

Abstract
Background
Merozoite proteins of the malaria parasites involved in the invasion of red blood cells are selected by host immunity and their diversity is greatly influenced by changes in malaria epidemiology. In the Greater Mekong Subregion (GMS), malaria transmission is concentrated along the international borders and there have been major changes in malaria epidemiology with Plasmodium vivax becoming the dominant species in many regions. Here, we aimed to evaluate the genetic diversity of P. vivax Duffy-binding protein gene domain II (pvdbp-II) in isolates from the eastern and western borders of Myanmar, and compared it with that from global P. vivax populations.
Methods
pvdbp-II sequences were obtained from 85 and 82 clinical P. vivax isolates from the eastern and western Myanmar borders, respectively. In addition, 504 pvdbp-II sequences from nine P. vivax populations of the world were retrieved from GenBank and used for comparative analysis of genetic diversity, recombination and population structure of the parasite population.
Results
The nucleotide diversity of the pvdbp-II sequences from the Myanmar border parasite isolates was not uniform, with the highest diversity located between nucleotides 1078 and 1332. Western Myanmar isolates had a unique R391C mutation. Evidence of positive natural selection was detected in pvdbp-II gene in P. vivax isolates from the eastern Myanmar area. P. vivax parasite populations in the GMS, including those from the eastern, western, and central Myanmar as well as Thailand showed low-level genetic differentiation (FST, 0.000–0.099). Population genetic structure analysis of the pvdbp-II sequences showed a division of the GMS populations into four genetic clusters. A total of 60 PvDBP-II haplotypes were identified in 210 sequences from the GMS populations. Among the epitopes in PvDBP-II, high genetic diversity was found in epitopes 45 (379-SIFGT(D/G)(E/K)(K/N)AQQ(R/H)(R/C)KQ-393, π = 0.029) and Ia (416-G(N/K)F(I/M)WICK(L/I)-424], Ib [482-KSYD(Q/E)WITR-490, π = 0.028) in P. vivax populations from the eastern and western borders of Myanmar.
Conclusions
The pvdbp-II gene is genetically diverse in the eastern and western Myanmar border P. vivax populations. Positive natural selection and recombination occurred in pvdbp-II gene. Low-level genetic differentiation was identified, suggesting extensive gene flow of the P. vivax populations in the GMS. These results can help understand the evolution of the P. vivax populations in the course of regional malaria elimination and guide the design of PvDBP-II-based vaccine.
Keywords: Plasmodium vivax, Duffy-binding protein, Genetic diversity, Myanmar border
Background
Malaria is a major public health concern in the Great Mekong Subregion (GMS) in Asia [1]. In this region, Myanmar has the heaviest malaria burden and contributes to over 50% of the malaria cases in the GMS [2–4]. In recent years, intensified malaria control efforts have resulted in substantial improvement of the malaria situation in most GMS countries. One noticeable change in malaria epidemiology is the emerging dominant status of P. vivax in most endemic areas of the GMS. The formation of liver hypnozoites in P. vivax patients responsible for relapses requires radical treatment that targets both asexual blood stages and liver-stage hypnozoites. Currently, chloroquine–primaquine (CQ–PQ) is the frontline therapy for P. vivax in all GMS countries [5]. However, degraded efficacy of CQ–PQ for P. vivax treatment has been notified in several sites in Myanmar [5–8]. This might be among the causes responsible for the recently increased P. vivax incidence in northeastern Myanmar bordering China’s Yunnan province [5, 9]. The development of an effective malaria vaccine is considered important for integrated malaria control. However, one of the main obstacles for a successful vaccine design to enable global protection is the extensive genetic diversity of vaccine candidate genes [10]. In this regard, to design an effective malaria vaccine, it is essential to determine the genetic diversity of the vaccine targets in parasite populations from different malaria-endemic areas.
Blood-stage replication of malaria parasites is the cause of clinical malaria, and vaccines targeting blood-stage parasites are designed to alleviate the clinical symptoms. The formation of tight junction between the Plasmodium merozoite and the host red blood cell (RBC) is central to the invasion process of the parasite. This process in P. vivax involves the Duffy-binding protein (PvDBP), a microneme protein, and the Duffy antigen receptor for chemokines (DARC) on the RBC membrane [11, 12]. Although P. vivax could infect DARC-negative individuals, these cases are rare; no alternative ligands for P. vivax binding to reticulocytes have been identified yet [13–16]. The presence of naturally-acquired antibodies to PvDBP in sera of residents from endemic area that block RBC invasion suggests that this protein is a potential target for effective antibody response [17–19]. Naturally-acquired binding-inhibitory antibodies directed against PvDBP contained both strain-specific and strain-transcending components and were associated with a reduced risk of clinical P. vivax malaria. Furthermore, these antibodies against PvDBP could block erythrocyte binding of PvDBP domain II (PvDBP-II) and inhibit merozoite invasion of erythrocytes [20–24], which justifies PvDBP as one of the most promising targets for blood-stage vaccine development against P. vivax.
PvDBP is a protein of 140 kDa and is divided into seven different regions (regions I–VII). The central binding motif necessary for DARC adhesion of PvDBP is mapped to a 170 amino-acid stretch located in the N-terminal cysteine-rich region (PvDBP-II, amino acids 291–460) [25]. Pvdbp-II encoding this protein domain also shows the highest degree of genetic diversity as compared to the rest of the pvdbp sequence. Although the cysteine residues are conserved in P. vivax populations, the remaining amino acids of PvDBP-II are highly polymorphic in P. vivax field isolates from endemic areas such as Brazil, Colombia, South Korea, Thailand and Myanmar, suggesting that this region is under positive natural selection [3, 26–29]. It has been reported that these polymorphic residues within the PvDBP-II region do not alter its capacity to bind DARC, but some of them affect the immune recognition of PvDBP, suggesting that genetic diversity may be responsible for immune evasion [30, 31]. Consequently, the polymorphic nature of pvdbp-II represents a major limitation in successful design of a protective vaccine. Therefore, understanding the nature and genetic polymorphism in pvdbp-II among P. vivax isolates from different geographic areas is important for the rational design of vaccines against vivax malaria.
In this study, the pvdbp-II sequences of P. vivax isolates from eastern and western Myanmar borders were analyzed for genetic diversity, natural selection, recombination, haplotype prevalence and population differentiation, and were compared with the global pvdbp-II sequences. Whereas limited polymorphism of pvdbp existed in the field P. vivax isolates from eastern and western Myanmar, the pvdbp-II region was found to be under natural selection. Besides, the observed low-level genetic differentiation among different P. vivax populations in the GMS, based on the pvdbp-II sequence analysis, supports a general design of an effective vaccine to cover these endemic areas.
Methods
Blood sample collection
A total of 163 and 95 blood samples were collected from patients with acute P. vivax malaria attending clinics in an eastern Myanmar border township (Laiza, Kachin State) in 2016 and a western Myanmar border township (Paletwa, Chin State) in 2017, respectively (Fig. 1). Plasmodium vivax infection was confirmed by microscopic examination of thin and thick blood smears. After obtaining written informed consent from participants and guardians in case of minors, finger-prick blood (~ 100 µl) was collected on Whatman 3M filters (Whatman, Shanghai, China), dried and individually stored at − 20 °C for subsequent use. This study was approved by the ethics committees from the Department of Medical Research, Myanmar Ministry of Health and Sports, the China Medical University, and the University of South Florida.
Fig. 1.

Map showing the distribution of sampling sites including Kachin State (eastern Myanmar), Mandalay Division (central Myanmar), Chin State (western Myanmar), and Thailand
PCR, cloning and sequencing of the pvdbp-II gene
Genomic DNA was extracted from dried blood spots using a QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The pvdbp-II sequence corresponding to nucleotides 715–1704 bp (amino acid positions 239–568 aa) in the Salvador I (Sal I) reference sequence (GenBank: M37514) was amplified by PCR using primers pvdbp-II-F (5′-ACC ACG ATC TCT AGT GCT ATT ATA-3′) and pvdbp-II-R (5′-ATT TGT CAC AAC TTC CTG AGT ATT-3′). PCR reactions included 1× KOD-Plus-Neo buffer, 200 μM dNTPs, 1 mM MgSO4, 250 nM of each primer, 0.4 units of KOD-Plus-Neo DNA polymerase (Toyobo, Shanghai, China), and 1.0 μl genomic DNA template. The PCR was performed using the following conditions: 95 °C for 5 min, 44 cycles at 95 °C for 30 s, 55 °C for 30 s, and 68 °C for 60 s, followed by extension at 68 °C for 5 min. The PCR products were analyzed by 1.2% agarose gel, purified with TaKaRa MiniBEST DNA Fragment Purification Kit (TaKaRa, Dalian, China), ligated into a pMD18-T plasmid vector (TaKaRa), and transformed into Escherichia coli DH5α competent cells (TaKaRa). Colony PCR was performed with the primers M13(-20)F (5′-TGT AAA ACG ACG GCC AGT-3′) and M13R (5′-CAG GAA ACA GCT ATG ACC-3′) to identify positive clones with the correct inserts. The colony PCR products having the correct DNA fragment were sequenced using the primers pvdbp-II-F and pvdbp-II-R at a commercial laboratory (Beijing Genomics Institute, Beijing, China). To verify the sequencing accuracy, at least two clones from each isolate were sequenced in both DNA strands. Nucleotide sequences are deposited in GenBank under accession numbers MN233407-MN233488 for the western Myanmar isolates and MN233489-MN233573 for the eastern Myanmar isolates.
Genetic diversity and natural selection
To estimate within-population diversity, the DNA sequence polymorphism analysis was performed on pvdbp-II sequences from 82 western 85 eastern Myanmar border parasite isolates, respectively. Sequence alignment was performed using the CLUSTAL W program in MEGA7.0 [32] and nucleotide diversity was analyzed using the DnaSP5.10.01 software [33]. To compare the genetic diversity of pvdbp-II gene with pvdbp-II sequences in global P. vivax isolates, we retrieved from GenBank pvdbp-II sequences from Asia: India (n = 100, FJ491142.1-FJ491241.1), Iran (n = 63, EU860428.1-EU860436.1, KF318358.1, KF318359.1, KF791921.1-KF791926.1) [17, 34, 35], central Myanmar (n = 12, JN255576.1-JN255587.1) [3], South Korea (n = 13, JN989472.1-JN989484.1) [28], Sri Lanka (n = 100, GU143914-GU143949, GU143950-GU143973, GU143974-GU144013) [36], and Thailand (n = 30, EF379127.1-EF379132.1, EF379134.1, EF368159.1-EF368180.1, EF219451.1) [29]; South America: Brazil (n = 122, EU812839-EU812960) [26] and Colombia (n = 17, U50575-U50590) [27]; and Oceania: Papua New Guinea (n = 96, AF289480-AF289483, AF289635-AF289653, AF469515-AF469602) [37]. The number of haplotypes (H), haplotype diversity (Hd), nucleotide diversity (π), number of segregating sites (S), the total number of mutations (η), the average number of nucleotide differences (k) and the corresponding standard deviation for the pvdbp-II fragments were calculated using the DnaSP [33]. Additionally, the π value was also estimated on a sliding window of 90 bases with a step size of 3 bp. The rates of synonymous (dS) and non-synonymous (dN) mutations were estimated and compared by the Z-test (P < 0.05) in MEGA 7.0 using the Nei & Gojobori’s method with Jukes & Cantor correction and 1000 bootstrap replications [38]. Under purifying selection, dN will be less than dS (dN/dS < 1), whereas under positive selection, dN will exceed dS (dN/dS > 1) [38]. The Tajima’s D-test [39], Fu and Li’s D* and F* tests [40] were used to test departures from the neutral theory of evolution, with the assumption that the population size was constant [41]. Sliding window plots with a window size of 90 bp and a step size of 3 bp were also performed for Tajima’s D and Fu and Li’s D* and F* tests implemented in DnaSP [33].
Recombination and linkage disequilibrium (LD)
The recombination parameter (C), which includes the effective population size and probability of recombination between adjacent nucleotides per generation, and the minimum number of recombination events (Rm) were measured using DnaSP [42]. LD between different polymorphic sites was computed in terms of the R2 index using DnaSP for the eastern and western Myanmar border isolates.
Genetic differentiation, haplotype network, and population genetic structure analysis
The genetic differences in populations was investigated evaluating the rate of fixation (FST) by analysis of molecular variance (AMOVA) implemented in ARLEQUIN v3.5.2.2 software [43]. The Median-Joining method in NETWORK v5.0.0.3 [44] was used to establish genealogical relationship among the global PvDBP-II haplotypes. STRUCTURE 2.3.4 was used to define the genetic structure of P. vivax parasite populations from the GMS based on the pvdbp-II [45]. The Bayesian approach was employed to identify the optimum number of clusters (K). All sample data were run for values K = 2–9 using the admixture ancestry model. Each run was implemented with a ‘burn-in’ period of 50,000 iterations and 100,000 Markov Chain Monte Carlo (MCMC) replications. The most likely number K in the data was estimated by calculating Delta K-values and identifying the K-value that maximizes the log probability of data, |ln’’(K)|. The most probable K-value was then calculated according to Evanno’s method by using the webpage interface STRUCTURE Harvester (http://taylor0.biology.ucla.edu/structureHarvester/). Distruct 1.1 was used to graphically display results produced by the STRUCTURE software [46].
Analysis of polymorphism associated with B- and T-cell epitopes
To evaluate the possibility that diversity in PvDBP-II within the eastern and western Myanmar border isolates may have arisen from host’s immune pressure, the genetic diversity in predicted or known B- and T-cell epitopes and MHC-binding regions in PvDBP-II was examined [26, 30]. Polymorphism of each region was analyzed by using DnaSP as described above.
Results
Pvdbp-II genetic diversity in the Myanmar border P. vivax populations
Out of the 258 P. vivax isolates collected from the eastern (n = 95) and western (n = 163) Myanmar borders, the 990 bp pvdbp-II gene fragment covering codons 239–568 was successfully amplified and sequenced in 85 and 82 samples, respectively. The 85 eastern border samples had 21 single nucleotide polymorphisms (SNPs), of which 4 are synonymous and 17 non-synonymous (Table 1). Among them, 20 are segregating sites, and 15 are parsimony informative. The 82 western border samples contained 16 SNPs, of which 15 are non-synonymous. All 16 SNPs are segregating sites, and 14 are parsimony informative. The overall haplotype diversity for the two sample sets was similar at 0.850 and 0.930 for the eastern and western border populations, respectively (Table 1). The observed pairwise nucleotide diversity for eastern and western Myanmar isolates is 0.006 and 0.004, respectively (Table 1). The highest peaks of nucleotide diversity within the pvdbp-II region were identified between nucleotide positions 1078–1332 (codons 360–444) (Fig. 2a). Meanwhile, 40% of the polymorphic amino acid sites were also accumulated within this segment (Fig. 2b). Sliding window plots of nucleotide diversity revealed π value ranging from 0 to 0.029 for the eastern isolates and from 0 to 0.021 for the western isolates, respectively (Fig. 2). These results indicated that the central segment for codons 360–444 of the pvdbp-II gene is more polymorphic than the rest of the pvdbp domain II.
Table 1.
Nucleotide diversity of pvdbp-II in Myanmar P. vivax isolates
| Region | n | S | η | NS | SP | k | π ± SD | H | Hd ± SD |
|---|---|---|---|---|---|---|---|---|---|
| Eastern Myanmar | 85 | 20 | 21 | 17 | 4 | 5.784 | 0.006 ± 0.000 | 16 | 0.850 ± 0.021 |
| Western Myanmar | 82 | 16 | 16 | 15 | 1 | 3.694 | 0.004 ± 0.000 | 25 | 0.930 ± 0.013 |
Note: The total sequenced region includes codons 239 to 568
Abbreviations: S, number of polymorphic (segregating) sites; η, the total number of mutations; NS, number of non-synonymous polymorphisms, SP, number of synonymous polymorphisms; k, the average number of nucleotide differences, π, pairwise nucleotide diversity; H, number of haplotypes; Hd, haplotype diversity
Fig. 2.
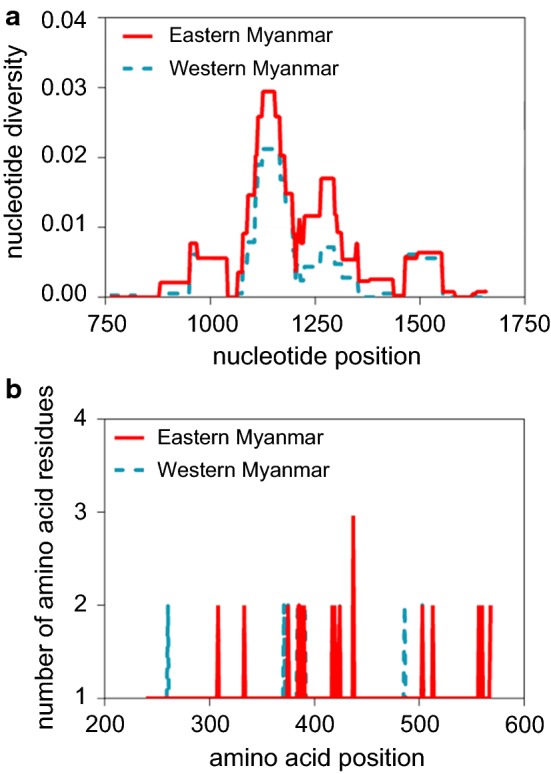
Sliding window plots of nucleotide diversity (a) and amino acid polymorphism (b) of pvdbp-II in eastern and western Myanmar populations. The analysis included 85 and 82 sequences from the eastern and western Myanmar borders, respectively. Nucleotide and amino acid positions are after the Salvador I strain
Most of the PvDBP-II amino acid changes were found in high frequencies (> 50%) in both populations, including L333F (eastern, 77.7%; western, 85.4%), D384G (eastern, 100%; western, 92.7%), R390H (eastern, 80.0%; western, 92.7%), N417K (eastern, 63.5%; western, 87.8%), and L424I (eastern, 77.7%; western, 85.4%) (Table 2). The variant E385K (eastern, 50.6%; western, 26.8%), K386N (eastern, 50.6%; western, 26.8%) and W437R (eastern, 62.4%; western, 2.4%) were observed with higher frequency in eastern Myanmar than those from western Myanmar isolates. It is interesting to note that within the commonly evaluated 299–503 aa region of PvDBP-II, 13 of the 14 non-synonymous changes in eastern Myanmar border isolates and western Myanmar isolates were previously reported, whereas the R391C (1.22 %) change was unique to western Myanmar isolates.
Table 2.
Nucleotide and amino acid changes of pvdbp-II among Myanmar isolates
| Nucleotide position | Codon | Wild-type | Mutant | Amino acid substitution | Frequency (%) | |
|---|---|---|---|---|---|---|
| Eastern Myanmar (n = 85) |
Western Myanmar (n = 82) |
|||||
| 778 | 260 | AAA | GAA | K - E | 0 | 1.22 |
| 924 | 308 | AGG | AGT | R - S | 10.59 | 2.44 |
| 997 | 333 | CTT | TTT | L - F | 50.59 | 50.00 |
| 1111 | 371 | AAA | GAA | K - E | 20.00 | 2.44 |
| 1123 | 375 | AAT | GAT | N - D | 44.71 | 21.95 |
| 1134 | 378 | CGC | CGT | R - R | 42.35 | 19.51 |
| 1151 | 384 | GAT | GGT | D - G | 100.00 | 92.68 |
| 1153 | 385 | GAA | AAA | E - K | 50.59 | 35.37 |
| 1158 | 386 | AAG | AAT | K - N | 50.59 | 26.83 |
| 1169 | 390 | CGT | CAT | R - H | 80.00 | 92.68 |
| 1251 | 417 | AAT | AAA | N - K | 63.53 | 87.80 |
| 1257 | 419 | ATA | ATG | I - M | 12.94 | 0 |
| 1270 | 424 | TTA | ATA | L - I | 77.65 | 85.37 |
| 1309 | 437 | TGG | CGG | W - R | 62.35 | 2.44 |
| 1392 | 464 | ATC | ATA | I - I | 11.76 | 0 |
| 1425 | 475 | CCA | CCC | P - P | 1.18 | 0 |
| 1456 | 486 | CAA | GAA | Q - E | 0 | 2.44 |
| 1508 | 503 | ATA | AAA | I - K | 47.06 | 48.78 |
| 1538 | 513 | ACG | AAG | T - K | 3.53 | 0 |
| 1669 | 557 | GCT | ACT | A - T | 1.18 | 0 |
| 1671 | 557 | GCT | GCC | A - A | 1.18 | 0 |
| 1679 | 560 | AAT | ATT | N - I | 1.18 | 0 |
| 1703 | 568 | GTC | GGG | V - G | 1.18 | 0 |
Notes: Number of the amino acid residues is according to Sal I sequence. Text highlighted in bold indicates non-synonymous mutations
Evidence of natural selection
The neutrality tests (Tajima’s D, and Fu and Li’s D*, and F*) did not identify significant deviation from zero in pvdbp-II in both border populations (Table 3). However, by zooming in specific regions via the sliding window analysis, both Fu and Li’s D* and F* tests detected significant negative values for the 1626–1659 bp region in the eastern Myanmar population (Fu and Li’s D* test: − 3.777, P < 0.05; Fu and Li’s F* test: − 3.694, P < 0.05), whereas Fu and Li’s F* test detected a significant positive value for the 1110–1176 bp region (1.987, P < 0.05). In addition, a significant positive Tajima’s D-value was found in the 1089–1197 bp region from the eastern border isolates (2.810, P < 0.01). All the three neutrality tests identified similar patterns in the western Myanmar population, albeit the P-values did not reach statistical significance (P > 0.05) (Fig. 3a–c).
Table 3.
Neutrality tests of pvdbp-II among Myanmar P. vivax isolates
| Region | n | dN ± SE | dS ± SE | dN/dS | D | D* | F* |
|---|---|---|---|---|---|---|---|
| Eastern Myanmar | 85 | 0.006 ± 0.002 | 0.004 ± 0.003 | 1.708 | 1.330 | − 0.841 | − 0.045 |
| Western Myanmar | 82 | 0.005 ± 0.001 | 0.001 ± 0.001) | 2.712* | 0.429 | 0.623 | 0.659 |
Abbreviations: dN/dS, the ratio of non-synonymous to synonymous mutations; D, Tajima’s D test; D*, Fu and Li’s D* value; F*, Fu and Li’s F* value; SE, standard error
*P < 0.05
Fig. 3.

Neutrality tests of pvdbp-II sequences from the eastern and western Myanmar isolates. Sliding window plots of Fu and Li’s D* (a), Fu and Li’s F* (b), and Tajima’s D (c) for the pvdbp-II sequence. Sites significantly departed from neutrality (P < 0.05, two-tailed) are indicated with circle symbols on the line. d Sliding window plots of dN/dS ratio for the pvdbp-II sequences. Nucleotide positions are after the Sal I sequence. Window length is 90 bp and step size is 3 bp
To determine whether selection has played any role in the evolution of pvdbp-II in the two parasite populations, we compared the dN and dS values (Table 3). In both populations, dN/dS > 1 was detected, but Fisher’s exact Z-test only showed significance for the western Myanmar isolates (P = 0.033). Sliding window plots of dN/dS for the pvdbp-II sequences showed a similar pattern with a significant excess of dN over dS detected at the 1116–1164 bp region (codons 372–388) in the western Myanmar population (Fig. 3d).
Recombination
In the global P. vivax populations except that from South Korea, which had recent vivax malaria resurgence, the minimum numbers of recombination events between adjacent polymorphic sites (Rm) were ≥ 5 (Table 4). The LD index R2 also declined across the analyzed region in eastern and western Myanmar populations, suggesting that intragenic recombination may also be a possible factor contributing to the increased diversity of pvdbp-II gene (Fig. 4). It is also noteworthy that the R2 value decreased much more rapidly in the western Myanmar parasites, suggesting a much large parasite population and higher level of intragenic recombination in western Myanmar.
Table 4.
Estimates of recombination events in pvdbp-II in global P. vivax populations
| Locality (n) | Ra | Rb | Rm |
|---|---|---|---|
| Eastern Myanmar (n = 85) | 0.011 | 7.3 | 5 |
| Western Myanmar (n = 82) | 0.021 | 13.9 | 7 |
| India (n = 100) | 0.024 | 16.1 | 10 |
| Iran (n = 63) | 0.014 | 9.4 | 5 |
| Central Myanmar (n = 11) | 0.191 | 128 | 7 |
| South Korea (n = 13) | 0.074 | 49.7 | 1 |
| Sri Lanka (n = 100) | 0.021 | 13.8 | 9 |
| Thailand (n = 30) | 0.152 | 102 | 7 |
| Brazil (n = 122) | 0.041 | 27.8 | 6 |
| Colombia (n = 16) | 0.071 | 47.9 | 6 |
| PNG (n = 96) | 0.017 | 11.4 | 8 |
Note: The analyzed region includes codons 292–516
Abbreviations: Ra, recombinant parameter between adjacent sites; Rb, recombinant parameter for the whole gene; Rm, minimum number of recombination events; n, total collected
Fig. 4.
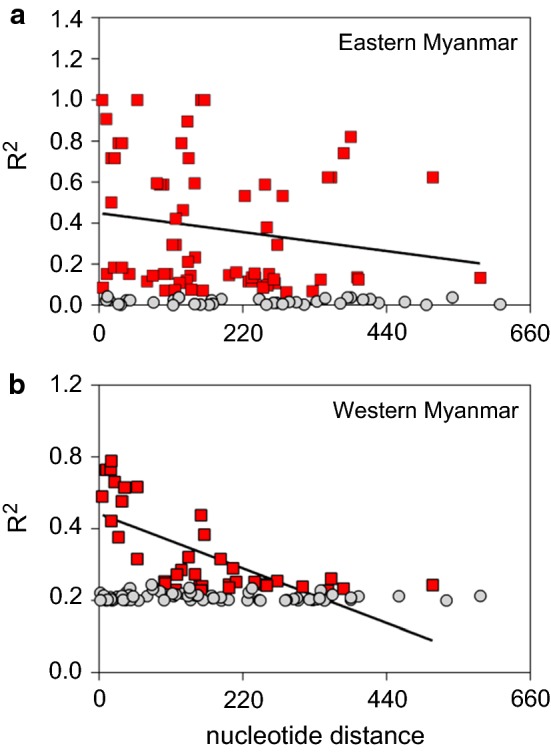
Linkage disequilibrium plots of R2 for pvdbp-II gene from eastern (a) and western (b) Myanmar isolates. Sites with significant LD (P <0.05) as detected by Fisher’s exact test are shown as red squares, whereas all others are shown as grey dots. Trace lines represent the regression lines
Population differentiation
FST values were calculated to assess the genetic differentiation of pvdbp-II among the global P. vivax populations. Though separated by more than 400 km, the eastern and western Myanmar P. vivax population had little genetic differentiation with an FST value of 0.067 (Table 5). Similarly, all P. vivax populations from the GMS had little genetic differentiation, whereas high levels of genetic differentiation were detected between the GMS populations and Asian Pacific population (PNG) and the South American population (Colombia).
Table 5.
Genetic differentiation (FST) of the pvdbp-II among 11 geographical populations
| Locality (n) | Eastern Myanmar | Western Myanmar | Central Myanmar | India | Iran | South Korea | Sri Lanka | Thailand | Brazil | Colombia |
|---|---|---|---|---|---|---|---|---|---|---|
| Eastern Myanmar (n = 85) | 0.001 | 0.151 | 0.000 | 0.000 | 0.002 | 0.000 | 0.057 | 0.000 | 0.000 | |
| Western Myanmar (n = 82) | 0.067 | 0.011 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| Central Myanmar (n = 12) | 0.033 | 0.096 | 0.026 | 0.000 | 0.003 | 0.009 | 0.877 | 0.020 | 0.000 | |
| India (n = 100) | 0.125 | 0.181 | 0.066 | 0.000 | 0.005 | 0.019 | 0.001 | 0.040 | 0.000 | |
| Iran (n = 63) | 0.225 | 0.285 | 0.113 | 0.045 | 0.000 | 0.000 | 0.000 | 0.019 | 0.000 | |
| South Korea (n = 13) | 0.187 | 0.212 | 0.155 | 0.096 | 0.172 | 0.014 | 0.001 | 0.002 | 0.000 | |
| Sri Lanka (n = 100) | 0.150 | 0.204 | 0.096 | 0.018 | 0.055 | 0.085 | 0.000 | 0.003 | 0.000 | |
| Thailand (n = 30) | 0.031 | 0.099 | 0.000 | 0.075 | 0.141 | 0.146 | 0.010 | 0.000 | 0.000 | |
| Brazil (n = 122) | 0.138 | 0.180 | 0.073 | 0.013 | 0.022 | 0.112 | 0.028 | 0.083 | 0.000 | |
| Colombia (n = 16) | 0.328 | 0.431 | 0.241 | 0.199 | 0.185 | 0.337 | 0.203 | 0.237 | 0.176 | |
| PNG (n = 96) | 0.261 | 0.310 | 0.197 | 0.169 | 0.176 | 0.213 | 0.171 | 0.179 | 0.168 | 0.239 |
Notes: The nucleotide positions 784–1458 of pvdbp-II were used for population differentiation analysis. FST values are shown below the diagonal and P-values are shown above the diagonal
Abbreviation: n, total collected
Relationship of parasites from the GMS
To identify the population structure of the GMS parasite populations, STRUCTURE analysis was performed using parasite populations from the eastern and western borders of Myanmar, central Myanmar (from the Mandalay region) and Thailand. Analysis of the |Ln’’(K)| curve, Delta K plot and the distribution of clusters amongst haplotypes indicated that the haplotypes were optimally grouped into four clusters (K = 4; Fig. 5). All four parasite populations from the GMS had admixed haplotypes, with the most common haplotypes found with a percentage of 79.1%, 76.1%, 81.2%, and 70.1% in western Myanmar border, eastern Myanmar border, central Myanmar and Thailand populations, respectively.
Fig. 5.

STRUCTURE analysis of PvDBP-II haplotypes. a |Ln’’(K)| plot. b Delta K Plot. c Clustering patterns of the PvDBP-II haplotypes (K = 4)
A haplotype network was constructed to establish the relationships among the pvdbp-II haplotypes from the four GMS P. vivax populations mentioned above. A total of 60 haplotypes were identified from 210 pvdbp-II sequences with haplotype prevalence ranging from 0.48 to 17.14%, of which 65% was represented by single parasite isolates. These haplotypes could be roughly grouped into four clusters (Fig. 6). Parasites with the Sal I reference haplotype (H1) had a 2.3% prevalence, and it was only detected in the western and central Myanmar parasite populations. Haplotype 4 (H4) was shared among all GMS populations, with an overall prevalence of 4.8%. Haplotypes 8, 28, 34, and 37 were shared between two of the four populations, whereas Haplotypes 15, 25, and 31 were shared among the eastern Myanmar, western Myanmar and Thai populations. Of these haplotypes, H25 has an observed frequency of 17.14%, and it is the dominant haplotype of the eastern Myanmar parasite population with a frequency of 29.41% (Fig. 6).
Fig. 6.

Network analysis of PvDBP-II haplotypes. Haplotypes composed of 224 amino acids were analyzed using the Median-Joining algorithm implemented in Network version 5.0.0.3 software. Pies represent the haplotypes and lines indicate connections between them. The size of each pie indicates the frequency of a particular haplotype. Colors of each pie indicate different countries. Four clusters are highlighted with dashed circles
Polymorphisms associated with B- and T-cell epitopes
To determine whether there is a link between positive natural selection and host immune pressure, we examined the genetic polymorphism in identified or predicted B- and T-cell epitopes and MHC-binding regions [26, 30]. All the predicted B- and T-cell epitopes and MHC binding regions were polymorphic, and had dN > dS (Table 6). Meanwhile, significant P-value of dN > dS was detected in epitopes 45 (P = 0.023), 48 (P = 0.028) and Ia (P = 0.036) of PvDBP-II protein by Z-test (Table 6). Particularly, high-level nucleotide diversity was found in epitopes 45 (π = 0.029) and Ia (π = 0.028), which contained polymorphic residues at 339, 340, 341, 345, and 346, as well as 372, 374, and 379, respectively. Though the Tajima’s D-values for the two epitopes were all positive, they were not significant. These results are consistent with the hypothesis that natural selection acts on epitopes in PvDBP-II and is responsible for the observed diversity of pvdbp-II [3, 26, 36].
Table 6.
Polymorphisms observed in each pvdbp-II epitope sequence
| Epitope name | Epitope | S | M | H | Hd ± SD | π ± SD | dN-dS | Tajima’s D |
|---|---|---|---|---|---|---|---|---|
| 5 | T/B | 1 | 1 | 2 | 0.124 ± 0.033 | 0.003 ± 0.007 | 0.003 | − 0.308 |
| 16 | T/B | 1 | 1 | 2 | 0.503 ± 0.000 | 0.012 ± 0.000 | 0.016 | 1.942 |
| 18 | B | 1 | 1 | 2 | 0.503 ± 0.000 | 0.011 ± 0.000 | 0.014 | 1.942 |
| 20 | T/B | 1 | 1 | 2 | 0.503 ± 0.000 | 0.015 ± 0.000 | 0.019 | 1.942 |
| 45 | B | 5 | 5 | 8 | 0.674 ± 0.024 | 0.029 ± 0.001 | 0.036* | 0.966 |
| 48 | B | 2 | 2 | 3 | 0.266 ± 0.039 | 0.007 ± 0.001 | 0.008* | − 0.333 |
| 66 | T | 1 | 1 | 2 | 0.273 ± 0.039 | 0.006 ± 0.001 | 0.008 | 0.576 |
| Ia | MHC class I | 3 | 3 | 4 | 0.491 ± 0.041 | 0.028 ± 0.003 | 0.034* | 0.757 |
| Ib | MHC class I | 1 | 1 | 2 | 0.024 ± 0.016 | 0.001 ± 0.001 | 0.001 | − 0.901 |
| Ic | MHC class I | 1 | 1 | 2 | 0.502 ± 0.005 | 0.012 ± 0.000 | 0.015 | 1.937 |
| IIa | MHC class II | 1 | 1 | 2 | 0.373 ± 0.034 | 0.012 ± 0.001 | 0.016 | 1.169 |
| IIb | MHC class II | 2 | 2 | 3 | 0.375 ± 0.042 | 0.015 ± 0.002 | 0.018 | 0.181 |
Notes: Epitope sequences are: 5 [299-VNNTDTNFH(R/S)DITFR-313]; 16 [321-LIYDAAVEGDLL(L/F)KL-335]; 18 [325-AAVEGDLL(L/F)KLNNYR-339]; 20 [329-GDLL(L/F)KLNNYRYNKD-343]; 45 [379-SIFGT(D/G)(E/K)(K/N)AQQ(R/H)(R/C)KQ-393]; 48 [385-(E/K)(K/N)AQQ(R/H)(R/C)KQWWNESK-399]; and 66 [421-ICK(L/I)NVAVNIEPQIY-435]. In silico predicted promiscuous epitopes: Ia [416-G(N/K)F(I/M)WICK(L/I)-424]; Ib [482-KSYD(Q/E)WITR-490]; Ic [497-VLSNKF(I/K)SVKNAEK-510]; IIa [408-YSVKKRLKG(N/K)-417]; and IIb [418-F(I/M)WICK(L/I)NV-426]
Abbreviations: S, number of polymorphic (segregating) sites; M, total number of mutations; H, number of haplotypes; Hd, haplotype diversity; π, observed average pairwise nucleotide diversity; dN, rate of non-synonymous mutations; dS, rate of synonymous mutations; B, B-cell epitope; T, T-cell epitope; SD, standard deviation
*P < 0.05
Discussion
Polymorphic antigens with the presence of variant haplotypes in different endemic areas complicate the development of an effective malaria vaccine. PvDBP is an essential ligand involved in the invasion of RBCs by the P. vivax parasites, and it appears to have similar drawbacks. Assessment of the level of diversity in the pvdbp-II region involved in binding to the Duffy antigen within and between populations helps guide the development of a PvDBP-based vaccine. In this study, we determined the genetic diversity and molecular evolution of pvdbp domain II in two Myanmar border populations and compared them with other parasite populations in the GMS and elsewhere in the world. Haplotype diversity (Hd) from the eastern (0.850) and western (0.930) Myanmar populations were at the similar level or slightly lower than other endemic areas such as PNG (0.936), Brazil (0.934), Colombia (0.985), India (0.921), Thailand (0.983) and Iran (0.944) [17, 27–29, 34, 35, 37]. In addition, the two highest peaks of nucleotide diversity within the pvdbp-II sequences in the Myanmar border parasite isolates were localized between Cysteine 5 and 7 of PvDBP-II, which was consistent with several previous reports [28, 30, 31, 47].
The D384G mutation is fixed in the eastern Myanmar border population and reached 92.68% in the western Myanmar isolates. D384G is also highly prevalent in isolates from central Myanmar (85.2%), Thailand (76.7%), Sri Lanka (94%), Iran (61.3%), and Brazil (85%), but it is not prevalent in PNG and Colombia [17, 27–29, 34, 35, 37]. Although eastern and western Myanmar isolates had similar amino acid changes with populations from central Myanmar and Thailand, some mutations found in the Thai isolates (S351C, I367T, S398T, T404R, Q433K and R436T) and central Myanmar isolates (I310L, K386Q, R391H, K455I, K473R, P475A, C477G, Q486E, R490K, D528G, V533M, K541T and A545V) were not identified in either eastern or western Myanmar isolates [3, 29]. It has been reported that amino acids 417, 437 and 503 are responsible for forming a discontinuous epitope of PvDBP-II, which was also the main target of inhibitory antibodies [48, 49]. In this regard, polymorphisms at these residues, either single or combined, could affect the binding efficiency of inhibitory antibodies to PvDBP and help the parasites to evade host immune attacks. All these three mutations were found in eastern and western Myanmar isolates. The N417K mutation was dominant with high frequencies in both eastern (63.5%) and western (87.8%) Myanmar, whereas the W437R mutation was found considerably high in the eastern (62%) but low (2%) in western Myanmar. The I503K variant was present at a similar prevalence in the eastern (47%) and western Myanmar (49%) populations. All these mutations suggest that antibody responses against PvDBP in these regions might be compromised.
The pvdbp-II has been found to be subject to strong selection in previous studies [17, 27–29, 34, 35, 37]. Although all neutrality tests and the dN/dS test did not identify significant deviation from neutral in the overall pvdbp-II region in the Myanmar parasite populations, sliding window analyses of Fu and Li’s F* test and Tajima’s D test did identify a region of pvdbp-II gene that might be under positive selection (e.g., codons 372–388). These results were further supported by the high degrees of polymorphism in the B- and T-cell epitopes in the global P. vivax populations [26]. In particular, high-level genetic diversity and positive selection were identified in peptide Ia, predicted to be exposed on the surface of the PvDBP protein [26]. Significant positive values observed for the neutrality tests for the codons 363–399 and codons 370–392 regions of pvdbp-II suggest a decrease in population size. This appears to be consistent with the scaling up of malaria control in these endemic areas, which has resulted in substantial declines of malaria incidence in recent years. However, the region of codons 542–553 did not follow the same trends with the Fu and Li’s D* test, which showed a significant negative value for this domain, suggesting an excess of singletons, probably generated by particular selective sweeps. Thereby, the codons 542–553 region of pvdbp-II may be necessary for maintaining the binding ability of PvDBP-II to DARC. Taken together, the current study supports the theory that strong balancing selection probably imposed by host immunity may have occurred in the pvdbp-II region in the GMS parasite populations.
The high value of the recombination parameters observed in global populations suggested that meiotic recombination might have occurred in PvDBP-II. Recombination contributes to genetic diversity of P. vivax and may also be the cause for the variation of pvdbp-II as reported in several previous studies [26, 50]. The existence of recombination events and the decline of LD index R2 with increasing distance between nucleotide sites support that meiotic recombination plays a role in generating diversity in pvdbp-II among the Myanmar parasite isolates. This result further corroborated previous reports from other P. vivax endemic regions [3, 17, 26–28, 36]. The relatively lower number of the recombination events observed in the eastern Myanmar border isolates might be correlated with decreased P. vivax transmission intensity as this region is moving towards malaria elimination.
The FST index was used to access population differentiation due to genetic structure, and FST values over 0.25 generally refer to highly differentiated populations [51]. Whereas major population differentiation was observed between geographically well-separated parasite populations (e.g. GMS vs Colombia, GMS vs PNG), little population differentiation was observed in parasite from the GMS, as evidenced by the low FST values. This may reflect extensive gene flow among these GMS parasite populations in the recent past, which is further boosted by intensive human migration between these GMS countries, especially in the border regions [2]. Both STRUCTURE and network analyses further confirmed the close relationships among the four GMS populations. The PvDBP-II haplotypes were grouped into four closely related clusters and are shared by all sites.
Genetic diversity of vaccine candidate antigens usually causes poor clinical efficacy of malaria vaccines [52, 53]. Thus, an effective PvDBP-II vaccine should include alleles that induce host immune responses that are sufficiently broad to cover the existing antigenic diversity. However, because of higher genetic diversity of P. vivax compared to P. falciparum, generating a broad cross-reactive immune response against highly polymorphic asexual stage antigens faces even greater challenges [54]. Our observation that only 15% of PvDBP-II haplotypes were shared among the four populations from the GMS suggests that antigenic diversity will need to be taken into account for a PvDBP-based vaccine. In addition, the Sal I PvDBP-II haplotype was restricted to western and central Myanmar populations at a low frequency (4.76%), indicating that a PvDBP-II vaccine designed based on this reference strain may not work in the GMS. It is noteworthy that the GMS parasite populations did show highly prevalent haplotypes (e.g. H25 at 17.14%), which may serve as the starting point for vaccine development.
Conclusions
This study shows that pvdbp gene is genetically diverse in both the eastern and western Myanmar P. vivax isolates, which is comparable to global P. vivax populations. Part of the pvdbp-II domain is under positive selection, while multiple recombination events further favor diversity. Little genetic differentiation identified among the four populations in the GMS suggests extensive gene flow between these areas. These findings provide important information for understanding the P. vivax population structure and its evolution in endemic areas of the GMS, and allow investigators to select dominant haplotypes for designing an effective blood-stage vaccine.
Acknowledgements
We would like to thank patients from the surrounding villages for participating in this study and providing the blood samples.
Abbreviations
- RBC
red blood cell
- DARC
Duffy antigen receptor for chemokines
- PvDBP-II
Plasmodium vivax Duffy-binding protein region II
- Sal I
Salvador I
- FST
fixation index
- LD
linkage disequilibrium
- Rm
recombination event
- dN
mean number of non-synonymous substitutions per non-synonymous site
- dS
mean number of synonymous substitutions per synonymous site
- PNG
Papua New Guinea
- MCMC
Markov Chain Monte Carlo
- SNP
single nucleotide polymorphism
Authors’ contributions
XZ, YC and LC designed the studies and drafted the manuscript. MTS and MPK participated in the collection of clinical parasite samples. YH, LW, CY and HF took part in the genomic DNA extraction, and PCR amplification. YH, GN and XZ participated in sequence analysis and writing of the manuscript. YC and LC revised the final manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health USA (U19AI089672).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. The pvdbp-II sequences of field isolates obtained in this study were submitted to the GenBank database under the accession numbers MN233407-MN233488 for the western Myanmar parasites and MN233489-MN233573 for the eastern Myanmar parasites.
Ethics approval and consent to participate
The study protocol was approved by the ethics committees from the Department of Medical Research, Myanmar Ministry of Health and Sports, China Medical University, and University of South Florida. Written informed consent/assent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yubing Hu, Email: 985573879@qq.com.
Lin Wang, Email: 370798306@qq.com.
Huguette Gaelle Ngassa Mbenda, Email: ngassambenda@mail.usf.edu.
Myat Thu Soe, Email: dr.myatthusoe@gmail.com.
Chunyun Yu, Email: 284670789@qq.com.
Hui Feng, Email: fenghuisy@yahoo.com.
Myat Phone Kyaw, Email: kyaw606@gmail.com.
Liwang Cui, Email: lcui@health.usf.edu.
Xiaotong Zhu, Email: xtzhu@cmu.edu.cn.
Yaming Cao, Email: ymcao@cmu.edu.cn.
References
- 1.WHO . World malaria report. Geneva: World Health Organization; 2015. [Google Scholar]
- 2.Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, et al. Malaria in the Greater Mekong Subregion: heterogeneity and complexity. Acta Trop. 2012;121:227–239. doi: 10.1016/j.actatropica.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ju HL, Kang JM, Moon SU, Kim JY, Lee HW, Lin K, et al. Genetic polymorphism and natural selection of Duffy binding protein of Plasmodium vivax Myanmar isolates. Malar J. 2012;11:60. doi: 10.1186/1475-2875-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray CJ, Ortblad KF, Guinovart C, Lim SS, Wolock TM, Roberts DA, et al. Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:1005–1070. doi: 10.1016/S0140-6736(14)60844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan L, Wang Y, Parker DM, Gupta B, Yang Z, Liu H, et al. Therapeutic responses of Plasmodium vivax malaria to chloroquine and primaquine treatment in northeastern Myanmar. Antimicrob Agents Chemother. 2015;59:1230–1235. doi: 10.1128/AAC.04270-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Htun MW, Mon NCN, Aye KM, Hlaing CM, Kyaw MP, Handayuni I, et al. Chloroquine efficacy for Plasmodium vivax in Myanmar in populations with high genetic diversity and moderate parasite gene flow. Malar J. 2017;16:281. doi: 10.1186/s12936-017-1912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nyunt MH, Han JH, Wang B, Aye KM, Aye KH, Lee SK, et al. Clinical and molecular surveillance of drug resistant vivax malaria in Myanmar (2009–2016) Malar J. 2017;16:117. doi: 10.1186/s12936-017-1770-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guthmann JP, Pittet A, Lesage A, Imwong M, Lindegardh N, Min Lwin M, et al. Plasmodium vivax resistance to chloroquine in Dawei, southern Myanmar. Trop Med Int Health. 2008;13:91–98. doi: 10.1111/j.1365-3156.2007.01978.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhou G, Lo E, Zhong D, Wang X, Wang Y, Malla S, et al. Impact of interventions on malaria in internally displaced persons along the China-Myanmar border: 2011–2014. Malar J. 2016;15:471. doi: 10.1186/s12936-016-1512-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takala SL, Plowe CV. Genetic diversity and malaria vaccine design, testing and efficacy: preventing and overcoming ‛vaccine resistant malariaʼ. Parasite Immunol. 2009;31:560–573. doi: 10.1111/j.1365-3024.2009.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JH, Sim BK, Dolan SA, Fang X, Kaslow DC, Miller LH. A family of erythrocyte binding proteins of malaria parasites. Proc Natl Acad Sci USA. 1992;89:7085–7089. doi: 10.1073/pnas.89.15.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnwell JW, Nichols ME, Rubinstein P. In vitro evaluation of the role of the Duffy blood group in erythrocyte invasion by Plasmodium vivax. J Exp Med. 1989;169:1795–1802. doi: 10.1084/jem.169.5.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan JR, Stoute JA, Amon J, Dunton RF, Mtalib R, Koros J, et al. Evidence for transmission of Plasmodium vivax among a duffy antigen negative population in western Kenya. Am J Trop Med Hyg. 2006;75:575–581. doi: 10.4269/ajtmh.2006.75.575. [DOI] [PubMed] [Google Scholar]
- 14.Menard D, Barnadas C, Bouchier C, Henry-Halldin C, Gray LR, Ratsimbasoa A, et al. Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc Natl Acad Sci USA. 2010;107:5967–5971. doi: 10.1073/pnas.0912496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurtz N, Mint Lekweiry K, Bogreau H, Pradines B, Rogier C, Ould Mohamed Salem Boukhary A, et al. Vivax malaria in Mauritania includes infection of a Duffy-negative individual. Malar J. 2011;10:336. doi: 10.1186/1475-2875-10-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oboh MA, Badiane AS, Ntadom G, Ndiaye YD, Diongue K, Diallo MA, et al. Molecular identification of Plasmodium species responsible for malaria reveals Plasmodium vivax isolates in Duffy negative individuals from southwestern Nigeria. Malar J. 2018;17:439. doi: 10.1186/s12936-018-2588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valizadeh V, Zakeri S, Mehrizi AA, Djadid ND. Non-allele specific antibody responses to genetically distinct variant forms of Plasmodium vivax Duffy binding protein (PvDBP-II) in Iranians exposed to seasonal malaria transmission. Acta Trop. 2014;136:89–100. doi: 10.1016/j.actatropica.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Zakeri S, Babaeekhou L, Mehrizi AA, Abbasi M, Djadid ND. Antibody responses and avidity of naturally acquired anti-Plasmodium vivax Duffy binding protein (PvDBP) antibodies in individuals from an area with unstable malaria transmission. Am J Trop Med Hyg. 2011;84:944–950. doi: 10.4269/ajtmh.2011.11-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceravolo IP, Bruna-Romero O, Braga EM, Fontes CJ, Brito CF, Souza JM, et al. Anti-Plasmodium vivax duffy binding protein antibodies measure exposure to malaria in the Brazilian Amazon. Am J Trop Med Hyg. 2005;72:675–681. doi: 10.4269/ajtmh.2005.72.675. [DOI] [PubMed] [Google Scholar]
- 20.Nicolete VC, Frischmann S, Barbosa S, King CL, Ferreira MU. Naturally acquired binding-inhibitory antibodies to Plasmodium vivax Duffy binding protein and clinical immunity to malaria in rural Amazonians. J Infect Dis. 2016;214:1539–1546. doi: 10.1093/infdis/jiw407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valizadeh V, Zakeri S, Mehrizi AA, Mirkazemi S, Djadid ND. Natural acquired inhibitory antibodies to Plasmodium vivax Duffy binding protein (PvDBP-II) equally block erythrocyte binding of homologous and heterologous expressed PvDBP-II on the surface of COS-7 cells. Med Microbiol Immunol. 2016;205:85–95. doi: 10.1007/s00430-015-0429-7. [DOI] [PubMed] [Google Scholar]
- 22.Michon P, Fraser T, Adams JH. Naturally acquired and vaccine-elicited antibodies block erythrocyte cytoadherence of the Plasmodium vivax Duffy binding protein. Infect Immun. 2000;68:3164–3171. doi: 10.1128/IAI.68.6.3164-3171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimberg BT, Udomsangpetch R, Xainli J, McHenry A, Panichakul T, Sattabongkot J, et al. Plasmodium vivax invasion of human erythrocytes inhibited by antibodies directed against the Duffy binding protein. PLoS Med. 2007;4:e337. doi: 10.1371/journal.pmed.0040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceravolo IP, Souza-Silva FA, Fontes CJ, Braga EM, Madureira AP, Krettli AU, et al. Inhibitory properties of the antibody response to Plasmodium vivax Duffy binding protein in an area with unstable malaria transmission. Scand J Immunol. 2008;67:270–278. doi: 10.1111/j.1365-3083.2007.02059.x. [DOI] [PubMed] [Google Scholar]
- 25.Ranjan A, Chitnis CE. Mapping regions containing binding residues within functional domains of Plasmodium vivax and Plasmodium knowlesi erythrocyte-binding proteins. Proc Natl Acad Sci USA. 1999;96:14067–14072. doi: 10.1073/pnas.96.24.14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sousa TN, Tarazona-Santos EM, Wilson DJ, Madureira AP, Falcao PR, Fontes CJ, et al. Genetic variability and natural selection at the ligand domain of the Duffy binding protein in Brazilian Plasmodium vivax populations. Malar J. 2010;9:334. doi: 10.1186/1475-2875-9-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ampudia E, Patarroyo MA, Patarroyo ME, Murillo LA. Genetic polymorphism of the Duffy receptor binding domain of Plasmodium vivax in Colombian wild isolates. Mol Biochem Parasitol. 1996;78:269–272. doi: 10.1016/S0166-6851(96)02611-4. [DOI] [PubMed] [Google Scholar]
- 28.Ju HL, Kang JM, Moon SU, Bahk YY, Cho PY, Sohn WM, et al. Genetic diversity and natural selection of Duffy binding protein of Plasmodium vivax Korean isolates. Acta Trop. 2013;125:67–74. doi: 10.1016/j.actatropica.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Gosi P, Khusmith S, Khalambaheti T, Lanar DE, Schaecher KE, Fukuda MM, et al. Polymorphism patterns in Duffy-binding protein among Thai Plasmodium vivax isolates. Malar J. 2008;7:112. doi: 10.1186/1475-2875-7-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cole-Tobian J, King CL. Diversity and natural selection in Plasmodium vivax Duffy binding protein gene. Mol Biochem Parasitol. 2003;127:121–132. doi: 10.1016/S0166-6851(02)00327-4. [DOI] [PubMed] [Google Scholar]
- 31.Xainli J, Adams JH, King CL. The erythrocyte binding motif of plasmodium vivax duffy binding protein is highly polymorphic and functionally conserved in isolates from Papua New Guinea. Mol Biochem Parasitol. 2000;111:253–260. doi: 10.1016/S0166-6851(00)00315-7. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 34.Babaeekho L, Zakeri S, Djadid ND. Genetic mapping of the duffy binding protein (DBP) ligand domain of Plasmodium vivax from unstable malaria region in the Middle East. Am J Trop Med Hyg. 2009;80:112–118. doi: 10.4269/ajtmh.2009.80.112. [DOI] [PubMed] [Google Scholar]
- 35.Valizadeh V, Zakeri S, Mehrizi AA, Djadid ND. Population genetics and natural selection in the gene encoding the Duffy binding protein II in Iranian Plasmodium vivax wild isolates. Infect Genet Evol. 2014;21:424–435. doi: 10.1016/j.meegid.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Premaratne PH, Aravinda BR, Escalante AA, Udagama PV. Genetic diversity of Plasmodium vivax Duffy Binding Protein II (PvDBPII) under unstable transmission and low intensity malaria in Sri Lanka. Infect Genet Evol. 2011;11:1327–1339. doi: 10.1016/j.meegid.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 37.Fazzini PF, Marchi F, Torrini S. Letter: Energy levels of commercial defibrillators. Am Heart J. 1975;90:409. doi: 10.1016/0002-8703(75)90336-1. [DOI] [PubMed] [Google Scholar]
- 38.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 39.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 42.Hudson RR. Estimating the recombination parameter of a finite population model without selection. Genet Res. 1987;50:245–250. doi: 10.1017/S0016672300023776. [DOI] [PubMed] [Google Scholar]
- 43.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 44.Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 45.Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9:1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. doi: 10.1046/j.1471-8286.2003.00566.x. [DOI] [Google Scholar]
- 47.Tsuboi T, Kappe SH, Al-Yaman F, Prickett MD, Alpers M, Adams JH. Natural variation within the principal adhesion domain of the Plasmodium vivax duffy binding protein. Infect Immun. 1994;62:5581–5586. doi: 10.1128/iai.62.12.5581-5586.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.VanBuskirk KM, Cole-Tobian JL, Baisor M, Sevova ES, Bockarie M, King CL, et al. Antigenic drift in the ligand domain of Plasmodium vivax duffy binding protein confers resistance to inhibitory antibodies. J Infect Dis. 2004;190:1556–1562. doi: 10.1086/424852. [DOI] [PubMed] [Google Scholar]
- 49.Sousa TN, Ceravolo IP, Fernandes Fontes CJ, Couto A, Carvalho LH, Brito CF. The pattern of major polymorphisms in the Duffy binding protein ligand domain among Plasmodium vivax isolates from the Brazilian Amazon area. Mol Biochem Parasitol. 2006;146:251–254. doi: 10.1016/j.molbiopara.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Martinez P, Suarez CF, Cardenas PP, Patarroyo MA. Plasmodium vivax Duffy binding protein: a modular evolutionary proposal. Parasitology. 2004;128:353–366. doi: 10.1017/S0031182003004773. [DOI] [PubMed] [Google Scholar]
- 51.Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002;11:155–165. doi: 10.1046/j.0962-1083.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- 52.Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, et al. A field trial to assess a blood-stage malaria vaccine. N Engl J Med. 2011;365:1004–1013. doi: 10.1056/NEJMoa1008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz L, Brown GV, Genton B, Moorthy VS. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar J. 2012;11:11. doi: 10.1186/1475-2875-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44:1046–1050. doi: 10.1038/ng.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article. The pvdbp-II sequences of field isolates obtained in this study were submitted to the GenBank database under the accession numbers MN233407-MN233488 for the western Myanmar parasites and MN233489-MN233573 for the eastern Myanmar parasites.