

Summary
Laboratory mice, while paramount for understanding basic biological phenomena, are limited in modeling complex diseases of humans and other free-living mammals. Because the microbiome is a major factor in mammalian physiology, we aimed to identify a naturally evolved reference microbiome to better recapitulate physiological phenomena relevant in the natural world outside the laboratory. Among 21 distinct mouse populations worldwide we identified a closely related wild relative to standard laboratory mouse strains. Its bacterial gut microbiome differed significantly from its laboratory mouse counterpart, and was transferred to and maintained in laboratory mice over several generations. Laboratory mice reconstituted with natural microbiota exhibited reduced inflammation and increased survival following influenza virus infection, and improved resistance against mutagen/inflammation-induced colorectal tumorigenesis. By demonstrating the host fitness-promoting traits of natural microbiota, our findings should enable the discovery of protective mechanisms relevant in the natural world and improve the modeling of complex diseases of free-living mammals.
Graphical Abstract
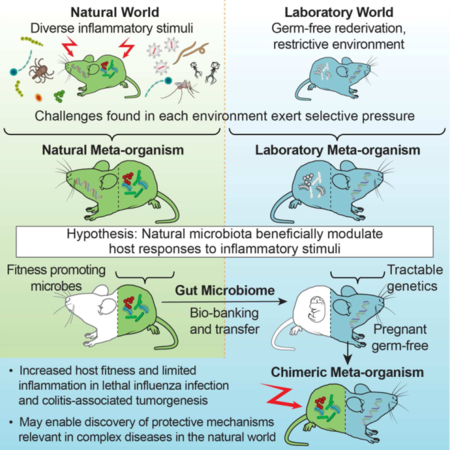
eTOC
Characterization of a wild mice reference microbiome opens a window of opportunity to understand how the gut microbiota affects aspects of host physiology that are important in the natural world, outside the laboratory.
Introduction
Laboratory mice have played a fundamental role in deciphering basic immunological mechanisms (Zinkernagel and Doherty, 1974a, b). Key advantages of the laboratory mouse model include low cost, availability of a wide variety of inbred strains, ease of genetic manipulation and standardized, specific-pathogen free (SPF) husbandry to increase reproducibility of experimental findings. At the same time, standard environmental conditions employed in mouse husbandry, including ambient temperature, day/night cycle, water treatment and diet are far removed from those found in the natural world outside of the laboratory environment. Further, laboratory mice are not exposed to many of the microbes that they would encounter or harbor in their natural habitat.
The mammalian phenotype is largely driven by the combination of host and microbiome genes, together known as the metagenome. The mammalian microbiome is the collection of all host-associated microorganisms, a complex and diverse ecosystem that is present on all epithelial barriers. It includes the bacterial microbiome, the archaeal microbiome, the virome (bacteriophages, eukaryotic viruses), the mycobiome (fungi) and the meiofauna (unicellular protozoa, helminthic worms) (Norman et al., 2014; Stappenbeck and Virgin, 2016), and is acquired through vertical transmission and environmental exposure. The microbiomes of free-living organisms (including wild mice and humans) co-evolved with their respective hosts over eons by natural selection creating a symbiotic host-microbe relationship integral to host physiology (Dethlefsen et al., 2007; Ley et al., 2008; McFall-Ngai, 2007; Sommer and Backhed, 2013). In contrast, the laboratory mouse microbiome evolved over much shorter periods in a sanitized and restrictive environment devoid of numerous microorganisms. These conditions were exacerbated by repeated and complete microbiome deletion via germ-free (GF) re-derivation and subsequent re-colonization in a restrictive laboratory environment.
We hypothesized that laboratory mice lack important symbiotic host-microbe interactions crucial for host physiology, including immunity, found in wild mice, which evolved under greater evolutionary pressure from infectious diseases and naturally occurring inflammatory immune stimuli. This may therefore limit the predictive utility of laboratory mice for modeling complex diseases of free-living mammals. Here, we examined how restoring the laboratory mouse gut microbiome to a natural gut microbiome found in wild mice affects host responses systemically in infectious diseases and locally in inflammation-associated neoplastic development.
Results
Wild Mus musculus domesticus from Maryland are close relatives to standard laboratory mouse strains
Laboratory mouse strains have a convoluted history but are ultimately derived from admixture between house mice of different subspecies. The ancestors of laboratory strains most likely originated in northern Europe and Japan (Beck et al., 2000; Nishioka, 1995; Yang et al., 2011). Wild mice, the relatives of laboratory mouse strains, live in close proximity to humans in their natural habitat and move from cultivated fields and wooded areas into barns and houses during cooler weather (Phifer-Rixey and Nachman, 2015). To identify a close wild living relative of standard laboratory mice, we trapped more than 800 wild mice in 8 geographically distinct locations (horse barns) in Maryland and the District of Columbia, USA during the fall of two consecutive years (Figure S1) and compared them with 21 wild mouse populations worldwide. We excluded barns with chemical pest control, which may alter the microbiome, and barns proximal to research facilities, as this could lead to contact with escaped laboratory mice and mingling of their microbiomes.
Based on appearance, we excluded other rodents such as deer mice (Peromyscus), which can be mistaken for house mice but are more closely related to rats. Since age and reproductive status modulate microbial composition, we developed a scoring system to classify the mice (Table S1). After exclusion of small immature animals, this selection process reduced the number of mice for this study to 101 from locations A, B and C (Figure S1), of which 98 were sexually mature (Table S1). Genotyping with a SNP microarray identified these mice unambiguously as Mus musculus domesticus and placed them in a clade with standard laboratory strains and mice from northern Europe (Figure 1A). Formal admixture analyses demonstrated that Maryland mice share a greater proportion of their genomes with standard laboratory strains (including C57BL/6 substrains) than do mice from 21 other wild mouse populations from Europe, Asia and the Americas (Figure 1B).
Figure 1. Wild Mus musculus domesticus from Maryland, USA are close Relatives to Standard Laboratory Mouse Strains in Particular C57BL/6 Mice from Commercial Vendors.
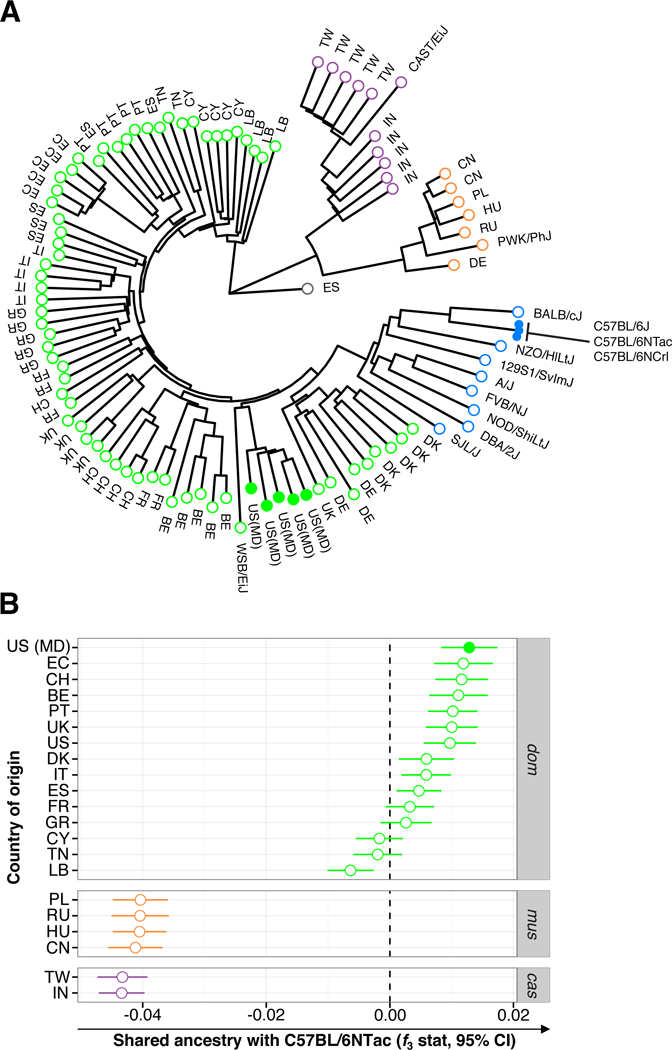
(A)Phylogenetic tree of wild mice from Maryland, USA (closed green circles) compared to other Mus musculus domesticus (open green circles; wild mice from countries with indicated 2-letter code, plus inbred strain WSB/EiJ), Mus musculus musculus (open orange circles; wild mice from countries with indicated 2-letter code, plus inbred strain PWK/PhJ), Mus musculus castaneus (open purple circles; wild mice from countries with indicated 2-letter code, plus inbred strain CAST/EiJ) and standard laboratory strains (blue). Each branch represents a single mouse. The tree is based on SNP genotypes and rooted using a single Mus spretus individual from Spain (grey circle labeled “ES”) as the outgroup. Outgroup f3 statistic; higher values indicate more shared ancestry between wild Mus musculus (dom, domesticus; mus, musculus and cas, castaneus) and C57BL/6 substrains (C57BL/6NTac, Taconic Biosciences; C57BL/6NCrl, Charles River; C57BL/6J, the Jackson Laboratory). Labels and colors as in panel A. CI, confidence interval.
Taken together, these data identify Mus musculus domesticus from Maryland as an appropriate source for an external natural microbiome reference for standard laboratory mouse strains.
Characterization of the Mus musculus domesticus bacterial gut microbiome
Next, we characterized the mucosal-associated and luminal bacterial microbiome of Mus musculus domesticus populations at locations A, B and C. The ileocecal microbiome was chosen because it is known to encompass much of the diversity within the gastrointestinal tract. The microbiome community structure of each population was assessed using principal coordinate analysis (PCoA) of unweighted (considering only operational taxonomic unit [OTU] presence and absence) and weighted (considering OTU abundance) pairwise UniFrac distances. As shown in Figures 2A and 2B and Figure S2, the gut microbiome of wild mice from geographically distinct locations clustered together. This microbiome similarity cannot be explained by recent migration between locations or by family structure within locations, because the majority of individuals either within or between locations were no more related than second cousins (Figure S3). The gut microbiome of wild mice sampled one year later clustered in the same region (Figure S2A and S2B).
Figure 2. The Laboratory Mouse Bacterial Gut Microbiome from Commercial Vendors is Significantly Different from that of their Wild Living Kin.
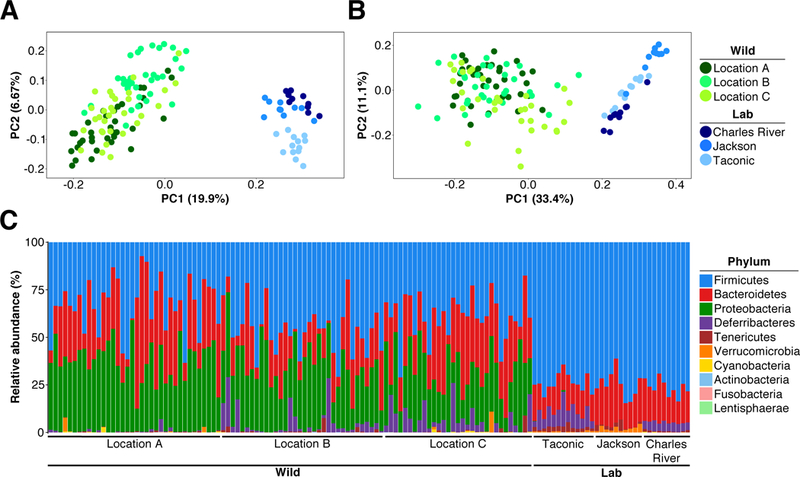
16S rRNA gene profiling data comparing the gut microbiome of Mus musculus domesticus from locations A, B and C in Maryland, USA (Wild) to that of C57BL/6 mice from Taconic Biosciences, Charles River and the Jackson Laboratory (Lab). (A) Unweighted UniFrac PCoA. (B) Weighted UniFrac PCoA. (C) Relative abundance at the rank of phylum.See also Figure S2, Figure S4 and statistics within Tables S2 and S3.
Importantly, the gut microbiome clusters from the wild mice were separate and significantly different from the gut microbiome clusters of C57BL/6 from Taconic Biosciences, Charles River and the Jackson Laboratory (P=0.0002 for each comparison, PERMANOVA; Figure 2A, 2B and Table S2). The unweighted pairwise UniFrac distances among gut communities from geographically distinct wild mouse locations were significantly decreased compared to distances between all wild and laboratory groups (P<0.0001 for each comparison, Kruskal-Wallis with FDR adjustment; Figure S2C: green versus orange).
The difference between the gut microbiome of wild-living Mus musculus domesticus and that of commercial C57BL/6 mice was also evident in the relative abundance of taxa at the rank of phylum (Figure 2C; Table S3). Specifically, there was significantly higher relative abundance of Bacteroidetes and Proteobacteria in wild mice compared to C57BL/6 mice (P<0.01 and P<0.0001 respectively, Mann-Whitney), and significantly lower relative abundance of Firmicutes, Tenericutes and Verrucomicrobia (P<0.0001 for each comparison, Mann-Whitney; Figure S2D and Table S3). Similar differences were also observed at the rank of order (Figure S2E; Table S4). We re-analyzed our data with a recently published 16S subOTU analysis algorithm (Callahan et al., 2016). In such subOTU analysis the sequences are compared with an error model and sequences which are statistically unlikely to be sequencing errors of each other are assigned to their own sequence variant, while sequences likely to be sequencing errors are “corrected” to a more abundant sequence variant. Importantly, subOTU analysis (Figure S4) confirmed the primary conclusions drawn from the OTU analysis (Figure 2, Figure S2).
Thus, despite being outbred and living in a varied environment, wild mice from Maryland displayed remarkable similarity in their bacterial gut microbiome. More importantly, the laboratory mouse bacterial gut microbiome from commercial vendors was significantly different from that of their wild living kin. We viably preserved the wild mouse gut microbiome for bio-banking and eventual engraftment into laboratory mice to compare the effects of wild and commercial mouse microbiomes on host responses to infectious and inflammatory diseases.
The Mus musculus domesticus bacterial gut microbiome can safely be transferred to and maintained in a laboratory mouse colony
Prior to transfer and engraftment of the natural gut microbiome we tested preserved biospecimens from the trapped wild mice for known mouse pathogens using polymerase chain reaction (PCR) and antibody detection. As expected, many wild mice were exposed to a variety of pathogens (Figure S3C). Similar to the microbiome results, the pathogen exposure history was comparable between mice from locations A, B and C (Figure S3C). Despite pathogen exposure, none of the examined and dissected wild mice showed gross pathological signs of disease. Moreover, pathogen-free wild mouse gut microbiota were readily found in the indigenous wild mouse population. Thus, we selected bio-banked gut microbiota from three sexually mature wild mice that matched the SPF criteria of our and most other animal facilities (Table S5). This allowed us to test our hypothesis, that natural gut microbiota have been shaped by evolutionary pressure and that they can confer host fitness promoting traits without the necessity and attendant risk of transferring known mouse pathogens into the animal facility.
Germ-free (GF) C57BL/6 mice were chosen as microbiome recipients, because they enable a more complete and unbiased engraftment compared to fully colonized or antibiotic-treated mice. GF recipients were 14 days pregnant on the first of three consecutive, daily oral gavages (Figure 3A) with pooled ileocecal material. This approach allowed vertical microbiome transfer to newborn pups enabling microbiota-mediated effects on the pups’ immune systems before and after birth (Gensollen et al., 2016; Gomez de Aguero et al., 2016). Hereafter we will refer to these mice as wild mouse microbiome-reconstituted (WildR). Re-screening of WildR mice verified their SPF status (Table S5), and no clinical signs of disease were observed in any WildR mouse. Whereas the transfer of unknown infectious agents cannot completely be ruled out, the high susceptibility of pregnant GF mice to infection and their unaffected health status post engraftment suggests the absence of disease causing pathogens. A second group of pregnant GF mice was reconstituted with the viably frozen gut microbiome of SPF C57BL/6 mice (hereafter referred to as LabR). In addition, barrier-raised C57BL/6 mice (hereafter referred to as Lab) were used to evaluate the efficacy of the engraftment procedure and to compare WildR and LabR phenotypes with the current laboratory mouse standard.
Figure 3. The Mus musculus domesticus Bacterial Gut Microbiome can be Transferred to Pregnant GF C57BL/6 Mice and Maintained in their Multigenerational Offspring.
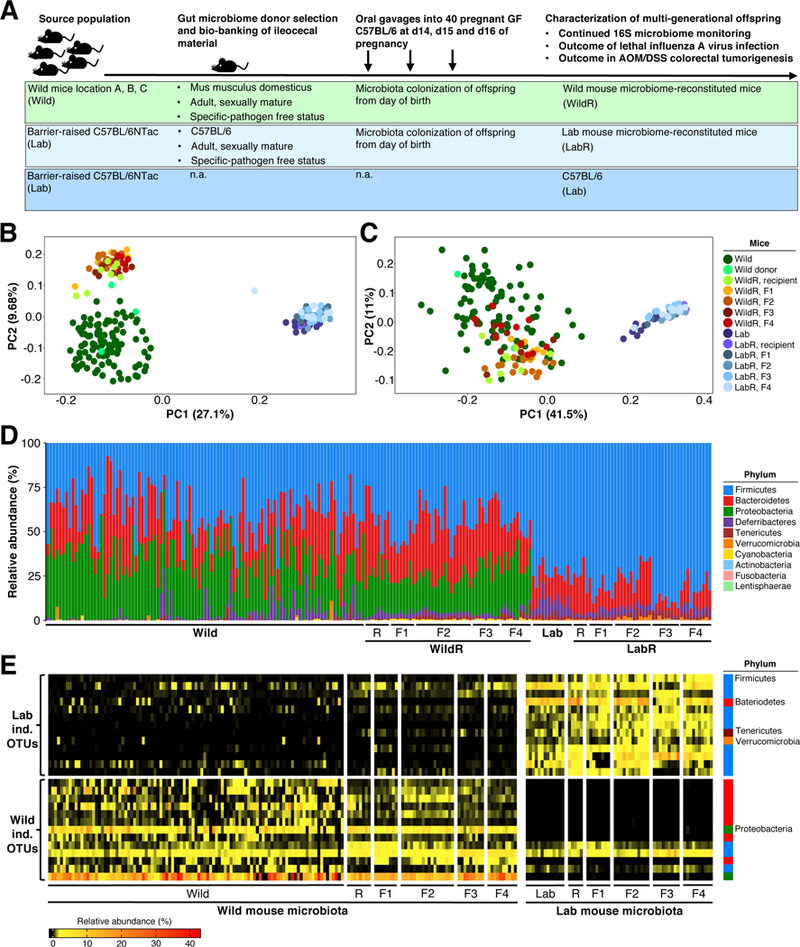
(A) Experimental strategy. (B – E) 16S rRNA gene profiling data comparing the gut microbiome of Mus musculus domesticus (Wild) or C57BL/6NTac (Lab), the recipient mice (WildR and LabR recipients [R]), and their subsequent generations (WildR F1, F2, F3, F4 and LabR F1, F2, F3, F4). (B) Unweighted UniFrac PCoA. (C) Weighted UniFrac PCoA. (D) Relative abundance at the rank of phylum. (E) Indicator species analysis identified bacterial OTUs of the investigated Wild and Lab gut microbiomes. Heatmap shows the relative abundances of Wild indicative and Lab indicative OTUs from all sampled generations.See also Figures S5, Figure S6 and Table S6.
Next, we characterized the bacterial gut microbiome of the reconstituted GF mothers and their F1, F2, F3 and F4 offspring. As shown in the PCoA in Figures 3B (unweighted UniFrac) and 3C (weighted UniFrac), the gut microbiome of LabR mice clustered together with that of Lab mice without major changes over subsequent generations (Figure 3B and 3C; Table S6). This indicates that the preservation and transfer protocol had no major effect on commensal community structure, confirming engraftment efficacy. PCoA of unweighted and weighted pairwise UniFrac distances illustrated that gut microbiomes of WildR mice clustered together without major changes over subsequent generations (Figure 3B and 3C; Table S6). This cluster was distinct but in close proximity to the original wild mouse cluster and significantly different from the overlapping LabR and Lab mouse microbiome clusters (P=0.0002, PERMANOVA). As shown in Figure S5A, unweighted pairwise UniFrac distances between corresponding WildR and LabR generations remained stable over time and were significantly greater (P<0.0001, Kruskal-Wallis with FDR adjustment, orange versus green and blue) than the stable UniFrac distances between subsequent WildR and LabR generations and their respective recipient mice. Assessment of taxa abundance in Wild, Lab, WildR and LabR generations confirmed the above observations at the rank of phylum (Figure 3D; Table S6) and order (Figure S5B; Table S7). The stability of the transferred wild mouse microbiome is intriguing, given the alterations in temperature, day/night cycle, diet and social structure.
We identified OTUs that were most indicative of the Lab and Wild group to track them through the LabR and WildR generations using an algorithm originally developed by Dufrene and Legendre in 1997 as an ecological tool (Dufrêne and Legendre, 1997) and recently employed by Seedorf et al. (Seedorf et al., 2014) to assess OTU persistence following environmental changes. Wild-indicative OTUs remained abundant throughout all WildR generations and were generally not found to be abundant in LabR mice (Figure 3E). Vice versa, the Lab-indicative OTUs remained abundant in all LabR generations and remained minor in the WildR group and its subsequent F1 to F4 generations. SubOTU analysis (Figure S6) confirmed the primary conclusions drawn from the OTU analysis (Figure 3, Figure S5).
As an alternate approach, we also performed whole shotgun metagenomics analysis. Across all samples, only a minor component of the non-host metagenome was assigned to archaea, fungi, protista and viruses (not shown). The top 50 genera with greatest variance between sample groups were all bacteria, and their relative log2 abundance patterns differed between Lab/LabR and Wild/WildR mouse groups (Figure 4).
Figure 4. The Mus musculus domesticus Bacterial Gut Microbiome can be Transferred to Pregnant GF C57BL/6 Mice.
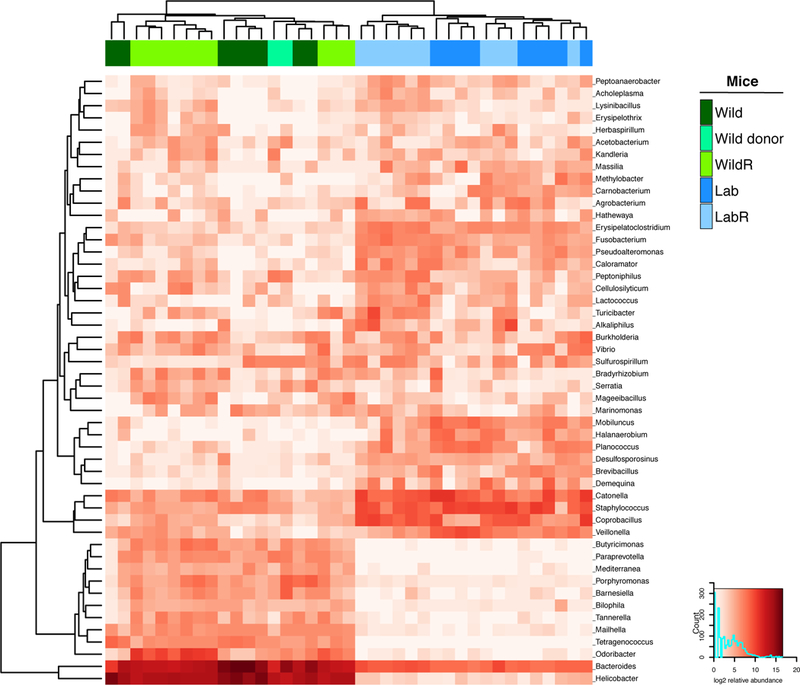
Analysis of whole shotgun metagenomic data comparing the gut microbiome of Mus musculus domesticus (Wild), C57BL/6NTac (Lab), WildR and LabR mice. The heatmap shows the top 50 genera with greatest variance between sample groups of log2 transformed relative abundance.
Taken together these data demonstrate that a wild mouse bacterial gut microbiome can be transferred to and stably maintained in the multigenerational offspring of a C57BL/6 laboratory mouse colony.
The Mus musculus domesticus gut microbiome promotes host fitness and survival of an otherwise lethal virus infection
Because infectious diseases exert much of the selection pressure encountered in the natural world, we evaluated whether the transfer of natural microbiota promotes the fitness of laboratory mice in the context of a viral challenge. Influenza A virus (IAV) mouse adapted A/PuertoRico/8/1934 H1N1 strain (PR8) was chosen for intranasal infection.
First, C57BL/6 mice from Taconic Biosciences, Charles River and the Jackson Laboratory were used to establish the clinically equivalent and lethal doses of 600 TCID50 for male mice and 400 TCID50 for female mice (Figure S7A), thereby normalizing for sex-related differences in susceptibility (Klein et al., 2012). These doses were then used in all experiments. Remarkably, while only 17% of LabR and Lab mice survived the intranasal IAV infection, 92% of WildR mice did (P<0.0001, log rank (Mantel-Cox) analysis; Figure 5A). WildR mice exhibited an initial weight gain with delayed and significantly reduced weight loss and disease severity compared to LabR and Lab mice (P<0.0001, comparing slopes of weight loss in a linear regression analysis; Figure 5B). Lung viral titers were ~10-fold lower for WildR than for LabR and Lab mice at day 3 post infection (P<0.01, Kruskal-Wallis with Dunn’s multiple comparison test; Figure 5C). Histopathological examination and scoring of lung tissue at the peak of clinical symptoms at day 7 post infection demonstrated significantly less bronchitis, bronchiolitis and alveolitis with less pyknotic bronchial epithelial cell death and degeneration in WildR than in LabR and Lab mice. Lung tissue of WildR mice also contained significantly lower numbers of inflammatory cells such as lymphocytes, macrophages and neutrophils. Microscopic bleeding and loss of aeration were also less frequently observed in WildR mice (P<0.05 for each score, One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval; Figure 5D and 5E). Overall, WildR mice presented with an infection more limited to the upper respiratory tract, with less inflammatory cell infiltration and with less immune-mediated pathology compared to LabR and Lab mice. These results were independently confirmed for both sexes (Figure S7B, C and D).
Figure 5. The Mus musculus domesticus Gut Microbiome Confers a Survival Advantage after Lethal IAV Infection.
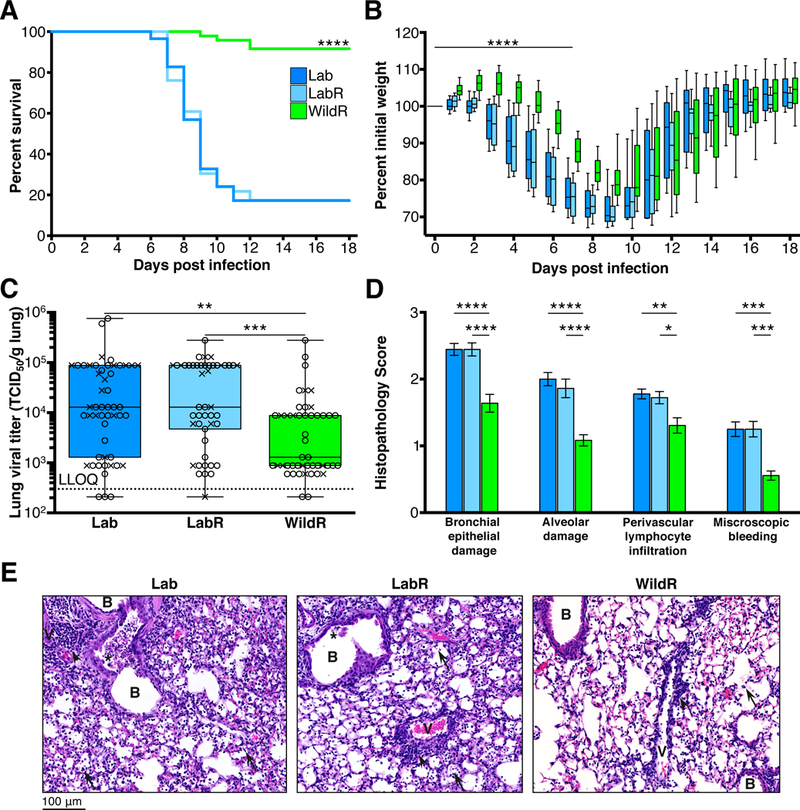
Female mice were inoculated with 400 TCID50 and male mice with 600 TCID50 of PR8. (A and B) Mice were monitored daily for 18 days and mice that lost 30% or more of their body weight were euthanized (Lab,n=58; LabR, n=46; WildR, n=48). (A) Kaplan Meier survival curves, ****P<0.0001 comparing WildR with either LabR or Lab by log rank (Mantel-Cox) analysis. (B) Weight loss curves, ****P<0.0001 comparing the slope of the weight loss (day 0 to day 7) of WildR to that of LabR or Lab in a linear regression analysis. Weight loss of LabR did not significantly differ from that of Lab. Median and IQR are presented. (C) Lung viral titer 3 days post IAV infection assessed via MDCK monolayers in 96-well plates using an antibody-based assay. Lab, n=51; LabR, n=49; WildR, n=48. Median and IQR are presented. (D) Histopathological scores 7 days post IAV infection. B: Bronchi; V: Vessels; arrows: lymphocytes and/or red blood cells in alveoli; arrowheads: perivascular lymphocyte infiltration; asterisks: bronchial epithelial cell death. Mean and SEM are presented. (E) Representative lung histology 7 days post IAV infection (original magnification 40x). Lab, n=18; LabR, n=18; WildR, n=18. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Significance was determined using parametric One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval (Gaussian model), or Kruskal-Wallis with Dunn’s multiple comparison test. All data shown are from three independent experiments using both female (X) and male (O) mice.See also Figure S7.
The clinical signs and viral replication kinetics pointed to critical involvement of early innate immune mediators in WildR mouse resistance to IAV. Analysis of lung tissue at day 4 post infection revealed significantly higher levels of multiple inflammatory cytokines, chemokines and growth factors in LabR and Lab than in WildR mice (Figure 6A). The type of hyper-activation and cytokine response seen in LabR and Lab is consistent with descriptions of cytokine storms associated with a lethal IAV infection (Kash et al., 2006; Kobasa et al., 2007; Tisoncik et al., 2012). In contrast, no typical cytokine storm signature was found in WildR mice and levels of anti-inflammatory cytokines such as IL-10 and IL-13, known to be protective during lethal IAV infection, were either the same or significantly elevated. Levels of pro-inflammatory cytokines were negatively correlated with levels of anti-inflammatory cytokines, indicating two distinct cytokine responses to the same infection in WildR versus LabR and Lab mice (Figure 6B). LabR and Lab mice exhibited similar cytokine profiles (Figure 6A and 6B), indicating that the exclusive engraftment of the gut microbiome into GF mice recapitulates both the immune response and the clinical outcome of laboratory mice.
Figure 6. The Mus musculus domesticus Gut Microbiome Affects the Cytokine Profile of Mice after Lethal IAV Infection.
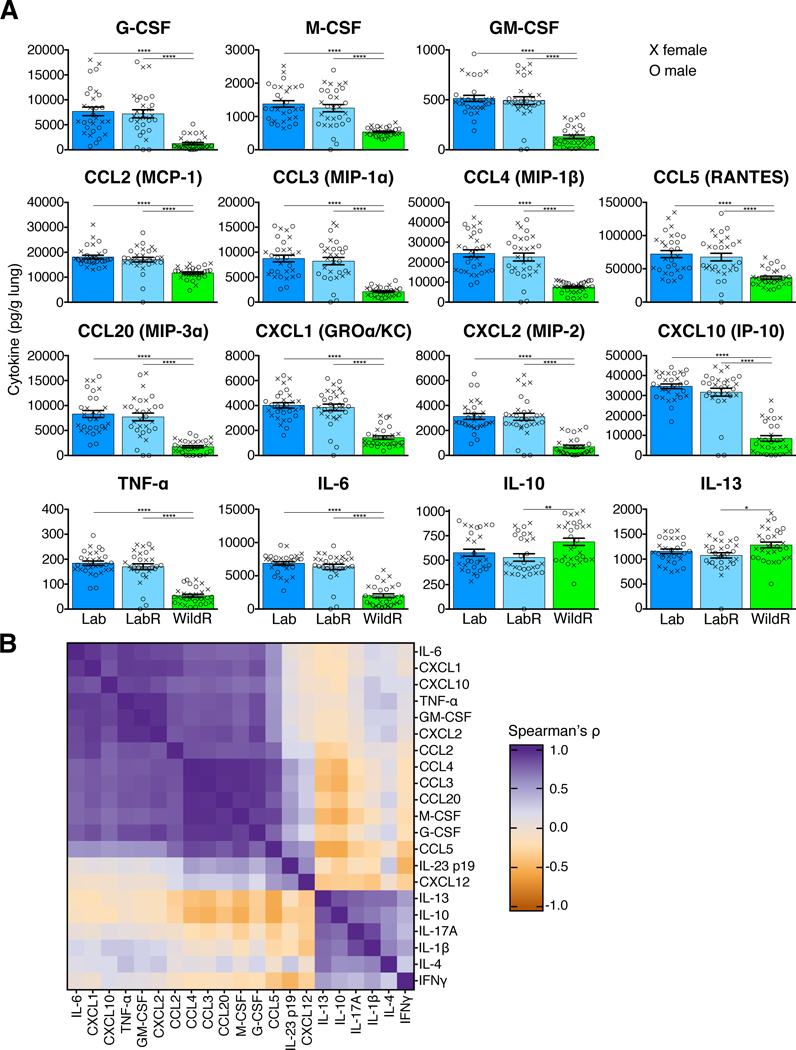
Female mice were inoculated with 400 TCID50 and male mice with 600 TCID50 of PR8. Cytokines, chemokines and growth factors were quantified in lung tissue at day 4 post IAV infection using a multiplex Luminex assay. (A) Univariate analysis of the 15 of 21 cytokines that were differentially expressed in WildR as compared to LabR and Lab. Mean and SEM are presented. (B) Correlation matrix (using Spearman’s rank-correlation) between measurements of all 21 cytokines, hierarchically clustered and rendered as a heatmap. Lab, n=29; LabR, n=30; WildR, n=30. *P<0.05, **P<0.01, ****P<0.0001. Significance was determined using parametric One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval (Gaussian model), or Kruskal-Wallis with Dunn’s multiple comparison test. All data shown are from three independent experiments using both female (X) and male (O) mice.
In summary these data indicate that the wild mouse gut microbiome confers traits that abrogate excessive inflammation and promote host fitness in the context of an otherwise lethal pulmonary viral infection.
The Mus musculus domesticus gut microbiome protects against mutagen- and inflammation-induced neoplastic development
To determine if the benefits conferred by a natural gut microbiome extend beyond an infectious disease to other diseases that include inflammatory stimuli we chose a model of mutagen- and inflammation-induced colorectal tumorigenesis. Similar to influenza A virus infection (Taubenberger and Morens, 2008), colorectal cancer also represents a significant disease burden in humans (Torre et al., 2015).
In the corresponding murine model, an intraperitoneal injection of the chemical mutagen azoxymethane (AOM) is followed by oral application of the colitis-inducing agent dextran sodium sulfate (DSS). This results in chronic inflammation and progression to dysplasia and adenocarcinoma, thereby creating a robust model of colitis-associated colorectal cancer (Tanaka et al., 2003). As shown in Figure 7A, weight loss of LabR mice did not significantly differ from that of Lab mice. In contrast, WildR mice exhibited significantly less inflammation-induced weight loss than LabR and Lab mice (P<0.001 and P<0.0001, respectively, repeated measures mixed model linear regression). Moreover, the WildR group displayed a significantly lower number (P<0.0001, Figure 7B-E) and surface area of colorectal tumors relative to LabR and Lab mice at the end of the study period (day 85) (P<0.0001, One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval; Figure 7B-E).
Figure 7. The Mus musculus domesticus Gut Microbiome Confers Protection from Colitis-Associated Tumorigenesis.
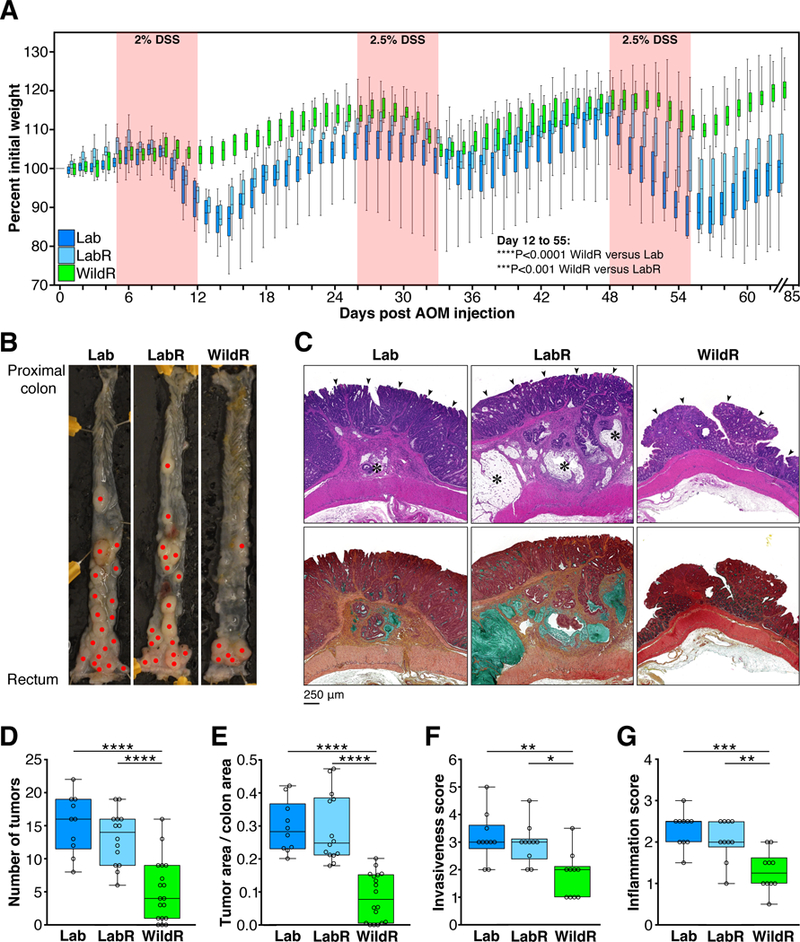
Clinical data from male mice that received a single intraperitoneal injection of AOM (10 mg/kg of body weight), followed by three 7 day cycles of 2–2.5% DSS in drinking water. Mice were monitored for weight loss throughout the time-course of the experiment and euthanized on day 85 to assess tumor burden. (A) Weight loss curves following AOM/DSS treatment. Lab, n=18; LabR, n=16; WildR, n=19. WildR vs. Lab, ****P<0.0001, and WildR vs. LabR, ***P<0.001 (repeated measures mixed model linear regression). Weight loss of LabR did not significantly differ from that of Lab. (B) Representative images of dissected colons (red dots indicate tumors). (C) Representative colon histology (original magnification 10x). Upper panel: HandE staining of longitudinal colon tumor sections; arrowheads: well differentiated adenocarcinoma in mucosa. LabR and Lab tumors invade the submucosa and muscular layers with moderate to severe inflammation; asterisks: mucinous nodules. Lower panel: Movat’s staining of serial sections of the same tumors as in the upper row images. Mucinous nodules (mucin stained in green) containing mucinous carcinoma cells and tubular adenocarcinoma lining are found in submucosa and muscle layers of LabR and Lab tumors. Lab, n=10; LabR, n=10; WildR, n=10. (D) Number of tumors. (E) Fraction of total colon area covered in tumors. Tumor burden was assessed using ImageJ software. (F) Invasiveness scores based on tumor location. (G) Inflammation score based on inflammatory cell infiltration. Median and IQR are presented. *P<0.5, **P<0.01, ***P<0.001, ****P<0.0001. Significance was determined using parametric One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval (Gaussian model). All data shown are from three independent experiments.
The tumors of LabR and Lab mice represented tubular adenoma, well differentiated tubular adenocarcinoma and mucinous carcinoma. The latter invaded the muscle layer and subserosa of the tissue and metastasis were found in small lymph nodes attached to the colon. In contrast, WildR mice exhibited fewer adenomas and adenocarcinomas and no mucinous carcinomas resulting in a significantly lower invasiveness score in WildR mice than in LabR and Lab mice (P<0.05 and P<0.01, respectively; Figure 7F). Consistent with the influenza model WildR mice also displayed significantly less inflammatory cell infiltration than LabR and Lab mice (P<0.01 and P<0.001, respectively, One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval; Figure 7G).
Collectively, the results demonstrate that the wild mouse gut microbiome confers traits that promote host fitness and limit inflammation in an infectious disease and in mutagen- and inflammation-induced neoplastic development.
Discussion
Our findings demonstrate that the bacterial gut microbiome of laboratory mice markedly differs from that of a genetically-similar wild population, Mus musculus domesticus from Maryland. They also establish that the wild mouse bacterial gut microbiota can be viably preserved, transferred to laboratory mice and maintained over at least several generations under vivarium conditions. Importantly, relative to the laboratory gut microbiota, the wild gut microbiota promoted host fitness and limited inflammatory responses in two diseases relevant to humans: influenza A virus pulmonary infection and mutagen- and inflammation-induced colorectal tumorigenesis.
It is well known that the bacterial gut microbiome affects disease outcomes in influenza virus infection (Abt et al., 2012; Ichinohe et al., 2011; Oh et al., 2014) and colorectal tumorigenesis models (Zackular et al., 2016; Zackular et al., 2013). However, previous studies depleted the gut microbiome of laboratory mice by either using broad-spectrum antibiotics and/or using GF mice leading to differential effects on host disease resistance. In contrast, we started out with the concept that important host fitness promoting traits are missing in the laboratory mouse. Indeed, transfer of the natural wild mouse gut microbiome to laboratory mice improved outcomes in both disease models tested in our study.
Natural microbiota appear to beneficially balance systemic and local inflammatory responses upon disease challenges. This is illustrated by decreased levels of pro- inflammatory and increased levels of anti-inflammatory cytokines and significantly less inflammatory cell infiltration along with higher survival rates during lethal influenza infection. It is further supported by lower inflammatory scores, reduced colitis-induced weight loss and tumor burden in the colorectal tumorigenesis model, consistent with the known impact of chronic inflammation on cancer development and progression (Lasry et al., 2016). The observed capability of the natural microbiota to modulate inflammatory responses, minimizing collateral damage while preserving beneficial anti-viral and tumor immunity, likely promotes host fitness in the natural world, with its burden of infectious diseases and other pro-inflammatory stimuli present in the environment.
Taken together, these data support our hypothesis, that the protective traits conferred by natural microbiota in free-living mammals were selected to promote host fitness and survival in the context of a challenging environment rife with infectious diseases and diverse naturally occurring mutagens (e.g. aflatoxins in agricultural products like grains) and inflammatory immune stimuli. In clear contrast, the microbiota of laboratory mice have been subject to limited selection, regarding such challenges. Thus, studying natural microbiota should facilitate the discovery of novel protective mechanisms relying upon natural host-microbe interactions that are absent in laboratory mice. In addition to shotgun metagenomics, future mechanistic studies should also employ targeted extraction and sequencing methods to more precisely assess nonbacterial organisms of the microbiome such as protists (Chudnovskiy et al., 2016), parasites (Howitt et al., 2016), fungi (Ackerman and Underhill, 2017), and viruses (Lim et al., 2016), as well as important trans-kingdom interactions (Pfeiffer and Virgin), which may contribute to the observed effects.
The mammalian phenotype is largely driven by the combination of host and microbial genes, together known as the metagenome. Our current study preserves the genetic tractability of laboratory mice and their associated tools but changes the microbial component of the metagenome to better match natural microbiota. Our approach should facilitate the development of animal models that better recapitulate complex natural physiological phenomena (e.g. pathophysiology). While recent studies have pursued this goal by exposing laboratory mice to selected pathogens (Reese et al., 2016) or co-housing with pathogen-infected pet store mice (Beura et al., 2016), we demonstrate that host fitness promoting traits relevant in the natural world can be conferred by a natural microbiota devoid of known mouse pathogens.
We propose that combining a complete natural mouse microbiome (gastrointestinal tract, lung, skin, vagina) with a defined population of naturally occurring murine pathogens will further improve the accuracy of mouse models in regards to recapitulating complex physiological phenomena relevant in the natural world outside of the laboratory environment. This approach should also facilitate the development of mouse models that more accurately reflect disease courses in humans. Given the wide-ranging effect of the microbiome on host physiology, it is likely that natural microbiota will additionally influence other aspects of laboratory mouse physiology. Beside immunological studies, this may benefit many research fields including endocrinology, metabolism and behavioral studies, with obvious applications to other model organisms.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barbara Rehermann (Rehermann@nih.gov.).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Barrier-raised SPF C57BL/6NTac mice and timed, 14 days pregnant, GF C57BL/6NTac mice (8–12 weeks of age) were acquired from Taconic Biosciences. 8–12 weeks old barrier-raised SPF C57BL/6 mice were also acquired from the Jackson Laboratory (C57BL/6J) and from Charles River (C57BL/6NCrl).
All mice were housed under a 12:12 light:dark cycle and gnotibiotic conditions in isolators (Park Bioservices) in an American Association for the Accreditation of Laboratory Animal Care (AALAC)-accredited animal facility at the National Institute for Diabetes, Digestive and Kidney Diseases (NIDDK). Animals were fed autoclaved rodent chow (NIH-31 open formula) and autoclaved tap water ad libitum. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by the NIDDK Animal Care and Use Committee. Non-randomized, gender-and age-matched mice between 8–12 weeks of age were used for all experiments. Investigators were not blinded unless otherwise noted.
Trapping and sampling of wild Mus musculus domesticus
Mus musculus domesticus were trapped in 8 geographically distinct horse stables throughout Maryland and the District of Columbia (USA) with the following GPS coordinates: location A, N 38° 59’ 26.05”, W 77° 3’ 38.78”; location B, N 39° 5’ 21.49”, W 77° 14’ 56.87”; location C, N 39° 9’ 11.25”, W 76° 52’ 13.55”; location D, N 39° 13’ 46.11”, W 77° 4’ 45.35”; location E, N 39° 12’ 18.51”, W 77 ° 6’ 3.70”; location F, N 38° 57’ 27.82”, W 77°3’ 3.64”; location G, N 39°8’ 13.95”, W 76° 58’ 29.94”; location H N 39°7’ 29.11”, W 76°59’ 8.23” from October-December 2014 and again in October 2015. Mice were trapped in autoclaved live animal aluminum traps (H. B. Sherman) with peanut butter as bait. Traps were placed after 8 pm and checked the following morning (8 am) to avoid long captivity times. Animals were pre-selected based on appearance to exclude other animals such as deer mice and young mice. The remaining mice were brought to the laboratory, photographed, examined for signs of disease, and assigned to an age class based on body weight, body length (tip of nose to base of tail) and developmental status of the reproductive tract (Extended Data Table 1a) (Chou et al., 1998; Gomez, 2008; Marshall, 1981; Smith et al., 1993). Mice were dissected to harvest tissues and microbiota (see below), and identified as Mus musculus domesticus based on phenotype and genotype (microarray genotyping, see below).
METHOD DETAILS
Pathogen screening
Blood drops dried on EZ-SPOT® (Charles River), body swabs, fecal pellets, and lung tissue were harvested according to Charles River sampling guidelines and screened for pathogens by PCR and Serology with the Mouse PRIATM (PCR Rodent Infectious Agent) Panel Surveillance Plus and the Serology Profile Assessment Plus by Charles River infectious agent testing (Charles River). A mouse was considered pathogen- exposed if it tested positive in at least one of these assays. Equivocal results were counted as positive. The fraction of positive mice was calculated and visualized using a heatmap created using the pheatmap (Kolde, 2015) and RColorBrewer (Neuwirth, 2014) packages in Rv3.3.0 (The R Foundation, https://cran.r-project.org/).
Harvest and storage of mouse organs, tissue, stool and microbial communities
To harvest viable mucosal-associated and luminal microbial communities from the terminal ileum (the distal 1.0 to 1.5 cm of the small intestine) and the cecum of wild mice and barrier-raised SPF C57BL/6NTac mice, animals were sacrificed and immediately transferred into an anaerobic chamber with an atmosphere composed of 75% nitrogen, 5% hydrogen, 20% carbon dioxide (Coy Lab Products). The abdomen was opened under sterile conditions. The terminal ileum and cecum were opened and manually extruded along the cephalocaudal axis to collect the fecal material into a 70 μm cell-strainer placed inside of a sterile petri dish. The residual ileocecal tissue was placed into a 2-ml cryovial with 1 ml of cryoprotectant (PBS containing 0.1% cysteine and 12% glycerol) and vortexed for one minute. Surgical instruments were rinsed in the same cryoprotectant to avoid the loss of residual fecal material. The collected material was weighed and then diluted at a 1:15 or 1:30 ratio (g collected material:ml cryoprotectant) including the cryoprotectant used for the ileocecal tissue wash. The resulting suspension was mashed through a 60 μm cell-strainer to remove insoluble particles, aliquotted into 2-ml internally threaded plastic screw-capped cryo-vials (Biocision) and sealed into Nunc® CryoFlex™ Tubing. Sealed cryovials were removed from the anaerobic chamber and subjected to controlled freezing (−1°C per minute) prior to transfer into liquid nitrogen for long-term liquid-phase storage.
Whole lung tissue, whole skin tissue (both ears), tail tissue and fecal pellets were snap-frozen on dry ice and stored in a −80°C freezer.
Microarray genotyping of wild-caught mice
Genomic DNA was isolated from cryopreserved tail clips from wild-caught mice using the Qiagen DNeasy Blood and Tissue kits according to the manufacturer’s instructions. High molecular weight DNA from a panel of standard laboratory mouse strains (Yang et al., 2011) was used as reference. All samples were genotyped using the Mega Mouse Universal Genotyping Array (MegaMUGA, 77K SNPs) and the GigaMUGA (143K SNPs) by Neogen/GeneSeek Inc (Lincoln, NE) (Morgan et al., 2016). The two array platforms were combined by retaining the 66,914 SNPs shared between them. Samples with more than 10% missing data across the 67K SNPs present on both platforms – a threshold that reliably identifies genotyping failures – were excluded from further analyses. Subsequent analyses were performed in R v3.2.2 (The R Foundation, https://cran.r-project.org/) using the argyle package (Morgan, 2016).
16S rRNA gene sequencing
16S rRNA gene sequencing methods were adapted from the methods developed for the Earth Microbiome Project (Caporaso et al., 2012; Caporaso et al., 2011) and Human Microbiome Project (Human Microbiome Project, 2012a, b). Briefly, bacterial genomic DNA was extracted using MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories). The 16S rDNA V4 region was amplified by PCR and sequenced in the MiSeq platform (Illumina) using the 2×250 bp paired-end protocol yielding pair-end reads. The primers used for amplification (515F-806R) contain adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products (Caporaso et al., 2012) on the Illumina MiSeq platform.
Whole shotgun metagenomic sequencing
Whole metagenomic DNA was extracted using MagAttract PowerMicrobiome DNA/RNA EP (Qiagen). DNA (1 ng) obtained from each sample was used for Illumina Nextera XT paired-end library construction which were then shotgun sequenced in the Illumina NextSeq Platform using Nextseq 500/550 High output kit V2 (Illumina).
Transfer of viable gastrointestinal microbiota
The viably preserved ileocecal microbial communities from terminal ileum and cecum of Mus musculus domesticus were transferred by three consecutive daily oral gavage procedures into timed pregnant GF mice. Cryopreserved ileocecal stocks were thawed in a 37°C water bath until a small piece of ice remained. Fecal stocks from three sexually mature, SPF Mus musculus domesticus mice with representative microbiomes were mixed so that each recipient GF mouse received the same 0.1 to 0.15 ml suspension on day 14, 15 and 16 of pregnancy. This ensured a natural transfer of the microbiota to the litter, thereby facilitating microbiota-mediated effects on the immune system of the F1 generation in utero and during birth, the neonatal period and puberty (mice referred to as WildR). A second group of time-mated pregnant GF mice served as a control and was reconstituted with viably preserved ileocecal microbial communities of barrier-raised SPF C57BL/6 mice (mice referred to as LabR).
Mice that received the same ileocecal fecal stocks were housed in the same isolator under gnotobiotic conditions (Park Bioservices). To maintain a homogenous microbial flora within a given group of mice, bedding was transferred within each group one to two times per week. 16S rRNA sequencing was performed as described above, starting with recipient former GF mothers, followed by all subsequent generations.
IAV challenge model
A/Puerto Rico 8/34 (PR8; H1N1) was propagated in embryonated chicken eggs. Viral titers were determined by standard 50% tissue culture infective dose (TCID50) assay on MDCK cells. Age- (8–12 weeks) and sex-matched C57BL/6, WildR, and LabR mice were anesthetized using isoflurane (induction chamber ParklandScientific Inner Dimension: 5.25” W x 3.5” H. Overall Dimensions: 7” W × 7” H.; oxygen flow rate 1.5 L/min; isoflurane flowrate 3.5L/min) and intranasally infected with PR8 in 25 μl PBS 0.1 % BSA. Female mice were intranasally inoculated with 400 TCID50 and male mice with 600 TCID50 PR8. Weight was monitored daily for 18 days and mice that lost 30% or more of their body weight were euthanized and scored as dead. Mice that lost less than 10% of their body weight were considered as insufficiently infected and excluded from the experiment (13.4% of all Lab mice, 11.5% of all LabR mice and 12.7% of all WildR mice).
Determination of IAV titers
IAV-infected mice were euthanized at day 3 post infection by intraperitoneal avertin injection and exsanguinated through the abdominal aorta. Whole lung tissue was collected using sterile instruments. Lungs were kept cold at all processing steps, weighed, suspended in PBS with Calcium and Magnesium at a 1:4 ratio (g:ml), snap-frozen on dry ice and stored at −80°C. Subsequently, lungs were homogenized in a 20% (wt/vol) suspension. After 15 minutes centrifugation at 3000 × g the clarified supernatant was titrated in quadruplicate on mycoplasma-free Madin-Darby canine kidney (MDCK, ATCC, VA) monolayers in 96-well plates. Twenty hours after incubation cells were washed with PBS and fixed in ice-cold MetOH for 10 min. Infected cells were detected by incubation with the anti-nucleoprotein mAb HB-65 for 2 hours followed by rat-anti-mouse-kappa HRP-conjugated (Southern Biotech), as previously described (Yewdell et al., 1981). Plates were developed using TMB substrate (KPL biomedical) and halted by the addition of 0.1N HCl. Wells with an OD450 reading >2× the non-infected cell controls were scored as positive. TCID50 was calculated as above and titers expressed as TCID50/g of tissue.
Cytokine analysis in lung tissue of IAV-infected mice
IAV-infected mice were euthanized at day 4 post infection, lungs were harvested, weighed and placed into Cell Lysis Buffer (Affymetrix eBioscience catalog number EPX- 99999–000) at a ratio of 1:5 (g:ml) according to manufacturer’s recommendations. To limit protein degradation, an additional 1 mM protease inhibitor phenylmethylsulfonyl fluoride (SIGMA-ALDRICH) was added to Cell Lysis Buffer right before use. Lungs were homogenized at 4°C for 2 min using a bead beater homogenizer at 25 Hz (Biospec Products). Homogenized samples were centrifuged at 16,000 × g for 10 min at 4°C, and the supernatant was transferred into a 1.5 ml Protein LoBind tube with Safe-Lock (Eppendorf), snap frozen on dry ice, and stored at −80°C. The supernatant was thawed on ice and further clarified by two additional centrifugation steps at 16,000 × g for 10 min and each time transferred into new Protein LoBind tubes. A customized multiplex cytokine/chemokine Luminex assay (RandD Systems) was performed according to manufacturer’s recommendations with samples plated in duplicates.
Histopathological examination of lung tissue of IAV-infected mice
IAV-infected mice were euthanized at day 7 post infection by intraperitoneal avertin injection. Whole lungs were inflated with cold PBS through the trachea, tied by thread, then harvested and fixed in 10% buffered formalin for over night followed by 70% ethanol. Paraffin blocks were made and serial step sections were processed. Every 10th slide was stained with hematoxylin and eosin (HE). All HE slides were digitally scanned on a Nanozoomer-XR digital slide scanner (Hamamatsu Photonics K. K., Japan) and lung pathology was evaluated and digitally stored (score 0–3 at intervals of 0.5). The pathologist was blinded with regards to the group definition and clinical outcome.
Colorectal tumorigenesis model
8–12-week-old male C57BL/6 mice (Lab), LabR and WildR mice received a single intraperitoneal injection of azoxymethane (AOM; 10 mg/kg of body weight) on day 0. This treatment was followed by three 7-day cycles of 2–2.5% dextran sodium sulfate (DSS) in drinking water (day 5–12; day 26–33; day 48–55). Mice were monitored for weight loss and euthanized on day 85 to assess colorectal tumor burden. Images of dissected colorectal tissue were analyzed using ImageJ software (NIH, MD), in a blinded manner with regards to the groups the samples were assigned to. For histological analysis, colorectal tissue was fixed in 10% buffered formalin for over night followed by 70% ethanol. Paraffin blocks were made and serial step sections were processed. Every 10th slide was stained with hematoxylin and eosin (HE) and Movat. All slides were digitally scanned on a Nanozoomer-XR digital slide scanner (Hamamatsu Photonics K. K., Japan) for semi-quantitative analysis of invasiveness and inflammation. Invasiveness was scored based on tumor location as follows: tumors located within lamina propria (score of 1), muscularis mucosae (score of 2), submucosa (score of 3), muscular propria (score of 4), subserosa or serosa (score of 5). Inflammation was scored based on inflammatory cell infiltration; 1=mild, 2=moderate, 3=severe.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details are indicated throughout the main text, in the Figure legends and within the supplemental Tables.
Phylogenetic analyses of wild-caught mice
The ancestry of wild-caught Maryland mice was determined by comparing their SNPs to those of a diverse collection of mice trapped around the world, including representatives of all three subspecies of Mus musculus and the outgroup Mus spretus (Didion et al., 2016) and a panel of twelve laboratory inbred strains. Because the content of the MUGA arrays is biased toward polymorphisms segregating in laboratory mice rather than in wild mice, we restricted our analysis to a subset of 28,284 SNPs, which are informative for ancestry at the subspecies level (Morgan et al., 2016). A distance matrix was computed by calculating the proportion of alleles shared identical-by-state (IBS) on the autosomes between all pairs of samples, and a tree was constructed from the matrix using the neighbor-joining method. The tree was rooted using Mus spretus as the outgroup.
To assess the degree of shared ancestry between C57BL/6NTac and the Maryland mice relative to other wild populations, we used the f3 statistic. Although originally derived as a test for admixture, the f3 statistic of the form f(outgroup; A, B) can be interpreted as a measure of the shared branch subtending populations A and B in a phylogeny rooted by the outgroup (Peter, 2016). The threepop utility in the TreeMix package (Pickrell and Pritchard, 2012) was used to calculate f(M. spretus; C57BL/6NTac, test population) for each wild population using 10,000 unlinked autosomal SNPs. Standard errors and Z-scores were obtained by the block jackknife over blocks of 250 SNPs.
Population structure of wild-caught mice
To assess the population structure within the Maryland mice, we imputed the observed allele frequency in place of missing values, dropped 20,303 monomorphic autosomal SNPs, and scaled the remaining columns of the genotype matrix to have zero mean and unit variance. PCA was performed on this matrix and the top three eigenvalue- eigenvector pairs were kept. KING v1.4 (Manichaikul et al., 2010) with options “--kinship -- ibs” were used to estimate kinship coefficients and to identify related individuals among the Maryland mice.
16S rRNA gene compositional analysis
The 16S rRNA gene pipeline data incorporates phylogenetic and alignment-based approaches to maximize data resolution (Buffington et al., 2016; Viladomiu et al., 2017). The 16S rRNA gene read pairs were demultiplexed based on the unique molecular barcodes, and reads were merged using an 8 bp seed, 0 mismatches over a 50bp overlap and an expected error filter of 0.05 with USEARCH v7.0.1090 (Edgar, 2013). 16S rRNA gene sequences were clustered into Operational Taxonomic Units (OTUs) at a similarity cutoff value of 97% using the UPARSE algorithm. OTUs were mapped at 97% sequence identity to an optimized version of the SILVA Database (version 128, https://www.arb-silva.de/documentation/release-128/)) (Quast et al., 2013) containing only the 16S v4 region to determine taxonomies. Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A rarefied OTU table from the output files generated in the previous two steps was used for downstream analyses using a visualization toolkit also developed at the CMMR named ATIMA (Agile Toolkit for Incisive Microbial Analyses).
ATIMA is an R (Team, 2014) software suite combining publicly available packages (i.e. APE and VEGAN) (Oksanen, 2017; Paradis et al., 2004) and purpose written code to import sample data and identify trends in taxa abundance, alpha-diversity, and beta-diversity with sample metadata. Significance of categorical variables are determined using the non-parametric Mann-Whitney test (Mann and Whitney, 1947) for two category comparisons or the Kruskal-Wallis test (Kruskall, 1952 #459} when comparing three or more categories. Correlation between two continuous variables is determined with R’s base “lm” function for linear regression models, where p-values indicate the probability that the slope of the regression line is zero. PCoA plots employ the Monte Carlo permutation test (Dwass, 1957) to estimate p-values. All p-values are adjusted for multiple comparisons with the FDR algorithm (Benjamini and Hochberg, 1995).
Pairwise PERMANOVA tests with false discovery rate adjustments for multiple comparisons were applied using the adonis function in the vegan (Oksanen et al., 2016) package to the unweighted and weighted UniFrac metric to determine differences in microbial community composition. Differences in taxonomic abundance and differences between pairwise unweighted UniFrac distances were assessed with the Kruskall-Wallis test with a false discovery rate adjustment for multiple comparisons in Rv3.3.0 (The R Foundation, https://cran.r-project.org/). Additionally, multiple other helper functions and graphing tools were utilized in the R environment (De Caceres and Legendre, 2009; Dowle and Srinivasan, 2016; McMurdie and Holmes, 2013; Wickham, 2007, 2009; Wilke, 2016).
Indicator species analysis (Dufrêne and Legrende, 1997; Seedorf et al., 2014), as implemented by the indicspecies package, was used to identify OTUs that were most indicative of the Wild or Lab group based upon both probability of occurrence and abundance in those groups. Those OTUs that were found to be significant indicators through permutation tests and subsequent False-Discovery-Rate adjustment were further screened to ensure that we identified highly indicative OTUs. Therefore, we removed those indicator OTUs that did not occur in at least 75% of the mice for which they were indicative (i.e. Wild or Lab) and we further removed indicator OTUs that were not observed at an average relative abundance of greater than 0.3% for the samples shown.
The DADA2 subOTU analysis was performed using DADA2 1.4.0 (Callahan et al., 2016) running on R 3.4.1. The resulting abundance table and taxonomic classification was loaded into R and analyzed and plotted using phyloseq 1.2.0 (McMurdie and Holmes, 2013). Statistical analysis of differentially abundant sequences and taxa were performed by DESeq2 1.16.1 (Love et al., 2014).
Whole shotgun metagenomics analysis pipeline
The raw paired-end Illumina reads obtained from the whole shotgun metagenome sequencing of each metagenomic sample were trimmed using Trimmomatic (Bolger et al., 2014) and aligned against the Mus musculus complete genome (GenBank Accession Number GCF_000001635.25) using Bowtie2 (Langmead and Salzberg, 2012) to filter out host reads, yielding between 1.2 and 7.2 Gbp of non-host data for each sample (mean=3.7 Gbp). These non-host reads were assembled de novo using MEGAHIT (Li et al., 2015) and the resulting contigs were taxonomically classified by Kraken (Wood and Salzberg, 2014) using a custom-built k-mer database containing whole genome sequences available in NCBI’s GenBank at the complete and scaffold levels for all bacteria, and at all levels for archaea, fungi, protozoa and viruses plus the Homo sapiens and Mus musculus genomes. The sequencing reads used as input for de novo assembly were then re-aligned back to the assembled contigs using Bowtie2 to gauge sequencing depths pertaining to each contig. Reads which were not assembled into contigs were then themselves classified by Kraken using the same database as for contigs. The total number of bases sequenced for each microbial taxon in each sample was obtained by adding the number of bases realigned onto contigs identified as belonging to that taxon to the number of non- assembled bases in reads identified as belonging to the same taxon. The relative abundance of each taxon in each metagenomic sample was then computed as the proportion of total non-host bases identified as belonging to a taxon either in contigs or non-assembled reads to the total number of non-host bases sequenced for that sample. A table with the relative abundances for all taxa for all samples, expressed in parts per million were then used as input for the metagenomeSeq package in R (Paulson et al.) to generate heatmaps representing the taxa with the greatest variance of abundance across sample groups. Only a minor part of the non-host metagenome was non-bacterial (0.092% ± 0.073 of all non-host bases) across all samples.
Statistical analysis of IAV and colorectal tumor data
Statistical differences in survival rate were determined by log rank (Mantel-Cox) analysis. A study with a sample size of 14 in each of 3 groups was calculated to have 90% power to detect a difference between 20% and 80% in survival. In the IAV model, the slope of the weight loss (day 0 to day 7 post IAV infection) of WildR mice was compared to that of LabR mice or Lab mice in a linear regression analysis using SAS Proc Mixed (version 9.4). In the model of AOM/DSS-induced colorectal tumors, changes in weight for days 12–55 for the 3 groups were compared using a repeated measures mixed model linear regression with an AR(1) covariance structure. The variances were different among the groups (Welch test, p<0.0001). The analysis therefore used separate variance estimates for each group. The studentized maximum modulus method was used to adjust for multiple comparisons when comparing the 3 groups. Lung viral titer and cytokine data of IAV-infected mice, and data on number of tumors and tumor area/colon area in AOM/DSS-treated mice were subjected to the D’Agostino-Pearson omnibus normality test. If a Gaussian model of sampling was satisfied, parametric One-Way ANOVA with Tukey multiple comparison test with 95% confidence interval were used. Otherwise, non- parametric t tests were used (Mann–Whitney U test for two groups, Kruskal–Wallis with Dunn’s multiple comparison test for more than two groups). Analyses were performed with GraphPad Prism 6.0f (GraphPad Software). Two-sided P values less than 0.05 were considered significant.
DATA AND SOFTWARE AVAILABILITY
SNP microarray data were deposited in the Zenodo repository with unique identifier 10.5281/zenodo.583615. 16S and shotgun metagenomics raw sequence data were deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA390686.
Supplementary Material
Highlights:
Wild mice from Maryland are close relatives to standard laboratory mouse strains
Gut microbiota of wild mice and laboratory mice differ significantly
Wild mouse gut microbiota can be banked and maintained in a laboratory mouse colony
Natural gut microbiota improves outcome in viral infection and tumorigenesis models
Acknowledgements
The authors thank Martin Toft and James Vitale, Taconic Biosciences, Inc. for providing timed pregnant GF C57BL/6 mice and commenting on the manuscript, and, together with Marc St. Clair, NIDDK for advice on gnotobiotic husbandry; Leigh Samsel, NHLBI, and Meghan Keane, NIDDK for technical assistance; Wuxing Yuan, Leidos Biomedical Research Inc., for processing samples for shotgun metagenomics; Elizabeth C. Wright, NIDDK, for statistical analysis. Yasmine Belkaid, NIAID and Christopher B. Buck, NCI for discussion. This study was funded by the intramural research programs of NIDDK, NIAID and NCI, NIH. Development and validation of the mouse genotyping was supported in part by NIH grant U19AI-100625 (FPMV) and APM was supported by F30 MH-103925. The authors declare that they have no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman AL, and Underhill DM (2017). The mycobiome of the human urinary tract: potential roles for fungi in urology. Ann Transl Med 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, and Fisher EM (2000). Genealogies of mouse inbred strains. Nat Genet 24, 23–25. [DOI] [PubMed] [Google Scholar]
- Benjamini, and Hochberg (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57 289–300. [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, and Costa- Mattioli M (2016). Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 165, 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108 Suppl 1, 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CW, Lee PF, Lu KH, and Yu HT (1998). A population study of house mice (Mus musculus castaneus) inhabiting rice granaries in Taiwan. Zool Stud 37, 201–212. [Google Scholar]
- Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, Remark R, Mogno I, Ng R, Gnjatic S, et al. (2016). Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 167, 444–456 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Caceres M, and Legendre P (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, and Relman DA (2007). An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion JP, Morgan AP, Yadgary L, Bell TA, McMullan RC, Ortiz de Solorzano L, Britton-Davidian J, Bult CJ, Campbell KJ, Castiglia R, et al. (2016). R2d2 Drives Selfish Sweeps in the House Mouse. Mol Biol Evol 33, 1381–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowle M, and Srinivasan A (2016). Data.table: extension of ‘data.frame’. R package version 1.10.0 (https://cran.r-project.org/package=data.table). [Google Scholar]
- Dufrêne M, and Legendre P (1997). Species assemblages and indicator species: The need for a flexible asymetrical approach. Ecol Monogr 67, 345–366. [Google Scholar]
- Dwass (1957). Modified Randomization Tests for Nonparametric Hypotheses. Ann Math Stat 28, 181–187. [Google Scholar]
- Edgar RC (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996–998. [DOI] [PubMed] [Google Scholar]
- Gensollen T, Iyer SS, Kasper DL, and Blumberg RS (2016). How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, et al. (2016). The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302. [DOI] [PubMed] [Google Scholar]
- Gomez MD (2008). A population study of house mice (Mus musculus) inhabiting different habitats in an Argentine urban area. Int Biodeter Biodegr 62, 270–273. [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. (2016). Tuft cells, taste- chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project, C. (2012a). A framework for human microbiome research. Nature 486, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project, C. (2012b). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, and Iwasaki A (2011). Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108, 5354–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, et al. (2006). Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein SL, Hodgson A, and Robinson DP (2012). Mechanisms of sex disparities in influenza pathogenesis. J Leukoc Biol 92, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, Hatta Y, Kim JH, Halfmann P, Hatta M, et al. (2007). Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445, 319–323. [DOI] [PubMed] [Google Scholar]
- Kolde R (2015). pheatmap: Pretty Heatmaps. R package version 1.0.8 [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasry A, Zinger A, and Ben-Neriah Y (2016). Inflammatory networks underlying colorectal cancer. Nat Immunol 17, 230–240. [DOI] [PubMed] [Google Scholar]
- Ley RE, Lozupone CA, Hamady M, Knight R, and Gordon JI (2008). Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6, 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu CM, Luo R, Sadakane K, and Lam TW (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. [DOI] [PubMed] [Google Scholar]
- Lim ES, Wang D, and Holtz LR (2016). The Bacterial Microbiome and Virome Milestones of Infant Development. Trends Microbiol 24, 801–810. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, and Chen WM (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, and Whitney (1947). On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann Math Stat 18, 50–60. [Google Scholar]
- Marshall JT (1981). Taxonomy. In The mouse in biomedical research, Foster HL, Small JD, and Fox JG, eds. (New York: Academic Press; ), pp. 17–21. [Google Scholar]
- McFall-Ngai M (2007). Adaptive immunity: care for the community. Nature 445, 153. [DOI] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP (2016). argyle: An R Package for Analysis of Illumina Genotyping Arrays. G3 (Bethesda) 6, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Fu CP, Kao CY, Welsh CE, Didion JP, Yadgary L, Hyacinth L, Ferris MT, Bell TA, Miller DR, et al. (2016). The Mouse Universal Genotyping Array: From Substrains to Subspecies. G3 (Bethesda) 6, 263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwirth E (2014). RColorBrewer: ColorBrewer Palettes. R package version 1.1–2 [Google Scholar]
- Nishioka Y (1995). The origin of common laboratory mice. Genome 38, 1–7. [DOI] [PubMed] [Google Scholar]
- Norman JM, Handley SA, and Virgin HW (2014). Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 146, 1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, Hakimpour P, Gill KP, Nakaya HI, Yarovinsky F, et al. (2014). TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J (2017). vegan: Community Ecology Package. R package version 2.4–2 [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, et al. (2016). vegan: community ecology package. R package version 2.4–1 (https://cran.r-project.org/package=vegan). [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Paulson JN, Talukder H, Pop M, and Bravo HC MetagenomeSeq: Statistical analysis for sparse high-throughput sequencing. In Bioconductor package: 1180. [Google Scholar]
- Peter BM (2016). Admixture, Population Structure, and F-Statistics. Genetics 202, 1485–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, and Virgin HW (2016). Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phifer-Rixey M, and Nachman MW (2015). Insights into mammalian biology from the wild house mouse Mus musculus. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, and Pritchard JK (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8, e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glockner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Burger MC, Pulendran B, Sekaly RP, Jameson SC, Masopust D, et al. (2016). Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host Microbe 19, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE, Smith MI, Simon GM, Scheffrahn RH, Woebken D, et al. (2014). Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 159, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Singleton GR, Hansen GM, and Shellam G (1993). A serologic survey for viruses and Mycoplasma pulmonis among wild house mice (Mus domesticus) in southeastern Australia. J Wildl Dis 29, 219–229. [DOI] [PubMed] [Google Scholar]
- Sommer F, and Backhed F (2013). The gut microbiota--masters of host development and physiology. Nature Rev Microbiol 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Stappenbeck TS, and Virgin HW (2016). Accounting for reciprocal host-microbiome interactions in experimental science. Nature 534, 191–199. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, and Mori H (2003). A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 94, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, and Morens DM (2008). The pathology of influenza virus infections. Annu Rev Pathol 3, 499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC (2014). R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria: ). [Google Scholar]
- Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, and Katze MG (2012). Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76, 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, and Jemal A (2015). Global cancer statistics, 2012. CA Cancer J Clin 65, 87–108. [DOI] [PubMed] [Google Scholar]
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. (2017). IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2007). Reshaping data with the reshape package. J Statistical Software 21, 1–20. [Google Scholar]
- Wickham H (2009). ggplot2: Elegant graphics for data analysis (New York, NY: Springer Verlag; ). [Google Scholar]
- Wilke CO (2016). cowplot: streamlined plot theme and plot annotation for ‘ggplot2’. R package version 0.6.2 (https://cran.r-project.org/package=cowplot). [Google Scholar]
- Wood DE, and Salzberg SL (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, Bonhomme F, Yu AH, Nachman MW, Pialek J, et al. (2011). Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Frank E, and Gerhard W (1981). Expression of influenza A virus internal antigens on the surface of infected P815 cells. J Immunol 126, 1814–1819. [PubMed] [Google Scholar]
- Zackular JP, Baxter NT, Chen GY, and Schloss PD (2016). Manipulation of the Gut Microbiota Reveals Role in Colon Tumorigenesis. mSphere 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, and Schloss PD (2013). The gut microbiome modulates colon tumorigenesis. MBio 4, e00692–00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM, and Doherty PC (1974a). Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature 251, 547–548. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, and Doherty PC (1974b). Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 248, 701–702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SNP microarray data were deposited in the Zenodo repository with unique identifier 10.5281/zenodo.583615. 16S and shotgun metagenomics raw sequence data were deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA390686.