

Abstract
Triple negative breast cancer (TNBC) is the most aggressive breast cancer subtype. Currently, no targeted treatment is available for TNBC, and the most common clinical therapy is tumor resection, which often promotes metastasis risks. Strong evidence suggests that the lymphatic metastasis is mediated by the C-C chemokine receptor type 7 (CCR7)/C-C motif chemokine ligand 21 crosstalk between tumor cells and the lymphatic system. It is hypothesized that CCR7 is a key immune modulator in the tumor microenvironment and the local blockade of CCR7 could effectively inhibit TNBC lymphatic metastasis. Accordingly, a plasmid encoding an antagonistic CCR7 affinity protein-CCR7 trap is delivered by tumor targeting nanoparticles in a highly metastatic 4T1 TNBC mouse model. Results show that CCR7 traps are transiently expressed, locally disrupt the signaling pathways in the tumor site, and efficiently inhibit TNBC lymphatic metastasis, without inducing immunosuppression as observed in systemic therapies using CCR7 monoclonal antibody. Significantly, upon applying CCR7 trap therapy prior to tumor resection, a 4T1 TNBC mouse model shows good prognosis without any further metastasis and relapse. In addition, CCR7 trap therapy efficiently inhibits the lymphatic metastasis in a B16F10 melanoma mouse model, indicating its great potential for various metastatic diseases treatment.
Keywords: CCR7, lymphatic metastasis, trap protein, triple negative breast cancer, tumor resection
1. Introduction
Breast cancer is the second leading deadly cancer for women. According to the American Cancer Society, in 2017 there were over 252 710 women in the United States diagnosed with breast cancer and more than 40 500 died.[1] Among all the different subtypes, TNBC, characterized by tumors that do not express estrogen receptor (ER), progesterone receptor (PR), or HER-2 genes, represents the most aggressive one, with poorer prognosis, higher recurrence rate, and higher metastasis incidence than any other non-TNBC subtypes.[2] Since mammary tumor cells lack the necessary receptors, common treatments such as hormone therapy and receptor-targeted drugs are ineffective for TNBC. The only approved systemic treatment is chemotherapy that shows short-lived response and significant toxicity. Currently, the most prevalent clinical therapy for TNBC is tumor resection. Although the surgery removes primary tumor and reduces the level of circulating tumor cells (CTCs), it also brings about high risks of hastening metastasis.[3] Presumably, many wound healing mediators produced postoperatively, including transforming growth factor beta (TGF-β) and fibroblast growth factor 2 (FGF-2), prominently increase the metastatic tumor growth.[4] Reports have shown that surgical trauma promoted CTCs implantation and growth, and accordingly accelerated tumor metastasis. In the clinic, most women with breast cancer do not die from primary tumor, but instead from the metastasis after resection.[5] Since metastasis after resection is the leading cause of mortality for TNBC, a preventive therapy prior to tumor resection is urgently needed.[6]
When TNBC metastasis occurs, mammary tumor cells frequently migrate through lymphatic system with the very first arrival at the sentinel lymph node (LN) and then to the distal sites.[7] Strong evidence suggests that the lymphatic metastasis is mediated by CCR7/CCL21 crosstalk between tumor cells and lymphatic endothelial cells (LECs).[8] CCR7 is a cell surface chemokine receptor, which is expressed by both tumor cells for lymphatic metastasis and various lymphoid tissues for immune responses. As one of the endogenous chemokine ligands for CCR7, CCL21, which is produced and secreted from LECs, binds CCR7 and recruits CCR7 positive tumor cells toward lymphatic system. Indeed, it has been reported that mammary tumor cells overexpressing CCR7 exhibited enhanced lymphatic metastasis due to the active chemotaxis between CCR7/CCL21 crosstalk.[9] Therefore, we hypothesized that CCR7/CCL21 is the key signaling pathway for TNBC lymphatic metastasis, and blockade of CCR7/CCL21 interaction is a promising strategy to prevent the lymphatic metastasis of TNBC.
To inhibit the CCR7/CCL21 crosstalk, development of small molecule antagonists has hardly shown any success, while therapeutic monoclonal antibodies (mAbs) are much more prospective. Since the efficacy of mAbs against cell surface-restricted chemokine receptors is presumably higher than those against delocalized secreted chemokines, the development of CCR7 mAb drugs has drawn some attention.[10] A CCR7 mAb therapy has been tested in a subcutaneous human mantle cell lymphoma mouse model. It was found that mice treated with CCR7 mAb developed substantially less metastasis to distant organs compared with untreated ones, suggesting the importance and effectiveness of blocking CCR7 for preventing tumor metastasis.[11] However, CCR7 is also expressed in various lymphoid tissues and activates B and T lymphocytes, and plays pivotal roles in multiple physiological processes, including the stimulation of dendritic cells (DCs) maturation and homing and trafficking of T cells to various secondary lymphoid organs.[12] The systemic application of CCR7 mAb could induce severe immunosuppressive side effect through blocking CCR7 expressed on dendritic cells or lymphocytes.[13] Therefore, a local and transient blockade of CCR7 in the TME would be of great value for the prevention of TNBC lymphatic metastasis without the unwanted side effects.
Herein, we describe an antibody-like affinity molecule named as CCR7 trap that antagonistically binds and disrupts the function of CCR7 (Scheme 1). The small size of the CCR7 trap allows for efficient tumor penetration and rapid clearance, which could presumably result in significantly reduced immune-related systemic toxicity. To achieve the local and transient expression in TME, we delivered a plasmid encoding CCR7 trap into the tumor site, by using a tumor targeting nanoparticle platform—lipid-protamine-DNA (LPD) nanoparticles (NPs).[14] Accordingly, CCR7 traps were produced by and secreted out of the tumor cells and took function locally at the TME (Scheme 1). We hypothesized that CCR7 trap therapy can be a good complement prior to tumor resection to efficiently prevent the TNBC lymphatic metastasis.
Scheme 1.

Mechanism of the locally expressed CCR7 trap blocking the metastasis of tumor cells into lymphatic system.
2. Results and Discussion
2.1. CCR7 Overexpression Plays a Key Role in the Metastatic Human Breast Cancer
Breast cancers fall into two major categories: invasive (≈90%) and noninvasive.[15] Noninvasive breast cancers take place in a specific location (lobule or duct) and remain in situ without spreading to surrounding tissues. However, without timely and proper treatments, noninvasive breast cancers are destined to become invasive over time, indicating a high metastatic potential of breast cancer even in the noninvasive stage.[16] When breast cancers turn into invasive, mammary tumor cells break through normal breast tissue barriers, invade nearby tissues, and metastasize through lymphatic system first to the sentinel LN and then to the distal sites.[7] It has been reported that the lymphatic metastasis of mammary tumor cells is mediated by the CCR7/CCL21 crosstalk between tumor cells and LECs.[8] We hypothesized that CCR7 is overexpressed on the tumor cell membrane when breast cancers are highly metastatic, regardless of the invasiveness. Figure 1a shows CCR7 is overexpressed on mammary tumor cells from both noninvasive and invasive human TNBC specimens. Based on where the breast cancer initiated from, invasive breast cancers are classified into invasive ductal carcinoma (IDC) and invasive lobular carcinoma (ILC). Both human IDC and ILC TNBC specimens showed dominant CCR7 overexpression on the tumor cell membrane (Figure 1b). Statistical analysis of CCR7 RSEM-normalized RNA-seq values from 1236 breast cancer patients in The Cancer Genome Atlas (TCGA) database is demonstrated in Figure 1c. Consistent with our hypothesis, CCR7 is significantly expressed in breast cancer patients’ tumor tissues, compared with normal tissues. When tumor is more metastatic, CCR7 overexpression level is even higher. These results support the conclusion that CCR7 overexpression plays a key role in the metastasis human breast cancer, indicating a promising therapeutic target to treat TNBC tumor metastasis.
Figure 1.
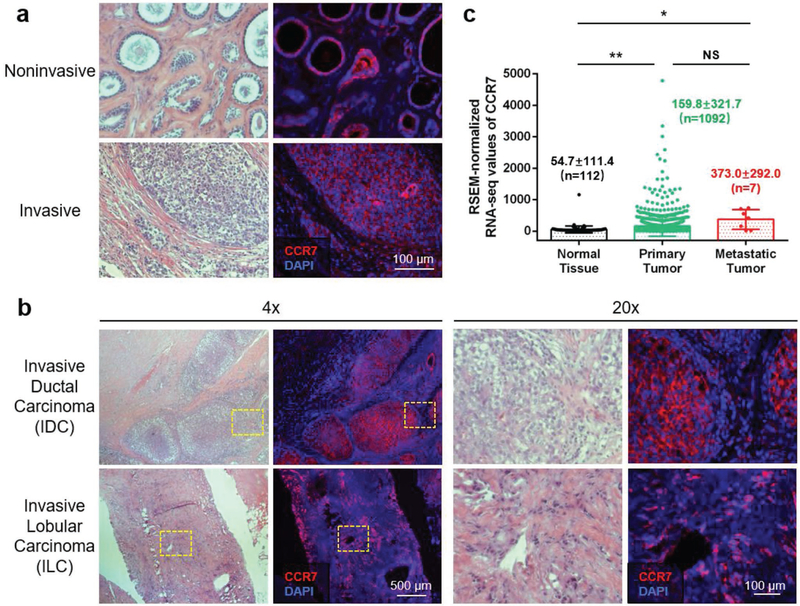
CCR7 overexpression plays a key role in the metastasis of human breast cancer. a) H&E staining (left panel) and immunofluorescence staining (right panel) for CCR7 expression in adjacent tumor sections from both invasive and noninvasive human TNBC specimens (red: CCR7, blue: nucleus). b) H&E staining (left panel) and immunofluorescence staining (right panel) for CCR7 expression in adjacent tumor sections from both IDC and ILC of human TNBC specimens at different magnification (red: CCR7, blue: nucleus). c) RSEM-normalized RNA-seq values of CCR7 in normal tissue, primary tumor, and metastatic tumor from 1236 breast cancer patient data from TCGA (NS: not significant, p ≥ 0.05;*, p < 0.05; **, p < 0.01).
2.2. Murine 4T1 Breast Cancer Is a Highly Metastatic Orthotopic TNBC Model with CCR7 Overexpression
To develop an orthotopic and spontaneous metastatic TNBC model, surgical orthotopic implantation (SOI) of histologically intact tumor tissue and cellular orthotopic injection (COI) of cell suspensions are two most widely applied strategies. The implanted tumor tissue architecture in the SOI model plays an important role in the initiation of primary tumor growth, invasion, and distant metastasis. On the one hand, primary tumors resulting from SOI are larger and much more locally invasive than primary tumors resulting from COI. Typically, SOI generates higher metastatic tumors than COI.[17] To obtain more clinically accurate tumor model, patient-derived orthotopic xenograft (PDOX) nude mouse model was developed with SOI of intact human cancer tissues. The subsequent metastatic behavior of the tumors in the PDOX mice closely correlates with tumors in patients. Currently, PDOX has already gained wide acceptance as the optimal method of creating more reliable animal model to study human cancers responding to non-immunotherapeutic agents, especially for treatments against metastasis.[18] However, the most significant drawback of PDOX as well as SOI for cancer study is the use of immunodeficient nude mice, which are unable to mount many types of immune responses requiring T cell function.[19] Since CCR7 is a key immune modulator in the TME and this specific study is closely immune-related, an orthotopic and spontaneous metastatic TNBC model, with COI of syngeneic tumor cell suspensions in healthy mice having a complete immune system, is the best option.
Murine 4T1 cell line is a typical TNBC cell line, which closely mimics human TNBC considering features including tumor location, growth progression, metastatic pattern, and immunogenicity.[20] 4T1 tumor is highly invasive and metastatic. Unlike most primary tumor models, 4T1 tumor can spontaneously metastasize from primary site in the mammary gland to the sentinel LNs as well as to multiple distal organs.[21] Epithelial–mesenchymal transition (EMT) provides a plausible explanation for epithelial cancers like 4T1 to be highly metastatic, which is a process that epithelial cells lose their cell polarity and cell–cell adhesion, acquiring invasive and migratory properties. To prove the concept, 4T1/FLuc-GFP cells were cultured in vitro and stained with antibodies against CCR7 and vimentin, a common marker of EMT. Obvious overexpression of both CCR7 and vimentin was observed (Figure S1, Supporting Information) in 4T1 cells, no matter with or without incubation of TGF-β, an inflammatory cytokine serving as a potent EMT inducer.[22] These results showed that 4T1 cells were always in the EMT status with a high level of CCR7 overexpression, indicating that 4T1 cells were highly metastatic, which was favorable for further establishment of a clinically relevant TNBC animal model.
To closely imitate human TNBC, 1 × 106 4T1/FLuc-GFP cells were orthotopically injected in the left abdominal mammary gland of female BALB/c mice as shown in Figure 2a. On day 10 after tumor inoculation, the primary tumor, sentinel LNs, and contralateral LNs were harvested and sectioned for further staining. Since the 4T1/FLuc-GFP cells were developed by stably transducing the wild-type 4T1 cells with lentivirus, the genome of transduced cells may be integrated with different copy numbers of GFP gene and hence demonstrates varied fluorescence intensity. Thus, the use of polyclonal 4T1/FLuc-GFP cells in the tissue sections resulted in different levels of GFP expression for each tumor cell. Immunofluorescence staining (IF) of tumor sections showed that both vimentin and CCR7 were prominently overexpressed in the 4T1 mammary tumor cells (Figure 2b, left panel), which were consistent with our findings in vitro. Further staining of tumor sections against the lymphatic vessel endothelial receptor 1 (LYVE-1), a typical marker of LEC, revealed the existence of lymphatic vessels within the tumor site (Figure 2b, right panel). Importantly, abundant CCL21, an easily diffusible small secretive chemokine expressed by LECs, was observed around the lymphatic vessels, indicating the metastasis potential mediated by the CCR7/CCL21 chemotaxis between tumor cells and lymphatic vessels.
Figure 2.
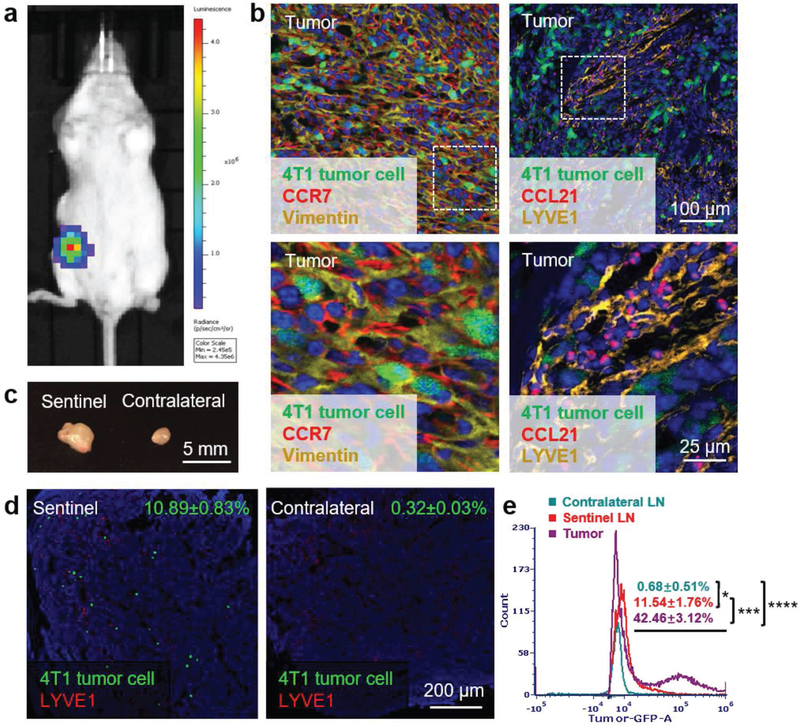
Murine 4T1 breast cancer is a highly metastatic orthotopic TNBC model with CCR7 overexpression. a) Bioluminescence imaging of 4T1 breast cancer orthotopic TNBC model. b) Immunofluorescence staining of CCR7 and vimentin expressed in tumor (left panel, green: 4T1 tumor cells, red: CCR7, yellow: vimentin, blue: nucleus); immunofluorescence staining of lymphatic vessel marker LYVE1 and cytokine CCL21 in tumor (right panel, green: 4T1 tumor cells, red: CCL21, yellow: LYVE1, blue: nucleus). c) Volume comparison of sentinel and contralateral LNs on day 10 after tumor inoculation. d) Immunofluorescence staining of 4T1 tumor cell metastasis in sentinel and contralateral LNs (green: 4T1 tumor cells, red: LYVE1, blue: nucleus). e) Flow cytometry measuring green fluorescence of 4T1 cells in tumor, sentinel and contralateral LNs (*p < 0.05; ***, p < 0.001; ****, p < 0.0001; n=6).
Figure 2c showed that the excised sentinel LN was nearly double the size of the contralateral one, which was probably due to the local inflammatory activity induced by tumor metastasis. Immunofluorescence staining of LN sections (Figure 2d) and the flow cytometry (Flow Cyt) data (Figure 2e) both demonstrated that there were numbers of mammary tumor cells migrating from tumor to the sentinel LN, while no tumor cell was detected in the contralateral LN. Thus, the orthotopic 4T1 breast cancer is a highly metastatic and clinically relevant TNBC animal model that can be used to mimic human TNBC for anticancer studies.
2.3. Preparation and Biodistribution of LPD NPs Loaded with CCR7 Trap pDNA
To disrupt CCR7-mediated signaling pathways in lymphatic metastasis, we engineered a CCR7 trap protein from an antibody that binds to the highly homologous murine and human CCR7 by grafting the related complementarity-determining regions (CDRs) into a murine scFv scaffold. The map of the CCR7 plasmid is shown in Figure S2 (Supporting Information). The target-binding affinity of the recombinant CCR7 trap was estimated by using fluorescent-labeled CCR7 trap with CCR7-expressing HuT78 cells via flow cytometry, resulting in an affinity around 227 × 10−9 M with CCR7-expressing cells (Figure 3a), while no binding was detected when various control cells were used. The biological activity of the CCR7 trap was investigated by suppressing the mCCL19-stimulated migration of CCR7-expressing HuT78 cells. As shown in Figure 3b, CCR7 trap decreased the migration of HuT78 cells stimulated with mCCL19 (100 ng mL−1), with an IC50 around 239 × 10−9 M. To efficiently and preferentially deliver the CCR7 trap pDNA into the tumor site, we used LPD NPs that were previously well-characterized (Figure 3c).[14] CCR7 trap pDNA was first condensed with the cationic protamine to form a complex with slight excess anions. This complex was then coated with preformulated cationic liposomes, composed of DOTAP, cholesterol, DSPE-PEG, and DSPE-PEG-AEAA. The LPD NPs showed a uniform size distribution by the dynamic light scattering (DLS) (Figure 3d), with polydispersity index of 0.198 and zeta potential of ≈40 mV.
Figure 3.

Characteristics of CCR7 trap and LPD NPs loaded with CCR7 trap pDNA. a). Binding affinity measurement of CCR7 trap to CCR7-expressing cells. b) In vitro suppression of CCR7-expressing cells’ migration via CCR7 trap. c) Scheme of the preparation process of LPD NPs loaded with CCR7 trap pDNA. d) Size distribution by DLS and TEM of LPD NPs loaded with CCR7 trap pDNA. e) Biodistribution of the DiD-labeled LPD NPs with (W/) or without (W/O) AEAA in different mouse organs including tumor (***, p < 0.001;****, p < 0.0001; n = 3).
Using transmission electron microscopy (TEM), LPD NPs appeared as compacted spheres of ≈95 nm in diameter, slightly smaller than the hydrated value measured by DLS (Figure 3d). Aminoethyl anisamide (AEAA) has been exploited in the Huang lab for tumor-targeted delivery of LPD NPs on many epithelial cancers overexpressing the sigma-1 receptor including 4T1 murine breast cancer.[14,23] The AEAA functionalized LPD NPs labeled with DiD mainly accumulated in the tumor as revealed with IVIS imaging system 24 h after intravenous (i.v.) injection, whereas nontargeted LPD NPs resulted in significantly less tumor accumulation (Figure 3e). Although there was some biodistribution observed in the liver and lung, the region of interest (ROI) values per gram weight were significantly lower than the value in the tumor. Moreover, although NPs of ≈100 nm in diameter are mainly internalized by Kupffer cells in the liver by phagocytosis, the trap gene expression in these cells is expected to be low because they are difficult to transfect.[24] In addition, no CCR7 expression has been reported in healthy liver and lung, where the CCR7 trap can hardly take function. Therefore, the CCR7 trap pDNA can be delivered selectively to the tumor site after circulation and transfected locally to produce abundant CCR7 trap, which provides a favorable condition for testing if trapping CCR7 prior to primary tumor resection would prevent lymphatic metastasis (see below).
2.4. CCR7 Trap Efficiently Inhibited Lymphatic Metastasis of 4T1 TNBC Tumor Cells
The therapeutic effect of LPD NPs loaded with CCR7 trap pDNA was tested in the orthotopic 4T1 TNBC animal model. At the early stage of tumor progression, phosphate buffered saline (PBS) and LPD NPs loaded with control pDNA (enhanced green fluorescence protein, or EGFP) as negative controls, as well as LPD NPs loaded with CCR7 trap pDNA were i.v. injected, respectively, and CCR7 mAb as a positive control was i.p. injected on day 5, 7, and 9 after tumor inoculation (Figure 4a). Modest tumor growth inhibition effect was observed in the CCR7 trap treatment group. At the endpoint on day 20, mice were humanly sacrificed, and tumor, sentinel LN, contralateral LN, all the major organs, and blood were harvested. Indeed, the lymphatic metastasis to the sentinel LN was remarkably inhibited by both CCR7 mAb and CCR7 trap therapies (Figure 4b), shown by bioluminescence imaging and LN size. The irregular shape of LN indicated the local inflammatory response induced by tumor metastasis. Immunofluorescence staining of LN sections (Figure 4c) and the quantitative flow cytometry data (Figure 4d) also demonstrated that the number of tumor cells in the sentinel LNs were efficiently reduced by CCR7 mAb and CCR7 trap therapies. These results confirmed that blocking CCR7 effectively inhibited the lymphatic metastasis of 4T1 TNBC tumor cells. This long-lasting inhibition effect for 4T1 TNBC tumor metastasis suggests a potential clinical application complementing tumor resection.
Figure 4.
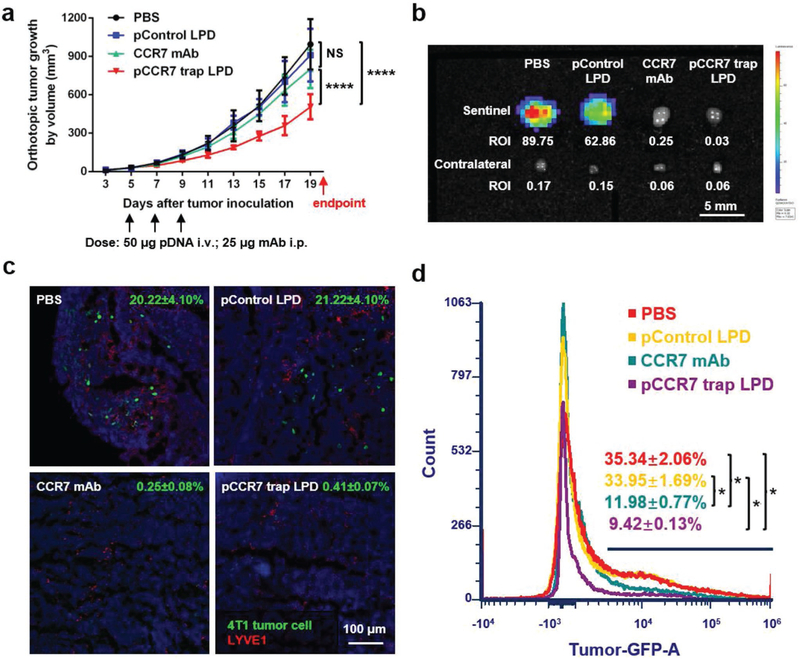
CCR7 trap efficiently inhibited lymphatic metastasis of 4T1 tumor cells. a) Tumor growth curve with three treatments on day 5, 7, and 9, respectively (NS: not significant, p ≥ 0.05;****, p < 0.0001; n = 6). b) Bioluminescence imaging of sentinel and contralateral LNs at the endpoint. c) Immunofluorescence staining of 4T1 tumor cell metastasis in sentinel LNs at the endpoint (green: 4T1 tumor cells, red: LYVE1, blue: nucleus). d) Flow cytometry measuring green fluorescence of 4T1 cells in sentinel LNs at the endpoint (*, p < 0.05; n = 6).
2.5. CCR7 Trap Induced No Immunosuppressive Side Effect Compared with CCR7 mAb
Considering that CCR7 is expressed in some specific immunocytes and plays an important role in stimulating dendritic cells maturation and homing and trafficking T cells, immune-related side effects on DC recruitment and T cell infiltration were examined by immunofluorescence staining and flow cytometry.[12,13] Indeed, compared with PBS and control pDNA groups, CCR7 mAb therapy induced obvious immunosuppressive effects with decreased DC recruitment in LN and CD8+ T cell infiltration in the tumor, while CCR7 trap group remained at a normal level (Figure 5). The observed immune-related side toxicity of CCR7 mAb can be attributed to its systemic distribution and relatively long circulatory half-life, which limits its antitumor efficacy in the treatment.[25] Therefore, the relatively transient and local expression of CCR7 trap in the tumor site showed obvious advantages to exempt from immunosuppressive side effect, consistent with what we observed using the same approach for the delivery of other chemokine traps.[26]
Figure 5.
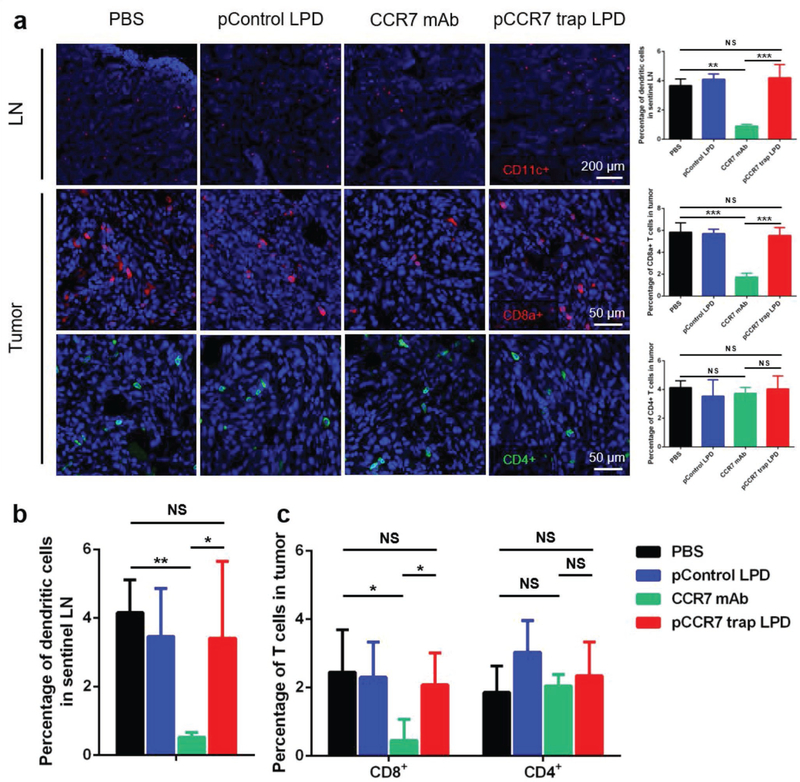
CCR7 trap did not influence DC recruitment in LN and T cell infiltration in the tumor. a) Immunofluorescence staining and quantification of DC recruitment in LN (higher panel, red: CD11c+, blue: nucleus) and T cell infiltration in tumor (lower two panels, red: CD8a+, green: CD4+, blue: nucleus; NS: not significant, p ≥ 0.05; **, p < 0.01; ***, p < 0.001; n = 6). b,c) Flow cytometry measuring DC (MCHII+CD11c+) recruitment in lymph node and CD8a+ T cells (CD45+CD8+) or CD4+ T cells (CD45+CD4+) in tumor (NS: not significant, p ≥ 0.05; *, p < 0.05; **, p < 0.01; n = 6).
The safety of these therapies was extensively evaluated by hematoxylin and eosin (H&E) staining of the major organs and blood tests for whole cell counts and serum biochemical markers. Compared to a full-length antibody that is around 150 kDa, this CCR7 trap has a MW around 28.5 kDa, which should have significantly reduced circulatory half-life while possessing comparable affinity and specificity. Therefore, the potential systemic side toxicity resulting from the long circulated full-length antibody is presumably greatly reduced. Indeed, we observed neither any obvious immune-related adverse events (irAEs) nor obvious pathological damages in the major organs regardless of CCR7-based therapies (Figure S3, Supporting Information), suggesting low side toxicity by using our approach. At the endpoint, no obvious pathological damage was observed in the major organs regardless of CCR7-based therapies (Figure S3, Supporting Information). Even in the spleen, where CCR7 positive immunocytes were normally located, there was no pathological change under different therapies. The major serum biochemical parameters, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) were within the normal ranges for all the groups, indicating no liver and kidney dysfunctions were induced by treatments (Figure S4, Supporting Information). The red blood cell (RBC) count also remained normal, suggesting no systemic anemia occurred. In the CCR7 mAb therapy, the white blood cell (WBC) count remarkably decreased, among which the percentage of lymphocyte was significantly decreased (Figure S4, Supporting Information), consistent with the results in Figure 5. Unlike the CCR7 mAb therapy, the whole blood cell counts remained in the normal range for the CCR7 trap therapy. Thus, compared with CCR7 mAb that showed immunosuppressive side effects by decreasing the lymphocyte counts, CCR7 trap therapy demonstrated a reliable systemic safety.
2.6. CCR7 Trap Downregulated the CCR7 Expression in 4T1 TNBC Tumor Cells
Interestingly, we found that the expression level of CCR7 was significantly decreased after the CCR7 trap therapy, as demonstrated in Figure 6a by the immunofluorescence staining of tumor tissues. This observation was further confirmed with Western blot (WB) analysis (Figure 6b). The CCR7 expression was correlated with the trap expression against the FLAG tag engineered at its C-terminus (Figure S2, Supporting Information). Results showed that the CCR7 trap molecules were transiently expressed in tumor from day 2 after treatments, but 8 d later the expression level was significantly reduced (Figure 6b). On the other hand, the CCR7 expression was significantly downregulated at day 2, but gradually recovered afterward. Thus, the CCR7 trap therapy can be applied 2 d prior to tumor resection, keeping the primary tumor at nonmetastatic status. The transient expression and long-lasting trapping effect guarantee CCR7 trap therapy as a potent preventive treatment prior to tumor resection, which efficiently inhibits the lymphatic metastasis induced by surgery (see below).
Figure 6.

CCR7 trap downregulated the CCR7 expression in 4T1 tumor cells. a) Immunofluorescence staining and quantification of CCR7 in tumor on day 2 after treatments (green: 4T1 tumor cells, red: CCR7, blue: nucleus; NS: not significant, p ≥ 0.05; ****, p < 0.0001; n = 3). b) Western blot and quantification of CCR7 expression in the tumor. Left panel: on day 2 after one treatment; right panel: on day 0, 2, 4, and 8 after one treatment of CCR7 trap therapy (NS: not significant, p ≥ 0.05; ***, p < 0.001; ****, p < 0.0001; n = 3). c) Western blot and quantification of CCR7, Erk1/2, and AP-1 expression in 4T1 and NMuMG cells after incubation with or without CCR7 trap protein (NS: not significant, p ≥ 0.05; ****, p < 0.0001; n = 3).
The mechanism of CCR7 downregulation was further assessed in vitro with both 4T1 tumor and NMuMG normal mammary gland epithelial cells (Figure 6c). It has been reported that the Erk1/2-AP-1-CCR7 is a key signaling pathway, through which CCR7 mediates the lymphatic metastasis of gallbladder cancer depending on the upstream Erk1/2 activation.[27] On the other hand, CCR7 also regulates tumor lymphangiogenesis and cell migration through activating the downstream Erk1/2 signaling pathway in breast cancer and squamous cell carcinoma of head and neck.[28] Thus, we hypothesized that the CCR7 trap may downregulate the CCR7 expression in 4T1 TNBC through a Erk1/2 involved feedback signaling pathway. CCR7 expression is positively controlled by Erk1/2 and AP-1, and Erk1/2 and AP-1 expression are also positively controlled by CCR7. Blocking CCR7 by trap would trigger downregulation of all three signaling proteins in the tumor cells. To test our hypothesis, both 4T1 and NMuMG cells were separately incubated in vitro with control protein (GFP) and CCR7 trap protein. As can be seen in Figure 6c, both Erk1/2 and AP-1 were also downregulated together with CCR7 in the trap-treated group, but not in the PBS or GFP-treated group. Significantly, downregulation of Erk1/2 and AP-1 and CCR7 by trap was only observed in 4T1 cells but not in NMuMG cells in which CCR7 expression was originally low. Thus, it appears that the action of the trap is amplified by the feedback signaling mechanism. This is a quite favorable situation considering the fact that in vivo trap activity might be limited by the overall efficiency of the nanoparticle-mediated gene delivery to the tumor. Also, the fact that such amplification mechanism is absent in normal cells would limit the extent of on-target, off-tumor effect of trap delivery.
2.7. CCR7 Trap Efficiently Inhibited Lymphatic Metastasis of 4T1 TNBC Tumor Cells Prior to Tumor Resection
As previously mentioned, the most prevalent clinical therapy for TNBC at present is surgical removal of the tumor. Although tumor resection effectively removes primary tumor and reduces CTC level in blood, it may also hasten the tumor metastasis by raising the levels of wound healing mediators.[3,4] Therefore, we hypothesized that the CCR7 trap therapy, which efficiently inhibited the lymphatic metastasis of 4T1 tumor cells, may be a prospective complementary treatment to surgical resection for TNBC. To further imitate the clinical treatment of TNBC, we began the therapeutic strategies as early as the tumor was just observed (on day 7). The breast cancer was at a very early stage on day 7, and presumably tumor cells had much weaker metastatic capability than those after day 15. With different therapeutic strategies, we treated the 4T1 TNBC animal model with PBS or CCR7 trap therapy before or after tumor resection, and accordingly checked the postoperative lymphatic metastasis. To mimic the clinical surgery, 4T1 tumor bearing mice underwent the tumor resection either prior to or post treatments on day 7 or 15 (green arrows in the figure) after tumor inoculation. Bioluminescence imaging before versus immediately after surgery confirmed the thorough excision of the tumor (Figure 7a). LPD NPs loaded with CCR7 trap pDNA or PBS were i.v. injected on day 9, 11, and 13 (black arrows in the figure) after tumor inoculation (Figure 7b). At the endpoint on day 35, mice were humanly sacrificed, and both the sentinel LN and contralateral LN were harvested. For groups with tumor removal before treatments on day 7, no matter which treatment was applied, the sentinel LNs were slightly larger in size than contralateral LNs (Figure 7c), revealing the inflammation status induced by tumor metastasis. Although no obvious bioluminescence signal was observed by IVIS imaging (Figure 7c), a few metastatic mammary tumor cells were still found in the sentinel LNs by fluorescence staining of sections (Figure 7d) and flow cytometry (Figure 7e). In the quantitative flow cytometry result, there is no significant difference between mice group of PBS after resection and mice group of PBS before resection, and the mice group of trap treatment before resection showed significantly less metastasis than the other three mice groups. Since the trap protein is mainly expressed by the tumor cells (Figure 3e), for animals with tumor removed before the trap treatment, there was no place for CCR7 trap molecules to be produced and take function. As a consequence, the implantation of CTCs in the sentinel LNs readily occurred. While for groups with tumor removal after treatments on day 15, when mice were injected with CCR7 trap pDNA in NPs, there was no metastasis observed in the sentinel LNs at the endpoint day (Figure 7c–e). However, when mice were treated with PBS, the sentinel LNs were nearly three times larger by diameter than contralateral LNs, with quite high signals for metastatic tumor cells (Figure 7c–e). These results strongly suggest that although the CCR7 trap expression was transient, its prevention function for tumor cell metastasis could last for quite a long time (>20 d). Therefore, applying the CCR7 trap therapy prior to tumor resection efficiently prevented the 4T1 tumor cell lymphatic metastasis, indicating a good prospect for the future TNBC clinical therapeutic application as a complementary treatment.
Figure 7.
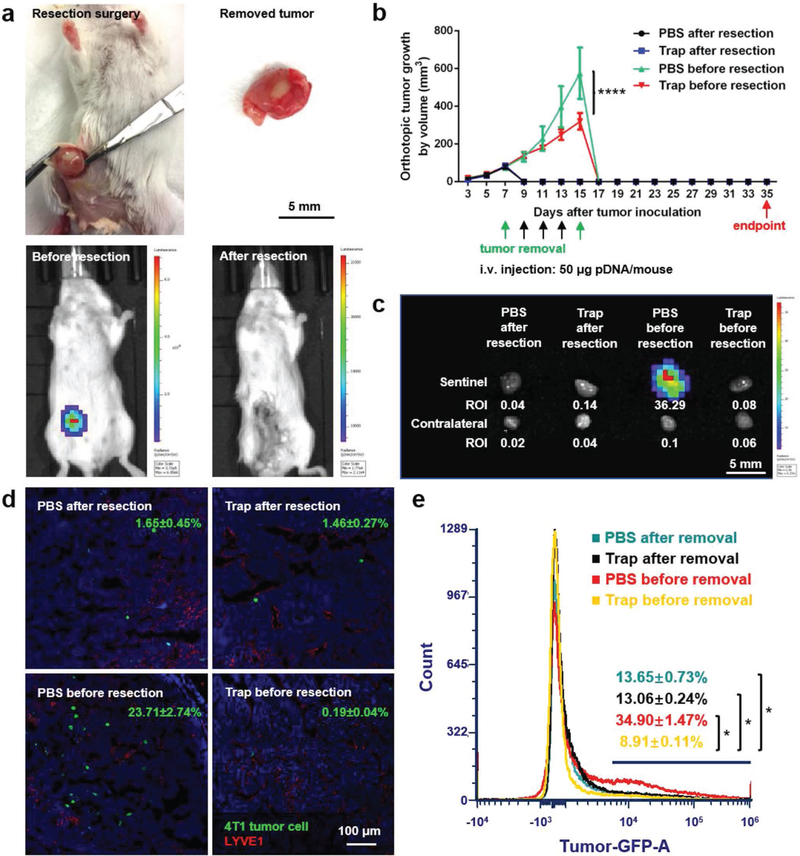
CCR7 trap efficiently inhibited lymphatic metastasis of 4T1 tumor cells prior to tumor resection. a) Photos of tumor resection on 4T1 breast cancer orthotopic model and the removed tumor (upper panel). Bioluminescence imaging before and after tumor resection of 4T1 breast cancer orthotopic model (lower panel). b) Tumor growth curve of the four groups of 4T1 TNBC mice: (1) PBS after resection (mice were treated with PBS on day 9, 11, and 13 (black arrows) after tumor removal surgery on day 7 (green arrow)); (2) Trap after resection (mice were treated with CCR7 trap LPD NPs on day 9, 11, and 13 (black arrows) after tumor removal surgery on day 7 (green arrow)); (3) PBS before resection (mice were treated with PBS on day 9, 11, and 13 (black arrows) before tumor removal surgery on day 15 (green arrow)); (4) Trap before resection (mice were treated with CCR7 trap LPD NPs on day 9, 11, and 13 (black arrows) before tumor removal surgery on day 15 (green arrow)) (NS: not significant, p ≥ 0.05; ****, p < 0.0001; n = 6). c) Bioluminescence imaging of sentinel LNs and contralateral LNs at the endpoint. d) Immunofluorescence staining of 4T1 tumor cell metastasis in sentinel LNs at the endpoint (green: 4T1 tumor cells, red: LYVE1, blue: nucleus). e) Flow cytometry measuring green fluorescence signals of 4T1 cells in sentinel LNs at the endpoint (*, p < 0.05; n = 6).
2.8. CCR7 Trap Efficiently Inhibited Lymphatic Metastasis of B16F10 Melanoma Tumor Cells
Reports have shown that, same as TNBC, the lymphatic metastasis of melanoma tumor cells are also mediated by the CCR7/CCL21 crosstalk.[29] Since the CCR7 trap therapy has demonstrated very favorable metastasis prevention effect in 4T1 TNBC mouse model, we hypothesized that it would have similar favorable outcome for the metastatic melanoma model. Thus, LPD NPs loaded with CCR7 trap pDNA were tested in the subcutaneous B16F10 melanoma animal model. At the early stage of tumor progression, PBS and LPD NPs loaded with control pDNA (EGFP) as negative controls, as well as LPD NPs loaded with CCR7 trap pDNA were i.v. injected, respectively, on day 9, 11, and 13 after tumor inoculation (Figure 8a). At the endpoint on day 22, mice were humanly sacrificed, and both the sentinel LN and contralateral LN were harvested. For negative control groups, obvious tumor cell metastasis in the sentinel, but not in the contralateral, LNs was observed. Metastasis involved LNs were characterized by the black color due to the melanin content in the tumor cells and the enlarged node size. While after treatment with CCR7 trap therapy, the lymphatic metastasis of B16F10 melanoma tumor cells to the sentinel LN was remarkably inhibited (Figure 8b). H&E staining of sentinel LN sections (Figure 8c) showed obvious metastatic B16F10 tumor cells (yellow circle), characterized by a larger size of tumor cell than lymphocyte, in PBS and control pDNA groups. However, after treatment with CCR7 trap, no obvious tumor metastasis was observed. Therefore, CCR7 trap therapy also worked well in inhibiting melanoma metastasis to LN.
Figure 8.

CCR7 trap efficiently inhibited lymph node metastasis of B16F10 melanoma cells. a) Tumor growth curve with three treatments on day 9, 11, and 13 (**, p < 0.01; n = 6). b) Volume comparison of LNs, B16F10 tumor cell metastasis of LNs, and proportion of metastasis positive mice at the endpoint. c) H&E staining of B16F10 tumor cell metastasis (yellow circle) in sentinel LNs at the endpoint.
3. Conclusion
Metastasis is responsible for up to 90% of cancer-associated mortality.[30] Developing an efficient approach for preventing tumor metastasis, especially the metastasis hastened by tumor resection, is urgently needed in the clinic. In this study, we showed the important role of CCR7 in TNBC lymphatic metastasis and proved that CCR7 trap therapy prior to tumor resection efficiently inhibited the postoperative lymphatic metastasis. We demonstrated that the efficient inhibition was possibly attributed to a combination of the following mechanisms: (1) tumor-targeted delivery of LPD NPs promised the local production and functioning of CCR7 trap (Figure 3e), greatly reducing the systemic immunosuppressive side effect; (2) relatively transient production of CCR7 trap conferred the lasting inhibitive effect (Figure 6b); (3) trap molecules not only blocking the CCR7/CCL21 crosstalk but also downregulating the CCR7 expression through a feedback signal pathway further facilitated the efficient inhibition for tumor metastasis (Figure 6c).
Thus, combining with the current most prevalent clinical therapy for TNBC surgical resection, CCR7 trap therapy prior to surgery showed great prospects to reduce the risk of tumor metastasis of TNBC, melanoma, and potentially other CCR7-mediated metastatic cancers.
4. Experimental Section
Materials:
The SUNBRIGHT DSPE-020PA (DSPE-PEG-NH2) was purchased from NOF America Corporation (White Plains, NY). 1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) was purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (CAS 57–88-5), 4-methoxybenzoyl chloride (CAS 100–07-2), 2-bromoethylamine hydrobromide (CAS 2576–47-8), N,N-diisopropylethylamine (CAS 7087–68-5), and acetonitrile (CAS 75–05-8) were purchased from Sigma-Aldrich (St. Louis, MO). The DSPE-PEG-AEAA was synthesized according to a previous publication.[31] The LPD NPs were prepared as previously reported.[32] DiD (Catalog No. D7757) was purchased from ThermoFisher Scientific (Waltham, MA). Recombinant human TGF-β1 protein (Catalog No. 240-B-002) was purchased from R&D Systems (Minneapolis, MN). D-luciferin (Catalog No. 556876) was purchased from BD Biosciences (Sparks, MD).
Antibodies:
CD197 (CCR7) monoclonal antibody (4B12) (Catalog No. 16–1971-85) was purchased from ThermoFisher Scientific (Waltham, MA) as the CCR7 mAb drug. All the other antibodies used for IF, Flow Cyt, and WB were listed below. Anti-CCR7 antibody [Y59] (Catalog No. ab32527), anti-vimentin antibody [RV202] (Catalog No. ab8978), goat anti-rabbit IgG H&L (Alexa Fluor 555) (Catalog No. ab150078), goat anti-mouse IgG H&L (Alexa Fluor 647) (Catalog No. ab150115), and donkey anti-goat IgG H&L (Alexa Fluor 555) (Catalog No. ab150130) were purchased from Abcam (Cambridge, MA). ERK 1/2 antibody (MK1) (Catalog No. sc-135900), goat anti-mouse IgG-HRP (Catalog No. sc-2031), and mouse anti-rabbit IgG-HRP (Catalog No. sc-2357) were purchased from Santa Cruz (Dallas, TX). Alexa Fluor 647 anti-mouse CD11c antibody (Catalog No. 117314), Alexa Fluor 594 anti-mouse CD8a antibody (Catalog No. 100758), and Alexa Fluor 647 anti-mouse CD4 antibody (Catalog No. 100426) were purchased from BioLegend (San Diego, CA). LYVE-1 antibody (ALY7) (Catalog No. NBP1–43411) was purchased from Novus Biologicals (Littleton, CO). Anti-AP-1 antibody (Catalog No. MBS2516982) was purchased from MyBioSource (San Diego, CA).
Cell Lines and Animals:
Murine 4T1/FLuc-GFP TNBC cell line and B16F10 melanoma cell line were cultured with RPMI-1640 medium (Gibco, Catalog No. 11875093) and Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Catalog No. 11965092), respectively, both added with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Murine NMuMG normal mammary gland epithelial cell line was cultured with DMEM (Gibco, Catalog No. 11965092), added with 10% FBS, insulin (10 mcg mL−1) and 1% penicillin/streptomycin. Culture condition for all these three cell lines was at 37 °C and 5% CO2. Six-week-old female BALB/c and C57BL/6 mice were purchased from Charles River Laboratories. All animal regulations and procedures were accepted by Institutional Animal Care and Use Committee, University of North Carolina at Chapel Hill.
Human TNBC Samples:
Paraffin-embedded biopsies of TNBC patients were kindly provided by The Second Hospital of Shanxi Medical University and Baoding First Central Hospital, China, according to an approved patient sample management protocol. Informed consent was obtained from patient before evaluation.
Analysis of CCR7 Expression in Human Breast Cancer Patients from TCGA:
The TCGA breast invasive carcinoma (BRCA) cohort data were downloaded from the Broad Institute Genome Data Analysis Centers (GDAC) Firehose database (http://gdac.broadinstitute.org/). The RNAseq data set called “illuminahiseq_rnaseqv2-RSEM_genes_normalized (MD5)” with release date 20160128 contained upper quartile normalized RSEM values summarized at the gene level. The total 1236 patient samples were split into three groups according to whether the tissue biopsy was retrieved from the normal tissue (112 samples), primary tumor (1092 samples), or metastatic tumor (7 samples). Data were analyzed with GraphPad Prism software. The p-value was calculated with ordinary one-way ANOVA.
Development and Tumor Removal Surgery of 4T1 TNBC Mouse Model, Development of B16F10 Melanoma Mouse Model:
BALB/c mice were inoculated with 1 × 106 4T1/FLuc-GFP tumor cells orthotopically in the mammary gland, with an appropriate injection volume (50 μL). When tumor began to develop, diameters (length and width) of tumor were measured with a vernier caliper every other day. Tumor volume was calculated as 0.5 × length × width2. To surgically remove the primary tumor, an incision was made on the skin around, and then tumor was cut away from the underlying normal tissue. At last the surgical wound was closed with sutures.
C57BL/6 mice were inoculated with 2 × 105 B16F10 tumor cells subcutaneously on lower flank area, with an appropriate injection volume (50 μL). When tumor began to develop, diameters (length, width, and height) of tumor were measured with a vernier caliper every other day. Tumor volume was calculated as 0.5 × length × width × height.
Construction of CCR7 Trap Gene and Expression of the Trap Protein:
The CCR7 trap was engineered through an antibody that binds to mouse/human CCR7 by grafting the CDRs into a murine scFv scaffold. Different versions of CCR7 traps, in which the VH and VL domains were linked through a (GGGGS)3 hinge, were tested and the one with the best affinity and specificity to mouse CCR7 was chosen for further optimization. Both the ORF codons and mRNA secondary structures were optimized using the GeneOptimizer program in Invitrogen.[33] To increase the solubility and facilitate the purification of the trap protein, a GKKGK motif as well as the negatively charged FLAG tag were introduced at the C-terminus. To facilitate the secretion of the trap protein to the TME, a strong secreting signaling peptide was engineered at the N-terminus of the trap as used previously.[26a] The map of the CCR7 trap plasmid is shown in Figure S2 (Supporting Information). The codon-optimized DNA sequences code for a signaling peptide, CCR7-binding domains, FLAG tag, and His (6×) tag, respectively, was synthesized. The resulting cDNA was cloned into pCDNA3.1 between Nhe I and Xho I sites and the accuracy was confirmed by DNA sequencing.
To express the trap protein, pcDNA3.1-CCR7 trap plasmid (≈50 μg) was diluted into Opti-MEM media (2.5 mL) in a 50 mL falcon tube followed by incubation at room temperature for 5 min. After mixing plasmid and lipofectamine 2000 solutions, Opti-MEM (25 mL) was supplemented and incubated at room temperature for 20 min. The mixture was then applied to 70–90% confluent 293T/17 cells in T175 flasks followed by cultivation for 3–5 d in a CO2 incubator at 37 °C. The supernatant was harvested and loaded to an IMAC Nickel-charged resin column (BIORAD, Hercules, CA) for protein purification. Recombinant trap protein was eluted with imidazole (200 × 10−3 M) in the binding buffer and dialyzed against PBS at 4 °C.
Trap Binding Affinity Measurement:
The affinity of CCR7 trap to CCR7 was estimated by binding with CCR7-expressing HuT78 cells, while Jurkat e.6 cells were used as a negative control. Cells were incubated with different concentrations of CCR7 trap and the binding was detected by flow cytometry (Attune NxT) using mouse anti-FLAG antibody and goat anti-mouse secondary antibody. Isotype controls and SYTOX AADvanced Ready Flow Reagent were used to gate cells. Experiments were repeated in triplicate and the data were fit using nonlinear least squares to estimate the binding affinity.
In Vitro Suppression of Migration via CCR7 Trap:
HuT78 cells were cultured in a transwell system. HuT78 cells were incubated with different concentrations of CCR7 trap in upper chamber and allowed to equilibrate for 2 h. mCCL19 (100 ng mL−1) was added to lower chamber and the percentage of migratory cells (migrated cells/nonmigrated cells) was counted by flow cytometry (Attune NxT) after 4 h. SYTOX AADvanced Ready Flow Reagent was used to gate cells. Experiments were repeated in triplicate and the data were fit using a four-parameter model to estimate IC50.
Biodistribution of LPD NPs Loaded with CCR7 Trap pDNA:
DiD-labeled LPD NPs were prepared with liposomes containing ≈0.1% of hydrophobic dye DiD. 4T1 TNBC model mice (n = 3) were i.v. injected with DiD-labeled LPD NPs, and then sacrificed after 24 h. Tumor and major organs were properly collected and subject to IVIS optical imaging system for fluorescent imaging and ROI quantification (excitation/emission = 644/665 nm). Data were analyzed with GraphPad Prism software. The p-value was calculated with ordinary one-way ANOVA.
Dosing Regimen, Tumor Growth Suppression, and Metastasis Inhibition:
4T1 TNBC mouse model: after tumor inoculation, once the tumor volume reached ≈30 mm3, mice were randomized into four groups (n = 6) with different treatments as follows: PBS (PBS group), LPD NPs loaded with control pDNA (EGFP) (control pDNA group), CCR7 monoclonal antibody (CCR7 mAb group), and LPD NPs loaded with CCR7 trap pDNA (CCR7 trap group). PBS group (200 μL PBS/mouse) and LPD NPs groups (50 μg plasmid/mouse) were i.v. injected and CCR7 mAb group (25 μg mAb/mouse) was i.p. injected on day 5, 7, and 9, respectively. Tumor size was monitored every 2 d. Mice were sacrificed before tumors reached 2000 mm3. At the endpoint on day 20, tumors, sentinel LNs, contralateral LNs, major organs, and blood samples were harvested and tested.
4T1 TNBC Mouse Model Underwent Tumor Removal Surgery:
After tumor inoculation, once the tumor volume reached ≈30 mm3, mice were randomized into four groups (n = 6) with different treatments as follows: PBS after tumor removal surgery (PBS after removal group), LPD NPs loaded with CCR7 trap pDNA after tumor removal surgery (trap after removal group), PBS before tumor removal surgery (PBS before removal group), and LPD NPs loaded with CCR7 trap pDNA before tumor removal surgery (trap before removal group). Tumor removal surgery was carried out on day 7 or 15. PBS group (200 μL PBS/mouse) and LPD NPs group (50 μg plasmid/mouse) were i.v. injected on day 9, 11, and 13, respectively. Tumor size was monitored every 2 d. Mice were sacrificed before tumors reached 2000 mm3. At the endpoint on day 35, tumors, sentinel LNs, and contralateral LNs were harvested and tested.
B16F10 Melanoma Mouse Model:
After tumor inoculation, once the tumor volume reached ≈200 mm3, mice were randomized into three groups (n = 6) with different treatments as follows: PBS (PBS group), LPD NPs loaded with control pDNA (control pDNA group), and LPD NPs loaded with CCR7 trap pDNA (CCR7 trap group). PBS group (200 μL PBS/mouse) and LPD NPs groups (50 μg plasmid/mouse) were i.v. injected on day 9, 11, and 13. Tumor size was monitored every 2 d. Mice were sacrificed before tumors reached 2000 mm3. At the endpoint on day 22, tumors, sentinel LNs, and contralateral LNs were harvested and tested.
All the data were analyzed with GraphPad Prism software. The p-value was calculated with ordinary two-way ANOVA.
Immunofluorescence Staining of Sections:
Staining was performed on paraffin-embedded sections (human TNBC samples) and frozen sections (tumor and LNs from 4T1 TNBC model mice). For paraffin-embedded sections, immunofluorescence staining was performed at first by deparaffinization and antigen retrieval. Afterward, both kinds of sections underwent a blocking by 0.5% goat serum for 1 h at room temperature. Slices were then incubated with primary antibodies for 1 h at 37 °C, followed by incubation with fluorescent secondary antibodies for 1 h at room temperature. At last, the nuclei were counterstained with DAPI. The well-stained slides were imaged under Zeiss LSM 710 Confocal Microscopy.
Flow Cytometry Assay:
4T1/FLuc-GFP tumor cell and lymphocyte populations (n = 6) were analyzed by Attune NxT Flow Cytometer. Briefly, single cells were collected from fresh LNs directly and from fresh tumor tissues after an incubation with collagenase A at 37 °C for 40 min. Lymphocytes were then stained with fluorescein-conjugated antibodies. Fluorescently stained beads and in vitro cultured 4T1/FLuc-GFP tumor cells were used as positive control. Data were analyzed with GraphPad Prism software. The p-value was calculated with ordinary one-way ANOVA.
Western Blot Assay:
Freshly harvested tumor tissues were analyzed by Western blot assay. Briefly, tumor tissues were first homogenized with radioimmunoprecipitation assay (RIPA) buffer and then centrifuged to get the supernatant. Afterward, protein concentrations of the supernatants were measured with bicinchoninic acid (BCA) assay. Western blot assay was then preformed with NuPAGE 10% Bis-Tris Protein Gels and MOPS running buffer at a loading amount of 70 μg protein/lane. Finally, the gels were stained with DAB and observed under the Bio-Rad Gel Imaging System.
Supplementary Material
Acknowledgements
The authors appreciate funding for this work from NIH grants CA198999 (to L.H.), CA157738 (to R.L.), and a grant from Eshelman Institute for Innovation (to L.H. and R.L.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The trap technology has been licensed to OncoTrap Inc. L.H. and R.L. are co-founders.
Contributor Information
Sai An, Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Karthik Tiruthani, Division of Chemical Biology and Medicinal Chemistry Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Ying Wang, Division of Chemical Biology and Medicinal Chemistry Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Ligeng Xu, Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Mengying Hu, Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Jingjing Li, Division of Chemical Biology and Medicinal Chemistry Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Wantong Song, Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Hongnan Jiang, Department of Breast Surgery, The Second Hospital of Shanxi Medical University, Taiyuan, Shanxi 030013, China.
Jirui Sun, Department of Pathology, Baoding First Central Hospital, Baoding, Hebei 071000, China.
Rihe Liu, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
Leaf Huang, Division of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
References
- [1].DeSantis CE, Ma J, Sauer AG, Newman LA, Jemal A, Ca-Cancer J. Clin 2017, 67, 439. [DOI] [PubMed] [Google Scholar]
- [2].Tseng LM, Hsu NC, Chen SC, Lu YS, Lin CH, Chang DY, Li H, Lin YC, Chang HK, Chao TC, Ouyang F, Hou MF, Neoplasma 2013, 60, 290. [DOI] [PubMed] [Google Scholar]
- [3].Chiarella P, Bruzzo J, Meiss RP, Ruggiero RA, Cancer Lett 2012, 324, 133. [DOI] [PubMed] [Google Scholar]
- [4].Demicheli R, Retsky MW, Hrushesky WJ, Baum M, Gukas ID, Ann. Oncol 2008, 19, 1821. [DOI] [PubMed] [Google Scholar]
- [5].Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL, Maceli AR, Dickopf S, Papachristou EK, D’Santos CS, Carey LA, Wilkinson JE, Harrell JC, Perou CM, Goodarzi H, Poulogiannis G, Hannon GJ, Nature 2018, 554, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lauria R, Perrone F, Carlomagno C, De Laurentiis M, Morabito A, Gallo C, Varriale E, Pettinato G, Panico L, Petrella G, Bianco AR, De Placido S, Cancer 1995, 76, 1772. [DOI] [PubMed] [Google Scholar]
- [7].a) Chua B, Ung O, Taylor R, Boyages J, ANZ J Surg 2001, 71, 723; [DOI] [PubMed] [Google Scholar]; b) Hinck L, Näthke I, Curr. Opin. Cell Biol 2014, 26, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pang MF, Georgoudaki AM, Lambut L, Johansson J, Tabor V, Hagikura K, Jin Y, Jansson M, Alexander JS, Nelson CM, Jakobsson L, Betsholtz C, Sund M, Karlsson MC, Fuxe J, Oncogene 2016, 35, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Li X, Sun S, Li N, Gao J, Yu J, Zhao J, Li M, Zhao Z, Cell. Physiol. Biochem 2017, 43, 531; [DOI] [PubMed] [Google Scholar]; b) Cunningham HD, Shannon LA, Calloway PA, Fassold BC, Dunwiddie I, Vielhauer G, Zhang M, Vines CM, Transl. Oncol 2010, 3, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Klarenbeek A, Maussang D, Blanchetot C, Saunders M, van der Woning S, Smit M, de Haard H, Hofman E, Drug Discovery Today: Technol 2012, 9, e237; [Google Scholar]; b) Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L, Front. Immunol 2015, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Somovilla-Crespo B, Alfonso-Pérez M, Cuesta-Mateos C, Carballo-de Dios C, Beltrán AE, Terrón F, Pérez-Villar JJ, Gamallo-Amat C, Pérez-Chacón G, Fernández-Ruiz E, Zapata JM, Muñoz-Calleja C, J. Hematol. Oncol 2013, 6, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Förster R, Davalos-Misslitz AC, Rot A, Nat. Rev. Immunol 2008, 8, 362. [DOI] [PubMed] [Google Scholar]
- [13].a) Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Förster R, Immunity 2004, 21, 279; [DOI] [PubMed] [Google Scholar]; b) Correale P, Rotundo MS, Botta C, Del Vecchio MT, Ginanneschi C, Licchetta A, Conca R, Apollinari S, De Luca F, Tassone P, Tagliaferri P, Clin. Cancer Res 2012, 18, 850. [DOI] [PubMed] [Google Scholar]
- [14].Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K, Zhang X, Hou L, Wang D, Liu R, Huang L, ACS Nano 2018, 12, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sharma GN, Dave R, Sanadya J, Sharma P, Sharma KK, J. Adv. Pharm. Technol. Res 2010, 1, 109. [PMC free article] [PubMed] [Google Scholar]
- [16].Cowell CF, Weigelt B, Sakr RA, Ng CK, Hicks J, King TA, Reis-Filho JS, Mol. Oncol 2013, 7, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) An Z, Jiang P, Wang X, Moossa AR, Hoffman RM, Clin. Exp. Metastasis 1999, 17, 265; [DOI] [PubMed] [Google Scholar]; b) Furukawa T, Kubota T, Watanabe M, Kitajima M, Hoffman RM, Int. J. Cancer 1993, 53, 608. [DOI] [PubMed] [Google Scholar]
- [18]. a) Fu X, Le P, Hoffman RM, Anticancer Res 1993, 13, 901; [PubMed] [Google Scholar]; b) Hoffman RM, Invest. New Drugs 1999, 17, 343; [DOI] [PubMed] [Google Scholar]; c) Hoffman RM, Nat. Rev. Cancer 2015, 15, 451. [DOI] [PubMed] [Google Scholar]
- [19].Hoffman RM, Molecular and Translational Medicine, Springer International Publishing AG, Cham, Switzerland: 2017. [Google Scholar]
- [20].Pizato N, Luzete BC, Kiffer LFMV, Corrêa LH, de Oliveira Santos I, Assumpção JAF, Ito MK, Magalhães KG, Sci. Rep 2018, 8, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pulaski BA, Ostrand-Rosenberg S, Current Protocols in Immunology, Wiley, Hoboken, NJ: 2001. [DOI] [PubMed] [Google Scholar]
- [22].Karlsson MC, Gonzalez SF, Welin J, Fuxe J, Mol. Oncol 2017, 11, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Goodwin TJ, Huang L, J. Controlled Release 2016, 243, 382. [DOI] [PubMed] [Google Scholar]
- [24].Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, J. Controlled Release 2016, 240, 332. [DOI] [PubMed] [Google Scholar]
- [25].a) Ryman JT, Meibohm B, CPT: Pharmacometrics Syst. Pharmacol 2017, 6, 576; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tabrizi M, Bornstein GG, Suria H, AAPS J. 2010, 12, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Goodwin TJ, Zhou Y, Musetti SN, Liu R, Huang L, Sci. Transl. Med 2016, 8, 364ra153; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Goodwin TJ, Shen L, Hu M, Li J, Feng R, Dorosheva O, Liu R, Huang L, Biomaterials 2017, 141, 260; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miao L, Li J, Liu Q, Feng R, Das M, Lin CM, Goodwin TJ, Dorosheva O, Liu R, Huang L, ACS Nano 2017, 11, 8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hong H, He C, Zhu S, Zhang Y, Wang X, She F, Chen Y, J. Exp. Clin. Cancer Res 2016, 35, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Tutunea-Fatan E, Majumder M, Xin X, Lala PK, Mol. Cancer 2015, 14, 35; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu FY, Safdar J, Li ZN, Fang QG, Zhang X, Xu ZF, Sun CF, Int. J. Oncol 2014, 45, 2502. [DOI] [PubMed] [Google Scholar]
- [29].a) Emmett MS, Lanati S, Dunn DB, Stone OA, Bates DO, Microcirculation 2011, 18, 172; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wiley HE, Gonzalez EB, Maki W, Wu MT, Hwang ST, JNCI J Natl. Cancer Inst 2001, 93, 1638; [DOI] [PubMed] [Google Scholar]; c) Takeuchi H, Fujimoto A, Tanaka M, Yamano T, Hsueh E, Hoon DS, Clin. Cancer Res 2004, 10, 2351. [DOI] [PubMed] [Google Scholar]
- [30].Chaffer CL, Weinberg RA, Science 2011, 331, 1559. [DOI] [PubMed] [Google Scholar]
- [31].Banerjee R, Tyagi P, Li S, Huang L, Int. J. Cancer 2004, 112, 693. [DOI] [PubMed] [Google Scholar]
- [32].Li S, Rizzo MA, Bhattacharya S, Huang L, Gene Ther 1998, 5, 930. [DOI] [PubMed] [Google Scholar]
- [33].Raab D, Graf M, Notka F, Schödl T, Wagner R, Syst. Synth. Biol 2010, 4, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.