

SUMMARY
Regulation of hematopoietic stem cells (HSCs) by bone marrow (BM) niches has been extensively studied; however, whether and how HSC subpopulations are distinctively regulated by BM niches remain unclear. Here, we functionally distinguished reserve HSCs (rHSCs) from primed HSCs (pHSCs) based on their response to chemotherapy and examined how they are dichotomously regulated by BM niches. Both pHSCs and rHSCs supported long-term hematopoiesis in homeostasis; however, pHSCs were sensitive but rHSCs were resistant to chemotherapy. Surviving rHSCs restored the HSC pool and supported hematopoietic regeneration after chemotherapy. The rHSCs were preferentially maintained in the endosteal region that enriches N-cadherin+ (N-cad+) bone-lining cells in homeostasis and post-chemotherapy. N-cad+ cells were functional bone and marrow stromal progenitor cells (BMSPCs), giving rise to osteoblasts, adipocytes, and chondrocytes in vitro and in vivo. Finally, ablation of N-cad+ niche cells or deletion of SCF from N-cad+ niche cells impaired rHSC maintenance during homeostasis and regeneration.
In Brief
Zhao et al. demonstrate that blood-forming stem cells have a back-up system in stressed condition. Both reserve and active stem cells are maintained in the bone marrow by specific niches. While the latter are chemo-sensitive, the former survive and restore stem cells, and thereby generate the blood system.
Graphical Abstract
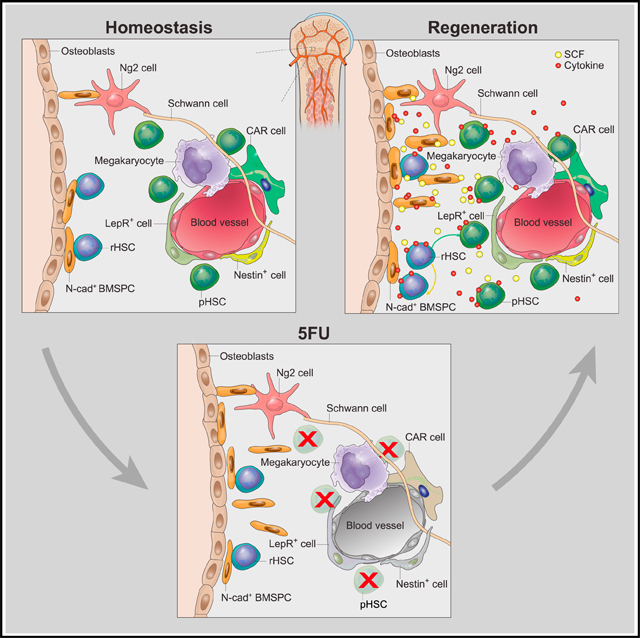
INTRODUCTION
Hematopoietic stem cells (HSCs) in bone marrow (BM) maintain homeostatic hematopoiesis throughout life and support regeneration after myeloablation (Weissman, 2000). Quiescent HSCs are resistant to DNA damage and outperform proliferative HSCs in hematopoietic regeneration in lethally irradiated mice (Fleming et al., 1993; Arai et al., 2004; Walter et al., 2015). However, a recent study showed that DNA damage is accumulated in quiescent HSCs, which have attenuated DNA repair and response pathways (Beerman et al., 2014). In fact, the majority of HSCs, despite their quiescence, are sensitive to DNA damage from chemotherapy, such as 5-fluorouracil (5FU) (Lerner and Harrison, 1990). The unresolved issue is how the hematopoietic system overcomes the consequence of myeloablation to regenerate the hematopoietic system. Given the remarkable heterogeneity of HSCs (Morita et al., 2010; Benz et al., 2012; Zhou et al., 2016; Itkin et al., 2016), the existence of a reserve HSC (rHSC) subpopulation was proposed, with the feature of drug resistance and the capacity to restore the HSC pool to overcome myeloablation (Haug et al., 2008; Li and Clevers, 2010; Wilson et al., 2008). Thus far, however, no functional evidence has been provided to support the existence of rHSCs in the blood system.
HSCs are preserved in complex BM niches for their maintenance and regeneration (Morrison and Scadden, 2014; Schofield, 1978; Li and Xie, 2005). In the past decades, multiple studies have uncovered the complexity of HSC BM niche components, including endosteal (inner bone surface) cells, sinusoidal endothelial cells, Cxcl12 abundant reticular (CAR) cells, Nestin+ and NG2+ perivascular cells, LepR+ and Prx-1+ mesenchymal stem and progenitor cells (MSPCs), non-myelinating Schwann cells, and megakaryocytes (Wei and Frenette, 2018). However, whether and how the BM niche complexity contributes to HSC heterogeneity regulation, especially under chemotherapeutic stress, remain largely unknown. Furthermore, the first HSC niche was initially identified as the spindle shaped N-Cadherin+ (N-cad+) cells in the endosteum of the trabecular bone (TB) region (Zhang et al., 2003; Calvi et al., 2003), but the nature and function of N-cad+ niche cells in BM remain unclear.
Herein, we have functionally defined the drug-resistant rHSC population and found that rHSCs are primarily maintained by the N-cad+ cells in the endosteal niche, especially under chemotherapeutic stress. We have further shown that N-cad+ cells are bone and marrow stromal progenitor cells (BMSPCs) and contribute to rHSC maintenance.
RESULTS
Reserve and Primed HSCs Are Functionally Distinguishable
To explore the rHSC subpopulation, we adapted a cell-surface marker, CD49b (Integrin α2), which can distinguish intermediate-term from permanently long-term HSCs (LT-HSCs) (Benveniste et al., 2010; Wagers and Weissman, 2005). Intriguingly, we found that a CD48−CD49b− subpopulation exists only in conventional LT-HSCs but not in short-term HSCs (ST-HSCs) or multipotent progenitor cells (MPPs). We proposed that the CD48−CD49b− LT-HSCs subpopulation enriches rHSCs and that the CD48−CD49b+ LT-HSCs subpopulation enriches primed HSCs (pHSCs), a transitional state between rHSCs and cycling ST-HSCs (Figure 1A). We performed a repopulation assay and found that both rHSCs and pHSCs supported hematopoiesis for up to 40 weeks after transplantation without significant difference (Figure 1B), consistent with a previous report (Benveniste et al., 2010). However, transplanted pHSCs had low efficiency in generating rHSCs (95.4% reduction) in recipients compared to transplanted rHSCs, which can generate both rHSCs and pHSCs, suggesting that rHSCs hierarchically preceded pHSCs (Figure 1C). Using the Scl-tTA-induced H2B-GFP label-retaining model, we found that rHSCs enriched 1.9-fold more H2B-GFPhigh cells compared to pHSCs (p = 0.0039), indicating that rHSCs divided less frequently over time compared to pHSCs (Figure 1D). Molecularly, we found that rHSCs had higher expression of HSC quiescence maintenance genes, such as Cdkn1c (p57), and Foxo1, whereas pHSCs had higher expression of cell-cycle activators such as Myc, Pcna, Ccng2, Cdk4, and Cdkn2c encoding G1-phase CDK4–6 inhibitor p18-Ink4c (Figure 1E). There was no significant difference in Ki67 expression observed between rHSCs and pHSCs (Figure 1E), indicating that Ki67 expression alone was insufficient to distinguish the two HSC subpopulations.
Figure 1. Functional Identification of rHSC Population.

(A) Fluorescence-activated cell sorting (FACS) of rHSCs, primed HSCs (pHSCs), ST-HSCs, and MPPs.
(B) Transplantation of rHSCs and pHSCs. Peripheral blood (PB) analysis for donor engraft cells shown at indicated weeks post-transplantation. The percentage of donor-derived B, T, and myeloid lineage cells shown at 20 weeks post-transplantation (n = 10).
(C) Donor-derived cells in recipient mice at 40 weeks post-transplantation from rHSCs or pHSCs.
(D) H2B-GFP LRCs in rHSCs and pHSCs at 130 days post-chasing (n = 4).
(E) Cell-cycle gene expression in rHSCs and pHSCs (n = 3 replicates from 20 mice).
(F) rHSCs and pHSCs transplanted mice received 5FU at 4 weeks post-transplantation. PB analysis for donor engraft cells shown at indicated weeks post-transplantation. The percentage of donor-derived B, T, and myeloid lineage cells shown at 20 weeks post-transplantation (n = 10).
(G) rHSCs and pHSCs at day 3 post-5FU (pool from 15 mice).
(H) DNA damage gene expression in rHSCs and pHSCs (n = 3 replicates from 20 mice).
(I) DNA damage genes in rHSCs (n = 20) and 5FU-rHSCs (n = 40). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars, SEM.
(J) Heatmap of stress-response genes in rHSCs, pHSCs, and 5FU rHSCs.
Because the functional definition for rHSCs is their drug-resistance capacity and being responsible for hematopoietic regeneration after myelosuppressive chemotherapy, we transplanted rHSCs or pHSCs into recipient mice and challenged the mice with 5FU at 4 weeks post-transplantation. As shown in Figure 1F, rHSCs were insensitive to 5FU treatment; however, pHSCs dramatically reduced their reconstitution ability (44% reduction at 20 weeks post-transplantation). Taken together, our data indicate that rHSCs were resistant, whereas pHSCs were sensitive to chemotherapy; furthermore, the former gave rise to the latter but rarely vice versa in the transplantation assay.
We further analyzed the direct consequence of acute 5FU challenge on rHSCs and pHSCs. As shown in Figure 1G, 69.4% of rHSCs survived, but only 4.34% of pHSCs remained at 3 days post-5FU. As early as 1 day post-5FU, we found apoptosis increased 3.2-fold more in pHSCs than in rHSCs in central marrow (CM), and 5-fold more in the endosteal region (Figure S1A). These data reveal that rHSCs may have a special stress-response program to overcome chemotherapeutic stress. To address this question, we analyzed the transcriptome profiling for DNA damage response genes in rHSCs, pHSCs, and rHSCs post-5FU treatment (5FU-rHSCs). We observed that, although rHSCs had attenuated DNA damage repair pathways compared to pHSCs during homeostasis (Figure 1H), most DNA repair pathways, such as DNA mismatch repair, nucleotide excision repair, base excision repair, and homologous recombination, were significantly activated in 5FU-rHSCs (Figure 1I). Stress-response genes, such as Gadd45 g, which regulate DNA damage response, apoptosis, and senescence (Liebermann and Hoffman, 2008), were higher in rHSCs than in pHSCs in homeostasis (Figure 1E). Furthermore, Cdc37 and Stip1 (Hop) that regulate the stress-response complex (HSP70 or HSP90) (Rodina et al., 2016) were upregulated in 5FU-rHSCs (1.8- ± 0.17-fold and 1.4 ± 0.1-fold, respectively) (Figure 1J). This finding partially explains how rHSCs survive and restore the HSC pool under chemotherapeutic stress.
Taken together, we functionally demonstrate the coexistence of pHSCs and rHSCs in BM. Even with their “quiescent” state and active DNA-repairing pathways, the majority of pHSCs are sensitive to chemotherapy, whereas rHSCs activate their DNA damage repair and stress-response pathways to survive chemotherapeutic stress and generate pHSCs to restore the HSC pool for hematopoietic regeneration after myelosuppressive chemotherapy.
Drug-Resistant rHSCs Predominantly Localize Adjacent to the N-Cad+ Stromal Cells in the Endosteal Region of BM
BM niche cells are key for stem cell maintenance and activity regulation (Morrison and Scadden, 2014; Li and Xie, 2005). To understand how rHSCs are protected from chemotherapeutic stress, we hypothesized that the rHSC subpopulation might be preserved in a specialized niche. To address this, we performed whole-mount immuno-staining to simultaneously detect the relative distribution of rHSCs and pHSCs with respect to the blood vessels (BVs), megakaryocytes (MKs), or bone (detected with second-harmonic generation, SHG) (Figures 2A, 2B, and S1B). Our quantification data showed that 43.4% ± 5.1% of rHSCs and 31.0% ± 5.9% of pHSCs were located within 5 μm from BVs (Figure 2C), and that 22.6% ± 6.1% of rHSCs and 24.6% ± 3.8% of pHSCs were located within 5 μm from MKs (Figure S1B), whereas rHSCs had no preference in association with BVs or MKs compared to randomly distributed HSC spots (Figures 2C and S1B), consistent with previous reports that the bulk of the HSC population resides in the perivascular or vascular niches (Acar et al., 2015; Chen et al., 2016; Zhao et al., 2014; Bruns et al., 2014). Interestingly, we noticed that 16.4% ± 6.3% of rHSCs compared to only 3.69% ± 3.4% of pHSCs were located within 5 μm from the bone surface (p = 0.00182) (Figure 2D). These data show that, although the majority of bulk HSCs were associated with BVs and MKs, rHSCs were localized significantly closer to the endosteal surface compared to pHSCs.
Figure 2. rHSCs Located by N-Cad+ Endosteal Region in BM Niche.

(A) Representative whole-mount images of sternum, with bone (white by SHG) and megakaryocytes (MKs) (yellow, distinguished by size, morphology, and CD41 expression). Green and white arrowheads denote phenotypic rHSCs (Lin−CD48−CD41−CD150+CD49b−) and pHSCs (Lin−CD48−CD41−CD150+CD49b+).
(B) Representative images of rHSCs, pHSCs, and 5FU rHSCs (arrows). Scale bar, 10 μm.
(C–E) Relative distance between rHSCs, pHSCs, and 5FU rHSCs to BVs (C), bone (D), or N-cad+ cells (E) (n = 51 rHSCs, n = 144 pHSCs, n = 102 5FU rHSCs in distance quantification of HSCs from BVs and bone; n = 40 rHSCs, n = 50 pHSCs, n = 34 5FU rHSCs in distance quantification of HSCs from N-cad+ cells). n = 3 mice for each distance quantification dataset.
(F) Representative whole-mount images of N-cad-CreERT; R26-ZsG mice induced by TMX for 24 h, co-stained with CD150, Lin, CD41, CD48, and CD49b as well as SHG signal (control and day 3 after 5FU).
(G) The rHSCs (arrows) localized near the N-cad+ bone surface after 5FU. Dashed-lined higher-power image (inset) shows one N-cad+ rHSC and one N-cad− rHSC.
(H) Absolute numbers of AnnexinV+ SytoxG− stromal cells in control and day 3 post-5FU (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars, SEM.
To test whether the endosteal region preserves rHSCs during chemotherapeutic stress, we examined the distribution of rHSCs at day 3 post-5FU treatment when pHSCs were largely eliminated (Figure 1G) (Haug et al., 2008). We found that 57.9% ± 6.8% of surviving rHSCs were preserved by the endosteal niche (within 5 μm from the bone surface), which is significantly enriched compared to randomly distributed HSC spots (p = 1.04 × 10−14). This was also a 3.5-fold enrichment compared to homeostatic rHSCs (p = 2.93 × 10−7) (Figure 2D). In contrast, no significant enrichment of surviving rHSCs was observed near BVs and MKs compared to randomly distributed HSC spots (Figures 2C and S1B). Previously, we demonstrated that N-cad+ bone-lining cells present in the endosteal region were in direct contact with long-term label retaining cells (LRCs) (Zhang et al., 2003; Xie et al., 2009). To further study the role of N-cad+ cells in relation to HSCs, we established the N-cad-tdTomato (N-cad-TdT) mouse line in which the Tomato+ cells report N-cad expression. Immunostaining revealed that N-cad-expressing cells exist in both the CM (N-cad-TdT(M)) and endosteal surface (N-cad-TdT(B)) (Figures S2A and S2B). Interestingly, we found that N-cad-TdT(B) cells typically resided near the CD31+ Endomucin+ endosteal type H capillaries (transition zone [TZ] vessels), which might provide a specific metabolic microenvironment and maintain perivascular osteoprogenitors (Kusumbe et al., 2014) (Figure S2C). In homeostatic condition, we found 56.7% ± 9.6% of rHSCs and 41.6% ± 6.4% of pHSCs were associated with N-cad+ cells. The percentage of N-cad+ cell-associated rHSCs notably increased at day 3 post-5FU compared to homeostatic rHSCs (76% ± 10.8% within 5 μm, p = 0.0114) (Figure 2E). The rHSCs were often found near the N-cad+ cells adjacent to the endosteal surface (Figures 2F and 2G; Video S1). In line with this finding, we also observed a 3.5-fold greater increase in apoptotic rHSCs post-5FU in CM than in the endosteal surface, suggesting that rHSCs in endosteum were less susceptible to apoptosis than in CM under 5FU stress (Figure S1A). Taken together, these data show that rHSCs were relatively preserved by N-cad+ stromal cells in the endosteal region compared to pHSCs, especially under 5FU stress.
To test the influence of myeloablative stress on different HSC niches, we performed an apoptosis assay and whole-mount staining at day 3 post-5FU stress. We found a 2.63-fold increase of apoptotic CD31+VE-cadherin+ cells, which was consistent with the dilated BV structure (Figures 2H and S2C). Disruption of vasculature after myeloablation has also been shown in previous reports (Dominici et al., 2009; Sugimura et al., 2012). We also observed a significant increase of apoptotic cells in other niche components in the perivascular region, including LepR-tdT cells (14.8-fold), Nestin-GFP cells (4.15-fold), Cxcl12-RFP cells (6.05-fold), and N-cad-TdT(M) (8.8-fold) (Figure 2H). However, apoptotic N-cad-TdT(B) cells showed no discernible increase post-5FU, and N-cad+ cells expanded in the endosteal region post-5FU (Figures 2H, S2C, and S2D).
We crossed the N-cad-CreERT mouse line with the tdTomato (R26-tdT) or ZsGreen (R26-ZsG) reporter mice (Figure S2E) and found an increase in proliferation of endosteal N-cad-derived cells (N-cad-CreERT(B)) post-5FU (3.35% ± 0.79% in homeostasis versus 5.73% ± 2.35% post-5FU) by bromodeoxyuridine (BrdU) incorporation. In contrast, CM N-cad-derived cells (N-cad-CreERT(M)) showed a 50% decrease in proliferation (Figure S3A). These data indicate that endosteal N-cad+ cells survived stress and expanded after chemotherapy (Figure S3B).
Overall, our data demonstrate that N-cad-TdT(B) endosteal stromal cells survive and expand, which in turn might contribute to rHSC protection under chemotherapeutic stress.
N-Cad+ Niche Cells Maintain Functional HSCs Including rHSCs in BM
Although N-cad+ stromal cells have been proposed as an HSC niche, direct evidence of the N-cad+ niche functionally supporting HSCs is still missing. To investigate the HSC niche role of N-cad+ cells, we generated N-cad-CreERT-induced DTR (encoding diphtheria toxin receptor) line (N-cad-CreERT; iDTR), in which N-cad+ cells were rendered sensitive to diphtheria toxin (DT) (Figure 3A). We observed efficient ablation of N-cad+ stromal cells in N-cad-CreERT; iDTR; R26-tdT treated with DT (Figure 3B). We next examined whether N-cad+ cell ablation affects HSCs in vivo. After ablating N-cad+ cells, we observed no significant change of cellularity in BM compared to the control (Figure 3C). However, the number of hematopoietic stem and progenitor cells (HSPCs) was dramatically reduced: rHSCs (65.0%), pHSCs (60.0%), ST-HSCs (59.6%), and MPPs (29.3%) (Figure 3D). This indicates that N-cad+ niche cells maintain the most primitive HSCs, including rHSCs. We further performed a transplantation assay and found that BM cells from N-cadCreERT; iDTR mice gave significantly lower levels of donor cell reconstitution (28.3% reduction at 20 weeks) (Figure 3E) with reduced myeloid cell production (24.5% to 17.1%) (Figure 3F). To further investigate whether N-cad+ niche cells contribute to maintaining the long-term self-renewal of HSCs, we conducted a secondary transplantation. We observed that BM cells from the N-cad-CreERT; iDTR mice had steeper reduction in donor cell reconstitution capacity in the secondary transplantation (40.5% reduction at 16 weeks) (Figure 3G), although they were capable of multilineage reconstitution (Figure 3H). Furthermore, we found that conditional knockout of Cxcl12 from N-cad+ stromal cells significantly reduced pHSCs (48%) and ST-HSCs (28.8%), but no significant reduction was observed in rHSCs. There was a slight, albeit insignificant, increase of MPPs (Figure S3C). Conditional deletion of SCF from N-cad+ stromal cells significantly reduced rHSCs (60%), pHSCs (38.6%), and ST-HSCs (53.7%) (Figure S3D). BM cells from N-cad-CreERT; Scf.f/f mice showed 60% decreased engraftment compared to control mice at 16 weeks post-transplantation of 5 × 104 BM cells (Figure S3E), though no significant reduction was observed when higher cell numbers were transplanted (data not shown). Notably, under 5FU stress, N-cad-CreERT; Scf.f/f mice had a more profound reduction of the HSC compartment, with 89.8% reduction in rHSCs, 90.5% reduction in pHSCs, 94.2% reduction in ST-HSCs, and 94.9% reduction in MPPs (Figure 3I). To further clarify the role of SCF from N-cad+ stromal cells in stressed condition, we quantified functional HSCs by limiting dilution (2.5 × 104, 5 × 104, 10 × 104) competitive repopulating unit (CRU) assays from 5FU-treated mice. We observed that functional HSCs dropped by 63% in N-cad-CreERT; Scf.f/f mice compared to the controls (Figure 3J). No significant lineage bias was detected in donor-derived cells (Figure 3K). These data indicate that SCF from N-cad+ stromal cells are critical for rHSCs maintenance especially under chemotherapeutic stress.
Figure 3. N-Cad+ Cells Maintain Functional HSCs in BM Niche.

(A) Protocol for DT injection to N-cad-CreERT; iDTR mice shown in (B)–(G). D, day.
(B) Efficiency of ablating N-cad+ cells.
(C and D) Absolute number of total nucleated cells (TNC) (C) and HSPCs (D) in the BM from N-cad-CreERT; iDTR mice post-TMX and DT injections (n = 5).
(E–H) Total engrafted donor cells measured in PB in 1° (E) and 2° (G) transplantation. Percentage of donor-derived B, T, and myeloid lineage cells in 1° (F) and 2° (H) transplantation measured at 16 weeks (n = 10).
(I) Absolute numbers of HSPCs in the BM from N-cad-CreERT; SCFf/f (n = 5) at 1 month post-TMX and 3 days after 5FU.
(J) CRU frequency determined using ELDA (extreme limiting dilution analysis).
(K) Percentage of donor-derived B, T, and myeloid lineage cells at 16 weeks post-transplantation (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars, SEM.
Overall, we provide functional evidence that N-cad+ niche cells preserve rHSCs under 5FU stress, partially by providing survival factors including SCF.
Transcriptome Analysis for Hematopoietic Cells and Their BM Niche Cells
To understand the molecular mechanisms governing how different niche cells contribute to the regulation of HSC subpopulations, we performed RNA sequencing on 4 sub-types of HSPCs during homeostasis and on rHSCs at day 3 post-5FU, as well as on 12 types of BM niche cells (Table S1).The BM niche cells were harvested from different niche zones of endosteum (B) and central BM (M) (Figures S4A and S4B). Our Pearson distance tree and principal-component analysis (PCA) data showed that rHSCs and pHSCs shared a unique transcriptome profile compared to ST-HSCs and MPPs (Figure 4A). Interestingly, rHSCs post-5FU appeared to be closer to pHSCs (Figure 4B), suggesting that surviving rHSCs are primed for activation to support subsequent hematopoietic regeneration post-chemotherapy.
Figure 4. Transcriptome Analysis for Hematopoietic Cells and Niche Cells.

(A–D) Pearson distance tree and PCA analysis for HSPCs (A and B) and niche cells (C and D).
(E) HSC signature gene expression in HSPCs.
(F) Niche signature gene expression in niche cells.
(G) GO term analysis for niche cells.
(H) Stromal cell development gene expression in niche cells from endosteal and perivascular zones.
(I) Differential expression of chemokine and growth factor receptors in rHSCs and 5FU-rHSCs. Fold change (FC) >2; false discovery rate (FDR) <0.05.
(J) Expression of selected cytokine, chemokine, and growth factors in endosteal N-cad+ cells in homeostasis and day 3 after 5FU from N-cad-CreERT; R26-tdT mice post-TMX induction.
We found that the rHSCs enriched most of the published HSC-specific markers, such as Slamf1 (CD150), H19, Ctnnal1 (α-catulin), Fgd5, Tek(Tie2), Procr (Epcr), Hoxb5, and Meg3 (Gtl2) (Qian et al., 2016; Chen et al., 2016; Sanjuan-Pla et al., 2013; Acar et al., 2015; Venkatraman et al., 2013). We also detected a higher percentage of EPCR+ rHSCs (48.8% ± 1.2%) compared to EPCR+ pHSCs (38.2% ± 0.8%) (Figure S4C). Progenitor signature genes such as CD34, CD48, Flt3 (Flk2), and Itga2 (CD49b) had lower expression in rHSCs (Gazit et al., 2014). Interestingly, rHSCs post-5FU upregulated expression of CXCR4, Ctnnal1 (α-catulin) (Park et al., 2002), Esam (Endothelial cell-Specific Adhesion Molecule), and CD150 (Slamf1), consistent with their known functions associated with the converted primed state (Figure 4E).
To confirm identity of different niche cells isolated, we examined signature genes in niche populations we isolated. We found that Pecam (CD31) and CDH5 (VE-cadherin) were enriched in Pecam-GFP+ endothelial cells. Pf4 and Adgre1 were enriched in MKs and macrophages (Macs) respectively. Cspg4 (NG2) was enriched in NG2-RFP cells. Nestin (Nes) was indeed expressed in endothelial (Pecam-GFP) and lowly expressed in Nestin-GFP cells as previously reported (Ding et al., 2012; Greenbaum et al., 2013). Interestingly, we found that several marker genes such as Kitl (SCF), LepR, Cdh2 (N-Cad), Cxcl12, and Pdgfrα were broadly expressed in perivascular niche cells (Figure 4F). N-cad-TdT(B) cells also had higher levels of SCF, Cxcl12, LepR, and Pdgfrα expression compared to other cells from the endosteal zone, such as Col2.3-GFP and NG2-RFP cells (Figure 4F). A PCA plot revealed that N-cad-TdT(B) cells positioned between N-cad-TdT(M) and Col2.3-GFP cells, while N-cadTdT(M) cells had a very similar transcriptome profile to other perivascular niche cells, including LepR, Cxcl12-RFP, and Nestin-GFP cells in both the Pearson distance tree (Figure 4C) and the PCA analysis (Figure 4D). These perivascular stromal cells are known to have mesenchymal stem cell (MSC) potential (Kfoury and Scadden, 2015). Indeed, N-cad+ stromal cells showed comparable clonogenic potential with other niche cells as measured by the colony forming unit-fibroblast (CFU-F) assay (Figure S5A). Consistent with an earlier study (Kunisaki et al., 2013), the Ng2-Cre-derived cells (Cspg4-Cre;R26-YFP) had a similar transcriptome profile to N-cad-TdT(M) cells and were predominantly in the perivascular region (Figures S4D–S4F). Interestingly, NG2-RFP cells from the NG2-DsR mouse line, which directly reports Ng2 expression, were predominantly restricted to the endosteum and had a very similar transcriptome profile to the Col2.3-GFP+ osteoblasts (Figures 4C, 4D, and S4G). In addition, Nestin-GFP cells isolated from the endosteal region had similar transcriptome profile to their marrow counterpart (Figure S4D).
Because N-cad+ cells had overlapped transcriptome profile to MSCs such as LepR+, Cxcl12-RFP, and Nestin-GFP cells, we next compared stromal development-related genes among niche cells (Figure 4H). We found that the Col2.3-GFP cells enriched osteoblast gene (Dmp1, Col1a1, Spp1, and Bglap) and progenitor cell gene Ly6a (SCA1) (Yang et al., 2014). Interestingly, both N-Cad-TdT(B) and N-Cad-TdT(M) enriched most of the MSPC genes such as Prrx1, Pdgfrβ, Pdgfrα, Sp7, Sox9, Gli, and Grem1 (Kfoury and Scadden, 2015). Grem1 was enriched in N-Cad-TdT(B) and all perivascular niche cells except NG2-RFP cells. Consistent with our previous observation that NG2-RFP cells mainly localized in the endosteum, they not only had higher levels of MSPC gene expression, such as Alcam, Sp7, Mcam, Sox9, and Gli1, but also enriched bone development genes (Dmp1, Bglap, Runx2, Col1a1) and chondrocyte-related genes (Col2a1 and Tnc) (Figure S4H). Nestin-GFP, Cxcl12-RFP, and LepR cells enriched MSPC genes including Prrx1, Pdgfrα, Itgb1, and Grem1. Among all perivascular niche cells, Cxcl12-RFP cells particularly enriched Itgav, and LepR+ cells greatly enriched Gla as well as several osteo-chondrocyte markers such as Atf4 and Tnc.
Interestingly, apart from expressing MSPC genes, N-Cad-TdT(M) and N-Cad-TdT(B) were enriched for chondrogenic (Sox9, Col2a1, and Tnc), adipogenic (Cebpα, Pparγ, Adipoq), and osteogenic (Col1a1, Runx2) genes, suggesting a tri-potential nature of N-cad+ stromal cells (Figures S4H and S4I). These results not only suggest the heterogeneity of the MSPCs in BM, but also provide evidence of molecular overlap of the stromal populations identified with different markers.
Our gene ontology (GO) term analysis showed that MKs, Macs, and Col2.3-GFP and Pecam-GFP cells highly enriched genes regulating RNA metabolism, protein metabolism, and translation activities (Figure 4G). This observation indicates that these cells are in a relatively activated and functional state and therefore are more differentiated, compared to N-cad-TdT cells and other perivascular zone cells, which are in a relatively lower metabolic state.
To investigate the role of N-cad+ cells in preserving rHSCs post-5FU stress, we compared the transcriptome of CM-derived (N-cad-CreERT(M)) and endosteal-derived N-cad+ (N-cad-CreERT(B)) cells at day 3 post-5FU (Figures S5B and S5C). Enriched GO terms in upregulated genes in N-cad-CreERT(B) cells included regulation of stress response, inflammatory response, and growth factors, suggesting the advantage of endosteal N-cad+ cells in withstanding chemotherapy. Indeed, we found that genes related with endochondral bone morphogenesis and osteoblast differentiation were enriched in N-cad-CreERT(B) cells, which is consistent with the expansion of N-cad+ cells near the bone surface post-5FU (Figures 2F, S2C, S2D, and S3A).
To understand how rHSCs are maintained by niche signals post-5FU, we analyzed the differentially expressed genes in rHSCs and 5FU-rHSCs (Figure 4I). We found that 40 receptors had more than 2-fold difference. Interestingly, within the 40 receptors, only 6 of them showed downregulation and 34 were upregulated in rHSCs under stress, including several chemokine receptors such as Cxcr2, Cxcr3, Cxcr4, Cxcr5, Cxcr6, and Cxcr8, indicating the microenvironment regulation of rHSCs post-5FU stress. To investigate how N-cad+ niche cells promote HSC regeneration after chemotherapy, we explored the differentially expressed genes in endosteal N-cad+ cells before and after 5FU (Figure 4J). Correspondent to chemokine receptors in rHSCs, we observed the increased expression of leukocyte chemoattractant genes such as Cxcl13 and Ccl24. These results indicate enhanced HSC mobilization for HSC recovery after 5FU. We further found that expression of genes promoting HSC survival and proliferation were elevated, such as Lif and Il23a that activate the Jak-Stat signaling cascade, stimulate interferon (IFN)-γ production, and regulate HSPC activity (Griseri et al., 2012; Baldridge et al., 2010), as well as Gdf15, which facilitates leukemia stem cell survival and proliferation under stress (Duan et al., 2014). Angpt4 expression was dramatically increased, which induces endothelial cell migration and potentially involves recovery of the vascularized endosteal niche (Lee et al., 2004). We also observed a slight increase of HSC maintenance genes, including Fgf21 and Igf2 (Yamauchi et al., 2006; Venkatraman et al., 2013). Intriguingly, we detected increased expression of the gene Trp53cor1 (linc-p21), which regulates the cycle-inhibitory gene p21cip1/waf1 and suppresses JunB (Cheng et al., 2000; Dimitrova et al., 2014). On the other hand, we found significantly downregulated chemotactic signal genes, including Ccl3, Ccl4, Ccl19, Cxcl2, and Cxcl5, suggesting these chemokines were not preferred by endosteal N-cad+ cells to help the stress resistance of HSCs. The expression of Dkkl1, an antagonist of the HSC-promoting Wnt/β-catenin signal (Fleming et al., 2008), was decreased in endosteal N-cad+ cells post-5FU, which might be a requisite for the activation of Wnt signaling for both HSC recovery and osteogenesis (Krishnan et al., 2006). The transcriptomic changes in both the rHSCs and endosteal N-cad+ cells reveal potential mechanisms for N-cad+ niche expansion prior to activation of rHSCs after acute stress.
To confirm the molecular overlap of N-cad+ stromal cells with multiple niche stromal cells, we performed in vivo lineage tracing and found that N-cad+ stromal cells gave rise to both Col2.3-GFP+ osteoblastic cells and perivascular cells (Figure S5D). More specifically, we observed that N-cad-CreERT lineage traced cells partially overlapped with Cxcl12-RFP and Nestin-GFP cells (Figure S5E). Moreover, N-cad-CreERT-derived cells were 98.3% and 97.9% positive for LepR and Pdgfrα expression, respectively (Figure S5F). These results further demonstrate that N-cad+ stromal cells represent a BMSPC population as suggested by the transcriptome analysis. The term BMSPC is used hereafter to avoid confusion with “MSC” (Sipp et al., 2018; Chan et al., 2018).
N-Cad+ Stromal Cells Give Rise to Osteoblasts, Chondrocytes, and Adipocytes In Vitro and In Vivo
To test the progenitor cell potential of N-cad+ stromal cells, we performed in vitro differentiation assay with N-cad-CreERT; R26-tdT mice and found that most CFU-F colonies were Tomato+(Tom+) (Figures 5A and 5B), which suggested that N-cad+ cells are the main source of BMSPCs with CFU-F activity. We found that Tom+ cells gave rise to osteoblasts (Alkaline phosphate+ [AP+]), adipocytes (Oil-red-O+), and chondrocytes (Aggrecan+ and Toluidine-blue+) in an in vitro differentiation assay (Figures 5C–5E).
Figure 5. In Vitro Differentiation and Localization of N-Cad+ Stromal Cells.

(A) Protocol.
(B) CFU-F colonies stained with Cell Trace with Tom+ cells in high magnification.
(C–E) In vitro differentiation culture of stromal cells from N-cad-CreERT; R26-tdT mice at day 21: (C) live Tom+ cells and AP staining; (D) live Tom+ cells and oil red O lipid staining; (E) Aggrecan and toluidine-blue-stained chondrocytes.
(F) Protocol for TMX induction.
(G) Diagram of TB and CB in femur.
(H) Tom+ cells (arrows) at indicated time post TMX. Black background area is outside the microscope tiling regions in stitched composite images. Scale bar, 50μm.
(I) Percentage of Tom+ cells in TB, CB, and CM at indicated time post-TMX (n = 2 in 1- and 2-week groups, n = 3 in 4- and 6-week groups).
*p < 0.05, **p < 0.01, ***p < 0.001. Error bars, SEM.
To characterize in vivo function of N-cad+ stromal cells, we first analyzed the dynamic anatomical distribution of N-cad-derived cells (Figures 5F and 5G). Interestingly, we detected Tom+ cells in metaphysis as early as 6 h post-Tamoxifen (TMX), but only very few cells in CM, suggesting that N-cad+ cells originate from the endosteal region (Figure 5H). We also found that the number of Tom+ cells increased by 2.2- ± 0.2-fold in TB from 1 to 2 weeks post-TMX and remained stable afterward; in compact bone (CB), Tom+ cells increased by 2.95- ± 0.2-fold at 2 weeks but declined at 6 weeks post-TMX, suggesting that N-cad+ cells in endosteal region remain quiescent after initial expansion. Moreover, Tom+ cells continuously increased in CM up to 6 weeks, suggesting the proliferating and differentiating of N-cad-derived cells in CM (Figure 5I).
Next, we performed an in vivo lineage tracing and observed that N-cad+ cells generated Col2.3-GFP+ osteoblasts in a time-dependent manner (Figures 6A, S6A and S6B). N-cad-derived osteoblasts were detected in the peri-trabecular region as early as 6 h post-TMX (Figure 6B) and in the CB at 14 h post-TMX (Figure 6C). Further analysis showed that at 6 h post-TMX, immature Tom+ Col2.3− cells (1.1% ± 0.2%) were 10 times more enriched in TB compared to CB (0.11% ± 0.04%) (Figures 6D and 6E). Consistently, more immature Tom+Col2.3− cells were enriched in TB than CB at 14 h and 2 weeks post-TMX (Figure 6F). At 4 weeks post-TMX, a large portion of Col2.3-GFP cells (72% ± 6%) were derived from N-cad+ cells (Figure 6G).
Figure 6. N-Cad+ Stromal Cells Give Rise to Osteoblasts and Adipocytes in Adult Mice.

(A) Representative images of N-cad-CreERT; R26-tdT; Col2.3-GFP mice at indicated time post-TMX showing Tom+Col2.3-GFP+ osteoblasts. Scale bar, 100 μm.
(B and C) High-power images of N-cad-CreERT; R26-tdT; Col2.3-GFP mice shown at 6 (B) and 14 h (C) post-TMX. Hollow arrows denote Tom+Col2.3GFP+ osteoblasts (yellow); solid arrows indicate that N-cad recombined in TB (i, ii, iv, v) and CB (iii, vi) (red) and cells potentially undifferentiated (Col2.3-GFP−). Scale bar, 50 μm.
(D and E) Image quantification of percentage of Tom+Col2.3-GFP+ and Tom+Col2.3-GFP− cells in TB (D) and CB (E) at indicated time post-TMX (n = 2).
(F) Image quantification of potentially undifferentiated Tom+Col2.3-GFP− cells in TB and CB (n = 2).
(G) Representative images of TB and CB in N-cad-CreERT; R26-tdT; Col2.3-GFP mice at 4 weeks post-TMX. Scale bar, 20 μm.
(H) Representative images of femur section in N-cad-CreERT; R26-tdT mice with Perilipin staining in periosteal region (i), TB (ii), and CM (iii) (arrows) at 4 weeks post-TMX.
(I) BODIPY staining showed the adiposome (green) inside the Tom+ Perilipin+ adipocyte (arrows). Scale bar, 20 μm.
(J and K) Image quantification of Tom+Perilipin+ adipocytes in TB (J) and periosteal region (K) at 6, 14, and 24 h and 2 and 4 weeks post-TMX (n = 2). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars, SEM.
We further observed that at 4 weeks post-TMX, N-cad+ stromal cells generated adipocytes in the CM and TB, particularly near the periosteal region (Figure 6H), and this was further confirmed by BODIPY lipid probe staining (Figure 6I). Interestingly, the frequency of N-cad-derived adipocytes increased quickly initially and declined later, as evidenced by the quantification that 39.3% ± 4.5%, 77.1% ± 6.7%, and 17.2% ± 3% of N-cad-derived cells were observed at 6, 14, and 24 h, respectively, after TMX induction (Figures 6J, 6K, and S6C–S6E), suggesting that adipocytes may have a frequent turnover rate. Collectively, our data demonstrate that N-cad+ stromal cells generate both osteoblast and adipocyte lineages in vivo during homeostasis in adult mice.
N-Cad+ Stromal Cells Give Rise to Chondrocytes during Development and Post-Injury
Chondrogenesis is active in fetal development and declines in adulthood (Sophia Fox et al., 2009; Raghunath et al., 2005). In mice with TMX induction at postnatal day 2 (P2), we detected no Tom signal in Aggrecan+ chondrocytes in the femur (Figure S7A), despite the Tom+ osteoblasts and perichondrocytes adjacent to the growth plate. We next started TMX induction in N-cad-CreERT; R26-tdT mice at embryonic day 12.5 (E12.5), when mesenchymal cells undergo condensation and chondrocyte differentiation in the fetal bone (Figure 7A). We found that the majority of undifferentiated Tom+ cells were located peripherally, with differentiated Tom+ chondrocytes located in the center of the rib at E14.5 (Figure 7B). Importantly, in TB at P2, we detected Tom+ chondrocytes in the columnar zone, where the secondary ossification center forms, and in the superficial zone, which is imperative to resist the sheer force for deeper layer protection (Figures 7C and 7D). Tom+ adipocytes were detected at 2 and 10 months after birth (Figures 7E and S7B), suggesting that N-cad + cells contribute to early and long-term adipogenesis during development. N-cad-derived Osteopontin+ cells were detected at 2 months after birth, but not at P2 (Figures 7F and S7C). These results demonstrate that embryonic N-cad-derived cells give rise to all three lineages. Particularly, N-cad+ cells efficiently formed chondrocytes from the embryonic stage, suggesting the primitive nature of N-cad+ BMSPCs.
Figure 7. N-Cad+ BMSPCs Give Rise to Chondrocytes during Development and Post-Injury.

(A) Protocol.
(B) Tom+ Aggrecan+ chondrocytes in rib at E14.5.
(C) Chondrocyte development in femur.
(D) Representative images of femur from 2-day-old N-cad-CreERT; R26-tdT mice with E12.5 TMX induction. Note the Tom+Aggrecan+ chondrocytes (arrows) in the articular surface (i) and the developing secondary ossification center (ii).
(E and F) Representative images of femur from 10- and 2-month-old N-cad-CreERT; R26-tdT mice with E12.5 TMX induction. N-cad+ cells differentiated to Perilipin+ adipocytes (E), Osteopontin+ hypertrophic chondrocytes (F) (i and ii), and osteoblasts (iii, iv, v, and vi).
(G) Protocol for femoral groove injury.
(H) Quantification of Tom+ chondrocytes in control and 3 weeks after injury.
(I) Representative images of distal femur from N-cad-CreERT; R26-tdT mice. Note the clustered Tom+ Aggrecan+ cells (arrows) at the knee surface of control (i) and 3 weeks after injury (ii).
(J) Alcian blue-hematoxylin-Orange G staining showing the Alcian blue+ chondrocytes in control (i) and at the injury site (ii).
We next investigated whether N-cad+ cells generate chondrocytes in adult mice post-injury. We surgically created a full thickness cartilage defect in the patella groove of the lower femur in N-cad-CreERT; R26-tdT mice that received TMX at E12.5 (Figure 7G). The knee joint was swollen due to articular cartilage damage in post-surgery mice (Figure S7D). At 2–3 weeks post-cartilage damage, we found Tom+ chondrocytes clustered in the callus at the damage site with a 2-fold increase compared to the control leg (Figures 7H and 7I). Alcian blue+ cells with a typical chondrocyte feature were dramatically increased in the injured region (Figure 7J), indicating that N-cad+ BMSPCs effectively differentiated into chondrocytes in response to injury.
Taken together, our data prove that N-cad-derived cells induced at early bone developmental stage (E12.5) can give rise to chondrocytes in adults and support cartilage regeneration in response to injury.
DISCUSSION
Reserve and pHSCs
As the most rigorously studied adult stem cell system, HSC heterogeneity has been widely explored. Some HSCs are maintained in an active cycling state while others are maintained in a quiescent state. The quiescence of HSCs has been shown to be functionally related with their long-term self-renewal capacity (Foudi et al., 2008; Wilson et al., 2008; Bernitz et al., 2016). HSC quiescence also correlates with their low metabolic activity, termed HSC dormancy or hibernation (Yamazaki et al., 2011; Takubo et al., 2013; Qian et al., 2016; Venkatraman et al., 2013). Paradoxically, despite their quiescence, the majority of HSCs cannot survive chemotherapeutic stress such as 5FU (Longley et al., 2003). In our study, we identify a small HSC subpopulation, rHSCs, that can survive 5FU stress in primary mice and after transplantation. In contrast, the other quiescent HSC subpopulation, pHSCs, which represent the majority of HSC pool, is chemotherapy sensitive. Most likely pHSCs reflect a transitional state between rHSCs and proliferating ST-HSCs (Li and Clevers, 2010). Though both rHSCs and pHSCs support long-term hematopoiesis, rHSCs not only survive chemotherapy, but also give rise to pHSCs, whereas pHSCs rarely give rise to rHSCs, in the transplantation model.
The survival of rHSCs from 5FU stress depends on both intrinsic and extrinsic properties. Intrinsically, despite an attenuated DNA repair system during homeostasis, rHSCs can quickly activate their DNA repair pathways and stress-response program, restore the HSC pool, and support subsequent hematopoietic regeneration. Additionally, rHSCs upregulate several CXC chemokine receptors and HSC-activation genes, such as Clec2c (CD69) and Bst2 (CD317) (Table S1) (Bujanover et al., 2018), for receiving signals from niche for their temporal activation, survival, mobilization, and expansion in response to stress. The pHSCs that account for the bulk of the HSC population reside predominantly in the perivascular and sinusoidal regions (Acar et al., 2015; Chen et al., 2016; Zhao et al., 2014; Bruns et al., 2014). Expression of CD49b (Integrin a2) in pHSCs might facilitate their interaction with BVs, which have high laminin expression, but the integrinα2-laminin interaction in BVs might be disrupted under 5FU stress (Yousif et al., 2013).
N-Cad+ Cells in the Endosteal Niche Preserve rHSCs
The extrinsic property of rHSCs to survive and/or to repopulate is dependent on their proximity to niche cells. The rHSCs in CM are more sensitive to 5FU-induced apoptosis than rHSCs in endosteum (3.5-fold increase) (Figure S1A). This can be partially explained by our observations that N-cad+ cells in endosteum provided a protective niche for rHSCs. This is consistent with the observations that deeply quiescent (H2B-GFP LRCs or BrdU-LRCs) HSCs were frequently detected in the endosteal region and even directly attached to the N-Cad+ bone lining cells located in the TB region (Zhang et al., 2003; Wilson et al., 2008). Of note, after transplantation, most of the HSCs lodged and homed to the TB area, and some HSCs were directly adjacent to N-cad+ cells (Xie et al., 2009; Lo Celso et al., 2009; Nilsson et al., 2001). Our whole-mount HSC staining was done in the sternum, which has abundant bone branches and extensions inside the marrow, similar to the TB region in the femur. A higher frequency of rHSCs in the marrow of sternum was observed compared to the CM of femur, suggesting that the TB structure preferentially hosts rHSCs (Figure S1C). Using two reporter mouse lines, we found that N-cad+ stromal cells were distributed in both the endosteal and perivascular regions and supported HSC maintenance, explaining why ablation of N-cad+ cells eliminated both pHSCs and rHSCs. Interestingly, N-cad+ BMSPCs appeared in the TB region as early as 6 h post-TMX, which explains our observation that N-cad+ cells were abundant in the TZ vessels, especially post-5FU. The TZ endothelial cells typically localized in the TB and mediated growth of vasculature and perivascular osteoprogenitors (Kusumbe et al., 2014). This observation suggests the role of N-cad+ cells in restoring the vascularized endosteal niche prior to HSC recovery. Indeed, we found that BV development and osteoblast differentiation-related genes were upregulated in N-cad+ cells after 5FU.
We previously reported that N-cad+ cells were osteoblastic progenitor cells, which support HSCs (Zhang et al., 2003). This observation was confirmed by subsequent studies (Arai et al., 2004; Sugiyama et al., 2006; Xie et al., 2009). We also showed that N-cad+ cells in the endosteal region were resistant, whereas Osx+ osteoblasts were sensitive, to chemotherapy (Sugimura et al., 2012). In this study, we compared the response of various niche cells, including N-cad+ cells, in CM perivascular and endosteal regions. We found that the BVs and perivascular cells, including N-cad-TdT(M), were profoundly disrupted, accounting for a large loss of pHSCs. However, endosteal N-cad+ cells survived and expanded, corresponding to the location where rHSCs were substantially enriched after chemotherapy.
Identity of N-Cad Stromal Cell and Its Role in HSC Maintenance
Although N-cad was the first niche marker identified in BM, the nature of N-cad+ stromal cells remains unclear. In past years, a debate has arisen regarding whether the endosteal region functions as an HSC niche, largely due to using Col2.3-GFP or Col2.3-Cre mouse models, which revealed that Col2.3-marked mature osteoblasts were dispensable for HSC maintenance (Ding and Morrison, 2013; Ding et al., 2012; Greenbaum et al., 2013).
In this study, we used molecular profiling, lineage tracing, and injury modeling to show that N-Cad+ stromal cells enrich immature BMSPCs with tri-lineage differentiation potential in vitro and in vivo. Earlier studies have shown that endochondral ossification is required for HSC niche formation and precedes the appearance of HSCs in the BM (Chan et al., 2009). Transient expansion of N-cad+ endosteal niche is required for HSC recovery after radio-myeloablation (Dominici et al., 2009). In this context, we found that N-cad+ cells not only gave rise to chondrocytes in fetal development, but also recapitulated endochondral ossification in the injured femur. These data further underscore the importance of N-cad+ BMSPCs in the development of HSC niches. Furthermore, N-cad+ stromal cells molecularly overlapped with several recently reported MSC types including LepR+, Nestin+, and CAR cells and expressed genes including Grem1, Gli, and Prrx1 enriching in MSCs (Crane et al., 2017). Our data strongly indicate that the long-standing controversy of HSC niche cell types may likely be due to different cell markers being used rather than to their cellular identities.
Here, we showed that N-cad marked BMSPCs developmentally give rise to osteoblasts (Col2.3+) and demonstrated that N-cad+ BMSPCs are essential for rHSC maintenance by depleting N-cad+ cells followed by BM transplantation. In addition, genetic deletion of Cxcl12 or SCF from N-cad+ cells affected HSC numbers. Particularly, SCF deletion from N-cad+ cells under stress led to a dramatic decrease of rHSCs and functional HSC number, indicating that N-cad+ stromal cells maintain HSCs by providing SCF, especially under chemotherapeutic stress. Furthermore, endosteal N-cad+ cells that survived 5FU stress selectively upregulated several genes related to HSC survival and proliferation, such as Gdf15 and Trp53cor1(lincp-21) genes. Gdf15 is required to mediate niche protection for drug resistance (Duan et al., 2014), suggesting a similar role of Gdf15 in the N-cad+ niche to protect the chemo-resistant rHSCs. Linc-p21 not only activates the gene p21cip1/waf1, which governs HSC quiescence and regulates apoptosis by cell-cycle arrest in response to stress (Gartel and Tyner, 2002), but also inhibits JunB expression, deletion of which favors LT-HSC expansion (Yoon et al., 2012; Passegué et al., 2004; Santaguida et al., 2009). Our RNA sequencing (RNA-seq) data also revealed that endosteal N-cad+ cells upregulated a series of stress and inflammatory response genes (Ccl11, Tak1, and Metrnl) (Rao et al., 2014; Lee et al., 2017; Kindstedt et al., 2017) as well as bone and vessel development genes (Mef2c, Efnb2, and Angpt4) (Foo et al., 2006; Arnold et al., 2007) to overcome the 5FU stress. Our observation is consistent with a previous report that, following chemotherapy or irradiation of mice, transient and reversible expansion of N-cad+ bone lining cells occurred prior to HSC restoration (Dominici et al., 2009).
In summary, we have shown that quiescence is not the only mechanism underlying drug resistance; instead, the intrinsic DNA-damage repair, stress-response program, and the extrinsic niche protection are all involved in rHSC protection under chemotherapeutic stress. In addition, we have demonstrated that N-cad+ stromal cells are functional BMSPCs, which protect primitive rHSCs from chemotherapeutic stress, facilitate restoration of the HSC pool, and support hematopoietic regeneration post-myeloablation.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACTONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Linheng Li (lil@stowers.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice used in this study were housed in the animal facility at the Stowers Institute for Medical Research (SIMR) and were handled according to SIMR and National Institutes of Health (NIH) guidelines. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the SIMR. C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J(iDTR), B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J(R26R-tdT), B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J(R26R-ZsG), B6.129X1t(ROSA)26Sortm1(EYFP)Cos/J, B6.Cg-Tg(Col1a1*2.3-GFP)1Rowe/J, Tg(Cspg4-DsRed.T1)1Akik/J, B6;FVB-Tg(Cspg4-cre)1Akik/J, B6.129(Cg)-Leprtm2(cre)Rck/J, Cxcl12tm2.1Sjm/J, Kitltm2.1Sjm/J (SCFf/f), and Cxcl12tm1.1Sjm/J (CXCL12f/f) mice were obtained from The Jackson Laboratory. Pecam-GFP mice were generated in SIMR with blastocyst injection of Pecam-GFP ES cells (gift from Victoria L. Bautch’s lab (Kearney et al., 2004)). The Nestin-GFP transgenic mouse line was previously described (Mignone et al., 2004). N-cad-CreERT, and N-cad-TdT mice were generated by Applied StemCell, Inc. The mCherry reporter in N-cad-CreERT and the Cre recombinase in N-cad-TdT are inactive (Refer to Figure S2). To induce N-cad-CreERT; R26-tdT mouse, TMX (Sigma) was injected intraperitoneally at 2mg per injection for 3 days. To induce N-cad-CreERT; R26-tdT at embryonic stage, a single dose of 1.5mg of TMX was injected intraperitoneally into the E12.5 pregnant dam. Cesarean section was performed at E19.5, and the neonatal mice were transferred to foster mice. To induce N-Cad+ cells ablation in N-cad-CreERT; iDTR mice, DT (Sigma) was injected intraperitoneally every other day at a dose of 50ng per gram body weight as indicated. 5FU (Sigma-Aldrich) was injected once in the tail vein at 150 μg per gram body weight. After 5FU injection, mice were analyzed as described in the text. To label Bromodeoxyuridine+ (BrdU+) proliferating cells, BrdU was injected intraperitoneally b.i.d. at a dose of 100mg per kg body weight. For analysis of endothelial cells, mice were intravenously injected with 10μg Alexa Fluor 647 conjugated anti-VE-cadherin antibody (BV13, eBioscience) per mouse. All mouse strains used in this study had a C57BL/6J genetic background. Animals were randomly included in the experiments according to genotyping results. Animal experiments were conducted in a blinded fashion with respect to the investigator. The numbers of animals used per experiment are stated in the Figure Legends.
METHOD DETAILS
BM and Bone Stromal Cell Isolation
To prepare BM stromal cell suspensions, the BM plug was gently flushed using PBS with 2% heat-inactivated bovine serum and digested with 1ml pre-warmed digestion solution (200U/ml DNase I (Sigma), 250μg/ml LiberaseDL (Roche) in HBSS plus Ca2+ and Mg2+) at 37°C for 20 min with gentle shaking. For bone stromal cell isolation, muscle and ligament attached were cleaned. Bone was cut into small pieces using a scissor in PBS with 2% heat-inactivated bovine serum and digested in bone digestion buffer (1.5mg/ml collagenase I dissolved in 8ml Dispase (5U/ml, STEMCELL Technologies) and 2ml HBSS with Ca2+ and Mg2+).
Antibodies and Flow Cytometry
For phenotype analysis, hematopoietic cells were harvested from BM (femur and tibia). Red blood cells were lysed using 0.16M ammonium chloride solution. For cell surface phenotyping, LT-HSCs were CD34−Flk2−Lineage−Sca-1+c-Kit+ (LSK); ST-HSCs were CD34+FLK2−LSK); and MPPS were CD34+FLK2+LSK. A lineage cocktail (Lin, phycoerythrin (PE)-Cy5) was used, including anti-CD3 (145–2C11), anti-CD4 (RM4–5), anti-CD8 (53–6.7), anti-Mac-1 (M1/70), anti-Gr1 (RB6–8C5), anti-B220 (RA3–6B2), anti-IgM (II/41) and anti-TER119 (TER-119) (100ng antibody cocktail per million BM cells, eBioscience). Monoclonal antibodies to SCA1 (D7, eBioscience), c-KIT (2B8, eBioscience), FLK2 (A2F10, eBioscience), CD34 (RAM34, eBioscience), CD48 (HM48–1, eBioscience), CD150 (TC15–12F12.2, BioLegend) and CD49b (HMα2, BioLegend) (all used as 50ng per million BM cells) were also used where indicated. For lineage analysis of peripheral blood, monoclonal antibodies to CD45.1 (A20, eBioscience), CD45.2 (104, eBioscience), CD3, B220, Mac-1 and Gr1 were used. 7-aminoactinomycin D (7-AAD) (A1310, Life technologies) was used to exclude dead cells. For stromal niche cell analysis, anti-CD45 (30-F11, eBioscience), anti-CD31 (390, eBioscience), anti-PDGFRα-biotin (APA5, eBioscience), anti-LepR-Bio (R&D), anti-CD51 (clone RM7-V, BioLegend) were used. Samples stained with biotin conjugated antibodies were washed with staining medium, then incubated with streptavidin brilliant violet 421™ (BioLegend, 1:500). For whole mount immunostaining, the following antibodies were use: anti-IgM (RMM-1, BioLegend), anti-Ter119 (TER-119, BioLegend), anti-Gr1 (RB6–8C5, BioLegend), anti-CD3 (17A2, BioLegend), anti-CD4 (RM4–5, eBioscience), anti-CD8a (53–6.7, eBioscience), anti-B220 (RA3–6B2, eBioscience), anti-CD11b (M1/70, BioLegend), anti-CD150 (TC15–12F12.2, BioLegend), anti-CD150 (mShad150, eBioscience), anti-49b (HM-α2, eBioscience), anti-49b (HM-α2, BioLegend), anti-CD41(MWReg30, eBioscience), anti-CD48 (HM48–1, eBioscience), anti-Endomucin (V.7C7.1) and Streptavidin Alexa Fluor 405 conjugate (Invitrogen). For apoptosis analysis, Pacific Blue Annexin V (640918, BioLegend) was used. For BrdU analysis, APC BrdU Flow Kit (51–9000019BK BD-PharMingen) was used. Cell sorting and analysis were performed using MoFlo (Dako), InFlux Cell Sorter (BD Biosciences), MACSQuant (Miltenyi Biotec), ZE5 Cell Analyzer (BioRad) or CyAn ADP (Dako) instruments. Data analysis was performed using FlowJo software.
Whole-mount Sternum HSC Immunostaining
Sternal bones were collected and transected with a surgical blade into 3–4 fragments. The fragments were bisected sagittally to expose BM cavity, fixed in 4% PFA, blocked/permeabilized in PBS containing 20% normal goat serum and 0.5% Triton X-100, and stained with primary antibodies for 3 days. The tissues were incubated with secondary antibodies for 2 hours (Bruns et al., 2014; Kunisaki et al., 2013). Fluorescence imaging was performed on a spinning-disk confocal microscope (UltraVIEW; PerkinElmer), including an inverted microscope (Axiovert 200 M; Carl Zeiss Microimaging, Jena, Germany), attached to a spinning-disk confocal system (CSU-X1; Yokogawa Corporation of America) and Orca-R2 camera (Hamamatsu) with Volocity acquisition software (PerkinElmer) and a 20x/0.8 Plan-Apochromat objective (Carl Zeiss). Images were collected as a series of optical sections, with a step size of 4μm. Images were collected in a tile pattern (overlap 10%) sufficient to cover the entire sample. Channels were collected sequentially. DAPI and Pacific Blue were excited using 405nm light (50mW diode laser, OEM), and PE was excited using 561nm light (50mW solid state laser, OEM), and each was collected using a multibandpass emission filter with 415nm-775nm and 580nm-650nm bands. FITC and ZsG were excited using 488nm light (50mW solid state laser, OEM) and collected using a multibandpass emission filter of 500nm-550nm, and APC was excited using 640nm light (50mW solid state laser, OEM) and collected using a multibandpass emission filter with 455nm-515nm and 660nm-750nm. Exposure times and laser powers were adjusted to compensate for staining variations. SHG imaging was performed immediately following fluorescence imaging. SHG images were collected on an LSM-780 laser scanning confocal microscope (Carl Zeiss) equipped with a QUASAR detection unit, a 10× 0.45 Plan-Apochromat objective (Carl Zeiss), and Zen 2012 acquisition software (v8.1.3, Carl Zeiss). Images were collected as a series of optical sections, with a step size of 8μm and a pixel size (1.32μm/pixel) an integer multiple of four times the fluorescence pixel size. SHG images were taken with a laser light wavelength of 900nm and collected at 371–420nm. As a reference for image alignment, images of CD150-PE were taken using the 561nm line of a DPSS laser (Melles Griot) and collected at 566–735nm concurrently with the SHG images. Images were collected in a tile pattern (no overlap) sufficient to cover the entire sample. Tiles were stitched into a complete 3D image using Zen software.
Images were analyzed using Fiji software (1.51 g National Institutes of Health). To align SHG and fluorescence images, fluorescence images were first background subtracted, and image tiles were stitched into a complete 3D image using the Grid/Collection stitching plugin (reference https://academic.oup.com/bioinformatics/article-abstract/25/11/1463/332497). Given that the transfer of the sample from the fluorescence microscope to the SHG microscope involved a small amount of sample rotation, it was necessary to realign the SHG and fluorescence images in 3 dimensions. Alignment of SHG and fluorescence image sets was carried out using a custom plugin available at http://research.stowers.org/imagejplugins. First, a minimum of 8 common landmarks were identified by visual inspection of both the SHG and fluorescence datasets. Next the Kabsch algorithm was used to find the best scaled rotation to transform the fluorescence coordinates to the SHG image coordinates. Finally, each 3D voxel in the fluorescence image was transformed to the corresponding SHG position, and trilinear interpolation was used the find the SHG intensity at that position to create the realigned composite image.
Image analysis was conducted by researchers unfamiliar with the hypotheses of the study. HSCs were identified by eye. Cells were considered negative for staining if the shape of the cell could not be discerned by eye, or if the shape of the cell formed a dark region in a field of positive signal. Distance measurements to niche components were made using Fiji and Microsoft Excel software. The locations of the HSC and nearest point of each of the three niche components were marked using point ROIs in Fiji, and locations were transferred to excel, where distances in 3D were then calculated using the 3D Pythagorean Theorem. To calculate distances for randomly distributed HSCs, random point ROIs were generated using a custom plugin in FIJI in the same images analyzed for observed HSCs. The randomly generated points were considered simulated HSCs if they appeared in a reasonable HSC location (as assessed by the presence of Lin+ cells in the surrounding regions). Niche distance measurements for simulated HSCs were then made in the same manner as for observed HSCs.
The statistical significance of differences in the distribution of distances was assessed by Kolmogorov-Smirnov analysis using Origin software. Statistical significance of changes in percentages of HSCs at 5μm were assessed using a Student’s t test in Microsoft Excel. Changes were considered significant if p < 0.05.
Final images shown in figures are maximum projections that have been background subtracted and contrast adjusted for clarity.
Transplantation and Repopulation Assay
One hundred sorted pHSCs or rHSC cells together with 1 × 105 CD45.1 rescue BM cells were transplanted into lethally irradiated (10 Gy) CD45.1 recipients. 2 × 105 CD45.2 BM cells from N-cad-CreERT; iDTR and control mice together with 2 × 105 CD45.1 rescue BM cells were transplanted into lethally irradiated (10 Gy) CD45.1 recipients. Every 4 weeks post transplantation, PB was collected from the submandibular vein. Hematopoietic repopulation was measured from donor-derived blood cells (CD45.2). The in vivo competitive repopulation unit (CRU) assay (Szilvassy et al., 1990) was performed by transplantation of 25,000, 50,000 and 100,000 total BM cells from control or N-cad-CreERT induced SCF KO mice with a fixed number of 2 × 105 CD45.1 rescue cells into groups of ten lethally irradiated (10 Gy) recipient mice (CD45.1). CRU frequency was measured using ELDA (Extreme Limiting Dilution Assay) software (Hu and Smyth, 2009), in which successful engraftment was defined as the presence of ≥ 1% CD45.2+ CD45.1− population. For 2° transplantation, the original, primary transplant recipients were sacrificed; BM was harvested from the femur and then transplanted mouse-to-mouse at a dosage of 1 × 106 cells per mouse.
RNA-Seq and Analysis
cDNA was generated from 1000 purified cells using SMARTer Ultra Low Input RNA kit (Clonetech), and library was generated by the Nextera XT DNA Library Preparation Kit (Illumina), followed by sequencing on an Illumina HiSeq2500 for 50bp single reads. Raw reads were demultiplexed into Fastq format allowing up to one mismatch using Illumina bcl2fastq2 v2.18. Reads were aligned to UCSC genome mm10 with TopHat v2.0.13, default parameters. FPKM values were generated using Cufflinks v2.2.1 with “-u –max-bundle-frags 100000000.” Read counts were generated using HTSeq-count with “-m intersection-nonempty.” Three to four replicates were sequenced for each population.
CFU-F Assay and In Vitro Differentiation
Cells were sorted directly into culture at a 96-well plate. The cultures were incubated at 37°C in a humidified atmosphere with 5% O2 and 10% CO2 for 7–10 days. Colonies were stained by CellTracker™ Green CMFDA (Life Technologies), and image was acquired by Celigo Imaging Cytometer (Nexcelom). For in vitro differentiation, clonally expanded Ncad-CreERT driven Tomato+ BM and bone stromal cells were isolated from CFU-F cultures by digesting with 0.25% Trypsin/EDTA, split into 3 aliquots, and seeded into separate cultures permissive for differentiation: StemPro Osteogenesis kit GIBCO A10072–01, Adipogenesis kit A10070–01 and Chondrogenesis differentiation kit A10071–01. The osteoblastic differentiation was assessed by VECTOR Red Alkaline Phosphatase; the adipogenic differentiation was detected by Oil Red O (Sigma); and the chondrogenic differentiation was detected by Toluidine blue (Sigma, 0.1g T Blue/100mL distilled water).
Bone Sectioning, Immunostaining and Imaging
Freshly isolated femurs were fixed in 4% paraformaldehyde overnight, followed by 1 to 3 days decalcification in 10% EDTA. For paraffin section, bone samples were processed with Sakura Tissue Tek VIP 5 Tissue Processor (Sakura America, Torrance, CA), and paraffin sections were cut in 5um thickness. Sections were deparaffinized with xylene, followed by Alcian Blue-Hematoxylin-Orange G staining. For frozen section, bone samples were processed with the CryoJane tape-transfer system. Sections were blocked with Power Block™ Universal Blocking Reagent for 30 min to 1 hr and then stained overnight with anti-BrdU (GE Healthcare RPN202 1: 100), rabbit-anti-Aggrecan (Millipore, 1:300), rabbit-anti-Perilipin (Cell Signaling, 1:300) and goat-anti-Osteopontin (R&D, 1:300). Donkey-anti-goat Alexa Fluor 488 and Donkey-anti-goat Alexa Fluor 647 were used as secondary antibodies (all from Invitrogen, 1:300). Antibodies were diluted with Antibody Diluent Solution (Invitrogen 00–3218). Slides were mounted with FLUORO-GEL (Electron Microscopy Science 1798510), and images were acquired with an Olympus slide scanner.
Femoral Groove Surgery
Mice were anesthetized with 2.5% isoflurane, and buprenorphine was administered for analgesia. The skin of the right leg was shaved and scrubbed with alcohol and iodine. A small incision was made in the skin, lateral to the knee joint. After sliding the skin medially for visualization, an internal incision was made medial to the patella, extending into the quadriceps muscle and along the patella tendon to release the tissue. The patella was subluxated laterally, and the distal femur was exposed. The subchondral bone was perforated using a microsaw to penetrate the articular cartilage at the knee joint. The extensor mechanism (quadriceps, patella tendon and patella) was returned to its original anatomical location. The internal incision was sutured with absorbable suture, and the skin sutured with non-absorbable suture.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, the definition of center, dispersion, and precision measures (mean ± SEM) and statistical significance are reported in the Figures and Figure Legends. Data are judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****). All statistical analyses were generated using GraphPad Prism 5.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the RNA-seq reported in this paper is GEO: GSE104887. Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1231.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD3 | Biolegend | Cat# 100243 |
| Armenian hamster monoclonal anti-CD3 | eBioscience | Cat# 15–0031-82 |
| Rat monoclonal anti-CD4 | eBioscience | Cat# 15–0042-82 |
| Rat monoclonal anti-CD4 | eBioscience | Cat# 13–0042-82 |
| Rat monoclonal anti-CD8a | eBioscience | Cat# 15–0081-82 |
| Rat monoclonal anti-CD8a | eBioscience | Cat# 36–0081-85 |
| Rat monoclonal anti-CD11b (Mac-1) | eBioscience | Cat# 15–0112-82 |
| Rat monoclonal anti-CD11b (Mac-1) | eBioscience | Cat# 25–0112-82 |
| Rat monoclonal anti-CD11b (Mac-1) | Biolegend | Cat# 101203 |
| Rat monoclonal anti-Ly6G/Ly6C (Gr1) | eBioscience | Cat# 35–5931-82 |
| Rat monoclonal anti-Ly6G/Ly6C (Gr1) | eBioscience | Cat# 47–5931-82 |
| Rat monoclonal anti-Ly6G/Ly6C (Gr1) | eBioscience | Cat# 13–5931-82 |
| Rat monoclonal anti-IgM | eBioscience | Cat# 15–5790-82 |
| Rat monoclonal anti-IgM | Biolegend | Cat# 406503 |
| Rat monoclonal anti-Ter119 | eBioscience | Cat# 15–5921-82 |
| Rat monoclonal anti-Ter119 | Biolegend | Cat# 116203 |
| Rat monoclonal anti-Ly-6A/E (Sca1) | eBioscience | Cat# 25–5981-82 |
| Rat monoclonal anti-CD117 (c-Kit) | eBioscience | Cat# 17–1171-82 |
| Rat monoclonal anti-CD135(Flt3/Flk2) | eBioscience | Cat# 13–1351-82 |
| Rat monoclonal anti-Cd34 | eBioscience | Cat# 11–0341-82 |
| Armenian Hamster monoclonal anti-CD48 | Biolegend | Cat# 103418 |
| Rat monoclonal anti-CD150 | Biolegend | Cat# 115904 |
| Rat monoclonal anti-CD150 | eBioscience | Cat# 12–1502-82 |
| Armenian Hamster monoclonal anti-CD49b | Biolegend | Cat# 103506 |
| Armenian Hamster monoclonal anti-CD49b | Biolegend | Cat# 103516 |
| Armenian Hamster monoclonal anti-CD49b | eBioscience | Cat# 11–0491-82 |
| Mouse monoclonal anti-CD45.1 | eBioscience | Cat# 15–0453-81 |
| Mouse monoclonal anti-CD45.2 | eBioscience | Cat# 11–0454-82 |
| Rat monoclonal anti-CD3 | eBioscience | Cat# 17–0032-82 |
| Rat monoclonal anti-B220 | eBioscience | Cat# 12–0452-82 |
| Rat monoclonal anti-B220 | eBioscience | Cat# 13–0452-82 |
| Rat monoclonal anti-CD45 | eBioscience | Cat# 17–0451-82 |
| Rat monoclonal anti-CD31 | eBioscience | Cat# 17–0311-82 |
| Rat monoclonal anti-PDGFRα-biotin | eBioscience | Cat# 13–1401-82 |
| Mouse monoclonal anti-LepR Biotinylated | R&D systems | Cat# BAF497 |
| Rat monoclonal anti-CD51-biotin | Biolegend | Cat# 104104 |
| Brilliant Violet 421 Streptavidin | Biolegend | Cat# 405225 |
| Rat monoclonal anti-CD41-biotin | eBioscience | Cat# MA1–82655 |
| Rat monoclonal anti-Endomucin | Abcam | Cat# Ab106100 |
| Rat monoclonal anti-Endomucin | SCBT | Cat# sc-65495 |
| Rat monoclonal anti-VE-cadherin | eBioscience | Cat # 17–1441-82 |
| Streptavidin, Alexa Fluor 405 conjugate | Invitrogen | Cat# S32351 |
| Mouse monoclonal anti-BrdU | GE Healthcare | Cat# RPN202 |
| Rabbit polyclonal anti-Aggrecan | Millipore | Cat# AB1031 |
| Rabbit polyclonal anti-Perilipin | Cell Signaling | Cat# 9349S |
| Goat polyclonal anti-Osteopontin | R&D systems | Cat# AF808 |
| Donkey-anti-goat Alexa Fluor 647 | Invitrogen | Cat# A-21447 |
| Donkey-anti-goat Alexa Fluor 488 | Invitrogen | Cat# A-11055 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 7-aminoactinomycin D (7-AAD) | Life technologies | Cat# A1310 CAS: 7240–37-1 |
| SYTOX Green | Life technologies | Cat# S34860 CAS: 67–68-5 |
| 5-Bromo-2′-deoxyuridine | Sigma | Cat# B9285 CAS: 59–14-3 |
| 5-Fluorouracil | Sigma | Cat# F6627 CAS: 51–21-8 |
| Diphtheria toxin | Sigma | Cat# D0564 |
| Deoxyribonuclease I | Sigma | Cat# D5025 CAS: RN 9003–98-9 |
| Collagenase I | Worthington | Cat# LS004197 |
| Dispase | STEMCELL Technologies | Cat# 07913 |
| Oil Red O | Sigma | Cat# O0625 CAS: 1320–06-5 |
| Toluidine blue | Sigma | Cat# 89640 CAS: 6586–04-5 |
| Liberase DL | Roche | Cat# 5466202001 |
| Critical Commercial Assays | ||
| Annexin A5 Apoptosis Detection Kit | Biolegend | Cat# 640928 |
| APC BrdU Flow Kit | BD PharMingen | Cat# 552598 |
| VECTOR Red Alkaline Phosphatase (Red AP) Substrate Kit | VECTOR LABORATORIES | Cat# SK-5100 |
| StemPro Osteogenesis Differentiation Kit | GIBCO | Cat# A1007201 |
| StemPro Chondrogenesis Differentiation Kit | Cat# A1007101 | |
| StemPro Adipogenesis Differentiation Kit | Cat# A1007001 | |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE104887 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J(iDTR) | The Jackson Laboratory | JAX: 007900 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J(R26R-tdT) | The Jackson Laboratory | JAX: 007914 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J(R26R-ZsG) | The Jackson Laboratory | JAX: 007906 |
| Mouse: B6.129X1t(ROSA)26Sortm1(EYFP)Cos/J | The Jackson Laboratory | JAX: 006148 |
| Mouse: B6.Cg-Tg(Col1a1*2.3-GFP)1Rowe/J | The Jackson Laboratory | JAX: 013134 |
| Mouse: B6.129X1t(ROSA)26Sortm1(EYFP)Cos/J | ||
| Mouse: Tg(Cspg4-DsRed.T1)1Akik/J | The Jackson Laboratory | JAX: 008241 |
| Mouse: B6;FVB-Tg(Cspg4-cre)1Akik/J | JAX: 008533 | |
| Mouse: Cxcl12tm2.1Sjm/J | The Jackson Laboratory | JAX: 022458 |
| Mouse: Cxcl12tm1.1Sjm/J | The Jackson Laboratory | JAX: 022457 |
| Mouse: B6.129(Cg)-Leprtm2(cre)Rck/J | The Jackson Laboratory | JAX: 008320 |
| Mouse: Kitltm2.1Sjm/J | The Jackson Laboratory | JAX: 017861 |
| Mouse: Tg(Nes-EGFP)33Enik | MGI | 5523870 |
| Mouse: Pecam-GFP | This paper | This paper |
| Mouse: N-cad-TdT | This paper | This paper |
| Mouse: N-cad-CreERT | This paper | This paper |
| Software and Algorithms | ||
| Grid/Collection stitching plugin | Preibisch et al., 2009 | https://academic.oup.com/bioinformatics/article/25/11/1463/332497 |
| ImageJ custom plugin | This paper | http://research.stowers.org/imagejplugins |
| Tophat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2010 | http://cole-trapnell-lab.github.io/cufflinks/ |
| Htseq-count | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/count.html |
| EdgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
Highlights.
rHSC and pHSCs are distinguished by their resistance or sensitivity to chemotherapy
pHSCs in the perivascular niche are largely eliminated by chemotherapy
rHSCs are maintained by N-cad+ cells and restore the HSC pool after chemotherapy
N-cad+ BMSPCs expand and produce SCF and other cytokines for rHSCs upon stress
ACKNOWLEDGMENTS
We thank P. Frenette for training on whole-mount immunostaining and T. Suda and F. Arial for scientific discussion and collaboration. We are grateful to M. Miller for model image modification; K. Tannen for proofreading and editing; M. Hembree, K. Zapien, D. Dukes, A. Moran, Y. Wang, M. Durnin, C. Semerad, A. Box, J. Park, J. Blanck, A. Perera, and A. Peak for technical support. We are grateful to L. Li lab members for scientific discussion and manuscript critical reading. M.Z. is supported by National Key Research and Development Program of China (2017YFA0103403, 2018YFA0107203). A.V. was supported by the Department of Biotechnology, Ministry of Science and Technology, Government of India, as an overseas associateship. J.W. is supported by NIH R01 AR05988 and NIH R01 DE018713. This work is mainly supported by Stowers Institute for Medical Research funding to L.L. (SIMR-1004) and in part by NIH National Cancer Institute Cancer Center Support Grant (P30 CA168524) to the University of Kansas Cancer Center.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, one table, and one video and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.093.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, and Morrison SJ (2015). Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, and Suda T. (2004). Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118, 149–161. [DOI] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, and Olson EN (2007). MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 12, 377–389. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Seita J, Inlay MA, Weissman IL, and Rossi DJ (2014). Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste P, Frelin C, Janmohamed S, Barbara M, Herrington R, Hyam D, and Iscove NN (2010). Intermediate-term hematopoietic stem cells with extended but time-limited reconstitution potential. Cell Stem Cell 6, 48–58. [DOI] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. (2012). Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 10, 273–283. [DOI] [PubMed] [Google Scholar]
- Bernitz JM, Kim HS, MacArthur B, Sieburg H, and Moore K. (2016). Hematopoietic stem cells count and remember self-renewal divisions. Cell 167, 1296–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, and Frenette PS (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujanover N, Goldstein O, Greenshpan Y, Turgeman H, Klainberger A, Scharff Y, and Gazit R. (2018). Identification of immune-activated hematopoietic stem cells. Leukemia 32, 2016–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846. [DOI] [PubMed] [Google Scholar]
- Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, Helms JA, Kuo CJ, Kraft DL, and Weissman IL (2009). Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 457, 490–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CKF, Gulati GS, Sinha R, Tompkins JV, Lopez M, Carter AC, Ransom RC, Reinisch A, Wearda T, Murphy M, et al. (2018). Identification of the human skeletal stem cell. Cell 175, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, Seita J, Sahoo D, Nakauchi H, and Weissman IL (2016). Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 530, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, and Scadden DT (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808. [DOI] [PubMed] [Google Scholar]
- Crane GM, Jeffery E, and Morrison SJ (2017). Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590. [DOI] [PubMed] [Google Scholar]
- Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, and Jacks T. (2014). LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell 54, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici M, Rasini V, Bussolari R, Chen X, Hofmann TJ, Spano C, Bernabei D, Veronesi E, Bertoni F, Paolucci P, et al. (2009). Restoration and reversible expansion of the osteoblastic hematopoietic stem cell niche after marrow radioablation. Blood 114, 2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, Zhou XC, Cai YJ, Li ZQ, Zhang F, et al. (2014). Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25, 778–793. [DOI] [PubMed] [Google Scholar]
- Fleming WH, Alpern EJ, Uchida N, Ikuta K, Spangrude GJ, and Weissman IL (1993). Functional heterogeneity is associated with the cell cycle status of murine hematopoietic stem cells. J. Cell Biol 122, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, and Scadden DT (2008). Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2, 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, and Adams RH (2006). Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124, 161–173. [DOI] [PubMed] [Google Scholar]
- Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, and Hock H. (2008). Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol 27, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel AL, and Tyner AL (2002). The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther 1, 639–649. [PubMed] [Google Scholar]
- Gazit R, Mandal PK, Ebina W, Ben-Zvi A, Nombela-Arrieta C, Silberstein LE, and Rossi DJ (2014). Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med 211, 1315–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griseri T, McKenzie BS, Schiering C, and Powrie F. (2012). Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin23-driven chronic intestinal inflammation. Immunity 37, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug JS, He XC, Grindley JC, Wunderlich JP, Gaudenz K, Ross JT, Paulson A, Wagner KP, Xie Y, Zhu R, et al. (2008). N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell 2, 367–379. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, Ledergor G, Jung Y, Milo I, Poulos MG, et al. (2016). Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JB, Kappas NC, Ellerstrom C, DiPaola FW, and Bautch VL (2004). The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 103, 4527–4535. [DOI] [PubMed] [Google Scholar]
- Kfoury Y, and Scadden DT (2015). Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 16, 239–253. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindstedt E, Holm CK, Sulniute R, Martinez-Carrasco I, Lundmark R, and Lundberg P. (2017). CCL11, a novel mediator of inflammatory bone resorption. Sci. Rep 7, 5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Bryant HU, and Macdougald OA (2006). Regulation of bone mass by Wnt signaling. J. Clin. Invest 116, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumbe AP, Ramasamy SK, and Adams RH (2014). Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Cho CH, Hwang SJ, Choi HH, Kim KT, Ahn SY, Kim JH, Oh JL, Lee GM, and Koh GY (2004). Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J. 18, 1200–1208. [DOI] [PubMed] [Google Scholar]
- Lee ES, Boldo LS, Fernandez BO, Feelisch M, and Harmsen MC (2017). Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF-β1. Sci. Rep 7, 42487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner C, and Harrison DE (1990). 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp. Hematol 18, 114–118. [PubMed] [Google Scholar]
- Li L, and Clevers H. (2010). Coexistence of quiescent and active adult stem cells in mammals. Science 327, 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, and Xie T. (2005). Stem cell niche: Structure and function. Annu. Rev. Cell Dev. Biol 21, 605–631. [DOI] [PubMed] [Google Scholar]
- Liebermann DA, and Hoffman B. (2008). Gadd45 in stress signaling. J. Mol. Signal 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Côté D, Rowe DW, Lin CP, and Scadden DT (2009). Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 457, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, and Johnston PG (2003). 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338. [DOI] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G. (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Morita Y, Ema H, and Nakauchi H. (2010). Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J. Exp. Med 207, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson SK, Johnston HM, and Coverdale JA (2001). Spatial localization of transplanted hemopoietic stem cells: Inferences for the localization of stem cell niches. Blood 97, 2293–2299. [DOI] [PubMed] [Google Scholar]
- Park B, Nguyen NT, Dutt P, Merdek KD, Bashar M, Sterpetti P, Tosolini A, Testa JR, and Toksoz D. (2002). Association of Lbc Rho guanine nucleotide exchange factor with alpha-catenin-related protein, alpha-catulin/CTNNAL1, supports serum response factor activation. J. Biol. Chem 277, 45361–45370. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagner EF, and Weissman IL (2004). JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119, 431–443. [DOI] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, and Tomancak P. (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P, He XC, Paulson A, Li Z, Tao F, Perry JM, Guo F, Zhao M, Zhi L, Venkatraman A, Haug JS, Parmely T, Li H, Dobrowsky RT, Ding WX, Kono T, Ferguson-Smith AC, and Li L. (2016). The Dlk1-Gtl2 locus preserves LT-HSC function by inhibiting the PI3K-mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell 18, 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunath J, Salacinski HJ, Sales KM, Butler PE, and Seifalian AM (2005). Advancing cartilage tissue engineering: The application of stem cell technology. Curr. Opin. Biotechnol 16, 503–509. [DOI] [PubMed] [Google Scholar]
- Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, Jedrychowski MP, Ruas JL, Wrann CD, Lo JC, et al. (2014). Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157, 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236. [DOI] [PubMed] [Google Scholar]
- Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, Braun BS, and Passegué E. (2009). JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 15, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield R. (1978). The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 4, 7–25. [PubMed] [Google Scholar]
- Sipp D, Robey PG, and Turner L. (2018). Clear up this stem-cell mess. Nature 561, 455–457. [DOI] [PubMed] [Google Scholar]
- Sophia Fox AJ, Bedi A, and Rodeo SA (2009). The basic science of articular cartilage: Structure, composition, and function. Sports Health 1, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, Haug JS, Peng L, Zhong XB, Suda T, and Li L. (2012). Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell 150, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, and Nagasawa T. (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. [DOI] [PubMed] [Google Scholar]
- Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, and Eaves CJ (1990). Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc. Natl. Acad. Sci. USA 87, 8736–8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. (2013). Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, and Pachter L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman A, He XC, Thorvaldsen JL, Sugimura R, Perry JM, Tao F, Zhao M, Christenson MK, Sanchez R, Yu JY, et al. (2013). Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature 500, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers AJ, and Weissman IL (2005). Differential expression of {alpha}2 integrin separates long-term and short-term reconstituting Lin-/loThy1.1lockit+Sca-1+ hematopoietic stem cells. Stem Cells 24, 1087–1094. [DOI] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552. [DOI] [PubMed] [Google Scholar]
- Wei Q, and Frenette PS (2018). Niches for hematopoietic stem cells and their progeny. Immunity 48, 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman IL (2000). Stem cells: Units of development, units of regeneration, and units in evolution. Cell 100, 157–168. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, Perko K, Alexander R, Schwartz J, Grindley JC, et al. (2009). Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 457, 97–101. [DOI] [PubMed] [Google Scholar]
- Yamauchi H, Hotta Y, Konishi M, Miyake A, Kawahara A, and Itoh N. (2006). Fgf21 is essential for haematopoiesis in zebrafish. EMBO Rep. 7, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, and Nakauchi H. (2011). Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, Teng Y, and Yang X. (2014). Osteogenic fate of hypertrophic chondrocytes. Cell Res. 24, 1266–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, and Gorospe M. (2012). LincRNA-p21 suppresses target mRNA translation. Mol. Cell 47, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousif LF, Di Russo J, and Sorokin L. (2013). Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh. Migr 7, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. (2003). Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841. [DOI] [PubMed] [Google Scholar]
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, and Li L. (2014). Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med 20, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Zhou F, Li X, Wang W, Zhu P, Zhou J, He W, Ding M, Xiong F, Zheng X, Li Z, et al. (2016). Tracing haematopoietic stem cell formation at single-cell resolution. Nature 533, 487–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Resources
The accession number for the RNA-seq reported in this paper is GEO: GSE104887. Original Data Repository at https://www.stowers.org/research/publications/LIBPB-1231.