

Summary
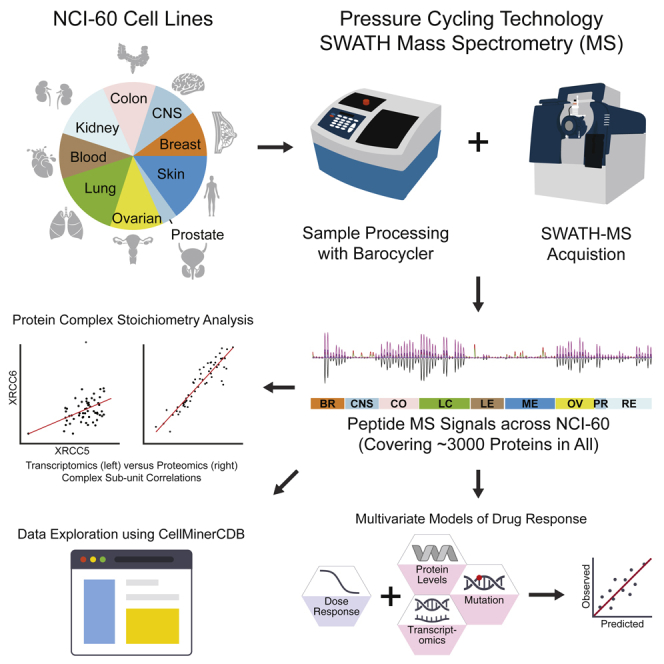
Here we describe a proteomic data resource for the NCI-60 cell lines generated by pressure cycling technology and SWATH mass spectrometry. We developed the DIA-expert software to curate and visualize the SWATH data, leading to reproducible detection of over 3,100 SwissProt proteotypic proteins and systematic quantification of pathway activities. Stoichiometric relationships of interacting proteins for DNA replication, repair, the chromatin remodeling NuRD complex, β-catenin, RNA metabolism, and prefoldins are more evident than that at the mRNA level. The data are available in CellMiner (discover.nci.nih.gov/cellminercdb and discover.nci.nih.gov/cellminer), allowing casual users to test hypotheses and perform integrative, cross-database analyses of multi-omic drug response correlations for over 20,000 drugs. We demonstrate the value of proteome data in predicting drug response for over 240 clinically relevant chemotherapeutic and targeted therapies. In summary, we present a novel proteome resource for the NCI-60, together with relevant software tools, and demonstrate the benefit of proteome analyses.
Subject Areas: Biological Sciences, Systems Biology, Proteomics, Cancer Systems Biology
Graphical Abstract
Highlights
-
•
High-quality NCI-60 proteotypes created using pressure cycling technology and SWATH-MS
-
•
Proteotypes improve drug response prediction in multi-omics regression analysis
-
•
∼3000 measured proteins allow investigation into protein complex stoichiometry
-
•
CellMinerCDB (discover.nci.nih.gov/cellminercdb) portal allows dataset exploration
Biological Sciences; Systems Biology; Proteomics; Cancer Systems Biology
Introduction
To date, forays into the molecular landscape of diseases, in particular cancers, have primarily focused on genomics and transcriptomics (Barretina et al., 2012, Cancer Genome Atlas Research Network et al., 2013, Garnett et al., 2012) due to the maturity and availability of high-throughput DNA- and RNA-based techniques. Protein-level measurements, although important for providing the granularity and detail necessary for personalized therapeutic decisions, are underutilized due to technical hurdles. Advances in data-dependent acquisition (DDA) mass spectrometry (MS) have permitted quantitative proteomic profiling of hundreds of tumor samples using multi-dimensional fractionated MS analyses of each sample (Mertins et al., 2016, Zhang et al., 2014, Zhang et al., 2016), demonstrating the added value of protein measurement in classifying tumors. Nevertheless, such DDA workflows suffer from relatively lower sample-throughput, higher sample consumption, and increased technical complexity relative to genomic analyses. These factors have precluded their routine use in clinically relevant applications (e.g. tumor classification and drug response prediction) at the speed and scale achieved by genomic and transcriptomic approaches (Barretina et al., 2012, Garnett et al., 2012, Rajapakse et al., 2018, Reinhold et al., 2019).
The NCI-60 human cancer cell line panel contains 60 lines from nine different tissue types. The NCI-60 have been molecularly and pharmacologically characterized with unparalleled depth and coverage, offering a prime in vitro model to further our understanding of cancer biology and cellular responses to anti-cancer agents (Monks et al., 2018, Reinhold et al., 2012, Reinhold et al., 2019, Shoemaker, 2006). Discoveries enabled by the NCI-60 in recent years include the development of the FDA-approved drugs, such as oxaliplatin for the treatment of colon cancers (Fojo et al., 2005), eribulin for metastatic breast cancers (Shoemaker, 2006), bortezomib for the treatment of multiple myeloma (Holbeck et al., 2010), and romidepsin for cutaneous T cell lymphomas (Bates et al., 2015), and development of the indenoisoquinoline class of non-camptothecin topoisomerase I inhibitors (Burton et al., 2018). The sensitivity of the NCI-60 to over 100,000 synthetic or natural compounds derived from a wide range of academic and industrial sources has been measured, constructing the most comprehensive open resource for cancer pharmacology. The NCI-60 remains actively used by many academic laboratories and drug companies to assess overall toxicity and drug response selectivity. In addition, many of the NCI-60 cell lines are widely used for cell biology and pharmacology (MCF-7, MDA-MB231, HCT116, HCT15, HT29, HL60, CCR-CEM, K562, etc.), and 55 and 44 of the NCI-60 cell lines overlap within larger cancer cell line databases GDSC and CCLE, respectively (Rajapakse et al., 2018), providing a unique and highly valuable resource for cross-comparisons.
The proteome of the NCI-60 cells has been analyzed previously by data-dependent analysis with a commonly used discovery MS technique (Gholami et al., 2013). This proteome dataset was obtained using a sophisticated two-dimensional peptide fractionation strategy. However, peptides and proteins were quantified without technical replicates (Gholami et al., 2013), making it difficult to evaluate quantitative accuracy. To achieve reproducible and high-throughput proteomic profiling while developing new technologies, we have developed a workflow (Guo et al., 2015, Shao et al., 2015) integrating pressure cycling technology (PCT) with SWATH-MS. PCT is an emerging sample preparation method that accelerates and standardizes sample preparation for proteomic profiling (Powell et al., 2012). SWATH-MS is an MS-based proteomic technique that consists of data-independent acquisition (DIA) and a targeted data analysis strategy with unique advantages over other MS-based proteomic methods (Gillet et al., 2012, Rost et al., 2014). With this technique, all MS-measurable peptides of a sample are fragmented and recorded recursively, and the resulting digital proteome maps can be used to reproducibly detect and quantify proteins across large numbers of samples without the need for isotope labeling. The integrated PCT-SWATH workflow thus significantly increases the sample throughput and data reproducibility, providing quantitative accuracy, while also reducing sample consumption to ca. 1 microgram of total peptide mass per sample (Guo et al., 2015, Shao et al., 2015).
Here, we describe the acquisition of proteome maps of the NCI-60 in duplicate by PCT-SWATH and make them available via the CellMiner portals (discover.nci.nih.gov/cellminercdb/ and discover.nci.nih.gov/cellminer), enabling interactive exploration and data download (Rajapakse et al., 2018). The techniques described in this report allowed the efficient acquisition of 120 proteome maps (within about 30 working days from sample preparation to SWATH data acquisition on a single instrument) with minimal sample requirement (ca. 1 microgram of total peptide mass). We focused on 3,171 SwissProt proteotypic proteins that were identified across all cell lines, generating a data matrix (120 proteomes vs. 3,171 proteins). Raw signals of each peptide and protein in each sample were curated and visualized with an expert system. The proteomic data expand the existing NCI-60 molecular landscapes (Holbeck et al., 2010, Monks et al., 2018, Rajapakse et al., 2018, Reinhold et al., 2012, Reinhold et al., 2019) and their integration with the larger databases from the Broad-MIT (CCLE, CTRP) and MGH-Sanger (GDSC) (Rajapakse et al., 2018), allowing systematic investigation of the complementarity among genomics, transcriptomics, and proteomics.
Results
Acquisition of the NCI-60 Proteome
We applied the PCT-SWATH workflow (Guo et al., 2015) to generate quantitative proteome maps of the NCI-60 cell lines in technical replicates, resulting in 120 SWATH maps with high reproducibility at the raw data level (Figure S1). Approximately 1 microgram peptide mass per sample was sufficient for analyses. The PCT-assisted sample preparation took about 18 working days and the SWATH-MS data acquisition about 12 working days. Thus, the entire process, from sample preparation to data acquisition, could be accomplished within 30 working days. This results from the elimination of multidimensional fractionation and the consequent processing of each sample using one barocycler per mass spectrometer, from which a single file per sample was acquired (Figure S1, Table S1). We have matched our cell line IDs with a previous publication from the Kuster group (Gholami et al., 2013) and corrected a few known errors in the cell line identifiers (Table S1). These cell lines were shuffled randomly to avoid bias from tissue types and minimize batch effects from PCT-assisted sample preparation. The two sets of technical replicates were acquired using SWATH-MS in different time periods to allow the evaluation of batch effects from the MS analysis. This approach constitutes an advance in sample-throughput compared with other cancer proteomic workflows of similar scale (Gholami et al., 2013, Mertins et al., 2016, Zhang et al., 2014, Zhang et al., 2016).
SWATH proteome maps contain fragment ion chromatograms from all MS-measurable peptides, albeit in a highly convoluted form. To interpret the SWATH maps, we built a human cancer cell line spectral library containing 86,209 proteotypic peptides, i.e. peptides that uniquely identify a specific protein from 8,056 SwissProt proteins (Table S1). Using this library and the OpenSWATH software (Rost et al., 2014), we identified 6,556 protein groups, covering 81% of the library (Figure S2). To avoid ambiguity of peptide/protein quantification, we limited our analyses to canonical and proteotypic peptides and proteins by excluding protein isoforms, un-reviewed protein sequences, peptide/protein sequence variants, and protein groups that could not be deconvoluted.
Development of DIA-expert for SWATH/DIA Data Curation
We evaluated the technical variation of each measurement through manual inspection of the OpenSWATH results based on the replicated measurement for each cell line. Observed missing values and technical variation were attributed to cell-type-specific interfering signals leading to invalid SWATH assays and the presence of irregular liquid chromatography (LC) and MS behavior of certain peptides. These phenomena have been observed previously in selected reaction monitoring (SRM)-based targeted proteomics studies (Picotti and Aebersold, 2012). To obtain high accuracy quantitative data, we developed an expert system, i.e. DIA-expert to refine the peptide identification and quantification (Figure S3).
The DIA-expert reads SWATH search results containing a q-value for each peptide identified in a sample and then selects the sample in which a peptide precursor is identified with the highest confidence among all samples (Figure S3). The selected sample then becomes the reference against which identification of the particular peptide in the other samples is evaluated. This step is iterated for each peptide precursor analyzed. Then, DIA-expert selects from the SWATH assay library the peptides identified for the specific sample set and proceeds to build a new library containing all the transitions for each peptide precursor. Extracted chromatograms for each precursor and its fragments are obtained. This initial transition set is used for subsequent transition refinement. We next applied empirical expert rules (Keller et al., 2002, Shao et al., 2015), including peak detection expert, reference sample expert, and peak group pairing expert. The software outputs a data matrix of quantities of each peptide in all samples and graphically presents the peak groups of curated peptide fragments used for generating the reported results. In contrast to typical SRM or SWATH/DIA analysis strategies, which apply the same few selected peptide fragments as indicators of peptide abundance in all cohort samples, the DIA-expert examines the sample-specific suitability of all peptide fragments and builds peptide abundance values based on ad hoc curated peptide fragments.
DIA-Expert-Curated Results of the NCI-60 Proteome
Excluding proteins/peptides that were not technically reproducible resulted in 22,554 proteotypic peptides from 3,171 proteins, with 8% missing values at the peptide level and 0.1% missing values at the protein level across all MS runs (Table S1). On average, seven peptide precursors and six unique peptide sequences were identified per protein. Several proteins were identified with more than 200 peptides (Figure 1B). The proteins excluded by DIA-expert may not be incorrect identifications but rather irreproducible quantifications due to either technical (for instance the signal-to-noise ratio) or biological issues (such as post-translational modifications or splicing variants). Improved computational methods may recover more information from this dataset.
Figure 1.

Acquisition of NCI-60 Proteotype
(A) Representative peptide signals as curated and visualized by the DIA-expert software.
(B–D) (B) Distribution of peptide precursors and peptides per protein. Overall coefficient of determination between technical replicates at the peptide level (C) and the protein level (D). Heatmap of the log10 transformed intensity of each peptide/protein in each cell line technical replicate.
(E) Dynamic range of the MS signals for 22,968 proteotypic peptides.
Most peptides for the 3,171 proteins were quantified in all cell lines at both MS1 and MS2 levels. Although the replicates show consistent quantification, different cell lines expressed variable levels of proteins. Two representative peptides are shown in Figure 1A. The coefficients of determination (R2) between technical replicates for the overall expression of peptides (Figure 1C) and proteins (Figure 1D) were 0.974 and 0.978, respectively, with a dynamic range over five orders of magnitude (Figure 1E). The DIA-expert provides the raw MS signals for each quantitative value, allowing visual inspection of the MS signal for every peptide in each sample. Increasing the minimal number of peptides identified per protein to 2, 3, or 4 resulted in fewer proteins quantified (2,200; 1,741; and 1,428 proteins, respectively). However, this did not substantially improve quantitative accuracy (Figure S4).
Characterization of the NCI-60 Quantitative Proteomes
The landscape of the 120 proteotypes is displayed in Figure 2A. Technical replicates of the quantified proteotypes were clustered using an unsupervised approach, confirming high quantitative accuracy. In most cases, the proteotypes are not strikingly different across different cancer cell lines, in sharp contrast with the distinct proteomes of tumor versus non-tumor kidney tissues (Guo et al., 2015). The median protein intensity coefficient of variation (CV) of the different cell lines was 48%. The CV demonstrated a low dependence on protein abundance, as evident from the distribution of its values for different expression level quantile groups of the measured proteins (Figure 2B).
Figure 2.

Characterizing the NCI-60 Quantitative Proteomes
(A) Heatmap overview of NCI-60 proteotype data matrix. Quantification of 3,171 Swiss-Prot proteins in 120 SWATH runs.
(B) Variation of protein expression for all proteins (All) and proteins in each abundance quantile group (from low abundance to high abundance).
(C) Density plot of correlations between pairs of random proteins versus pairs of proteins within a complex.
(D) Expression ratio variation of protein complexes in the NCI-60. The x axis shows the average Pearson correlation of each protein complex across the NCI-60. The y axis shows the average abundance of proteins in a complex.
(E) Snapshot image obtained with CellMinerCDB (Rajapakse et al., 2018) protein (left image) and mRNA (right image) expression of XRCC6/KU70 and XRCC5/KU80.
(F) The correlation densities for protein pairs derived from the same complex are significantly shifted relative to those from random pairs across different resources. In each plot, the light blue density is for correlation values from random protein pairs. Densities associated with specific resources for protein complexes or stable protein interactions are indicated with different colors. The relatively lighter resource-colored density plot shows the distribution of correlation values for true protein interactors, whereas the darker one is derived from random pairs present in that database. The upper panel shows correlation values for the measured protein quantities, whereas the lower one corresponds to the mRNA levels of the same proteins. The vertical dashed red line indicates a value of the Spearman's coefficient of correlation of 0.75.
We then compared the data with the previously reported DDA-MS proteomic data for the NCI-60 (Gholami et al., 2013). Whereas the DDA data reported a comparable number of IPI protein groups per cell line as the SwissProt proteotypic protein number from this SWATH dataset (Table S2), the SWATH data exhibited a much higher degree of consistency (Figure S5) and better quantitative accuracy (Figures S6–S32).
Accessibility of the NCI-60 Proteotypes
To enable easy data access, visualization, and comparison with other NCI-60 datasets, we have incorporated the SWATH data into the CellMiner databases and web application (Rajapakse et al., 2018, Reinhold et al., 2012, Shankavaram et al., 2009). This allows direct downloads of the data, as well as direct comparative and integrative analyses with other molecular and pharmacological data, (e.g. sensitivity of each cell line to over 20,000 compounds) and the inspection of specific genes, up to 150 per query. The detailed instructions for using this resource are provided in Figure S33 and at the project websites (discover.nci.nih.gov/cellminer and discover.nci.nih.gov/cellminercdb). Figure 2E shows snapshots of data queries for KU70 versus KU80 protein and transcript expression levels (XRCC6 and XRCC5, respectively). Raw and processed data matrices of the NCI-60 proteotype have also been deposited in public databases, including PRIDE (Jones et al., 2006) and ExpressionArray (Brazma et al., 2003).
Insights from a Quantitative Comparison of Protein versus Transcript Expression
Because of the extensive prior characterization of the NCI-60 transcriptome (Monks et al., 2018, Rajapakse et al., 2018, Reinhold et al., 2012), we were able to correlate protein and gene expression for each of the 3,171 proteins quantified across the NCI-60. Table S3 shows that some proteins exhibit a high correlation with their transcript, indicating the transcripts are the main drivers of protein expression. Correlations and cell line identification can be readily checked with CellMinerCDB (see Figures S34–S46). The most highly correlated proteins include MARCKSL1 (myristyolated alanine-rich C kinase; r = 0.93), LGALS3 (galectin 3; r = 0.90), and ITGB1 (integrin-β1; r = 0.88) (Table S3 and Figure S34). PARP1 (poly(ADP-ribose)polymerase 1; r = 0.77) and CDK2 (cyclin-dependent kinase 2; r = 0.58) also showed high correlation. High correlations would be expected for structural proteins and proteins with short half-lives. By contrast, some protein levels are not correlated with their transcripts (Table S3). These include TP53 (r = 0.14), TOP1 (r = 0.13), TOP2B (r = 0.09), and DHX9 (RNA helicase A; r = - 0.1; see Figure S35C). Such proteins are likely primarily regulated by post-transcriptional modifications and protein turnover.
From a translational and omic viewpoint, these results indicate that the proteins exhibiting high correlation with transcripts could be indirectly assessed by transcriptome analyses, including RNA-Seq, whereas transcriptome analyses are insufficient for the proteins that are not consistently correlated with their transcripts. In these cases, proteomic analyses, including those enabled by the SWATH-proteome, are most useful to phenotype samples. Our analyses and the CellMinerCDB tools provide insight into identifying such proteins (Table S3).
Protein Complex Predictions Based on Stoichiometry at the Protein Levels across the NCI-60
A unique benefit of proteomic data, compared with genomic and transcriptomic data, is its capacity to reveal the abundance of protein complexes and their stoichiometry (Ori et al., 2016). Our measurements included 101 predicted protein complexes comprising 1,045 proteins (Table S4) from a curated resource (Ori et al., 2016). Significantly high Pearson correlation coefficients for pairs of proteins that are part of a complex further supported the quantitative accuracy of our data matrix (Figure 2C). This was also reflected by the conserved stoichiometry of stable protein complexes, such as prefoldins (PFN1, PFN6, PFN4, and PFN5), transcription complexes (FUS, EWSR1, and DHX9), DNA repair complexes (KU70 and KU80; Figure 2E), replication and chromatin complexes, as well as membrane protein complexes (catenins and EPCAM) (Figure 2D, with additional examples in the next section).
We further investigated whether this trend was present when we used different public resources to assign protein complexes. We compared interacting protein pairs or proteins assigned to complexes according to (1) a curated CORUM database of mammalian protein complexes, (2) annotations for stable interactions in the Reactome database, (3) high-quality interaction partners in the STRING database, (4) known and modeled interactions in the Interactome3D based on available protein structures, (5) interaction pairs observed in at least three affinity purification-mass spectrometry experiments (APMS, see Methods), or (6) a small set of curated and annotated protein complexes available from the EMBL-EBI Complex Portal (Figure 2F, top panel). Compared with random protein pairs, the correlation of the measured protein quantities for the annotated interaction partners was strongly shifted to higher values (p value <1.1 x 10−10, Wilcoxon test). Of interest, the shift also reflected the confidence of protein complex assignments with the mean values of 0.43, 0.24, 0.21, 0.21, 0.19, and 0.13 for protein pairs from the EMBL-EBI complex portal, CORUM, Interactome3D, Reactome, STRING databases, and APMS studies, respectively. In addition, for almost all resources, a shoulder with overrepresented negative correlations (r ≤ −0.5) was visible.
Next, we performed the same analysis, substituting transcriptomics data for protein quantities for the same protein pairs (Figure 2F, bottom panel). The co-expression of the interacting protein pairs was verified by the positive shift of correlation values for mRNA quantities. However, this shift was smaller compared with the proteomic comparisons (Figure 2F), and the right-skewed shoulder reflecting overrepresentation of highly correlating protein interactors was absent using mRNA levels (demarcated by the red vertical dashed line in Figure 2F). Moreover, the left shoulder corresponding to negatively correlated protein pairs (r < −0.5) at the protein level disappeared when using mRNA levels (Figure 2F). Correlations of expression values for protein interaction partners often reflect a preserved stoichiometry of protein complexes. Our comparison of mRNA and protein quantities across the NCI60 demonstrates the benefits of proteomics data for detecting protein-protein interactions.
Examples of Stoichiometric Protein Complexes
KU70 and KU80 (XRCC6 and XRCC5, respectively) form a heterodimer critical for DNA recombination, immune system maturation, DNA repair, and resistance to radiotherapy and chemotherapy. Figure 2E shows the high correlation between KU70 and KU80 protein levels across the NCI-60. Remarkably, this correlation is not seen using mRNA measurements (Figures 2E and S36), indicating that the expression of Ku is tightly regulated by post-transcriptional mechanisms independent of cancer types. Indeed, KU80 is degraded when not bound to KU70 (Chang et al., 2016, Kanungo, 2010).
Another example of a small protein complex stoichiometrically regulated across the NCI-60 is the heterotrimeric RP1/2/3 complex, which is critically important for coating single-stranded DNA during replication and repair. Using the CellMinerCDB “Compare Patterns” tool with RPA3 as the “identifier” and selecting “swa” as “Data Type” yields RPA2 as top correlate (p = 0.75; r = 6.58 × 10−12), followed by the two subunits of the MCM replicative helicase MCM5 and MCM7 (p = 0.63; r = 1 × 10−7) (Figure S37). Such highly significant correlations are not observed for the corresponding transcripts (Figure S38). We also tested proteins co-expressed with PCNA, the essential cofactor for replicative DNA polymerase processivity. Because PCNA is also included in the small number of proteins determined by reverse phase proteomic array (RPPA) (Nishizuka et al., 2003), we were able to establish the reproducibility of the SWATH measurements by plotting PCNA protein expression with SWATH vs. RPPA using CellMinerCDB (r = 0.63; p = 1 × 10−7; Figure S39). Repeating the CellMinerCDB “Compare Patterns” with PCNA, we found protein co-expression of PCNA with MCM3 and notably with FEN1, the replicative nuclease for the maturation of Okazaki fragments (Figure S38), both of which are biologically logical. To our knowledge, the stoichiometric relationship of PCNA with FEN1 has not been reported previously.
Other hypotheses and correlations can be readily found by users with the CellMinerCDB Compare Patterns tool. For instance, the large subunit of ribonucleotide reductase (RRM1) is highly correlated with the purine metabolic enzyme PAICS by SWATH (0.76, p value <1.1e-10). Notable instances detailed below also include the two RNA binding proteins involved in mRNA splicing, DHX9 and FUS, and the nucleosome remodeling complex NuRF and β-catenin (CTNNB1).
Both DHX9 (RNase A) and FUS (Fused in sarcoma and associated with liposarcoma and amyotrophic lateral sclerosis [AML]) are stoichiometrically coregulated across the NCI-60 with highly significant correlations at the protein levels (r = 0.81; p = 4.7 × 10−15) even more than at the transcript levels (r = 0.42; p = 0.001) (Figure S35). Looking further at the cells co-expressing FUS and DHX9 transcripts across the larger MGH-Sanger (GDSC) database (Garnett et al., 2012) using CellMinerCDB (Rajapakse et al., 2018) confirmed the coregulation of these two RNA binding genes across 986 cell lines (r = 0.52; p = 1.8e-70), with highest expression in leukemia, lymphomas, and small cell lung cancer cell lines (Figure S40).
For large protein complexes, the nucleosome remodeling deacetylase (NuRD) complex (Basta and Rauchman, 2015) provides a notable example of protein complex stoichiometry. NuRD consists of at least 11 proteins (Figures S41 and S47B), including the two retinoblastoma binding proteins RBB7 and RBB4, the two metastasis-associated proteins MTA3 and MTA1, the two histone deacetylases HDAC2 and HDAC1, the three methyl-CpG-binding proteins GATAD2B, GATAD2A, and MBD3, and the chromodomain helicase CHD4. All of them show a high stoichiometric correlation across the NCI-60 at the protein levels, as determined by SWATH (Figures S41 and S42). Similarly, we found stoichiometric correlation across the NCI-60 for β-catenin (CTNNB1) and its membrane-associated family members CTNND1, CTNNA1, CTNNA2, as well as EPCAM, all of which are involved in cell-cell interactions (Figures S43 and S44). Together, these examples illustrate the potential value of SWATH analyses to explore and predict stoichiometric protein complexes.
Google-Map-Based Visualization of Cancer Signaling Pathways
Our NCI-60 proteotypes cover 648 proteins in the Atlas of Cancer Signaling Networks (ACSN), a manually curated pathway database presenting published biochemical reactions involved in cancer using a Google-Maps-style visualization (Figure S48) (Kuperstein et al., 2015). When mapping the mean protein expression per cancer type, we found that, in different cell types, multiple pathways, including apoptosis, cell survival, motility, and DNA repair, displayed a similar pattern, consistent with the fact that immortal cancer cells retain cancer hallmarks in tissue culture (Hanahan and Weinberg, 2011). An example of a proteotypic pattern is the delta isoenzyme of protein kinase C, i.e. PRKCD, involved in cancer progression and a drug target (Mackay and Twelves, 2007). In agreement with PRKCD downregulation in renal clear cell carcinoma lines (Engers et al., 2000), PRKCD stood out in our visualization, with significantly lower protein expression in the NCI-60 renal carcinoma cells, relative to the average expression across the NCI-60 panel.
We also tested cellular pathways using ROMA (Representation and quantification Of Module Activities) (Martignetti et al., 2016) (Figure S48), a gene-set-based quantification algorithm. This approach revealed substantial diversity of pathway activity between different proteotypes as evidenced by two-tailed t-tests of activity scores (p value < 0.05). When mapping activity scores onto ACSN, some tissue specificities were revealed, with particular cell line proteotypes displaying distinct patterns. For instance, the activity of apoptosis (with both caspases and apoptosis genes modules) was found significantly higher in ovarian cell lines (Table S5). Although there are only two prostate cancer cell lines in the panel, our analysis was able to highlight modules including “AKT-mTOR” and “Apoptosis,” whose differential activity can be attributed to HSP90AA1 and PRDX. The latter protein has been independently reported to be overexpressed in prostate tumors (Ummanni et al., 2012).
Drug Response Predictions
The SWATH proteotypes covered 105 established protein targets for FDA-approved anti-cancer compounds, 661 protein targets annotated in DrugBank (Law et al., 2014) (including 68 drug-metabolizing enzymes, 5 drug carriers, and 15 drug transporters), 694 proteins linked with human diseases (Law et al., 2014, Uhlen et al., 2015), 58 protein kinases, 2 topoisomerases (TOP1 and TOP2β), and 9 tubulins. Some kinases were found to be broadly expressed with high abundance across cell lines, including MST4 and WNK1 (Figure S49), consistent with previous reports regarding their abundance (Huang et al., 2007, Lin et al., 2001). Other kinases were highly expressed in specific cell lines, for example, EGFR in the breast cancer cell line MDA-MB468, ERBB2 in the SKOV3 ovarian cell line, and CDK6 in leukemia MOLT4 cells, in agreement with previous studies using antibody-based methods (Uhlen et al., 2015, Xu et al., 2005). TOP1, TOP2β, and tubulins tended to be expressed across cell lines, consistent with their ubiquitous functions.
To assess drug response prediction, we used two main methods, an automated regression-based pipeline and a complementary interactive analysis for developing regression models (a functionality easily accessible to readers using CellMinerCDB, discover.nci.nih.gov/cellminercdb). In both cases, drug response was predicted as a weighted sum of selected feature values, where the signs and magnitudes of the feature weights indicate the direction and strength of feature influence, respectively. For the automated pipeline, we used the elastic net regression algorithm to select model features. The interactive approach involved examination of individual features that correlate with drug response (i.e. univariate models), along with features selected using the LASSO algorithm. These were integrated with experimentally established features and then assessed using the univariate and multivariate (regression models) analysis tools of the CellMinerCDB website.
Complementarity of the SWATH Proteotype with Genomic Measurement for Drug Response Predictions
First, we investigated the utility of the SWATH-based proteotype with existing genomic and transcriptomic features for 158 FDA-approved or investigational compounds in CellMiner (Luna et al., 2015, Rajapakse et al., 2018, Reinhold et al., 2012, Shankavaram et al., 2009) (Figure 3A, Table S6). A number of these compounds have been screened multiple times as they were submitted independently to the NCI-DTP and/or served as positive controls. Drugs are given unique NSC (National Service Center) identifiers for each submission to the DTP NCI-60 screen (Reinhold et al., 2012), and 47 of the compounds are represented by multiple replicates. For instance, doxorubicin is represented by two independent NSC numbers, 123127 and 759155, with Pearson correlation of 0.962 (p = 6 × 10−34), validating data reproducibility. Each of the compounds is categorized by mechanism-of-action annotation (Figure 3A). The largest group of drugs is DNA damaging agents (including DNA alkylating and cross-linking agents, DNA synthesis and topoisomerase inhibitors).
Figure 3.

Prediction of Drug Responsiveness Using the NCI-60 Proteome
(A) Workflow for drug responsiveness prediction. Drug mechanism categories are shown.
(B) Distribution of predictive power (Pearson's correlation of cross-validation predicted vs. observed response) for 240 compounds using all molecular features (All) versus common features (Common) available for all molecular data types.
(C) Distribution of predictive power for molecular features (i.e. gene expression and mutation profiles) with and without the SWATH proteotype.
(D) Pearson correlation coefficient distribution of drug responsiveness predicted with and without SWATH data. p value for Kolmogorov–Smirnov test (two-sided, two-sample) was computed.
(E) Venn diagram of drugs successfully modeled using elastic net with the SWATH data containing 3,171 proteins (SW3171) and the DDA data based on iBAQ (DDA-iBAQ) and LFQ (DDA-LFQ).
(F) Venn diagram of protein predictors using the SWATH and DDA datasets.
(G) Distribution of predictive improvement using the SWATH proteotype within each mechanism class.
(H) Predictive power of different omics data combinations for the activity of 20 FDA-approved compounds based on elastic net modeling of the drug response. Each row indicates compound-specific results using gene expression and mutation input data alone or in combination with proteomic abundances; each column represents a compound. The color indicates the predictive power, measured by Pearson correlation of cross-validation predicted and observed drug response values. The top and bottom 10 drugs by difference of the absolute value of predictive power are shown. Columns specifying compound-specific response prediction accuracies are sorted by mechanism of action and whether the inclusion of the SWATH data improved the overall model.
Using the elastic net algorithm (Barretina et al., 2012, Garnett et al., 2012, Rajapakse et al., 2018), we developed multivariate linear models to predict the NCI-60 response for each compound based on selected genomic, transcriptomic, and SWATH proteomic features. The Pearson's correlation between observed drug response values and leave-one-out cross-validation-predicted response values was applied to evaluate the performance of each predictive dataset. As different numbers of features were measured for each omics dataset, two strategies were adopted in the modeling analyses. In one case, we used all omics features, with and without the SWATH proteotype, as inputs to evaluate their general performance (see Methods). In the second, we selected 1,566 features that were available for all three molecular data types (denoted as common features). In both cases, we obtained models for 224 (93%) of the drugs (with adequate numbers of responsive lines). The predictive power achieved with all features was slightly higher than that obtained using the common features for all three data types (Figure 3B); a likely reason for this is that the latter excluded some genomic and transcriptomic features not detected at the protein level. Here, we focus on the analysis derived from all available molecular features (Figures 3H and S50, Table S7).
We identified several validated predictors for drug response. For instance, mRNA expression of SLFN11 was the most dominant indicator of sensitivity to a number of DNA-targeted compounds (including FDA-approved drugs spanning platinum drugs, topoisomerase inhibitors, alkylating agents, PARP inhibitors, and DNA synthesis inhibitors), in agreement with recent reports (Barretina et al., 2012, Rajapakse et al., 2018, Zoppoli et al., 2012) (Table S7). This pipeline generated models for 224 compounds (Figure S50, Table S7). The results of these models, summarized in Figures 3G and 3H, show that predictive improvement using SWATH data was achieved across the mechanisms of actions analyzed. Given the relatively small sample size, it was not surprising that accurate predictive models could not be found for every drug (Figure 3G), particularly those with limited numbers of responsive cell lines amid a diversity of cancer types. The SWATH-MS-derived proteotypes displayed a higher percentage of predictive features than mutations and transcripts. Twelve percent of SWATH features were selected in one or more predictive models, whereas the corresponding proportions were 2% for mutation features and 6% for transcript expression features (Table S7). The responsiveness of 49 screened drugs (22%) was best predicted with SWATH data, and 83 compounds (37%) were best predicted by combining SWATH data with transcripts and mutational data.
Through an examination of predictive gain by mechanism-of-action (Figure 3G), we made a few notable observations. Out of six HDAC inhibitors, four showed improvement using the SWATH feature set making it the most improved mechanism category; this observation should be taken with caution given the limited number of compounds. Kinase inhibitors were one of the least improved categories, but this observation should be revisited using future phospho-proteomic datasets, which would include additional markers of kinase regulation (Ardito et al., 2017, Johnson, 2009). Lastly, the diverse set of compounds in the “Other” category also showed predictive improvement with the SWATH features and merits further future study. The remaining categories show a largely even distribution in predictive improvement. Next, we compared the distributions of predictive power as measured by Pearson correlation coefficients for models derived with and without the inclusion of SWATH features. The SWATH-included distribution is wider, but spans a comparable range of (higher-accuracy-associated) positive correlations relative to the strictly genomic feature-based distribution (Figures 3C and 3D). Based on these analyses, we conclude the complementarity value of the SWATH proteotype with genomic features.
Exploration of the SWATH data can also reveal secondary predictors of drug response. The protein kinase inhibitor vemurafenib (VEM, NSC 761431) yielded a multivariate model where the most prominent SWATH feature was the expression level of LAMTOR3. Although the BRAF V600E mutation is a highly significant predictor of vemurafenib activity in the NCI-60 (Abaan et al., 2013), we speculate that LAMTOR3 may be a secondary drug response predictor, although further studies outside of the scope of the current work are necessary for validation. LAMTOR3 (MP1) is part of an endosomal scaffolding complex interacting with components of the RAF/MEK/ERK mitogenic signaling pathway. LAMTOR3 binds MEK1 and ERK1, facilitating activation of the latter protein (Schaeffer et al., 1998). Elevated LAMTOR3 protein expression was correlated with vemurafenib resistance (r = 0.44), consistent with the hypothesis that LAMTOR3 has the capacity to enhance RAF/MEK/ERK pathway signaling downstream of RAF. Increased protein expression of LAMTOR3 was observed in two BRAF mutant cell lines, SK-MEL-5 and LOXIMVI, which are relatively resistant to vemurafenib (Abaan et al., 2013). By contrast, the two cell lines with the lowest LAMTOR3 protein expression (MALME-3M and HT29) were notably among the most sensitive to vemurafenib.
We also compared the predictive power of the DDA data from the literature (Gholami et al., 2013) with the SWATH data. Although the DDA data were able to generate multivariate models for a comparable number of drugs (Figure 3E), the number of selected protein expression predictors was lower than in the SWATH data, with some overlap (Figure 3F). The DDA datasets (Gholami et al., 2013) analyzed using iBAQ and LFQ algorithms displayed moderate overlap with each other in terms of selected model predictors (Figure 3F).
Examples of Novel SWATH Predictors for NCI-60 Drug Responses Using CellMinerCDB
Our automated analysis along with our interactive exploration with CellMinerCDB (Rajapakse et al., 2018) produced multiple predictors with plausible drug response associations. For instance, ABCC4 was a SWATH predictor for resistance to alkylating agents, including chlorambucil (NSC 3088), uracil mustard (NSC 34462), and nitrogen mustard (NSC 762) in highly ranked models (by predictive power), consistent with its established role as a drug efflux pump (Borst and Elferink, 2002). Across molecular features (mutation, transcript, or protein expression), 14 ATP-binding cassette family transporters were predictive of sensitivity to 51 compounds. P-glycoprotein (encoded by ABCB1), which mediates resistance to a broad range of anticancer agents (Robey et al., 2018), predicted resistance to widely used chemotherapeutic anticancer drugs including doxorubicin (r = 0.38; p = 0.003; Figure S45B) and taxol (r = 0.43; p = 0.00086; Figure S45D), as well as to the HDAC inhibitor romidepsin (depsipeptide; r = 0.41; p = 0.0015; Figure S45C) and the HSP90 inhibitor alvespimycin. ABCB1 protein expression across the NCI-60 was also positively correlated with its transcripts (r = 0.44; p = 0.00044; Figure S45A). Of note, the BCR-ABL inhibitor nilotinib was correlated with the ABC transporter protein (ABCF1) (Table S7). These results confirm the importance of measuring ABC transporters to optimize the use of anticancer agents and warrant further investigation.
Another negatively weighted SWATH predictor is CTNND1 for several compounds targeting DNA, including daunorubicin (NSC 756717), valrubicin (NSC 246131), and carmustine (NSC 409962). CTNND1 encodes δ-catenin, which promotes cell survival through activation of the WNT signaling pathway (Tang et al., 2016). Inhibition of apoptosis (Chen et al., 2001) plausibly confers drug resistance in cells with high CTNND1 protein. In addition, as discussed previously and shown in Figure S43, CTNND1 is stoichiometrically correlated with β-catenin (CTNNB1), α-catenins (CTNNA1 and CTNAA2), and EPCAM, the epithelial cell adhesion molecule, indicating a potential role of plasma membrane signaling in cellular response to DNA-targeted agents. Consistent with this hypothesis and the potential predictive value of β-catenin, analysis performed in CellMinerCDB showed significant negative correlation with etoposide (r = −0.518, p = 0.000032), topotecan (r = −0.3, p = 0.02; Figure 4), melphalan (r = 0.534, p = 1.34 × 10−6), chlorambucil (r = −0.526, p = 1.85 × 10−6), and cisplatin (r = −0.366, p = 0.00254; Figure S48). EPCAM expression was also significantly predictive of cisplatin resistance (r = −0.44, p = 0.00047; Figure S44) as was EPCAM promoter methylation (r = −0.52, p = 2 × 10−5; Figure S44).
Figure 4.

Pedictive Protein Biomarkers for Topotecan (NSC609699) Activity
(A) The snapshots from discover.nci.nih.gov/CellMinerCDB show results obtained with the “Multivariate Analyses” tool of CellMinerCDB using topotecan as “Response Identifier” for the query.
(B and C) Plots of the observed response values for topotecan (NSC 609699) (y axis) versus the 10-fold cross-validation predicted response values (x axis), using SWATH measurements for predictors (B) and gene expression-based predictors (C).
As noted above, CellMinerCDB (discover.nci.nih.gov/CellMinerCDB) allows biologically plausible drug response correlates to be integrated within exploratory multivariate regression models. Figures 4 and S44 provide two examples using SWATH measurements. In the first, cisplatin is used as the response variable. The top response determinants are β-catenin and EPCAM, as discussed above (Figure S44). In the second example, the top predictive response determinants for the clinical topoisomerase I inhibitor topotecan are POLD1, RNASEH2B, and BAX (Figure 4A). POLD1 is the large subunit of the replicative polymerase δ and RNaseH2B one of the subunits of RNaseH2, which removes ribonucleotides misincorporated during DNA synthesis. The higher sensitivity of cells with high POLD1 and high RNaseH2 is likely reflective of hyperactive replication, which determines response to TOP1 inhibitors such as topotecan (Pommier et al., 2016). The value of β-catenin (CTNNB1) as a negative (resistance) predictor can be related to its anti-cell death activity (see prior section). Conversely, the identification of BAX, the mitochondrial pro-apoptotic effector, as a positive predictive (sensitivity) determinant at the protein level (Figure 4A) is consistent with apoptotic propensity shaping the response to DNA damaging drugs. Together, Figures 4B and 4C show this to be a predictive multivariate model at the protein, but not the RNA level, incorporating established features associated with replication stress and apoptosis.
Discussion
Complementarity of protein and transcript data (Liu et al., 2016, Mertins et al., 2016, Zhang et al., 2014, Zhang et al., 2016) can be expected to reveal new biological insights that are not apparent from the commonly used mutation and transcriptome profiles and which could be applied to enhance precision medicine. However, due to technical limitations, the acquisition of proteomic cohort datasets has been challenging. Here, using the NCI-60 cell line compendium, we demonstrate the ability of the PCT-SWATH proteomic technique to consistently quantify over 3,000 proteins across each of the NCI-60 cell lines measured in duplicate with a realistic turnaround. The data were acquired in approximately 30 working days on a single mass spectrometer, and for each sample measurement ca. 1 microgram of total peptide mass was consumed. This has been enabled by the pressure cycling technology, which minimizes sample consumption and the data-independent MS data acquisition using SWATH-MS (Guo et al., 2015).
Because of their extensive omic annotation and drug databases including over 21,000 publicly accessible compounds, the NCI-60 are a unique platform for testing new technologies and exploring determinants and predictors of drug response (Abaan et al., 2013, Monks et al., 2018, Rajapakse et al., 2018, Reinhold et al., 2012, Reinhold et al., 2019, Zoppoli et al., 2012). In addition, most of the NCI-60 cell lines are widely used for cell biology and pharmacology (MCF-7, MDA-MB231, HCT116, HCT15, HT29, HL60, CCR-CEM, and K562 to name a few). Hence, providing MS data for over 3,000 proteins and making the data available and easy to mine through CellMinerCDB will provide a unique resource for the scientific community worldwide. This is especially true because of the overlap between the cell lines in the NCI-60 and the Broad-MIT (CCLE/CTRP) and the MGH-Sanger (GDSC) (55 and 44 of the 60 cell lines, respectively) (Rajapakse et al., 2018). The proteome of the NCI-60 has been previously measured by sample fractionation and DDA-MS analysis of over 1,000 fractionated samples (Gholami et al., 2013). However, in this early study with basic MS technology, data acquisition for each cell line required an average of about 29 h MS instrument time. By contrast, our SWATH analyses required about 140 min MS instrument time, demonstrating the feasibility of extensive human proteotypes with a throughput almost comparable with genomic and transcriptomic analyses and its potential translation to tumor material in the future. Our integration of the SWATH data into CellMinerCDB (discover.nci.nih.gov/cellminercdb) also enables the user to readily check the SWATH data for concordance with a small number of proteins measured by RPPA (Nishizuka et al., 2003) (see the example for PCNA in Figure S38).
Two aspects of our workflow ensure robust and quantitatively accurate protein expression measurements. First, we obtained technical duplicates for the entire set of NCI-60 proteotypes. This was feasible due to the unparalleled high sample-throughput of the PCT-SWATH methodology, which is gaining popularity in proteomic profiling of clinical specimens. Additionally, we developed an expert system software to further curate peptide and protein identification and quantification. Applying stringent criteria, 3,171 proteins were included for further analyses and are made available through CellMiner (discover.nci.nih.gov/cellminer and discover.nci.nih.gov/cellminercdb). The raw MS signal for each of the quantified proteins, in each cell line, was inspected by the expert system, simulating manual inspection, and is available for visual inspection in the Supplemental Information. Because the NCI-60 cell lines are widely used in cell biology, we anticipate the broad utility of these highly curated proteomic data. Additionally, our rapid proteotype acquisition pipeline using PCT-SWATH requires little biological material, making it suitable for clinical settings and in precision medicine efforts (Guo et al., 2015, Shao et al., 2015, Shao et al., 2016).
Compared with other omics data, proteotypes offer unique insights into the coordinated expression of protein complexes (Dudley et al., 2005, Fraser and Plotkin, 2007, Ori et al., 2016, Wang et al., 2012), which are dysregulated in many diseases, especially cancer (Le, 2015). Our high-quality proteomic data allowed a systematic investigation of the composition of 101 protein complexes in 60 cell lines. We expect that this represents a proof-of-principle for a generic, high-throughput approach, applicable to larger cancer cell line databases and clinical specimens (Guo et al., 2015), for exploring biological networks and the association of defective protein complexes with diseases and drug responses. In addition, using CellMinerCDB (discover.nci.nih.gov/cellminercdb) casual users can test the stoichiometric expression of proteins belonging to small complexes. Several examples are provided in the manuscript including dimeric complexes (XRCC6/KU70 and XRCC5/KU80; Figures 2E and S36), trimeric complexes (RPA1, RPA2 and RPA3), larger complexes such as the chromatin remodeling NuRD complex (Figures S37, S41, and S47), the β-catenin plasma membrane complexes (Figure S43), and replication complexes including MCM helicases, RPAs, PCNA, RFC4, RFC2, and FEN1 (Figures S37–S39), as well as stoichiometrically coordinated RNA binding protein complexes, such FUS and the RNase A DHX9 (Figures S35 and S40).
The NCI-60 panel has enabled many landmark discoveries, and often emerging technologies are first tested on this panel due to its diversity and depth of surrounding knowledge (Abaan et al., 2013, Barretina et al., 2012, Garnett et al., 2012, Reinhold et al., 2019, Shoemaker, 2006, Weinstein, 2012). Each cancer cell line in the NCI-60 has been tested against tens of thousands of compounds, including the FDA-approved and investigational drugs featured in our analyses. With the addition of the SWATH proteomic data, the NCI-60 remains positioned as one of the most comprehensive models for cancer research and drug discovery. The NCI-60 uniquely enabled our thorough, integrative analysis of different molecular profiles (genomic, transcriptomic, and proteomic) in predicting drug responsiveness. Our findings strengthen the body of work, highlighting the importance of integrative omics approaches in understanding drug mechanisms, and establish the benefit of large-scale proteomic measurements. Therefore, we expect our study to become a seminal work in the area of pharmacoproteomics, the benefit of which will grow with anticipated expansion of sample size, proteomic coverage including extension to phosphoproteomic expression, as well as extension to mouse models (Gao et al., 2015) and human specimens (Guo et al., 2015).
The existing SWATH data specifically enabled the use of advanced analysis techniques to produce multivariate models of drug response. Examples of multivariate analyses using CellMinerCDB are provided for topotecan and cisplatin (Figures 4 and S44) with predictive protein biomarkers including the sensitivity signature of the proapoptotic protein BAX and a resistance signature centered around β-catenin. Yet, it has been challenging to identify such signatures, and the combination of proteomic, transcriptomic, and mutation data will likely be necessary to generate predictive signatures for precision medicine. Likely, a limitation at the current technical level is the number of proteins identified, and their skewing toward the higher expressed proteins. As technology improves, and a broader group of proteins is identified; it can be anticipated that the predictive utility of the protein data will increase rapidly.
Effort was put into making our work publicly available and easily accessible through data submission to the NCI-60 CellMiner database and an accompanying R package, rcellminer (Luna et al., 2015). We expect that the analyses developed, including those based on the widely used LASSO and elastic net methods, will continue to evolve and enable future studies on additional datasets and phenotypes. Although the strengths of these methods over other related methods have been previously described (Jang et al., 2014, Papillon-Cavanagh et al., 2013), the resulting models still require careful scrutiny by individual researchers. The interpretation of the models developed here and by others using our pipeline, should be guided by understanding of the biological activities of the associated predictors in the context of the mechanisms of action for the input drugs.
Limitations of the Study
This study and the resulting dataset invite further investigation of this unique proteomic data resource. The analysis of protein complexes in the current study highlights the value of mining beyond transcriptomic data, at the functionally critical proteomic level. The proteomic data of this study were acquired by the SWATH-MS technology, a massively parallel targeting method, at an early stage in its development. Over the last years the technology has rapidly advanced in terms of the proteomic depth that it can achieve. Our observation of the common lack of correlation between mRNA and protein expression has been similarly made in tumor samples across multiple tissue types (Kosti et al., 2016). Understanding these differences should help drive future studies in the development of mathematical and experimental models that leverage these -omics datasets effectively. Future investigations considering different approaches to handling the data, providing increased penetrance in the number of genes assessed, extending the functional implications of the data, and assessing perturbed proteomes in these cells will push back the current limitations of the field. Additionally, we have strived to make our work publicly available and easily accessible through data submission to the NCI-60 CellMiner databases (http://discover.nci.nih.gov/and http://discover.nci.nih.gov/cellminercdb) and an accompanying R package, rcellminer (Luna et al., 2015).Although the strengths of these methods over other related methods have been previously described (Jang et al., 2014, Papillon-Cavanagh et al., 2013), the resulting models still require scrutiny. The interpretation of the models developed here, and by others using our pipeline, should be guided by an understanding of the biological activities of the associated predictors in the context of the mechanisms of action and survival assays used for the input drugs.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Margot Sunshine who developed the early versions of CellMiner and Jeffrey Wang within the CCR CellMiner team for developing the current versions of CellMiner and CellMinerCDB and the NCI-DTP team (Dr. Jerry Collins and Dr. James H. Doroshow) for sharing the drug data and supporting for data acquisition from the DTP to the CellMiner team. We also thank Emanuel Gonçalves for comments on the manuscript. The work was supported by the SystemsX.ch project PhosphoNetX PPM (to R.A.), the Swiss National Science Foundation (grant no. 3100A0-688 107679 to R.A.), the European Research Council (grants no. ERC-2008-AdG 233226 and ERC-20140AdG 670821 to R.A.), the Ruth L. Kirschstein National Research Service Award (grant no. F32 CA192901 to A.L.), the National Resource for Network Biology (NRNB) from the National Institute of General Medical Sciences (NIGMS) (grant no. P41 GM103504 to C.S.), the Center for Cancer Research, Intramural Program of the National Cancer Institute (grant no. Z01 BC006150 to Y.P), the Wellcome Trust Award (102696 to M.J.G.), the Westlake Startup Grant (to T.G.), Zhejiang Provincial Natural Science Foundation of China (grant No. LR19C050001 to T.G.), Hangzhou Agriculture and Society Advancement Program (grant No. 20190101A04 to T.G.), National Natural Science Foundation of China (General Program) (grant No. 81972492 to T.G.), and National Science Fund for Young Scholars (grant No. 21904107 to Y.Z.). A.B.E was supported by the SystemsX.ch project TbX and the National Institutes of Health project Omics4TB Disease Progression (U19 AI106761). We thank An Guo for helping in preparing the graphics.
Author Contributions
T.G., R.A., and Y.P. designed and coordinated the project. C.C.K. processed the samples. L.G., C.C.K., and T.G. acquired the SWATH data. T.G. performed the SWATH data interpretation and benchmarking with help from C.C.K., and the expert system analysis with help from C.X., U.S. A.L., V.N.R., Z.W., and Y.P. performed the drug response prediction analysis and developed the reproducible research infrastructure, with critical inputs from M.P.M., J.S.R., M.J.G., S.V., W.C.R., C.S., and Y.P.. L.C. and L.M. performed the pathway analysis. A.L., V.N.R., W.C.R., S.V., and Y.P. integrated the SWATH data into rcellminer and CellMiner. A.O., M.I., R.C., C.A., M.B., and A. B.E. performed the protein complex analysis, with help from A.L., Z.W., Y.C., V.N.R., C.S., Y.S., Y.Z., Y.P.. P.Q. and Q.Z. contributed to the data analysis. W.L., H.G., R.B., J.Z., H.Z., and Y.S. performed the validation and PRM experiments. T.G., A.L., V.N.R., and Y.P. wrote the manuscript with inputs from all co-authors. R.A., Y.P., and T.G. supervised the project.
Declaration of Interests
R.A. holds shares of Biognosys AG, which operates in the field covered by the article. The research groups of R.A. and T.G. are supported by SCIEX, which provides access to prototype instrumentation, and Pressure Biosciences Inc., which provides access to advanced sample preparation instrumentation. All remaining authors declare no competing interests.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.059.
Contributor Information
Tiannan Guo, Email: guotiannan@westlake.edu.cn.
Yves Pommier, Email: yves.pommier@nih.gov.
Ruedi Aebersold, Email: aebersold@imsb.biol.ethz.ch.
Data and Code Availability
The NCI-60 SWATH datasets and SWATH assay library has been deposited in PRIDE. Project Name: NCI60 proteome by PCT-SWATH; Project accession: PXD003539.
Reviewer account details:
Username: reviewer15254@ebi.ac.uk
Password: dWdyptzf
The protein data matrix has also been deposited in ArrayExpress. Project accession: E-PROT-2. Project title: Proteomic profiling of NCI60 cell lines from Cancer Cell Line Encyclopedia.
Reviewer account details:
Username: Reviewer_E-PROT-2
Password: gdgywGco
The protein data matrix is also accessible in CellMiner website (Reinhold et al., 2012) and R package rcellminer (Luna et al., 2015).
Supplemental Information
(A) Information for PCT-SWATH analysis of the NCI-60 cells.
(B) List of peptide precursors appeared in the library.
(C) Quantitative values of 22,554 peptide precursors in the NCI-60 cells in duplicates.
(D) Quantitative values of 3,171 proteins in the NCI-60 cells in duplicates.
(E) Averaged protein intensity in the NCI-60 cells.
This table shows the count of IPI protein group number from the DDA dataset quantified using iBAQ and LFQ algorithms 1 and the count of SwissProt proteotypic proteins from the SWATH data as reported in this data (Gholami et al., 2013a).
Average Abundance (log10) means the averaged log 10 scaled protein abundance signal for proteins in a complex. Standard Deviation means the standard deviation of log 10 scaled protein abundance signal for proteins in a complex. Average Pearson Correlation means the averaged Pearson correlation value for each pair of proteins in a complex.
(A) The modules that show a significant dispersion are reported here.
(B) A t test is performed for cell lines from one cancer type vs. all other cancer cell lines.
(A) Exome data of the NCI-60 cells.
(B) Log2 scaled mRNA expression data of the NCI-60 cells.
(C) Common features at three different levels, i.e. DNA, mRNA, and protein.
References
- Abaan O.D., Polley E.C., Davis S.R., Zhu Y.J., Bilke S., Walker R.L., Pineda M., Gindin Y., Jiang Y., Reinhold W.C. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardito F., Giuliani M., Perrone D., Troiano G., Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review) Int. J. Mol. Med. 2017;40:271–280. doi: 10.3892/ijmm.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehar J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta J., Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015;165:36–47. doi: 10.1016/j.trsl.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates S.E., Eisch R., Ling A., Rosing D., Turner M., Pittaluga S., Prince H.M., Kirschbaum M.H., Allen S.L., Zain J. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. Br. J. Haematol. 2015;170:96–109. doi: 10.1111/bjh.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P., Elferink R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- Brazma A., Parkinson H., Sarkans U., Shojatalab M., Vilo J., Abeygunawardena N., Holloway E., Kapushesky M., Kemmeren P., Lara G.G. ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003;31:68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J.H., Mazcko C.N., LeBlanc A.K., Covey J.M., Ji J.J., Kinders R.J., Parchment R.E., Khanna C., Paoloni M., Lana S.E. NCI Comparative Oncology Program testing of non-camptothecin indenoisoquinoline topoisomerase i inhibitors in naturally occurring canine lymphoma. Clin. Cancer Res. 2018;24:5830–5840. doi: 10.1158/1078-0432.CCR-18-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.W., Nam H.Y., Kim H.J., Moon S.Y., Kim M.R., Lee M., Kim G.C., Kim S.W., Kim S.Y. Effect of beta-catenin silencing in overcoming radioresistance of head and neck cancer cells by antagonizing the effects of AMPK on Ku70/Ku80. Head Neck. 2016;38(Suppl 1):E1909–E1917. doi: 10.1002/hed.24347. [DOI] [PubMed] [Google Scholar]
- Chen S., Guttridge D.C., You Z., Zhang Z., Fribley A., Mayo M.W., Kitajewski J., Wang C.Y. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley A.M., Janse D.M., Tanay A., Shamir R., Church G.M. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol. Syst. Biol. 2005;1 doi: 10.1038/msb4100004. 2005 0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers R., Mrzyk S., Springer E., Fabbro D., Weissgerber G., Gernharz C.D., Gabbert H.E. Protein kinase C in human renal cell carcinomas: role in invasion and differential isoenzyme expression. Br. J. Cancer. 2000;82:1063–1069. doi: 10.1054/bjoc.1999.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fojo T., Farrell N., Ortuzar W., Tanimura H., Weinstein J., Myers T.G. Identification of non-cross-resistant platinum compounds with novel cytotoxicity profiles using the NCI anticancer drug screen and clustered image map visualizations. Crit. Rev. Oncol. Hematol. 2005;53:25–34. doi: 10.1016/j.critrevonc.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Fraser H.B., Plotkin J.B. Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol. 2007;8:R252. doi: 10.1186/gb-2007-8-11-r252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Korn J.M., Ferretti S., Monahan J.E., Wang Y., Singh M., Zhang C., Schnell C., Yang G., Zhang Y. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami A.M., Hahne H., Wu Z.X., Auer F.J., Meng C., Wilhelm M., Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Gillet L.C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Kouvonen P., Koh C.C., Gillet L.C., Wolski W.E., Rost H.L., Rosenberger G., Collins B.C., Blum L.C., Gillessen S. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015;21:407–413. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Holbeck S.L., Collins J.M., Doroshow J.H. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol. Cancer Ther. 2010;9:1451–1460. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.L., Cha S.K., Wang H.R., Xie J., Cobb M.H. WNKs: protein kinases with a unique kinase domain. Exp. Mol. Med. 2007;39:565–573. doi: 10.1038/emm.2007.62. [DOI] [PubMed] [Google Scholar]
- Jang I.S., Neto E.C., Guinney J., Friend S.H., Margolin A.A. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. Pac. Symp. Biocomput. 2014;19:63–74. [PMC free article] [PubMed] [Google Scholar]
- Johnson L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009;37:627–641. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- Jones P., Cote R.G., Martens L., Quinn A.F., Taylor C.F., Derache W., Hermjakob H., Apweiler R. PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 2006;34:D659–D663. doi: 10.1093/nar/gkj138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanungo J. Exogenously expressed human Ku70 stabilizes Ku80 in Xenopus oocytes and induces heterologous DNA-PK catalytic activity. Mol. Cell Biochem. 2010;338:291–298. doi: 10.1007/s11010-009-0363-3. [DOI] [PubMed] [Google Scholar]
- Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kosti I., Jain N., Aran D., Butte A.J., Sirota M. Cross-tissue analysis of gene and protein expression in normal and cancer tissues. Sci. Rep. 2016;6:24799. doi: 10.1038/srep24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I., Bonnet E., Nguyen H.A., Cohen D., Viara E., Grieco L., Fourquet S., Calzone L., Russo C., Kondratova M. Atlas of cancer signalling network: a systems biology resource for integrative analysis of cancer data with google maps. Oncogenesis. 2015;4:e160. doi: 10.1038/oncsis.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., Maciejewski A., Arndt D., Wilson M., Neveu V. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D.H. A novel method for identifying disease associated protein complexes based on functional similarity protein complex networks. Algorithms Mol. Biol. 2015;10:14. doi: 10.1186/s13015-015-0044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.L., Chen H.C., Fang H.I., Robinson D., Kung H.J., Shih H.M. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene. 2001;20:6559–6569. doi: 10.1038/sj.onc.1204818. [DOI] [PubMed] [Google Scholar]
- Liu Y., Beyer A., Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- Luna A., Rajapakse V.N., Sousa F.G., Gao J., Schultz N., Varma S., Reinhold W., Sander C., Pommier Y. rcellminer: exploring molecular profiles and drug response of the NCI-60 cell lines in R. Bioinformatics. 2015;32:1272–1274. doi: 10.1093/bioinformatics/btv701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay H.J., Twelves C.J. Targeting the protein kinase C family: are we there yet? Nat. Rev. Cancer. 2007;7:554–562. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- Martignetti L., Calzone L., Bonnet E., Barillot E., Zinovyev A. ROMA: representation and quantification of module activity from target expression data. Front. Genet. 2016;7:18. doi: 10.3389/fgene.2016.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks A., Zhao Y., Hose C., Hamed H., Krushkal J., Fang J., Sonkin D., Palmisano A., Polley E.C., Fogli L.K. The NCI transcriptional pharmacodynamics workbench: a tool to examine dynamic expression profiling of therapeutic response in the NCI-60 cell line panel. Cancer Res. 2018;78:6807–6817. doi: 10.1158/0008-5472.CAN-18-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka S., Charboneau L., Young L., Major S., Reinhold W.C., Waltham M., Kouros-Mehr H., Bussey K.J., Lee J.K., Espina V. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc. Natl. Acad. Sci. U S A. 2003;100:14229–14234. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A., Iskar M., Buczak K., Kastritis P., Parca L., Andres-Pons A., Singer S., Bork P., Beck M. Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol. 2016;17:47. doi: 10.1186/s13059-016-0912-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon-Cavanagh S., De Jay N., Hachem N., Olsen C., Bontempi G., Aerts H.J., Quackenbush J., Haibe-Kains B. Comparison and validation of genomic predictors for anticancer drug sensitivity. J. Am. Med. Inform. Assoc. 2013;20:597–602. doi: 10.1136/amiajnl-2012-001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P., Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Pommier Y., Sun Y., Huang S.N., Nitiss J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016;17:703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell B.S., Lazarev A.V., Carlson G., Ivanov A.R., Rozak D.A. Pressure cycling technology in systems biology. Methods Mol. Biol. 2012;881:27–62. doi: 10.1007/978-1-61779-827-6_2. [DOI] [PubMed] [Google Scholar]
- Rajapakse V.N., Luna A., Yamade M., Loman L., Varma S., Sunshine M., Iorio F., Sousa F.G., Elloumi F., Aladjem M.I. CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience. 2018;10:247–264. doi: 10.1016/j.isci.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Sunshine M., Liu H.F., Varma S., Kohn K.W., Morris J., Doroshow J., Pommier Y. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499–3511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C., Varma S., Sunshine M., Elloumi F., Ofori-Atta K., Lee S., Trepel J.B., Meltzer P.S., Doroshow J.H., Pommier Y. RNA sequencing of the NCI-60: integration into CellMiner and CellMiner CDB. Cancer Res. 2019;79:3514–3524. doi: 10.1158/0008-5472.CAN-18-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey R.W., Pluchino K.M., Hall M.D., Fojo A.T., Bates S.E., Gottesman M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer. 2018;18:452–464. doi: 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost H.L., Rosenberger G., Navarro P., Gillet L., Miladinovic S.M., Schubert O.T., Wolskit W., Collins B.C., Malmstrom J., Malmstrom L. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- Schaeffer H.J., Catling A.D., Eblen S.T., Collier L.S., Krauss A., Weber M.J. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Shankavaram U.T., Varma S., Kane D., Sunshine M., Chary K.K., Reinhold W.C., Pommier Y., Weinstein J.N. CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics. 2009;10:277. doi: 10.1186/1471-2164-10-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S., Guo T., Gross V., Lazarev A., Koh C.C., Gillessen S., Joerger M., Jochum W., Aebersold R. Reproducible tissue homogenization and protein extraction for quantitative proteomics using micropestle-assisted pressure-cycling technology. J. Proteome Res. 2016;15:1821–1829. doi: 10.1021/acs.jproteome.5b01136. [DOI] [PubMed] [Google Scholar]
- Shao S., Guo T., Koh C.C., Gillessen S., Joerger M., Jochum W., Aebersold R. Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry. Proteomics. 2015;15:3711–3721. doi: 10.1002/pmic.201500161. [DOI] [PubMed] [Google Scholar]
- Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Tang B., Tang F., Wang Z., Qi G., Liang X., Li B., Yuan S., Liu J., Yu S., He S. Overexpression of CTNND1 in hepatocellular carcinoma promotes carcinous characters through activation of Wnt/beta-catenin signaling. J. Exp. Clin. Cancer Res. 2016;35:82. doi: 10.1186/s13046-016-0344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Ummanni R., Barreto F., Venz S., Scharf C., Barett C., Mannsperger H.A., Brase J.C., Kuner R., Schlomm T., Sauter G. Peroxiredoxins 3 and 4 are overexpressed in prostate cancer tissue and affect the proliferation of prostate cancer cells in vitro. J. Proteome Res. 2012;11:2452–2466. doi: 10.1021/pr201172n. [DOI] [PubMed] [Google Scholar]
- Wang Q., Liu W., Ning S., Ye J., Huang T., Li Y., Wang P., Shi H., Li X. Community of protein complexes impacts disease association. Eur. J. Hum. Genet. 2012;20:1162–1167. doi: 10.1038/ejhg.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J.N. Drug discovery: cell lines battle cancer. Nature. 2012;483:544–545. doi: 10.1038/483544a. [DOI] [PubMed] [Google Scholar]
- Xu H., Yu Y., Marciniak D., Rishi A.K., Sarkar F.H., Kucuk O., Majumdar A.P. Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol. Cancer Ther. 2005;4:435–442. doi: 10.1158/1535-7163.MCT-04-0280. [DOI] [PubMed] [Google Scholar]
- Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M.C., Zimmerman L.J., Shaddox K.F., Kim S. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E., Zhou J.Y., Petyuk V.A., Chen L., Ray D. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppoli G., Regairaz M., Leo E., Reinhold W.C., Varma S., Ballestrero A., Doroshow J.H., Pommier Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. U S A. 2012;109:15030–15035. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Information for PCT-SWATH analysis of the NCI-60 cells.
(B) List of peptide precursors appeared in the library.
(C) Quantitative values of 22,554 peptide precursors in the NCI-60 cells in duplicates.
(D) Quantitative values of 3,171 proteins in the NCI-60 cells in duplicates.
(E) Averaged protein intensity in the NCI-60 cells.
This table shows the count of IPI protein group number from the DDA dataset quantified using iBAQ and LFQ algorithms 1 and the count of SwissProt proteotypic proteins from the SWATH data as reported in this data (Gholami et al., 2013a).
Average Abundance (log10) means the averaged log 10 scaled protein abundance signal for proteins in a complex. Standard Deviation means the standard deviation of log 10 scaled protein abundance signal for proteins in a complex. Average Pearson Correlation means the averaged Pearson correlation value for each pair of proteins in a complex.
(A) The modules that show a significant dispersion are reported here.
(B) A t test is performed for cell lines from one cancer type vs. all other cancer cell lines.
(A) Exome data of the NCI-60 cells.
(B) Log2 scaled mRNA expression data of the NCI-60 cells.
(C) Common features at three different levels, i.e. DNA, mRNA, and protein.
Data Availability Statement
The NCI-60 SWATH datasets and SWATH assay library has been deposited in PRIDE. Project Name: NCI60 proteome by PCT-SWATH; Project accession: PXD003539.
Reviewer account details:
Username: reviewer15254@ebi.ac.uk
Password: dWdyptzf
The protein data matrix has also been deposited in ArrayExpress. Project accession: E-PROT-2. Project title: Proteomic profiling of NCI60 cell lines from Cancer Cell Line Encyclopedia.
Reviewer account details:
Username: Reviewer_E-PROT-2
Password: gdgywGco
The protein data matrix is also accessible in CellMiner website (Reinhold et al., 2012) and R package rcellminer (Luna et al., 2015).