

Abstract
Abstract Histone deacetylase 1 (HDAC1) regulates transcription by deacetylating histones. In addition to histones, several non-histone proteins are HDAC1 substrates, which verified a role for HDAC1 beyond epigenetics. Unfortunately, identification of nonhistone substrates has been largely serendipitous, making full characterization of HDAC1 functions difficult. To overcome this challenge, inactive “trapping” mutants were recently developed to identify HDAC1 substrates. To optimize substrate trapping, the relative trapping abilities of 17 inactive HDAC1 mutants was assessed. HDAC1 H141A, F150A and C151A showed strong binding to substrates LSD1 and p53. Interestingly, each mutant preferentially trapped a different substrate. By combining several inactive mutants, the trapping strategy will facilitate discovery of new HDAC1 substrates and shed light on the variety of HDAC1-related functions in cell biology.
Keywords: Histone Deacetylase, substrate trapping mutants
Graphical Abstract
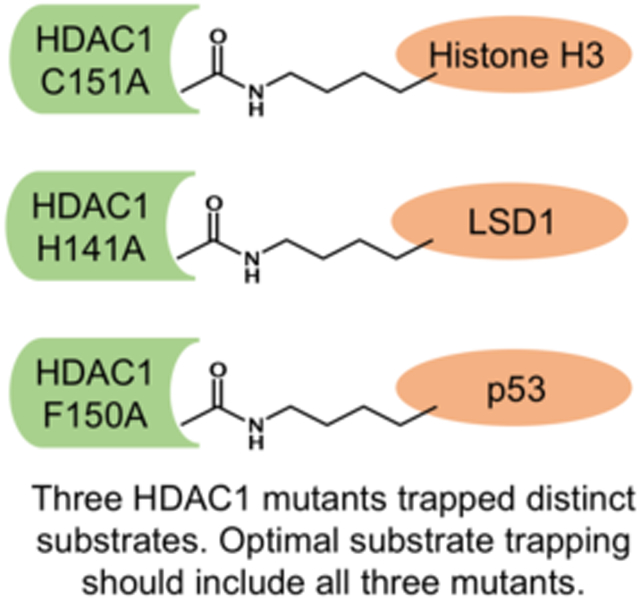
To trap a substrate: Trapping mutants have been valuable tools to discover histone deacetylase 1 (HDAC1) substrates and uncover novel biological mechanism. Here, three optimal mutants were identified, each with preference to a different substrate. Future trapping should include all three mutants for efficient substrate discovery.
Introduction
The eukaryotic genome is packaged into nucleosomes, where genomic DNA is bound by an octamer of histone proteins. Gene expression from genomic DNA involves multiple layers of regulation, including the modification of histone proteins by phosphorylation, methylation, and acetylation.[1] In the case of acetylation, histone deacetylase (HDAC) proteins regulate the acetylation of histones by removing acetyl groups from ε-N-acetyl lysine amino acids of histone proteins.[2] Deacetylation of histones by HDAC proteins promotes chromatin condensation and induces transcriptional repression.[3] Importantly, HDAC-regulated transcription is associated with several diseases, such as asthma, arthritis, neurodegenerative diseases, and cancer, thus making them important drug targets.[2] In fact, four HDAC inhibitors are clinically approved as anti-cancer drugs. Vorinostat (SAHA or suberoylanilide hydroxamic acid) and Romidepsin (depsipeptide) are approved for the treatment of cutaneous T cell lymphoma, whereas Belinostat (PXD101) and Panabinostat (LBH-589) are used for the treatment of peripheral T cell lymphoma and multiple myeloma, respectively.[4] With key roles in transcriptional regulation and disease, HDAC proteins are actively studied.
The HDAC family is comprised of 18 members belonging to four major classes based on their homology to yeast proteins.[3, 5] While most HDAC proteins are associated with diseases, this study focuses on one isoform, HDAC1, due to its anomalous expression in multiple diseases, including cancer.[6] The HDAC1 homolog in yeast is the transcriptional regulator protein Rpd3, suggesting that HDAC1 is a player in transcription regulation.[5] In fact, the role of HDAC1-mediated deacetylation of histones in regulation of transcription has been well characterized in mammalian systems.[7]
Recent proteomics analyses have identified a wide range of acetylated non-histone proteins.[8] Importantly, acetylation influences protein structure and function,[9] akin to other post-translational modification such as phosphorylation.[10] The presence of acetylated proteins in human cells implicate HDAC proteins in the deacetylation of substrates outside of histones. If HDAC proteins deacetylate non-histone proteins, they likely play a larger role in human cell biology beyond epigenetics. However, among the long list of acetylated proteins, the number of verified HDAC substrates remains considerably short. For example, only five substrates of HDAC1 have been identified, namely histones, p53, E2F1, LSD1 and Eg5.[9b-d, 11] Historically, identification of nonhistone substrates has been largely serendipitous due to the absence of a systematic substrate identification tool. Without a full characterization of the substrate profiles of HDAC proteins, the many biological functions of HDAC proteins in cell biology is likely incomplete.
The traditional method for isolating non-histone substrates of HDAC proteins involves immunoprecipitation of the HDAC-substrate complex. Unfortunately, immunoprecipitation is problematic for substrate isolation because enzymatically active wild type (WT) HDAC proteins bind substrates transiently (Figure 1A), resulting in loss of the substrate during enrichment (Figure 1A). To overcome the problem of transient enzyme-substrate interaction, previous studies from our lab developed a simple method to identify HDAC substrates using inactive trapping mutants.[11] Catalytically inactive mutants are expected to bind with longer residence time to substrates due to the lack of catalysis (Figure 1B), allowing isolation by immunoprecipitation. Using inactive mutants, we successfully identified demethylase LSD1 as a substrate of HDAC1 and revealed a novel cross talk between HDAC1 and LSD1 to regulate gene expression.[11b] Eg5 (Kinesin – like protein 11, KIF11) was also identified as a HDAC1 substrate using trapping, which revealed a new role of HDAC1 in mitotic progression through Eg5 acetylation.[11a] More recently, the trapping strategy was improved by incorporating proteomics-based mass spectrometry (MS) analysis, which allowed discovery of many non-histone substrates of HDAC1 in a single study.[12] These prior studies document the value of mutant trapping to identify and validate new substrates of HDAC1, which will facilitate the full characterization of HDAC cell biology and assist in development of more effective drugs targeting HDAC proteins.
Figure 1.

HDAC1 substrate trapping. A) Catalytically active WT HDAC binds substrates transiently, which makes purification and identification of HDAC1 substrate challenging. B) An inactive HDAC mutant will bind substrates with longer residence time, which makes purification and identification of bound substrates possible. C) HEK293 cells were transiently transfected with expression plasmids for FLAG-tagged wild-type or mutant HDAC1 (HDAC1-F) before treatment with SAHA (10 μM) for 24 hr to induce robust protein acetylation. After lysis, expressed HDAC1-Flag proteins were immunoprecipitated (IP) with the FLAG antibody, separated by SDS-PAGE, and immunoblotted with FLAG or acetyl-histone H3 lysine 9 (Ac-H3K9) antibodies. As a control, lysates prior to immunoprecipitation were separated by SDS-PAGE and immunoblotted with acetyl-H3K9 antibody to assure equal protein content and loading. Figure 1C was reprinted from reference 11a, with copyright permission from Elsevier.[11a]
Prior HDAC1 trapping work predominantly utilized the inactive C151A mutant of HDAC1.[11-12] However, the Pflum lab has created roughly 70 mutants of HDAC1 to probe its enzymatic function and mechanism.[13] Among these HDAC1 mutants, almost all were catalytically inactive. Despite the availability of many inactive mutants of HDAC1, only three (H141A, F150A, and C151A) were tested in earlier work for trapping properties against the known substrate of HDAC1, histone 3 (H3).[11a] We found that only two out the three mutants (F150A and C151A) trapped H3 at acetyl-lysine 9 (Ac-H3K9, Figure 1C, lanes 4 and 5 compared to lanes 2 and 3).[11a] Interestingly, the H141A HDAC1 mutant did not trap Ac-H3K9, similar to WT HDAC1 (Figure 1C, compare lane 3 to lane 2). The variable ability of each mutant to trap the Ac-H3K9 substrate suggested that each inactive mutant has unique substrate binding properties. Therefore, we reasoned that screening additional mutants could identify an optimal mutant that would trap substrates more efficiently.
Here, we screened 17 inactive HDAC1 mutants with mutations located in various regions of the active site to identify optimal substrate trapping mutants. Two known substrates of HDAC1, p53 and LSD1,[9b, 9c, 11b] were used as test substrates to assess stable binding by the HDAC1 mutants. Strong data showed that each HDAC1 mutant preferably trapped a different substrate. The data suggest that an optimized trapping method would use multiple mutants to uncover new substrates, which is a critical first step to fully characterize the role of HDAC1 proteins in cellular activities.
Results and Discussion
To identify optimal trapping mutants of HDAC1 for discovery of substrates, we screened inactive mutants with single point amino acid mutations located in various regions of the HDAC1 active site. The crystal structure of HDAC1 contains a narrow and hydrophobic 11Å active site channel where the substrate binds in proximity to the catalytic metal (Figure 2).[14] An adjacent 14Å internal cavity at the base of the active site channel acts as a release hatch for the acetate byproduct of deacetylation.[13b, 14a, 15] Amino acids that reside in the 11 Å channel of HDAC1 include conserved catalytic residues (H140 and H141) and hydrophobic amino acids that line the channel (G149, F150, and L276).[13a] In addition, several amino acids (H28, P29, E98, D99, and F205) reside near the solvent-exposed surface of the active-site channel where they are not expected to promote catalysis directly, although their proximity to the active site could potentially influence substrate binding. Prior activity profiling of 11 Å active site channel mutants documented that alanine mutation of most active site amino acids dramatically reduced activity, with only two mutants maintaining high activity (E98A and Y204A).[13a] Based on their low activities and locations in the active site channel, eight mutants with alanine mutation of residues in the 11 Å active site channel were selected for this trapping study (H28A, P29A, D99A, H141A, F150A, G149A, F205A, and L276A, Figure 2).
Figure 2.

HDAC1 trapping mutants. Crystal structure image of HDAC1 (PDB: 4BKX)[14b] where the seventeen amino acids in the 11 Å active site channel (green) and 14 Å internal cavity (yellow) of HDAC1 that were alanine mutated are shown as ball and stick structures. The HDAC1 structure is shown as grey mesh. The metal ion required for catalysis is shown as a white sphere. Image created with the Pymol molecular graphics system (version 1.3, Schrödinger, LLC).
Amino acids that reside in the 14Å internal cavity include polar residues that assist in releasing the acetate byproduct of deacetylation from the active site (Y23, Y24, R34, and C151, Figure 2).[13b, 14a, 15] The close proximity of these four amino acids to the 11 Å active site channel make them candidate trapping mutants. Importantly, mutation of these four amino acids to alanine resulted in dramatic reduction in catalytic activities. The critical catalytic amino acid (Y303, Figure 2) resides in the cleft between the 14Å internal cavity and the 11 Å active site channel and its mutation results in catalytic inactivity. Finally, eight other amino acids that make up the 14Å internal cavity displayed minimal activity (M30, I35, R36, and F109, Figure 2),[13b] making them also trapping mutant candidates. Based on their low activities, we selected to study nine mutants with alanine mutation of amino acids in the 14Å internal cavity (Y23A, Y24A, M30A, R34A, I35A, R36A, F109A, C151A and Y303A, Figure 2).
Identification of the optimal trapping mutant of LSD1
In prior work using the substrate trapping mutant strategy, LSD1 was identified as a novel substrate of HDAC1.[11b] LSD1 was successfully trapped by the HDAC1 C151A mutant.[11b] Here, we screened the selected eight 11 Å active site channel and nine 14 Å internal cavity mutants of HDAC1 for binding and immunoprecipitation of LSD1 with the goal of identifying the best trapping mutant. The C151A mutant was included as a positive control.
For the binding screen, HEK293 cells were transfected with Flag-tagged wild type or mutant HDAC1 expression constructs, grown for 48h, and then treated with SAHA (10 μM), a pan HDAC inhibitor,[16] for an additional 24 h to increase global acetylation of cellular proteins. After lysis, HDAC proteins were immunoprecipitated via the Flag tag and eluted, followed by separation of bound proteins by SDS-PAGE, and immunobloting with FLAG and LSD1 antibodies. Among the eight 11Å active site channel mutants, the H141A mutant immunoprecipitated higher levels of LSD1 compared to wild type HDAC1 (Figure 3A, lane 6 vs lane 2). In contrast, the rest of the 11Å active site mutants bound the same background level of LSD1 as wild type (Figure 3A, lanes 2-5 and 7-10). The H141 residue is a catalytic amino acid necessary for activation of water for hydrolysis of the acetyl-lysine amide bond.
Figure 3.

Screen of seventeen HDAC1 mutants for LSD1 trapping. Wild type (WT) HDAC, along with mutants with single point mutation of residues in the 11 Å active site channel (A), 14 Å channel (B), or selected mutants from both (C), were expressed as Flag-tagged proteins (HDAC1-F) in HEK293 cells. Cells were also incubated with SAHA (10 μM) for 24 hr to increase robust acetylation. After lysis, proteins in the lysates were immunoprecipitated with anti-FLAG agarose beads, separated by SDS-PAGE, and western blot analyzed with Flag (top) or LSD1 (bottom) antibodies. As an expression control, proteins in lysates were also separated by SDS-PAGE and visualized with a LSD1 antibody. Repetitive trials are provided in supplementary Figures S1-S3.
Among the 14Å internal cavity mutants, seven out of the nine mutants immunoprecipitated higher levels of LSD1 compared to wild type HDAC1 (Figure 3B, compare lanes 4-10 with lane 2). Among these seven trapping mutants, R36A, F109A, and C151A immunoprecipitated the most LSD1 (Figure 3B, lanes 8, 9, and 10). The high level of LSD1 trapping by the C151A mutant is consistent with our prior work utilizing C151A to identify LSD1 as an HDAC1 substrate.[11b] C151 is located near the 11Å active site and likely assists in the removal of the acetate byproduct of deacetylation. In contrast, R36A and F109A are located more distant from the 11Å active site channel and perhaps play a structural role.
Considering the 17 HDAC1 mutants screened, eight showed some trapping abilities (Figure 3A and 3B). To select the optimal trap for LSD1, the eight trapping mutants were rescreened together. Comparing all eight mutants, HDAC1 H141A immunoprecipitated the highest levels of LSD1 compared to wild type HDAC1 and the other mutants (Figure 3C, compare lane 10 to the others). In prior studies, LSD1 was identified as a substrate through trapping with the C151A mutant.[11b] Consistent with this prior work, the C151A mutant trapped LSD1 (Figure 3B, lane 10), although not at as high a level as the H141A mutant (Figure 3C, lane 11). The remaining six mutants bound similar background levels of LSD1 as the wild type (Figure 3C, lanes 2-9 and 11). In total, the mutant screening established the H141A and C151A mutants as the optimal substrate traps for LSD1.
Identification of the optimal trapping mutant of p53
Acetylated p53 protein was identified as the first non–histone substrate of HDAC1.[9c] As the second test substrate, p53 was used to identify the optimal trapping mutants of HDAC1. Here again the eight 11Å active site channel and nine 14Å internal cavity mutants as Flag-tagged fusion proteins were expressed in HEK293 cells in the presence of SAHA to induce robust cellular acetylation. After washing and lysis, HDAC1 and trapped substrates were immunoprecipitated via the Flag tag, separated by SDS-PAGE, and immunoblotted with FLAG and p53 antibodies.
Among the 11Å active site channel mutants, HDAC1 F150A immunoprecipitated a higher level of p53 compared to wild type HDAC1 or the other mutants (Figure 4A, compare lanes 8 to the others). The fact that the H141A mutant trapped the highest level of LSD1 documents that different substrates bind the active site of mutant proteins with distinct interactions. Crystallographic data of the HDAC1 homolog HDLP from Aquifex aeolicus,[14a] human HDAC2,[17] human HDAC6,[18] and human HDAC8[19] document that the phenyl groups of the F150 and F205 residues sandwiches the hydrophobic carbon chain of the inhibitor SAHA to form the most narrow segment of the active site. The fact that only the F150A mutant, but not the F205A mutant, showed strong p53 binding shows that each residue plays a unique role in substrate binding.
Figure 4.

Screen of seventeen HDAC1 mutants for p53 trapping. Wild type (WT) HDAC1, along with mutants with single point mutation of residues in the 11 Å active site channel (A), 14 Å channel (B), or selected mutants from both (C), were expressed as Flag-tagged proteins (HDAC1-F) in HEK293 cells. Cells were also incubated with SAHA (10 μM) for 24 hr to increase robust acetylation. After lysis, proteins in the lysates were immunoprecipitated with anti-FLAG agarose beads, separated by SDS-PAGE, and western blot analyzed with Flag (top) or p53 (bottom) antibodies. As an expression control, proteins in lysates were also separated by SDS-PAGE and visualized with a p53 antibody. Repetitive trials are provided in the supplementary Figures S4-S6.
The p53 trapping assay was next used to screen the 14Å internal cavity mutants. Among the nine mutants, HDAC1 F109A and C151A immunoprecipitated higher levels of p53 compared to wild type and the other mutant HDAC1 (Figure 4B, compare lanes 9 and 10 to the others). Both F109A and C151A mutants also trapped LSD1 effectively (Figure 3B), suggesting similar influence on substrate binding. Unlike the screen with LSD1, the screen with p53 only identified three high binding mutants among the 17 tested.
Further screening for p53 trapping was performed only with the three best mutants from the individual screening of 11Å active site channel and 14Å internal cavity mutants. All three mutants, HDAC1 F109A, F150A, and C151A, bound higher levels of p53 compared to wild type HDAC1 (Figure 4C, compare lanes 3-5 to lane 2). Among the three, the F150A mutant immunoprecipitated the highest levels of p53 compared to wild type HDAC1 and other mutants (Figure 4C, compare lane 4 to the others). The results identified that F150A was the optimal substrate trap for p53.
Discussion
Prior studies showed that thousands of proteins in cells are acetylated,[20] acetylation influences protein function,[9] and HDAC proteins deacetylate substrates in addition to histones.[9b-d, 11] Despite the likelihood that HDAC protein deacetylate many substrates to regulate their function, few non-histone substrates of HDAC proteins are known. Without a full characterization of the substrate profiles of HDAC proteins, the role of HDAC proteins in cellular events remains understudied. Historically, identification of non-histone substrates has been largely serendipitous. Recent introduction of substrate trapping mutants of HDAC proteins to discover new substrates has resulted in the identification of a number of unanticipated substrates.[11] By identifying non-histone substrates, HDAC proteins were linked to cellular processes in addition to gene expression.[9b-d, 11] The trapping strategy has the potential to uncover novel mechanisms of action of HDAC proteins, which will lead to a better understanding of the role of acetylation in biological events.
Substrate trapping was developed by testing only three single point mutants of HDAC1.[11a] The histone H3 substrate was screened in a gel-based binding assay with HDAC1 H141A, F150A, and C151A mutants, and only F150A and C151A bound H3 to a greater extent than wild type HDAC1 (Figure 1C).[11a] The fact that only two out of three mutants tested trapped H3 suggested that each HDAC1 mutant displays a distinct preference for substrate binding. Given our prior work reporting a wide variety of mutants with alanine mutation of residues in both the 11Å active site channel and 14Å internal cavity of the HDAC1 active site,[13] we hypothesized that a thorough screening of many HDAC1 mutants would reveal an optimal substrate trapping mutant for high efficiency substrate discovery.
In this study, HDAC1 mutants with alanine mutation of residues in both the 11Å active site channel and 14Å internal cavity (Figure 2) were tested as substrate traps against known substrates LSD1 and p53. The results from the LSD1 binding assay identified the HDAC1 H141A mutant as the optimal substrate trap for LSD1. According to the proposed substrate deacetylation mechanism of HDAC protein based on crystallographic and computational evidence,[14a, 18, 21] and also supported by HDAC1 mutagenesis,[7, 13a] H141 acts as proton donor during the deacetylation reaction. The H141A mutant was shown previously to maintain only 20% activity compared to the WT, yet bind to associated proteins, suggesting that the global HDAC1 structure is not disrupted upon mutation.[7, 13a] The fact that mutation of a catalytic amino acid created the optimal substrates trap for LSD1 suggests that residues in the active site channel required for substrate interaction must remain unaltered for retention of acetylated LSD1 binding.
In contrast to the LSD1 binding data, the optimal substrate trap for p53 was the HDAC1 F150A mutant. Prior mutagenesis of the 150 position in HDAC1 documented that F150 is uniquely required to maintain enzymatic activity.[13a] Kinetics data with immunoprecipitated HDAC1 from mammalian cells[22] using a fluorescence assay and a p53-derived peptide substrate revealed that wild type and F150A HDAC1 displayed similar Km values of 88 ± 3 and 66 ± 2 μM, respectively,[13a] which is similar to previous kinetics measurements using a different peptide substrate.[23] In contrast, the F150A mutant demonstrated a 12-fold reduced Vmax value compared to the wild type protein, which is consistent with activity data showing that the F150A mutant displays only 11% of wild type activity.[13a] In total, the kinetics data indicate that loss of catalysis, but not substrate binding, is primarily responsible for the low activity of the of the F150A mutant.
Phosphatases, like HDAC proteins, are enzymes that remove a post-translational modification. In the case of phosphatases, the phosphoryl groups from phosphorylated amino acids are removed. Substrate trapping was first developed to identify the substrates of several phosphatases,[24] including protein tyrosine phosphatase 1B (PTP1B).[25] Two widely used PTP1B mutants are C215S and D181A, where the mutated amino acids are located in the active site of PTP1B and involved in the catalysis.[25] Kinetic data with the C215S and D181A mutants documented similar or even reduced Km, but drastically reduced Vmax,[25] similar to the HDAC1 F150A mutant.[13a] Although kinetics analysis with mammalian-expressed HDAC1 is low throughput and not amenable to screening, the available data suggests that trapping with HDAC1 mutants is successful due to a loss in catalysis without loss of substrate binding efficiency.
Crystallographic analysis of the inactive Y306F mutant of HDAC8 bound to a peptide substrate documented binding interactions both inside and outside of the active site channel.[26] In particular, a conserved aspartate on the outside rim of the HDAC8 active site made contacts with the backbone of the peptide substrates to stabilize binding. Based on this HDAC8 structure, binding of the acetyllysine residue in the active site could influence interactions of the substrate outside of the active site. Extending these HDAC8 results to HDAC1, we spectulate that the binding orientation of acetyllysine in the active sites of the different mutants could alter interactions of the substrate protein outside of the active site, resulting in different substrate preferences.
Results from this binding screen suggest that there is no single optimal HDAC1 mutant trap for all substrates. Instead three mutants- H141A, F150A and C151A- each bind optimally to different substrates. Therefore, these studies suggest that all three mutants should be used to isolate and identify unanticipated HDAC1 substrates. In future studies, we will explore using all three mutants simultaneously for optimal trapping. Given the high sequence conservation of H141, F150, and C151 among the eleven metal-dependent HDAC isoforms (Figure S7), trapping can be applied to other HDAC isoforms, which is currently being explored in our lab. Substrate trapping offers a powerful and unbiased approach to identify unexpected HDAC substrates and HDAC-related function, which has the potential to reveal new mechanisms in cell biology and disease.
Experimental Section
Expression Plasmids, Cell Culture, and Transient Transfections.
HDAC1 single point mutants were previously generated in the pBJ5 expression vector.[13] HEK293 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (Life technologies) and 1% antibiotic/ antimycotic (Hyclone) at 37 °C in a 5% CO2 incubator. Jetprime reagent (VWR) was used for transfection of pBJ5-HDAC wild type or mutant plasmid DNA (5 μg) into HEK293 cells (2 × 107) at 70% confluency. After a 48 h growth period, cells were treated with SAHA (10 μM in growth media containing <2% DMSO) for an additional 24 h to induce robust acetylation of cellular proteins. Cells were harvested by centrifugation and washed with DPBS (Hyclone, Dulbecco’s Phosphate Buffered Saline, 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), and either used immediately or stored at −80 °C as a cell pellet.
Cell lysis
Cells after transfection, generated as described above (2 × 107 cells), were lysed in lysis buffer (500 μL; 50 mM Tris-Cl at pH 8.0, 150 mM NaCl, 10% glycerol, and 0.5% triton-X100) containing 1× protease inhibitor cocktail (GenDEPOT)) at 4°C for 30 min with rotation. The supernatant was collected using centrifugation at 13.2 × 103 rpm for 15 min at 4°C. Total protein concentration was determined using Bradford assay (BioRad, #5000205). Cell lysates were stored at −80°C or used immediately.
Immunoprecipitation, and Substrate Trapping
Prior to immunoprecipitation, anti-Flag M2 affinity agarose beads (20 μL bead slurry, Sigma-Aldrich) were washed twice with cold TBS (Tris Buffered Saline, 500 μL; 20 mM Tris-Cl at pH 8.0, 150 mM NaCl). Lysates containing either wild type or mutant HDAC-Flag expressed proteins (1 mg total protein) were incubated with prewashed anti-Flag M2 agarose beads at 4°C overnight with rotation. After immunoprecipitation, beads were washed three times with lysis buffer (1 mL) containing high salt (500 mM NaCl). For the LSD1 binding assay, bound proteins were eluted with SDS buffer (20 μL; 100 mM Tris-Cl at pH 6.8, 4% SDS, 20% glycerol, 0.008% bromophenol blue) by boiling at 95°C for 5 min. For the p53 binding assay, bound proteins were eluted with TBS containing 3xFlag peptide (APEXBIO; 40 μL; 0.25 mg mL−1 in TBS) for 30 min at 4°C. The eluted proteins were mixed with SDS loading dye (10 μL; SDS buffer containing 10% v/v β-mercaptoethanol) and boiled at 95°C for 2 min. As controls, lysates without immunoprecipitation (50 μg) were denatured with SDS loading dye (5 μL) by boiling at 95°C for 2 min. Proteins were separated by 10% SDS-PAGE, transferred to PVDF membrane (Immobilon P, Fischer Scientific), and immunoblotted with monoclonal FLAG® M2 (F3165, Sigma), LSD1 (L4418, Sigma) or p53 (sc-126, SantaCruz Biotechnology) antibodies.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R01GM121061. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank Wayne State University for funding and R. Andrade, K. Herath, J. Knoff, Y. Zhang for comments on the manuscript..
References
- [1].Khorasanizadeh S, Cell 2004, 116, 259–272. [DOI] [PubMed] [Google Scholar]
- [2].Kramer OH, Gottlicher M, Heinzel T, Trends Endocrinol. Metab. 2001, 12, 294–300. [DOI] [PubMed] [Google Scholar]
- [3].Haberland M, Montgomery RL, Olson EN, Nat Rev Genet 2009, 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4] a.Warrell RP Jr., He LZ, Richon V, Calleja E, Pandolfi PP, J. Natl. Cancer. Inst. 1998, 90, 1621–1625; [DOI] [PubMed] [Google Scholar]; b Grant S, Easley C, Kirkpatrick P, Nat Rev Drug Discov 2007, 6, 21–22; [DOI] [PubMed] [Google Scholar]; c Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R, Molecular cancer therapeutics 2003, 2, 721–728; [PubMed] [Google Scholar]; d Laubach JP, Moreau P, San-Miguel JF, Richardson PG, Clinical cancer research : an official journal of the American Association for Cancer Research 2015, 21, 4767–4773. [DOI] [PubMed] [Google Scholar]
- [5].Yang XJ, Seto E, Nat Rev Mol Cell Biol 2008, 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6] a.Hayashi A, Horiuchi A, Kikuchi N, Hayashi T, Fuseya C, Suzuki A, Konishi I, Shiozawa T, International Journal of Cancer 2010, 127, 1332–1346; [DOI] [PubMed] [Google Scholar]; b Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN, Prostate 2004, 59, 177–189; [DOI] [PubMed] [Google Scholar]; c Kawai H, Li H, Avraham S, Jiang S, Avraham HK, International Journal of Cancer 2003, 107, 353–358. [DOI] [PubMed] [Google Scholar]
- [7].Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, Schreiber SL, Proc. Natl. Acad. Sci. U S A 1998, 95, 3519–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M, Science 2009, 325, 834–840. [DOI] [PubMed] [Google Scholar]
- [9] a.Spange S, Wagner T, Heinzel T, Kramer OH, Int J Biochem Cell Biol 2009, 41, 185–198; [DOI] [PubMed] [Google Scholar]; b Luo J, Su F, Chen D, Shiloh A, Gu W, Nature 2000, 408, 377–381; [DOI] [PubMed] [Google Scholar]; c Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, Wu CW, The Journal of biological chemistry 2000, 275, 20436–20443; [DOI] [PubMed] [Google Scholar]; d Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T, The EMBO journal 2000, 19, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hunter T, Cell 1995, 80, 225–236. [DOI] [PubMed] [Google Scholar]
- [11] a.Nalawansha DA, Gomes ID, Wambua MK, Pflum MKH, Cell Chem Biol 2017, 24, 481–492 e485; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nalawansha DA, Pflum MK, ACS chemical biology 2017, 12, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nalawansha DA, Zhang Y, Herath K, Pflum MKH, ACS chemical biology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13] a.Weerasinghe SVW, Estiu G, Wiest O, Pflum MKH, J. Med. Chem. 2008, 51, 5542–5551; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wambua MK, Nalawansha DA, Negmeldin AT, Pflum MK, Journal of medicinal chemistry 2014, 57, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14] a.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Pavletich NP, Nature 1999, 401, 188–193; [DOI] [PubMed] [Google Scholar]; b Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JWR, Mol. Cell 2013, 51, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL, J. Med. Chem. 2004, 47, 3409–3417. [DOI] [PubMed] [Google Scholar]
- [16].Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, B Jensen P, Lichenstein HS, Sehested M, The Biochemical journal 2008, 409, 581–589. [DOI] [PubMed] [Google Scholar]
- [17].Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, Jong RD, O'Connell S, Grimshaw CE, Navre M, Gangloff AR, Bioorg Med Chem Lett 2010, 20, 3142–3145. [DOI] [PubMed] [Google Scholar]
- [18].Hai Y, Christianson DW, Nature chemical biology 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19] a.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang B-C, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW, Structure 2004, 12, 1324–1334; [DOI] [PubMed] [Google Scholar]; b Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, Francesco RD, Gallinari P, Steinkuhler C, Marco SD, Proc Natl Acad Sci U S A 2004, 101, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Scholz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, Jensen LJ, Streicher W, McCarthy AR, Westwood NJ, Lain S, Cox J, Matthias P, Mann M, Bradner JE, Choudhary C, Nature biotechnology 2015, 33, 415–423. [DOI] [PubMed] [Google Scholar]
- [21].Chen K, Zhang X, Wu YD, Wiest O, Journal of the American Chemical Society 2014, 136, 11636–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li J, Staver MJ, Curtin ML, Holms JH, Frey RR, Edalji R, Smith R, Michaelides MR, Davidsen SK, Glaser KB, Life Sciences 2004, 74, 2693–2705. [DOI] [PubMed] [Google Scholar]
- [23] a.Riester D, Hildmann C, Grunewald S, Beckers T, Schwienhorst A, Biochemical and biophysical research communications 2007, 357, 439–445; [DOI] [PubMed] [Google Scholar]; b Schultz BE, Misialek S, Wu J, Tang J, Conn MT, Tahilramani R, Wong L, Biochemistry 2004, 43, 11083–11091. [DOI] [PubMed] [Google Scholar]
- [24].Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML, Methods 2005, 35, 44–53. [DOI] [PubMed] [Google Scholar]
- [25].Flint AJ, Tiganis T, Barford D, Tonks NK, Proc Natl Acad Sci U S A 1997, 94, 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S, EMBO Rep 2007, 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.