

Abstract
Reagents that activate the signaling adaptor stimulator of interferon genes (STING) suppress experimentally induced autoimmunity in murine models of multiple sclerosis and arthritis. In this study, we evaluated STING agonists as potential reagents to inhibit spontaneous autoimmune type I diabetes (T1D) onset in non‐obese diabetic (NOD) female mice. Treatments with DNA nanoparticles (DNPs), which activate STING when cargo DNA is sensed, delayed T1D onset and reduced T1D incidence when administered before T1D onset. DNP treatment elevated indoleamine 2,3 dioxygenase (IDO) activity, which regulates T‐cell immunity, in spleen, pancreatic lymph nodes and pancreas of NOD mice. Therapeutic responses to DNPs were partially reversed by inhibiting IDO and DNP treatment synergized with insulin therapy to further delay T1D onset and reduce T1D incidence. Treating pre‐diabetic NOD mice with cyclic guanyl‐adenyl dinucleotide (cGAMP) to activate STING directly delayed T1D onset and stimulated interferon‐αβ (IFN‐αβ), while treatment with cyclic diguanyl nucleotide (cdiGMP) did not delay T1D onset or induce IFN‐αβ in NOD mice. DNA sequence analyses revealed that NOD mice possess a STING polymorphism that may explain differential responses to cGAMP and cdiGMP. In summary, STING agonists attenuate T1D progression and DNPs enhance therapeutic responses to insulin therapy.
Keywords: indoleamine 2,3 dioxygenase; stimulator of interferon genes; type I diabetes
Abbreviations
- 1‐MT
1‐methyl‐[d]‐tryptophan
- 2′3′‐cGAMP
cyclic 2′,3′‐cyclic guanosine monophosphate‐adenosine monophosphate
- cdiGMP
3′,5′‐cyclic dimeric guanosine monophosphate
- CDNs
cyclic dinucleotides
- DNPs
DNA nanoparticles
- IDO
indoleamine 2,3 dioxygenase
- NOD
non‐obese diabetic mouse
- STING
stimulator of interferon genes
- Treg cells
Foxp3‐lineage regulatory CD4 T cells
Introduction
The immunostimulatory properties of DNA are commonly exploited to enhance immune responses to vaccines.1 The discovery of an array of cytosolic DNA sensors that activate the signaling adaptor stimulator of interferon genes (STING) broadens the utility of using DNA to improve vaccine efficacy, as these DNA sensors are expressed by most mammalian cells and detect many forms of DNA.2, 3 In contrast, the microbial DNA sensor Toll‐like receptor 9 is expressed only by innate immune cells specialized to sense microbial infections. Recent studies linked some systemic autoimmune syndromes to defective DNA degradation and gain‐of‐function STING mutations.4, 5, 6, 7 Moreover, mice with defective DNA‐degrading enzymes succumbed to spontaneous, lethal, hyper‐inflammatory syndromes due to constitutive STING activation and sustained type I interferon (IFN‐I) production that incited autoimmunity.8, 9
Paradoxically, DNA also regulates immunity and autoimmunity as Toll‐like receptor 9 ablation in lupus‐prone mice accelerated autoantibody production.10 Moreover, immune regulatory responses to apoptotic cells were abolished in mice lacking STING, and systemic administration of DNA nanoparticles (DNPs) to mice suppressed T‐cell immunity via STING.11, 12 Regulatory responses to DNPs were dependent on STING/IFN‐I signaling to induce indoleamine 2,3 dioxygenase (IDO) enzyme activity in dendritic cells that activated Foxp3‐lineage regulatory CD4 T (Treg) cells. Consistent with these findings, DNPs also attenuated experimental autoimmune arthritis (AIA) and encephalitis (EAE) in mice and therapeutic responses were dependent on STING/IFN‐I signaling to induce IDO.12, 13 Cyclic dinucleotides (CDNs) that bind to STING directly to induce IFN‐I production also attenuated EAE by inducing IDO.13 In the current study, we evaluated DNPs and CDNs as potential reagents to inhibit progression and onset of autoimmune type I diabetes (T1D) in susceptible non‐obese diabetic (NOD) female mice.
Methods
Mice
NOD/LtJ female mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in a specific pathogen‐free (SPF) facility. The Institutional Animal Care and Use Committee approved all procedures before performing experiments.
Blood glucose level and T1D onset
Blood glucose levels (BGL) were monitored twice a week with a Bayer Contour Next EZ Glucose Meter and strips (Bayer, Whippany, NJ). Diabetes was defined as two consecutive blood glucose level readings of 250 mg/dl.
DNA nanoparticles
The DNPs were prepared as described previously.13 For some experiments, insulin B chain from bovine pancreas (Sigma, St Louis, MO; cat. no. I6383) was dissolved in HCl 0.1 m, diluted to 100 μg/200 μl/injection and mixed with DNPs (200 μl) before injection. Acidified glucose solution (5%) was used as vehicle.
Cyclic dinucleotides
The CDNs with canonical [3′,5′‐cyclic dimeric guanosine monophosphate (3′,3′‐cdiGMP)] or non‐canonical [cyclic 2′,3′‐cyclic guanosine monophosphate‐adenosine monophosphate (2′,5‐cGAMP)] linkages between saccharide moieties were synthesized (YH), dissolved in saline and injected into NOD mice (intravenously).
IDO inhibition
Mice were treated with IDO pathway inhibitor 1‐methyl‐[d]‐tryptophan (1MT; supplied by NewLink Genetics Inc., Ames, IA) in drinking water (2 g/l) as described previously.13 Oral 1MT treatment was started 1 day before initial DNP treatments and was continued until experiment end‐points.
Type I IFN bioassay
Serum was collected to assess IFN‐αβ levels as described elsewhere.14 In brief, serially diluted serum samples were incubated overnight with NCTC929 cells and 2 mg/ml anti‐IFN‐γ monoclonal antibody (clone XMG1.2; BD Biosciences, San Jose, CA), and then infected with vesicular stomatitis virus. After 3 days, viable cells were stained with crystal violet and quantified by light absorption at 595 nm using a plate reader.
IDO enzyme activity
IDO activity was estimated in tissues from NOD mice as described previously.12 In brief, tissues were homogenized in phosphate‐buffered saline at a protein concentration of 50–100 mg/ml. Cell‐free homogenates were added to IDO enzyme cocktails and kynurenine generated after 2 hr was measured by high‐performance liquid chromatography.
Cytokine assays
Spleens from NOD female mice were snap‐frozen and homogenized in RIPA‐1640 buffer (Sigma; cat. no. R0278) containing EDTA/EGTA (5 mm/5 mm) and protease inhibitors (Problock™, Goldbio, St Louis, MO; cat. no. GB‐334‐20), then centrifuged (10 000 g) and the protein concentration was measured (Coomassie Protein Assay Reagent, Thermo Scientific, Waltham, MA; cat. no. 1856209) and adjusted to 500 µg/ml before using a multiplex (26‐plex) assay to detect mouse cytokines and chemokines (Affymetrix, Santa Clara, CA; cat. no. EPX260‐26088‐91). Data were acquired using Luminex 200 reader and bio‐plex manager software (Bio‐Rad, Hercules, CA).
DNA sequencing
The STING cDNA (B6) sequence was obtained from the Ensemble Genome Browser (www.ensemble.org). RNA from NOD spleen was extracted using TRIzol (Invitrogen, Carlsbad, CA) and cDNA was generated using Superscript III Reverse Transcriptase (Invitrogen). The coding region of STING cDNA with some flanking sequence was amplified using AccuPrime Taq DNA polymerase (Invitrogen) and sequenced (GRU Genomics Core laboratory, Georgia Cancer Center, Augusta University, Augusta, Georgia).
Statistical analyses
Data are expressed as means ± SEM. Statistical significance was evaluated using the Log‐rank test (for T1D incidence) or two‐tailed Student’s t‐test (all other analyses). Statistical tests were performed with PRISM software (GraphPad, San Diego, CA), with significance defined as *P < 0·05, **P < 0·01 and ***P < 0·001.
Results
DNPs delay T1D onset and reduce T1D incidence in NOD mice
Pre‐diabetic NOD female mice (age 4 weeks) were treated with DNPs for 4 weeks as described in Methods. Previously, we showed that polyethylenimine or cargo DNA alone had no impact on incidence or severity of EAE in B6 mice.13 Blood glucose levels were monitored weekly to detect T1D onset, defined as consecutive hyperglycemic readings (>250 mg/dl). As expected, T1D began to manifest from age 16 weeks in vehicle‐treated NOD mice and T1D incidence rose to 90% when mice were 35 weeks old (Fig. 1a). DNP treatments delayed T1D progression significantly, as all mice were disease‐free until age 27 weeks and T1D incidence at age 35 weeks was reduced (Fig. 1a). Hence, treating NOD female mice in the early pre‐diabetic period extended the T1D‐free period and reduced T1D incidence.
Figure 1.
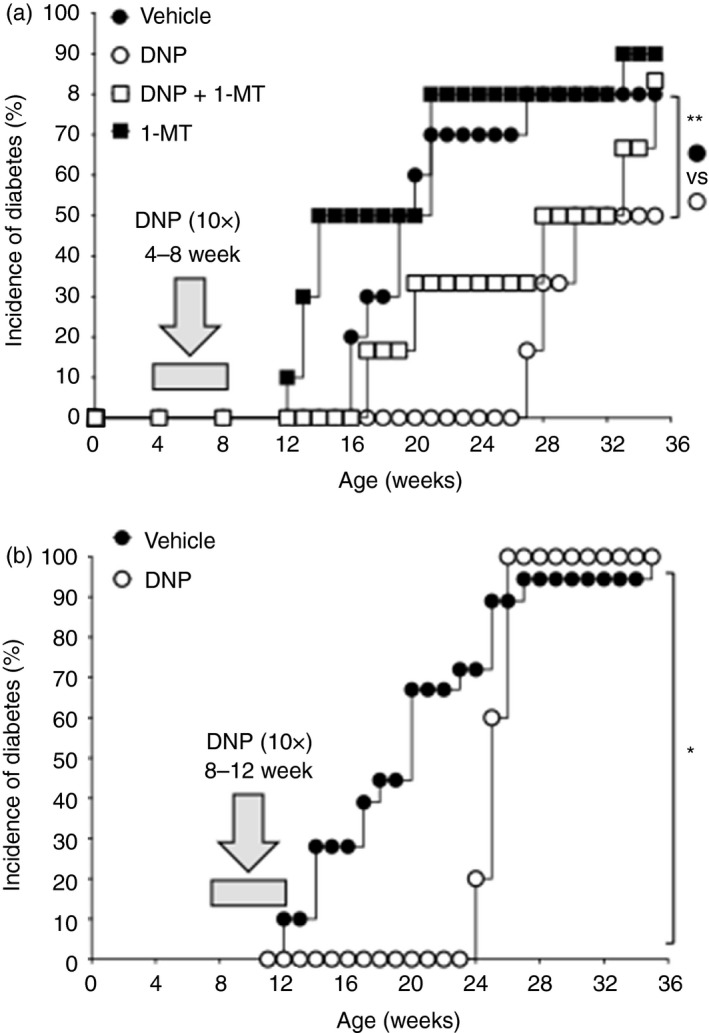
DNA nanoparticle (DNP) treatment inhibits type 1 diabetes (T1D) progression in non‐obese diabetic (NOD) mice. (a) Pre‐diabetic NOD female mice were given DNP treatment or vehicle (open or closed circles) from age 4–8 weeks. Blood glucose levels (BGL) were monitored until mice were age 35 weeks or at T1D onset (BGL > 250 mg/dl) and T1D incidence was plotted. Other mice were given oral 1‐methyl‐[d]‐tryptophan (1‐MT) only (closed squares) or DNPs and 1‐MT (open squares) and BGL was monitored (n = 9 to n = 12/group). (b) Pre‐diabetic NOD female mice (age 8 weeks) were given DNPs or vehicle for 4 weeks. BGLs were monitored until age 35 weeks or at T1D onset, and T1D incidence was plotted. (n = 9 to n = 12/group). Data are representative of two independent experiments in which comparable outcomes were obtained. Statistical significance was evaluated by Log‐rank test, **P < 0·01, *P < 0·05.
Therapeutic responses to DNPs are partially IDO‐dependent
To assess if IDO mediated therapeutic responses to DNPs, female NOD mice were given drinking water containing the IDO pathway inhibitor 1‐MT15 from age 4 weeks. Onset of T1D manifested from age 17 weeks in ~30% of DNP‐treated mice given 1‐MT, an onset time comparable with control NOD mice and earlier than in mice treated with DNPs only (Fig. 1a). 1‐MT treatment also abolished reduced T1D incidence following DNP treatment. Some NOD mice (~50%) given 1‐MT alone developed T1D faster than mice given vehicle, though this effect was not statistically significant (Fig. 1a). Hence, DNP therapeutic responses were reversed partially by 1‐MT, indicating that optimal therapeutic responses to DNPs depended on IDO activity.
DNPs inhibit T1D progression in older NOD female mice
Next, we assessed the effects of administering DNPs to older pre‐diabetic NOD mice from age 8–12 weeks. Vehicle‐treated mice developed T1D from age 12 weeks (Fig. 1b). In contrast, mice treated with DNPs (see Methods) remained T1D free until age 24 weeks (Fig. 1b). However, T1D developed rapidly after 24 weeks and T1D incidence was comparable with that in vehicle‐treated mice (>90%) 2 weeks later.
DNPs stimulate IFN‐I and IDO expression in NOD mice
Previously, we reported that DNPs stimulated STING‐dependent IFN‐I production, which induced IDO‐dependent regulatory phenotypes in a subset of dendritic cells from C57BL/6 (B6) mice.11, 12 To assess if NOD female mice responded similarly, pre‐diabetic NOD female mice age 8 weeks were treated once with DNPs or vehicle, and levels of bioactive IFN‐I in serum and IDO enzyme activity in tissues were evaluated after 3 hr and 24 hr, respectively. DNP treatment stimulated rapid increase in serum IFN‐I (Fig. 2a) and levels of induced IFN‐I were comparable with levels observed in DNP‐treated B6 mice.12 DNP treatment also stimulated IDO enzyme activity in pancreas, spleen and pancreatic lymph nodes of NOD mice (Fig. 2b–d), as IDO activity was threefold to sixfold higher than basal levels in comparable tissues from vehicle‐treated mice. The DNPs stimulated IFN‐I production and increased IDO activity in lymphoid and pancreatic tissues of NOD mice.
Figure 2.
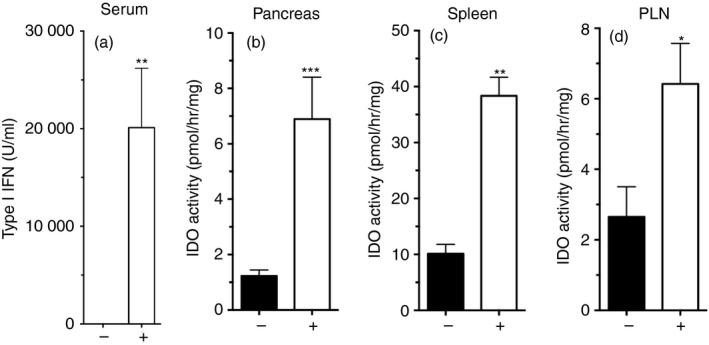
DNA nanoparticle (DNP) treatment induces interferon type I (IFN‐I) and indoleamine 2,3‐dioxygenase (IDO) activity in non‐obese diabetic (NOD) mice. Pre‐diabetic NOD female mice (age 8 weeks) were treated with DNP or vehicle and 24 hr later levels of serum IFN‐I (a) and IDO activity in pancreas (b), spleen (c) and pancreatic lymph nodes (d, PLN) were determined. (n = 3–6/group). Data are representative of five independent experiments. Statistical significance was determined by two‐tailed unpaired Student’s t‐test; *P < 0·05, **P < 0·01, ***P < 0·001.
DNP and insulin therapy synergize to inhibit T1D progression and reduce incidence
As previous studies have shown the adjuvant effects of insulin B chain (InsB), and peptides derived from InsB, in combination with immunomodulatory therapies to promote robust anti‐diabetogenic responses,16, 17, 18, 19, 20 we next tested if co‐treatments with InsB and DNPs synergized in late‐stage, pre‐diabetic NOD female mice relative to monotherapies with InsB or DNPs. NOD mice (age 12 weeks) were treated with vehicle, DNPs, InsB or DNPs + InsB, and monitored to assess T1D onset. DNP treatment from age 12–20 weeks delayed T1D onset until mice were 23 weeks old (Fig. 3a, open diamonds) and T1D incidence was slightly (but not significantly) lower than in vehicle‐treated mice at experiment end‐points (45 weeks). Systemic InsB treatment alone did not delay T1D onset, though T1D incidence was slightly (but not significantly) lower relative to vehicle‐treated control mice (Fig. 3a) at experiment end‐points. In contrast, combined treatment with DNP + InsB delayed T1D onset by 4–6 weeks, and 50% of NOD mice in this group remained T1D free until experiment end‐points at age 45 weeks (Fig. 3a). Hence, combined treatments with DNPs and InsB further attenuate T1D progression and reduce T1D incidence relative to monotherapies using these reagents.
Figure 3.
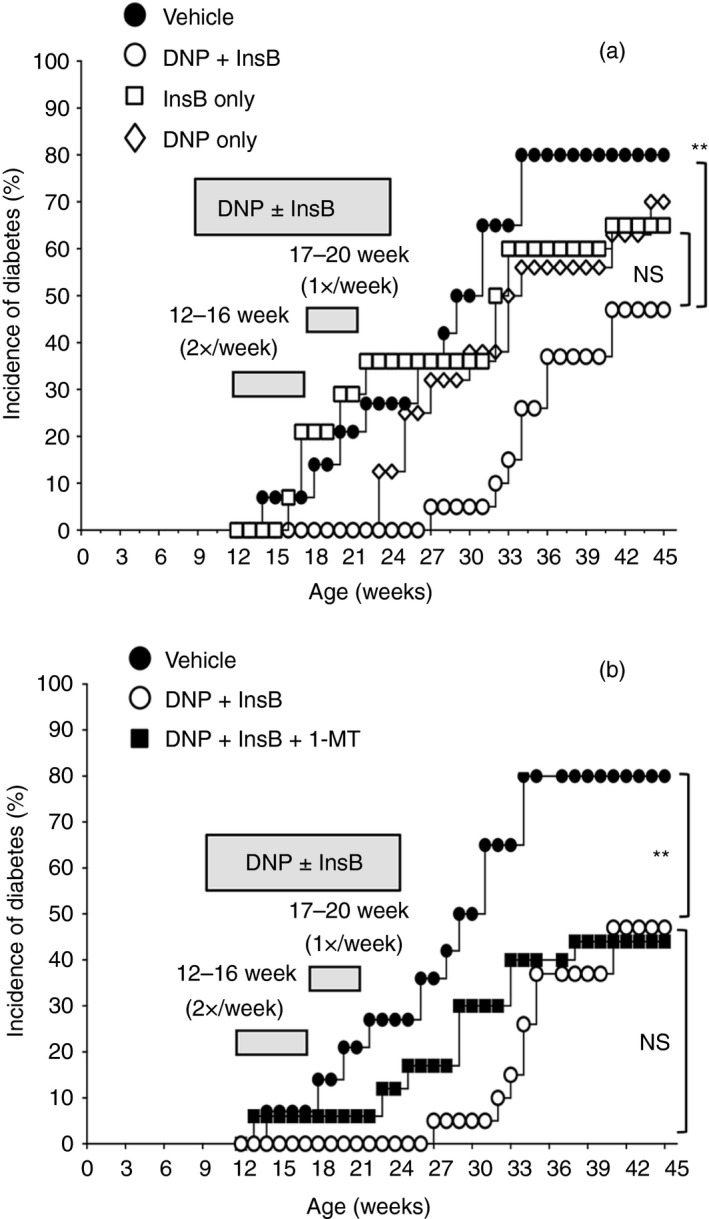
DNA nanoparticle (DNP) and insulin treatments synergize to restrain type 1 diabetes (T1D) progression and reduce incidence in non‐obese diabetic (NOD) mice. (a) Pre‐diabetic NOD female mice (age 12 weeks) were given vehicle (closed circles), DNP plus insulin B chain (InsB) treatment (open circles), InsB (squares), or DNPs only (diamonds) from age 12–20 weeks. (b) Other NOD mice were given oral 1‐methyl‐[d]‐tryptophan (1‐MT) plus InsB plus DNP (closed squares). Blood glucose levels (BGLs) were monitored until mice were age 45 weeks or at T1D onset (BGL > 250 mg/dl) and T1D incidence was plotted (n = 20/group). Two data sets from panel (a) are replicated in panel (b) to facilitate comparison. Data are representative of two independent experiments that generated comparable outcomes. Statistical significance was evaluated using the Log‐rank test, **P < 0·01, NS, not significant.
We also evaluated the effects of inhibiting IDO on therapeutic responses to combined DNP + InsB treatments. Oral 1‐MT given from age 12 weeks partially reversed the therapeutic effects of combined DNP + InsB treatments on T1D progression, as T1D onset started to manifest earlier (from age 23 weeks) relative to DNP + InsB‐treated mice (Fig. 3b). However, 1‐MT treatment did not increase T1D incidence in DNP + InsB‐treated mice, suggesting that therapeutic responses to combined DNP + InsB treatment prevented T1D progression through an IDO‐independent mechanism in some NOD mice.
CDNs that activate STING slow T1D progression in NOD mice
Cyclic dinucleotides are bioactive molecules that bind to the signaling adaptor STING in many mammalian cell types to stimulate IFN‐I production.21, 22, 23, 24 Microorganisms such as Listeria monocytogenes synthesize the canonical CDN cdiGMP. The mammalian cytosolic DNA sensor cyclic GAMP synthase (cGAS) generates a different (non‐canonical) CDN isoform, cGAMP, when DNA is sensed.24 Importantly, these archetypical microbial and mammalian CDNs have distinct phosphodiester linkages, c[G(2′5′)pG(2′5′)p for cdiGMP and c[G(2′5′)pA(3′5′)p] for cGAMP.22 Like DNPs, systemic cdiGMP treatment attenuated EAE progression and severity in B6 mice by way of an immune regulatory pathway dependent on STING/IFN‐I signaling to induce IDO in dendritic cells, which activated Treg cells.11, 13 To test if CDNs attenuated T1D progression, pre‐diabetic NOD female mice (age 8 weeks) were treated two or three times weekly with synthetic cdiGMP or cGAMP (200 µg, intravenously) for 6 weeks (16 treatments total) and T1D onset was monitored thereafter. Unexpectedly, cdiGMP treatment had no effect on onset or incidence of T1D in NOD mice relative to vehicle‐treated (saline) mice (Fig. 4a). In contrast, cGAMP treatments delayed T1D progression and onset by 4–6 weeks and T1D incidence remained lower in cGAMP‐treated mice than in vehicle‐treated mice until age >30 weeks (Fig. 4b); however, T1D incidence was comparable in these groups at experiment end‐points. The short courses of cGAMP treatment slowed T1D progression but did not reduce T1D incidence. In contrast, cdiGMP treatment did not impact T1D progression, onset or incidence in NOD mice.
Figure 4.
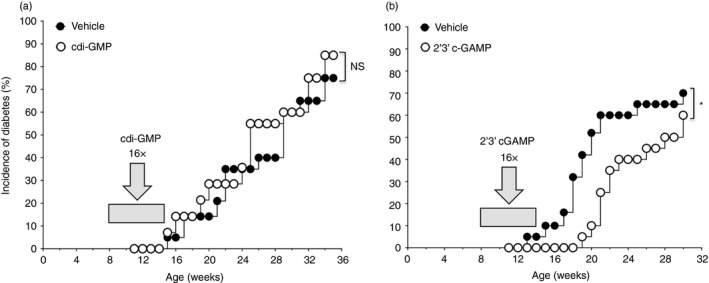
Some cyclic dinucleotide (CDN) isoforms modify the course of type 1 diabetes (T1D) in non‐obese diabetic (NOD) mice. (a) Pre‐diabetic NOD female mice (n = 20, age 8 weeks) were treated with cdiGMP (150 µg/mouse) or saline two or three times per week (16× total from age 8‐14 weeks) and T1D onset was monitored. (b) As in (a), except that mice were treated with cGAMP (150 µg/mouse) or saline and monitored for T1D onset. Log‐rank test, *P < 0·05, NS, not significant. Data in panel (a) are representative of two independent experiments that yielded comparable outcomes.
NOD female mice respond differently to distinct CDN isoforms
To further investigate why NOD mice responded differently to cGAMP and cdiGMP, we evaluated innate immune responses to CDN isoforms. Pre‐diabetic NOD female mice (age 8 weeks) were treated with cdiGMP or cGAMP (200 μg, intravenously) and cytokine and chemokine levels in serum or homogenized spleen were assessed after 3 hr. As expected, copious amounts of IFN‐I were detected in serum from mice treated with cGAMP, whereas no IFN‐I was detected in serum from untreated mice (Fig. 5a). In contrast, IFN‐I was barely detectable in serum from NOD mice treated with cdiGMP (Fig. 5a). This profound defect in cdiGMP responsiveness did not extend to other pro‐inflammatory responses as levels of tumor necrosis factor‐α, interleukin‐6 and some chemokines were all elevated in spleen, though levels of interleukin‐6 and CCL4 were ~50% lower than levels detected in spleen from mice treated with cGAMP (Fig. 5b–d). As for tumor necrosis factor‐α, cGAMP and cdiGMP induced expression of the pro‐inflammatory chemokines CCL2, CCL3, CCL5, CCL7, CXCL1 and CXCL10 to comparable levels in NOD spleen (see Supplementary material, Fig. S1). Hence, non‐canonical cGAMP elicited rapid and robust innate immune responses in NOD mice, whereas NOD mice exhibited a selective defect in rapid IFN‐I production, a signature downstream response to STING activation, following treatment with the canonical microbial CDN cdiGMP.
Figure 5.
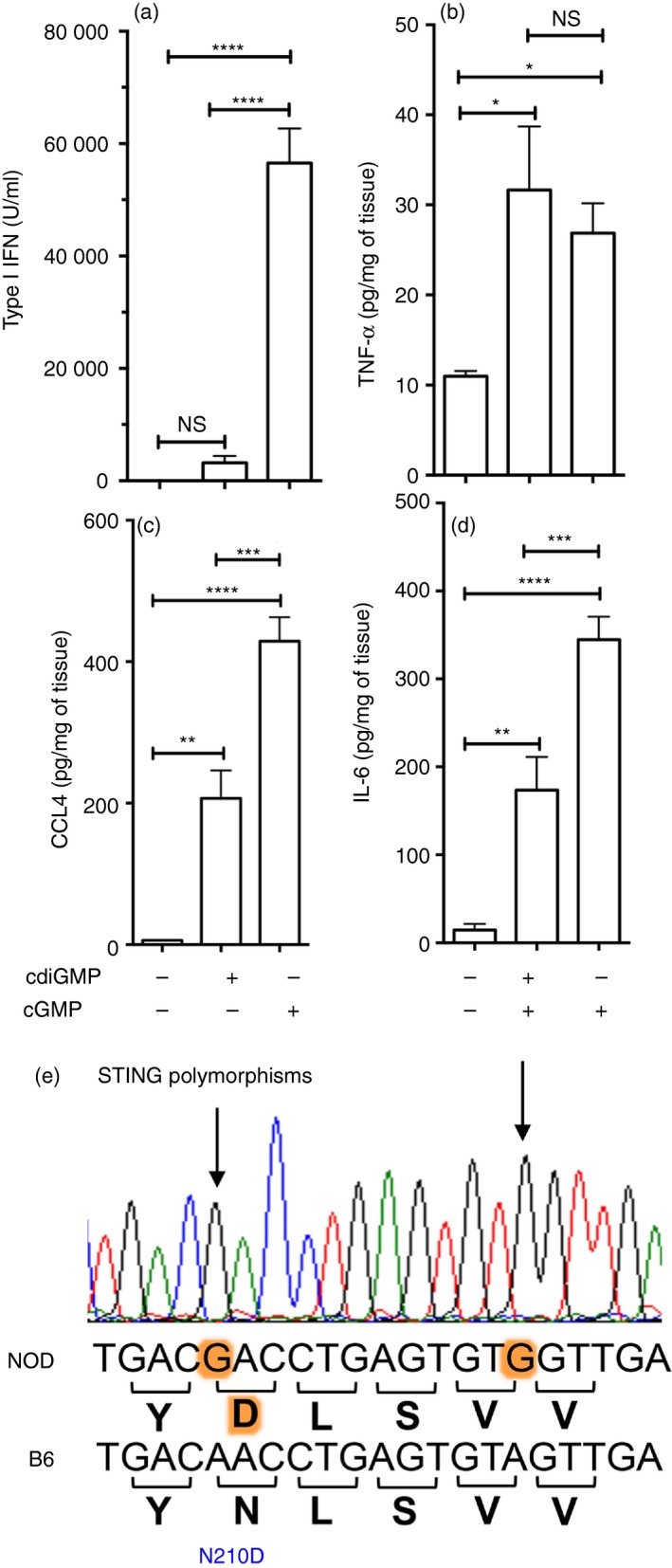
Non‐obese diabetic (NOD) mice exhibit distinct responses to different cyclic dinucleotide (CDN) isoforms. (a–d). Pre‐diabetic NOD female mice (age 8 weeks) were treated with cdiGMP, cGAMP or vehicle and 3 hr later levels of serum interferon type I (IFN‐I) (a), and tumor necrosis factor‐α (TNF‐α), interleukin‐6 (IL‐6) and CCL4 (b–d) in spleen were determined. Data are representative of five independent experiments (n = 5/group). Statistical significance was evaluated by one‐way analysis of variance, followed by the Bonferroni multiple comparison test; *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001 NS, not significant. (e) Trace showing part of the STING DNA sequence from NOD mice compared with B6 mice. Polymorphisms are highlighted.
A STING polymorphism in NOD mice
To seek potential genetic explanations for defective functional responses to cdiGMP, we sequenced the STING gene (encoded by the TMEM173 gene) from NOD mice and compared the DNA sequence obtained with the published STING gene sequence from B6 mice (www.ensemble.org). The entire coding region of the STING gene was identical between B6 and NOD mice, except for two polymorphic A→G (B6→NOD) transitions (Fig. 5e). One base transition at position 213 is silent and has no effect on the amino acid coding sequence, but the other base transition at position 210 encodes an amino acid change from asparagine (N) to aspartic acid (D) in B6 and NOD mice, respectively. This polymorphism represents a radical change in amino acid side chains from basic (N) to acidic (D).
Discussion
Major goals of this study were to evaluate two distinct reagent classes that activate the signaling adaptor STING as potential treatments to inhibit T1D progression in susceptible NOD female mice. The rationale for this approach was based on our previous studies showing that DNP or CDN treatments inhibited T‐cell immunity, and attenuated AIA and EAE in mice.11, 12, 13 Our findings reveal that DNP or CDN treatments attenuated T1D progression and that DNP treatments also reduced T1D incidence significantly when administered to pre‐diabetic NOD mice. Hence, as in mouse models of AIA and EAE, STING agonists stimulate immune regulatory pathways in NOD mice that delay, and in some mice prevent, autoimmune‐mediated destruction of pancreatic islet cells leading to T1D onset.
STING promotes or regulates immunity contingent on several key factors, including the type of cells responsive to STING‐activating signals, the downstream effects of STING activation, and the route of administration of reagents that activate STING. The innate immune adjuvant effects of STING emerged from observations that viral DNA and canonical CDNs from microbial sources (e.g. cdiGMP from L. monocytogenes) were sensed in infected cells to activate STING and incite host immunity via IFN‐I signaling and nuclear factor‐κB activation.21, 25 This paradigm is further reinforced by studies on mice with defects in DNA catabolism, which succumbed to lethal hyper‐inflammatory and autoimmune syndromes due to sustained STING activation, which incited constitutive IFN‐I production.8, 9 Defective DNA catabolism and gain‐of‐function STING mutations have also been linked to higher risk of developing some clinical autoimmune syndromes.4, 5, 26, 27 These observations provide a strong rationale for evaluating CDNs, which bind and activate STING, as potential vaccine adjuvants.28, 29 On the other hand, T‐cell regulatory responses mediated by STING have been reported in several systems, including L. monocytogenes‐infected mice,30 MRLlpr mice susceptible to SLE‐like systemic autoimmunity,31 and systemic DNP and CDN treatments that stimulated STING/IFN‐I signaling to induce IDO in dendritic cells, which in turn activated Treg cells to suppress T‐cell immunity and autoimmunity.11, 12, 13, 32
Short (4 weeks) DNP treatment courses administered to early‐ or late‐stage pre‐diabetic NOD female mice (age 4–8 weeks) delayed T1D onset for 11–12 weeks, about three times longer than the period of DNP treatment. These outcomes suggested that DNPs regulated autoimmunity directed against islet antigens in all treated NOD mice. DNPs prevented T1D onset in ~30% of NOD mice treated early (from age 4 weeks), but DNPs did not prevent T1D progression when administered later (from age 8 weeks). Hence, DNP treatments reinforced defective tolerance to protect a subset of NOD mice from T1D when mice were treated before disease onset.
Systemic administration of InsB, which did not modify T1D progression per se, synergized with DNP treatment to further impede T1D onset and reduce T1D incidence when administered to late‐stage pre‐diabetic NOD mice, relative to mice treated with DNPs only. Insulin therapy and Treg cell activation using anti‐CD3 monoclonal antibodies are potential treatments for recent‐onset T1D patients to slow or prevent progression into full blown T1D, though outcomes from large clinical trials were encouraging but not conclusive.33, 34 In NOD mice, subcutaneous treatment with InsB peptide (B9:23) plus incomplete Freund’s adjuvant induced Treg cells and controlled late‐stage pre‐diabetes via interleukin‐10 and IFN‐γ.17 However, the route of insulin administration is critical for therapeutic responses to manifest.35 This and other patient‐specific variables that may modify responses to insulin therapy will complicate treatments for recent‐onset T1D patients. Insulin administration combined with DNP or CDN treatments offer alternative and versatile approaches to suppress islet‐specific autoimmunity, as DNP cargo DNA can be engineered to encode diabetogenic peptide epitopes and CDNs can be conjugated with such peptides to enhance antigen‐specific responses to control T1D progression.
Regarding the regulatory mechanism induced by DNPs, systemic DNP treatment stimulated rapid IFN‐I production and elevated IDO enzyme activity in the spleens of NOD mice and these responses were comparable with those elicited by DNPs in B6 mice.12 These responses suggest that IFN‐I‐stimulated IDO induction is not defective in NOD mice, despite previous reports of transient defects in endogenous IDO expression in young NOD female mice age 3–4 weeks.36 DNPs also stimulated IDO activity in pancreas and in lymph nodes draining the pancreas of pre‐diabetic NOD mice, suggesting that this T‐cell regulatory pathway may be an appropriate target to regulate autoimmunity against islet autoantigens such as insulin. In humans, IDO is constitutively expressed in β cells, whereas IDO expression is reduced to absent as T1D develops.37 This suggests that IDO may be an important factor to maintain immune homeostasis in the pancreas. As oral 1‐MT dosing only partially reversed therapeutic responses to DNPs in NOD mice, DNP‐induced IDO activity contributed to, but did not fully account for, therapeutic responses to DNPs. Previously, we reported that T‐cell regulatory responses to DNPs and CDNs depended on STING/IFN‐I signaling to induce IDO.11, 12, 13 Moreover, therapeutic responses mediated by DNPs in two induced autoimmunity models (AIA and EAE) were abolished in mice lacking intact IDO1 genes or in B6 mice given the oral IDO inhibitor 1‐MT. In these models of autoimmune syndromes IDO activity was essential for therapeutic responses to manifest in response to DNP treatment.
STING signaling was essential for regulatory responses to manifest in AIA and EAE models following DNP treatments. As NOD mice with induced genetic defects in STING were unavailable, we treated NOD mice with CDNs to test if these STING‐activating reagents also induced therapeutic responses that delayed T1D onset. As for DNPs, treatments with non‐canonical cGAMP, produced by the mammalian DNA sensor cGAS,24 delayed T1D onset significantly but did not reduce T1D incidence, suggesting that therapeutic responses that attenuate T1D progression in NOD mice are mediated by STING. However, we also uncovered a selective defect in innate immune responsiveness of NOD mice to distinct CDN isoforms because canonical cdiGMP, produced by microorganisms such as L. monocytogenes, had no discernable effect on the course of T1D in NOD mice. Functional responses to these two CDN isoforms revealed a profound but selective defect in IFN‐I production in response to cdiGMP treatment in NOD mice. Moreover, sequence analyses of the STING gene (encoded by the TMEM173 gene) in NOD mice provide a potential explanation for this selective functional defect, as a single base transition encoding a radical amino acid change was found in STING genes from NOD mice, This NOD‐specific STING polymorphism may impact CDN binding to STING or STING binding to downstream signaling molecules such as TANK‐binding kinase 1, IFN regulatory factor 3 and nuclear factor‐κB.
In summary, the findings reported in this study show that two distinct classes of reagents that activate STING regulate T‐cell immunity‐attenuated autoimmune disease progression in susceptible NOD female mice and therapeutic responses to STING agonists were in part mediated by IDO. These findings provide further evidence that STING signaling can regulate autoimmunity, and suggest that DNPs and CDN treatments could be optimized to improve therapies designed to slow, reverse or prevent T1D progression and onset.
Disclosures
ALM receives licensing income from NewLink Genetics Inc.
Author contributions
HL and ALM conceived and designed the study and wrote the manuscript. HL, ES, LH, RO, PRC and RP contributed to data generation and analyses and provided scientific advice and comments on the manuscript. All authors reviewed and critiqued the manuscript before submission.
Supporting information
Figure S1. Chemokine responses induced by cyclic dinucleotides in NOD mice. Eight‐week‐old pre‐diabetic NOD female mice were treated with cdiGMP, 2′3′‐cGAMP or vehicle (saline) and 3 hr later, levels of the chemokines indicated were determined in spleen homogenates. Experiments were repeated three times and data are representative of comparable outcomes obtained, n = 5/group. Data were analyzed by one‐way analysis of variance, followed by the Bonferroni multiple comparison test; *P < 0·05, **P < 0·01, ***P < 0·001, NS, not significant.
Acknowledgements
We thank Gabriela Pacholczyk and Janice Randall for expert technical assistance. We thank the NewLink Genetics Corporation for the generous gift of 1‐MT and Yoshihiro Hayakawa (Toyota Japan) for synthesizing the CDNs used in this study. This study was supported by grants to ALM from NIAID (AI103347) and the Carlos and Marguerite Mason Trust. HL was supported by a postdoctoral research fellowship from the Juvenile Diabetes Research Foundation (grant no. 3‐2013‐219).
References
- 1. van den Berg JH, Nuijen B, Schumacher TN, Haanen JB, Storm G, Beijnen JH et al. Synthetic vehicles for DNA vaccination. J Drug Target 2010; 18:1–14. [DOI] [PubMed] [Google Scholar]
- 2. Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology 2013; 218:1312–21. [DOI] [PubMed] [Google Scholar]
- 3. Lemos H, Huang L, McGaha TL, Mellor AL. Cytosolic DNA sensing via the stimulator of interferon genes adaptor: Yin and Yang of immune responses to DNA. Eur J Immunol 2014; 44:2847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 371:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC et al. Inherited STING‐activating mutation underlies a familial inflammatory syndrome with lupus‐like manifestations. J Clin Invest 2014; 124:5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J et al. Clinical and molecular phenotype of Aicardi–Goutieres syndrome. Am J Hum Genet 2007; 81:713–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2006; 443:998–1002. [DOI] [PubMed] [Google Scholar]
- 8. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon‐dependent autoimmune disease. Immunity 2012; 36:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA‐dependent inflammatory disease. Proc Natl Acad Sci USA 2012; 109:19386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K et al. TLR9 regulates TLR7‐ and MyD88‐dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010; 184:1840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol 2013; 191:3509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B et al. Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. J Immunol 2012; 188:4913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GS, Li L et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol 2014; 192:5571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tyring S, Fleischmann WR Jr, Baron S. A convenient microassay for cytolysis and cytostasis. Methods Enzymol 1986; 119:574–9. [DOI] [PubMed] [Google Scholar]
- 15. Lewis HC, Chinnadurai R, Bosinger SE, Galipeau J. The IDO inhibitor 1‐methyl tryptophan activates the aryl hydrocarbon receptor response in mesenchymal stromal cells. Oncotarget 2017; 8:91914–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akbarpour M, Goudy KS, Cantore A, Russo F, Sanvito F, Naldini L et al. Insulin B chain 9–23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag‐specific FoxP3+ Tregs. Sci Transl Med 2015; 7:289ra81. [DOI] [PubMed] [Google Scholar]
- 17. Fousteri G, Dave A, Bot A, Juntti T, Omid S, von Herrath M. Subcutaneous insulin B:9‐23/IFA immunisation induces Tregs that control late‐stage prediabetes in NOD mice through IL‐10 and IFNγ . Diabetologia 2010; 53:1958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bresson D, Fousteri G, Manenkova Y, Croft M, von Herrath M. Antigen‐specific prevention of type 1 diabetes in NOD mice is ameliorated by OX40 agonist treatment. J Autoimmun 2011; 37:342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarikonda G, Sachithanantham S, Manenkova Y, Kupfer T, Posgai A, Wasserfall C et al. Transient B‐cell depletion with anti‐CD20 in combination with proinsulin DNA vaccine or oral insulin: immunologic effects and efficacy in NOD mice. PLoS ONE 2013; 8:e54712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC et al. Anti‐CD3 and nasal proinsulin combination therapy enhances remission from recent‐onset autoimmune diabetes by inducing Tregs. J Clin Invest 2006; 116:1371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 2013; 14:19–26. [DOI] [PubMed] [Google Scholar]
- 22. Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA et al. Structure–function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 2013; 154:748–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu J, Sun L, Chen X, Du F, Shi H, Chen C et al. Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339:826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339:786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barber GN. STING‐dependent signaling. Nat Immunol 2011; 12:929–30. [DOI] [PubMed] [Google Scholar]
- 26. Dittmar M, Woletz K, Kahaly GJ. Reduced DNASE1 gene expression in thyroid autoimmunity. Horm Metab Res 2013; 45:257–60. [DOI] [PubMed] [Google Scholar]
- 27. Martinez‐Valle F, Balada E, Ordi‐Ros J, Bujan‐Rivas S, Sellas‐Fernandez A, Vilardell‐Tarres M. DNase1 activity in systemic lupus erythematosus patients with and without nephropathy. Rheumatol Int 2010; 30:1601–4. [DOI] [PubMed] [Google Scholar]
- 28. Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, Demartino JA et al. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cell Immunol 2012; 278:113–9. [DOI] [PubMed] [Google Scholar]
- 29. Dubensky TW Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING‐activating cyclic dinucleotide adjuvants. Ther Adv Vaccines 2013; 1:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Archer KA, Durack J, Portnoy DA. STING‐dependent type I IFN production inhibits cell‐mediated immunity to Listeria monocytogenes . PLoS Pathog 2014; 10:e1003861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci USA 2015; 112:E710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res 2016; 76:2076–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti‐CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol 2013; 149:268–78. [DOI] [PubMed] [Google Scholar]
- 34. Chatenoud L, Waldmann H. CD3 monoclonal antibodies: a first step towards operational immune tolerance in the clinic. Rev Diabet Stud 2012; 9:372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hutchings P, Cooke A. Protection from insulin dependent diabetes mellitus afforded by insulin antigens in incomplete Freund's adjuvant depends on route of administration. J Autoimmun 1998; 11:127–30. [DOI] [PubMed] [Google Scholar]
- 36. Grohmann U, Fallarino F, Bianchi R, Orabona C, Vacca C, Fioretti MC et al. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J Exp Med 2003; 198:153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anquetil F, Mondanelli G, Gonzalez N, Rodriguez Calvo T, Zapardiel Gonzalo J, Krogvold L et al. Loss of IDO1 expression from human pancreatic beta‐cells precedes their destruction during the development of Type 1 diabetes. Diabetes 2018; 67:1858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemokine responses induced by cyclic dinucleotides in NOD mice. Eight‐week‐old pre‐diabetic NOD female mice were treated with cdiGMP, 2′3′‐cGAMP or vehicle (saline) and 3 hr later, levels of the chemokines indicated were determined in spleen homogenates. Experiments were repeated three times and data are representative of comparable outcomes obtained, n = 5/group. Data were analyzed by one‐way analysis of variance, followed by the Bonferroni multiple comparison test; *P < 0·05, **P < 0·01, ***P < 0·001, NS, not significant.