

Abstract
Mitochondrial dysfunction is considered as a crucial contributory factor in cardiac pathology. This has highlighted the therapeutic potential of targeting mitochondria to prevent or treat cardiac disease. Mitochondrial dysfunction is associated with aberrant electron transport chain activity, reduced ATP production, an abnormal shift in metabolic substrates, ROS overproduction and impaired mitochondrial dynamics. This review will cover the mitochondrial functions and how they are altered in various disease conditions. Furthermore, the mechanisms that lead to mitochondrial defects and the protective mechanisms that prevent mitochondrial damage will be discussed. Finally, potential mitochondrial targets for novel therapeutic intervention will be explored. We will highlight the development of small molecules that target mitochondria from different perspectives and their current progress in clinical trials.
Linked Articles
This article is part of a themed section on Mitochondrial Pharmacology: Featured Mechanisms and Approaches for Therapy Translation. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.22/issuetoc
Abbreviations
- ∆Ψ
membrane potential
- AAV
adeno‐associated virus serotypes
- ANT
adenine nucleotide translocase
- Apaf1
apoptotic protease activator‐1
- ATF2
activating transcription factor 2
- CsA
cyclosporine A
- CPT1
carnitine palmitoyltransferase‐1
- CREB
cAMP response element‐binding protein
- CypD
cyclophilin D
- Drp1
dynamin‐1‐like‐protein
- EMRE
essential MCU regulatory element
- ER
endoplasmic reticulum
- ERR
oestrogen‐related receptors
- ETC
electron transport chain
- FAO
fatty acid β‐oxidation
- Fis1
fission‐protein 1
- GPX
glutathione peroxidase
- I/R
ischaema reperfusion
- IMM
inner mitochondrial membrane
- LC3
light chain 3
- Letm1
Leucine zipper and EF‐hand containing transmembrane protein 1
- MCU
mitochondrial calcium uniporter
- MCUR
MCU regulator
- MEF2
myocyte enhancer factor 2
- Mfn1/2
mitofusin 1/2
- Mff
mitochondrial fission factor
- MICU1/2
mitochondrial calcium uptake1/2
- Mid49/51
mitochondrial elongation factor 1 and 2
- MLKL
mixed lineage kinase domain‐like protein
- MnSOD
mitochondrial antioxidant manganese SOD
- mPTP
mitochondrial permeability transition pore
- mtDNA
mitochondrial DNA
- NCLX
Na+/Ca2+/Li+ exchanger
- NR
nicotinamide riboside
- NRF
nuclear respiratory factors
- O₂−
superoxide
- OMM
outer mitochondrial membrane
- Opa1
optic atrophy 1
- OSCP
oligomycin sensitivity‐conferring protein
- PA
phosphatidic acid
- PDHK
pyruvate dehydrogenase kinase
- PE
phosphatidylethanolamine
- PGC1α
PPARγ coactivator 1
- PS
phosphatidylserine
- PTM
posttranslational modification
- RaM
rapid mode of Ca2+ uptake
- RIPK1
receptor‐interacting kinase‐1
- SIRT1/2/3
sirtuin 1/2/3
- SLC25A23
solute carrier family 25 member 3
- SMAC
second mitochondria‐derived activator of caspase
- SNO
S‐Nitrosylation
- TCA
tricarboxylic acid cycle
- TFAM
mitochondrial transcription factor A
- VDAC
voltage‐activated anion channel
- XIAP
X‐linked inhibitor of apoptosis protein
Introduction
The heart permanently consumes large quantities of energy, predominantly for contraction and ion transport purposes. However, its capacity to store energy is unexpectedly low. Hence, to maintain this high energy flux, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 must be constantly and rapidly synthesized. In cardiac myocytes, mitochondria occupy approximately one third of the cell volume reflecting the high energy demands of these cells. Mitochondria produce more than 95% of the ATP in the myocardium; in addition, mitochondria also play important roles in regulating redox status, calcium homeostasis and lipid synthesis. It is therefore not surprising that mitochondrial dysfunction has been strongly linked to the development of cardiomyopathy and an increased risk of heart failure (Murphy et al., 2016).
Mitochondrial functions in the heart
The powerhouse in cardiac cells
To provide a constant supply of energy, and at the same time adapt to stimuli, mitochondria transform different substrates available into ATP. This substrate plasticity accommodates changes in the environment, such as oxygen and nutrient availability in blood, and rapid changes in workload, such as during exercise. Fatty acids are the preferred substrate, accounting for 60–90% of the myocardium's energy supply (Murphy et al., 2016). Once fatty acids are taken from the bloodstream, they are transported into the mitochondria for fatty acid β‐oxidation (FAO). In this set of reactions, the long‐chain fatty acids are broken down into acetyl‐CoA, which then enters the tricarboxylic acid cycle (TCA), also known as Kreb's cycle. Electron carriers, FADH2 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4487, produced during the TCA cycle transfer electrons into the electron transport chain (ETC). This, in turn, drives protons into the intermembrane space to activate the ATP synthase while consuming oxygen. In contrast, glucose is first transformed into http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4809 in the cytosol, a process denominated glycolysis. The mitochondrial complex pyruvate dehydrogenase converts pyruvate into acetyl‐CoA, in turn feeding the TCA cycle and ETC (Figure 1). Fatty acid and glucose metabolisms regulate one another in a negative feedback cycle, known as the Randle cycle, which ensures optimal use of the resources available at all times (Hue and Taegtmeyer, 2009). Other energy substrates, such as ketones, amino acids and lactate, can also be oxidized to sustain the TCA cycle and produce ATP (Murphy et al., 2016). Their contribution to overall myocardial energetics at rest is minor but becomes important under different stress conditions.
Figure 1.
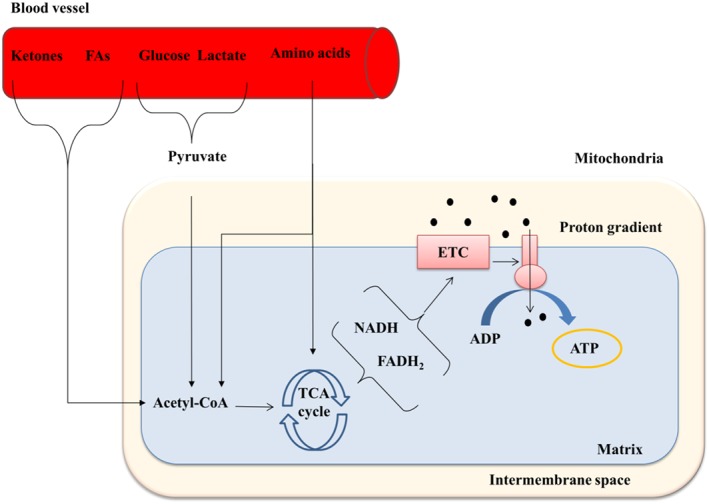
The main mitochondrial function is energy production. Mitochondria transform different substrates into ATP by driving the TCA cycle that supplies electron carriers. The electron carriers, NADH and FADH2 then stimulate the ETC to maintain a constant flux of protons towards the intermembrane space. The gradient generated by this flux is the force that powers ATP synthase. Mitochondrial substrates do not enter directly into the TCA cycle. Fatty acids and ketones are oxidized inside the mitochondria and broken into acetyl‐CoA. Glucose and lactate are converted into pyruvate in the cytosol, and pyruvate is converted into acetyl‐CoA in the mitochondrial matrix. Amino acids can supply not only acetyl‐CoA but also other TCA intermediates.
Regulation of redox status
Cardiovascular diseases are closely associated with oxidative stress triggered by elevated levels of ROS. The accumulation of ROS in cells is a primary cause of mitochondrial damage and dysfunction. Indeed, mitochondria are the main cellular source of ROS. Electrons from coenzymes (NADH and FADH2) are tightly coupled to oxidative phosphorylation for ATP synthesis and are driven to oxygen by the ETC to release water. Nevertheless, about 0.2–2% of electrons leak from the ETC and are aberrantly transferred to O2, resulting in the formation of superoxide (O2 −). Electron leakage happens at all three complexes I, II and III. Superoxide releases from complexes I and II into the matrix and from complex III to the intermembrane space and the matrix (Murphy et al., 2016).
In mitochondria, ROS production is counterbalanced by efficacious detoxification defence. In the matrix compartment, superoxide can be further transformed into http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2448 by the mitochondrial antioxidant manganese SOD (MnSOD, SOD2) (Bhattacharyya et al., 2014). MnSOD‐deficient mice die 10 days postnatally and exhibit cardiomyopathy. Inhibition of MnSOD activities in isolated cardiomyocytes also increases the levels of apoptosis and hypertrophy (Gustafsson and Gottlieb, 2008). Moreover, H2O2 can either cross mitochondrial membranes freely or can be further detoxified to water by other mitochondrial antioxidant enzymes, such as glutathione peroxidase (GPX) in the presence of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6737 (Bhattacharyya et al., 2014).
At low levels, ROS can serve as a signalling molecule to modify signalling proteins and have functional consequences (Murphy et al., 2016). On the contrary, during cardiovascular diseases, overproduction of ROS is accompanied by reduced antioxidant capacity and oxidative stress. For example, MnSOD and GPX activities have been shown to be reduced significantly in animal models of congestive heart failure and myocardial infarction (Gustafsson and Gottlieb, 2008). Excessive ROS results in the shutdown of energy production, increased cell death and irreversible oxidative damage to mitochondrial DNA (mtDNA) and altered gene expression. Consequently, this results in the development and progression of cardiac dysfunction.
Homeostasis of calcium
Mitochondria are also responsible for regulating the transport of ions, and their numerous ion channels and exchangers may be important for protecting cells. The most important ones are the selective K+ and Ca2+ channels, the Na+/H+ exchanger and the K+/H+ exchanger. The correct function of these ion channels is required to preserve mitochondrial membrane potential, which supports the production of ATP and redox regulation (Murphy et al., 2016).
Among these ions, Ca2+ plays a central role in the regulation of many cellular functions including metabolism and the effects of transcription‐modifying enzymes (Stefani et al., 2016). In cardiomyocytes, excitation‐contraction coupling during contraction is accompanied by a surge in cytosolic calcium, which leads to ensuing calcium removal from cytosol during subsequent relaxation. As they contain several different ion channels and transporters, mitochondria are capable of sensing cytosolic calcium signals and mediating calcium sequestrion from the cytosol into the mitochondrial matrix. Ca2+ is able to easily diffuse through the outer mitochondrial membrane (OMM) via voltage‐dependent anion channels (VDACs) (Arruda and Hotamisligil, 2015). In contrast, the inner mitochondrial membrane (IMM) is impermeable to ions, emphasizing the need to identify mitochondrial Ca2+ transporter proteins. The identification of the mitochondrial calcium uniporter (MCU) on IMM as a central mediator of mitochondrial calcium influx has made the field more attractive and challenging (Baughman et al., 2011; Stefani et al., 2011). MCU is a 40 kDa protein comprised of two coiled‐coil domains and two transmembrane helices separated by a 9‐amino acid loop and is characterized by its low affinity for binding Ca2+. Despite that, siRNA‐mediated knockdown of MCU led to an attenuated mitochondrial calcium uptake (Baughman et al., 2011; Stefani et al., 2011). Isolated mitochondria from MCU‐deleted mice demonstrated attenuated calcium uptake and a decreased level of calcium in the mitochondrial matrix (Pan et al., 2013). Further studies revealed the other two core‐components of the MCU complex: MCUb and essential MCU regulatory element (EMRE). MCUb, a 33 kDa protein, shares 50% homology with MCU and is capable of disrupting MCU‐mediated Ca2+ permeability; whilst EMRE is a 10 kDa protein and absolutely pivotal for the assembly of the MCU complex (Stefani et al., 2016).
In addition to the structural components, MCU activities are tightly controlled by two regulatory proteins: mitochondrial calcium uptake 1 (MICU1) and mitochondrial calcium uptake 2 (MICU2). These proteins form a heterodimer that is linked with MCU via EMRE. Several studies have implied that MICU1 and MICU2 function as Ca2+ sensors by closing MCU pores when cytosolic [Ca2+] is low. MICU1‐ablation in mitochondria led to matrix Ca2+ overload and less efficient Ca2+ uptake (Liu et al., 2016; Stefani et al., 2016). Therefore, as a MCU gatekeeper, MICU1/2 prevent excessive accumulation of Ca2+ within the mitochondrial matrix, preserving proton motive force. Additionally, MCU regulator 1 (MCUR1) and the mitochondrial nucleotide transporter http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=206#1078 have also recently been reported to regulate calcium homeostasis. Although deletion of both MCUR1 and SLC25A23 results in reduced MCU‐mediated Ca2+ intake, the precise mechanisms underlying the regulation of MCU activities remains unknown (Stefani et al., 2016).
MCU‐independent and rapid mechanisms have been studied. In contrast to MCU, these routes mediate faster Ca2+ uptake than MCU due to a high affinity for Ca2+ and are inhibited by high Ca2+ concentrations (Ryu et al., 2010). Among these is the rapid mode of Ca2+ uptake (RaM), which was initially recognized in isolated heart and liver mitochondria. Although the kinetic mode of RaM has been demonstrated to occur only transiently during the beginning of calcium pulses, its molecular identity has remained unclear (Ryu et al., 2010). Moreover, mitochondrial ryanodine receptors are IMM proteins that are widely accepted as an alternative for rapid Ca2+ import (Ryu et al., 2010). Recently, a mitochondrial protein leucine zipper and EF‐hand containing transmembrane protein 1 (Letm1), originally known as the K+/H+ exchanger, has been demonstrated as an important mitochondrial Ca2+/H+ antiporter, which extrudes H+ from the matrix in exchange for Ca2+. One study has demonstrated that in vitro deletion of Letm1 leads to impaired Ca2+ uptake and reduced ATP production, whereas in vivo ablation of Letm1 is lethal (Jiang et al., 2013).
Mitochondrial Ca2+ influx is counteracted by the IMM http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=202#1050, which is able to extrude Ca2+ in exchange for Na+ and Li+. Mice with cardiac‐specific deletion of NCLX have increased lethality due to severe cardiac remodelling, which are attributed to mitochondrial Ca2+ overload along with augmented ROS production (Luongo et al., 2017). Interestingly, NCLX activity has been reported to serve in a reverse manner to take Ca2+ into the mitochondrial matrix; therefore, more studies are required to unravel this concept (Stefani et al., 2016).
Lipid synthesis
Mitochondria also have a crucial role in lipid homeostasis. Most lipids are synthesized in the endoplasmic reticulum (ER); however, some major components of the mitochondrial membranes are synthesized in IMM from imported lipids. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3638) and phosphatidic acid (PA) are imported from the ER membrane to the OMM through membrane contact sites called mitochondrial‐associated ER membranes. The lipid precursors are then transferred across the intermembrane space by protein complexes, such as the ubiquitin specific peptidase 1/2‐mitochondrial distribution and morphology protein 35 complex, or the PRELID1‐TRIAP1 complex in humans (Mesmin, 2016). When reaching the IMM, phosphatidylserine decarboxylase transforms PS to phosphatidylethanolanime (PE); meanwhile, PA goes through a series of reactions and is converted to the phospholipid cardiolipin. Both phospholipids are especially abundant in IMM and are essential for cristae structure. PE plays a role in membrane protein folding (Bogdanov and Dowhan, 1999) but, more importantly, has been observed to be necessary for efficient mitochondrial respiration (Tasseva et al., 2013). Moreover, cardiolipin has been found to be distributed towards the OMM through mitochondrial contact sites that hold the IMM and OMM close together and maintain structural stability. The transport of cardiolipin to the OMM has been observed to be increased in response to stress, serving as a binding site for signalling molecules in mitophagy (Chu et al., 2013).
Mitochondrial alterations in cardiac diseases
Substrate shifts
Although fatty acids are the main substrate for ATP production in the myocardium, it must rely on different substrates to respond to and compensate for stress conditions. For example, during strenuous exercise, lactate consumption has been observed to significantly increase to cover the demand for rapid and higher energy production (Brown et al., 2017). In pathology, the substrate balance is initially shifted towards a temporarily more efficient profile, but later, this shift results in a deficiency in ATP that contributes to heart failure (Figure 2). In high‐fat‐diet‐induced hypertrophy and diabetes, an increase in FAO is observed during early stages, whereas pressure overload and ischaemia favour glucose metabolism (Kolwicz et al., 2013). The increase in either situation strongly inhibits the utilization of the other substrates by the Randle cycle, thereby reducing the myocardium's ability to process different substrates. For example, non‐diabetic hypertrophy models relying on glucose oxidation develop insulin resistance, which limits substrate plasticity (Nikolaidis et al., 2004). In contrast, diabetic models with a high FAO rate accumulate lipid metabolites that generate mitochondrial dysfunction, a process called lipotoxicity (Bayeva et al., 2013b). In advanced stages of the disease, both fatty acid and glucose metabolisms are reduced. The myocardium turns to ketone utilization for ATP production but still faces energy deprivation (Brown et al., 2017). The myocardium requires the ability to process different substrates in order to maintain proper function.
Figure 2.
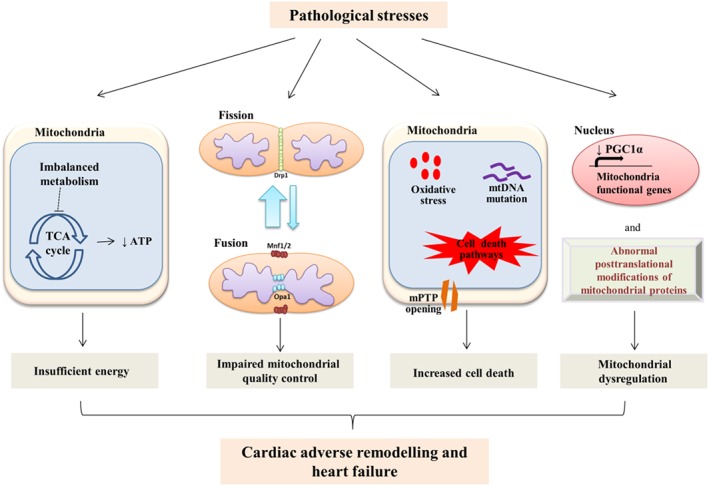
Mitochondrial dysregulation under pathological stresses. Sustained pathological stresses result in alterations in mitochondrial structure and function through different mechanisms, eventually leading to cardiac pathological remodelling and heart failure. Firstly, aberrant metabolic substrate preference in the heart impairs oxidative phosphorylation and ATP production, resulting in energy insufficiency. Secondly, an imbalance between mitochondrial fission and fusion induces the accumulation of fragmented mitochondria and impaired mitochondrial quality control, which ultimately disrupts the mitochondrial network and functions. Thirdly, excessive ROS production, increased mtDNA damages, activated cell death pathways and mPTP opening are the main causes of cell death. Finally, a dysregulation of transcription factors and abnormal posttranslational modification of mitochondrial function‐related genes impairs mitochondrial biogenesis.
Alterations in mitochondrial dynamics
The balance between mitochondrial fission and fusion maintains its normal dynamics under physiological conditions, which contributes largely to the regulation of cell survival and cellular metabolism. In addition, well‐controlled mitochondrial dynamics are critical for the maintenance of mitochondrial morphology and distribution, which is strictly regulated based on cellular demands (Murphy et al., 2016). Mitochondrial fission is activated during cell division to generate healthy daughter mitochondria through replicative fission for higher energy demand. Mitochondrial fission is initiated by the recruitment and oligomerization of the GTPase from the dynamin family, dynamin‐1‐like‐protein (Drp1), onto the OMM to form helical rings encircling mitochondria. Upon the hydrolysis of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1742, the ring‐like structure constricts to split the OMM and IMM (Ong et al., 2015). In addition, several mitochondrial outer membrane proteins including mitochondrial fission protein‐1 (Fis1), mitochondria fission factor (Mff) and mitochondrial elongation factor 1 and 2 (MiD49 and MiD51) are required to mediate the assembly of the mitochondrial fission machinery. Mff and MiD49/51 are proposed as the bona fide Drp1 receptors to recruit Drp1 onto the OMM, whereas the specific roles of Fis1 are still controversial. Although early studies, using Fis1 overexpression and inhibition, support pivotal roles of Fis1 during mitochondrial fission, a follow‐up investigation in cancer cell lines shows that Fis1 deletion does not impair Drp1 recruitment and mitochondrial morphology (reviewed in detail by Chan, 2012). However, mitochondrial fusion is enhanced in response to pathological stress to prevent damage to mtDNA and mitochondrial proteins. This process is regulated by members of the dynamin protein family, namely, mitofusin 1/2 (Mfn1/2) and optic atrophy 1 (Opa1), which work in collaboration to mediate fusion of both OMM and IMM (Ong et al., 2015).
Rat and human failing hearts exhibit mitochondrial fragmentation and decreased levels of fusion proteins while fission protein levels are increased (Chen et al., 2009; Javadov et al., 2011). These data might indicate the predominance of fission over fusion during heart failure by which the heart function is worsened (Figure 2). Alterations in mitochondrial dynamic proteins trigger excessive ROS generation, abnormal enzymatic activities and impaired calcium homeostasis, eventually leading to cardiac dysfunction. For example, Drp1 ablation results in elongated mitochondria and dilated cardiomyopathy; whereas mice with deletion of Mfn1/2 develop eccentric hypertrophy along with the accumulation of fragmented mitochondria (Song et al., 2015b; Song et al., 2017). Additionally, hearts from heterozygous Opa1+/− mice display decreased fractional shortening and reduced cardiac output associated with impaired mitochondrial function (Chen et al., 2012). Consistently, knockdown of Opa1 or Drp1 reduces oxygen consumption and the rate of ATP synthesis (Chen et al., 2005; Benard et al., 2007). Further study has identified that stress‐induced metalloendopeptidase OMA1 deactivates Opa1 activity, which leads to mitochondrial fragmentation, cardiac metabolic shift towards glucose utilization and dilated cardiomyopathy (Wai et al., 2015). Therefore, a balance between mitochondrial fission and fusion proteins is required to maintain cardiac function.
Impairments in the quality control of mitochondria
Mitochondria have developed mechanisms to control and maintain their quality at appropriate levels through the balance of biogenesis and mitophagy. In contrast to mitochondrial biogenesis, which facilitates repopulation with new healthy mitochondria through fission–fusion cycle, mitophagy is a cellular process that specifically identifies and eliminates damaged and dysfunctional mitochondria. Mitophagy is mediated by the cytosolic E3 ubiquitin ligase Parkin and the mitochondrial kinase http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2161. Activation of the PINK1–Parkin pathway is tightly controlled and regulated by the mitochondrial membrane potential (∆Ψ), which is a driving force for ATP synthesis (Murphy et al., 2016). In healthy and hyperpolarized mitochondria, PINK1 activity is maintained at a low level, whereas damaged mitochondria with abnormal ∆Ψ display inhibited PINK1 degradation, leading to its accumulation and stabilization at OMM. In addition, Wang et al., 2018 demonstrated that http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1542 isoforms are required to phosphorylate PINK1 at Ser495, facilitating its stabilization and induction of mitophagy in response to transverse aortic constriction‐induced chronic heart failure. PINK1 recruits Parkin from the cytosol, allowing it to ubiquitylate mitochondrial proteins. As a result, Parkin‐mediated polyubiquitinated proteins function as receptors for protein‐1 light chain 3 (LC3) on the autophagosome, hence promoting autophagosomal engulfment of the depolarized mitochondria and commencing mitophagy (Shirihai et al., 2015). This pathway contributes largely to the mitochondrial quality control mechanism. Recently, a communicating mechanism between PINK1 and Parkin has been elucidated, which provides a link between Mfn2 and mitophagy. A deficiency in Mnf2 induces the accumulation of Parkin in cytosol, which results in reduced levels of mitophagy (Song et al., 2015b). Parkin‐mediated mitophagy is also closely associated with mitochondrial fission. Parkin is up‐regulated in Drp1‐ablated hearts, which leads to hyper‐mitophagy and dilated cardiomyopathy, while concomitant ablation of Parkin attenuated the pathological phenotypes caused by a deficiency in Drp1 (Song et al., 2015a).
Besides the conventional PINK1–Parkin pathway, PINK1‐independent mitophagy is considered to be a generalized mitochondrial autophagy. Several OMM proteins such as http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2844&familyId=910&familyType=OTHER#OtherNames/adenovirus E1B 19 kDa protein‐interacting protein 3‐like/mitochondrial receptor Nip3‐like protein X (BNIP3L/NIX), Bcl‐2/adenovirus E1B 19 kDa protein‐interacting protein 3 (BNIP3) and FUN14 domain containing 1 (FUNDC1) have been shown to function as receptors for LC3 to trigger mitochondrial autophagy (Shirihai et al., 2015). Recently, Bhujabal et al. (2017) expanded the repertoire of PINK1‐independent mitochondrial autophagy by an anti‐apoptotic protein, FKBP8, which is able to interact with LC3 to induce autophagosomal engulfment. In addition, McWilliams et al., 2018 have pointed out that mitophagy happens preferentially through PINK1‐independent mechanisms at physiological conditions in high‐energy‐demanding tissues.
To date, several reports have focused on the protective roles of mitophagy in the heart in response to stress. For instance, hearts subjected to ex vivo ischaemia/reperfusion (I/R) injury showed enhanced mitophagy (Moyzis et al., 2015). PINK1‐deficient hearts are more susceptible to ex vivo I/R injury and develop hypertrophy in response to pressure overload more rapidly (Billia et al., 2011; Lee et al., 2011). Furthermore, the infarct size after I/R injury was exacerbated due to loss of PINK1 (Siddall et al., 2013). Additionally, Parkin‐deficient mice accumulate dysfunctional mitochondria and exhibit increased mortality following a myocardial infarction (Kubli et al., 2013). Interestingly, the translocation of Parkin to mitochondria was observed in ischaemic preconditioning (IPC), which was abolished in Parkin‐knockout mice (Huang et al., 2011). Although mitophagy has been studied for several decades, further studies are required to investigate whether mitophagy will be a promising target for cardioprotection.
mtDNA mutations
mtDNA remains a key element of mitochondrial function. It encodes 37 essential mitochondrial proteins (POLG), including tRNAs, rRNAs and subunits of the enzyme complexes in ETC. mtDNA replication is performed by DNA polymerase γ, and its spontaneous mutation rate has been estimated to be much higher than replication of the nuclear genome. The frequency of a spontaneous pathogenic mtDNA mutation is estimated to be as high as 1 in 5000 individuals; therefore, it is not surprising that even if most of the severe mutations are selectively eliminated from the maternal germline in a few generations, some others can be conserved and result in cardiomyopathy (Fan et al., 2008). Moreover, the crosstalk between mitochondrial and nuclear genomes is necessary for normal mitochondrial performance and, ultimately, for optimal cardiac function. For example, a particular mutation in the nuclear gene, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=206#1062, leads to hypertrophy or critical dilated cardiomyopathy depending on the patient's particular haplogroup (Strauss et al., 2013). In mice with ANT mutation, cardiomyopathy was associated with age‐related accumulation of somatic mtDNA mutations (Narula et al., 2011). Therefore, the interaction of mtDNA mutation with nuclear DNA mutation can exacerbate cardiac pathology.
Regulation of mitochondrial biogenesis and metabolism
Mitochondrial transcriptional network
Transcriptional regulation of mitochondrial genes
In response to greater energy expenditure, mitochondrial biogenesis is triggered within cells, leading to enrichment in their mitochondrial mass and number to increase ATP production. The process is a critical facilitator for mitochondrial homeostasis and metabolic enzyme activities, and it is intricately regulated at the transcriptional level by genes encoding mitochondrial proteins. A widely recognized master regulator of mitochondrial gene transcription in the nucleus is PPARγ coactivator 1 (PGC1α), which has been continuously identified as a key element in the progression of cardiomyopathy to heart failure (Rowe et al., 2010). Despite not having independent transcriptional activity, PGC1α regulates a transcriptional network integrated by nuclear respiratory factors (NRFs), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=622s), and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=86 to control substrate utilization, mitochondrial biogenesis and dynamics.
Cardiac function is dependent on this transcriptional network properly regulating mitochondrial function. Cardiac‐specific E3 ligase mitsugumin 53 overexpression in mice, which up‐regulates http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=593 expression, strongly promotes FAO and results in diabetic cardiomyopathy (Liu et al., 2015). In hypertrophied mouse hearts induced by angiotensin II infusion, reduced mitochondrial expression of PGC1α, ERR and mitochondrial transcription factor A (TFAM) correlated with mitochondrial damage and increased mitophagy (Dai et al., 2011). Of note, transcriptional factors mediating mitochondrial genes are required for fine‐tuned regulation. For example, different mouse models with global or cardiac‐specific deletion of PGC1α, PPAR and ERR have been generated, resulting in the development of cardiomyopathy, although murine models with constitutive overexpression of PGC1 and PPAR are associated with increased mitochondrial biogenesis, which also leads to cardiomyopathy and heart failure (Rowe et al., 2010). Additionally, NRFs bind to the promoters of ETC genes and mitochondrial transcription factor family genes (TFAM and TFB), which are in charge of mtDNA transcription and translation; therefore, achieving necessary crosstalk between nuclear and mitochondrial genomes. NRF (Ristevski et al., 2004) and TFAM (Larsson et al., 1998) deletions were embryonically lethal and displayed mtDNA depletion. However, TFAM overexpression did preserve mitochondrial content and function after myocardial infarction, improving cardiac function (Ikeuchi et al., 2005).
Transcriptional and posttranslational regulation of PGC1α
PGC1α expression and activity are constantly modified by extracellular stimuli and intracellular conditions. First of all, its expression is mostly regulated by transcriptional factors. In muscles, myocyte enhancer factor 2 (MEF2), cAMP response element‐binding protein (CREB) and activating transcription factor 2 (ATF2) increase PGC1α expression by binding to its promoter. They are independently activated or repressed by different signalling cascades. For example, during exercise, enhanced intracellular calcium initiates calcineurin A and Ca2+/calmodulin‐dependent protein kinase pathways, which subsequently activate MEF2 and CREB (Handschin et al., 2003). The MAPK http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1499 phosphorylates MEF2 and ATF2 to stimulate mitochondrial biogenesis in response to exercise (Akimoto et al., 2005). Under high‐fat‐diet stress, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2093‐MEF2 pathway is active to maintain PGC1α expression for preserving mitochondrial integrity (Liu et al., 2017). Of note, a depressed PGC1α transcriptional network is a feature of heart failure induced by pressure overload (Garnier et al., 2003) or myocardial infarction, and it correlates with a reduced mitochondrial respiratory capacity, even when mitochondrial mass is not decreased, which is observed in human hypertrophic myocardium and ischaemic‐induced failing heart (Pisano et al., 2016) (Figure 2).
Secondly, PGC1α is strongly regulated at the posttranslational level as well. Its activity is affected by phosphorylation, acetylation, methylation and ubiquitination. The ‘energy sensor', http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1540, is activated by high ADP/ATP levels to phosphorylate PGC1α (Jäger et al., 2007). It is not clear how, but the ensuing increase in transcriptional activity and expression of PGC1α induced by AMPK is possibly through an auto‐regulatory effect (Handschin et al., 2003). This positive feedback amplifies the control of AMPK over PGC1α. In fact, AMPK stimulation preserves cardiac contractility after myocardial infarction by promoting PGC1α expression and augmenting mitochondrial respiration (Gundewar et al., 2009). PGC1α phosphorylation is also regulated by p38 (Adams et al., 1998). Acetylation of PGC1α is predominantly mediated by amino acid synthesis 5; whereas its increased activity via deacetylation is regulated by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2707). A down‐regulation of SIRT1 was observed in human failing or ageing hearts (Lu et al., 2014). Additionally, preconditioning before I/R injury increased SIRT1 expression associated with an up‐regulated antioxidant capacity and diminished apoptosis (Hsu et al., 2010). The roles of methylation and ubiquitination of PGC1α in the heart are still under study.
Posttranslational modifications (PTMs) of mitochondrial proteins
The correct crosstalk between the nucleus, cytosol and mitochondria is carried out by PTMs of mitochondrial proteins by acetylation, phosphorylation and nitrosylation. First, acetylation of mitochondrial enzymes modulates FAO, ATP production, antioxidant response and apoptosis in response to nutrient availability and cardiac stress signals (Murphy et al., 2016). In addition to SIRT1 found in the nucleus, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2708 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=848#2709 are NAD+‐dependent deacetylases predominantly located in the cytosol and mitochondria respectively. Cardiomyocyte‐specific SIRT3 expression has been shown to protect the heart against pathological hypertrophy through activation of FoxO3‐dependent antioxidant genes (Sundaresan et al., 2009). Meanwhile, its deficiency resulted in increased vulnerability to I/R due to inhibition of MnSOD (Porter et al., 2014) and opening of the mitochondrial permeability transition pore (mPTP) (Parodi‐Rullán et al., 2017).
Secondly, phosphorylation of mitochondrial proteins plays an essential role in mitochondrial function, such as protein transporting, enzyme activation and substrate shift. AMPK phosphorylates a wide selection of enzymes to promote ATP synthesis. For example, it phosphorylates http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=255#1264 which stimulates FAO; moreover, it also enhances glucose metabolism by promoting http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=165#878 translocation and phosphorylating phosphofructokinase 2. With its diversity of targets, it is not surprising that AMPK manipulation has produced conflicting results; nevertheless, it is generally agreed that mild AMPK activation has cardioprotective effects and its deletion impairs energy metabolism (Zaha and Young, 2012). The http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=608), branched chain α‐ketoacid dehydrogenase kinase (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1939), and phosphatase are also known to control substrate shifts by phosphorylation of mitochondrial proteins. However, more than 150 proteins were identified to possess phosphorylation sites in cardiac mitochondria (Deng et al., 2011) suggesting future research will uncover further regulatory systems mediated by phosphorylation.
Thirdly, S‐nitrosylation (SNO) is the covalent attachment of NO to a cysteine residue. The mechanisms controlling this process are yet to be clarified; nevertheless, it has been observed that mitochondrial proteins are prone to be nitrosylated for their function. More importantly, SNO has been intimately linked to I/R injury and heart failure. Protein SNO was found to be decreased after I/R (Lima et al., 2010) and, in contrast, was increased during IPC. Augmented SNO resulting in improved cardiac function is associated with a preserved complex I respiration rate and ATPase function (Sun et al., 2007; Prime et al., 2009).
Mechanisms of mitochondria‐mediated cell death in cardiac diseases
Cell death is a hallmark characteristic of heart diseases, and mitochondria contribute largely to cardiomyocyte death in response to pathological stresses. Distinct types of cell deaths including apoptosis, necrosis and necroptosis have been recognized during the progression of cardiac diseases (Figure 2).
Apoptosis
Mitochondria are a well‐known regulator of intrinsic cardiomyocyte apoptosis. In response to death signals and ROS accumulation, mitochondria facilitate the release of apoptogens, such as cytochrome c, Omi/HtrA2, second mitochondria‐derived activator of caspase (SMAC or DIABLO) and apoptosis‐inducing factor (AIF) into cytosol from the mitochondrial intermembrane space (Murphy et al., 2016). Once in the cytosol, these apoptogens bind to apoptotic protease activator‐1 (Apaf1) to mediate assembly of the apoptosome for procaspase‐9 recruitment and activation (McIlwain et al., 2015). The initiator http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1625 then cleaves inactive procaspase dimers to form active executioner http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1623. Upon activation, these executioner caspases can activate other caspases to induce a magnitude of caspase activation within the cell. Finally, these activated endoproteases take part in degrading structural proteins and enzymes to bring about cellular apoptotic responses, such as DNA fragmentation, membrane shrinkage and the formation of apoptotic bodies, for subsequent phagocytosis (Murphy et al., 2016).
The integrity of OMM is regulated by both anti‐apoptotic (Bcl‐2, Bcl‐XL) and pro‐apoptotic members (Bax, Bak) of the Bcl‐2 protein family. In response to apoptotic signals, Bax and Bak bind to BH‐3 proteins, such as Bim or truncated Bid, to assist their conformational changes. Once activated, cytosolic Bax translocates to OMM where it undergoes both homo‐oligomerization and hetero‐oligomerization with Bak. Meanwhile, anti‐apoptotic proteins, such as Bcl‐2 and Bcl‐XL, prevent Bax and Bad activation through direct interactions with Bim and truncated Bid in the cytosol (Murphy et al., 2016). Overall, the ratio between apoptotic and pro‐apoptotic protein levels within a cell determines OMM permeability and cytochrome c release to the cytosol.
Unlike other cell types, cardiomyocytes are highly resistant to caspase‐dependent apoptosis, which suggests the importance of caspase‐independent apoptotic mechanisms. This is due to a high expression level of the endogenous http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2790) in cardiomyocytes, which prevents Apaf1 activity and caspase activation even in the presence of apoptotic stimuli (Potts et al., 2005). Moreover, under severe oxidative stress, mitochondrial serine protease HtrA2/Omi translocate from mitochondria to cytosol, where they promote apoptosis through protease‐dependent degradation of XIAP and caspase activation (Liu, 2005). By contrast, http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=889 (IAPs), expressed in the heart as caspase 9 inhibitors, limit I/R‐induced apoptosis in isolated perfused mouse hearts (Chua et al., 2007).
Alterations in mitochondrial morphology also contribute to apoptotic responses in the heart. For example, angiotensin II‐induced hypertensive hearts demonstrate a downregulation of SIRT1, thus allowing p53 acetylation and Drp1‐dependent mitochondrial fission, from which mitochondrial fragmentation eventually results in cardiomyocyte apoptosis (Qi et al., 2018). Additionally, a down‐regulation of cardiac dual‐specificity protein phosphatase 1 in acute I/R injury activates mitochondrial fission and cardiomyocyte apoptosis (Jin et al., 2018). I/R injury can also bring about cardiac microcirculation collapse by facilitating Mff expression, which promotes the apoptosis of endothelial cells in the cardiac microcirculation through increased mPTP opening and cytochrome c release to the cytosol (Zhou et al., 2017). Taken together, morphological alterations in mitochondria are critical causes for stimulating apoptosis in cardiac diseases.
Necrosis
In contrast to apoptosis, necrosis is a passive and unregulated form of cell death that is distinguished by distinctive mitochondrial alterations, such as mitochondrial swelling, loss of ATP and cellular membrane damage. As a result, cell integrity is severely disrupted and cellular contents are released that eventually lead to secondary inflammatory responses, with the potential for pathological consequences. A mechanism through which mitochondria mainly contribute to necrosis during pathological conditions is permeability transition, the opening of high‐conductance and long‐lasting mPTP on IMM.
Under physiological conditions, IMM is almost impermeable to all chemicals except a few ions and metabolites, which is crucial for the maintenance of the pH gradient and mitochondrial ∆Ψ. Under pathological conditions, calcium overload and oxidative stress are key factors that induce mPTP opening on IMM. In contrast, it is inhibited by low pH, adenine nucleotides and pharmacological inhibitors such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1024 (CsA) (Halestrap et al., 2004). Increased mPTP opening causes loss of IMM integrity followed by ∆Ψ dissipation across IMM, which reduces the efficiency of ATP synthesis and induces necrotic cell death. However, upon mPTP opening, only ions, water and other solutes up to 1.5 kDa enter/exit the matrix. Proteins, due to their high MW, are retained in the matrix. Since the concentration of ions (Ca2+) in the matrix is less than in the cytoplasm, ions enter the matrix through pores upon mPTP induction. As a result, high colloidal osmotic pressure increases water influx into the matrix, and hence eliciting colloid osmotic pressure. Consequently, the IMM expands and swells in order to resist this pressure. However, the OMM is unable to expand, which ultimately causes mitochondrial membrane rupture and leads to cellular necrosis. In addition, OMM rupture might secondarily cause the release of apoptogens, which activate downstream apoptotic signalling via apoptosome assembly and caspase activation (Kwong and Molkentin, 2015). Beyond cell death, mPTPs opening has been proposed to be involved in ROS‐induced ROS release (RIRR), a positive feedback mechanism in which mitochondria increase their own ROS production in response to an elevated level of ROS (Gustafsson and Gottlieb, 2008).
In spite of considerable efforts, the molecular composition and regulation of mPTP has become an area of controversy and remains uncertain. Several studies have identified at least three proteins, cyclophilin D (CypD), VDAC and ANT as critical facilitators for mPTP opening (Murphy et al., 2016). CypD is a matrix mitochondrial enzyme, which was first reported to be involved in mPTP function through the investigation of an immunosuppressive drug CsA (Crompton et al., 1988). Nowadays, CypD is accepted as an undisputed and indispensable regulator of mPTP, which has been verified using CypD‐deficient mice, which show less sensitivity to Ca2+‐ and ROS‐induced mPTP opening (Baines et al., 2005; Nakagawa et al., 2005). Moreover, pharmacological inhibition of CypD or genetically disrupted expression of CypD ameliorated mPTP opening and cell death and protected against I/R injury and muscular dystrophy (Palma et al., 2009; Alam et al., 2015). However, to‐date the precise mechanism whereby CypD regulates mPTP remains unknown. ANT, a 32 kDa IMM protein, is responsible for importing ADP into mitochondrial matrix in exchange for ATP, while VDAC localizes mainly on OMM to form a pore for low‐specific passage of small hydrophilic molecules (<5 kDa) (Kwong and Molkentin, 2015). Pulldown experiments elucidate that ANT and VDAC are components involved in a pore formation of mPTP via interaction with CypD (Crompton et al., 1998). Together with other proteins, such as hexokinase in the cytosol, translocator protein on OMM, and creatine kinase in the intermembrane space, a complex of mPTP, is formed (Brenner and Moulin, 2012). However, genetic ablation of any of these genes resulting in increases in Ca2+‐ and oxidative stress‐sensitive thresholds of mPTP does not block mPTP opening under various Ca2+ levels, suggesting that they are not essential for the function of mPTP (Baines et al., 2007; Kwong and Molkentin, 2015).
Furthermore, PiC, an IMM protein which transports organic phosphate (Pi) into the mitochondrial matrix, is a newly suggested potential candidate component of the mPTP due to its ability to trigger mPTP opening and being a CypD binding partner (Kwong and Molkentin, 2015). Although a partial deletion of PiC via siRNA does not successfully block mPTP opening, cardiac‐specific deletion of PiC protected hearts against I/R injury by blunting Ca2+ overload‐induced mPTP opening, suggesting it regulates mPTP through altering mitochondrial matrix Pi levels (Kwong et al., 2014; Kwong and Molkentin, 2015). Recently, mitochondrial http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=156#803 has also emerged as a component of mPTP. Giorgio et al. (2013) initially showed that the binding of CypD to an oligomycin sensitivity‐conferring protein (OSCP) subunit of the peripheral stalk of ATP synthase led to a modulation of ATP synthase activity. Moreover, a purified dimer of F1F0 ATP synthase reconstitutes to form a Ca2+‐activated channel in lipid bilayers, which harbour activity resembling mPTP (Giorgio et al., 2013). A further study by Alavian et al. (2014) demonstrated that deletion of the c‐subunit of ATP synthase results in the prevention of Ca2+‐induced mPTP opening and protects against Ca2+‐ and ROS‐induced cell death in cultured neurons. However, two recent findings by the Walker group have challenged these concepts by creating various clonal cells that harboured deletions of genes coding for OSCP and c‐subunits, which showed that the absence of either of these components does not prevent permeability transition in human mitochondria (He et al., 2017a; He et al., 2017b). Taken together, these findings provide compelling evidence that F1F0 ATP synthase contributes to mPTP function, but the underlying mechanisms require clarification.
Several studies have indicated that the opening of mPTP occurs predominantly during various cardiac diseases. During ischaemia, the anaerobic condition lowers cytosolic pH to acidic values by metabolizing pyruvate into lactic acid, which inhibits mPTP opening (Gustafsson and Gottlieb, 2008). On the other hand, most of the Ca2+ pumping channels are blunted due to lack of ATP, and a sudden rise in Ca2+ concentration primes for prolonged mPTP opening during I/R injury (Kwong and Molkentin, 2015). During heart failure, impairments in Ca2+ homeostasis, ATP depletion and up‐regulated ROS could favour mPTP opening, which might also be a key factor contributing to pump failure. CypD−/− mice displayed improved mortality and reduced infarct size in response to prolonged I/R injury, whereas mice with CypD overexpression had mitochondrial dysfunction and cell death (Baines et al., 2005; Lim et al., 2010). Despite considerable efforts to address the beneficial effects of inhibiting mPTP, Elrod et al. (2010) demonstrated that CypD‐deficient mice exhibit more severe cardiac hypertrophy and fibrosis with reduced function in response to pressure overload. The following experiments showed that these CypD‐deficient hearts had accumulations of Ca2+ within the mitochondrial matrix and altered cardiomyocyte metabolism, thereby reducing metabolic flexibility and facilitating the onset of heart failure. These results indicate the importance of mPTP in the regulation of cardiac metabolism through maintaining Ca2+ homeostasis, which might be beneficial during the compensation stage of cardiac hypertrophy (Elrod et al., 2010). Although mPTP opening contributes significantly to the progression of heart failure, further studies are required to fully explore the molecular structure and regulation of mPTP in order to develop novel therapeutic strategies for treating heart disease.
Necroptosis
Recently, the conventional dogma of necrosis in cardiac disease is changing with a concept of programmed and regulated necrosis, termed necroptosis. Under pathological conditions, the initiation of necroptosis is highly associated with an inflammatory response through activation of the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074 signalling pathway. This is initiated by the binding of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2189) to http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2191, causing oligomerization and autophosphorylation of RIP3 to form necrosomes. The activated necrosomes in turn mediate phosphorylation of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2106) and its translocation to the plasma membrane to form pores, eventually disrupting the integrity of the cell membrane and promoting necrosis (Galluzzi et al., 2017).
In addition, Wang et al., 2012 showed that mitochondrial fragmentation is a mediator of necroptosis. Upon TNF‐α induction, the mitochondria phosphatase PGAM5, a part of RIPK1‐RIPK3‐MLKL complexes, becomes activated leading to the recruitment and dephosphorylation of Drp1. Consequently, excessive GTPase activity of Drp1 results in an increased mitochondrial fragmentation, which is required for early events of necroptosis. However, further investigation indicated that activation of PGAM5 is cardioprotective against necroptosis in acute stress by independently activating PINK1‐mediated mitophagy. Cardiomyocytes from Pgam5 −/− mice showed an accumulation of damaged mitochondria, having greater levels of necroptosis and were more susceptible to IR injury (Lu et al., 2016). These data reveal different facets of the role of mitochondria in necroptosis.
Numerous studies have indicated the role of necroptosis in heart diseases, including myocardial infarction and heart failure. In patients with failing hearts, increased levels of TNF‐α lead to activation of RIPK1‐RIPK3‐mediated necroptosis. Moreover, several necroptotic markers including RIPK1, RIPK3 and phosphor‐MLKL become significantly increased in human end‐stage heart failure (He et al., 2009). Several animal models have also confirmed the existence of necroptosis and its crucial role during the development of heart failure, and necroptosis inhibition seems to be beneficial and alleviate cardiac dysfunction. For example, pharmacological inhibition of RIPK1 activity by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9750 prevented necroptosis and ameliorated I/R injury in mice and rats (Anbin et al., 2014). Inhibition of RIPK1 and RIPK3 also prevents cardiomyocyte necroptosis‐mediated cell death in response to hypertrophy‐induced lipotoxicity stress (Zhao et al., 2016). Thus, understanding the pathological role of necroptosis sheds light on a new strategy to treat heart disease; however, the molecular mechanisms underlying necroptosis in the pathogenesis of heart diseases are needed to be fully delineated.
Therapeutic approaches targeting mitochondria
Given the important roles of mitochondria in cardiomyocyte homeostasis and cardiac function, mitochondrial‐based therapies have been extensively studied. These therapeutic approaches include stimulators of mitochondrial biogenesis, metabolic modulators, ROS scavengers, mPTP inhibitors and gene therapy by targeting the deregulated mitochondrial‐related genes (Figure 3). Each strategy will be further discussed as follows.
Figure 3.
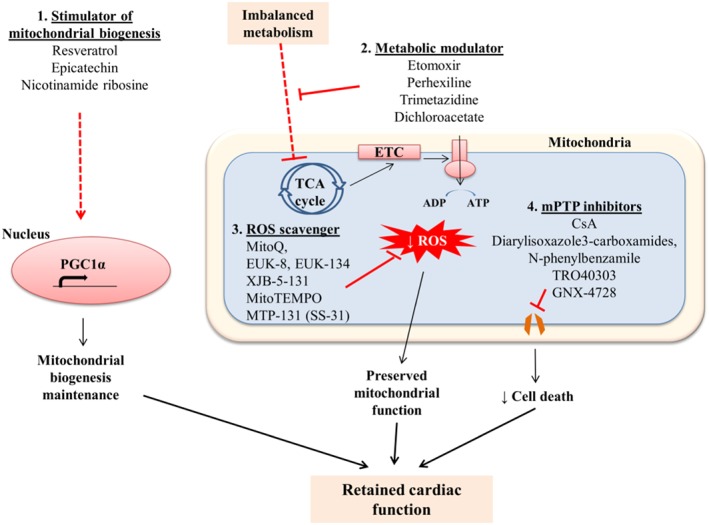
Preserving mitochondrial biogenesis and function as therapeutic approaches in cardiac diseases. The potential therapeutic strategies aim to facilitate mitochondrial biogenesis and maintain mitochondrial function to prevent cardiac dysfunction, including (i) enhancing mitochondrial biogenesis; (ii) retaining mitochondrial metabolism by limiting fatty acid import and oxidation while improving glucose utilization; (iii) reducing ROS production by ROS scavengers; and (iv) inhibiting mPTP by the use of mPTP inhibitors.
Stimulators of mitochondrial biogenesis
The first therapeutic approach focuses on improving mitochondrial biogenesis and thus mitochondrial function, which results in the preservation of cardiac function. Polyphenol compounds such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8741 and epicatechin activate SIRT1/3 to promote AMPK and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249 signalling, which are responsible for regulating mitochondrial biogenesis through the activation of mitochondrial transcription factors, PGC1α and NRFs (Brown et al., 2017). The beneficial effects of resveratrol have also been proven in recent studies on coronary artery disease, atherosclerosis, hypertension and myocardial infarction (Berman et al., 2017). In a 3‐month randomized, double‐blind, placebo‐controlled clinical trial, resveratrol, 10 mg·day−1, was shown to improve systolic function and reduce LDL‐cholesterol and platelet aggregation in patients with myocardial infarction (Bonnefont‐Rousselot, 2016). However, its short half‐life of 8–14 min within the body is the main challenge to deliver an effective dose in humans. Therefore, the development of other derivatives with longer half‐lives would offer better treatment for patients with myocardial infarction. Meanwhile, catechins are the key regulators of systemic blood pressure, atherosclerosis, platelet activation and thrombosis formation (Mangels and Mohler, 2017). The 25 year Zutphen Elderly Study has indicated that dietary epicatechin, mainly found in cocoa, black and green teas, lowers mortality in elderly Dutch men with prevalent cardiovascular disease (Dower et al., 2016). Two large clinical trials, namely, the COCOA‐BP trial and the FLAVASCULAR study, are currently underway to assess the effect of catechin‐rich food on regulating systemic BP (NCT02764203; NCT02013856).
In addition to these compounds, another natural compound nicotinamide riboside (NR) can be utilized as a diet supplement to enhance NAD+ availability, leading to SIRT1 activation. The up‐regulated deacetylase activity of SIRT1 in turn improves mitochondrial function via PGC1α cascade (Viscomi et al., 2015). A NR‐supplemented diet has been shown to improve NAD+ levels, thus attenuating the progression to heart failure in both mouse models of dilated cardiomyopathy and of cardiac hypertrophy (Diguet et al., 2017). A recent clinical trial has demonstrated that 6 weeks of NR supplementation induces a significant increase in blood NAD+ concentration and was well‐tolerated in healthy middle‐aged people (Martens et al., 2018). Another double‐blind clinical trial is currently underway to investigate the safety and tolerability of NR in patients with clinically stable systolic heart failure (NCT03423342).
Metabolic modulators
As mentioned earlier, myocardial energy metabolism undergoes dramatic alterations in glycolysis, FAO capacity and ATP production under pathological stresses. To explore potential therapeutic strategies by reserving cardiac energy metabolism, inhibition of fatty acid uptake, inhibition of FAO and stimulation of glucose oxidation are currently under investigation.
The first strategy aims mainly to limit the adverse effects of accumulated fatty acids in the heart and thus cardiac lipotoxicity through inhibiting the key fatty acid importer carnitine palmitoyltransferase‐1 (CPT1). CPT1 is mainly responsible for mitochondrial import of fatty acids, thus it determines the balance between fatty acid and glucose oxidation within the cardiomyocyte mitochondrial matrix. CPT1 inhibition by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9089 and perhexiline, which have been shown to improve cardiac output and the exercise capacity of patients with heart failure, but their use remains limited due to side effects such as peripheral neuropathy and hepatoxicity (Lee et al., 2005; Holubarsch et al., 2007).
The second strategy is achieved using the pharmacological compound trimetazidine, an inhibitor of long‐chain 3‐ketoacyl CoA thiolase, which is involved in the last step of FAO in mitochondria (Kantor et al., 2000). In ischaemic hearts, trimetazidine can restore the balance between glycolysis and glucose oxidation to preserve energy production with limited oxygen supply. Furthermore, this compound also exerts other beneficial effects, such as the induction of pyruvate dehydrogenase in the TCA cycle, inhibition of hypoxia/reperfusion injury and inhibition of cardiac fibrosis (McCarthy et al., 2016). Trimetazidine is currently used for treating angina in more than 100 countries and has been shown to improve cardiac function and the quality of life in patients with both non‐ischaemic and ischaemic heart failure (Gao et al., 2011). A combination of trimetazidine and β‐blockers for angina treatment has also been tested in three different clinical trials, which have shown that 12 week trimetazidine administration in conjunction with β‐blockers could increase time to onset of angina pain and exercise capacity compared to the group treated with only β‐blockers (Vitale et al., 2013). Moreover, a small clinical trial involving 64 patients with stable angina revealed that a combination of trimetazidine and calcium channel blockers reduces the number of angina attacks without adverse haemodynamic effects (Manchanda and Krishnaswami, 1997). In addition to angina treatment, trimetazidine also offers clinical benefits in patients with diabetes mellitus, coronary artery disease and heart failure, although these effects require confirmation in larger clinical trials (Dézsi, 2016).
The third strategy focuses on enhancing myocardial glucose oxidation to meet increased cardiac glycolysis in the failing heart through use of the pharmacological agent http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4518, a PDK inhibitor (Bersin and Stacpoole, 1997). Inhibition of PDK thus reduces its inhibitory effect on pyruvate dehydrogenase, a key enzyme involved in pyruvate oxidation, thus allowing increased glucose oxidation in ischaemic cardiomyocytes. Using a rat model, dichloroacetate pretreatment retains cardiac function and glucose oxidation in response to ventricular fibrillation‐induced ischaemic. However, some studies challenged that, via the Randle cycle, increased glucose metabolism could lead to reduced fatty acid metabolism; thus, the accumulation of lipid metabolites such as triglycerides, diglycerides and ceramide might cause cardiac lipotoxicity (Fillmore and Lopaschuk, 2013). Therefore, this strategy requires further investigation and validation.
ROS scavengers
Reducing ROS production is a theoretical strategy to treat cardiac pathological remodelling and heart failure. However, the clinical implications need to be further inspected. In a large clinical trial involving 765 middle‐aged men, often‐used antioxidants such as vitamin E or C failed to reduce the risk of cardiovascular events, suggesting that these compounds might not get to the site of ROS generation (Sesso et al., 2008). So far, MitoQ, a ROS scavenger linked to triphenylphosphonium, is the best characterized mitochondria‐targeted antioxidant. By accumulating in the negatively charged mitochondrial matrix, lipophilic triphenylphosphonium cations of MitoQ effectively recruit CoQ10 to the mitochondrial matrix in order to improve ETC efficiency and reduce ROS production (Bayeva et al., 2013a). However, depolarization of mitochondrial matrix induced by the accumulation of cationic triphenylphosphonium could potentially interfere with mitochondrial function in the heart (Bayeva et al., 2013a). In various preclinical studies, MitoQ appears to be protective in animal models with hypertension, I/R injury and pressure overload stress (Ribeiro et al., 2018). In a small clinical trial involving 55 middle‐aged and older adults, MitoQ treatment reduced aortic stiffness, plasma oxidized LDL and improved vascular endothelial function (Rossman et al., 2018). However, the uptake of MitoQ is dependent on the ∆Ψ, which is dramatically disrupted in human failing hearts (Bayeva et al., 2013a). This remains the key challenge for the therapeutic application of MitoQ for heart failure.
In addition to MitoQ, other ROS‐scavenging compounds have been rigorously tested in different heart disease models. For example, EUK‐8, a SOD and catalase mimetic, has been shown to protect mouse hearts against pressure overload‐induced heart failure and dilated cardiomyopathy (Empel et al., 2006; Kawakami et al., 2009). EUK‐134, a more lipophilic derivative of EUK‐8, blunts hypertrophic responses in H9C2 cardiac cell lines (Purushothaman and Nair, 2016). EUK‐134 also prevents diaphragm muscle weakness in the rat model of monocrotalin‐induced pulmonary hypertension (Himori et al., 2017). Also, XJB‐5‐131, an antioxidant which scavenges electrons that have leaked from the ETC, blocks the oxidative stress response and improves mitochondrial respiratory function in aged rat skeletal muscle and aged I/R stressed hearts (Escobales et al., 2014; Javadov et al., 2015). Moreover, MitoTEMPO, a mitochondrial‐targeted ROS scavenger, ameliorates hypertension and enhances endothelial NO production in mice infused with angiotensin II (Dikalova et al., 2010). However, since studies on the above compounds are mostly based on transgenic animal models, further clinical investigation into their safety and efficacy is required.
The IMM phospholipid, cardiolipin, is important for regulating ETC activities, mitochondrial membrane transporters and ROS generation. To overcome non‐selective delivery of other ROS scavengers, small mitochondrial‐targeted peptides have recently been developed to enter the mitochondrial matrix independently of membrane potential. MTP‐131 (also known as SS‐31 or Bendavia), a novel pharmacological compound targeting cardiolipin, protects the heart against I/R injury in rabbit and sheep models (Kloner et al., 2012); it also prevents pressure overload‐induced heart failure through suppression of cardiac proteomic remodelling and cardiomyocyte necrosis (Dai et al., 2013; Nickel et al., 2015). Although MTP‐131 appears to be well‐tolerated in phase I and II clinical trials, the phase 2a EMBRACE STEMI study has indicated that treatment with MTP‐131 at a dose of 0.05 mg·kg−1·h−1 cannot reduce myocardial infarct size in STEMI patients undergoing percutaneous coronary intervention (Gibson et al., 2016). Meanwhile, in a double‐blind, placebo‐controlled, ascending‐dose trial involving patients suffering from heart failure with reduced ejection fraction, MTP‐131 treatment at a dose of 0.25 mg·kg−1·h−1 significant decreases left ventricular end‐diastolic volume and end‐systolic volume (Daubert et al., 2017). Therefore, further studies are essential to assess its effect for the treatment of specific heart diseases.
mPTP inhibitors
Inhibition of mPTP presents a promising therapeutic strategy to prevent cardiomyocyte death and mitochondrial dysfunction. CsA, a CypD inhibitor, has been previously shown to protect the Langendorff‐perfused hearts from re‐oxygenation and perfusion injury (Griffiths and Halestrap, 1993). CsA administration also enhances mitochondrial respiratory function and ∆Ψ in isolated cardiomyocytes from dogs with heart failure (Sharov et al., 2007). In a phase II clinical trial, administration of CsA immediately before percutaneous coronary intervention reduces infarct size in patients with acute anterior ST‐segment elevation myocardial infarction (STEMI) (Piot et al., 2008). However, in a following phase III trial involving 970 patients with STEMI, intravenous injection of CsA failed to improve clinical outcome or prevent adverse ventricular remodelling at 1 year compared to the group with placebo treatment (Cung et al., 2015). The failure of CsA treatment is likely due to its inhibition of CypD, which might disturb CypD‐dependent cardiac mitochondrial Ca2+ homeostasis and cellular energy metabolism (Javadov et al., 2017). Of note, this compound also displays severe side effects such as immunosuppression and adverse effects on renal pathology (Brown et al., 2017), plausibly making CsA application an inappropriate treatment for heart diseases.
With the interest in suppressing mPTP‐induced cell death in ischaemic hearts, new mPTP blockers have recently been developed to selectively inhibit mPTP opening with few adverse effects on CypD. For example, diarylisoxazole3‐carboxamides, as potent inhibitors of mPTP, increase Ca2+ retention in mitochondria and prevent mitochondrial swelling in zebrafish muscle (Roy et al., 2015). N‐phenylbenzamide, a compound also identified as a mPTP‐specific blocker through high‐throughput screening, inhibits mitochondrial swelling in mouse liver mitochondria and HeLa cells (Roy et al., 2016). Furthermore, cinnamic anilides have been developed as a novel class of mPTP inhibitors, which exert protective effects in the rabbit acute myocardial infarction model (Fancelli et al., 2014). Chronic treatment with GNX‐4728, a cinnamic anilide compound, can also be a potential treatment for amytrophic lateral sclerosis in mice (Martin et al., 2014). However, in a recent MITOCARE clinical trial, another mPTP inhibitor TRO40303 failed to show efficacy in limiting cardiac injury in patients with STEMI (Atar et al., 2015). Taken together, further studies on these newly discovered mPTP blockers are required to comprehensively explore their therapeutic potential for treating heart diseases.
Gene therapy
Mitochondrial dysfunction in cardiac disease and metabolic syndromes are highly associated with a down‐regulation of key proteins responsible for mitochondrial biogenesis and function such as PGC1α and NRFs (discussed in ‘Mitochondrial transcriptional network' section). Gene therapy is a novel approach to restore the expression of critical proteins responsible for mitochondrial function via adeno‐associated virus serotypes (AAVs) delivery system. AAVs are small, non‐pathogenic viruses with a single‐strand DNA genome. One major advantage of using recombinant AAVs for research is that the transduced gene does not integrate into the host's genome and the transgene expression level is controllable by adjusting the amount of virus injected (Viscomi et al., 2015). Among AAV serotypes, AAV9 can efficiently deliver a transgene under the control of a cardiac‐specific promoter into cardiomyocytes. Liu et al. (2017) reported that ERK5 is a requisite for sustaining PGC1α expression and the restoration of ERK5 expression by the AAV9 system ameliorates mitochondrial function and prevents high‐fat‐diet‐induced cardiomyopathy.
Conclusion
Mitochondrial biogenesis in the myocardium is closely related to cardiac physiological function. However, myocardial mitochondrial function is damaged under various stresses, leading to pathological cardiac remodelling and heart failure. Mitochondrial dysfunction contributes to the development of cardiomyopathies through energy deprivation, the accumulation of ROS and cell death. Designing therapeutic strategies targeted to maintain mitochondrial function has been challenging due to the different responses observed that depend on the aetiology of the disease. Nonetheless, the continuous advancement in our knowledge of the molecular basis underlying mitochondrial biogenesis in physiological and pathological conditions is being pursued in order to discover novel therapeutic targets for heart diseases.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by the British Heart Foundation (PG/14/71/31063, PG/14/70/31039 and FS/15/16/31477). We thank Miss Fay Pu (Medical School, The University of Edinburgh) for proofreading the manuscript.
Nguyen, B. Y. , Ruiz‐Velasco, A. , Bui, T. , Collins, L. , Wang, X. , and Liu, W. (2019) Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. British Journal of Pharmacology, 176, 4302–4318. 10.1111/bph.14431.
Contributor Information
Xin Wang, Email: xin.wang@manchester.ac.uk.
Wei Liu, Email: wei.liu@manchester.ac.uk.
References
- Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB et al (1998). Enhanced Gα signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. PNAS 95: 10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB et al (2005). Exercise stimulates Pgc‐1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem. American Society for Biochemistry and Molecular Biology 280: 19587–19593. [DOI] [PubMed] [Google Scholar]
- Alam MR, Baetz D, Ovize M (2015). Cyclophilin D and myocardial ischemia–reperfusion injury: a fresh perspective. J Mol Cell Cardiol 78: 80–89. [DOI] [PubMed] [Google Scholar]
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H‐A, Licznerski P et al (2014). An uncoupling channel within the c‐subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci 111: 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbin Z, Xiaogang M, Lin L, Yunjie T, Yanli H, Yanli L et al (2014). Necrostatin‐1 inhibits Hmgb1‐IL‐23/IL‐17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl Int 27: 1077–1085. [DOI] [PubMed] [Google Scholar]
- Arruda AP, Hotamisligil GS (2015). Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab 22: 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atar D, Arheden H, Berdeaux A, Bonnet JL, Carlsson M, Clemmensen P et al (2015). Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST‐elevation myocardial infarction: MITOCARE study results. Eur Heart J 36: 112–119. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA et al (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007). Voltage‐dependent anion channels are dispensable for mitochondrial‐dependent cell death. Nat Cell Biol 9: 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y et al (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayeva M, Gheorghiade M, Ardehali H (2013a). Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 61: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayeva M, Sawicki KT, Ardehali H (2013b). Taking diabetes to heart – deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc 2: e000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T et al (2007). Mitochondrial bioenergetics and structural network organization. J Cell Sci 120: 838–848. [DOI] [PubMed] [Google Scholar]
- Berman AY, Motechin RA, Wiesenfeld MY, Holz MK (2017). The therapeutic potential of resveratrol: a review of clinical trials. NPJ Precision Oncology 1: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersin RM, Stacpoole PW (1997). Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am Heart J 134 (5 Pt 1): 841–855. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE (2014). Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 94: 329–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S et al (2017). FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO reports 18: 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak WT (2011). PTEN‐inducible kinase 1 (PINK1)/Park6 is indispencable for normal heart function. PNAS 108: 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W (1999). Lipid‐assisted protein folding. J Biol Chem. American Society for Biochemistry and Molecular Biology 274: 36827–36830. [DOI] [PubMed] [Google Scholar]
- Bonnefont‐Rousselot D (2016). Resveratrol and cardiovascular diseases. Nutrients 8 pii: E250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Moulin M (2012). Physiological roles of the permeability transition pore. Circ Res 111: 1237–1247. [DOI] [PubMed] [Google Scholar]
- Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR et al (2017). Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14: 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280: 26185–26192. [DOI] [PubMed] [Google Scholar]
- Chen L, Gong Q, Stice JP, Knowlton AA (2009). Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu T, Tran A, Lu X, Tomilov AA, Davies V et al (2012). OPA1 mutation and late‐onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J Am Heart Assoc 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA et al (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CC, Gao J, Ho YS, Xiong Y, Xu X, Chen Z et al (2007). Overexpression of IAP‐2 attenuates apoptosis and protects against myocardial ischemia/reperfusion injury in transgenic mice. Biochim Biophy Acta ‐ Molecular Cell Research 1773: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A (1988). Inhibition by cyclosporin A of a Ca2+‐dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255: 357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM (1998). Cyclophilin‐D binds strongly to complexes of the voltage‐dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: 729–735. [DOI] [PubMed] [Google Scholar]
- Cung TT, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D et al (2015). Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 373. [DOI] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves‐Cintrón M, Chen T et al (2011). Mitochondrial oxidative stress mediates angiotensin II‐induced cardiac hypertrophy and Galphaq overexpression‐induced heart failure. Circ Res 108: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L et al (2013). Global proteomics and pathway analysis of pressure‐overload‐induced heart failure and its attenuation by mitochondrial‐targeted peptides. Circ Heart Fail 6: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS et al (2017). Novel mitochondria‐targeting peptide in heart failure treatment: a randomized, placebo‐controlled trial of elamipretide. Circ Heart Fail 10: e004389. [DOI] [PubMed] [Google Scholar]
- Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P et al (2011). Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol Cell Proteomics: MCP. American Society for Biochemistry and Molecular Biology 10: M110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dézsi CA (2016). Trimetazidine in practice: review of the clinical and experimental evidence. Am J Ther 23: e871–e879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N et al (2017). Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W et al (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dower JI, Geleijnse JM, Hollman PCH, Soedamah‐Muthu SS, Kromhout D (2016). Dietary epicatechin intake and 25‐y risk of cardiovascular mortality: the Zutphen Elderly Study. Am J Clin Nutr 104: 58–64. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA et al (2010). Cyclophilin D controls mitochondrial pore‐dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120: 3680–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empel VPM, Bertrand AT, van Oort RJ, van der Nagel R, Engelen M, van Rijen HV et al (2006). EUK‐8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload‐induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol 48. [DOI] [PubMed] [Google Scholar]
- Escobales N, Nuñez RE, Jang S, Parodi‐rullan R, Sacher JR, Skoda EM et al (2014). Mitochondria‐targeted ROS scavenger improves post‐ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol 77: 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE et al (2008). A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319: 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancelli D, Abate A, Amici R, Bernardi P, Ballarini M, Cappa A et al (2014). Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia‐reperfusion injury protective effect in vivo . J Med Chem 57: 5333–5347. [DOI] [PubMed] [Google Scholar]
- Fillmore N, Lopaschuk GD (2013). Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim Biophys Acta ‐ Molecular Cell Research. Elsevier B.V. 1833: 857–865. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Chan FKM, Kroemer G (2017). Necroptosis: mechanisms and relevance to disease. Ann Rev Pathol: Mechanisms of Disease 12: 103–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Ning N, Niu X, Hao G, Meng Z (2011). Trimetazidine: a meta‐analysis of randomised controlled trials in heart failure. Heart 97: 278–286. [DOI] [PubMed] [Google Scholar]
- Garnier A, Fortin D, Deloménie C, Momken I, Veksler V, Ventura‐Clapier R (2003). Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CM, Giugliano RP, Kloner RA, Bode C, Tendera M, Jánosi A et al (2016). EMBRACE STEMI study: a phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP‐131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J 37: 1296.1–1296.121303. [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M et al (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci 110: 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP (1993). Protection by Cyclosporin A of ischemia/reperfusion‐induced damage in isolated rat hearts. J Mol Cell Cardiol 25: 1461–1469. [DOI] [PubMed] [Google Scholar]
- Gundewar S, Calvert JW, Jha S, Toedt‐Pingel I, Ji SY, Nunez D et al (2009). Activation of AMP‐activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res 104: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson ÅB, Gottlieb RA (2008). Heart mitochondria: gates of life and death. Cardiovasc Res 77: 334–343. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Javadov SA (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection. Cardiovasc Res 61: 372–385. [DOI] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM (2003). An autoregulatory loop controls peroxisome proliferator‐activated receptor gamma coactivator 1α expression in muscle. Proc Natl Acad Sci U S A 100: 7111–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Carroll J, Ding S, Fearnley IM, Walker JE (2017a). Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci 114: 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE (2017b). Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci 114: 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L et al (2009). Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐α. Cell 137: 1100–1111. [DOI] [PubMed] [Google Scholar]
- Himori K, Abe M, Tatebayashi D, Lee J, Westerblad H, Lanner JT et al (2017). Superoxide dismutase/catalase mimetic EUK‐134 prevents diaphragm muscle weakness in monocrotalin‐induced pulmonary hypertension. PLoS One 12: e0169146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubarsch CJF, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P et al (2007). A double‐blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci 113: 205–212. [DOI] [PubMed] [Google Scholar]
- Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N et al (2010). Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 122: 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA (2011). Preconditioning involves selective mitophagy mediated by parkin and p62/SQSTM1. PLoS One 6: e20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H (2009). The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297: E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T et al (2005). Overexpression of mitochondrial transcription factor A ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 112: 683–690. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St‐Pierre J, Spiegelman BM (2007). AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1alpha. Proc Natl Acad Sci U S A 104: 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov S, Rajapurohitam V, Kilić A, Hunter JC, Zeidan A, Said Faruq N et al (2011). Expression of mitochondrial fusion‐fission proteins during post‐infarction remodeling: the effect of NHE‐1 inhibition. Basic Res Cardiol 106: 99–109. [DOI] [PubMed] [Google Scholar]
- Javadov S, Jang S, Rodriguez‐reyes N, Rodriguez AE, Hernandez JS, Krainz T et al (2015). Mitochondria‐targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 6: 39469–39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov S, Jang S, Parodi‐Rullán R, Khuchua Z, Kuznetsov AV (2017). Mitochondrial permeability transition in cardiac ischemia–reperfusion: whether cyclophilin D is a viable target for cardioprotection? Cell Mol Life Sci 74: 2795–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clish CB, Clapham DE (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf‐Hirschhorn syndrome. Proc Natl Acad Sci 110: E2249–E2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S et al (2018). DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff‐required mitochondrial fission and Bnip3‐related mitophagy via the JNK pathways. Redox Biol 14: 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor P, Lucien A, Kozak R, Lopaschuck G (2000). The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhiniting mitochondrial long chain 3 ketoacyl coenzyme a thiolase. Circ Res 86: 580–588. [DOI] [PubMed] [Google Scholar]
- Kawakami S, Matsuda A, Sunagawa T, Noda Y, Kaneko T, Tahara S et al (2009). Antioxidant, EUK‐8, prevents murine dilated cardiomyopathy. Circ J 73: 2125–2134. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ et al (2012). Reduction of ischemia/reperfusion injury with bendavia, a mitochondria‐targeting cytoprotective peptide. J Am Heart Assoc 1: e001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolwicz SC, Purohit S, Tian R (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK et al (2013). Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X et al (2014). Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 21: 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Molkentin JD (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 21: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M et al (1998). Mitochondrial transcription factor A is necessary for mtDNA maintance and embryogenesis in mice. Nat Genet 18: 231–236. [DOI] [PubMed] [Google Scholar]
- Lee L, Campbell R, Scheuermann‐Freestone M, Taylor R, Gunaruwan P, Williams L et al (2005). Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short‐term use of a novel treatment. Circulation 112: 3280–3288. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee HY, Hanna RA, Gustafsson AB (2011). Mitochondrial autophagy by Bnip3 involves Drp1‐mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. AJP: Heart and Circulatory Physiology 301: H1924–H1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SY, Hausenloy DJ, Arjun S, Price AN, Davidson SM, Lythgoe MF et al (2010). Mitochondrial cyclophilin‐D as a potential therapeutic target for post‐myocardial infarction heart failure. J Cell Mol Med 15: 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS (2010). S‐Nitrosylation in cardiovascular signaling. Circ Res 106: 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y et al (2015). Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation‐activated receptor. Circulation 131: 795–804. [DOI] [PubMed] [Google Scholar]
- Liu H‐R (2005). Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation 111: 90–96. [DOI] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM et al (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16: 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ruiz‐Velasco A, Wang S, Khan S, Zi M, Jungmann A et al (2017). Metabolic stress‐induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signalling. Nat Commun 8: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HI, Huang TH, Sung PH, Chen YL, Chua S, Chai HY et al (2016). Administration of antioxidant peptide SS‐31 attenuates transverse aortic constriction‐induced pulmonary arterial hypertension in mice. Acta Pharmacol Sin 37: 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TM, Tsai JY, Chen YC, Huang CY, Hsu HL, Weng CF et al (2014). Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci 21: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S et al (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545: 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchanda SC, Krishnaswami S (1997). Combination treatment with trimetazidine and diltiazem in stable angina pectoris. Heart 78: 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangels DR, Mohler ER (2017). Catechins as potential mediators of cardiovascular health. Arterioscler Thromb Vasc Biol 37: 757–763. [DOI] [PubMed] [Google Scholar]
- Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB et al (2018). Chronic nicotinamide riboside supplementation is well‐tolerated and elevates NAD+ in healthy middle‐aged and older adults. Nat Commun 9: 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Fancelli D, Wong M, Niedzwiecki M, Ballarini M, Plyte S et al (2014). GNX‐4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci 8: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CP, Mullins KV, Kerins DM (2016). The role of trimetazidine in cardiovascular disease: beyond an anti‐anginal agent. Eur Heart J ‐ Cardiovascular Pharmacotherapy 2: 266–272. [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW (2015). Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 7 pii: a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava‐Garriga L, Ball G, Singh F, Barini E et al (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B (2016). Mitochondrial lipid transport and biosynthesis: a complex balance. J Cell Biol 214: 9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyzis AG, Sadoshima J, Gustafsson ÅB (2015). Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol 308: H183–H192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN et al (2016). Mitochondrial function, biology, and role in disease. Circ Res 118: 1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H et al (2005). Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658. [DOI] [PubMed] [Google Scholar]
- Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J et al (2011). Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. J Am Coll Cardiol Img 4: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel AG, Von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J et al (2015). Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 22: 472–484. [DOI] [PubMed] [Google Scholar]
- Nikolaidis LA, Sturzu A, Stolarski C, Elahi D, Shen YT, Shannon RP (2004). The development of myocardial insulin resistance in conscious dogs with advanced dilated cardiomyopathy. Cardiovasc Res 61: 297–306. [DOI] [PubMed] [Google Scholar]
- Ong SB, Kalkhoran SB, Cabrera‐Fuentes HA, Hausenloy DJ (2015). Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol 763: 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Tiepolo T, Angelin A, Sabatelli P, Maraldi NM, Basso E et al (2009). Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum Mol Genet 18: 2024–2031. [DOI] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y et al (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. Nature Publishing Group 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi‐Rullán RM, Chapa‐Dubocq X, Rullán PJ, Jang S and Javadov S (2017). High sensitivity of SIRT3 deficient hearts to ischemia‐reperfusion is associated with mitochondrial abnormalities., Frontiers in pharmacology. Frontiers Media SA, 8. [DOI] [PMC free article] [PubMed]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N et al (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481. [DOI] [PubMed] [Google Scholar]
- Pisano A, Cerbelli B, Perli E, Pelullo M, Bargelli V, Preziuso C et al (2016). Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end‐stage ischemic heart failure. Cardiovasc Pathol 25: 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM (2014). SIRT3 deficiency exacerbates ischemia‐reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol 306: H1602–H1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Vaughn AE, McDonough H, Patterson C, Deshmukh M (2005). Reduced Apaf‐1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J Cell Biol 171: 925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC et al (2009). A mitochondria‐targeted S‐nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia‐reperfusion injury. Proc Natl Acad Sci U S A 106: 10764–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman S, Nair RR (2016). Mitoprotective antioxidant EUK‐134 stimulates fatty acid oxidation and prevents hypertrophy in H9C2 cells. Mol Cell Biochem 420: 185–194. [DOI] [PubMed] [Google Scholar]
- Qi J, Wang F, Yang P, Wang X, Xu R, Chen J et al (2018). Mitochondrial fission is required for angiotensin II‐induced cardiomyocyte apoptosis mediated by a Sirt1‐p53 signaling pathway. Front Pharmacol 9: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JRF, Dabkowski ER, Shekar KC, O'Connell KA, Hecker PA, Murphy MP (2018). MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med 117: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristevski S, O'Leary DA, Thornell AP, Owen MJ, Kola I, Hertzog PJ (2004). The ETS transcription factor GABP is essential for early embryogenesis. Mol Cell Biol 24: 5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman MJ, Santos‐Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL et al (2018). Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe GC, Jiang A, Arany Z (2010). PGC‐1 coactivators in cardiac development and disease. Circ Res 107: 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Šileikytė J, Schiavone M, Neuenswander B, Argenton F, Aubé J et al (2015). Discovery, synthesis, and optimization of diarylisoxazole‐3‐carboxamides as potent inhibitors of the mitochondrial permeability transition pore. ChemMedChem 10: 1655–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Šileikytė J, Neuenswander B, Hedrick MP, Chung TDY, Aubé J et al (2016). N‐phenylbenzamides as potent inhibitors of the mitochondrial permeability transition pore. ChemMedChem 11: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu SY, Beutner G, Dirksen RT, Kinnally KW, Sheu SS (2010). Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett 584: 1948–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, Macfadyen J et al (2008). Vitamins E and C in the prevention of cardiovascular disease in men. JAMA 300: 2123–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov VG, Todor A, Khanal S, Imai M, Sabbah HN (2007). Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol 42: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirihai OS, Song M, Dorn GW (2015). How mitochondrial dynamism orchestrates mitophagy. Circ Res 116: 1835–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR et al (2013). Loss of PINK1 increases the heart's vulnerability to ischemia‐reperfusion injury. PLoS One 8: e62400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Gong G, Burelle Y, Gustafsson ÅB, Kitsis RN, Matkovich SJ et al (2015a). Interdependence of parkin‐mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res 117: 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, Dorn GW (2015b). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Franco A, Fleischer JA, Zhang L, Dorn GW (2017). Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab 26: 872–883.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani D, Raffaello A, Teardo E, Szabó I, Rizzuto R (2011). A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani D, Rizzuto R, Pozzan T (2016). Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85: 161–192. [DOI] [PubMed] [Google Scholar]
- Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P et al (2013). Severity of cardiomyopathy associated with adenine nucleotide translocator‐1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci 110: 3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E (2007). Preconditioning results in S‐nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 101: 1155–1163. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP (2009). Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a‐dependent antioxidant defense mechanisms in mice. J Clin Investig 119: 2758–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE (2013). Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem 288: 4158–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Zeviani M (2015). Emerging concepts in the therapy of mitochondrial disease. Biochim Biophys Acta ‐ Bioenergetics 1847: 544–557. [DOI] [PubMed] [Google Scholar]
- Vitale C, Spoletini I, Malorni W, Perrone‐Filardi P, Volterrani M, Rosano GMC (2013). Efficacy of trimetazidine on functional capacity in symptomatic patients with stable exertional angina – the VASCO‐angina study. Int J Cardiol 168: 1078–1081. [DOI] [PubMed] [Google Scholar]
- Wai T, García‐Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P et al (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350: aad0116. [DOI] [PubMed] [Google Scholar]
- Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L et al (2018). AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res 122: 712–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148: 228–243. [DOI] [PubMed] [Google Scholar]
- Zaha VG, Young LH (2012). AMP‐activated protein kinase regulation and biological actions in the heart. Circ Res 111: 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X et al (2016). Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxid Med Cell Longev. Hindawi Publishing Corporation 2016: 1451676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P et al (2017). Mff‐dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS‐mediated cardiolipin oxidation and HK2/VDAC1 disassociation‐involved mPTP opening. J Am Heart Assoc 6: e005328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov (Internet). Identifier NCT02764203. Chocolate Consumption and Blood Pressure (COCOA‐BP): A Wearable Devices Pilot Trial (COCOA‐BP). Available at https://clinicaltrials.gov/ct2/show/NCT02764203 (accessed 7/6/2018)
- ClinicalTrials.gov (Internet). Identifier NCT02013856. The Effects of Apple Derived Flavanols on Cardiovascular Disease Risk (FLAVASCULAR Study). Available at https://clinicaltrials.gov/ct2/show/NCT02013856 (accessed 7/6/2018)
- ClinicalTrials.gov (Internet). Identifier NCT03423342. Nicotinamide Riboside in Systolic Heart Failure. Available at https://clinicaltrials.gov/ct2/show/NCT03423342 (accessed 7/6/2018)