

Abstract
Understanding of the appropriate regulation of enzymatic activities of histone‐modifying enzymes remains poor. The lysine methyltransferase, SETDB1, is one of the enzymes responsible for the methylation of histone H3 at lysine 9 (H3K9) and plays a key role in H3K9 trimethylation‐mediated silencing of genes and retrotransposons. Here, we reported that how SETDB1's enzymatic activities can be regulated by the nuclear protein, ATF7IP, a known binding partner of SETDB1. Mechanistically, ATF7IP mediates SETDB1 retention inside the nucleus, presumably by inhibiting its nuclear export by binding to the N‐terminal region of SETDB1, which harbors the nuclear export signal motifs, and also by promoting its nuclear import. The nuclear localization of SETDB1 increases its ubiquitinated, enzymatically more active form. Our results provided an insight as to how ATF7IP can regulate the histone methyltransferase activity of SETDB1 accompanied by its nuclear translocation.
Keywords: ATF7IP, histone lysine methylation, nuclear localization, SETDB1, ubiquitination
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Post-translational Modifications, Proteolysis & Proteomics; Transcription
Nuclear protein ATF7IP promotes the nuclear localization of the H3K9 methyltransferase SETDB1. The nuclear retention of SETDB1 increases its ubiquitination, which enhances its enzymatic activities.
Introduction
Epigenetic regulation of transcription is mediated, in part, by chemical modifications on histone proteins 1, 2, 3. Most of these histone modifications are mediated by enzymatic reactions. Methylation is an important epigenetic modification of histones, catalyzed by various kinds of enzymes (e.g., lysine methyltransferases and demethylases), which have been identified in the past two decades 4. Identification of histone‐modifying enzymes has facilitated numerous studies aimed at deciphering the function of each histone modification and to determine as to how histone marks are established at specific regions of the genome. For an in‐depth understanding of the histone modification‐dependent gene regulation, it is important to understand the regulation of histone‐modifying enzymes.
Mammalian SETDB1 (also known as ESET and KMT1E) is an enzyme that catalyzes H3K9me3 5, which is generally considered as a repressive mark for transcription 6. SETDB1 plays a role in diverse developmental processes, including early development 7, neurogenesis 8, immune cell development 9, 10, 11, 12, germ cell development 13, 14, 15, chondrocyte and osteoblast differentiation 16, 17, and adipogenesis 18. Moreover, in mouse embryonic stem cells (mESCs), loss of SETDB1 has been reported to result in severe growth defects and de‐repression of various kinds of endogenous retroviruses (ERVs), including IAP, MmERVK10c, and MusD, concomitant with a decrease in H3K9me3 marks on the loci 19, 20. SETDB1 also represses ERVs in several other cell types besides mESCs 9, 10, 11, 13, 21, 22. In pro‐B cells, de‐repression of ERVs, including MLV, by the loss of SETDB1 leads to unfolded protein response and apoptosis 10. SETDB1 enables acute myeloid leukemia cell lines to avoid the dsRNA‐induced interferon response via silencing of ERVs 21. Therefore, proper regulation of SETDB1 can contribute to the silencing of ERVs and might have biological significance.
As of date, several mechanisms for the regulation of SETDB1 have been proposed. SETDB1 harbors motifs for nuclear export signal (NES) and nuclear localization signal (NLS), suggesting that it shuttles between the nucleus and cytoplasm 23. In fact, overexpressed SETDB1 showed cytoplasmic localization and treatment with leptomycin B (LMB), a CRM1‐dependent nuclear export inhibitor, partly induced its nuclear accumulation 23, 24. Moreover, SETDB1 has been speculated to be a target for proteasomal degradation in the nucleus; however, the underlying molecular mechanism and physiological significance remain unknown 24, 25. These two regulatory mechanisms can control the level of nuclear SETDB1. In addition, in a recent study, it was shown that SETDB1 is mono‐ubiquitinated and this modification positively regulates the enzymatic activity of SETDB1 26, 27. SETDB1 is a SET domain‐type H3K9 methyltransferase and requires the SET domain, with two adjacent pre‐SET and post‐SET domains, for its enzymatic activity 28. The SET domain of SETDB1 contains a large insertion, which is the site for mono‐ubiquitination 26, 27. Although the mechanism underlying the enhanced catalytic activity of mono‐ubiquitinated SETDB1 is still unknown, ubiquitination has been shown to be crucial for SETDB1‐mediated silencing of ERVs in mESCs 26.
With regard to the binding partners of SETDB1, several groups have reported that the nuclear protein ATF7IP (also known as MCAF1 or AM) 29, 30, 31 is an important regulator of SETDB1 and plays a pivotal role in its function 5, 18, 25, 30, 32. In human cells, loss of ATF7IP has been shown to exhibit a similar effect on the genome‐wide transcriptome and H3K9 trimethylation (H3K9me3) levels as was observed after the loss of SETDB1 25. Moreover, CRISPR‐gRNA‐based knock‐out (KO) or knock‐down (KD) of Atf7ip in wild‐type (WT) or Dnmt1/3a/3b triple KO mESCs resulted in reactivation of SETDB1‐regulated ERVs, such as IAP, MmERVK10c, and MusD, and decreased the H3K9me3 levels at the loci 32, 33, suggesting that ATF7IP also plays a role in SETDB1‐mediated silencing of ERVs. However, the underlying mechanism of ATF7IP‐mediated regulation of SETDB1 remains unclear. In a pioneering study, it was proposed that ATF7IP facilitates SETDB1‐mediated conversion of H3K9me2 to H3K9me3 by an unknown mechanism 5. However, in another report, it was argued that ATF7IP does not enhance the catalytic activity of SETDB1 in in vitro 34. Another notion for the in vivo regulation of SETDB1 by ATF7IP in human cells is that it contributes to the stability of SETDB1 in the nucleus 25. However, reduction of H3K9me3 on the SETDB1‐target loci has been commonly recognized in different ATF7IP depletion experiments 18, 25, 32, although, the levels of SETDB1 on the target ERV loci are maintained in the Atf7ip KD mESCs 32. We, therefore, re‐examined the role(s) of ATF7IP in the regulation of SETDB1 in our experimental system in the present study.
Results
ATF7IP plays a crucial role in SETDB1‐target retroelement silencing and H3K9me3 in mESCs
We previously described the establishment of Atf7ip KO mESCs 35. As described in Fig EV1, the two independent Atf7ip KO mESC clones, TT#2‐5 and TT#2‐12, showed similar de‐repression of the reporter retrovirus, MSCV‐GFP, which was integrated into the genome and was silenced through the SETDB1 pathway 19, 33. RT–qPCR analysis clearly showed that not only the exogenous MSCV‐GFP, but also other SETDB1‐target ERVs, IAP, MmERVK10c, and MusD 19, 20 were derepressed in the Atf7ip KO mESCs (Fig 1A). Furthermore, the levels of H3K9me3 on these SETDB1‐target retroelements were significantly diminished (Fig 1B). These data are consistent with the previous findings 32, 33. The total amount of SETDB1 was not reduced much in the Atf7ip KO mESCs (Fig 1C). Although ATF7IP might contribute to the stability of SETDB1 in the nucleus, as suggested previously 25, our Western blot data suggested other roles of ATF7IP with regard to the regulation of SETDB1 and its function.
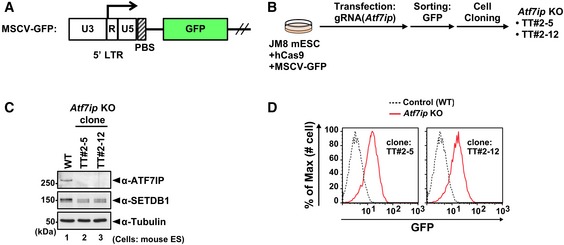
Figure EV1. Establishment and characterization of Atf7ip KO mESCs (related to Fig 1).
-
AThe structure of MSCV‐GFP reporter construct. PBS: primer binding site.
-
BEstablishment of Atf7ip KO cell lines by CRISPR/Cas9 technology.
-
CConfirmation of a complete loss of ATF7IP protein and comparable expression of SETDB1 in the parental WT and established Atf7ip KO cell lines, TT#2‐15 and TT#2‐12, by WB analysis.
-
DFlow cytometric analysis shows that Atf7ip KO cell lines increase the expression of MSCV‐GFP reporter.
Source data are available online for this figure.
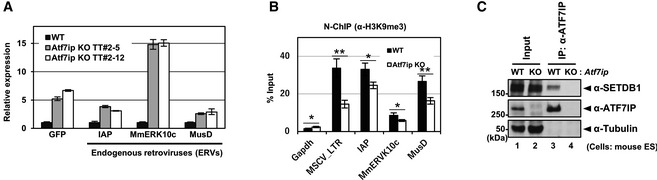
Figure 1. ATF7IP represses the SETDB1‐regulated exogenous provirus reporter and ERVs in mESCs.
-
AAtf7ip KO cells show increased expression of SETDB1‐regulated ERVs and the provirus reporter, MSCV‐GFP (Fig EV1B), as evidenced by RT–qPCR analysis. RNA expression was normalized to Hprt expression and is shown relative to the level in WT cells. Data are mean ± SD; n = 3, technical replicates.
-
BAtf7ip KO mESCs (TT#2‐12) show decreased H3K9me3 at the LTR of the SETDB1‐regulated reporter and the ERVs, as evidenced by Native ChIP followed by qPCR analysis. Gapdh was used as a negative control. Data are mean ± SEM; n = 5, independent experiments. *P < 0.05 and **P < 0.01 by paired t‐test.
-
CSETDB1 cellular amount is mostly maintained in Atf7ip KO mESCs. Endogenous SETDB1‐ATF7IP interaction is validated by anti‐ATF7IP antibody co‐IP experiment with anti‐ATF7IP antibody.
Source data are available online for this figure.
ATF7IP regulates the nuclear localization of SETDB1
Since Atf7ip KO mESCs showed little difference in the abundance of SETDB1 protein compared to that in the parental WT cells (Figs 1C and EV5B), we examined whether ATF7IP regulates the nuclear localization of SETDB1. We performed immunofluorescence (IF) analysis using anti‐SETDB1 antibody in WT and Atf7ip KO ESCs. Consistent with the results of a previous study 36, SETDB1 was mainly localized to the nucleus with some nuclear foci in WT mESCs (Fig 2A). The loss of ATF7IP decreased the nuclear signal of SETDB1 and enhanced its cytoplasmic signal (Fig 2A; quantification in Figs 2B and EV2A). SETDB1 still sustained and rather increased nuclear foci formation in Atf7ip KO cells (Fig 2A; quantification in Fig 2C and D). Anti‐SETDB1 antibody specificity was validated by conditional KO of Setdb1 (Fig EV2B) and the difference of nuclear SETDB1 immunostaining signals between WT and Atf7ip KO ESCs was confirmed by the mixed‐cell immunostaining analysis (Fig EV2C and D).
Figure EV5. Characterization of SETDB1 d1 and K885 mutant, and SETDB1 stability in Atf7ip KO treated with LMB (related to Figs 4 and 5).
-
AV5‐SETDB1 d1 mutant expression with or without 3xF‐ATF7IP co‐expression in HEK293T cells as shown in Fig 4B.
-
BMeasurement of total SETDB1 signals of WT, Atf7ip KO, and Atf7ip KO treated with LMB shown in Fig 5C top panel. Data from three different samples in a single experiment. Error bars: SD.
-
CHEK293T cells were transfected with V5‐SETDB1 K885 mutant and either empty or 3xF‐ATF7IP expressing vector. Over 50 transfected cells per sample were analyzed. The representative data of two independent experiments are shown. Scale bar: 10 μm.
-
DHEK293T cells were transfected with 3xF‐ATF7IP and either empty, V5‐SETDB1 WT, or K885 mutant. After V5‐IP, the bound 3xF‐ATF7IP was detected by WB analysis.
Source data are available online for this figure.
Figure 2. ATF7IP controls SETDB1 nuclear localization.
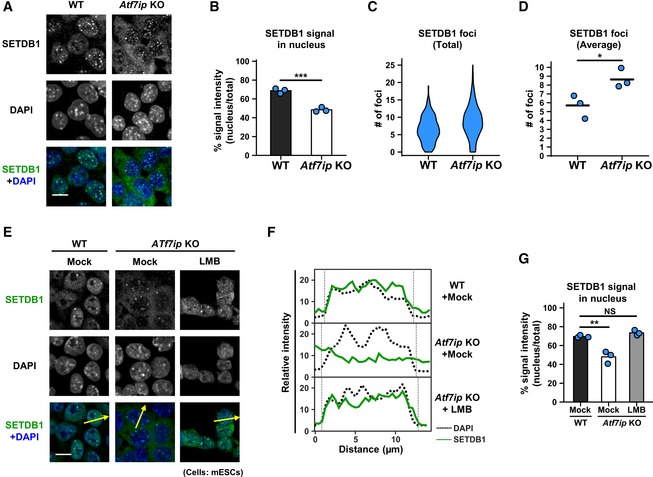
-
AIF analysis shows preferential cytoplasmic localization of SETDB1 in Atf7ip KO mESCs. Representative projected images are shown, and the quantitative analyses are shown in (B–D). Scale bar: 10 μm.
-
BSETDB1 signal in the nucleus that was determined by DAPI staining was calculated. The mean from three independent experiments is shown as a bar graph with jittered points indicating the average % intensity of each experiment. Over 100 cells were analyzed per sample per experiment. ***P < 0.001 by unpaired Student's t‐test.
-
C, DSETDB1 foci in the nucleus that was determined by DAPI staining were calculated. Violin plot for SETDB1's foci numbers from all the cells analyzed in three independent experiments is shown in (C). The mean from the three independent experiments is shown as a bar with jittered points indicating the average number of each experiment in (D). Over 100 cells were analyzed per sample per experiment. *P < 0.05 by unpaired Student's t‐test.
-
EIF analysis shows preferential cytoplasmic localization of SETDB1 in Atf7ip KO mESCs. Representative projected images are shown. Arrows indicate the region used for line‐plot analysis. Cells were treated with 10 ng/ml LMB or mock for 5 h before analysis. Scale bar: 10 μm.
-
FThe line‐plot analysis for (E). Relative intensities of SETDB1 signal were measured by ImageJ.
-
GThe quantitative analysis for (E) is shown. SETDB1 signal in the nucleus that was determined by DAPI staining was calculated. The mean from three independent experiments is shown as a bar graph with jittered points indicating the average % intensity of each experiment. Over 100 cells were analyzed per sample per experiment. NS: P > 0.05, **P < 0.01 vs. “Mock, WT” group by Dunnett's test.
Figure EV2. Reduction of nuclear SETDB1 in Atf7ip KO mESCs (related to Fig 2).
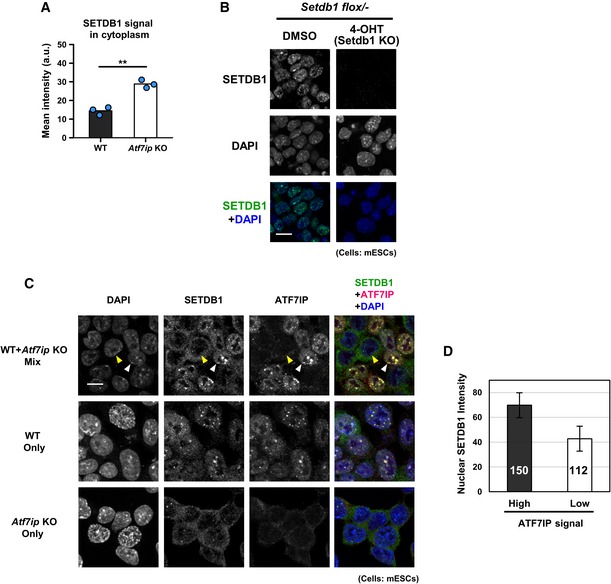
-
ASETDB1 signal in the cytoplasm that is shown in Fig 2A was calculated. The mean from three independent experiments is shown as a bar graph with jittered points indicating the average % intensity of each experiment. Over 100 cells were analyzed per sample per experiment. **P < 0.001 by unpaired Student's t‐test.
-
BValidation of anti‐SETDB1 antibody used in this study by IF analysis. Conditional Setdb1 KO mESCs show no signal after 4‐OHT treatment to deplete SETDB1. Scale bar: 7.33 μm.
-
CImmunostaining of ATF7IP as shown in Fig 2A was conducted using a mixture of WT and Atf7ip KO mESCs (1:1) (top) in addition to WT (middle) or Atf7ip KO (bottom) alone. The yellow and white arrowhead indicates AFP7IP signal high and low cell which are supposedly WT and Atf7ip KO cell, respectively. Scale bar: 10 μm.
-
DSETDB1 signal in the nucleus of ATF7IP high and low cells as shown in (C) top panel was calculated. The mean with SD is shown as a bar graph. Over 100 of each cell type (150 and 112 for ATF7IP high and low cells, respectively) was analyzed. Error bars: SD.
Since the overexpressed SETDB1 is retained in the cytoplasm and its nuclear localization is partially induced by treatment with LMB 23, we hypothesized that the nuclear protein ATF7IP antagonizes the nuclear export of SETDB1 and that insufficient ATF7IP abundance results in the translocation of SETDB1 from the nucleus to the cytoplasm. To test this, we treated Atf7ip KO cells with LMB for 5 h to inhibit the nuclear export of SETDB1 23. By IF analysis, we observed that the LMB treatment restored the nuclear signal of SETDB1 in Atf7ip KO ESCs (Fig 2E; line‐plot analysis in Fig 2F; quantification in Fig 2G).
To determine whether the regulation of SETDB1 localization by ATF7IP occurs in other species, we inactivated ATF7IP in human HEK293T cells and examined cellular localization of SETDB1 in such a background. As shown in Fig EV3A–C, we established two ATF7IP KO 293T cell lines by CRISPR/Cas9, in which both alleles of exon 5 of ATF7IP were deleted and no ATF7IP production was confirmed. At first, we examined endogenous SETDB1 cellular localization in WT and ATF7IP KO HEK293T cells (Fig 3A). In contrast to WT mESCs, SETDB1 in WT HEK293T cells was mostly detected in both cytoplasm and nucleus with some showing slightly higher abundance of SETDB1 in nucleus. In ATF7IP KO cells, SETDB1 highly accumulated in cytoplasm as seen in Atf7ip KO ESCs. If mouse ATF7IP is transiently expressed in ATF7IP KO 293T cells, SETDB1 mostly localizes in nucleus. Endogenous SETDB1 in WT HEK293T cells was also highly enriched in the nucleus by ectopic expression of mouse ATF7IP (Fig 3B). Next, we treated WT and ATF7IP KO HEK293T cells with LMB for 5 h. Interestingly, while LMB treatment enhanced SETDB1 nuclear accumulation in WT cells, only marginal SETDB1 nuclear accumulation could be observed in the ATF7IP KO HEK293T cells (Figs 3C and D, and EV3D). These results indicated that (i) ATF7IP regulates SETDB1 nuclear localization in human, (ii) the relative level of ATF7IP might be low and not sufficient for complete SETDB1 nuclear accumulation in 293T cells, and (iii) ATF7IP might play a role in SETDB1 nuclear accumulation other than inhibition of nuclear export such as shielding SETDB1 from degradation in the nucleus 25 or inducing nuclear import of SETDB1 in HEK293T cells.
Figure EV3. Establishment and characterization of ATF7IP KO HEK293T cells (related to Fig 3).
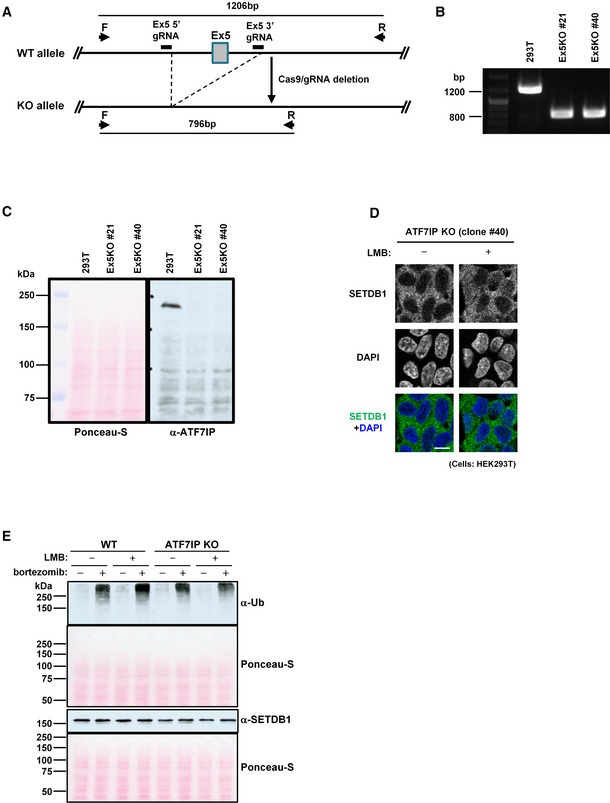
-
AATF7IP KO strategy by the CRISPR/Cas9 system. If Exon 5(Ex5) of ATF7IP is deleted between the 5′ and 3′ gRNA target sequences, PCR with primer F and R amplifies about 800‐bp DNA fragment from the KO allele vs. 1,200 bp from WT allele.
-
BPCR validation result of two independent ATF7IP KO HEK293T cell clones, #21 and #40.
-
CConfirmation of no ATF7IP protein expression in two ATF7IP KO HEK293T cell clones, #21 and #40. Ponceau‐S staining of same blotted membrane used for ATF7IP detection. The representative data of two independent experiments are shown.
-
DSETDB1 cellular localization in another ATF7IP KO HEK239T cell clone, #40 without or with LMB treatment for 5 h. The representative data of multiple independent experiments are shown. Scale bar: 10 μm.
-
EImpact of bortezomib treatment on cellular protein stability. Same number of WT or ATF7IP KO HEK293T cells was aliquoted into the 1.5‐ml tubes and untreated or treated with LMB (10 ng/ml) or/and bortezomib (100 nM) for 5 h before analysis which is same condition as used for Fig 3E. Ponceau‐S staining of same blotted membrane used for poly‐Ub and SETDB1 detection, respectively. The representative data of two independent experiments are shown.
Source data are available online for this figure.
Figure 3. ATF7IP may have a role in SETDB1 import in HEK293T cells.
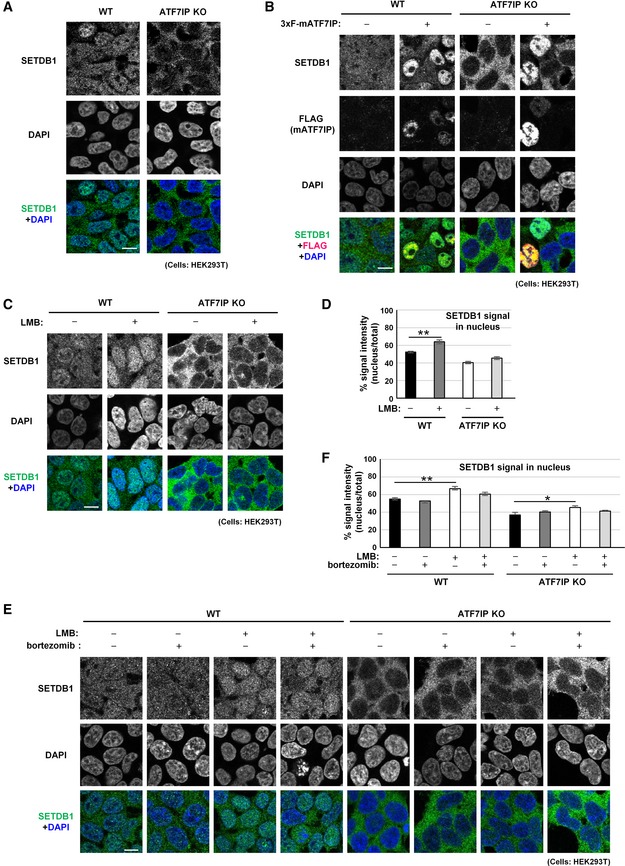
-
AATF7IP KO HEK293T cells show endogenous SETDB1 cytoplasmic localization phenotype. Endogenous SETDB1 in HEK293T cells shows cytoplasm + nuclear localization profile. Scale bar: 10 μm.
-
BOver‐production of exogenous mouse SETDB1 induces endogenous SETDB1 nuclear localization in both WT and ATF7IP KO HEK293T cells. The representative data of two independent experiments is shown. Scale bar: 10 μm.
-
CLMB treatment enhances SETDB1 nuclear accumulation in WT HEK293T cells, but no significant impact for ATF7IP KO HEK293T cells. Cells were treated with 10 ng/ml of LMB for 5 h and then analyzed with immunofluorescent staining. The representative data of three independent experiments are shown. Scale bar: 10 μm.
-
DSETDB1 signal in the nucleus shown in (C) was calculated. The mean from three independent experiments is shown as a bar graph ± SEM; n = 3. Over 50 cells were analyzed per sample per experiment. **P < 0.01 by Student's t‐test (two‐sided test).
-
EFive hours of proteasome inhibitor bortezomib treatment does not have much impact on SETDB1 relative nuclear accumulation in ATF7IP KO HEK293T cells. The representative data of three independent experiments are shown. Scale bar: 10 μm.
-
FSETDB1 signal in the nucleus shown in (E) was calculated. The mean from three independent experiments is shown as a bar graph ± SEM; n = 3. Over 50 cells were analyzed per sample per experiment. Statistics comparison was only shown between without and with drug treatment in WT and ATF7IP KO cells. *P < 0.05 and **P < 0.01 by Dunnett's method.
To examine how protein degradation by the ubiquitin‐proteasome pathway is crucial for the poor SETDB1 accumulation phenotype observed in the LMB‐treated ATF7IP KO HEK293T cells, we added the proteasome inhibitor, bortezomib, in the LMB experiments. Bortezomib treatment for 5 h suppressed the proteasome action and enhanced cellular levels of ubiquitinated proteins (Fig EV3E). Furthermore, total amount of SETDB1 also slightly increased in bortezomib‐treated ATF7IP KO, but not WT HEK293T cells (Fig EV3E), which was consistent with the previous findings 25. However, bortezomib did not increase relative intensities of SETDB1 nuclear signals in the LMB‐treated ATF7IP KO HEK 293T cells (Fig 3E and F). Insignificant accumulation of SETDB1 in the dual‐treated ATF7IP KO HEK293T cells supports the idea that ATF7IP contributes to SETDB1 nuclear import.
N‐terminal region of SETDB1 contributes to ATF7IP interaction and its cellular localization
We then asked whether the interaction between ATF7IP and SETDB1 is required for the ATF7IP‐dependent nuclear localization of SETDB1. For this question, we tried to identify the region of SETDB1 involved in interaction with ATF7IP (Fig EV4) and mapped the N terminus of SETDB1 (residues 1–109) as the major region involved in this interaction (Fig EV4 d13). The region is partially consistent with the ATF7IP ortholog‐binding region of the SETDB1 ortholog in Drosophila (Appendix Fig S1) 37. We also observed that the deletion mutant of SETDB1 (residues 110–1,308), named d1, had severely impaired ATF7IP binding (Fig EV4B).
Figure EV4. Mapping analyses for the identification of ATF7IP‐interacting region of SETDB1 (related to Fig 4).
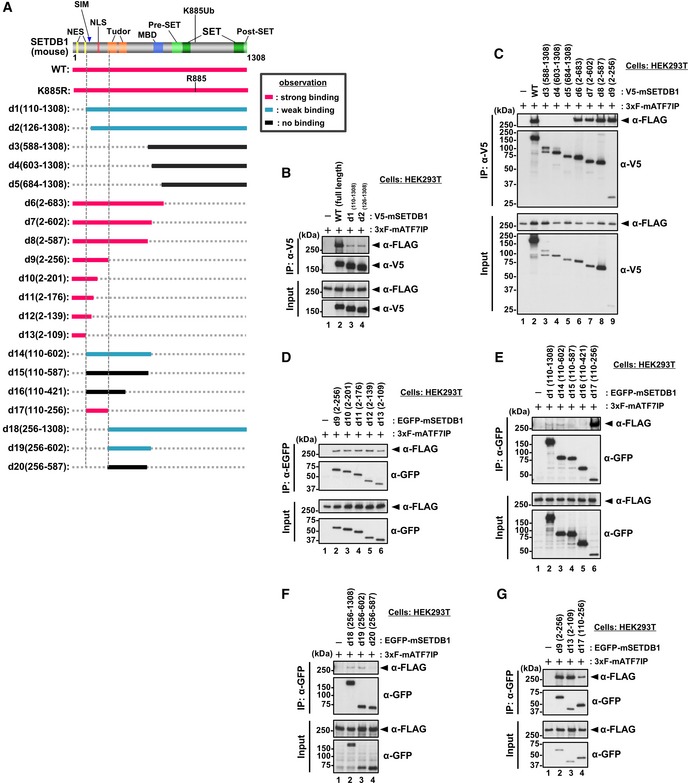
-
ASummary of the co‐IP experiments. After the transfection, V5‐SETDB1 was immune‐purified with anti‐V5 antibody and used for WB analysis. Location of estimated NES and NLS motifs is shown 23.
-
B–GHEK293T cells were co‐transfected with 3xFLAG‐ATF7IP and V5‐tagged or EGFP‐fused SETDB1 WT or deletion mutants as shown in (A). After IP with anti‐V5 antibody (B and C) or anti‐GFP antibody (D–G), the bound ATF7IP was detected by WB analysis. The residues 2–109 (d13) of SETDB1 are mostly sufficient for the interaction with ATF7IP (G).
Source data are available online for this figure.
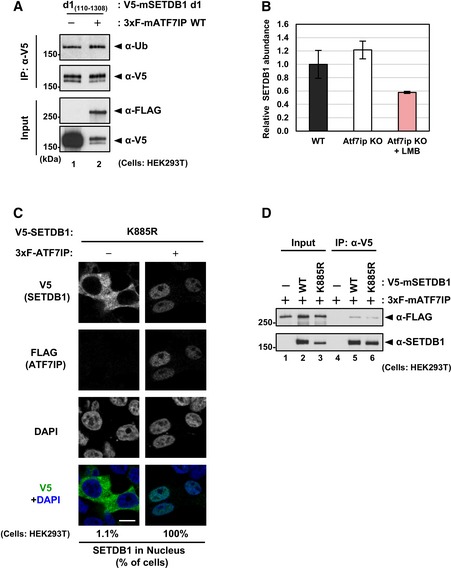
When exogenous mouse V5‐tagged SETDB1 was transiently expressed in HEK293T WT cells, it showed cytoplasmic localization (Fig 4A left panel), as reported previously 23. When the SETDB1 co‐transfected with 3xFLAG‐ATF7IP, it showed nuclear localization in most of the co‐transfected cells (98.7%) (Fig 4A middle panel). Interestingly, if the d1 mutant of V5‐SETDB1 was transiently expressed in HEK293T cells (Fig EV5A), certain population of transfected cells exhibited both cytoplasmic and nuclear localization profiles for SETDB1 (Fig 4B −3xF‐ATF7IP panels). Furthermore, co‐transfection of FLAG‐ATF7IP could not induce further enhancement of nuclear d1‐positive population of cells (Fig 4B +3xF‐ATF7IP panels). As shown in Fig EV4A, two NES motifs of SETDB1 exist in the N‐terminal ATF7IP interaction region, which is lost in the d1 mutant. Therefore, cellular localization phenotypes of the d1 mutant suggest that ATF7IP interaction is essential for ATF7IP‐mediated SETDB1 nuclear localization and further indicated the possibility that binding of ATF7IP to the N‐terminal region of SETDB1 interferes with the NES‐mediated nuclear export.
Figure 4. N‐terminal region of SETDB1 contributes to its cellular localization.
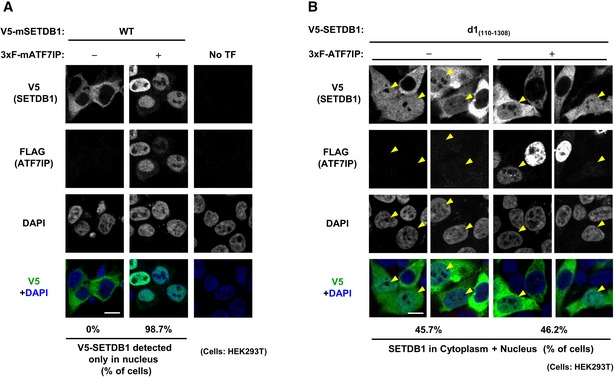
-
AHEK293T cells were transfected with V5‐SETDB1 and either empty or 3xF‐ATF7IP expressing vector. Over 50 transfected cells analyzed per sample were analyzed. The representative data of two independent experiments are shown. Scale bar: 10 μm.
-
BHEK293T cells were transfected with V5‐SETDB1 d1 mutant and either empty or 3xF‐ATF7IP expressing vector. Over 50 transfected cells per sample were analyzed. The representative data of two independent experiments were shown. Scale bar: 10 μm. The yellow arrowheads indicate the cells that show nuclear (and cytoplasmic)‐localization of introduced exogenous mSETDB1.
ATF7IP‐mediated SETDB1 nuclear localization increases its ubiquitination level
Under the similar conditions as in the co‐transfection experiments (Fig 4A), we examined the expression levels of exogenous SETDB1 and ATF7IP in HEK293T cells by Western blot analysis and found a change in the doublet banding pattern of V5‐SETDB1: transfection of V5‐SETDB1 alone resulted in a doublet band, whereas co‐transfection with 3xFLAG‐ATF7IP was mostly associated with the appearance of the upper band of V5‐SETDB1 (Fig 5A, lane 1 vs. 2). SETDB1 has been shown to be subjected to mono‐ubiquitination and the upper band corresponded to its ubiquitinated form 26, 27. Anti‐Ub antibody analysis of the immunoprecipitated (IPed) V5‐SETDB1 samples confirmed that the upper band, the intensity of which was increased upon co‐transfection with 3xF‐ATF7IP, was indeed the ubiquitinated form (Fig 5A, lane 1 vs. 2). Moreover, V5‐SETDB1 K885R mutant that contained a lysine‐to‐arginine substitution at position 885, which corresponds to the ubiquitinatable residue in mouse SETDB1, showed a single lower band when probed with anti‐V5 antibody and no reactivity with anti‐Ub antibody (Fig 5A, lane 1 vs. 3), suggesting that the observed ubiquitination occurred at the K885 residue. Although the K885R mutant, co‐expressed with 3xF‐ATF7IP, showed no ubiquitination signal, it displayed a very slight shift of the upper band compared to that observed for V5‐SETDB1 K885R without co‐transfection with 3xF‐ATF7IP (Fig 5A lane 3 vs. 4); this suggested that ATF7IP may induce other unknown modification(s) in SETDB1. Similar to the WT SETDB1, the K885R mutant binds to 3xFLAG‐ATF7IP and shows nuclear localization when co‐transfected with 3xFLAG‐ATF7IP (Fig EV5C and D).
Figure 5. ATF7IP‐mediated SETDB1 nuclear localization enhances SETDB1 ubiquitination.
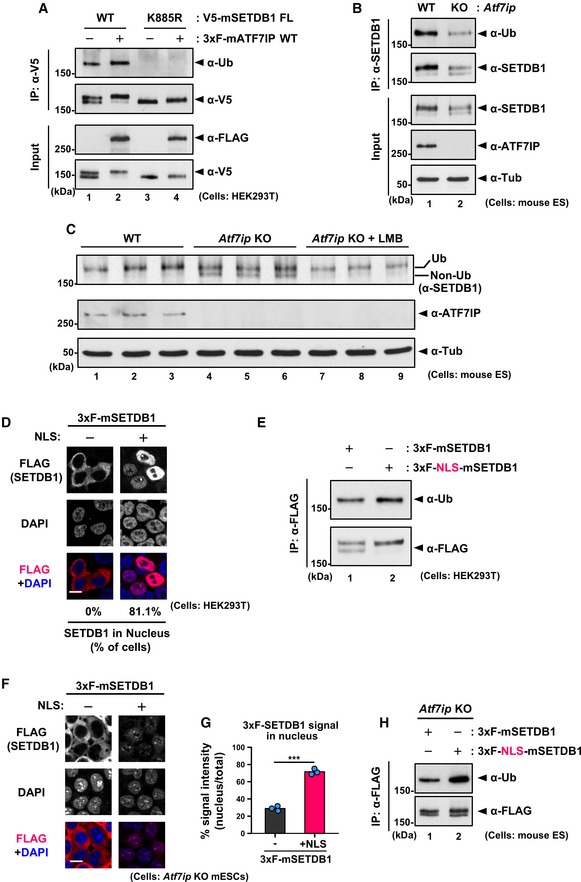
-
ACo‐transfection with ATF7IP augments the K885 ubiquitination of SETDB1. HEK293T cells were transfected as in Fig 4A. After the transfection, V5‐SETDB1 was immune‐purified with anti‐V5 antibody and used for WB analysis.
-
BATF7IP regulates the ubiquitination of SETDB1 in mESCs. Endogenous SETDB1 was immune‐purified from WT or Atf7ip KO mESCs cells and used for WB analysis.
-
CLMB treatment increased the relative intensity of the upper ubiquitinated upper band of SETDB1 in Atf7ip KO mESCs. Cells were treated with 10 ng/ml LMB or mock for 5 h before analysis.
-
DHEK293T cells were transfected with either 3xF‐ or 3xF‐NLS‐SETDB1. Over 50 transfected cells per sample were analyzed. The representative data of two independent experiments are shown. Scale bar: 10 μm.
-
ENuclear localization itself promotes the ubiquitination of SETDB1. HEK293T cells were transfected with 3xFLAG‐ or 3xFLAG‐NLS‐SETDB1. After FLAG‐IP, the purified SETDB1s were analyzed by WB.
-
F, GIF analysis was performed with Atf7ip KO mESCs stably expressing 3xF‐ or 3xF‐NLS‐SETDB1. Representative images are shown in (F), and the quantitative analysis is shown in (G). Scale bar: 10 μm. Addition of NLS increases nuclear localization of SETDB1 in Atf7ip KO mESCs. The mean from three independent experiments is shown as a bar graph with jittered points indicating the average % intensity of each experiment. Over 50 cells were analyzed per sample per experiment. ***P < 0.001 by unpaired Student's t‐test.
-
HNuclear localization promotes the ubiquitination of SETDB1 in Atf7ip KO mESCs. Atf7ip KO mESCs expressing 3xF‐ or 3xF‐NLS‐SETDB were used for FLAG‐IP, and the purified SETDB1s were analyzed by WB.
Source data are available online for this figure.
We also examined the banding pattern of SETDB1 in Atf7ip KO mESCs. As shown in Fig 5B and C, the relative intensity of the lower band, which is supposedly the non‐ubiquitylated form of SETDB1, increased in Atf7ip KO mESCs compared to that in the WT cells. Indeed, we confirmed that upper band was the ubiquitylated form, which indicated that the level of SETDB1 mono‐ubiquitination was decreased in the Atf7ip KO mESCs (Fig 5B SETDB1 IP panels). Since LMB treatment resulted in the nuclear accumulation of SETDB1 (Fig 2E), we examined whether the inhibition of nuclear export could lead to enhanced ubiquitination of SETDB1. Although SETDB1 abundance in the LMB‐treated Atf7ip KO mESCs was reduced compared to those in untreated WT and KO cells (Fig EV5B), LMB treatment decreased the relative intensity of the lower band in the Atf7ip KO mESCs (Fig 5C) suggesting that the nuclear accumulation of SETDB1 by LMB increased its ubiquitination level. The LMB‐induced SETDB1 reduction might be caused by degradation of SETDB1 in the nucleus, as reported previously 24, 25. In conclusion, our results indicated that ATF7IP positively regulates the ubiquitination of SETDB1 by retaining it into the nucleus.
Finally, we examined whether nuclear localization itself is sufficient for the enhanced ubiquitination of SETDB1. We generated an SETDB1 expression vector by adding an NLS from c‐myc (3xFLAG‐NLS‐SETDB1) and confirmed the nuclear localization profile of 3xFLAG‐NLS‐SETDB1, transiently expressed in HEK293T cells (Fig 5D). Furthermore, the nuclear localized 3xFLAG‐NLS‐SETDB1 exhibited increased ubiquitination level (Fig 5E). We also examined ubiquitination status for NLS‐tagged d1 mutant and found that NLS‐d1 also localized in the nucleus and increased its ubiquitination level compared to non‐tagged d1 (Appendix Fig S2). To rule out the possibility of the involvement of endogenous ATF7IP in these findings in HEK293T cells, we established Atf7ip KO mESCs expressing either 3xFLAG‐SETDB1 or 3xFLAG‐NLS‐SETDB1 and confirmed the preferential cytoplasmic or nuclear localization of these, respectively, as expected (Fig 5F and G). It should be noted that we observed a weak staining of the NLS‐SETDB1 compared to that of the non‐tagged SETDB1 in Atf7ip KO mESCs stable line, suggesting the instability of SETDB1 in the nucleus in the absence of ATF7IP 25. We again observed that the relative abundance of ubiquitinated form increased in the cells expressing 3xFLAG‐NLS‐SETDB1 in comparison with that in the cells expressing SETDB1 without exogenous NLS (Fig 5H), supporting the notion that nuclear localization itself promotes SETDB1 ubiquitination.
Discussion
In this study, we found that nuclear localization of SETDB1 augments its ubiquitinated form. The ubiquitylated form increases the histone methyltransferase activity 26, 27, suggesting that SETDB1 is enzymatically activated upon nuclear translocation to ensure the H3K9me3‐mediated silencing. SETDB1 can be mono‐ubiquitylated by UBE2E enzymes, independent of E3 ligase, and is deubiquitylated by USP2A and USP17 26. It has also been reported that endogenous UBE2E enzymes are predominantly detected in the nucleus 38, consistent with our findings. We also demonstrated that ATF7IP is a crucial regulator for this process and positively regulates the nuclear localization of SETDB1. Nuclear localization of ATF7IP and the LMB treatment phenotype of the Atf7ip KO mESCs strongly support the notion that ATF7IP antagonizes the nuclear export of SETDB1, even though LMB induces nuclear retention of many other proteins and may indirectly accumulate SETDB1 in the nucleus. As indicated, ATF7IP binding to the N terminus region of SETDB1, in which the NES motifs exist, might also actively interfere with the NES‐mediated nuclear export of SETDB1. Interestingly, LMB treatment for 5 h induced only marginal accumulation of SETDB1 in ATF7IP KO HEK293T cells, whereas the treatment induced clear nuclear accumulation of SETDB1 in WT HEK293T cells. Since proteasome inhibition could not enhance relative nuclear signal of SETDB1 in ATF7IP KO HEK293T cells, we suggest that ATF7IP may also play a role in nuclear import of SETDB1. In addition, a slight increase of total SETDB1 amount by proteasome inhibition in ATF7IP KO HEK293T cells suggests a role of ATF7IP in SETDB1 stability, which is consistent with the previous finding 25. Based on the findings of this study and previous findings 26, 27, we illustrated a model for ATF7IP‐mediated SETDB1 regulation (Fig 6). Multiple types of regulations of SETDB1 by ATF7IP may explain loss‐of‐function phenotypes for ATF7IP, which are similar to the phenotypes observed after the loss of SETDB1 25. Further detailed studies would be required to further elucidate the complex ATF7IP roles in SETDB1 regulation.
Figure 6.
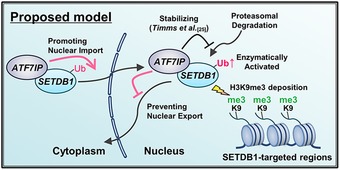
Proposed model of ATF7IP's roles on SETDB1 regulation.
We found that ATF7IP controls SETDB1 nuclear localization in both human and mouse. In Drosophila, an ATF7IP ortholog, Wde, was shown to be required for nuclear localization of Egg, a SETDB1 ortholog 37. In addition, recent studies have shown that LIN‐65, a protein resembling ATF7IP, also regulates the nuclear localization of the SETDB1 ortholog in Caenorhabditis elegans 39, 40. We, therefore, surmised that our finding regarding the ATF7IP‐mediated regulation of SETDB1 ubiquitination by nuclear translocation might be a conserved and common mechanism across various species. In fact, nuclear localization of SETDB1 also increases its ubiquitination levels in ovarian somatic cells of Drosophila 41. The residues 1–109 of mouse SETDB1 were mostly responsible for the binding of ATF7IP (Fig EV4). However, interestingly, a previous mapping analysis in Drosophila identified the residues 366–521 in an ortholog of SETDB1 as the binding region to the ATF7IP ortholog, which share only partial similarity with the residues 1–110 of mouse SETDB1 37. In addition, the residues 110–256 of mouse SETDB1 also share partial similarity with the 366–521 of the Drosophila SETDB1. These results suggest that the separated regions could bind to ATF7IP. To further understand as to how SETDB1 binds to ATF7IP, structural analysis of the SETDB1–ATF7IP complex must be conducted.
Finally, our results could provide a potential explanation for the discrepancy between two previous studies on the regulation of methyltransferase activity of SETDB1 by ATF7IP 5, 34. In both these studies, SETDB1 that was produced in insect cells was used. Furthermore, SETDB1 or the SETDB1–ATF7IP complex was produced by expression of SETDB1 alone or by co‐expression of the two proteins in the insect cells, respectively. The enzymatic activity of SETDB1 was compared in the two samples in in vitro. ATF7IP was observed to enhance the enzymatic activity of SETFB1 in a previous study (SETDB1‐mediated conversion of H3K9me2 to H3K9me3) 5. However, in these studies, the ubiquitination status of each SETDB1 sample was not known. Since the level of co‐expressed ATF7IP influences the ubiquitination level of SETDB1, if there was a difference in the expression of ATF7IP and SETDB1 in insect cells in the two studies, the enzymatic activity of SETDB1, which is activated by ubiquitination, would be different. Thus, we speculated that the ubiquitination level of SETDB1 in the complex was higher in the ATF7IP co‐expressed condition shown in the study by Wang et al 5, but not in the study conducted by Basavapathruni et al 34, and that the difference in the ubiquitination status between the two studies could be the reason for inconsistent results. In support of this idea, the addition of ATF7IP after the purification did not increase the enzymatic activity of SETDB1 in in vitro as shown in the study conducted by Wang et al 5. Further examination would be required for proving this hypothesis.
Taken together, our findings provided a comprehensive insight that links the regulation of localization and enzymatic activity of SETDB1 with ATF7IP, complements the previous findings by others, and contributes to the understanding of the regulation of histone‐modifying enzymes.
Materials and Methods
Cell culture and DNA transfection
mESCs and HEK293T cells were cultured as described previously 35. The mESC lines used in this study as follows: JM8 constitutively hCas9 infected with MSCV‐GFP 33 and its derivatives, Atf7ip KO (clone name: TT#2‐5 and TT#2‐12) 35; TT2 33#6 Setdb1 flox/‐ conditional KO mESCs 19. Stably transfected cell lines were established by the piggyBac transposon system. DNA transfection was performed using Lipofectamine 2000 (Invitrogen, USA; for mESCs) or Polyethylenimine Max (Polyscience, Inc., USA; for HEK293T cells), according to the manufacturer's instructions. For transient transfection experiments with HEK293T cells, cells were seeded at the day before transfection. At 24–48 h after the transfection, the transfected cells were used for each analysis. For nuclear export and proteasomal degradation inhibition experiments, cells were treated with 10 ng/ml LMB (L2931, Sigma‐Aldrich) and/or 100 nM bortezomib (021‐18901, Wako) for 5 h before analysis.
Plasmids
Full‐length mouse ATF7IP expression vector was described previously 35. Full‐length SETDB1 cDNA was amplified from the SETDB1‐expression vector 19, and the amplicon was inserted into pPB‐CAG‐V5‐IRES.puromycine vector, pPB‐CAG‐3xFLAG‐IRES.puromycine vector, and pPB‐CAG‐3xFLAG‐NLS‐IRES.puromycine vector that contains an NLS sequence of c‐myc (“PAAKRVKLD”), by In‐Fusion technology. All the mutant vectors were produced by over‐lapping PCR with primers harboring mutations. For making ATF7IP KO HEK293T cells by CRISPR/Cas9, gRNA of ATF7IP was subcloned into pL‐CRISPR.EFS.tRFP 42 or pKLV2‐U6.gRNA(Bbs1)‐PGK.puro‐BFP 43. The detailed information of plasmids and used primers is given in supplemental file (Table EV1).
Establishment of ATF7IP KO HEK293T cells
For inactivation of ATF7IP in HEK293T cells, we designed gRNA on 5′ upstream and 3′ downstream of exon 5 of ATF7IP (in intron) and synthesized 5′ upstream and 3′ downstream of exon 5 gRNA oligonucleotide was subcloned into the gRNA expression vector, pL‐CRISPR.EFS.tRFP which also expresses hCas9 and RFP 42 and pKLV2‐U6.gRNA(Bbs1)‐PGK.puro‐BFP which also expresses BFP 43, respectively. Two days after transfection of these two gRNA expression vectors, RFP+BFP double‐positive population was sorted out. To isolate ATF7IP KO HEK293T cell clones, limiting dilution was performed and each single clone was examined for exon 5 deletion by PCR. Deletion of exon 5 which encodes amino acids 549–597 of human ATF7IP induces also frame shift mutation and it is expected to produce no functional products from this allele. By PCR screening, we identified two homozygous exon 5 deletion mutant (ATF7IP KO) clones, #21 and #40. We mostly used #21 in this study.
Western blot and quantitative PCR analysis
Both analyses were performed as described previously 35. The antibodies used in this study are described in the Antibodies section. To obtain relatively clear doublet bands of SETDB1, samples were separated in 6% acrylamide mini‐gels with 10 wells and run for 50 min at 200 V constant. The primers used in qPCR analyses are described in supplemental file (Table EV1).
Immunoprecipitation
For immunoprecipitation (IP), cells were lysed with the normal‐lysis buffer (50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Nonidet P‐40, 1 mM PMSF, 1× protease inhibitor cocktail), mid‐lysis buffer (50 mM Tris–HCl at pH 7.5, 300 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Nonidet P‐40, 1 mM phenylmethanesulfonyl fluoride (PMSF), 1× protease inhibitor cocktail; Nacalai Tesque, Japan), or high‐salt lysis buffer (50 mM Tris–HCl at pH 7.5, 500 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Nonidet P‐40, 1 mM PMSF, 1 × protease inhibitor cocktail; Nacalai Tesque, Japan). After centrifugation at 14,000 × g for 10 min, the supernatants were incubated with anti‐FLAG affinity gel (Sigma‐Aldrich, USA) or antibody‐conjugated Protein A and Protein G Dynabeads mix for at least 1 h to overnight at 4°C. The resin was then washed five times with the lysis buffer and eluted by 2× Laemmli sample buffer. Equivalent amounts of the input and the precipitates were subjected to standard WB analysis.
Antibodies
Following antibodies used for this study: anti‐α‐tubulin (clone B‐5‐1‐2, Sigma‐Aldrich); anti‐FLAG M2 antibody (F3165, Sigma‐Aldrich for Western blot; F7425, Sigma‐Aldrich for IF analysis); anti‐MCAF1/ATF7IP (anti‐MCAF 44; ab84497, Abcam); anti‐SETDB1/ESET (CP10377, CELL APPLICATIONS, for Western blot and IF analysis; 11231‐1‐AP, Proteintech for IP assay); anti‐V5 (R960‐25, Thermo Fisher which is same as #46‐0705, Life technology); anti‐H3K9me3 (2F3, CMA318) 45; anti‐mono‐ and poly‐Ub (clone FK2, purchased from Nippon Bio‐test laboratory).
Immunostaining
Immunostaining was performed as described previously with some minor modifications 46. mESCs (8.0 × 104 cells) were seeded on 8‐well chamber (192‐008, WATSON) that was pre‐coated with 10 μg/ml of laminin for at least 2 h at 37°C. After overnight culture, the cells were washed with PBS twice and fixed with 4% paraformaldehyde for 20 min at room temperature (RT). After fixation, the cells were permeabilized with 0.2% Triton X‐100 in PBS for 10 min at RT and were then incubated with 3% BSA/0.2% Tween‐20 in 4xSSC for 30 min at RT and with primary antibody for additionally 2 h at RT. After washing twice with 4xSSC, the cells were incubated with secondary antibodies conjugated with Alexa Fluor for 1 h at RT, washed with 4xSSC twice, and finally mounted with ProLong Diamond Antifade Mountant with DAPI (P36961, Thermo Fisher Scientific). HEK293T cells were seeded on cover glass that was pre‐coated with gelatin for overnight at 37°C. Images were obtained using a confocal microscope (FV3000, Olympus, Japan) and analyzed by Image J (1.50i). Data graphics were generated by R (3.4.1).
ChIP assay
For native ChIP analysis, 2 × 106 cells were suspended in 50 μl Buffer I (15 mM Tris–HCl, pH 7.5, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 0.3 M sucrose, 0.5 mM DTT, 1 mM PMSF, and 1× protease inhibitor cocktail). For lysis, equal amount of Buffer II (50 μl Buffer I plus 5 μl 10% NP‐40) was added to the cell suspensions, and after 10‐min incubation on ice, 800 μl Buffer III (15 mM Tris–HCl, pH 7.5, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 1.2 M sucrose, 0.5 mM DTT, 1 mM PMSF, and 1× protease inhibitor cocktail) was then added to them. After centrifugation at 7,300 × g for 10 min at 4°C, the pellets (chromatin) were re‐suspended in 200 μl MNase buffer (50 mM Tris–HCl, pH 7.5, 0.32 M sucrose, 4 mM MgCl2, 1 mM CaCl2, and 1 mM PMSF) and were then digested with 0.3 U micrococcal nuclease (Takara) at 37°C for 15 min. The digestion was stopped by the addition of 10 μl of 0.5 M EDTA. After centrifugation at 14,000 × g for 10 min at 4°C, the supernatants were 10‐fold diluted with incubation buffer (20 mM Tris–HCl, pH 7.5, 50 mM NaCl, 5 mM EDTA, 0.01% NP‐40, and 1 mM PMSF). For input samples, 80 μl of the obtained nucleosomes was used. For IP samples, 800 μl of the obtained nucleosomes was incubated with either anti‐H3K9me3‐conjugated magnetic beads (Dynabeads M‐280 Sheep anti‐mouse IgG, Invitrogen) at 4°C for overnight. After sequential washes with Wash buffer A (50 mM Tris–HCl, pH 7.5, 75 mM NaCl, 10 mM EDTA, 0.01% NP‐40), B (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.01% NP‐40), and C (50 mM Tris–HCl, pH 7.5, 175 mM NaCl, 10 mM EDTA, 0.01% NP‐40), the bound DNA was recovered and analyzed by qPCR.
FACS analysis and cell sorting
The analysis of GFP expression for the reporter retrovirus MSCV‐GFP integrated in the genome and sorting of the GFP‐positive population is carried out with FACSAria II (DB). We also sorted out GFP + BFP double‐positive populations for establishment of ATF7IP KO HEK293T cells after introduction of two gRNA expression vectors.
Statistics
For statistical comparisons in replicate experiments between two groups, Student's two‐tailed t‐test was carried out by Excel 2016. For multiple comparisons to a control, Dunnett's test was carried out by R (3.4.1).
Author contributions
TT conceived the idea, designed and performed the experiments, analyzed data, wrote the draft of the paper. CS performed the experiments. YS supervised the study, designed and performed the experiments, and participated in the data interpretation, conceptualization, and writing the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 5
Acknowledgements
We thank Dr. Mitsuyoshi Nakao (Kumamoto University) who generously provided anti‐ATF7IP antibody, Dr. Hiroshi Kimura (Tokyo Institute of Technology) who generously shared the hybridoma for anti‐H3K9me3, Drs. Shingo Kose, Takeshi Mizuno, Ken Matsumoto for their experimental advices, Mr. Kenji Ohtawa (RIKEN) for flow cytometric analysis and cell sorting, and Shinkai laboratory members for their support. We also thank Dr. Naoko Imamoto for critically reading this article, Drs. Mikiko C. Siomi and Kaoru Sato and Mr. Ken Ohsumi (University of Tokyo) for communication before publication and sharing their data of Drosophila SETDB1. This research was supported by KAKENHI (18H03991 and 18H05530) and a RIKEN internal research fund for Y.S.
EMBO Reports (2019) 20: e48297
See also: https://doi.org/10.15252/embr.201948296 (December 2019)
and https://doi.org/10.15252/embr.201949262 (December 2019)
References
- 1. Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- 2. Marmorstein R (2001) Protein modules that manipulate histone tails for chromatin regulation. Nat Rev Mol Cell Biol 2: 422–432 [DOI] [PubMed] [Google Scholar]
- 3. Badeaux AI, Shi Y (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol 14: 211–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alam H, Gu B, Lee MG (2015) Histone methylation modifiers in cellular signaling pathways. Cell Mol Life Sci 72: 4577–4592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang H, An W, Cao R, Xia L, Erdjument‐Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y (2003) mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell 12: 475–487 [DOI] [PubMed] [Google Scholar]
- 6. Mozzetta C, Boyarchuk E, Pontis J, Ait‐Si‐Ali S (2015) Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol 16: 499–513 [DOI] [PubMed] [Google Scholar]
- 7. Dodge JE, Kang YK, Beppu H, Lei H, Li E (2004) Histone H3‐K9 methyltransferase ESET is essential for early development. Mol Cell Biol 24: 2478–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan SL, Nishi M, Ohtsuka T, Matsui T, Takemoto K, Kamio‐Miura A, Aburatani H, Shinkai Y, Kageyama R (2012) Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 139: 3806–3816 [DOI] [PubMed] [Google Scholar]
- 9. Collins PL, Kyle KE, Egawa T, Shinkai Y, Oltz EM (2015) The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc Natl Acad Sci USA 112: 8367–8372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pasquarella A, Ebert A, Pereira de Almeida G, Hinterberger M, Kazerani M, Nuber A, Ellwart J, Klein L, Busslinger M, Schotta G (2016) Retrotransposon derepression leads to activation of the unfolded protein response and apoptosis in pro‐B cells. Development 143: 1788–1799 [DOI] [PubMed] [Google Scholar]
- 11. Takikita S, Muro R, Takai T, Otsubo T, Kawamura YI, Dohi T, Oda H, Kitajima M, Oshima K, Hattori M et al (2016) A histone methyltransferase ESET is critical for T cell development. J Immunol 197: 2269–2279 [DOI] [PubMed] [Google Scholar]
- 12. Koide S, Oshima M, Takubo K, Yamazaki S, Nitta E, Saraya A, Aoyama K, Kato Y, Miyagi S, Nakajima‐Takagi Y et al (2016) Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood 128: 638–649 [DOI] [PubMed] [Google Scholar]
- 13. Liu S, Brind'Amour J, Karimi MM, Shirane K, Bogutz A, Lefebvre L, Sasaki H, Shinkai Y, Lorincz MC (2014) Setdb1 is required for germline development and silencing of H3K9me3‐marked endogenous retroviruses in primordial germ cells. Genes Dev 28: 2041–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eymery A, Liu Z, Ozonov EA, Stadler MB, Peters AH (2016) The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development 143: 2767–2779 [DOI] [PubMed] [Google Scholar]
- 15. Kim J, Zhao H, Dan J, Kim S, Hardikar S, Hollowell D, Lin K, Lu Y, Takata Y, Shen J et al (2016) Maternal Setdb1 is required for meiotic progression and preimplantation development in mouse. PLoS Genet 12: e1005970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang L, Lawson KA, Teteak CJ, Zou J, Hacquebord J, Patterson D, Ghatan AC, Mei Q, Zielinska‐Kwiatkowska A, Bain SD et al (2013) ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev Biol 380: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lawson KA, Teteak CJ, Gao J, Li N, Hacquebord J, Ghatan A, Zielinska‐Kwiatkowska A, Song G, Chansky HA, Yang L (2013) ESET histone methyltransferase regulates osteoblastic differentiation of mesenchymal stem cells during postnatal bone development. FEBS Lett 587: 3961–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, Tanaka T, Tsutsumi S, Nakao M, Doi T et al (2015) H3K4/H3K9me3 bivalent chromatin domains targeted by lineage‐specific DNA methylation pauses adipocyte differentiation. Mol Cell 60: 584–596 [DOI] [PubMed] [Google Scholar]
- 19. Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464: 927–931 [DOI] [PubMed] [Google Scholar]
- 20. Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M et al (2011) DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8: 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D et al (2017) Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol 216: 3535–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kato M, Takemoto K, Shinkai Y (2018) A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat Commun 9: 1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho S, Park JS, Kang YK (2013) Regulated nuclear entry of over‐expressed Setdb1. Genes Cells 18: 694–703 [DOI] [PubMed] [Google Scholar]
- 24. Tachibana K, Gotoh E, Kawamata N, Ishimoto K, Uchihara Y, Iwanari H, Sugiyama A, Kawamura T, Mochizuki Y, Tanaka T et al (2015) Analysis of the subcellular localization of the human histone methyltransferase SETDB1. Biochem Biophys Res Comm 465: 725–731 [DOI] [PubMed] [Google Scholar]
- 25. Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ (2016) ATF7IP‐mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep 17: 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun L, Fang J (2016) E3‐independent constitutive monoubiquitination complements histone methyltransferase activity of SETDB1. Mol Cell 62: 958–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishimoto K, Kawamata N, Uchihara Y, Okubo M, Fujimoto R, Gotoh E, Kakinouchi K, Mizohata E, Hino N, Okada Y et al (2016) Ubiquitination of lysine 867 of the human SETDB1 protein upregulates its histone H3 lysine 9 (H3K9) methyltransferase activity. PLoS One 11: e0165766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ III (2002) SETDB1: a novel KAP‐1‐associated histone H3, lysine 9‐specific methyltransferase that contributes to HP1‐mediated silencing of euchromatic genes by KRAB zinc‐finger proteins. Genes Dev 16: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ichimura T, Watanabe S, Sakamoto Y, Aoto T, Fujita N, Nakao M (2005) Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem 280: 13928–13935 [DOI] [PubMed] [Google Scholar]
- 30. Minkovsky A, Sahakyan A, Rankin‐Gee E, Bonora G, Patel S, Plath K (2014) The Mbd1‐Atf7ip‐Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenetics Chromatin 7: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waterfield M, Khan IS, Cortez JT, Fan U, Metzger T, Greer A, Fasano K, Martinez‐Llordella M, Pollack JL, Erle DJ et al (2014) The transcriptional regulator Aire coopts the repressive ATF7ip‐MBD1 complex for the induction of immunotolerance. Nat Immunol 15: 258–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson PJ, Dulberg V, Moon KM, Foster LJ, Chen C, Karimi MM, Lorincz MC (2015) hnRNP K coordinates transcriptional silencing by SETDB1 in embryonic stem cells. PLoS Genet 11: e1004933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fukuda K, Okuda A, Yusa K, Shinkai Y (2018) A CRISPR knockout screen identifies SETDB1‐target retroelement silencing factors in embryonic stem cells. Genome Res 28: 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Basavapathruni A, Gureasko J, Porter Scott M, Hermans W, Godbole A, Leland PA, Boriack‐Sjodin PA, Wigle TJ, Copeland RA, Riera TV (2016) Characterization of the enzymatic activity of SETDB1 and its 1:1 complex with ATF7IP. Biochemistry 55: 1645–1651 [DOI] [PubMed] [Google Scholar]
- 35. Tsusaka T, Kikuchi M, Shimazu T, Suzuki T, Sohtome Y, Akakabe M, Sodeoka M, Dohmae N, Umehara T, Shinkai Y (2018) Tri‐methylation of ATF7IP by G9a/GLP recruits the chromodomain protein MPP8. Epigenetics Chromatin 11: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yeap LS, Hayashi K, Surani MA (2009) ERG‐associated protein with SET domain (ESET)‐Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenetics Chromatin 2: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koch CM, Honemann‐Capito M, Egger‐Adam D, Wodarz A (2009) Windei, the Drosophila homolog of mAM/MCAF1, is an essential cofactor of the H3K9 methyl transferase dSETDB1/Eggless in germ line development. PLoS Genet 5: e1000644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hans F, Fiesel FC, Strong JC, Jackel S, Rasse TM, Geisler S, Springer W, Schulz JB, Voigt A, Kahle PJ (2014) UBE2E ubiquitin‐conjugating enzymes and ubiquitin isopeptidase Y regulate TDP‐43 protein ubiquitination. J Biol Chem 289: 19164–19179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mutlu B, Chen HM, Moresco JJ, Orelo BD, Yang B, Gaspar JM, Keppler‐Ross S, Yates JR III, Hall DH, Maine EM et al (2018) Regulated nuclear accumulation of a histone methyltransferase times the onset of heterochromatin formation in C. elegans embryos. Sci Adv 4: eaat6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Delaney CE, Methot SP, Guidi M, Katic I, Gasser SM, Padeken J (2019) Heterochromatic foci and transcriptional repression by an unstructured MET‐2/SETDB1 co‐factor LIN‐65. J Cell Biol 218: 820–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Osumi K, Sato K, Murano K, Siomi H, Siomi MC (2019) Essential roles of Windei and nuclear monoubiquitination of Eggless/SETDB1 in transposon silencing. EMBO Rep 20: e48296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, Ebert BL (2014) Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR‐Cas9 genome editing. Nat Biotechnol 32: 941–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koike‐Yusa H, Li Y, Tan EP, Velasco‐Herrera Mdel C, Yusa K (2014) Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol 32: 267–273 [DOI] [PubMed] [Google Scholar]
- 44. Fujita N, Watanabe S, Ichimura T, Ohkuma Y, Chiba T, Saya H, Nakao M (2003) MCAF mediates MBD1‐dependent transcriptional repression. Mol Cell Biol 23: 2834–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayashi‐Takanaka Y, Yamagata K, Wakayama T, Stasevich TJ, Kainuma T, Tsurimoto T, Tachibana M, Shinkai Y, Kurumizaka H, Nozaki N et al (2011) Tracking epigenetic histone modifications in single cells using Fab‐based live endogenous modification labeling. Nucleic Acids Res 39: 6475–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferry L, Fournier A, Tsusaka T, Adelmant G, Shimazu T, Matano S, Kirsh O, Amouroux R, Dohmae N, Suzuki T et al (2017) Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol Cell 67: 550–565 e5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 5