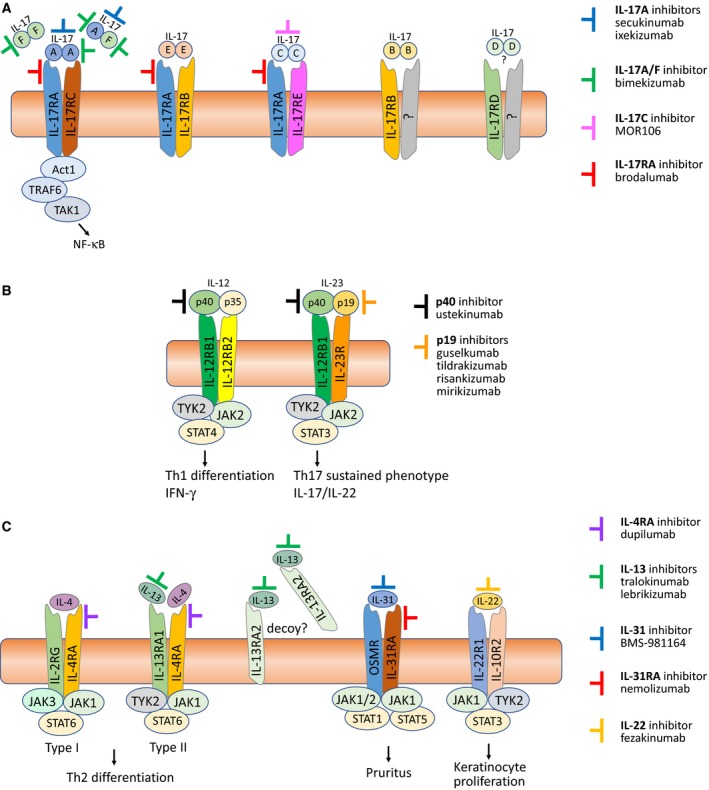
(A) Targeting the IL‐17 family of cytokines and their receptors. The six members of the IL‐17 cytokine family (IL‐17A/B/C/D/E/F) are shown as functional, disulfide‐linked homodimers, as well as the IL‐17A/F heterodimer 175. Also shown are their respective, heterodimeric receptors, each consisting of different combinations of five homologous receptor subunits (IL‐17RA/RB/RC/RD/RE). IL‐17A, IL‐17F (homodimers) and IL‐17A/F (heterodimer) signal through the IL‐17RA/RC receptor complex, IL‐17E (also known as IL‐25) via IL‐17RA/RB, IL‐17C via IL‐17RA/RE, while IL‐17B and IL‐17D signal via yet to be determined receptors. Indicated are also monoclonal antibodies that target either the cytokines or the IL‐17RA receptor subunit. Because IL‐17RA is common to signaling via IL‐17A/F/C/E/AF, blocking it will inhibit the downstream activities of all five IL‐17 dimers. IL‐17A/F and IL‐17RA inhibitors have already shown substantial effect in PSO, and currently, the IL‐17C inhibitor MOR106 is being tested in a Phase II clinical trial in moderate to severe AD 269. (B) Targeting IL‐12 and IL‐23. IL‐12 (p40/p35) and IL‐23 (p40/p19) are heterodimers that share the same p40 subunit. IL‐12 binds to the IL‐12Rβ1/β2 heterodimeric receptor and stimulates JAK2‐TYK2 to phosphorylate mainly STAT4, inducing IFN‐γ and a Th1 immune response. IL‐23 binds to the IL‐12Rβ1/IL‐23R heterodimeric receptor, and also induces JAK2‐TYK2 to phosphorylation, but primarily of STAT3, leading to Th17 signaling and release of IL‐17A/F and IL‐22 270. Because the p40 subunit is common to both IL‐12 and IL‐23, targeting it will inhibit the effects of both cytokines 271, while the p19‐specific antagonists target only the ‘master’ regulator of Th17 development, IL‐23 12. (C) Targeting IL‐4/IL‐13, IL‐31, and IL‐22. The two homologous cytokines, IL‐4 and IL‐13, drive type 2 inflammation and share many biological activities 272, the main differences being in their receptor interaction: The IL‐4R Type I receptor consists of the IL‐4RA and common‐gamma chain (IL‐2RG) subunits, and has IL‐4 as its exclusive ligand, while the IL‐4R Type II receptor is composed of the IL‐4RA and IL‐13RA1 chains, and binds both IL‐4 and IL‐13. The single‐chain IL‐13RA2 receptor is thought to function as a decoy receptor as it seems to lack the ability to induce intracellular signaling 273. As illustrated, targeting the common IL‐4RA subunit will inhibit the effects of both IL‐4 and IL‐13 signaling. IL‐31 signals via a heterodimer consisting of IL‐31RA and the oncostatin M receptor (OSMR), which is also common to oncostatin M (OSM), a member of the homologous IL‐6 superfamily 274. The IL‐31 receptor is found on sensory neurons in the dorsal root ganglia, where the itch sensation originates, which is why targeting IL‐31 by e.g. nemolizumab can potentially disrupt the itch–scratch cycle of pruritic diseases like AD 275. IL‐22 signals through the heterodimeric IL‐22R1/IL‐10R2 receptor and induces epidermal hyperplasia in AD, which is why the IL‐22 antagonist fezakinumab shows some promise in treatment of severe AD 161.
Different forms of PSO are associated with different pathways; chronic plaque psoriasis (also known as psoriasis vulgaris, the most common form) is dominated by the above‐mentioned IL‐23/Th17 pathway, while acute, erythrodermic psoriasis is characterized by Type I interferon (IFN‐α) producing plasmacytoid dendritic cells, and pustular psoriasis is associated with the IL‐36/IL‐1 pathway and accumulation of neutrophils15 179. This heterogeneity in immunopathogenesis highlights the complexity of psoriasis, as well as provides guidance – by identification of biomarkers reflecting the different endotypes – for novel and optimized targeted therapies. These therapies include promising new modalities, such as bispecific (e.g. blocking both TNF‐α and IL‐1716) 180, 181, and even trispecific antibodies 182, as well as vaccines 183. A compilation of these new and emerging treatment options for psoriasis can be found in Table 2.
The basic characteristics of psoriasis are summarized in Box 4, of which the following are of particular relevance for personalized medicine:
Comorbidities
Because PSO is a systemic disease associated with multiple severe comorbidities, including psoriatic arthritis (PsA) and cardiovascular disease (CVD) 122, targeted treatment of e.g. the IL‐23/Th17 pathway may not only reverse the cutaneous manifestations of the disease, but also the systemic, inflammatory comorbidities.
Genomics
GWAS (genome‐wide association studies) have already identified more than 60 risk loci, including several psoriasis susceptibility regions (PSORS), most of which contain immune system related genes 137. The increasing amount of genomic data may allow for identification of new variants (endotypes) of PSO, possibly predict who will develop the disease, identify responders to specific drugs, and guide further development of targeted therapies.
Epigenomics
EWAS (epigenome‐wide association studies) on PSO are emerging 184 and a recent study on 39 Indian PSO patients suggested that differential DNA methylation (comparing lesional to non‐lesional skin) can regulate the expression of key genes involved in the pathogenesis of PSO 185. In addition to DNA methylation, histone modification, specifically methylation of H3K27 and H3K4, showed some promise as pharmacoepigenetic biomarkers in a study of psoriasis patients’ response to biologics 186. Common for both of the above cases is that larger independent validation cohorts are needed to confirm the initial findings. Finally, several inflammation‐associated miRNAs, such as miR‐146a, miR‐21, miR‐31, miR‐221, and miR‐222, are consistently found to be upregulated in PSO skin 187, 188 and may be useful as disease activity biomarkers.
Transcriptomics
Analysis of the mRNA profiles of lesional, non‐lesional, and healthy skin has identified more than 2000 differentially expressed genes (DEGs), many of which may serve as potential biomarkers for disease progression and response to therapy 189. For a compilation of such studies with links to the actual data, please see Table 1.
Table 1.
Selected omics studies on psoriasis (PSO) and atopic dermatitis (AD)
| GEO ID | Year | Dx | Focus (# samples) | Technology | Reference |
|---|---|---|---|---|---|
| GSE16161 | 2009 | AD, PSO, NN | AD‐LS (9), PSO‐LS (15), NN (9) | HG‐U133_Plus_2 | 277 |
| GSE32924 | 2011 | AD, NN | LS (13), NL (12), NN (8) | HG‐U133_Plus_2 | 145 |
| GSE27887 | 2011 | AD | UVB, LS, NL, w0/w12, 10 pts. (35) | HG‐U133_Plus_2 | 157 |
| GSE36842 | 2012 | AD, NN | Acute/chronic, LS, NL, NN, 10 pts (39) | HG‐U133_Plus_2 | 196 |
| GSE75890 | 2016 | AD, PSO, NN | Mild ex‐/intrinsic, AD (14), PSO (9), NN (8) | HG 2.1 ST | 278 |
| GSE60709 | 2014 | AD, NN |
Epidermal shave, LS (12), NL (7), NN (14) DNA methylation, skin and blood |
Illumina HT‐12V3.0 Infinium 27K |
223 |
| GSE107361 | 2018 | AD | Infants/adults, LS (39), NL (40), NN (29) | HG‐U133_Plus_2 | 212 |
| GSE58558 | 2014 | AD | Cyclosporin A, LS, NL, w0/w2/w12 (109) | HG‐U133_Plus_2 | 158 |
| GSE59294 | 2014 | AD | Dupilumab, LS, NL, w0/w4 (40) | HG‐U133_Plus_2 | 159 |
| GSE120721 | 2015 | AD, NN | LCM, LS (15), NL (15), NN (22), epi/dermis | HG‐U133_Plus_2 | 222 |
| GSE65832 | 2015 | AD | RNA‐seq, LS (20), NL (20) | Illumina GA IIx | 279 |
| GSE81119 | 2017 | ‘AD’ mice | Mouse models of inflammation and ‘AD’ (37) | MG 1.0 ST | 280 |
| NA | 2018 | AD | Tape strip RNA‐seq, LS (11), NL (18), NN (13) | Ion Torrent | 213 |
| GSE120899 | 2018 | AD | Apremilast, LS, NL, w0/w12 (59) | HG‐U133_Plus_2 | Not published? |
| GSE99802 | 2018 | AD | Fezakinumab, LS, NL, w0/w4/w12 (302) | HG‐U133_Plus_2 | 161 |
| GSE121212 | 2019 | AD, PSO, NN | RNA‐seq, AD (27LS, 27NL), PSO (28LS, 27NL), NN (38) | Illumina GA | 334 |
| GSE14905 | 2008 | PSO, NN | LS (33), NL (28), NN (21) | HG‐U133_Plus_2 | 143 |
| GSE13355 | 2009 | PSO, NN | LS (58), NL (58), NN (64) | HG‐U133_Plus_2 | 281 |
| GSE31037 | 2011 | PSO, NN | miRNA, LS (24), NL (23), NN (20) | Illumina GA IIx | 282 |
| GSE30999 | 2012 | PSO, NN | LS (85), NL (85) | HG‐U133_Plus_2 | 283 |
| GSE26866 | 2012 | PSO | LCM, LS (20), NL (17), epi/dermis, | HG‐U133_A 2.0 | 284 |
| GSE11903 | 2009 | PSO | Etanercept, LS, NL, w0/1/2/4/12 (89) | HG‐U133_Plus_2 | 149 |
| GSE31652 | 2012 | PSO | Ixekizumab, LS, w0/w4 (30) | HG‐U133_Plus_2 | 150 |
| GSE55201 | 2014 | PSO, NN | Ixekizumab, blood, LS, NN, w0/w2 (81) | HG‐U133_Plus_2 | 285 |
| GSE51440 | 2014 | PSO | Guselkumab, LS, NL, w0/w1/w12 (59) | HG‐U133_Plus_PM | 152 |
| GSE53552 | 2014 | PSO | Brodalumab, LS, w0/w1/w2/wq6 (99) | HG‐U133_Plus_2 | 151 |
| GSE69967 | 2016 | PSO | Tofacitinib, LS, NL, d0/1/3/w1/2/4/12 (95) | HG‐U133_Plus_2 | 286, 287 |
| GSE54456 | 2014 | PSO, NN | RNA‐seq, LS (92), NN (82) | Illumina GA | 288 |
| GSE57225 | 2014 | PSO‐AD/ACD | PSO (23), AD (10), ECZ (13), NL (16) | SurePrint G3 8x60K | 289 |
| GSE63741 | 2016 | PSO, AD, other | AD‐LS, PSO‐LS, ACD, LP, NN (30 each) | PIQOR 2.0 | 290 |
| GSE80047 | 2016 | PSO, PPP(P) | PPP (3), PPPP (6), PSO (10), NN (31) | HG‐U133_Plus_PM | 291 |
| GSE79704 | 2017 | PSO, GPP, NN | GPP‐LS (32), PSO‐LS (12), NN (20) | HG 2.1 ST | 292 |
| GSE73894 | 2017 | PSO, NN | DNA methylation, LS (135), NL (41), NN (62) | Infinium 450k | 293 |
| GSE115797 | 2018 | PSO | DNA methylation, LS (24), NL (24) | Infinium 450k | 185 |
ACD, allergic contact eczema; Dx, diagnosis; d, day; epi, epidermis; ECZ, eczema (non‐atopic); GPP, generalized pustular psoriasis; LCM, laser‐capture microdissection; LP, lichen planus; PPP, palmoplantar pustulosis; PPPP (palmoplantar pustular psoriasis); PSO‐AD, patients co‐affected by both PSO and AD; w, week.
Microbiome
The cutaneous microbiome has been suggested as a factor that could trigger the immune system and initiate development of psoriasis 107, but as to date, the few and mainly descriptive data have been inconclusive. A recent analysis of the gut microbiome of 52 PSO patients suggested a specific ‘psoriatic core intestinal microbiome’ that differed from what is found in healthy subjects 190, but since the latter (healthy) data were pulled from the Human Microbiome Project, the analysis is confounded (with study), and calls for confirmation by a direct comparison of PSO patients with age and gender‐matched healthy controls. To establish – or rebut – a possible causative link between the microbiome (and its modulation by antibiotics, pre‐ or probiotics), psoriasis pathogenesis, and therapeutic effect, prospective, longitudinal intervention studies are needed. Also note that current microbiome analyses focus on taxonomic characterization (composition of the microbial community) rather than on functional, integrative studies involving metatranscriptomics and metabolomics, which eventually may enable in‐depth understanding of the dynamics of the microbiome 111.
Atopic dermatitis
Atopic dermatitis is the most common chronic, relapsing inflammatory skin disorder, characterized by intense itch (pruritus), redness (erythema), and eventually, thickening (lichenification) of the skin due to chronic rubbing. It affects 10–25% of all children, most with onset before 2 years of age, and 2–10% of adults 123, with wide regional variation 125, and with a prevalence that has more than doubled over the last 50 years 127. Due to its chronic and pruritic nature, AD adversely affects the quality of life (QoL) of most patients, in particular due to sleep disturbance and skin infections, and is also often followed by other atopic diseases, such as food allergy, asthma and allergic rhinitis, known as the ‘atopic march’ 132. Note, however, that <10% of AD patients travel the full atopic march (i.e. clinical manifestation of all four comorbidities) and that the risk is highest in the early‐onset persistent AD phenotype 191, 192.
The pathogenesis of AD is complex (illustrated in Fig. 2) and multifactorial as it involves genetic, immunologic, and environmental factors 193, including a defective skin barrier, permissive for entry of allergens that trigger inflammation, immune dysregulation with increased numbers of T‐cells and dendritic cells (DCs) and high levels of inflammatory molecules, and alterations in the cutaneous microbiome with overgrowth of Staphylococcus aureus. AD was first identified as a Th2 (IL‐4, IL‐13, IL‐31) driven disease 194, and later found to have also a Th22 (IL‐22) component 195 as well as variable Th17 and Th1 immune activation, the latter more pronounced in chronic AD 196.
Figure 2.
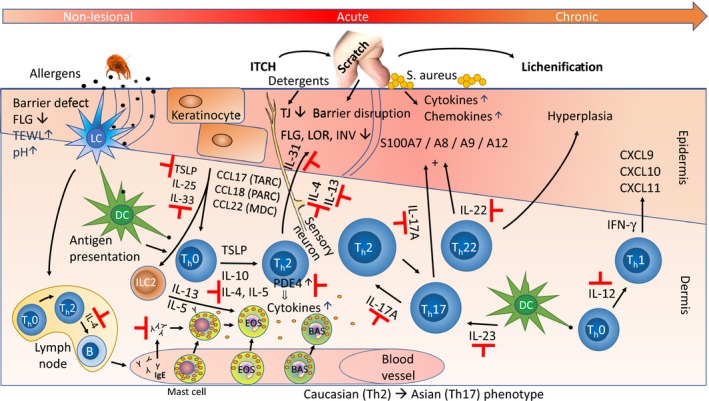
Pathways in the pathogenesis of AD. Epidermal barrier defects, which are partly due to FLG mutations, are associated with increased trans‐epidermal water loss [TEWL), increased skin pH, and penetration of epicutaneous allergens, such as dust mite debris. When the allergens encounter antigen‐presenting epidermal Langerhans cells (LCs, for an excellent review of the interplay between LCs and the epidermis, see Clayton et al. 259] and dermal dendritic cells (DCs), this causes immune activation and recruitment of inflammatory cells, including ILC2 [type 2 innate lymphoid cells 260] and type 2 helper T‐cells (Th2) that produce and release IL‐4, IL‐5, IL‐13, and IL‐31. These cells are considered part of the skin‐associated lymphoid tissue (SALT), the immunologically active cutaneous microenvironment, a concept which was proposed already in 1983 by Streilein 261. IL‐4 and IL‐13 suppress expression of terminal differentiation genes (such as FLG, LOR, INV), and also of tight junction (TJ) genes 208 leading to barrier disruption, while IL‐31 also acts directly on sensory neurons, triggering the itch–scratch cycle. This further damages the epidermis, increasing the risk of penetration by pathogens like Staphylococcus aureus. The stressed keratinocytes release TSLP, IL‐25, and IL‐33 that also drive Th2 differentiation. The Th2 cytokines induce IgE production in B cells and subsequently, release of inflammatory mediators (e.g. histamine) from activated (IgE bound) mast cells, basophils, and eosinophils. Th22 cells release IL‐22, which causes epidermal hyperplasia, and also, in synergy with IL‐17 – released from Th17 cells – induces expression of a subset of S100 family proteins. Acute AD lesions are characterized by a Th2 skewed (Th2, Th17, Th22) response, while chronic AD, which is often lichenified (thickened) by chronic scratching, progressively activates the Th1 axis with IL‐12 release, IFN‐γ expression and induction of chemokines (like CXCL9/CXCL10/CXCL11). Indicated with ⊣ are targets of approved and emerging drugs (see Table 2 for a detailed list). Figure modified, mainly from Vakharia & Silverberg 262, based on the original by Leung 2000 263 and 2004 264. For other representations, see Noda et al. 134, Paller et al. 265, Weidinger et al. 9, Lee et al. 9, 266, and Brunner et al. 267, 268.
Compared to PSO, both our molecular disease understanding and treatment options for AD are lagging some 10–15 years behind 197. For example, the first FDA‐approved biologic for treatment of moderate‐to‐severe AD, namely the much touted IL‐4Ra inhibitor dupilumab, reports response rates in the range of 44–52% EASI 7517 200, comparable to the rather moderate PASI75 response rates of the first generation TNF‐α targeting antibodies. Thus, there is still room for improvement and for development of even more efficacious targeted therapies; therapies that are tailored to the remaining subset(s) of severe AD patients, who may benefit from a personalized, endotype‐specific, treatment.
One reason that AD is a less‐mature field is the high diversity of the atopic landscape, with a wide spectrum of clinical manifestations ranging from localized nummular lesions to generalized exfoliative erythroderma in the most severe cases 132. Adding to the complexity of the clinical picture are the many possible categorizations of AD, such as:
infantile/childhood/adolescent/adult stages
early onset/late onset
transient/persistent
acute/subacute/chronic
mild/moderate/severe
intrinsic (low IgE, 20%)/extrinsic (high IgE, 80%)
African/Asian/European American phenotypes
± comorbidities: food allergy/asthma/rhinitis/infections
± genetic risk factors: e.g. FLG mutations
± environmental risk factors: multiple (microbiome/exposome)
± response to a given treatment
Though some of the categories are overlapping (e.g. all infantile stages are early onset), most of them can be combined (e.g. early/late onset × transient/persistent × mild/severe × low/high IgE × ethnicity × comorbidity × ±FLG mutations), resulting in thousands of possible composite classifications. This does not in itself pose a problem, because most of the above features are phenotypic and therefore relatively easy to record. No, what we would like to understand are the underlying disease endotypes. In other words: which molecular features and pathways characterize the different subtypes of AD 201, and can we identify relevant endotype‐specific biomarkers that can predict disease trajectories and guide choice and intensity of treatment? That is the question, and a difficult one indeed, because of the both heterogeneous and complex nature of AD, being the result of multiple genetic, environmental, and immunologic factors. This is reflected in our inadequate understanding of the pathogenesis of AD (outlined in Fig. 2), and the ongoing discussion of whether it is an ‘outside‐in’ (disruption of the epidermal barrier triggers the immune system) or an ‘inside‐out’ (inflammation causes the barrier dysfunction) disease 193, 202. But it is not really an either–or question, because current evidence speaks in favor of both the above hypotheses, which are therefore not mutually exclusive. Genetic evidence has established that loss‐of‐function mutations in FLG, the gene encoding filaggrin, an important structural protein in the stratum corneum of the epidermis 203, 204, are the major predisposing factors for AD 138. This has been confirmed by twin studies 140, and GWAS data 141, showing that FLG mutations, which are present in about 10% of the population, could stratify for the early‐onset persistent subphenotype in children 154. Between 20 and 50% of moderate‐to‐severe AD patients carry FLG mutations 136, so this AD subset fits well with the outside‐in hypothesis for initiation of AD. But what then, about the other half of AD patients who do not harbor any FLG mutations? In these patients, it is plausible that immune dysregulation results in secondary epidermal barrier disruption, in line with the inside‐out hypothesis. Or, alternatively, a combination of other genetic, epigenetic, immunological, and environmental factors – the exposome and microbiome included – may trigger and determine the course of AD, in which case, such compound endotypes may be difficult to tease out.
Still, it is beyond doubt that FLG mutation positive AD patients constitute a ‘true’ endotype, and therefore should be treated accordingly, preferably with the aim of reestablishing and maintaining an intact skin barrier as early as possible. This is necessary to prevent allergic sensitization and with this, development of asthma and allergic rhinitis. Ideally, one would like to perform prenatal diagnostics, i.e. WGS on the fetus’ DNA in order to identify all possible – not only skin disease related – genetic risk factors even before birth. Alternatively, and perhaps more feasibly, WGS of the newborn can provide the same information, albeit a little later. In case mutations in skin barrier genes (like FLG) are detected, early intervention schemes can be applied, such as use of emollients soon after birth 205. In best case, such a personalized preventive strategy may hinder development of AD and its comorbidities (the atopic march) altogether.
Besides FLG mutations, around 100 genes have been identified as AD associated in various studies 139, 141, 142. If one performs a functional enrichment analysis of these genes, it appears that the majority of them are related to inflammation and cytokine activity (Fig. 4), highlighting the potential importance of immune signaling and T‐cell activation in development of AD.
Figure 4.

STRING 276 network representation of 105 genes reported to have genetic associations to AD. The genes have been compiled from three publications: Paternoster et al. 2015 (31 loci) 141, Al‐Shobaili et al. 2016 (49 genes) 139, and Liang et al. 2016 (63 genes) 142. The clustering was performed with the ‘MCL inflation parameter’ = 3. The red cluster in the middle of the network represents the cytokine activity enriched gene set. The top enriched biological processes, molecular functions, and KEGG pathways are shown in the table below the network.
What remains is the detailed analysis of the many possible gene–gene and gene–environment interactions that define the complex endotypes of AD. Here, two genetic variants of particular interest will be mentioned:
CD207: encodes langerin, a pattern recognition receptor expressed in epidermal Langerhans cells (LCs) and involved in antigen‐processing and presentation to T‐cells. Defects in langerin function could therefore have implications for cutaneous immunity, in particular with respect to susceptibility to skin infections by viruses and bacteria like S. aureus 141.
CLDN1: encodes claudin‐1, a tight junction (TJ) protein, expressed by keratinocytes in the stratum granulosum layer of the epidermis and important for maintaining an intact epidermal barrier. In a study from 2011, AD patients (n = 5) were found to have markedly lower expression of CLDN1 compared to healthy controls 206. This finding has been replicated in other studies, showing that CLDN1 expression can be downregulated by IL‐33 via the STAT3 pathway in keratinocytes 207, via IL‐13 in bronchial epithelial tissue 208, and interestingly, that CLDN1 expression can be restored both in human keratinocytes and a murine model of AD by application of the proteasome inhibitor bortezomib 209.
An intriguing link between tight junction function, the recent 2–3‐fold rise in AD prevalence, and the increased use of detergents has been proposed by Dr. Cezmi A. Akdis and colleagues. They demonstrated that even trace concentrations (10−6 vol/vol) of commercial detergents were able to directly disrupt tight junctions between keratinocytes in culture and thereby potentially compromise epidermal barrier integrity, thus increasing the risk of allergen penetration and inflammation 210. Although this variant of the hygiene hypothesis is compelling, it remains to be reproduced in an in vivo setting on full thickness skin to determine if the detergents can actually penetrate the protective, outermost stratum corneum layer of the epidermis.
Gene expression analysis – from molecular pathology to targeted therapy
An important information source on the molecular pathology of AD (and any other skin disease) is transcriptomics analysis performed on skin biopsies. Optimally, the biopsies are obtained from site‐matched lesional and non‐lesional AD skin and from healthy – age and gender‐matched – controls. This enables both intra‐individual comparisons (paired analysis of samples from the same subject) and comparisons between the diseased and healthy population. Notably, individual gene expression patterns may expose not only overall disease signatures, but also the heterogeneity (endotypes and sub‐endotypes) of AD, and of the healthy population. This point is illustrated in Fig. 5, which is a re‐analysis of the transcriptomic profile of AD reported by Suárez‐Fariñas et al. 145.
Figure 5.
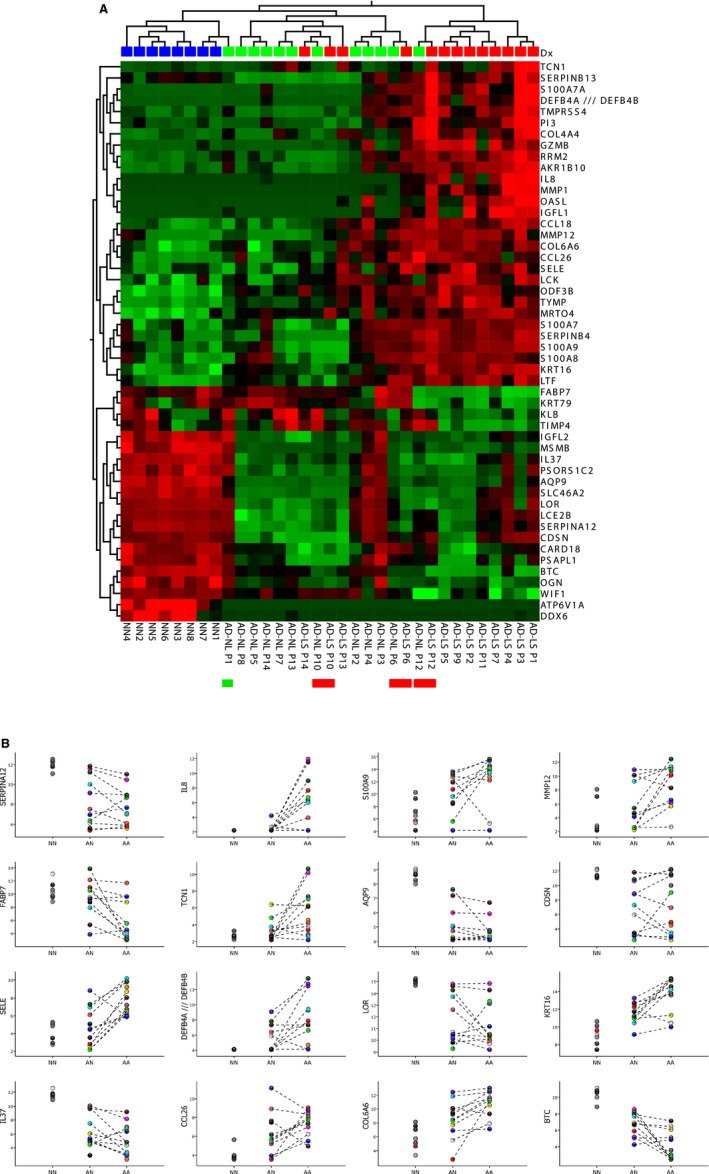
Gene expression analysis of lesional and non‐lesional skin biopsies from 13 AD patients and from eight healthy controls. (A) Heat‐map and two‐way unsupervised hierarchical clustering based on the 50 most variable genes between the three groups (non‐protein coding and orfs (open reading frames) removed). The samples cluster primarily according to disease (AD samples to the right and NN samples in the left cluster) and histology (LS to the right and NL in the middle cluster). What one also can see, is that three of the AD sample pairs (P12, P6, and P10, indicated by red bars below the heat map) cluster together, that is: there are only minor differences between the LS and NL samples from the same patient; the NL P12 sample is ‘lesional’‐like (clusters with the other LS samples), while the three ‘middle‐group’ (having overall low expression of most of the 50 DEG) LS samples (P13, P14, P10) appear more ‘non‐lesional’ like. One NL sample (P1, indicated with a green bar below the heat‐map) clusters with the normal (NN) group, and thus, this AD patient does not appear to have the ‘molecular scar’ typical of non‐lesional AD skin. The colors in the heat‐map signify high (red) or low (green) expression of the particular gene across samples (z‐scaled values). (B) Scatter plots of 16 selected genes, illustrating both the differences between lesional (AA), non‐lesional (AN), and healthy control (NN) samples, and the variability within the groups, revealing the heterogeneity of both the diseased and ‘normal’ (healthy) population. The Y‐axis are log2‐transformed expression values (detection limit: 2–4, saturating concentrations: around 15). The samples are colored according to individual, and the dotted lines connect samples (non‐lesional and lesional) originating from the same individual. All the data used for this illustration can be accessed in GEO by its accession number, GSE32924 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse32924).
The heat‐map in Fig. 5A is based on the top‐50 DEG, most of which are inflammatory or epidermis associated, and shows that the three histologies (healthy, non‐lesional, lesional) separate, and also, that the separation is not perfect, as several of the lesional (LS) and non‐lesional (NL) samples co‐cluster, which probably reflects the wide disease spectrum of AD. This heterogeneity is further emphasized when one looks at the individual genes (Fig. 5B), which display a striking variability in expression, both between and within groups. For example, IL8 (CXCL8, an inflammatory chemokine, involved in neutrophil activation), MMP12 (expressed by macrophages, degrades elastin), TCN1 (highly expressed in neutrophils), and DEFB4A/B (defensin, expressed by neutrophils and keratinocytes, induced by inflammation) all vary widely in expression, from undetectable to saturating concentrations in AD skin. Also, LOR (loricrin) and CDSN (corneodesmosin), which both are terminal differentiation genes, and believed to be downregulated in AD skin, are seen to have highly variable expression, ranging from undetectable (CDSN) to the high levels also found in normal skin. Also note, that not only AD skin but also healthy skin varies extensively in expression of some of the genes shown, such as MMP12, S100A9, and COL6A6, illustrating the variation within the normal population. The high variability, in particular in AD skin, most likely reflects both the extent of disease – the more inflamed skin, the higher the expression of inflammatory genes – and its pathogenesis, where multiple pathways, some associated with keratinocyte defects, some with immune system dysfunction, may coexist, and where proper (and probably problematic) identification of the underlying, dominant disease endotype eventually may guide targeted therapy.
We and others –‐ in particular the laboratories of Emma Guttman‐Yassky at Mount Sinai and James Krueger at Rockefeller University, both in New York, have generated a number of gene expression studies on skin have deposited transcriptomics data on AD and PSO in the public Gene Expression Omnibus Database GEO 211 for further analysis. Table 1 summarizes a selection of AD and PSO studies of interest and also links to the respective datasets.
A closer look at the above table reveals that several of the aforementioned categorization aspects of AD have already been addressed by transcriptomics analyses, including:
Early‐onset AD in children vs adult AD
The skin transcriptome of 19 young children (with no known FLG mutations) with early‐onset AD was compared to that of age‐matched healthy controls, as well as to adult AD patients/controls. In common for both child and adult AD patients were alterations in lipid metabolism and tight junction associated genes as well as Th2‐mediated inflammation 212. In addition, the pediatric patients displayed significant Th17/Th22 polarization, but neither Th1 activation nor downregulation of epidermal differentiation complex genes, which are characteristic features of adult AD. Still, larger cohorts are needed to take ethnic differences (the above study included Asian, African, Hispanic, and Caucasian patients, the former three ethnicities with only 1–2 matching controls) and subgroups with FLG mutations into account. However, because obtaining skin biopsies from children is challenging, in such studies other, less invasive techniques, such as tape‐stripping 213 or blood‐based biomarkers 214, 215, are preferable.
Acute vs chronic AD
Here, sequential biopsies were obtained from 10 patients in their acute and chronic phase of AD. Acute lesions were characterized by a marked increase (compared to non‐lesional skin) in expression of epidermal differentiation complex (EDC) genes, in particular S100A7/A8/A9, which are associated with Th2 (IL‐4, IL‐13, IL‐31) and Th22 (IL‐22) cytokine activation 196. When progressing to chronic lesions this Th2/Th22 axis was further activated followed by an increase in Th1‐associated products, such as CXCL9/10/11 (see Fig. 2). In terms of treatment selection, this could point toward targeting Th2/Th22 pathways in acute AD.
The effect of ethnicity on AD has been shown in Asian (Japanese and Korean) patients, who in general display a more psoriasiform AD phenotype and significantly higher Th17/Th22 activation as assessed by cytokine expression (IL‐17A, IL‐19, IL‐22, S100A12), compared to European American AD patients 212, 216. This can have implications for the choice of treatment, as the selective blockade of IL‐17/IL‐22 pathways could be indicated in the Asian, ‘psoriasis‐like’ immune phenotype. It will be interesting to see if the reported Japanese/Korean AD phenotype extends to the larger Chinese and Indian populations, and also, to include migration studies (investigating Asian American, Asian European, as well as local Asian AD) to evaluate the genetic, epigenetic, and environmental (exposome/microbiome) effects on the development of AD. The above considerations of course also apply to other non‐European ethnic groups, including the African population, who is likely to have yet other genetic susceptibilities, as recently reviewed by Kaufman et al. 217 and Brunner et al. 218.
A transcriptomic treatment response signature has been obtained in several clinical intervention studies, such as ultraviolet B (UVB) phototherapy 157, cyclosporin A 158, dupilumab 159, and fezakinumab 161. In these studies, pre‐ and post‐treatment skin biopsies were obtained from both lesional (AD‐LS) and non‐lesional (AD‐NL) skin. The number of differentially expressed genes (DEGs) between AD‐LS and AD‐NL was found to be lower post‐treatment compared to pre‐treatment, indicating a normalization of the AD disease signature, including suppression of Th1, Th2, and Th22 inflammatory markers 157. However, because not all genes improve, even after successful clinical remission as assessed by SCORAD, they are defined as comprising a residual disease genomic profile (RDGP) 219. The RDGP concept was originally introduced when the treatment of psoriasis with etanercept resulted in the resolution of disease and normalization of many, but not all psoriasis‐related genes 220. A subset of 248 genes did not return to baseline (or rather: exhibited less than 75% improvement after treatment) and could be indicative both of incomplete suppression of inflammation – leaving room for improvement – and for a ‘molecular scar’ intrinsic to the disease. Whether the latter represents different endotypes with implications for disease progression and treatment response remains to be determined.
A meta‐analysis derived AD (MADAD) signature identified 595 AD‐associated DEGs across four publicly available transcriptomics studies 146, and a subset of the most discriminatory of these genes was shown to be applicable as a robust standardized measure of treatment effect in the abovementioned UVB, cyclosporin A, and dupilumab studies. Since the MADAD reference transcriptome captures both immunological (inflammatory genes, cytokines, T‐cell receptor signaling) and barrier defect (epidermal differentiation, lipid metabolism) genes, it may be used for future evaluation of therapeutic response.
All of the above transcriptomic studies have been performed on full thickness punch biopsies of the skin, which is organized in three main layers: the epidermis, the dermis, and the subcutaneous fat (hypodermis). The epidermis contains >90% keratinocytes at different differentiation levels, a few melanocytes, Langerhans cells (LCs), Merkel cells, α‐dendritic cells, and inflammatory cells. The dermis mainly consists of extracellular matrix proteins, primarily collagen fibers produced by fibroblasts, and also dendritic cells/macrophages, mast cells, various unactivated/activated T‐cell subsets, plasma cells, hair follicles, sweat glands, sebaceous and apocrine glands, and endothelial cells 221. Thus, when we analyze gene expression in whole skin, the resulting average signal will reflect both the cellular distribution, such as the epidermis‐to‐dermis ratio, which is known to vary both in AD and PSO, as well as any up‐ or down‐regulation of differentially expressed genes. Furthermore, in a homogeneous assay the compartmental localization of gene expression is lost and also the expression of low‐abundance genes may become undetectable because of dilution. One way to generate a more refined skin transcriptome is to apply LCM (laser capture microdissection), which enables separation of the skin into its dermal and epidermal components. Such a – and so far only – study based on paired lesional and non‐lesional samples from five AD patients showed that indeed the dermal and epidermal transcriptomes differ, and also, that the AD signature could be expanded with some 1000 DEGs due to the increased signal‐to‐noise ratio, when working with separate compartments 222. Some of the DEGs now detectable by LCM included IL22, TSLP, IL34, CCL22, CCL26, CLDN4, and CLDN8 (the latter two involved in tight junction (TJ) formation). One reason why LCM is not applied routinely is that it is very labor intensive. Therefore, other methods need to be considered for separating the dermal and epidermal signals. One such method is based on epidermal shaves for transcriptomic profiling 223, another is tape stripping 213. A quick comparison of the top‐50 LS/NL DEGs from each of the three studies shows that they have only little overlap (Fig. 6). This may be partly due to different patient populations, different skin sampling technologies, and different RNA quantification platforms (Affymetrix microarrays, Illumina arrays, Ion Torrent sequencing). In particular, the tape‐stripping experiment displayed some unexpected findings, such as large differences in the expression of numerous keratin‐associated protein (KRTAP) genes (being more than 50‐fold downregulated in LS vs NL epidermis), which could suggest differences in the presence of hair.
Figure 6.
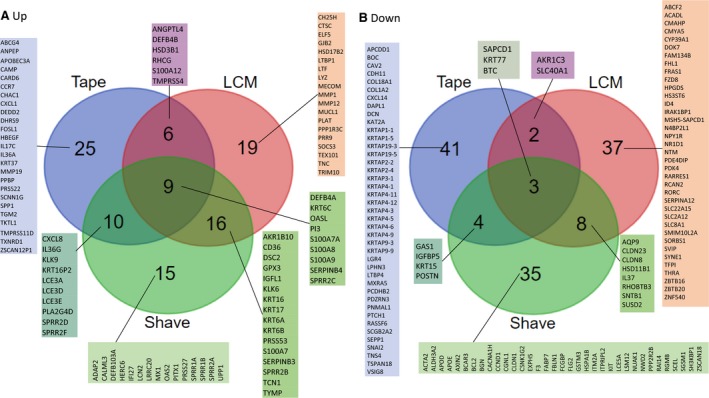
Comparison of three studies, assessing the epidermal transcriptomic profile of AD‐LS vs AD‐NL skin. (A) Top‐50 up DEG (LS vs NL). (B) Top‐50 down DEG (LS vs NL). The three studies included are Tape: Tape‐stripped skin (not in GEO) 213; LCM: GSE120721 222; Shave: GSE60709 223. For the genes that are up in LS/NL epidermis, the overlap in DEG between LCM and epidermal shave is 50% (25 out of 50 genes). Fewer genes are in common between the three studies for the down DEG. For the tape‐stripping study, interestingly, many KRTAP genes appear as lower expressed in LS skin. Since KRTAP genes are associated with the hair shaft, this could suggest that there is less hair in the LS epidermis region.
Microbiome
Both the gut and skin microbiome have been implicated in the pathogenesis of AD.
The gut microbiome is associated with the hygiene hypothesis proposed by Strachan in 1989, who noticed that the number of older children in a household had a striking inverse correlation to the prevalence of hay fever and eczema in their younger siblings 128. Thus, he hypothesized that smaller family size leads to less unhygienic contact with older siblings and thus, reduced the opportunity for cross infection that supposedly has a protective effect on development of eczema. In addition, higher microbial endotoxin exposure from farm animals was associated with protection against development of allergy, as suggested in the ‘Eat Dirt’ article by Weiss 224. Finally, because the gut microbiome is important for the prenatal–early‐life maturation of our immune system 225, it has been intensively studied in relation to allergic diseases, and perturbations in the infant gut microbiome have been linked to the risk of developing AD 226. However, several probiotic and prebiotic intervention trials have been conducted, and although some protective effect against AD was observed, the differences were small, and no clear gut microbiome–AD association could be demonstrated 227.
The cutaneous microbiome, on the other hand, is clearly associated with AD, as a vast majority (>90%) of AD patients have their skin colonized with Staphylococcus aureus, compared to only 5% of healthy controls 132. In addition, S. aureus positive AD patients seem to have more severe disease, higher levels of Type 2 biomarkers (CCL17, POSTN), allergen sensitization (IgE), and barrier dysfunction (higher TEWL) than non‐colonized AD controls 228. Also, in a prospective study on children with AD, an increase in the proportion of S. aureus and a concomitant decrease in bacterial diversity preceded worsening (flare) of AD 229. To address the question as to whether the observed colonization of AD skin by S. aureus is potentially driving the disease or merely an epiphenomenon, several intervention trials have been carried out. Such studies applying either antiseptics or antibiotics have demonstrated that a reduction in the level of S. aureus is indeed followed by a reduction in AD severity, further supporting a causal relationship between S. aureus colonization of skin and AD 227. Additionally, use of emollients in infants at risk for developing AD caused a decrease in skin pH and an increase in bacterial diversity, which may partly explain the preventative effects of emollients 230.
A mechanistic link to the pathogenesis of AD is suggested by the colonization of AD skin by toxigenic S. aureus strains that produce superantigens (SA), which drive the development of a Th2 immune response, and activate Langerhans cells (LCs) and cutaneous dendritic cells (DCs) that bridge innate and adaptive immunity 231. Moreover, mast cell degranulation is directly stimulated by the S. aureus δ‐toxin (δ‐hemolysin), which also promotes IgE production and Th2‐mediated inflammation 232. Conversely, Th2 cells enable S. aureus colonization due to IL‐4 and IL‐13 mediated inhibition of antimicrobial peptides (AMPs) and of terminal differentiation proteins important for skin barrier integrity 233. And in turn, S. aureus amplifies the inflammatory response by stimulating release of IL‐4, IL‐13, IL‐22, IL‐17, IL‐31, and IgE, thus closing the vicious circle 234. It will be interesting to see whether the topical application of protective commensal skin bacteria, such as coagulase negative Staphylococcus strains, can inhibit the growth of S. aureus and eventually, lead to a cure of AD 235 (Box 5).
Box 5. Technical considerations – what to sample and what to measure?
As the eyes are said to be the mirror of our soul, so is the skin a two‐way mirror,18 reflecting both our inner and outer environment 237. Therefore, the cutaneous inflammation observed in AD reflects both inherent skin barrier and immune system dysregulation (endotype) as well as the effect of external factors (exposome), such as allergens, bacterial toxins, detergents, and other irritants.
Fortunately, the skin is easily accessible, and as such, straightforward to sample and study by various techniques, from imaging (phenotype) to ‐omics analyses (molecular pathology) on biopsies. Standard 3–4 mm biopsies, however, damage the skin and are considered too invasive for routine use on children, who represent the majority of AD patients, which is why alternative sampling technologies should be considered. One alternative, is to take smaller, 1 mm ‘mini’ biopsies (for which commercial punches are available) with only minimal scarring, allowing for multiple biopsies to be sampled, but which still require the application of local anesthetics, as otherwise, the procedure is painful. Another, less invasive option is to apply tape stripping to remove the stratum corneum, enabling quick sampling of multiple epidermal layers. A third, and non‐invasive technology is the procedure of skin surface washings that within 30 min sampling time allows for quantification of stratum corneum associated cytokines 237.
Blood sampling is of course obvious, but first requires identification of adequate biomarkers 215. The advantage of a blood sample is that it can integrate the disease signature, and that it contains several subsets of inflammatory cells that can be identified, separated, and analyzed by flow cytometry. A disadvantage of blood‐based biomarkers is the dilution effect; if local skin inflammation is the dominating disease feature, it may be difficult to translate into a blood‐based biomarker.
Other sampling sites include urine 238 and exhaled breath condensate, the latter containing markers of airway inflammation, which has been reported in children with AD 239.
A note on the analysis of skin biopsies: when we take a full thickness skin biopsy, it contains a heterogeneous mixture of cells, which are being homogenized before DNA, RNA, or protein is extracted for further analysis. Thus, the sample is blended into a ‘cellular smoothie’, where the individual cell characteristics are evened out or even lost. Thus, what we see, when we analyze the gene expression pattern from such a homogenized biopsy, is a snapshot in time and space; it is an average signal from many (millions) individual cells and several cell types, but where the individual, cell‐specific signals are averaged out. The problem is that we do not know the distribution of different cell types in the sample. Thus, we cannot tell whether the actual – average – signal measured is due to activation or inhibition of specific genes (up‐ or down‐regulation of gene expression), or whether it is due to redistribution of compartments. For example, FLG and LOR are highly expressed in the epidermis and are often seen to decrease following perturbation. But whether the observed differential gene expression is due to inhibition, or whether it is because of thinning of the epidermis (thus, decreasing the epidermis to dermis ratio) remains unknown. One way of solving the problem is by deconvolution of the cellular compartments, i.e. estimating the percentage of epidermis, dermis, and other skin compartments based on tissue‐ and cell type‐specific expression patterns 240, 241, 242. This ‘housekeeping’ approach may work in a well‐defined system, but such a system is rarely well defined. The cell‐specific signals can be identified by laser capture microdissection (LCM), which enables separation of the dermis from the epidermis signal, and thus increases the signal‐to‐noise ratio compared to that of a full thickness biopsy 222. LCM is rarely applied, however, mainly because it is a very laborsome technique, and also because it can affect the actual state of the cells 243. Another, more high‐throughput technique, is separation by e.g. microfluidics or flow cytometry into single cells followed by single‐cell sequencing, for example, by drop‐seq, made open source by Steve McCarroll et al. (http://mccarrolllab.org/dropseq/). Thus, instead of a cellular smoothie, we now have a cellular fruit salad, where the characteristics of each individual piece of fruit are retained 244.19
How precise is precision medicine?
Usually, differences between diseased and healthy tissue are quantitative rather than qualitative. That is: a given target or pathway is rarely exclusive to just a single disease, tissue, or cell type. For example, cytokines and their receptors are expressed at highly variable levels across cell types and conditions, and while certain cells, such as Th2 cells, express and release high levels of IL‐4 and IL‐13, this is also the case for basophils and ILC2 cells 245. And conversely: the expression of cytokine receptors, e.g. the IL‐4Ra subunit, is not confined to just a single‐cell type, like keratinocytes (which themselves secrete numerous cytokines that act as both autocrine and paracrine mediators). Therefore, targeting the IL‐4 receptor pathway with a specific antibody like dupilumab may cause adverse effects – such as the conjunctivitis reported in 14–19% of AD patients treated with dupilumab 246. Of course, this concern also applies to any other targeted treatment, which is why it is important to evaluate and prioritize both the most relevant disease drivers, pathways, and targets (druggability considerations) as well as to take potential off‐target and on‐target adverse effects into consideration.
Finally, to evaluate the efficacy of novel medicine approaches and to identify the most important disease pathways, head‐to‐head comparisons against other targeted treatments are useful. A notable example of such a head‐to‐head comparison is the recent ECLIPSE study including 1048 moderate–severe PSO patients. In this Phase 3 study, the long‐term efficacy and safety of the IL‐17A inhibitor secukinumab was compared to that of the IL‐23 (p19) inhibitor guselkumab: at week 48, PASI90 was reached for 70% of patients on secukinumab, and for 85% of patients on guselkumab, while the PASI100 responses were 48% and 58%, respectively 247. Thus, for this patient group, the newer‐generation IL‐23 inhibitor demonstrated superior long‐term efficacy over the IL‐17A inhibitor.
In another head‐to‐head PSO study, the IL‐23 (p19) inhibitor risankizumab was compared to the dual IL‐12/23 inhibitor ustekinumab, and already at week 4 the p19 inhibitor showed a more pronounced effect than ustekinumab as assessed by molecular (RNA‐seq transcriptomics) and histopathologic profiling 153.
In a third PSO study, the effect of an anti‐IFN‐γ antibody was investigated, and although IFN‐γ was blocked at the molecular level, clinical efficacy could not be demonstrated 248. This – obviously – implies that it is not sufficient to demonstrate the molecular effect of target inhibition, if the target is not central for driving the disease.
Personalized medicine – strategies and candidates
Targeting signaling pathways in inflammatory skin diseases can be approached at different cellular levels: extracellularly, at the receptor level, either targeting the cytokine itself (e.g. tralokinumab for IL‐13), or its receptor (e.g. dupilumab for IL‐4RA) by monoclonal antibodies (or other biologics/biosimilars, such as nanobodies) or, alternatively, intracellularly, blocking the downstream signaling of e.g. the JAK‐STAT pathway (Fig. 3B‐C) by small‐molecule inhibitors of one or more of the four JAKs: JAK1, JAK2, JAK3, and TYK2 249.
Currently, several JAK inhibitors – both systemic and topical –with different selectivities are in clinical development for treatment of inflammatory diseases (see Table 2 for these and other compounds), including filgotinib (JAK1), upadacitinib (JAK1), abrocitinib (JAK1), ruxolitinib & baricitinb (JAK1/2), tofacitinib (JAK1/3), BMS‐986165 (TYK2), ASN002 (TYK2/SYK), and delgocitinib (JTE‐052, pan‐JAK). Most of these candidates show promising efficacy and overlapping systemic safety profiles with increased risk of opportunistic virus infections and cytopenias 250. Therefore, to avoid the latter adverse effects of systemic treatment, topical formulations should also be (and are) considered.
Table 2.
Future and emerging targeted treatment options for atopic dermatitis (AD) and psoriasis (PSO)
| Target | Drug | Literature reference | clinicaltrials.gov referencea | Indications | Clinical phase, status |
|---|---|---|---|---|---|
| AhR (agonist) |
Tapinarof (t)b (GSK2894512) |
294, 295 |
AD PSO |
Ph.II, completed Ph.III, withdrawn |
|
| CCL20 | GSK3050002 (i) | 296 | NCT02671188 | PsA | Ph.I, withdrawn |
| CD125 (IL5RA) | Benralizumab (sc) | 297 | NCT03563066 | AD | Ph.II, recruiting |
| H4R | ZPL‐389 (o) | 294 |
AD PSO |
Ph.II, recruiting Ph.II, completed |
|
| IgE |
Omalizumab (sc) QGE031 (sc) |
298 |
AD pediatric AD |
Ph.IV, active, Ph.II, completed |
|
| IL‐1a | Bermekimab (sc) | NCT03496974 | AD | AD: Ph.2, recruiting | |
| IL‐4Ra | Dupilumab (sc) (Dupixent) | 200, 299 |
FDA approved |
AD pediatric | Ph.III, enrolling |
| IL‐5 | Mepolizumab (sc) | 300 | NCT03055195 | AD | Ph.I, terminated |
| IL‐12B (p40) | Ustekinumab (Stelara) (sc) |
AD PSO |
Ph.II, completed Ph.III, pediatric PSO |
||
| IL‐13 | Tralokinumab (sc) | 304 | NCT03587805 | AD | Ph.III, recruiting |
| IL‐13 | Lebrikizumab (sc) | 299, 305 | NCT03443024 | AD | Ph.II, active |
| IL‐17A | Ixekizumab (sc) | 306 | NCT03073200 | PSO | Ph.III, pediatric PSO |
| IL‐17A | Secukinumab (sc) |
AD PSO |
Ph.II, completed Ph.III, completed |
||
| IL‐17A/IL‐17F | Bimekizumab (sc) | 307 | NCT03598790 | PSO | Ph.III, recruiting |
| IL‐17C | MOR106 (iv) | 308 | NCT03568071 | AD | Ph.II, recruiting |
| IL‐17RA | Brodalumab (sc) | 174 | NCT03403036 | PSO | Ph.IV, completed |
| IL‐22 | Fezakinumab (sc) | 161 | NCT01941537 | AD | Ph.II, active |
| IL‐23 (p19) |
Guselkumab (sc) Tildrakizumab (sc) |
PSO |
Ph.III, completed Ph.III, active |
||
| IL‐23 (p19) |
Risankizumab (sc) Mirikizumab (sc) |
PSO |
Ph.III, active Ph.III, recruiting |
||
| IL‐31 | BMS‐981164 (sc) | 312 | NCT01614756 | AD | Ph.I, completed |
| IL‐31RA | Nemolizumab (sc) | 275 | NCT03100344 | AD | Ph.II, completed |
| IL‐33 | Etokimab (sc) (ANB020) | NCT03533751 | AD | Ph.II, recruiting | |
| IL‐36R | ANB019 (sc) | NCT03619902 | PSO (GPP) | Ph.II, recruiting | |
| JAK1 |
Upadacitinib (o) Abrocitinib (o) (PF‐04965842) |
AD |
Ph.III, recruiting Ph.III, recruiting |
||
| JAK1/2 | Ruxolitinib (t) |
AD PSO AA Vitiligo |
Ph.III, recruiting Ph.II, completed Ph.II, terminated Ph.II, recruiting |
||
| JAK1/2 | Baricitinib (o) | 317, 318 |
AD PSO |
Ph.III, active Ph.II, completed |
|
| JAK1/3 | Tofacitinib (o,t) |
AD PSO AA |
Ph.II, completed Ph.III, completed Ph.II, active |
||
| JAK1/TYK2 | PF‐06700841 (o) | 320 | NCT02969018 | PSO | Ph.II, completed |
| JAK1/2/3, TYK2 |
Delgocitinib (t) (JTE‐052) |
321 | NCT03725722 | AD | Ph.II, recruiting |
| NK‐1R (TACR1) | Serlopitant (o) | 322 | NCT02975206 | AD, pruritus | Ph.II, completed |
| NK‐1R (Substance P) | Tradipitant (o) | 322 | NCT03568331 | AD, pruritus | Ph.III, recruiting |
| OX40 | GBR 830 (sc) | 323 | NCT03568162 | AD | Ph.II, recruiting |
| OX40 | KHK4083 (i) | NCT03096223 | AD | Ph.I, completed | |
| PDE4 | Apremilast (o) |
AD PSO |
Ph.II, completed Ph.III, completed |
||
| PDE4 | Crisaborole (t) |
AD PSO |
Ph.III, completed Ph.II, completed |
||
| PDE4 | OPA‐15406 (t) | 328 | NCT02068352 | AD | Ph.II, completed |
| RIP1 kinase | GSK2982772 (o) | 329 | NCT02776033 | PSO | Ph.II, completed |
| ROR‐γ | ESR‐114 (t) | NCT03630939 | PSO | Ph.II, recruiting | |
| SYK/JAK | ASN002 (o) | NCT03531957 | AD | Ph.II, recruiting | |
| TNF‐α | Infliximab (i) | 169 | NCT00686595 | PSO, PsA | Ph.IV, completed |
| TNF‐α |
Adalimumab (Humira) (sc) Etanercept (sc) |
330 | PSO |
Ph.III, completed Ph.IV, completed |
|
| TNF‐α/IL‐17A | ABT‐122 (sc) | 331 | NCT02349451 | PsA | Ph.II, completed |
| TNF‐α/IL‐17A | COVA322 (i) | 183, 332 | NCT02243787 | PSO | Ph.I, terminated (safety) |
| TrkA | CT327/SNA‐120 (t) | 322 | NCT01808157 | AD, pruritus | Ph.II, completed |
| TRPV1 | PAC‐14028 (t) | 333 | NCT02748993 | AD, pruritus | Ph.II, completed |
| TSLP |
Tezepelumab (sc) (AMG 157) |
323 | NCT00757042 | AD | Ph.I, completed |
| TYK2 | BMS‐986165 (o) | 178 | NCT03624127 | PSO, PsA | Ph.III, recruiting |
Sorted according to molecular target. Monoclonal antibody drugs can be identified by their names, which all end with ‘‐mab’.
The list is not exhaustive. For up‐to‐date information on the clinical trials, please see https://clinicaltrials.gov
(t) topical; (iv) intravenously; (o) oral; (sc) subcutaneously.
Finally, new treatment modalities include those based on vaccination and allergen‐specific immunotherapy. The latter is effective in treating allergies and involves de‐sensitization via repeated exposure to increasing doses of allergens, but the effect on AD is still unresolved 251. Vaccination against S. aureus could in principle eliminate this pathogenic factor from susceptible AD patients, and several clinical trials are ongoing to evaluate the effect of active and passive vaccine candidates on AD 252.
Conclusion
Historically, we have moved from ignorance, superstition (or act of God) and metaphysics, to a rational (Hippocratic), physical approach to personalized medicine, driven by major progress in technology; this has enabled us to zoom in, both on the cellular and the molecular (the omics revolution) basis of disease. And now – in the post‐genomic era, we are able to integrate the multiple levels of information: from molecular‐level genome, epigenome, metabolome, and proteome data, to higher level physiome, exposome, microbiome, and interactome data 253. Thus, we are aiming at a modern, systemic (systems biology), holistic disease understanding, where the gap between diagnostics and treatment options is steadily closing.
Oncology is leading the way in precision medicine 254, though for a critical review of precision oncology, see Brock and Huang 255. Immunology is catching up with asthma ahead, already linking phenotypes and endotypes to targeted therapy 256, and as a natural extension of this development, inflammatory skin diseases follow suit, with PSO ahead of AD 197.
In principle, chronic inflammatory skin diseases, like PSO and AD, and also alopecia and vitiligo, are (currently) incurable, but they do respond to treatment. They also, in particular AD, comprise complex and heterogeneous underlying endotypes, which are good candidates for a personalized medicine strategy. Thus, in order to apply endotype‐driven strategies for stratification and personalized medicine, it is necessary first to identify and understand these endotypes. Hopefully, such understanding can be obtained via an integrative, multi‐omics approach resulting in discovery of molecular biomarkers, both prognostic and predictive, for assessing the likelihood of comorbidity, disease progression, and response to novel, targeted treatments (Fig. 7). Additionally, and following the vision of P4 medicine being both personalized and participatory, patients will have the opportunity to monitor the health of their skin by using mobile apps 257 that in real‐time (by use of artificial intelligence (AI) and cloud‐based deep learning) can perform image analysis, evaluate the degree of treatment response, and eventually, recommend to stop, continue, or change the treatment, essentially enabling truly individualized medicine (Box 6).
Figure 7.
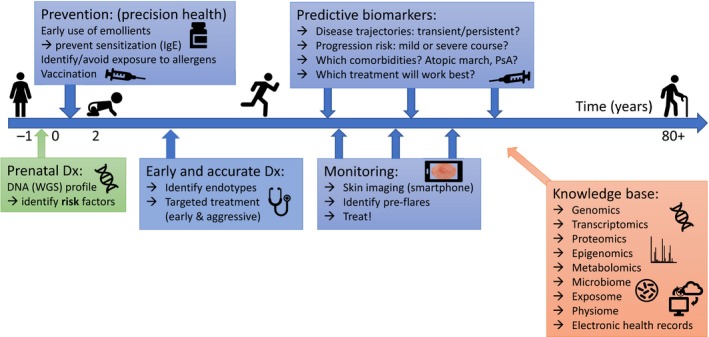
The vision of applied personalized medicine in inflammatory skin diseases. Risk factors (such as FLG mutations in AD) can be identified before birth, enabling preventive measures (such as use of emollients) in early childhood. Identification of endotypes can guide targeted treatment, and a combination of predictive biomarkers and skin monitoring (aided by machine learning, including AI, integrating the information knowledge base) may help identify pre‐flares and optimal time and type of treatment.
Box 6. Key messages.
As our mechanistic understanding of inflammatory skin conditions like PSO and AD increases, so does the potential for personalized treatment and prevention. In particular, because PSO and AD are both complex and heterogeneous diseases with variable course, treatment response, and hard to predict comorbidities, they pose paradigmatic obvious cases for a personalized medicine approach.
Inflammatory skin diseases are currently incurable, but not intractable.
Ideally, personalized management of PSO and AD is patient‐centric, i.e. taking the individual's needs into concern. Targeted treatment of the skin with emollients and topical corticosteroids may be sufficient to control disease in mild to moderate cases. Targeted, antibody‐based therapies have revolutionized the treatment of severe PSO and AD, and still more efficient (more patients reaching PASI 100/EASI 100) and safe medicines are in development.
Targeted therapies need to be tailored to the endotypes of AD, and thus, depend on identification of relevant biomarkers of the underlying pathways that drive the disease.
The move from personalized medicine to precision health can be achieved by early intervention (‘treat early and hard’) and prevention (see Fig. 7). Strategies include vaccination and avoidance of triggering factors in predisposed individuals, who can be identified even before birth by genotyping their DNA.
The author would like to acknowledge Hanne Norsgaard, Paola Lovato, Jakob Felding, and Witte Rush Koopman Jr. for insightful comments and suggestions for improvements, Adrian Ewald and Mette Vesterager for rhetorical sparring, and Michala Litman for lending a scratching hand (Fig. 2).
Litman T. Personalized medicine—concepts, technologies, and applications in inflammatory skin diseases. APMIS 2019; 127: 386–424.
Notes
And for less than 400 EUR. At this price for WGS, direct‐to‐consumer genomics is a reality.
and is extremely useful for stratifying according to pharmacokinetics (responders/non‐responders, fast/slow metabolizers, etc.), risk factors and targets [such as the around 300 driver genes and 3400+ driver mutations identified in cancer, crucial for precision oncology 30], and an absolute prerequisite for pharmacogenomics.
Endotype: a subtype of a disease or a subgroup of a population defined by a shared, underlying disease mechanism 31. Diseases like asthma 32 and AD 33 encompass several endotypes (for example, those with or without filaggrin mutations), each with their characteristic, underlying pathophysiology. This is in contrast to the Phenotype: the appearance of a disease or an individual, i.e. its observable features or traits (size, shape, pattern, color, behavior, etc.). The phenotype is determined by the sum of interactions between genotype, epigenetic factors, and the environment. Endophenotype: Also known as an ‘intermediate phenotype’ is a quantitative, biologic characteristic that lies between the phenotype and genotype, but is mechanistically closer to the disease than its clinical phenotype; in this context, it corresponds to a biomarker profile 34.
Again, Hippocrates should be recognized for introducing the central concept of prognostics, about which he remarked: ‘he will manage the cure best who foresees what is to happen from the present state of matters. For it is impossible to make all the sick well.’ 39
Sometimes such identification of responders (theratypes) to therapy is also called theratyping 41.
or ‘panoromic’, a term coined by Eric Topol to emphasize the wide‐angle view of multiple approaches 22, although one could argue that it is not only a wide, but also a deep view across layers.
less than 200 EUR, including strand‐specific library preparation and 30 million paired‐end reads per sample
The same consideration applies to ‘standard’ gene expression analysis, where the observed differential gene expression between two samples can be due to both redistribution of different compartments and to actual up‐ or down‐regulation of gene expression.
This EWAS acronym is not to be confused with the epigenome‐wide association studies mentioned before.
The principles for personalized medicine outlined for PSO and AD also apply to other inflammatory skin diseases, including allergic conditions such as contact dermatitis and urticaria, and autoimmune diseases like vitiligo, alopecia areata, and lichen planus.
Based on a world population estimate of 7.7 billion 124 of which 26% are children of age 0–14.
Because genetic changes take much longer (evolutionary) time to manifest. In addition, increased disease awareness (such as access to the Internet) may also partly explain the increase in prevalence.
According to opentargets.org
PASI 75 is a response rate and indicates the percentage of patients who have reached a 75% or more improvement (reduction) in their Psoriasis Area and Severity Index (PASI) score compared to baseline. PASI is a quantitative, composite measure of the severity and extent of psoriatic lesions taking into account erythema, thickness, and scaling of the lesions as well as percentage area affected. It is the most widely used tool in clinical trials for assessing psoriasis severity, although alternative, simpler to apply approaches, such as the Physician Global Assessment (PGA), have recently been proposed 170. As treatments become more effective, higher response rates are often reported, including PASI 90 and even PASI 100 (complete clearance of all disease) 171.
Note, however, that accumulation of neutrophils is not limited to pustular forms of psoriasis, as Munro's microabscesses containing large collections of neutrophils in the stratum corneum of psoriasis vulgaris are considered a hallmark of PSO.
Albeit promising, the dual IL‐17A/TNF inhibitors have been terminated in development due to safety concerns.
EASI 75 is the percentage of patients who have reached a 75% or more reduction in their Eczema Area and Severity Index (EASI) score compared to baseline, in this case after 16 weeks of treatment. Other commonly used clinical measures of AD severity include SCORAD (Severity Scoring of Atopic Dermatitis, which includes both objective physician estimates of disease severity as well as subjective patient estimates of itch and sleep loss), and IGA (Investigator's Global Assessment), and because the three scores complement each other, it has been recommended to apply at least two independent assessment schemes for a reliable evaluation of AD 198. What is not captured by the above scores is the actual disease burden on QoL (quality of life) of AD patients, which is why several questionnaire‐based schemes have been proposed for a standardized QoL assessment, including Patient‐Oriented Eczema Measure (POEM), Dermatology Life Quality Index (DLQI), ItchyQOL, and 5‐dimensions (5‐D) itch scales, all of which show good validity and internal consistency 199. Another important aspect when assessing the clinical efficacy of treatment is the placebo effect, which is notoriously high in AD, and which should therefore always be compared to the drug effect. For example, EASI 50 for the placebo arm was 35% compared to 85% for dupilumab in an early‐phase 12‐week study 200.
And a very large mirror indeed; while the conservative estimate of skin surface area is 2 m2, then, if one takes the presence of hair follicles, sweat, and sebaceous glands into account, the total skin area – and thus the interface for interaction with the skin microbiome – is probably closer to 30 m2, as pointed out by Richard Gallo 236.
Please see this inspiring TED talk by Steve McCarroll for the fruit analogy: https://www.ted.com/talks/steve_mccarroll_how_data_is_helping_us_unravel_the_mysteries_of_the_brain
References
- 1. Atherton DJ. Topical corticosteroids in atopic dermatitis. BMJ 2003;23:942–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vail J. Pharmacogenomics: the end of trial‐and‐error medicine? Int J Pharm Compd 2007;11:59–65. [PubMed] [Google Scholar]
- 3. Morgan P, Brown DG, Lennard S, Anderton MJ, Barrett JC, Eriksson U, et al. Impact of a five‐dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov 2018;17:167–81. [DOI] [PubMed] [Google Scholar]
- 4. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med 2005;353:1711–23. [DOI] [PubMed] [Google Scholar]
- 5. van der Velden VH. Glucocorticoids: mechanisms of action and anti‐inflammatory potential in asthma. Mediators Inflamm 1998;7:229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baida G, Bhalla P, Yemelyanov A, Stechschulte LA, Shou W, Readhead B, et al. Deletion of the glucocorticoid receptor chaperone FKBP51 prevents glucocorticoid‐induced skin atrophy. Oncotarget 2018;9:34772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dhar S, Seth J, Parikh D. Systemic side‐effects of topical corticosteroids. Indian J Dermatol 2014;59:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guttman‐Yassky E, Krueger JG. Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol 2017;48:68–73. [DOI] [PubMed] [Google Scholar]
- 9. Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers 2018;4:984. [DOI] [PubMed] [Google Scholar]
- 10. Griffiths CEM, van de Kerkhof P, Czarnecka‐Operacz M. Psoriasis and atopic dermatitis. Dermatol Ther 2017;7(Suppl 1):31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bieber T. Atopic dermatitis 2.0: from the clinical phenotype to the molecular taxonomy and stratified medicine. Allergy 2012;67:1475–82. [DOI] [PubMed] [Google Scholar]
- 12. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 2017;140:645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibson WM. Can personalized medicine survive? Can Fam Physician 1971;17:29–88. [PMC free article] [PubMed] [Google Scholar]
- 14. Arnold RM, Forrow L. Rewarding medicine: good doctors and good behavior. Ann Intern Med 1990;113:794–8. [DOI] [PubMed] [Google Scholar]
- 15. Abrahams E, Silver M. The history of personalized medicine In: Gordon E, Koslow S, editors. Integrative Neuroscience and Personalized Medicine. England, UK: Oxford University Press, 2010: 3–16. [Google Scholar]
- 16. Lamberti MJ, Awatin J. Mapping the landscape of patient‐centric activities within clinical research. Clin Ther 2017;39:2196–202. [DOI] [PubMed] [Google Scholar]
- 17. National Research Council (US) . Committee on a framework for developing a new taxonomy of disease. Glossary Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, DC: National Academies Press, 2011. [PubMed] [Google Scholar]
- 18. Siest G. Systems medicine, stratified medicine, personalized medicine but not precision medicine. Drug Metabol Drug Interact 2014;29:1–2. [DOI] [PubMed] [Google Scholar]
- 19. Strebhardt K, Ullrich A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat Rev Cancer 2008;8:473–80. [DOI] [PubMed] [Google Scholar]
- 20. Pirmohamed M. Pharmacogenetics and pharmacogenomics. Br J Clin Pharmacol 2001;52:345–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Genetics Home Reference . What is pharmacogenomics? [Internet]. Genetics Home Reference. Available from: https://ghr.nlm.nih.gov/primer/genomicresearch/pharmacogenomics [cited 2018 Sep 22].
- 22. Topol EJ. Individualized medicine from prewomb to tomb. Cell 2014;157:241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trusheim MR, Berndt ER, Douglas FL. Stratified medicine: strategic and economic implications of combining drugs and clinical biomarkers. Nat Rev Drug Discov 2007;6:287–93. [DOI] [PubMed] [Google Scholar]
- 24. Dias D, Paulo Silva Cunha J. Wearable Health devices‐vital sign monitoring, systems and technologies. Sensors 2018;18:pii: E2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flores M, Glusman G, Brogaard K, Price ND, Hood L. P4 medicine: how systems medicine will transform the healthcare sector and society. Per Med 2013;10:565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reardon S. US tailored‐medicine project aims for ethnic balance. Nature 2015;523:391–2. [DOI] [PubMed] [Google Scholar]
- 27. Lemke JR, Kernland‐Lang K, Hörtnagel K, Itin P. Monogenic human skin disorders. Dermatology 2014;229:55–64. [DOI] [PubMed] [Google Scholar]
- 28. Litman T, Druley TE, Stein WD, Bates SE. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci 2001;58:931–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017;551:327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018;174:1034–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agache I, Rogozea L. Endotypes in allergic diseases. Curr Opin Allergy Clin Immunol 2018;8:177–83. [DOI] [PubMed] [Google Scholar]
- 32. Lötvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 2011;127:355–60. [DOI] [PubMed] [Google Scholar]
- 33. Carson CG, Rasmussen MA, Thyssen JP, Menné T, Bisgaard H. Clinical presentation of atopic dermatitis by filaggrin gene mutation status during the first 7 years of life in a prospective cohort study. PLoS ONE 2012;7:e48678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bieber T, D'Erme AM, Akdis CA, Traidl‐Hoffmann C, Lauener R, Schäppi G, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: where are we, and where should we go? J Allergy Clin Immunol 2017;139:S58–64. [DOI] [PubMed] [Google Scholar]
- 35. Kearns JT, Lin DW. Improving the specificity of PSA screening with serum and urine markers. Curr Urol Rep 2018;19:80. [DOI] [PubMed] [Google Scholar]
- 36. Palmirotta R, Lovero D, Cafforio P, Felici C, Mannavola F, Pellè E, et al. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Ther Adv Med Oncol 2018;10:1758835918794630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Agache I, Akdis CA. Endotypes of allergic diseases and asthma: an important step in building blocks for the future of precision medicine. Allergol Int 2016;65:243–52. [DOI] [PubMed] [Google Scholar]
- 38. ICD‐11 [Internet]. Available from: https://icd.who.int/ [cited 2018 Sep 27].
- 39. Adams F. The Genuine Works of Hippocrates. London: The Sydenham Society, 1849. [Google Scholar]
- 40. FDA‐NIH Biomarker Working Group . Prognostic biomarker BEST (Biomarkers, EndpointS, and other Tools) Resource [Internet]. Silver Spring, MD: Food and Drug Administration; 2016. [PubMed] [Google Scholar]
- 41. Kucuksezer UC, Ozdemir C, Akdis M, Akdis CA. Precision/personalized medicine in allergic diseases and asthma. Arch Immunol Ther Exp 2018;66:431–42. [DOI] [PubMed] [Google Scholar]
- 42. Dong AN, Tan BH, Pan Y, Ong CE. Cytochrome P450 genotype‐guided drug therapies: an update on current states. Clin Exp Pharmacol Physiol 2018;45:991–1001. [DOI] [PubMed] [Google Scholar]
- 43. Barbarino JM, Whirl‐Carrillo M, Altman RB, Klein TE. PharmGKB: a worldwide resource for pharmacogenomic information. Wiley Interdiscip Rev Syst Biol Med 2018;10:e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ovejero‐Benito MC, Muñoz‐Aceituno E, Reolid A, Saiz‐Rodríguez M, Abad‐Santos F, Daudén E. Pharmacogenetics and pharmacogenomics in moderate‐to‐severe psoriasis. Am J Clin Dermatol 2018;19:209–22. [DOI] [PubMed] [Google Scholar]
- 45. Huang Y, Fong Y. Identifying optimal biomarker combinations for treatment selection via a robust kernel method. Biometrics 2014;70:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ralfkiaer U, Hagedorn PH, Bangsgaard N, Løvendorf MB, Ahler CB, Svensson L, et al. Diagnostic microRNA profiling in cutaneous T‐cell lymphoma (CTCL). Blood 2011;118:5891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marstrand T, Ahler CB, Ralfkiaer U, Clemmensen A, Kopp KL, Sibbesen NA, et al. Validation of a diagnostic microRNA classifier in cutaneous T‐cell lymphomas. Leuk Lymphoma 2014;55:957–8. [DOI] [PubMed] [Google Scholar]
- 48. Augello CJ, Noll JM, Distel TJ, Wainright JD, Stout CE, Ford BD. Identification of novel blood biomarker panels to detect ischemic stroke in patients and their responsiveness to therapeutic intervention. Brain Res 2018;1698:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Y, Zhang Y, Johnson A, Venable A, Griswold J, Pappas D. Combined CD25, CD64, and CD69 biomarker panel for flow cytometry diagnosis of sepsis. Talanta 2019;191:216–21. [DOI] [PubMed] [Google Scholar]
- 50. Kumar D, Gupta A, Mandhani A, Sankhwar SN. Metabolomics‐derived prostate cancer biomarkers: fact or fiction? J Proteome Res 2015;14:1455–64. [DOI] [PubMed] [Google Scholar]
- 51. Thijs JL, Strickland I, Bruijnzeel‐Koomen CAFM, Nierkens S, Giovannone B, Csomor E, et al. Moving toward endotypes in atopic dermatitis: identification of patient clusters based on serum biomarker analysis. J Allergy Clin Immunol 2017;140:730–7. [DOI] [PubMed] [Google Scholar]
- 52. Redenšek S, Dolžan V, Kunej T. From genomics to omics landscapes of Parkinson's disease: revealing the molecular mechanisms. OMICS 2018;22:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol 2016;17:451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet 2018;19:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet 2018;93:508–19. [DOI] [PubMed] [Google Scholar]
- 56. Ortiz V, Yu M. Analyzing circulating tumor cells one at a time. Trends Cell Biol 2018;28:764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang L, Ma F, Chapman A, Lu S, Xie XS. Single‐cell whole‐genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet 2015;16:79–102. [DOI] [PubMed] [Google Scholar]
- 58. Ichinohe T, Miyama T, Kawase T, Honjo Y, Kitaura K, Sato H, et al. Next‐generation immune repertoire sequencing as a clue to elucidate the landscape of immune modulation by host‐gut microbiome interactions. Front Immunol 2018;9:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Auer H, Newsom DL, Kornacker K. Expression profiling using Affymetrix genechip microarrays. Methods Mol Biol 2009;509:35–46. [DOI] [PubMed] [Google Scholar]
- 60. McGettigan PA. Transcriptomics in the RNA‐seq era. Curr Opin Chem Biol 2013;17:4–11. [DOI] [PubMed] [Google Scholar]
- 61. Spies D, Ciaudo C. Dynamics in transcriptomics: advancements in RNA‐seq time course and downstream analysis. Comput Struct Biotechnol J 2015;13:469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nguyen QH, Pervolarakis N, Nee K, Kessenbrock K. Experimental considerations for single‐cell RNA sequencing approaches. Front Cell Dev Biol 2018;6:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dasgupta S, Bader GD, Goyal S. Single‐cell RNA sequencing: a new window into cell scale dynamics. Biophys J 2018;115:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc 2015;10:442–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three‐dimensional intact‐tissue sequencing of single‐cell transcriptional states. Science 2018;361:pii: eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Strell C, Hilscher MM, Laxman N, Svedlund J, Wu C, Yokota C, et al. Placing RNA in context and space ‐ methods for spatially resolved transcriptomics. FEBS J 2018. 10.1111/febs.14435 [DOI] [PubMed] [Google Scholar]
- 67. Tost J. A translational perspective on epigenetics in allergic diseases. J Allergy Clin Immunol 2018;142:715–26. [DOI] [PubMed] [Google Scholar]
- 68. Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis 2018;5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rivera CM, Ren B. Mapping human epigenomes. Cell 2013;155:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Costa‐Pinheiro P, Montezuma D, Henrique R, Jerónimo C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics 2015;7:1003–15. [DOI] [PubMed] [Google Scholar]
- 71. Luchenko VL, Litman T, Chakraborty AR, Heffner A, Devor C, Wilkerson J, et al. Histone deacetylase inhibitor‐mediated cell death is distinct from its global effect on chromatin. Mol Oncol 2014;8:1379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 2004;101:2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nygaard S, Jacobsen A, Lindow M, Eriksen J, Balslev E, Flyger H, et al. Identification and analysis of miRNAs in human breast cancer and teratoma samples using deep sequencing. BMC Med Genomics 2009;2:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Martens‐Uzunova ES, Jalava SE, Dits NF, van Leenders GJLH, Møller S, Trapman J, et al. Diagnostic and prognostic signatures from the small non‐coding RNA transcriptome in prostate cancer. Oncogene 2012;31:978–91. [DOI] [PubMed] [Google Scholar]
- 75. Gaedcke J, Grade M, Camps J, Sokilde R, Kaczkowski B, Schetter AJ, et al. The rectal cancer microRNAome ‐ microRNA expression in rectal cancer and matched normal mucosa. Clin Cancer Res 2012;18:4919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Søkilde R, Vincent M, Møller AK, Hansen A, Høiby PE, Blondal T, et al. Efficient identification of miRNAs for classification of tumor origin. J Mol Diagn 2014;16:106–15. [DOI] [PubMed] [Google Scholar]
- 77. Lindahl LM, Besenbacher S, Rittig AH, Celis P, Willerslev‐Olsen A, Gjerdrum LMR, et al. Prognostic miRNA classifier in early‐stage mycosis fungoides: development and validation in a Danish nationwide study. Blood 2017;131:759–70. [DOI] [PubMed] [Google Scholar]
- 78. Bork‐Jensen J, Scheele C, Christophersen DV, Nilsson E, Friedrichsen M, Fernandez‐Twinn DS, et al. Glucose tolerance is associated with differential expression of microRNAs in skeletal muscle: results from studies of twins with and without type 2 diabetes. Diabetologia 2015;58:363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zibert JR, Løvendorf MB, Litman T, Olsen J, Kaczkowski B, Skov L. MicroRNAs and potential target interactions in psoriasis. J Dermatol Sci 2010;58:177–85. [DOI] [PubMed] [Google Scholar]
- 80. Huang R‐Y, Li L, Wang M‐J, Chen X‐M, Huang Q‐C, Lu C‐J. An exploration of the role of MicroRNAs in psoriasis: a systematic review of the literature. Medicine 2015;94:e2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Løvendorf MB, Skov L. miRNAs in inflammatory skin diseases and their clinical implications. Expert Rev Clin Immunol 2015;11:467–77. [DOI] [PubMed] [Google Scholar]
- 82. Rożalski M, Rudnicka L, Samochocki Z. MiRNA in atopic dermatitis. Postepy Dermatol Alergol 2016;33:157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang L, Peng D, Sood AK, Dang CV, Zhong X. Shedding light on the dark cancer genomes: long noncoding RNAs as novel biomarkers and potential therapeutic targets for cancer. Mol Cancer Ther 2018;17:1816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Elling R, Chan J, Fitzgerald KA. Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol 2016;46:504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rogers S, Girolami M, Kolch W, Waters KM, Liu T, Thrall B, et al. Investigating the correspondence between transcriptomic and proteomic expression profiles using coupled cluster models. Bioinformatics 2008;24:2894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hausser J, Zavolan M. Identification and consequences of miRNA‐target interactions–beyond repression of gene expression. Nat Rev Genet 2014;15:599–612. [DOI] [PubMed] [Google Scholar]
- 87. Petsalaki E, Helbig AO, Gopal A, Pasculescu A, Roth FP, Pawson T. SELPHI: correlation‐based identification of kinase‐associated networks from global phospho‐proteomics data sets. Nucleic Acids Res 2015;43 W1:W276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Peng B, Li H, Peng X‐X. Functional metabolomics: from biomarker discovery to metabolome reprogramming. Protein Cell 2015;6:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rattray NJW, Hamrang Z, Trivedi DK, Goodacre R, Fowler SJ. Taking your breath away: metabolomics breathes life in to personalized medicine. Trends Biotechnol 2014;32:538–48. [DOI] [PubMed] [Google Scholar]
- 90. Hao L, Greer T, Page D, Shi Y, Vezina CM, Macoska JA, et al. In‐depth characterization and validation of human urine metabolomes reveal novel metabolic signatures of lower urinary tract symptoms. Sci Rep 2016;6:30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Røpke MA, Alonso C, Jung S, Norsgaard H, Richter C, Darvin ME, et al. Effects of glucocorticoids on stratum corneum lipids and function in human skin—A detailed lipidomic analysis. J Dermatol Sci 2017;88:330–8. [DOI] [PubMed] [Google Scholar]
- 92. Lauc G, Pezer M, Rudan I, Campbell H. Mechanisms of disease: the human N‐glycome. Biochim Biophys Acta 2016;1860:1574–82. [DOI] [PubMed] [Google Scholar]
- 93. Almeida A, Kolarich D. The promise of protein glycosylation for personalised medicine. Biochim Biophys Acta 2016;1860:1583–95. [DOI] [PubMed] [Google Scholar]
- 94. Russell A, Adua E, Ugrina I, Laws S, Wang W. Unravelling immunoglobulin G Fc N‐glycosylation: a dynamic marker potentiating predictive, preventive and personalised medicine. Int J Mol Sci 2018;19:pii: E390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Houle D, Govindaraju DR, Omholt S. Phenomics: the next challenge. Nat Rev Genet 2010;11:855–66. [DOI] [PubMed] [Google Scholar]
- 96. Robinson PN. Deep phenotyping for precision medicine. Hum Mutat 2012;33:777–80. [DOI] [PubMed] [Google Scholar]
- 97. Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul‐Forestier I, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 2014;42(Database issue):D966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Graham S, McDonald L, Wasiak R, Lees M, Ramagopalan S. Time to really share real‐world data? F1000Res 2018;7:1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jensen AB, Moseley PL, Oprea TI, Ellesøe SG, Eriksson R, Schmock H, et al. Temporal disease trajectories condensed from population‐wide registry data covering 6.2 million patients. Nat Commun 2014;5:4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Turnbaugh PJ, Ley RE, Hamady M, Fraser‐Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007;449:804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ 2018;361:k2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bull MJ, Plummer NT. Part 1: The human gut microbiome in health and disease. Integr Med 2014;13:17–22. [PMC free article] [PubMed] [Google Scholar]
- 103. Rosenfeld CS. Microbiome disturbances and autism spectrum disorders. Drug Metab Dispos 2015;43:1557–71. [DOI] [PubMed] [Google Scholar]
- 104. ElRakaiby M, Dutilh BE, Rizkallah MR, Boleij A, Cole JN, Aziz RK. Pharmacomicrobiomics: the impact of human microbiome variations on systems pharmacology and personalized therapeutics. OMICS 2014;18:402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Doestzada M, Vila AV, Zhernakova A, Koonen DPY, Weersma RK, Touw DJ, et al. Pharmacomicrobiomics: a novel route towards personalized medicine? Protein Cell 2018;9:432–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013;341:295–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Langan EA, Griffiths CEM, Solbach W, Knobloch JK, Zillikens D, Thaçi D. The role of the microbiome in psoriasis: moving from disease description to treatment selection? Br J Dermatol 2018;178:1020–7. [DOI] [PubMed] [Google Scholar]
- 108. Pascal M, Perez‐Gordo M, Caballero T, Escribese MM, Lopez Longo MN, Luengo O, et al. Microbiome and allergic diseases. Front Immunol 2018;9:1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kong HH, Segre JA. The molecular revolution in cutaneous biology: investigating the skin microbiome. J Invest Dermatol 2017;137:e119–22. [DOI] [PubMed] [Google Scholar]
- 110. Thaiss CA. Microbiome dynamics in obesity. Science 2018;362:903–4. [DOI] [PubMed] [Google Scholar]
- 111. Aguiar‐Pulido V, Huang W, Suarez‐Ulloa V, Cickovski T, Mathee K, Narasimhan G. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol Bioinform Online 2016;12(Suppl 1):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang L, Chen SJ. Environment, genome and cancer. C R Acad Sci III 2001;324:1085–91. [DOI] [PubMed] [Google Scholar]
- 113. Wild CP. The exposome: from concept to utility. Int J Epidemiol 2012;41:24–32. [DOI] [PubMed] [Google Scholar]
- 114. Sharma A, Gilbert JA. Microbial exposure and human health. Curr Opin Microbiol 2018;44:79–87. [DOI] [PubMed] [Google Scholar]
- 115. Bessonneau V, Pawliszyn J, Rappaport SM. The saliva exposome for monitoring of individuals’ health trajectories. Environ Health Perspect 2017;125:077014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Steckling N, Gotti A, Bose‐O'Reilly S, Chapizanis D, Costopoulou D, De Vocht F, et al. Biomarkers of exposure in environment‐wide association studies ‐ Opportunities to decode the exposome using human biomonitoring data. Environ Res 2018;164:597–624. [DOI] [PubMed] [Google Scholar]
- 117. Patel CJ. Analytical complexity in detection of gene variant‐by‐environment exposure interactions in high‐throughput genomic and exposomic research. Curr Environ Health Rep 2016;3:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang P, Xu L. The internet of things (IoT): informatics methods for IoT‐enabled health care. J Biomed Inform 2018;87:154–6. [DOI] [PubMed] [Google Scholar]
- 119. Swan M. The quantified self: fundamental disruption in big data science and biological discovery. Big Data 2013;1:85–99. [DOI] [PubMed] [Google Scholar]
- 120. Ristevski B, Chen M. Big data analytics in medicine and healthcare. J Integr Bioinform 2018;15:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Griffiths CEM, van der Walt JM, Ashcroft DM, Flohr C, Naldi L, Nijsten T, et al. The global state of psoriasis disease epidemiology: a workshop report. Br J Dermatol 2017;177:e4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Grozdev I, Korman N, Tsankov N. Psoriasis as a systemic disease. Clin Dermatol 2014;32:343–50. [DOI] [PubMed] [Google Scholar]
- 123. Sacotte R, Silverberg JI. Epidemiology of adult atopic dermatitis. Clin Dermatol 2018;36:595–605. [DOI] [PubMed] [Google Scholar]
- 124. World Population Clock: 7.7 Billion People (2018) ‐ Worldometers [Internet]. Available from: http://www.worldometers.info/world-population/ [cited 2018 Sep 21].
- 125. Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab 2015;66(Suppl 1):8–16. [DOI] [PubMed] [Google Scholar]
- 126. Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med 2014;4:pii: a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thomsen SF. Epidemiology and natural history of atopic diseases. Eur Respir J 2015;2:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Strachan DP. Hay fever, hygiene, and household size. BMJ 1989;299:1259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Karimkhani C, Dellavalle RP, Coffeng LE, Flohr C, Hay RJ, Langan SM, et al. Global skin disease morbidity and mortality: an update from the global burden of disease study 2013. JAMA Dermatol 2017;153:406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Steinke S, Zeidler C, Riepe C, Bruland P, Soto‐Rey I, Storck M, et al. Humanistic burden of chronic pruritus in patients with inflammatory dermatoses: Results of the European Academy of Dermatology and Venereology Network on Assessment of Severity and Burden of Pruritus (PruNet) cross‐sectional trial. J Am Acad Dermatol 2018;79 457–63:e5. [DOI] [PubMed] [Google Scholar]
- 131. van de Kerkhof PCM, Nestlé FO. Psoriasis In: Bolognia JL, Schaffer JV, Cerroni L, editors. Dermatology. Amsterdam, Netherlands: Elsevier, 2017: 138–60. [Google Scholar]
- 132. McAleer MA, O'Regan GM, Irvine AM. Atopic dermatitis In: Bolognia JL, Schaffer JV, Cerroni L, editors. Dermatology. Amsterdam, Netherlands: Elsevier, 2017: 208–26. [Google Scholar]
- 133. Bisgaard H, Simpson A, Palmer CNA, Bønnelykke K, McLean I, Mukhopadhyay S, et al. Gene‐environment interaction in the onset of eczema in infancy: filaggrin loss‐of‐function mutations enhanced by neonatal cat exposure. PLoS Med 2008;5:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Noda S, Krueger JG, Guttman‐Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol 2015;135:324–36. [DOI] [PubMed] [Google Scholar]
- 135. Lønnberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, Thomsen SF. Heritability of psoriasis in a large twin sample. Br J Dermatol 2013;169:412–6. [DOI] [PubMed] [Google Scholar]
- 136. Elmose C, Thomsen SF. Twin studies of atopic dermatitis: interpretations and applications in the filaggrin era. J Allergy 2015;2015:902359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Capon F. The genetic basis of psoriasis. Int J Mol Sci 2017;18:pii: E2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Palmer CNA, Irvine AD, Terron‐Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss‐of‐function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006;38:441–6. [DOI] [PubMed] [Google Scholar]
- 139. Al‐Shobaili HA, Ahmed AA, Alnomair N, Alobead ZA, Rasheed Z. Molecular genetic of atopic dermatitis: an update. Int J Health Sci 2016;10:96–120. [PMC free article] [PubMed] [Google Scholar]
- 140. Thomsen SF, Elmose C, Szecsi PB, Stender S, Kyvik KO, Backer V, et al. Filaggrin gene loss‐of‐function mutations explain discordance of atopic dermatitis within dizygotic twin pairs. Int J Dermatol 2016;55:1341–4. [DOI] [PubMed] [Google Scholar]
- 141. Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi‐ancestry genome‐wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015;47:1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Liang Y, Chang C, Lu Q. The genetics and epigenetics of atopic dermatitis‐filaggrin and other polymorphisms. Clin Rev Allergy Immunol 2016;51:315–28. [DOI] [PubMed] [Google Scholar]
- 143. Yao Y, Richman L, Morehouse C, de los Reyes M, Higgs BW, Boutrin A, et al. Type I interferon: potential therapeutic target for psoriasis? PLoS ONE 2008;3:e2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Tian S, Krueger JG, Li K, Jabbari A, Brodmerkel C, Lowes MA, et al. Meta‐analysis derived (MAD) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS ONE 2012;7:e44274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Suárez‐Fariñas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles K, Cardinale I, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol 2011;127 954–64:e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ewald DA, Malajian D, Krueger JG, Workman CT, Wang T, Tian S, et al. Meta‐analysis derived atopic dermatitis (MADAD) transcriptome defines a robust AD signature highlighting the involvement of atherosclerosis and lipid metabolism pathways. BMC Med Genomics 2015;8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Witte E, Kokolakis G, Witte K, Philipp S, Doecke W‐D, Babel N, et al. IL‐19 is a component of the pathogenetic IL‐23/IL‐17 cascade in psoriasis. J Invest Dermatol 2014;134:2757–67. [DOI] [PubMed] [Google Scholar]
- 148. Solberg SM, Sandvik LF, Eidsheim M, Jonsson R, Bryceson YT, Appel S. Serum cytokine measurements and biological therapy of psoriasis ‐ Prospects for personalized treatment? Scand J Immunol 2018;88:e12725. [DOI] [PubMed] [Google Scholar]
- 149. Zaba LC, Suárez‐Fariñas M, Fuentes‐Duculan J, Nograles KE, Guttman‐Yassky E, Cardinale I, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL‐17 signaling, not immediate response TNF genes. J Allergy Clin Immunol 2009;124 1022–10:e1–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Krueger JG, Fretzin S, Suárez‐Fariñas M, Haslett PA, Phipps KM, Cameron GS, et al. IL‐17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol 2012;130:145–54. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Russell CB, Rand H, Bigler J, Kerkof K, Timour M, Bautista E, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti‐IL‐17 receptor monoclonal antibody. J Immunol 2014;192:3828–36. [DOI] [PubMed] [Google Scholar]
- 152. Sofen H, Smith S, Matheson RT, Leonardi CL, Calderon C, Brodmerkel C, et al. Guselkumab (an IL‐23‐specific mAb) demonstrates clinical and molecular response in patients with moderate‐to‐severe psoriasis. J Allergy Clin Immunol 2014;133:1032–40. [DOI] [PubMed] [Google Scholar]
- 153. Visvanathan S, Baum P, Vinisko R, Schmid R, Flack M, Lalovic B, et al. Psoriatic skin molecular and histopathological profiles following treatment with risankizumab versus ustekinumab. J Allergy Clin Immunol 2018. [Epub ahead of print]. 10.1016/j.jaci.2018.11.042 [DOI] [PubMed] [Google Scholar]
- 154. Paternoster L, Savenije OEM, Heron J, Evans DM, Vonk JM, Brunekreef B, et al. Identification of atopic dermatitis subgroups in children from 2 longitudinal birth cohorts. J Allergy Clin Immunol 2018;141:964–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Thijs JL, de Bruin‐Weller MS, Hijnen D. Current and future biomarkers in atopic dermatitis. Immunol Allergy Clin North Am 2017;37:51–61. [DOI] [PubMed] [Google Scholar]
- 156. Tamagawa‐Mineoka R, Okuzawa Y, Masuda K, Katoh N. Increased serum levels of interleukin 33 in patients with atopic dermatitis. J Am Acad Dermatol 2014;70:882–8. [DOI] [PubMed] [Google Scholar]
- 157. Tintle S, Shemer A, Suárez‐Fariñas M, Fujita H, Gilleaudeau P, Sullivan‐Whalen M, et al. Reversal of atopic dermatitis with narrow‐band UVB phototherapy and biomarkers for therapeutic response. J Allergy Clin Immunol 2011;128 583–93:e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Khattri S, Shemer A, Rozenblit M, Dhingra N, Czarnowicki T, Finney R, et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J Allergy Clin Immunol 2014;133:1626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hamilton JD, Suárez‐Fariñas M, Dhingra N, Cardinale I, Li X, Kostic A, et al. Dupilumab improves the molecular signature in skin of patients with moderate‐to‐severe atopic dermatitis. J Allergy Clin Immunol 2014;134:1293–300. [DOI] [PubMed] [Google Scholar]
- 160. GEO Accession viewer [Internet]. Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120899 [cited 2018 Nov 1].
- 161. Brunner PM, Pavel AB, Khattri S, Leonard A, Malik K, Rose S, et al. Baseline IL‐22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol 2018;143:142–54. [DOI] [PubMed] [Google Scholar]
- 162. Moscaliuc ML, Heller MM, Lee ES, Koo J. Goeckerman therapy: a very effective, yet often forgotten treatment for severe generalized psoriasis. J Dermatolog Treat 2013;24:34–7. [DOI] [PubMed] [Google Scholar]
- 163. Wollenberg A, Schnopp C. Evolution of conventional therapy in atopic dermatitis. Immunol Allergy Clin North Am 2010;30:351–68. [DOI] [PubMed] [Google Scholar]
- 164. Griffiths CE, Powles AV, Leonard JN, Fry L, Baker BS, Valdimarsson H. Clearance of psoriasis with low dose cyclosporin. Br Med J 1986;293:731–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Eedy DJ, Burrows D, Bridges JM, Jones FG. Clearance of severe psoriasis after allogenic bone marrow transplantation. BMJ 1990;300:908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin‐23 rather than interleukin‐12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003;421:744–8. [DOI] [PubMed] [Google Scholar]
- 167. Lowes MA, Kikuchi T, Fuentes‐Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 2008;128:1207–11. [DOI] [PubMed] [Google Scholar]
- 168. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 2004;199:125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL‐23/IL‐17 signaling pathway and the treatment of psoriasis. J Immunol 2018;201:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Walsh JA, Jones H, Mallbris L, Duffin KC, Krueger GG, Clegg DO, et al. The Physician Global Assessment and Body Surface Area composite tool is a simple alternative to the Psoriasis Area and Severity Index for assessment of psoriasis: post hoc analysis from PRISTINE and PRESTA. Psoriasis (Auckl) 2018;8:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Carrera CG, Dapavo P, Malagoli P, Naldi L, Arancio L, Gaiani F, et al. PACE study: real‐life Psoriasis Area and Severity Index (PASI) 100 response with biological agents in moderate‐severe psoriasis. J Dermatolog Treat 2018;29:481–6. [DOI] [PubMed] [Google Scholar]
- 172. Campa M, Mansouri B, Warren R, Menter A. A review of biologic therapies targeting IL‐23 and IL‐17 for use in moderate‐to‐severe plaque psoriasis. Dermatol Ther 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Canavan TN, Elmets CA, Cantrell WL, Evans JM, Elewski BE. Anti‐IL‐17 Medications Used in the Treatment of Plaque Psoriasis and Psoriatic Arthritis: A Comprehensive Review. Am J Clin Dermatol 2016;17:33–47. [DOI] [PubMed] [Google Scholar]
- 174. Menter A, Sobell J, Silverberg JI, Lebwohl M, Rastogi S, Pillai R, et al. Long‐term efficacy of brodalumab for the treatment of moderate‐to‐severe psoriasis: data from a pivotal phase 3 clinical trial. SKIN J Cutaneous Med 2018;2 S1 10.25251/skin.2.supp.9 [DOI] [Google Scholar]
- 175. Brembilla NC, Senra L, Boehncke W‐H. The IL‐17 family of cytokines in psoriasis: IL‐17A and beyond. Front Immunol 2018;9:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Blair HA. Brodalumab: a review in moderate to severe plaque psoriasis. Drugs 2018;78:495–504. [DOI] [PubMed] [Google Scholar]
- 177. Sawyer LM, Cornic L, Levin LÅ, Gibbons C, Møller AH, Jemec GB. Long‐term efficacy of novel therapies in moderate‐to‐severe plaque psoriasis: a systematic review and network meta‐analysis of PASI response. J Eur Acad Dermatol Venereol 2019;33:355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Papp K, Gordon K, Thaçi D, Morita A, Gooderham M, Foley P, et al. Phase 2 trial of selective tyrosine Kinase 2 inhibition in psoriasis. N Engl J Med 2018;379:1313–21. [DOI] [PubMed] [Google Scholar]
- 179. Conrad C, Gilliet M. Psoriasis: from pathogenesis to targeted therapies. Clin Rev Allergy Immunol 2018;54:102–13. [DOI] [PubMed] [Google Scholar]
- 180. Torres T, Romanelli M, Chiricozzi A. A revolutionary therapeutic approach for psoriasis: bispecific biological agents. Expert Opin Investig Drugs 2016;25:751–4. [DOI] [PubMed] [Google Scholar]
- 181. Santibanez JF, Bjelica S. Novel patents targeting interleukin‐17A; implications in cancer and inflammation. Recent Pat Anticancer Drug Discov 2018;13:133–44. [DOI] [PubMed] [Google Scholar]
- 182. Wu X, Demarest SJ. Building blocks for bispecific and trispecific antibodies. Methods 2018;154:3–9. [DOI] [PubMed] [Google Scholar]
- 183. Campa M, Menter A. A review of emerging IL‐17 inhibitors in the treatment of psoriasis focusing on preclinical through phase II studies. Expert Opin Investig Drugs 2016;25:1337–44. [DOI] [PubMed] [Google Scholar]
- 184. Pollock RA, Abji F, Gladman DD. Epigenetics of psoriatic disease: a systematic review and critical appraisal. J Autoimmun 2017;78:29–38. [DOI] [PubMed] [Google Scholar]
- 185. Chandra A, Senapati S, Roy S, Chatterjee G, Chatterjee R. Epigenome‐wide DNA methylation regulates cardinal pathological features of psoriasis. Clin Epigenetics 2018;10:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Ovejero‐Benito MC, Reolid A, Sánchez‐Jiménez P, Saiz‐Rodríguez M, Muñoz‐Aceituno E, Llamas‐Velasco M, et al. Histone modifications associated with biological drug response in moderate‐to‐severe psoriasis. Exp Dermatol 2018;27:1361–71. [DOI] [PubMed] [Google Scholar]
- 187. Løvendorf MB, Mitsui H, Zibert JR, Røpke MA, Hafner M, Dyring‐Andersen B, et al. Laser capture microdissection followed by next‐generation sequencing identifies disease‐related microRNAs in psoriatic skin that reflect systemic microRNA changes in psoriasis. Exp Dermatol 2015;24:187–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hawkes JE, Nguyen GH, Fujita M, Florell SR, Callis Duffin K, Krueger GG, et al. microRNAs in Psoriasis. J Invest Dermatol 2016;136:365–71. [DOI] [PubMed] [Google Scholar]
- 189. Suárez‐Fariñas M, Shah KR, Haider AS, Krueger JG, Lowes MA. Personalized medicine in psoriasis: developing a genomic classifier to predict histological response to Alefacept. BMC Dermatol 2010;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Codoñer FM, Ramírez‐Bosca A, Climent E, Carrión‐Gutierrez M, Guerrero M, Pérez‐Orquín JM, et al. Gut microbial composition in patients with psoriasis. Sci Rep 2018;8:3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Tham EH, Leung DY. Mechanisms by which atopic dermatitis predisposes to food allergy and the atopic march. Allergy Asthma Immunol Res 2019;11:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Belgrave DCM, Granell R, Simpson A, Guiver J, Bishop C, Buchan I, et al. Developmental profiles of eczema, wheeze, and rhinitis: two population‐based birth cohort studies. PLoS Med 2014;11 10:e1001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Leung DYM, Guttman‐Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol 2014;134:769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Reinhold U, Kukel S, Goeden B, Neumann U, Kreysel HW. Functional characterization of skin‐infiltrating lymphocytes in atopic dermatitis. Clin Exp Immunol 1991;86:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Eyerich K, Novak N. Immunology of atopic eczema: overcoming the Th1/Th2 paradigm. Allergy 2013;68:974–82. [DOI] [PubMed] [Google Scholar]
- 196. Gittler JK, Shemer A, Suárez‐Fariñas M, Fuentes‐Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012;130:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. No DJ, Amin M, Egeberg A, Thyssen JP, Wu JJ. Atopic dermatitis 2017: where we were 10‐15 years ago in psoriasis. J Dermatolog Treat 2018;29 1:100–1. [DOI] [PubMed] [Google Scholar]
- 198. Bożek A, Reich A. Assessment of Intra‐ and Inter‐Rater Reliability of Three Methods for Measuring Atopic Dermatitis Severity: EASI, Objective SCORAD, and IGA. Dermatology 2017;233:16–22. [DOI] [PubMed] [Google Scholar]
- 199. Patel K, Singam V, Vakharia PP, Chopra R, Sacotte R, Patel N, et al. Measurement properties of three assessments of burden used in atopic dermatitis in adults. Br J Dermatol 2018. [Epub ahead of print]. 10.1111/bjd.17243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gooderham MJ, Hong HC‐H, Eshtiaghi P, Papp KA. Dupilumab: a review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol 2018;78(3S1):S28–36. [DOI] [PubMed] [Google Scholar]
- 201. Muraro A, Lemanske RF Jr, Hellings PW, Akdis CA, Bieber T, Casale TB, et al. Precision medicine in patients with allergic diseases: airway diseases and atopic dermatitis‐PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 2016;137:1347–58. [DOI] [PubMed] [Google Scholar]
- 202. Silverberg NB, Silverberg JI. Inside out or outside in: does atopic dermatitis disrupt barrier function or does disruption of barrier function trigger atopic dermatitis? Cutis 2015;96:359–61. [PubMed] [Google Scholar]
- 203. McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol 2013;131:280–91. [DOI] [PubMed] [Google Scholar]
- 204. Brown SJ, McLean WHI. One remarkable molecule: filaggrin. J Invest Dermatol 2012;132(3 Pt 2):751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Chalmers JR, Haines RH, Mitchell EJ, Thomas KS, Brown SJ, Ridd M, et al. Effectiveness and cost‐effectiveness of daily all‐over‐body application of emollient during the first year of life for preventing atopic eczema in high‐risk children (The BEEP trial): protocol for a randomised controlled trial. Trials 2017;18:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2011;127:773–86. e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Ryu W‐I, Lee H, Bae HC, Jeon J, Ryu HJ, Kim J, et al. IL‐33 down‐regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J Dermatol Sci 2018;90:313–22. [DOI] [PubMed] [Google Scholar]
- 208. Sugita K, Steer CA, Martinez‐Gonzalez I, Altunbulakli C, Morita H, Castro‐Giner F, et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL‐13 in asthmatic patients. J Allergy Clin Immunol 2018;141:300–10. e11. [DOI] [PubMed] [Google Scholar]
- 209. Kim Y‐E, Cho N, Cheon S, Kim KK. Bortezomib, a proteasome inhibitor, alleviates atopic dermatitis by increasing claudin 1 protein expression. Biochem Biophys Res Commun 2017;493:744–50. [DOI] [PubMed] [Google Scholar]
- 210. Xian M, Wawrzyniak P, Rückert B, Duan S, Meng Y, Sokolowska M, et al. Anionic surfactants and commercial detergents decrease tight junction barrier integrity in human keratinocytes. J Allergy Clin Immunol 2016;138:890–3. e9. [DOI] [PubMed] [Google Scholar]
- 211. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol 2016;1418:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Brunner PM, Israel A, Zhang N, Leonard A, Wen H‐C, Huynh T, et al. Early‐onset pediatric atopic dermatitis is characterized by T2/T17/T22‐centered inflammation and lipid alterations. J Allergy Clin Immunol 2018;141:2094–106. [DOI] [PubMed] [Google Scholar]
- 213. Dyjack N, Goleva E, Rios C, Kim BE, Bin L, Taylor P, et al. Minimally invasive skin tape strip RNA sequencing identifies novel characteristics of the type 2‐high atopic dermatitis disease endotype. J Allergy Clin Immunol 2018;141:1298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Wen H‐C, Czarnowicki T, Noda S, Malik K, Pavel AB, Nakajima S, et al. Serum from Asian patients with atopic dermatitis is characterized by T2/T22 activation, which is highly correlated with nonlesional skin measures. J Allergy Clin Immunol 2018;142:324–8. e11. [DOI] [PubMed] [Google Scholar]
- 215. Thijs JL, Strickland I, Bruijnzeel‐Koomen CAFM, Nierkens S, Giovannone B, Knol EF, et al. Serum biomarker profiles suggest that atopic dermatitis is a systemic disease. J Allergy Clin Immunol 2018;141:1523–6. [DOI] [PubMed] [Google Scholar]
- 216. Noda S, Suárez‐Fariñas M, Ungar B, Kim SJ, de Guzman Strong C, Xu H, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol 2015;136:1254–64. [DOI] [PubMed] [Google Scholar]
- 217. Kaufman BP, Guttman‐Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups‐Variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol 2018;27:340–57. [DOI] [PubMed] [Google Scholar]
- 218. Brunner PM, Guttman‐Yassky E. Racial differences in atopic dermatitis. Ann Allergy Asthma Immunol 2018. [Epub ahead of print]. 10.1016/j.anai.2018.11.015 [DOI] [PubMed] [Google Scholar]
- 219. Czarnowicki T, Krueger JG, Guttman‐Yassky E. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol Pract 2014;2:371–9; quiz 380–1. [DOI] [PubMed] [Google Scholar]
- 220. Suárez‐Fariñas M, Fuentes‐Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease‐related genes. J Invest Dermatol 2011;131:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Shimizu H. Shimizu's Dermatology. Hoboken, NJ: John Wiley & Sons, 2017: 664. [Google Scholar]
- 222. Esaki H, Ewald DA, Ungar B, Rozenblit M, Zheng X, Xu H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Allergy Clin Immunol 2015;135:153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Rodríguez E, Baurecht H, Wahn AF, Kretschmer A, Hotze M, Zeilinger S, et al. An integrated epigenetic and transcriptomic analysis reveals distinct tissue‐specific patterns of DNA methylation associated with atopic dermatitis. J Invest Dermatol 2014;134:1873–83. [DOI] [PubMed] [Google Scholar]
- 224. Weiss ST. Eat dirt–the hygiene hypothesis and allergic diseases. N Engl J Med 2002;347:930–1. [DOI] [PubMed] [Google Scholar]
- 225. Laforest‐Lapointe I, Arrieta M‐C. Patterns of early‐life gut microbial colonization during human immune development: an ecological perspective. Front Immunol 2017;8:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lee M‐J, Kang M‐J, Lee S‐Y, Lee E, Kim K, Won S, et al. Perturbations of gut microbiome genes in infants with atopic dermatitis according to feeding type. J Allergy Clin Immunol 2018;141:1310–9. [DOI] [PubMed] [Google Scholar]
- 227. Brüssow H. Turning the inside out: the microbiology of atopic dermatitis. Environ Microbiol 2016;18:2089–102. [DOI] [PubMed] [Google Scholar]
- 228. Simpson EL, Villarreal M, Jepson B, Rafaels N, David G, Hanifin J, et al. Patients with atopic dermatitis colonized with staphylococcus aureus have a distinct phenotype and endotype. J Invest Dermatol 2018;138:2224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012;22:850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Glatz M, Jo J‐H, Kennedy EA, Polley EC, Segre JA, Simpson EL, et al. Emollient use alters skin barrier and microbes in infants at risk for developing atopic dermatitis. PLoS ONE 2018;13:e0192443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Novak N, Gros E, Bieber T, Allam J‐P. Human skin and oral mucosal dendritic cells as “good guys” and “bad guys” in allergic immune responses. Clin Exp Immunol 2010;161:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz‐Planillo R, Hasegawa M, et al. Staphylococcus δ‐toxin induces allergic skin disease by activating mast cells. Nature 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 2002;347:1151–60. [DOI] [PubMed] [Google Scholar]
- 234. Oliva M, Renert‐Yuval Y, Guttman‐Yassky E. The, “omics” revolution: redefining the understanding and treatment of allergic skin diseases. Curr Opin Allergy Clin Immunol 2016;16:469–76. [DOI] [PubMed] [Google Scholar]
- 235. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against and are deficient in atopic dermatitis. Sci Transl Med 2017;9:pii: eaah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Gallo RL. Human skin is the largest epithelial surface for interaction with microbes. J Invest Dermatol 2017;137:1213–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Portugal‐Cohen M, Kohen R. Non‐invasive evaluation of skin cytokines secretion: an innovative complementary method for monitoring skin disorders. Methods 2013;61:63–8. [DOI] [PubMed] [Google Scholar]
- 238. Assfalg M, Bortoletti E, D'Onofrio M, Pigozzi R, Molinari H, Boner AL, et al. An exploratory (1) H‐nuclear magnetic resonance metabolomics study reveals altered urine spectral profiles in infants with atopic dermatitis. Br J Dermatol 2012;166:1123–5. [DOI] [PubMed] [Google Scholar]
- 239. Peroni DG, Bodini A, Corradi M, Coghi A, Boner AL, Piacentini GL. Markers of oxidative stress are increased in exhaled breath condensates of children with atopic dermatitis. Br J Dermatol 2012;166:839–43. [DOI] [PubMed] [Google Scholar]
- 240. Bandyopadhyay S, Connolly SE, Jabado O, Ye J, Kelly S, Maldonado MA, et al. Identification of biomarkers of response to abatacept in patients with SLE using deconvolution of whole blood transcriptomic data from a phase IIb clinical trial. Lupus Sci Med 2017;4:e000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Chen S‐H, Kuo W‐Y, Su S‐Y, Chung W‐C, Ho J‐M, Lu HH‐S, et al. A gene profiling deconvolution approach to estimating immune cell composition from complex tissues. BMC Bioinformatics 2018;19(Suppl 4):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Abbas AR, Wolslegel K, Seshasayee D, Modrusan Z, Clark HF. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS ONE 2009;4:e6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Laczmanska I, Sasiadek M, Laczmanski L. The comparison between molecular tumour profiling in microdissected and surgical tissue samples. Anticancer Res 2018;38:1415–8. [DOI] [PubMed] [Google Scholar]
- 244. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, et al. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat Immunol 2018;19:1286–98. [DOI] [PubMed] [Google Scholar]
- 246. Shen E, Xie K, Jwo K, Smith J, Mosaed S. Dupilumab‐induced follicular conjunctivitis. Ocul Immunol Inflamm 2018. [Epub ahead of print]. 10.1080/09273948.2018.1533567 [DOI] [PubMed] [Google Scholar]
- 247. MorphoSys's Licensee Janssen Announces Data from the Phase 3 Head‐to‐Head Study ECLIPSE Demonstrating Superiority of Tremfya(R) vs. Cosentyx(R) as Measured by PASI 90 at Week 48 in the Treatment of Plaque Psoriasis [Internet]. MorphoSys AG. 2018. Available from: https://www.morphosys.com/media-investors/media-center/morphosyss-licensee-janssen-announces-data-from-the-phase-3-head-to [cited 2018 Dec 31].
- 248. Harden JL, Johnson‐Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, et al. Humanized anti‐IFN‐γ (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol 2015;135:553–6. [DOI] [PubMed] [Google Scholar]
- 249. Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK‐STAT biology: the present and future of jakinibs. J Leukoc Biol 2018;104:499–514. [DOI] [PubMed] [Google Scholar]
- 250. He H, Guttman‐Yassky E. JAK inhibitors for atopic dermatitis: an update. Am J Clin Dermatol 2018. [Epub ahead of print]. 10.1007/s40257-018-0413-2 [DOI] [PubMed] [Google Scholar]
- 251. Tam HH, Calderon MA, Manikam L, Nankervis H, Núñez IG, Williams HC, et al. Specific allergen immunotherapy for the treatment of atopic eczema: a Cochrane systematic review. Allergy 2016;71:1345–56. [DOI] [PubMed] [Google Scholar]
- 252. Clowry J, Irvine AD, McLoughlin RM. Next‐generation anti‐Staphylococcus aureus vaccines: a potential new therapeutic option for atopic dermatitis? J Allergy Clin Immunol 2018;143:78–81. [DOI] [PubMed] [Google Scholar]
- 253. Ghosh D, Bernstein JA, Khurana Hershey GK, Rothenberg ME, Mersha TB. Leveraging multilayered “OMICS” data for atopic dermatitis: a road map to precision medicine. Front Immunol 2018;9:2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Janiaud P, Serghiou S, Ioannidis JPA. New clinical trial designs in the era of precision medicine: an overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat Rev 2018;73:20–30. [DOI] [PubMed] [Google Scholar]
- 255. Brock A, Huang S. Precision oncology: between vaguely right and precisely wrong. Cancer Res 2017;77:6473–9. [DOI] [PubMed] [Google Scholar]
- 256. Chung KF. Precision medicine in asthma: linking phenotypes to targeted treatments. Curr Opin Pulm Med 2018;24:4–10. [DOI] [PubMed] [Google Scholar]
- 257. Greis C, Meier Zürcher C, Djamei V, Moser A, Lautenschlager S, Navarini AA. Unmet digital health service needs in dermatology patients. J Dermatolog Treat 2018;29:643–7. [DOI] [PubMed] [Google Scholar]
- 258. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T‐cell autoantigen in psoriasis. Nat Commun 2014;5:5621. [DOI] [PubMed] [Google Scholar]
- 259. Clayton K, Vallejo AF, Davies J, Sirvent S, Polak ME. Langerhans Cells‐Programmed by the Epidermis. Front Immunol 2017;8:1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Martinez‐Gonzalez I, Ghaedi M, Steer CA, Mathä L, Vivier E, Takei F. ILC2 memory: recollection of previous activation. Immunol Rev 2018;283:41–53. [DOI] [PubMed] [Google Scholar]
- 261. Streilein JW. Skin‐associated lymphoid tissues (SALT): origins and functions. J Invest Dermatol 1983;80(Suppl):12s–6s. [DOI] [PubMed] [Google Scholar]
- 262. Vakharia PP, Silverberg JI. Monoclonal Antibodies for Atopic Dermatitis: Progress and Potential. BioDrugs 2017;31:409–22. [DOI] [PubMed] [Google Scholar]
- 263. Leung DY. Atopic dermatitis: new insights and opportunities for therapeutic intervention. J Allergy Clin Immunol 2000;105:860–76. [DOI] [PubMed] [Google Scholar]
- 264. Leung DYM, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest 2004;113 5:651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Paller AS, Kabashima K, Bieber T. Therapeutic pipeline for atopic dermatitis: end of the drought? J Allergy Clin Immunol 2017;140:633–43. [DOI] [PubMed] [Google Scholar]
- 266. Lee DE, Clark AK, Tran KA, Shi VY. New and emerging targeted systemic therapies: a new era for atopic dermatitis. J Dermatolog Treat 2018;29:364–74. [DOI] [PubMed] [Google Scholar]
- 267. Brunner PM, Guttman‐Yassky E, Leung DYM. The immunology of atopic dermatitis and its reversibility with broad‐spectrum and targeted therapies. J Allergy Clin Immunol 2017;139(4S):S65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Brunner PM, Leung DYM, Guttman‐Yassky E. Immunologic, microbial, and epithelial interactions in atopic dermatitis. Ann Allergy Asthma Immunol 2018;120:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. A Study to Assess Efficacy, Safety, Tolerability and Pharmacokinetics (PK)/Pharmacodynamics (PD) of MOR106 in Subjects With Moderate to Severe Atopic Dermatitis ‐ Full Text View ‐ ClinicalTrials.gov [Internet]. Available from: https://clinicaltrials.gov/ct2/show/NCT03568071 [cited 2018 Oct 27]
- 270. Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J‐L, Cooper AM, et al. IL‐12 and IL‐23 cytokines: from discovery to targeted therapies for immune‐mediated inflammatory diseases. Nat Med 2015;21:719–29. [DOI] [PubMed] [Google Scholar]
- 271. Koutruba N, Emer J, Lebwohl M. Review of ustekinumab, an interleukin‐12 and interleukin‐23 inhibitor used for the treatment of plaque psoriasis. Ther Clin Risk Manag 2010;6:123–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Silverberg JI, Kantor R. The Role of Interleukins 4 and/or 13 in the Pathophysiology and Treatment of Atopic Dermatitis. Dermatol Clin 2017;35:327–34. [DOI] [PubMed] [Google Scholar]
- 273. Daines MO, Tabata Y, Walker BA, Chen W, Warrier MR, Basu S, et al. Level of expression of IL‐13R alpha 2 impacts receptor distribution and IL‐13 signaling. J Immunol 2006;176:7495–501. [DOI] [PubMed] [Google Scholar]
- 274. Hermanns HM. Oncostatin M and interleukin‐31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015;26:545–58. [DOI] [PubMed] [Google Scholar]
- 275. Kabashima K, Furue M, Hanifin JM, Pulka G, Wollenberg A, Galus R, et al. Nemolizumab in patients with moderate‐to‐severe atopic dermatitis: randomized, phase II, long‐term extension study. J Allergy Clin Immunol 2018;142:1121–30. e7. [DOI] [PubMed] [Google Scholar]
- 276. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality‐controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res 2017;45 D1:D362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Guttman‐Yassky E, Suárez‐Fariñas M, Chiricozzi A, Nograles KE, Shemer A, Fuentes‐Duculan J, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol 2009;124:1235–44. e58. [DOI] [PubMed] [Google Scholar]
- 278. Martel BC, Litman T, Hald A, Norsgaard H, Lovato P, Dyring‐Andersen B, et al. Distinct molecular signatures of mild extrinsic and intrinsic atopic dermatitis. Exp Dermatol 2016;25:453–9. [DOI] [PubMed] [Google Scholar]
- 279. Suárez‐Fariñas M, Ungar B, Correa da Rosa J, Ewald DA, Rozenblit M, Gonzalez J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol 2015;135:1218–27. [DOI] [PubMed] [Google Scholar]
- 280. Ewald DA, Noda S, Oliva M, Litman T, Nakajima S, Li X, et al. Major differences between human atopic dermatitis and murine models, as determined by using global transcriptomic profiling. J Allergy Clin Immunol 2017;139:562–71. [DOI] [PubMed] [Google Scholar]
- 281. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappaB pathways. Nat Genet 2009;41:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Joyce CE, Zhou X, Xia J, Ryan C, Thrash B, Menter A, et al. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum Mol Genet 2011;20:4025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Suárez‐Fariñas M, Li K, Fuentes‐Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate‐to‐severe psoriasis. J Invest Dermatol 2012;132:2552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Mitsui H, Suárez‐Fariñas M, Belkin DA, Levenkova N, Fuentes‐Duculan J, Coats I, et al. Combined use of laser capture microdissection and cDNA microarray analysis identifies locally expressed disease‐related genes in focal regions of psoriasis vulgaris skin lesions. J Invest Dermatol 2012;132:1615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Wang CQF, Suárez‐Fariñas M, Nograles KE, Mimoso CA, Shrom D, Dow ER, et al. IL‐17 induces inflammation‐associated gene products in blood monocytes, and treatment with ixekizumab reduces their expression in psoriasis patient blood. J Invest Dermatol 2014;134:2990–3. [DOI] [PubMed] [Google Scholar]
- 286. Krueger J, Clark JD, Suárez‐Fariñas M, Fuentes‐Duculan J, Cueto I, Wang CQ, et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: a randomized phase 2 study. J Allergy Clin Immunol 2016;137:1079–90. [DOI] [PubMed] [Google Scholar]
- 287. Correa da Rosa J, Kim J, Tian S, Tomalin LE, Krueger JG, Suárez‐Fariñas M. Shrinking the psoriasis assessment gap: early gene‐expression profiling accurately predicts response to long‐term treatment. J Invest Dermatol 2017;137:305–12. [DOI] [PubMed] [Google Scholar]
- 288. Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, et al. Transcriptome analysis of psoriasis in a large case‐control sample: RNA‐seq provides insights into disease mechanisms. J Invest Dermatol 2014;134:1828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Quaranta M, Knapp B, Garzorz N, Mattii M, Pullabhatla V, Pennino D, et al. Intraindividual genome expression analysis reveals a specific molecular signature of psoriasis and eczema. Sci Transl Med 2014;6:244ra90. [DOI] [PubMed] [Google Scholar]
- 290. D'Erme AM, Wilsmann‐Theis D, Wagenpfeil J, Hölzel M, Ferring‐Schmitt S, Sternberg S, et al. IL‐36γ (IL‐1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol 2015;135:1025–32. [DOI] [PubMed] [Google Scholar]
- 291. Bissonnette R, Suárez‐Fariñas M, Li X, Bonifacio KM, Brodmerkel C, Fuentes‐Duculan J, et al. Based on Molecular Profiling of Gene Expression, Palmoplantar Pustulosis and Palmoplantar Pustular Psoriasis Are Highly Related Diseases that Appear to Be Distinct from Psoriasis Vulgaris. PLoS ONE 2016;11:e0155215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Xing X, Liang Y, Sarkar MK, Wolterink L, Swindell WR, Voorhees JJ, et al. IL‐17 responses are the dominant inflammatory signal linking inverse, erythrodermic, and chronic plaque psoriasis. J Invest Dermatol 2016;136:2498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Zhou F, Wang W, Shen C, Li H, Zuo X, Zheng X, et al. Epigenome‐wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol 2016;136:779–87. [DOI] [PubMed] [Google Scholar]
- 294. Vakharia PP, Silverberg JI. New therapies for atopic dermatitis: additional treatment classes. J Am Acad Dermatol 2018;78(3S1):S76–83. [DOI] [PubMed] [Google Scholar]
- 295. Smith SH, Jayawickreme C, Rickard DJ, Nicodeme E, Bui T, Simmons C, et al. Tapinarof is a natural Ahr agonist that resolves skin inflammation in mice and humans. J Invest Dermatol 2017;137:2110–9. [DOI] [PubMed] [Google Scholar]
- 296. Bouma G, Zamuner S, Hicks K, Want A, Oliveira J, Choudhury A, et al. CCL20 neutralization by a monoclonal antibody in healthy subjects selectively inhibits recruitment of CCR6 cells in an experimental suction blister. Br J Clin Pharmacol 2017;83:1976–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Molfino NA, Gossage D, Kolbeck R, Parker JM, Geba GP. Molecular and clinical rationale for therapeutic targeting of interleukin‐5 and its receptor. Clin Exp Allergy 2012;42:712–37. [DOI] [PubMed] [Google Scholar]
- 298. Holm JG, Agner T, Sand C, Thomsen SF. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol 2017;56:18–26. [DOI] [PubMed] [Google Scholar]
- 299. Kaufman BP, Alexis AF. Biologics and small molecule agents in allergic and immunologic skin diseases. Curr Allergy Asthma Rep 2018;18:55. [DOI] [PubMed] [Google Scholar]
- 300. Gandhi NA, Bennett BL, Graham NMH, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov 2016;15:35–50. [DOI] [PubMed] [Google Scholar]
- 301. Pan Y, Xu L, Qiao J, Fang H. A systematic review of ustekinumab in the treatment of atopic dermatitis. J Dermatolog Treat 2018;29:539–41. [DOI] [PubMed] [Google Scholar]
- 302. Yiu ZZ, Warren RB. Ustekinumab for the treatment of psoriasis: an evidence update. Semin Cutan Med Surg 2018;37:143–7. [DOI] [PubMed] [Google Scholar]
- 303. Thaçi D, Blauvelt A, Reich K, Tsai T‐F, Vanaclocha F, Kingo K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol 2015;73:400–9. [DOI] [PubMed] [Google Scholar]
- 304. Wollenberg A, Howell MD, Guttman‐Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti‐IL‐13 mAb. J Allergy Clin Immunol 2018;143:135–41. [DOI] [PubMed] [Google Scholar]
- 305. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taïeb A, et al. Efficacy and safety of lebrikizumab (an anti‐IL‐13 monoclonal antibody) in adults with moderate‐to‐severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo‐controlled phase II trial (TREBLE). J Am Acad Dermatol 2018;78:863–71. e11. [DOI] [PubMed] [Google Scholar]
- 306. Griffiths CEM, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐3): results from two phase 3 randomised trials. Lancet 2015;386:541–51. [DOI] [PubMed] [Google Scholar]
- 307. Papp KA, Merola JF, Gottlieb AB, Griffiths CEM, Cross N, Peterson L, et al. Dual neutralization of both interleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12‐week randomized, double‐blinded, placebo‐controlled phase 2b trial. J Am Acad Dermatol 2018;79:277–86. e10. [DOI] [PubMed] [Google Scholar]
- 308. Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, et al. Neutralization of IL‐17C Reduces Skin Inflammation in Mouse Models of Psoriasis and Atopic Dermatitis. J Invest Dermatol 2018;138:1555–63. [DOI] [PubMed] [Google Scholar]
- 309. Machado Á, Torres T. Guselkumab for the treatment of psoriasis. BioDrugs 2018;32:119–28. [DOI] [PubMed] [Google Scholar]
- 310. Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 2018;392:650–61. [DOI] [PubMed] [Google Scholar]
- 311. Chan TC, Hawkes JE, Krueger JG. Interleukin 23 in the skin: role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther Adv Chronic Dis 2018;9:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Hamann CR, Thyssen JP. Monoclonal antibodies against interleukin 13 and interleukin 31RA in development for atopic dermatitis. J Am Acad Dermatol 2018;78(3S1):S37–42. [DOI] [PubMed] [Google Scholar]
- 313. Cotter DG, Schairer D, Eichenfield L. Emerging therapies for atopic dermatitis: JAK inhibitors. J Am Acad Dermatol 2018;78(3S1):S53–62. [DOI] [PubMed] [Google Scholar]
- 314. Cinats A, Heck E, Robertson L. Janus kinase inhibitors: a review of their emerging applications in dermatology. Skin Therapy Lett 2018;23:5–9. [PubMed] [Google Scholar]
- 315. Almutairi N, Nour TM, Hussain NH. Janus kinase inhibitors for the treatment of severe alopecia areata: an open‐label comparative study. Dermatology 2019;235:130–6. [DOI] [PubMed] [Google Scholar]
- 316. Joshipura D, Alomran A, Zancanaro P, Rosmarin D. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib: A 32‐week open‐label extension study with optional narrow‐band ultraviolet B. J Am Acad Dermatol 2018;78:1205–7. e1. [DOI] [PubMed] [Google Scholar]
- 317. Guttman‐Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate‐to‐severe atopic dermatitis: a phase 2 parallel, double‐blinded, randomized placebo‐controlled multiple‐dose study. J Am Acad Dermatol 2018. [Epub ahead of print]. 10.1016/j.jaad.2018.01.018 [DOI] [PubMed] [Google Scholar]
- 318. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate‐to‐severe psoriasis. Br J Dermatol 2016;174:1266–76. [DOI] [PubMed] [Google Scholar]
- 319. Azevedo A, Torres T. Tofacitinib: a new oral therapy for psoriasis. Clin Drug Investig 2018;38:101–12. [DOI] [PubMed] [Google Scholar]
- 320. Banfield C, Scaramozza M, Zhang W, Kieras E, Page KM, Fensome A, et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of a TYK2/JAK1 inhibitor (PF‐06700841) in healthy subjects and patients with plaque psoriasis. J Clin Pharmacol 2018;58:434–47. [DOI] [PubMed] [Google Scholar]
- 321. Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE‐052, a Janus kinase inhibitor, in Japanese adult patients with moderate‐to‐severe atopic dermatitis: a phase II, multicentre, randomized, vehicle‐controlled clinical study. Br J Dermatol 2018;178:424–32. [DOI] [PubMed] [Google Scholar]
- 322. Bonchak JG, Swerlick RA. Emerging therapies for atopic dermatitis: TRPV1 antagonists. J Am Acad Dermatol 2018;78(3S1):S63–6. [DOI] [PubMed] [Google Scholar]
- 323. Tidwell WJ, Fowler JF Jr. T‐cell inhibitors for atopic dermatitis. J Am Acad Dermatol 2018;78(3S1):S67–70. [DOI] [PubMed] [Google Scholar]
- 324. Abrouk M, Farahnik B, Zhu TH, Nakamura M, Singh R, Lee K, et al. Apremilast treatment of atopic dermatitis and other chronic eczematous dermatoses. J Am Acad Dermatol 2017;77:177–80. [DOI] [PubMed] [Google Scholar]
- 325. Pincelli C, Schafer PH, French LE, Augustin M, Krueger JG. Mechanisms underlying the clinical effects of apremilast for psoriasis. J Drugs Dermatol 2018;17:835–40. [PubMed] [Google Scholar]
- 326. Yosipovitch G, Gold LF, Lebwohl MG, Silverberg JI, Tallman AM, Zane LT. Early relief of pruritus in atopic dermatitis with crisaborole ointment, a non‐steroidal, phosphodiesterase 4 inhibitor. Acta Derm Venereol 2018;98:484–9. [DOI] [PubMed] [Google Scholar]
- 327. Lee EB, Lebwohl MG, Wu JJ. Treatment of psoriasis with crisaborole. J Dermatolog Treat 2018. [Epub ahead of print]. 10.1080/09546634.2018.1480747 [DOI] [PubMed] [Google Scholar]
- 328. Zebda R, Paller AS. Phosphodiesterase 4 inhibitors. J Am Acad Dermatol 2018;78(3S1):S43–52. [DOI] [PubMed] [Google Scholar]
- 329. Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, et al. Discovery of a first‐in‐class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem 2017;60:1247–61. [DOI] [PubMed] [Google Scholar]
- 330. Chima M, Lebwohl M. TNF inhibitors for psoriasis. Semin Cutan Med Surg 2018;37:134–42. [DOI] [PubMed] [Google Scholar]
- 331. Mease PJ, Genovese MC, Weinblatt ME, Peloso PM, Chen K, Othman AA, et al. Phase 2 study of ABT‐122, a TNF‐ and IL‐17A‐targeted dual variable domain immunoglobulin, in psoriatic arthritis with inadequate methotrexate response. Arthritis Rheumatol 2018;70:1778–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Silacci M, Lembke W, Woods R, Attinger‐Toller I, Baenziger‐Tobler N, Batey S, et al. Discovery and characterization of COVA322, a clinical‐stage bispecific TNF/IL‐17A inhibitor for the treatment of inflammatory diseases. MAbs 2016;8:141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Yun J‐W, Seo JA, Jeong YS, Bae I‐H, Jang W‐H, Lee J, et al. TRPV1 antagonist can suppress the atopic dermatitis‐like symptoms by accelerating skin barrier recovery. J Dermatol Sci 2011;62:8–15. [DOI] [PubMed] [Google Scholar]
- 334. Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic dermatitis is an IL‐13 dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]