

Abstract
Deciphering the pathophysiologic events in prion diseases is challenging, and the role of posttranslational modifications (PTMs) such as glypidation and glycosylation remains elusive due to the lack of homogeneous protein preparations. So far, experimental studies have been limited in directly analyzing the earliest events of the conformational change of cellular prion protein (PrPC) into scrapie prion protein (PrPSc) that further propagates PrPC misfolding and aggregation at the cellular membrane, the initial site of prion infection, and PrP misfolding, by a lack of suitably modified PrP variants. PTMs of PrP, especially attachment of the glycosylphosphatidylinositol (GPI) anchor, have been shown to be crucially involved in the PrPSc formation. To this end, semisynthesis offers a unique possibility to understand PrP behavior in vitro and in vivo as it provides access to defined site‐selectively modified PrP variants. This approach relies on the production and chemoselective linkage of peptide segments, amenable to chemical modifications, with recombinantly produced protein segments. In this article, advances in understanding PrP conversion using semisynthesis as a tool to obtain homogeneous posttranslationally modified PrP will be discussed.
Keywords: glycosylphosphatidylinositol (GPI) anchor, membrane interaction, prion protein (PrP), protein semisynthesis
The key pathophysiologic event in prion diseases is based on a conformational change of cellular (PrPC) into scrapie prion protein (PrPSc) and is closely linked to posttranslational modifications (PTMs). Semisynthesis offers a unique opportunity to study the impact of PTMs on prion conversion, transmission, and pathogenicity, which is the major focus of this review.
Abbreviations
- aa
amino acid
- Aβ
amyloid‐β
- AD
Alzheimer disease
- ADAM
a disintegrin and metalloproteinase
- Adgrg6
adhesion G protein–coupled receptor G6
- AFM
atomic force microscopy
- AL
amyloid light chain
- ALS
amyotrophic lateral sclerosis
- ATPase
adenosine 5′‐triphosphatase
- BSE
bovine spongiform encephalopathy
- CBD
chitin‐binding domain
- CCV
clathrin‐coated vesicle
- CD
circular dichroism
- Cdc‐42
cell division control protein 42 homolog
- CDP
chronic demyelinating polyneuropathy
- CFC
cell‐free conversion
- CJD
Creutzfeld‐Jakob disease
- CNS
central nervous system
- cryo‐EM
cryo electron microscopy
- CWD
chronic wasting disease
- DOPC
1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine
- DRM
Detergent‐resistant membrane
- E coli
Escherichia coli
- eQuIC
enhanced quaking‐induced conversion
- EPL
expressed protein ligation
- ER
endoplasmic reticulum
- ESR
electron spin resonance
- FFI
fatal familial insomnia
- FTD
frontotemporal dementia
- GPI AP
glycosylphosphatidylinositol anchored protein
- GSS
Gerstmann‐Sträussler‐Scheinker syndrome
- Hsp
heat shock protein
- KD
knockdown
- Kd
dissociation constant
- LC
light chain
- LRP1
low‐density lipoprotein receptor‐related protein 1
- MESNA
sodium 2‐mercaptoethanesulfonate
- MSA
multiple system atrophy
- Mxe
mycobacterium xenopi gyrA
- NCL
native chemical ligation
- NMR
nuclear magnetic resonance
- OR
octapeptide repeats
- ORF
open reading frame
- PAc
phenacyl
- PD
Parkinson disease
- PDB
protein data bank
- PEG
polyethyleneglycol
- PI‐PLC
phosphatidylinositol‐specific phospholipase C
- PIRIBS
parallel in‐register intermolecular β‐sheet
- PK
proteinase K
- PMCA
protein misfolding cyclic amplification
- POPG
1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐(1′‐rac‐glycerol) (sodium salt)
- PPO
polyethyleneglycol polyamide oligomer
- PrP
prion protein
- PrPC
cellular prion protein
- PrPres
(PK‐)resistant prion protein
- PrPSc
scrapie prion protein
- PTM
posttranslational modification
- PTS
protein trans‐splicing
- QuIC
quaking‐induced conversion
- RT‐QuIC
real‐time quaking‐induced conversion
- ScN2a
Scrapie‐infected mouse neuroblastoma cells
- SHa
Syrian hamster
- SPPS
Solid phase peptide synthesis
- S‐QuIC
standard quaking‐induced conversion
- TEV
tobacco etch virus
- ThT
thioflavin T
- TSEs
transmissible spongiform encephalopathies
1. PRION DISEASES
Prion diseases or transmissible spongiform encephalopathies (TSEs) are incurable, neurodegenerative disorders affecting humans and animals.1 They include scrapie of sheep and goats, bovine spongiform encephalopathy (BSE) of cattle, chronic wasting disease (CWD) of cervids, and several human diseases such as kuru, Creutzfeld‐Jakob disease (CJD), Gerstmann‐Sträussler‐Scheinker syndrome (GSS), and fatal familial insomnia (FFI). The disease progression is accompanied by the loss of cognitive skills and neuronal dysfunction and can be of inherited sporadic or iatrogenic origin.2, 3 The central pathophysiologic event is ascribed to the conformational change of the cellular prion protein (PrPC) into scrapie prion protein (PrPSc) that then not only propagates further PrPC misfolding in neighboring cells but can also infect other organisms.4 Identification of the infective pathogen of prion diseases and its proof of transmissibility started in the 1950s. By ending cannibalism within the Fore tribe in Papua New Guinea, the transmission of kuru could be stopped. Experiments with transferring brain samples of these kuru victims into primates induced spongiform encephalopathies.5 Due to its infective property, the pathogen was first assumed to be of nucleic acid‐based, viral nature. However, the application of ultraviolet and ionizing irradiation failed to inactivate the agent, leading to the “protein‐only hypothesis” by Griffith in 1967.6, 7 Eventually in 1982 the term “prion” defining a small proteinaceous infectious particle was introduced by Prusiner during the course of discovering the prion protein (PrP).8, 9 PrP 27‐30 corresponding to the protease‐resistant core of PrPSc with an apparent molecular mass of 27 to 30 kDa was isolated by enriching fractions from Syrian hamster (SHa) brain for scrapie infectivity.10, 11, 12, 13 Successful Edman degradation paved the way for subsequent molecular cloning studies of the PrP gene.14, 15, 16 The linkage of PrPSc to prion diseases was recognized as an important feature of the protein, together with its role in transmission and pathogenesis of these illnesses.17 Thus, the main focus of elucidating prion pathogenicity is assigned to PrP. Understanding the key features in prion diseases can serve as paradigm for other neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), Alzheimer disease (AD), and Parkinson disease (PD), that are characterized by misfolded proteins “prionoids” sharing the aggregation properties but being not strictly infectious.18, 19, 20 As it happens, the latter statement might not be entirely correct. Recent prion research reported the discovery of α‐synuclein prions21 in multiple system atrophy (MSA) and iatrogenic AD with evidence of transmissibility of amyloid‐β (Aβ),22 hence highlighting the need to understand prion transmission and toxicity even more.
2. PROPERTIES AND STRUCTURES OF THE PrP
2.1. Cellular prion protein
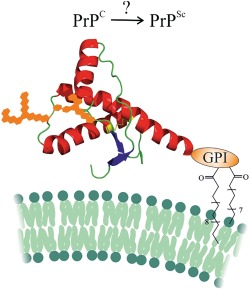
High expression levels of PrPC are found in the central nervous system (CNS), but it exists in other cell types and tissues, such as lymphoid organs, as well.23, 24, 25, 26 Accessing the gene‐encoding SHaPrPC, Prnp,14, 27 entailed its further identification in numerous other species and illustrated a highly conserved sequence.28, 29 The entire open reading frame (ORF) is contained within a single exon and primarily translates into a protein composed of 254 amino acids (aas).30, 31 The first 22 aas reflect an N‐terminal signal sequence for PrP entering the secretory pathway. Upon its cleavage, glycosylation at asparagine residues and formation of a disulfide bond occur in the endoplasmic reticulum (ER). Lastly, cleavage of the C‐terminal signal sequence facilitates the attachment of the glycosylphosphatidylinositol (GPI) anchor, providing mature, posttranslationally modified PrP at the outer leaflet of the cell membrane, typical for glycosylphosphatidylinositol anchored proteins (GPI APs).32 Interestingly, PrP can be found in three topologic forms at the ER. Apart from the fully translocated PrP, two transmembranal types occur with the N‐ or C‐terminus facing the ER lumen, denoted as NtmPrP or CtmPrP, respectively.33, 34 Normally, NtmPrP and CtmPrP only comprise a small portion of PrPC, whereas an excess of CtmPrP induces neurotoxicity. Neuronal cell death is caused in the absence of PrPSc formation, obviously by an aberrant metabolism of PrPC. PrPC mislocalization represents another mechanistic possibility for prion toxicity next to the alteration of PrPC‐mediated signaling and PrP‐derived oligomeric species.23 First structural studies on PrPC isolated from brains of SHas demonstrated a predominantly α‐helical content.35 As these measurements agreed well with subsequent spectroscopic data of recombinant PrP, accessible in larger amounts, it was considered an appropriate surrogate in biochemical experiments,36, 37, 38, 39 as well as in solving nuclear magnetic resonance (NMR) and crystal structures of PrP.40, 41, 42, 43, 44, 45, 46, 47 The PrP structure comprises an unstructured N‐terminal (aa 23‐120) and a globular C‐terminal part (aa 121‐231) (Figure 1).
Figure 1.
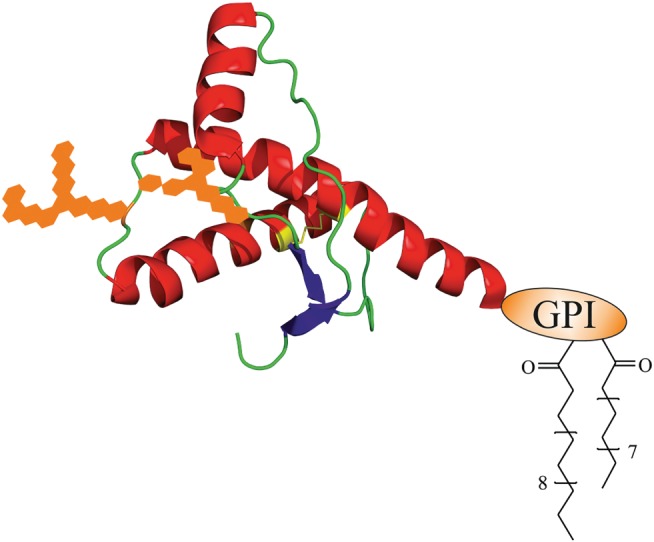
Tertiary structure of cellular prion protein (PrPC) with posttranslational modifications (PTMs). The structure is based on nuclear magnetic resonance (NMR) measurements of human PrPC (aa 23‐230) by Zahn et al (PDB 1QLZ)47
In more detail, the N‐terminal segment consists of a nonapeptide (PQGGGGWGQ) followed by four octapeptide (PHGGGWGQ) repeats (OR) with a high affinity for copper,48, 49, 50, 51 and other divalent cations,52 adjacent to a charged cluster (CC) or polybasic region (Figure 2). Noteworthy, the configuration of the copper binding region in hPrP (aa 23‐231) has been determined combining different experimental methods by using synthetic octapeptide and tetraoctapeptide as well as full‐length hPrP.53, 54, 55, 56 Depending on the concentration of the metal and pH, the OR region is capable to bind up to four copper ions in distinct coordination geometries.50, 57 Current estimates for dissociation constant (K d) values vary betweeen the micromolar and femtomolar range.50 The central hydrophobic domain (HD), comprising of aas 113 to 135, serves as a transmembrane domain33 and includes a palindromic region (AGAAAAGA, aa 113‐120) thought to be important in the PrPC‐PrPSc conversion.58, 59 Within the C‐terminal region, three α‐helices (aa 144‐154, 175‐193, and 200‐219), with two of them connected by a disulfide bond,60 and a small antiparallel β‐sheet (aa 128‐131 and 161‐164) are present. As posttranslational modifications (PTMs), a C‐terminal GPI anchor linked to serine 231 and two N‐linked glycosylation sites at asparagines 181 and 197 exist. PrPC can occur in nonglycosylated, monoglycosylated, and diglycosylated forms.61, 62 Variations in glycan structures attached to PrP may be differentially distributed depending on the areas of the CNS.63 Molecular dynamics simulations indicate that the N‐linked oligosaccharides located at two helices within the structured region of PrP contribute to its stabilization in generating a negative electrostatic field covering the helical surface,64 thus impacting strain diversity and prion infection.65, 66, 67, 68 The C‐terminal GPI anchor tethers PrP to the outer leaflet of the plasma membrane.69
Figure 2.
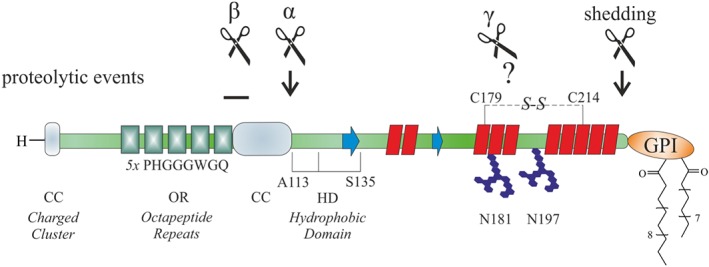
Schematic outline of the primary cellular prion protein (PrPC) structure. Residue numbers correspond to hPrPC
It has been postulated that mutations in the Prnp gene facilitate the pathogenic process by destabilizing the tertiary structure of PrPC. More than 30 mutations in Prnp could be linked to inherited prion diseases.70 In affecting the primary sequence of PrP, concomitant changes in its 3D structure may arise, and not cause, but influence a person's risk of developing a disease. Indeed, thermodynamic measurements of mutated PrP variants indicated destabilizing effects only for some of them.71 For example by comparing the wild‐type variant to the E200K mutant almost identical structures resulted, but major perturbations of the surface electrostatic potential were found. This suggests that these defects cause abnormalities in PrP interactions and should be considered as key determinants in the misfolding process.72
Moreover, it has been speculated that methionine oxidation in PrPC plays a destabilizing role and supports spontaneous conversion into PrPSc. Wolschner et al73 found a strong proaggregation behavior for hPrPC with oxidized methionine residues and a variant with methionine replaced by hydrophilic methoxinine as a stable substitute for oxidation‐sensitive methionine. These findings suggest a pivotal role of oxidative stress in PrP conversion.
2.2. Scrapie prion protein
PrPSc is the toxic, misfolded isoform of PrP. It is, as PrPC, encoded by the Prnp gene and exhibits identical PTMs, but distinct structural, biochemical, and physiological features.13 Despite a large interest in elucidating the structure of PrPSc, there are only limited data about its molecular details available.74 To date, obtaining a high‐resolution structure of PrPSc has been impaired by its insolubility, propensity to aggregate, and heterogeneity. Structural variations, such as differences in the glycosylation patterns, suggested to correlate with biochemical changes, including the extent of the proteinase K (PK) resistance, the electrophoretic mobility of the proteolytic fragments, and the conformational stability, depend on the distinct strains and complicate the determination of PrPSc structure.75 Besides, in agreement with discussions from the Prion 2018 round tables,76 the diversity of PrP assemblies implicates that there may be no single PrPSc structure. Data generated by biochemical and physical methods, such as spectroscopy analysis, electron microscopy, and limited proteolysis, have led to several 3D structural models. Govaerts and colleagues suggested that left‐handed β‐helices assembled into trimers, also known as the 4‐rung β‐solenoid model.77 Based on electron spin resonance (ESR) measurements, Cobb et al proposed a parallel in‐register intermolecular β‐sheet (PIRIBS) architecture where PrPSc consists of β‐strands and short turns and/or loops with no residual α‐helices.78 Still, so far, all models display discrepancies with experimental data.79
Notably, cryo electron microscopy (cryo‐EM) is a technique providing high‐resolution structures.80, 81 By rapidly cooling samples, proteins can be observed in their native state. In 2016, Wille and colleagues82 employed cryo‐EM to analyze GPI anchorless PrP 27‐30 amyloid fibrils. PrP 27‐30 was purified from brains of transgenic mice infected with prions. Further inoculation of wt mice with the purified GPI anchorless PrP 27‐30 confirmed the development of typical neurological signs of prion disease. The structure of GPI anchorless PrP 27‐30 amyloid fibrils was found to agree with a 4‐rung β‐solenoid architecture; 3D reconstruction revealed two distinct protofilaments and an average molecular height of approximately 17.7 Å. However, Collinge, Wadsworth, and coworkers83 studied the structural features distinguishing infectious ex vivo mammalian prions from noninfectious fibrillar assemblies generated in vitro. Applying cryo‐EM and atomic force microscopy (AFM) measurements noninfectious recombinant PrP fibrils were identified as 10‐nm‐wide single fibers with a double helical repeating substructure, agreeing with the structure described by Wille and colleagues.82 Prion‐infected transgenic mice replicate prions, but they mainly develop PrP amyloid plaques, which are not seen in prion‐inoculated wt mice.84, 85 Caughey and coworkers86, 87, 88 have described two morphologically distinct PrP fibril assemblies in prion‐infected transgenic mice. Therefore, considering the lower infecitivity titer of PrP 27‐30 in the studies of Wille and colleagues,82 it appears that the more abundant, single nonglycosylated PrP fibrils, corresponding essentially to recombinant PrP, has been described rather than the infectious glycosylated PrP rods. Collinge, Wadsworth, and coworkers83 characterized infectious PrP rods, 20 nm in width, that contained two fibers, each with a double helical repeating substructure separated by a central gap of 8 to 10 nm. The gap between the paired fibers consists of irregularly structured material compositionally distinct to the protein surface. Thus, it was proposed as a location of the N‐linked glycans of PrP. The structure of the infectious PrP rods differentiates them from all other protein assemblies so far studied in neurodegenerative diseases. This includes characterizations by cryo‐EM of tau filaments from AD89 and monoclonal immunoglobulin light chain (LC) fibrils from amyloid light‐chain (AL) amyloidosis.90 To date, cryo‐EM studies of tau and AL represent the only structural data of fibrils directly extracted from human tissue under pathologic conditions. For tau‐paired helical and straight filaments could be identified with cores made of two identical protofilaments that adopt a combined cross‐β/β‐helix structure. AL fibrils were found to be helical with a single protofilament showing a cross‐β architecture. It is widely accepted that during the PrPC‐PrPSc conversion, the β‐strand content increases vastly91, 92 and the PK resistance of the “folded core” (aa approximately 90‐231), as well.9, 14 Whereas PrPC is dominated by α‐helices, monomeric, soluble, and highly susceptible to proteolytic digestion, PrPSc contains predominantly β‐sheets (>43%),92 aggregates into amyloid fibrils,93 is insoluble in detergents and partially resistant to proteolysis.35, 94 These biochemical differences between the PrP isoforms appear to be associated with the changes of the secondary structure in PrPSc.
3. PHYSIOLOGY OF THE PrP
3.1. Function of PrPC
Although the relevance of PrPC in TSEs is widely accepted, its physiological function remains enigmatic. Studies with PrP knockout mice have failed on this regard. Transgenic mice lacking PrP were found to develop normally.95, 96 A multitude of functions has been ascribed to PrP in different tissues, cells, and experimental settings, although not always without controversy or questionable reproducibility. Among others, PrPC has been connected to developmental processes,97 cell adhesion,98, 99 neurite outgrowth, synapse formation,100, 101, 102, 103, 104 neuroprotection,105, 106, 107 and regulation of the circadian rhythm.108 Moreover, there is evidence for PrP contributing to myelin maintenance,109, 110, 111, 112 cellular homeostasis of divalent cations,113, 114, 115 and signaling events.116, 117, 118 A more detailed discussion can be found in reviews by the group of Aguzzi.109, 119 Recently, other functions have been attributed to PrPC, that is acting as a receptor for the aggregated proteins Aβ oligomers120, 121, 122 and α‐synuclein.123 By mediating the uptake of Aβ and α‐synuclein, PrPSc is unable to replicate in their presence.
Already 10 years ago, it was suspected that reports on the function of PrP represent just specific aspects of a more complex physiological role of PrPC.23 Causes for the functional diversity of PrPC might not only be its alternating transient binding partners in different cellular locations but also its proteolytic processing.124, 125 For once, PrP fragmentation may inhibit association with some binding partners while possibly allowing new interactions with others. Then again, the cleaved products may act as soluble ligands facilitating protein interactions over large distances. These findings contribute to the biological complexity of the physiological function of PrP. In fact, four different but highly conserved cleavage events have been significantly characterized (Figure 2).126, 127, 128 During transport to the cell surface, α‐cleavage results in a soluble flexible N1‐ and a globular membrane‐bound C1 part. Myelin maintenance has been initially linked to this C1 part derived from α‐cleavage. Mice expressing PrP mutants not able to undergo α‐cleavage suffered from chronic demyelinating polyneuropathy (CDP).110, 111, 129 Interestingly, recent work found a specific ligand role of the flexible N1 part towards the G‐protein coupled receptor Adgrg6, promoting myelin homeostasis.112 During shedding, PrPC is released from the plasma membrane by a disintegrin and metalloproteinase (ADAM) enzyme, namely, ADAM10, in a glycosylated form without the GPI anchor and designated as “shed PrP.”130 Although definite functions of “shed PrP” are not known to date, the shedding process regulates the membrane levels of PrPC and thus its functions at the cell surface. Similar to α‐synuclein,123 recent work by Jarosz‐Griffiths et al131 found that the toxicity and cellular binding of Aβ oligomers can be reduced by shedding of PrPC, thereby pointing towards a contribution as a receptor in AD. Moreover, PrPC is expressed in immune cells as well, particularly on mast cells.132 Upon activation of these cells, the PrPC shedding process is enhanced, proposing PrP involvement in the inflammatory mast cell response. Under pathological conditions and in response to oxidative stress, incidences of β‐cleavage occurring around aa position 90 are increased.133, 134 Lastly, γ‐cleavages restricted to unglycosylated PrP generate a large soluble N3 and a short C3 part by taking place in a region between aas 170 and 200. While prevalence and relevance of this cleavage requires further investigation, increased amounts of C3 in CJD brain samples suggest a pathophysiological role.135
Despite multiple evidence of PrP in physiological processes, the functional diversity based on its manifold binding partners and proteolytic fragments complicate an exact definition of its physiological function. Yet successful elucidation of pathways and roles of PrP could help to understand its linkage to toxicity in prion diseases and to other neurodegenerative diseases.136
3.2. Trafficking of PrPC
As the PrP function is closely intertwined with the cellular compartments where the protein is located, having a closer look at trafficking may assist in elucidating its involvement in pathological and physiological processes. PrPC is tethered via its GPI anchor to the outer leaflet of the plasma membrane.69 In 1993, data by Shyng et al137 revealed constitutive cycling of PrPC between the cell surface and endocytic compartments on varying times scales dependent on the cell line, as demonstrated in later work.138 From the cellular membrane, PrPC can enter the cell via multiple pathways, mediated mainly by the unstructured N‐terminal domain.139, 140 Evidence for a cooperation between clathrin138, 141, 142 and rafts143, 144, 145 in the internalization of PrPC was found.146 Clathrin is a large, oligomeric protein assembling into lattice structures on the inner surface of the plasma membrane. Thereby, it causes the membrane to invaginate and pinch off to form clathrin‐coated vesicles (CCVs), which can then fuse with other intracellular organelles.147 Although a clathrin‐dependent internalization might appear unusual since PrP lacks a cytoplasmic domain necessary for the direct interaction with clathrin and the adaptor protein, GPI APs can indeed enter the clathrin‐dependent pathway upon interaction with transmembrane proteins possessing a clathrin‐coated pit internalization signal.144 Moreover, the endocytosis of PrPC was found to be associated with the low‐density lipoprotein receptor‐related protein 1 (LRP1)142, 148 that belongs to a receptor family of cell‐surface transmembrane proteins capable of binding a variety of ligands and internalizing via clathrin‐coated pits.149, 150 As a nonclassical clathrin‐independent pathway, the raft‐dependent internalization route distinguishes caveolae‐dependent and caveolae‐independent endocytosis.151 Caveolae are membrane invaginations, originating from the oligomerization of caveolins, their integral coat proteins, and are considered to be specialized raft domains.152, 153 Due to the presence of PrPC in caveolae‐like domains154, 155 and its colocalization with caveolin‐1 (cav‐1),143, 156 the involvement of caveolae in PrPC endocytosis had been suggested earlier. To this end, Sarnataro et al157 could provide evidence that the raft‐mediated pathway is not affected by caveolin expression. Still, PrPC internalization was found to be impacted by cholesterol depletion and activation of the cell division control protein 42 homolog (Cdc‐42), a member of the Rho family of GTPases being specifically involved in clathrin‐independent endocytosis of GPI APs.158 Additionally Sarnataro et al157 reported that in coimmunoprecipitation studies of clathrin and PrPC, the latter remained associated with detergent‐insoluble microdomains. This fact supports a cooperation between rafts and clathrin in the internalization process. PrPC susceptibility to various endocytic pathways could also be the basis for its neuroprotective and neurodegenerative functions.
4. MECHANISM OF PrPC‐PrPSc CONVERSION
To date, despite considerable knowledge about the characteristics of the infective prion pathogen, its mechanism of replication and the molecular pathways leading to neurodegeneration are largely unknown. There is evidence from in vitro and transgenic mouse studies that the conversion to PrPSc implicates PrPC‐PrPSc interactions.84, 159, 160, 161, 162, 163 The rate of PrPSc formation and disease progression appears to be directly proportional to the level of PrPC expression, indicated by PrP knockout mice not propagating scrapie infectivity and transgenic mice heterozygous for a disrupted PrP gene requiring prolonged incubation times upon prion inoculation.164, 165, 166 In agreement with the “protein‐only hypothesis,” these findings have raised two models explaining prion replication (Figure 3). The template‐directed refolding model by Prusiner167 proposes that a high‐energy barrier prevents the spontaneous PrPC‐PrPSc conversion. Upon interaction, monomeric PrPSc induces PrPC to convert into PrPSc. However, until now, there is no experimental evidence for the existence of a stable PrPSc monomer.168 PrPSc seeds in this prion propagation process are not considered essential. Alternatively, in the more accredited seeded nucleation model by Jarrett and Lansbury,169 a reversible thermodynamic equilibrium between PrPC and PrPSc is postulated. In the presence of stable oligomeric PrPSc aggregates, the conversion from PrPC to PrPSc is favored, thus making PrPSc aggregates (seeds) inevitable for prion spread. Fragmentation of PrPSc aggregates increases the number of nuclei capable of recruiting further PrPSc. In fact, these soluble oligomers produced during the PrP amyloid aggregation have emerged as the primary neurotoxic species, supporting the seeded nucleation model.170, 171, 172
Figure 3.
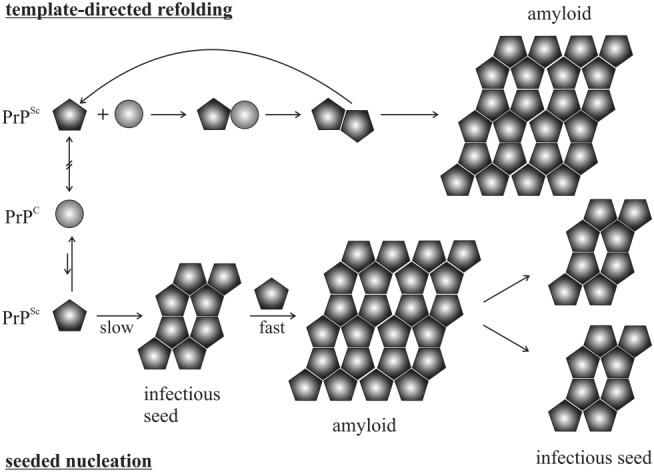
Models for the conversion of cellular prion protein (PrPC) into scrapie prion protein (PrPSc). The model for template‐directed refolding (top) and seeded nucleation (bottom) are depicted. The figure was modified from Aguzzi and Calella23
Ultimately, evidence for a direct PrPC‐PrPSc interaction in the conversion to PrPSc came from in vitro systems. Pioneering studies from Caughey and colleagues173 succeeded within a cell‐free conversion (CFC) assay in the generation of protease‐resistant (res), radioactive PrPres from mixed PrPC substrate and an excess of unlabeled PrPSc. This in vitro PrPres propagation recapitulates the species and strain specificity of prion transmission in vivo.173, 174 Mechanistically, it has identified structural factors underlying the species barrier and optimal conditions for the PrPres formation.66, 175, 176 The ability to generate PrPres not only from purified but also recombinant protein177 provides a unique opportunity to study prion propagation. CFC assays can be used as screening experiments as they have the potential to identify compounds directly inhibiting the PrPC‐PrPSc interaction or its subsequent conversion.178, 179 Still, the proportionally large amount of PrPSc seeds required to drive the CFC assay (PrPSc:PrPC = 50:1) has prevented it from generating de novo infectivity.180
A more efficient method for mimicking the autocatalytic replication of PrPSc was provided by Soto and colleagues38 in affording a larger than 10‐fold increase in PrPres with the usage of a 1:100 ratio of PrPSc to PrPC. By subjecting scrapie‐infected and normal brain homogenate to the so‐called protein misfolding cyclic amplification (PMCA) procedure, PrPres is amplified in cycles of sonication and incubation. Successive rounds of PMCAs and fragmentation of PrPSc rise the available amounts of replication‐competent species.181, 182, 183 Thus, with automation, this assay offers a promising diagnostic tool in pre‐symptomatic blood screening,184, 185 and eventually, it has facilitated the detection of de novo infectivity in hamsters.38 However, the levels of infectivity still remain lower than with a similar quantity of brain‐derived PrPSc, and the usage of complex brain homogenate itself represents an obstacle in thoroughly elucidating the conversion and association of infectivity with PrPres.186 Besides, the distinct efficiency differences between the CFC and PMCA assays applying purified and crude brain‐derived PrPSc proposed that cellular accessory factors are involved in the generation of PrPres. In fact, polyanionic molecules were identified as factors present in the brain homogenate that contribute to the conversion efficiency.187 Ongoing development of PMCA assays aiming to detect and early diagnose TSEs has led to the quaking‐induced conversion (QuIC) method.188 Sonication is replaced by reproducible and easier controllable shaking during the prion amplification process, which enables the application of standardized protocols. This accomplishment is reflected by the multiple variations currently available, such as standard (S‐QuIC), real‐time (RT‐QuIC), and enhanced QuIC (eQuIC).189, 190, 191, 192
Apart from the autocatalytic propagation of PrPSc, another crucial hallmark of the PrPC‐PrPSc conversion is the de novo generation of infectivity. However, when inoculated into animals, PrP fibrillar assemblies can range from being biologically inert to fully infectious, pathogenic, and transmissible in subsequent passages.37, 76, 193, 194, 195 Legname and coworkers196 inoculated transgenic mice expressing truncated PrPC (aa 89‐231) with amyloid fibrils formed from recombinant PrP (aa 89‐230). The outcomes were low infectious titers and the affection of only that single line of transgenic mice Tg9949, probably due to the high expression and truncation of the transgene sequence enhancing the susceptibility to prion infection within the mice.75 Hence, according to Supattapone,197 these highly concentrated samples of PrP amyloid fibrils are not suitable in mimicking the infectious properties of PrPSc. In contrast, Wang et al39 succeeded in the formation of infectious de novo recombinant PrP amyloid fibrils associated with neurological signs in wild‐type mice after approximately 130 days. Here, PrPres was formed in PMCA assays in the presence of negatively charged lipids, namely, 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐(1′‐rac‐glycerol) (sodium salt) (POPG). In their earlier work,198 they had shown that POPG promotes the conversion to PrPres under physiological conditions. In further studies, Wang et al199 could attribute a crucial role in the PrP‐lipid interaction to the highly conserved middle region of PrP that induced conformational change.
4.1. PrPC‐PrPSc conversion in cells
The findings regarding PrPres formation in the presence of POPG39 support the possibility of the plasma membrane being the cellular localization of PrPSc formation as a posttranslational event. At this position, contact between endogenous PrPC and exogenous PrPSc can easily occur.200, 201, 202, 203, 204 This is supported by the finding that by releasing PrPC from the cell surface or interrupting its transport to the plasma membrane prevents the formation of PrPSc.205, 206, 207 More precisely, both PrP isoforms were found to be associated with rafts.208, 209, 210, 211 These are defined as highly dynamic microdomains wherein specific lipids stabilize larger lipid platforms and compartmentalize cellular processes at the membrane.212 Impairing the integrity of the cholesterol‐enriched rafts associated with PrP by lowering the intracellular levels of cholesterol reduced the formation of PrPSc in infected cells.213 Moreover, PrPC‐ and PrPSc‐associated rafts were found to have distinct characteristics, as they can be separated from each other by solubilization and flotation on density gradients.208 According to Campana et al,200 this proposes that either the types of raft or the membrane association of each isoform has different characteristics. However, Baron et al209 illustrated that the PrPSc‐PrPC conversion only takes place in the presence of fused PrPSc‐ and PrPC‐containing membranes, suggesting that the two PrP isoforms need to be inserted into contiguous membranes. Alternatively, rafts were proposed to stabilize PrP in its conformation via a direct interaction with cholesterol. Thus, changes in the local lipid environment can mediate PrP conformation.214 Studies on model lipid bilayers regarding the impact of the PrP‐lipid interaction on structure and affinity of PrP support the idea that predominantly α‐helical PrPC is stabilized upon binding to raft membranes, whereas binding to negatively charged lipid (nonraft) membranes leads to an increased β‐sheet content.215 Interestingly, the PrP‐raft association is mediated by the GPI anchor213, 216 and the N‐terminal region of PrP.217 Unlike for a typical GPI‐anchored protein, for PrP, this raft association occurs already earlier in the secretory pathway and appears to be involved in the maturation and folding process of PrPC.218, 219 Alternatively to the plasma membrane, the formation of PrPSc was suggested to involve additional cellular places. Immediately after PrP internalization, the PrPC‐PrPSc conversion may occur in the endolysosomal compartment,205 in the Golgi apparatus and/or the ER following retrograde transport.220, 221 In infected cells, stimulation of retrograde transport towards the ER leads to an increase in PrPSc formation from PrPC precursor,222 suggesting that the ER may represent an amplification compartment for PrPSc.223 Participation of the endocytic pathway is indicated by PrPSc accumulation in the late endosomes.205, 211, 224, 225 Still, as demonstrated by Goold et al,201 the plasma membrane is the initial site of prion conversion and consequently of most interest in studying the earliest events in prion infection and PrP misfolding.
4.2. Impact of GPI anchor on PrPC‐PrPSc conversion
Typically, PrP is attached to membranes by its GPI anchor (Figure 6A).69 A better understanding of the interplay between membranes, GPI‐anchored PrP, and PrPC‐PrPSc conversion is provided by work from Baron and Caughey.209, 210 First, they studied the conditions necessary for PrPres formation of PrP associated with detergent‐resistant membranes (DRMs).209 Based on that, in CFC assays, Baron and Caughey210 investigated the impact of GPI‐anchoring of PrP associated with model membranes on PrPres formation. PrP was isolated by immunoprecipitation from mammalian cell lines expressing GPI‐anchored and anchorless PrP, respectively.173, 226, 227 GPI‐anchored PrP bound to liposomes could not be converted to PrPres upon exposure to exogenous PrPres in microsomes until phosphatidylinositol‐specific phospholipase C (PI‐PLC) was added or the combined membrane fractions were treated with a membrane‐fusing agent. These findings indicate for the initiation and propagation of PrPSc that at the membrane surface, an insertion of PrPSc into the host cell membrane is necessary for the conversion. Whereas if the conversion occurred extracellularly, PrPC needed to be released from the cell membrane. In contrast, anchorless PrP bound to liposomes was converted to PrPres without any treatments necessary. Hence, contradictory to PrP conversion occurring at the cellular membrane,205, 206, 207 only the membrane‐associated form containing PrP attached to a GPI anchor could resist the conversion induced by exogenous PrPres. Moreover, Chesebro et al84 found that anchorless PrP results in infectious amyloid disease but without typical clinical TSE. Scrapie infection of transgenic mice lacking GPI‐anchored PrPC leads to a formation of amyloid plaques in contrast to nonamyloid deposits, typically observed in wild‐type mice. Although neuropathological lesions were induced, clinical manifestations were minimal. Surprisingly, the combined expression of anchorless and wild‐type PrP accelerated the onset of clinical disease. This suggests that GPI‐anchored PrP may be critically involved in the pathogenesis of prion diseases.228 Overall, the findings mentioned above indicate a major contribution of the GPI anchor in the toxicity of the PrPC‐PrPSc conversion.
Figure 6.
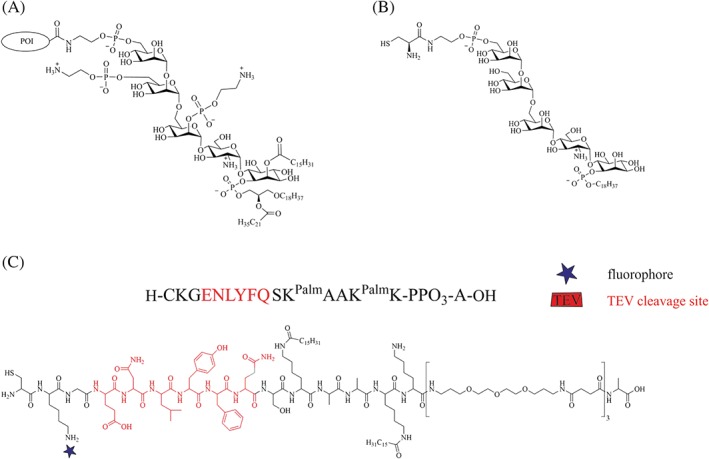
Structures of a native and artificial glycosylphosphatidylinositol (GPI) anchor together with a GPI anchor mimic. A shows a common structure of a native GPI anchor from human erythrocyte acetylcholinesterase. 226 B is a chemically synthesized GPI from Silva et al 227 with a cysteine residue for native chemical ligation (NCL) reactions. C shows the structure of the GPI anchor‐mimicking peptide used for the semisynthesis of lipidated PrP constructs 228
5. TOWARDS THE ELUCIDATION OF PrP CONVERSION
Recombinant PrP is an appropriate surrogate for PrPC, as determined by spectroscopic measurements, including circular dichroism (CD), that eventually facilitated solving the NMR and crystal structures of PrP.40, 41, 42, 43, 44, 45, 46, 47 However, it can be a suitable representative only under certain conditions, including thioflavin T (ThT) fluorescence‐based following of the aggregation process. Even though with this method, insights into the characteristics and kinetics of in vitro fibril formation have been gained,232 just recently, the molecular basis of PrP replication was established in detail by applying a single‐molecule fluorescence methodology to characterize individual aggregates. With total internal reflection fluorescence (TIRF) microscopy, Klenerman and colleagues233, 234 studied fibril fragmentation and elongation of individual murine PrP aggregates from seeded aggregation in vitro. PK‐resistant PrP fibrils elongated until length‐dependent fragmentation resulted in PK‐sensitive fragments. This method allowed direct observation of heterogeneous, transient, metastable oligomers during aggregation, found to be the most infectious PrP particles.235 Additionally, a spreading model for aggregate propagation through the brain could be predicted, and a framework was established to start determining the main factors that control the rate of prion spreading in animals. In 2011, Goold et al201 analyzed a PrP knockdown (KD) neuroblastoma cell line expressing epitope‐tagged PrPC upon infection with exogenous PrPSc. After facing the limitation of immunological differentiation between PrPSc and PrPC expressed on the recipient cell from cell lines susceptible to prion infection, epitope‐tagged PrPC appeared an elegant solution. However, several previous attempts had failed in generating PrP molecules capable of prion conversion, probably due to the sequence sensitivity in this process, particularly in certain key regions of the PrP molecule.236, 237, 238, 239 Eventually, out of eight different constructs, Goold et al201 succeeded with a PrP‐224AlaMYC construct, in which the tag is inserted within the C‐terminal domain. A detailed analysis of the cells shortly after prion exposure demonstrated that PrPSc is formed on the plasma membrane. Furthermore, PI‐PLC treatment effectively removed PrPC from the plasma membrane of PrP‐224AlaMYC cells and reduced the generation of PrPSc. However, immunostaining is only feasible on fixed cells and impedes dynamic studies revealing molecular details involved in the PrP conversion and propagation processes. ThT‐based detection of preformed PrP amyloid fibrils applied in a cellular environment cannot exclude ThT binding to other structures unrelated to amyloid and guarantee binding to all aggregates, as the binding mechanism is not fully understood yet.
To this end, labeling of PrP with an organic fluorophore is required for dynamic studies in cells. Recombinant PrP differs compared with PrPC in a complete lack of PTMs causing distinct infectivity and membrane interaction characteristics. These properties can be mainly ascribed to the GPI anchor, tethering PrP to the cellular membrane, as demonstrated in various studies.84, 209, 210, 231, 240, 241, 242 Advances in semisynthetic strategies based on solid‐phase peptide synthesis (SPPS),243 protein engineering, native chemical ligation (NCL),244 and expressed protein ligation (EPL)245, 246 have facilitated access to homogeneous membrane‐anchored labeled PrP variants that allow to directly observe the biophysical properties of PrP upon interaction with the cellular membrane.
6. SEMISYNTHESIS STRATEGIES FOR PrP
To date, the majority of studies on the function and structure of PrP have been carried out with recombinant protein lacking all PTMs, including the GPI anchor, or with heterogeneous protein preparations isolated from mammalian cell lines.247, 248, 249 Still, there have been attempts towards generating defined membrane‐anchored PrP. Glockshuber250 and Pinheiro251 with colleagues applied similar strategies, in which thiol‐reactive lipids were attached to the C‐terminus of recombinant PrP carrying a cysteine. However, further application of these PrP constructs in cell‐based assays may be impeded by potential side reactions with thiol‐containing compounds or internal cysteines of PrP, as the lipids were attached via disulfide bonds. Different strategies were utilized by Baskakov,252 Baldwin,253 and Moroder254 with coworkers: Baskakov and colleagues252 applied maleimide chemistry to introduce a myristoyl chain at the C‐terminus of genetically engineered PrP(S230C). This modification did not alter the structure of the protein. Interestingly, an increasing affinity of PrP for the cell membrane and a decreased extent of fibrillization was found. Baldwin253 and coworkers chemically synthesized several PrP segments, including a 106 residue “mini‐prion” (PrP106) by connecting PrP (aa 90‐141) to PrP (aa 178‐231) via a native peptide bond using NCL,244 a selective reaction that links an unprotected peptide containing a C‐terminal α‐thioester to another peptide with an N‐terminal cysteine. A membrane anchor made of a lipophilic myristoyl chain was introduced at the C‐terminus of shorter PrP peptides via an orthogonally side‐chain–protected lysine. Immunofluorescence analysis indicated that only myristylated PrP peptides could be targeted to the cell surface. The group of Moroder applied click and ligation chemistry to obtain lipidated peptides corresponding to the C‐terminal PrP segment (aa 214‐231).254 Confocal images of HeLa cells revealed a direct transfer of fluorescently labeled lipopeptides to the cellular membrane. Thus, lipopeptides can be used as mimics of the GPI anchor's ability to attach PrP to the cell membrane. A similar strategy with regard to using a lipidated peptide as a GPI‐mimicking membrane anchor was pursued by Becker et al, when starting to develop semisynthetic strategies, based on EPL and protein trans‐splicing (PTS) (Figure 4), to access to different posttranslationally modified PrP variants (Figure 5).228, 241, 255, 256, 257, 258, 259, 260, 261
Figure 4.
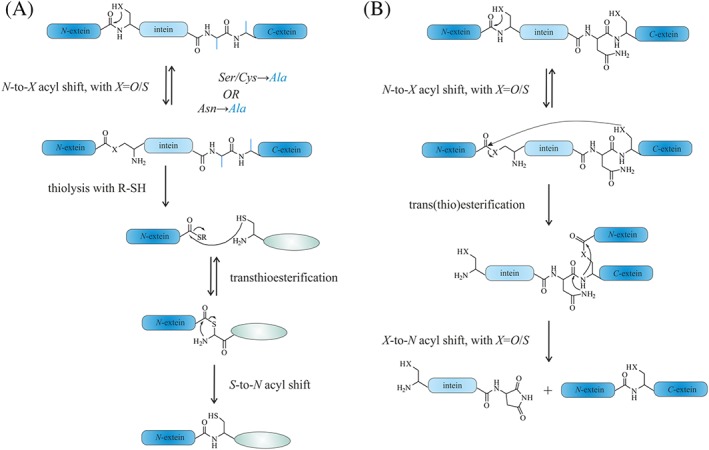
Mechanism of the expressed protein ligation (EPL) reaction (A) and protein trans‐splicing (PTS) (B)
Figure 5.
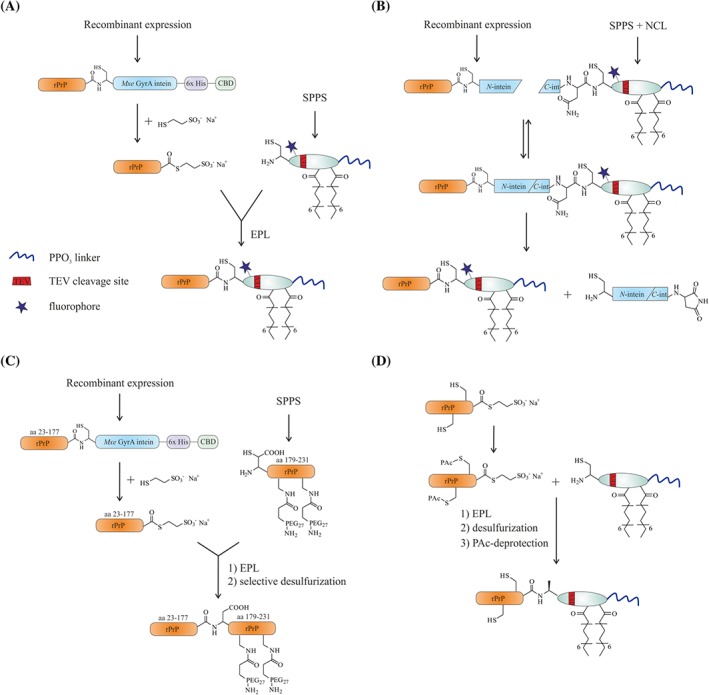
Semisynthetic strategies for prion protein (PrP) variants, developed in the Becker laboratory.228, 255, 256 Via an expressed protein ligation (EPL)‐ (A) and a protein trans‐splicing (PTS)‐based (B) approach, PrP variants equipped with a glycosylphosphatidylinositol (GPI) anchor mimic can be obtained. With the strategy displayed in C, PrP variants modified with monodisperse PEG chains as mimics of N‐glycans can be accessed. Selective desulfurization of the introduced cysteine following EPL (A) is depicted in D
In the EPL245, 246 reaction, a protein thioester, obtained by cleaving a fusion protein consisting of the protein of interest (POI) and an intein, is linked to a chemically synthesized peptide243 containing an N‐terminal cysteine in a reaction similar to NCL.244 An initial transesterifaction leads to formation of a thioester linking the recombinant and synthetic PrP segments, and a subsequent irreversible S → N acyl shift establishes the amide bond at the ligation site (Figure 4A). The recombinant protein α‐thioesters can be accessed using engineered inteins. Inteins are self‐processing protein segments, which mediate protein splicing.245, 262 In the course of this intramolecular process, the intein excises itself and joins the C‐ and N‐terminal flanking protein segments (C‐ and N‐extein). In more detail, a nucleophilic side chain, namely, a hydroxy or thiol group for serine and threonine or cysteine residues, accomplishes an N → O or N → S acyl shift. Then, in a trans‐(thio)esterification, the N‐extein gets attached to a conserved N‐terminal serine or cysteine of the C‐extein. The instable branched intermediate is resolved via an intramolecular rearrangement involving a conserved asparagine residue of the intein producing an intein with a C‐terminal succinimide, and an O → N or S → N acyl shift resulting in ligated exteins with a native bond at the ligation site.262, 263 Mutations of the C‐terminal asparagine of the intein and the N‐terminal cysteine, threonine, or serine residue of the C‐extein to alanine block the splicing process and only allow the initial N → S acyl shift, which enables the generation of a protein α‐thioester by addition of an excess of a thiol, such as sodium 2‐mercaptoethanesulfonate (MESNA), to trap the protein thioester.264 PTS is a process that relies on the assembly of two divided segments of inteins, so‐called split inteins, to form a functional intein. Upon assembly of the split inteins, PTS occurs and links the N‐ and C‐exteins in a similar sequence of events as described above (Figure 4B).265
The generation and biophysical characterization of PrP constructs containing a GPI anchor mimic started more than 10 years ago in the Becker laboratory with work described in Olschewski et al.228 Two strategies based on the EPL approach provided PrPPalm, an N‐terminally truncated PrP variant (T_PrP [aas 90‐231]) that is missing the unfolded N‐terminal domain (aa 23‐89) and modified at the C‐terminus with chemically synthesized membrane anchor peptides (Figure 5). At that time, the protease‐resistant PrP fragment comprising residues aa 90‐231 had been considered as the structure crucially involved in TSEs. The GPI anchor mimics (Figure 6C) feature two palmitoyl modifications (Palm) that induce a high affinity towards DOPC liposomes and locate PrP in its native conformation to the detergent‐resistant domains (DRMs) of cell membranes.266 A tobacco etch virus (TEV) protease recognition site (ENLYFQ) facilitates controlled release of PrP from the membrane, a polyethyleneglycol (PEG) polyamide oligomer (PPO) functions as solubilization tag to handle the palmitoylated peptides in aqueous buffers,267 and a fluorescent dye can be incorporated for tracking of the semisynthetic PrP in vitro and in vivo (Figure 6C).
One of the initial EPL‐based strategies relies on the expression of PrP in fusion with the Mxe GyrA intein and a combination of two affinity tags, namely, a His tag and a chitin‐binding domain (CBD) in Escherichia coli (E coli). Cleavage of this construct is achieved with an excess of thiol to generate PrP with a C‐terminal thioester. This PrP‐thioester is incubated with the GPI anchor‐mimicking peptides and gives the C‐terminally modified PrP (denoted as PrPPalm, Figure 5A). A second strategy is based on PTS by expressing PrP fused to the N‐terminal segment of the DnaE split intein (DnaEN) and chemical synthesis of its C‐terminal segment (DnaEC, 36 aa) linked to the GPI anchor‐mimicking peptides by a prior NCL reaction.268 Both DnaE segments spontaneously associate when folded and form a functional intein, which excises itself to give the desired modified PrP with its C‐terminal membrane anchor (Figure 5B). Aggregation assays based on PK resistance and ThT binding269 revealed an extended lag time for vesicle‐attached PrPPalm with respect to conversion into PrPres and fibril formation than for PrP in control experiments. Further, binding to zwitterionic DOPC liposomes indicated a very strong membrane interaction for PrPPalm in contrast to PrP. Transfer of PrPPalm onto neuronal cells gave rise to similar patterns observed for native PrPC by immunostaining. Together with extraction experiments of the cell membrane, this provided proof that soluble PrPPalm is attached to detergent‐resistant domains (DRMs) similar to wild‐type PrPC with a native GPI anchor.
Next, Becker et al257 developed a synthetic strategy for the preparation of PrP with a native, homogeneous GPI anchor that can also be applied for other GPI‐anchored proteins. A challenge lies here in the chemical diversity of GPI anchors on the same protein. Different glycoforms of native PrP GPI anchors have been reported with the exact linkage positions and anomeric configuration of the oligosaccharide branches not defined. At the same time, details about the lipids attached to these GPI anchors are not fully clear (Figure 6A).270 In view of this structural uncertainty, a core GPI pseudopentasaccharide, containing three mannose (Man), a glucoseamine (GlcN), and an inositol (Ino) glycan connected in an α‐Man‐(1 → 2)‐α‐Man‐(1 → 6)‐α‐Man‐(1 → 4)‐α‐GlcN‐(1 → 6)‐myo‐Ino way, was selected. The incorporation of a cysteine residue on the 2‐aminoethyl phosphate moiety of the GPI backbone prior to global deprotection provided a synthetic, cysteine‐tagged GPI anchor suitable for NCL reactions (Figure 6B). In a following EPL reaction, PrP with a C‐terminal thioester was linked to this synthetic GPI anchor. Analysis of the secondary structure of PrP attached to the synthetic GPI revealed that the CD curves are indistinguishable from the spectra of PrP and comparable with the spectra of PrPC. Moreover, the CD spectra were found to agree with the spectra of PrPPalm. This observation confirms the successful application of the GPI anchor‐mimicking peptides (Figure 6C) as an alternative to circumvent the elaborate synthesis of a GPI anchor (Figure 6B). Even though the synthesis of the GPI anchor succeeded, it remains a challenge to provide sufficient amounts for subsequent experiments and extension to other proteins. Isolating mostly homogeneous, cysteine‐carrying GPI anchors from natural sources could help to avoid this problem, and first steps have been made towards this goal by using yeast as an expression system for GPI‐anchored proteins, from which the GPI anchor is proteolytically released and purified.271 GPI‐anchored PrP was also found to quantitatively bind to DOPC vesicles. This emphasizes the contribution of GPI anchors in the membrane association of PrP.257 Noteworthy, the group of Silva et al is also working on intein‐based semisynthesis schemes to obtain homogeneous GPI‐anchored proteins, including PrP, using synthetic GPI anchors.272, 273, 274
A major limitation of obtaining semisynthetic PrP variants by EPL lies in the series of denaturation and renaturation steps required to obtain functional PrP‐intein fusion constructs due to expression into inclusion bodies in E. coli. The subsequent folding steps required for PrPPalm and GPI‐anchored PrP also limits the overall yield of EPL reactions. Deposition in inclusion bodies in E. coli is probably due to misprocessing of newly generated PrP and the overproduction that impedes proper folding, including the formation of the structurally important disulfide bridge.60, 275 Hence, to improve the semisynthetic access to posttranslationally modified PrP Chu and Becker261 developed a strategy for soluble expression of PrP‐intein constructs in E coli. Ultimately, the overexpression of a PrP‐intein construct N‐terminally fused to the ATPase domain of heat shock protein (Hsp) 70 DnaK chaperone gave high quantities of soluble PrP. This approach offers an alternative way to produce PrP‐thioester for subsequent EPL reactions but also requires an additional step for removing the N‐terminal ATPase domain by using sortase A.
With robust semisynthetic strategies established, the critical membrane attachment of PrP was studied by Chu et al241 using three PrP variants, including full‐length FL_PrP (aa 23‐231), central hydrophobic region deleted ΔCR_PrP (aa 23‐231 with ∆105‐125) and N‐terminally truncated T_PrP (aa 90‐231), all equipped with a C‐terminal membrane anchor. Interactions of the lipidated PrP constructs with phospholipid membranes demonstrated binding modes distinct from the nonmodified PrPs and impacts on the biochemical and conformational properties of PrP. Whereas nonmodified PrPs showed a conversion into β‐sheet–enriched structures upon interaction with anionic POPG vesicles, lipidated ΔCR_PrP and T_PrP retained their α‐helical structure and lipidated FL_PrP partially converted into random coil. Evidence indicating pore formation of lipidated ΔCR_PrP was found in fluorescence‐based assays and supported by patch clamp electrophysiological measurements of cells transfected with lipidated ΔCR_PrP. ΔCR_PrP was previously found to be neurotoxic in vivo. Yet, expressed in cultured cells, it is identically localized as wild‐type PrP. Thus, altered binding interactions had been suggested to cause the deleterious signaling pathways.276, 277 Based on these results, critical roles for both C‐terminal membrane attachment and the N‐terminal domain of PrP have been suggested.
PTMs in PrP comprise not only a C‐terminal GPI anchor but also N‐glycosylation of two asparagine residues278 at positions 181 and 197. Different prion strains and prion‐related diseases (TSEs) possess distinct glycosylation patterns of PrP. Studying the influence of these PTMs in prion pathogenesis has not been forthcoming mainly due to the confusing complexity and heterogeneity of these glycans.62, 279 Shi et al280 reported a strategy based on linking three segments of murine PrP, in which a recombinant PrP segment (aa 90‐177S) was ligated with two synthetic peptide segments (aa 178‐212 and aa 213‐230). This strategy was aimed at introduction sugars into PrP but did not fully succeed. Shortly after that, Araman et al255 demonstrated a semisynthetic approach to generate PrP variants modified with monodisperse PEG chains as mimics of N‐glycans that are similar in size and molar mass (Figure 5C). A new EPL strategy was established to achieve this, in which expressed PrP (aa 23‐177) with a C‐terminal thioester is used in EPL reactions with PEGylated, synthetic peptides (aa 179‐231). Selective desulfurization of the β‐mercapto‐aspartate at the ligation site gives homogeneous PEGylated full‐length PrP constructs. Interestingly, in vitro aggregation was completely abrogated for all PEGylated PrP constructs under conditions at which wild‐type PrP aggregated. Furthermore, the addition of only 10% of PEGylated PrP completely blocked aggregation of wild‐type PrP. This has raised the question if large N‐glycans interfere with aggregation in vivo. Recently, Mishra et al281 introduced lactosyl and mannobiosyl glycans in huPrP (aa 90‐231) at positions 181 and 197 via Asn to Cys mutations. In agreement with our results, they found that glycosylated PrPs are less prone to spontaneous fibril nucleation. Such a strategy raises the question if added cysteine residues influence PrP structure by disulfide shuffeling and if this affects the modification reaction used by Mishra et al.281
A similar question can arise from the cysteine residues introduced during the EPL reactions described above as in our previous approach depicted in Figure 5A. The introduced ligation site cysteine at the C‐terminus of T_PrP (aa 90‐231) was left undesulfurized, which could potentially be problematic for the folding of PrP. To finally prove that such an additional C‐terminal cysteine residue does not influence PrP folding, we employed a strategy recently introduced by Matveenko et al,256 in which the two native cysteines in recombinant PrP (aa 23‐231) are protected by a phenacyl (PAc) protecting group. This protection allowed selective desulfurization of the introduced cysteine following EPL (Figure 5D). Comparing PrP variants containing a cysteine at the ligation site and an alanine (by CD) proved that the introduced cysteine did not disturb the folding to native PrP.
7. CONCLUSION AND OUTLOOK
Based on the continuous progress in protein (semi)synthesis, access to homogeneous, posttranslationally modified PrP variants was facilitated over the past decade and a set of differently modified variants could be characterized with respect to their biophysical and conformational properties, including their interaction with membranes. Semisynthetic PrP variants have the potential to shed light on the crucial steps in PrP conversion, transmission, and pathogenicity, eg., by allowing for direct observation of the protein at the cellular membrane. Understanding these key features in prion diseases can further serve as paradigm for other neurodegenerative diseases.
ACKNOWLEDGEMENTS
SH is grateful for support by Wien Kultur.
Biographies
Stefanie Hackl received her master degree in chemistry from the University of Vienna in 2014. She has recently finished her joint PhD at the University of Natural Resources and Life Sciences (BOKU, Vienna), Austrian Institute of Technology (AIT, Vienna) and Nanyang Technological University (NTU, Singapore) under the supervision of Prof Christian Becker. She was working on the semisynthesis and trafficking of prion protein variants. Her current and future research interests include site‐specific protein modifications, membrane biophysics, and neurodegenerative diseases.

Christian F.W. Becker studied chemistry at the University of Dortmund, Germany and received his PhD in 2001 from the same university. He was a postdoctoral fellow with Gryphon Therapeutics from 2002 to 2003 and became group leader at the MPI in Dortmund in 2004. In 2007 he was appointed as professor at TU München. In 2011 he became Professor and Head of the Institute of Biological Chemistry at the University of Vienna. His group focuses on protein and peptide chemistry to address biomedical and biotechnological challenges.

Hackl S, Becker CFW. Prion protein—Semisynthetic prion protein (PrP) variants with posttranslational modifications. J Pep Sci. 2019;25:e3216 10.1002/psc.3216
This review covers parts of the lecture of Prof Christian Becker at the 34th European Peptide Symposium, 4 to 9 September 2016, Leipzig, Germany, where he received the Leonidas Zervas Award of the European Peptide Society.
REFERENCES
- 1. Weissmann C. Molecular genetics of transmissible spongiform encephalopathies. J. Biol. Chem. 1999;274(1):3‐6. [DOI] [PubMed] [Google Scholar]
- 2. Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RSG, Budka H. Mutations of the Prion Protein Gene. J. Neurol. 2002;249(11):1567‐1582. 10.1007/s00415-002-0896-9 [DOI] [PubMed] [Google Scholar]
- 3. Wadsworth JD, Collinge J. Molecular pathology of human prion disease. Acta Neuropathol. 2011;121(1):69‐77. 10.1007/s00401-010-0735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aguzzi A, Weissmann C. Prion diseases. Haemophilia. 1998;4(4):619‐627. [DOI] [PubMed] [Google Scholar]
- 5. Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a kuru‐like syndrome to chimpanzees. Nature. 1966;209(5025):794‐796. [DOI] [PubMed] [Google Scholar]
- 6. Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214(5090):764‐766. [DOI] [PubMed] [Google Scholar]
- 7. Griffith JS. Self‐replication and scrapie. Nature. 1967;215(5105):1043‐1044. [DOI] [PubMed] [Google Scholar]
- 8. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136‐144. [DOI] [PubMed] [Google Scholar]
- 9. Prusiner SB. Nobel Lecture: Prions. Proc. Natl. Acad. Sci. U. S. A. 1998;95(23):13363‐13383. 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218(4579):1309‐1311. [DOI] [PubMed] [Google Scholar]
- 11. Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. Further purification and characterization of scrapie prions. Biochemistry. 1982;21(26):6942‐6950. [DOI] [PubMed] [Google Scholar]
- 12. Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell. 1984;38(1):127‐134. [DOI] [PubMed] [Google Scholar]
- 13. Basler K, Oesch B, Scott M, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46(3):417‐428. 10.1016/0092-8674(86)90662-8 [DOI] [PubMed] [Google Scholar]
- 14. Oesch B, Westaway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27‐30 protein. Cell. 1985;40(4):735‐746. [DOI] [PubMed] [Google Scholar]
- 15. Meyer RK, McKinley MP, Bowman KA, et al. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. U. S. A. 1986;83(8):2310‐2314. 10.1073/pnas.83.8.2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc. Natl. Acad. Sci. U. S. A. 1986;83(17):6372‐6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prusiner SB. Prions and neurodegenerative diseases. N. Engl. J. Med. 1987;317(25):1571‐1581. 10.1056/NEJM198712173172505 [DOI] [PubMed] [Google Scholar]
- 18. Leighton PL, Allison WT. Protein misfolding in prion and prion‐like diseases: reconsidering a required role for protein loss‐of‐function. J. Alzheimers Dis. 2016;54(1):3‐29. 10.3233/jad-160361 [DOI] [PubMed] [Google Scholar]
- 19. Verma A. Prions, prion‐like prionoids, and neurodegenerative disorders. Ann. Indian Acad. Neurol. 2016;19(2):169‐174. 10.4103/0972-2327.179979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aguzzi A, Lakkaraju AK. Cell biology of prions and prionoids: a status report. Trends Cell Biol. 2016;26(1):40‐51. 10.1016/j.tcb.2015.08.007 [DOI] [PubMed] [Google Scholar]
- 21. Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for α‐synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U. S. A. 2015;112(38):E5308‐E5317. 10.1073/pnas.1514475112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaunmuktane Z, Mead S, Ellis M, et al. Evidence for human transmission of amyloid‐β pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247‐250. 10.1038/nature15369 [DOI] [PubMed] [Google Scholar]
- 23. Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol. Rev. 2009;89(4):1105‐1152. 10.1152/physrev.00006.2009 [DOI] [PubMed] [Google Scholar]
- 24. Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am. J. Pathol. 1986;122(1):1‐5. [PMC free article] [PubMed] [Google Scholar]
- 25. Linden R, Martins VR, Prado MA, et al. Physiology of the prion protein. Physiol. Rev. 2008;88(2):673‐728. 10.1152/physrev.00007.2007 [DOI] [PubMed] [Google Scholar]
- 26. Herms J, Tings T, Gall S, et al. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999;19(20):8866‐8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chesebro B, Race R, Wehrly K, et al. Identification of scrapie prion protein‐specific mRNA in scrapie‐infected and uninfected brain. Nature. 1985;315(6017):331‐333. [DOI] [PubMed] [Google Scholar]
- 28. Schatzl HM, Da Costa M, Taylor L, Cohen FE, Prusiner SB. Prion protein gene variation among primates. J. Mol. Biol. 1995;245(4):362‐374. [DOI] [PubMed] [Google Scholar]
- 29. Wopfner F, Weidenhofer G, Schneider R, et al. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999;289(5):1163‐1178. 10.1006/jmbi.1999.2831 [DOI] [PubMed] [Google Scholar]
- 30. Puckett C, Concannon P, Casey C, Hood L. Genomic structure of the human prion protein gene. Am. J. Hum. Genet. 1991;49(2):320‐329. [PMC free article] [PubMed] [Google Scholar]
- 31. Kretzschmar HA, Stowring LE, Westaway D, et al. Molecular cloning of a human prion protein cDNA. DNA. 1986;5(4):315‐324. [DOI] [PubMed] [Google Scholar]
- 32. Tatzelt J, Winklhofer KF. Folding and misfolding of the prion protein in the secretory pathway. Amyloid. 2004;11(3):162‐172. [DOI] [PubMed] [Google Scholar]
- 33. Hegde RS, Mastrianni JA, Scott MR, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279(5352):827‐834. [DOI] [PubMed] [Google Scholar]
- 34. Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402(6763):822‐826. 10.1038/45574 [DOI] [PubMed] [Google Scholar]
- 35. Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha‐helices into beta‐sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U. S. A. 1993;90(23):10962‐10966. 10.1073/pnas.90.23.10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timmes AG, Moore RA, Fischer ER, Priola SA. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS One. 2013;8(7):e71081 10.1371/journal.pone.0071081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang F, Wang X, Orrú CD, et al. Self‐propagating, protease‐resistant, recombinant prion protein conformers with or without in vivo pathogenicity. PLoS Pathog. 2017;13(7):e1006491 10.1371/journal.ppat.1006491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121(2):195‐206. 10.1016/j.cell.2005.02.011 [DOI] [PubMed] [Google Scholar]
- 39. Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132‐1135. 10.1126/science.1183748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hornemann S, Korth C, Oesch B, et al. Recombinant full‐length murine prion protein, mPrP(23‐231): purification and spectroscopic characterization. FEBS Lett. 1997;413(2):277‐281. [DOI] [PubMed] [Google Scholar]
- 41. Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121‐231). Nature. 1996;382(6587):180‐182. 10.1038/382180a0 [DOI] [PubMed] [Google Scholar]
- 42. Calzolai L, Lysek DA, Perez DR, Guntert P, Wuthrich K. Prion protein NMR structures of chickens, turtles, and frogs. Proc. Natl. Acad. Sci. U. S. A. 2005;102(3):651‐655. 10.1073/pnas.0408939102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Donne DG, Viles JH, Groth D, et al. Structure of the recombinant full‐length hamster prion protein PrP(29‐231): the N terminus is highly flexible. Proc. Natl. Acad. Sci. U. S. A. 1997;94(25):13452‐13457. 10.1073/pnas.94.25.13452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. James TL, Liu H, Ulyanov NB, et al. Solution structure of a 142‐residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. U. S. A. 1997;94(19):10086‐10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lysek DA, Schorn C, Nivon LG, et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc. Natl. Acad. Sci. U. S. A. 2005;102(3):640‐645. 10.1073/pnas.0408937102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lopez Garcia F, Zahn R, Riek R, Wuthrich K. NMR structure of the bovine prion protein. Proc. Natl. Acad. Sci. U. S. A. 2000;97(15):8334‐8339. 10.1073/pnas.97.15.8334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zahn R, Liu A, Luhrs T, et al. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U. S. A. 2000;97(1):145‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stöckel J, Safar J, Wallace AC, Cohen FE, Prusiner SB. Prion protein selectively binds copper (II) Ions. Biochemistry. 1998;37(20):7185‐7193. 10.1021/bi972827k [DOI] [PubMed] [Google Scholar]
- 49. Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc. Natl. Acad. Sci. U. S. A. 1999;96(5):2042‐2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walter ED, Chattopadhyay M, Millhauser GL. The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry. 2006;45(43):13083‐13092. 10.1021/bi060948r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miura T, Sasaki S, Toyama A, Takeuchi H. Copper reduction by the octapeptide repeat region of prion protein: pH dependence and implications in cellular copper uptake. Biochemistry. 2005;44(24):8712‐8720. 10.1021/bi0501784 [DOI] [PubMed] [Google Scholar]
- 52. Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J. Am. Chem. Soc. 2007;129(50):15440‐15441. 10.1021/ja077146j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Renner C, Fiori S, Fiorino F, et al. Micellar environments induce structuring of the N‐terminal tail of the prion protein. Biopolymers. 2004;73(4):421‐433. 10.1002/bip.20015 [DOI] [PubMed] [Google Scholar]
- 54. Mentler M, Weiss A, Grantner K, et al. A new method to determine the structure of the metal environment in metalloproteins: investigation of the prion protein octapeptide repeat Cu(2+) complex. European biophysics journal: EBJ. 2005;34(2):97‐112. 10.1007/s00249-004-0434-z [DOI] [PubMed] [Google Scholar]
- 55. del Pino P, Weiss A, Bertsch U, et al. The configuration of the Cu2+ binding region in full‐length human prion protein. European biophysics journal: EBJ. 2007;36(3):239‐252. 10.1007/s00249-006-0124-0 [DOI] [PubMed] [Google Scholar]
- 56. Weiss A, Del Pino P, Bertsch U, et al. The configuration of the Cu2+ binding region in full‐length human prion protein compared with the isolated octapeptide. Veterinary microbiology. 2007;123(4):358‐366. 10.1016/j.vetmic.2007.04.008 [DOI] [PubMed] [Google Scholar]
- 57. Burns CS, Aronoff‐Spencer E, Legname G, et al. Copper coordination in the full‐length, recombinant prion protein. Biochemistry. 2003;42(22):6794‐6803. 10.1021/bi027138+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Acevedo‐Morantes CY, Wille H. The Structure of Human Prions: From Biology to Structural Models—Considerations and Pitfalls. Viruses. 2014;6(10):3875‐3892. 10.3390/v6103875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc‐PrPC complex that leads to prion propagation. J. Biol. Chem. 2005;280(29):27236‐27243. 10.1074/jbc.M413441200 [DOI] [PubMed] [Google Scholar]
- 60. Maiti NR, Surewicz WK. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J. Biol. Chem. 2001;276(4):2427‐2431. 10.1074/jbc.M007862200 [DOI] [PubMed] [Google Scholar]
- 61. Haraguchi T, Fisher S, Olofsson S, et al. Asparagine‐linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989;274(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 62. Rudd PM, Endo T, Colominas C, et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U. S. A. 1999;96(23):13044‐13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DeArmond SJ, Qiu Y, Sanchez H, et al. PrPc glycoform heterogeneity as a function of brain region: implications for selective targeting of neurons by prion strains. J. Neuropathol. Exp. Neurol. 1999;58(9):1000‐1009. 10.1097/00005072-199909000-00010 [DOI] [PubMed] [Google Scholar]
- 64. Gready JE, Zuegg J. Molecular dynamics simulation of human prion protein including both N‐linked oligosaccharides and the GPI anchor. Glycobiology. 2000;10(10):959‐974. 10.1093/glycob/10.10.959 [DOI] [PubMed] [Google Scholar]
- 65. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383(6602):685‐690. 10.1038/383685a0 [DOI] [PubMed] [Google Scholar]
- 66. Priola SA, Lawson VA. Glycosylation influences cross‐species formation of protease‐resistant prion protein. EMBO J. 2001;20(23):6692‐6699. 10.1093/emboj/20.23.6692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tuzi NL, Cancellotti E, Baybutt H, et al. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol. 2008;6(4):e100 10.1371/journal.pbio.0060100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baskakov IV, Katorcha E, Makarava N. Prion strain‐specific structure and pathology: a view from the perspective of glycobiology. Viruses. 2018;10(12). 10.3390/v10120723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51(2):229‐240. 10.1016/0092-8674(87)90150-4 [DOI] [PubMed] [Google Scholar]
- 70. Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 2001;24(1):519‐550. 10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- 71. Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J. Biol. Chem. 1998;273(47):31048‐31052. [DOI] [PubMed] [Google Scholar]
- 72. Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sonnichsen FD. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J. Biol. Chem. 2000;275(43):33650‐33654. 10.1074/jbc.C000483200 [DOI] [PubMed] [Google Scholar]
- 73. Wolschner C, Giese A, Kretzschmar HA, Huber R, Moroder L, Budisa N. Design of anti‐ and pro‐aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc. Natl. Acad. Sci. U. S. A. 2009;106(19):7756‐7761. 10.1073/pnas.0902688106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Diaz‐Espinoza R, Soto C. High‐resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 2012;19(4):370‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Colby DW, Prusiner SB. De novo generation of prion strains. Nat. Rev. Microbiol. 2011;9(11):771‐777. 10.1038/nrmicro2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baskakov IV, Caughey B, Requena JR, Sevillano AM, Surewicz WK, Wille H. The prion 2018 round tables (I): the structure of PrP (Sc). Prion. 2019;13(1):46‐52. 10.1080/19336896.2019.1569450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left‐handed β‐helices into trimers. Proc. Natl. Acad. Sci. U. S. A. 2004;101(22):8342‐8347. 10.1073/pnas.0402254101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in‐register β‐structure. Proc. Natl. Acad. Sci. U. S. A. 2007;104(48):18946‐18951. 10.1073/pnas.0706522104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Requena JR, Wille H. The structure of the infectious prion protein: experimental data and molecular models. Prion. 2014;8(1):60‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Henderson R, Unwin PNT. Three‐dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257(5521):28‐32. 10.1038/257028a0 [DOI] [PubMed] [Google Scholar]
- 81. Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high‐resolution electron cryo‐microscopy. J. Mol. Biol. 1990;213(4):899‐929. 10.1016/s0022-2836(05)80271-2 [DOI] [PubMed] [Google Scholar]
- 82. Vázquez‐Fernández E, Vos MR, Afanasyev P, et al. The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLoS Pathog. 2016;12(9):e1005835 10.1371/journal.ppat.1005835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Terry C, Harniman RL, Sells J, et al. Structural features distinguishing infectious ex vivo mammalian prions from non‐infectious fibrillar assemblies generated in vitro. Sci. Rep. 2019;9(1):376‐376. 10.1038/s41598-018-36700-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308(5727):1435‐1439. 10.1126/science.1110837 [DOI] [PubMed] [Google Scholar]
- 85. Chesebro B, Race B, Meade‐White K, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6(3):e1000800 10.1371/journal.ppat.1000800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sim VL, Caughey B. Ultrastructures and strain comparison of under‐glycosylated scrapie prion fibrils. Neurobiol. Aging. 2009;30(12):2031‐2042. 10.1016/j.neurobiolaging.2008.02.016 [DOI] [PubMed] [Google Scholar]
- 87. Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. Parallel in‐register intermolecular β‐sheet architectures for prion‐seeded prion protein (PrP) amyloids. J. Biol. Chem. 2014;289(35):24129‐24142. 10.1074/jbc.M114.578344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baron GS, Hughson AG, Raymond GJ, et al. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry. 2011;50(21):4479‐4490. 10.1021/bi2003907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547(7662):185‐190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Swuec P, Lavatelli F, Tasaki M, et al. Cryo‐EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019;10(1):1269 10.1038/s41467-019-09133-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gasset M, Baldwin MA, Fletterick RJ, Prusiner SB. Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proc. Natl. Acad. Sci. U. S. A. 1993;90(1):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie‐associated protein PrP 27‐30 in water by infrared spectroscopy. Biochemistry. 1991;30(31):7672‐7680. [DOI] [PubMed] [Google Scholar]
- 93. Wille H, Bian W, McDonald M, et al. Natural and synthetic prion structure from X‐ray fiber diffraction. Proc. Natl. Acad. Sci. U. S. A. 2009;106(40):16990‐16995. 10.1073/pnas.0909006106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Riesner D. Biochemistry and structure of PrPC and PrPSc. Br. Med. Bull. 2003;66(1):21‐33. 10.1093/bmb/66.1.21 [DOI] [PubMed] [Google Scholar]
- 95. Bueler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell‐surface PrP protein. Nature. 1992;356(6370):577‐582. 10.1038/356577a0 [DOI] [PubMed] [Google Scholar]
- 96. Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994;8(2‐3):121‐127. 10.1007/bf02780662 [DOI] [PubMed] [Google Scholar]
- 97. Halliez S, Passet B, Martin‐Lannerée S, et al. To develop with or without the prion protein. Front. Cell Dev. Biol. 2014;2(58). 10.3389/fcell.2014.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Malaga‐Trillo E, Solis GP, Schrock Y, et al. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7(3):e55 10.1371/journal.pbio.1000055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schmitt‐Ulms G, Legname G, Baldwin MA, et al. Binding of neural cell adhesion molecules (N‐CAMs) to the cellular prion protein. J. Mol. Biol. 2001;314(5):1209‐1225. 10.1006/jmbi.2000.5183 [DOI] [PubMed] [Google Scholar]
- 100. Graner E, Mercadante AF, Zanata SM, et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. Mol. Brain Res. 2000;76(1):85‐92. [DOI] [PubMed] [Google Scholar]
- 101. Amin L, Nguyen XTA, Rolle IG, et al. Characterization of prion protein function by focal neurite stimulation. J. Cell Sci. 2016;129(20):3878‐3891. 10.1242/jcs.183137 [DOI] [PubMed] [Google Scholar]
- 102. Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005;169(2):341‐354. 10.1083/jcb.200409127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 2005;95(5):1373‐1386. 10.1111/j.1471-4159.2005.03469.x [DOI] [PubMed] [Google Scholar]
- 104. Martins VR, Beraldo FH, Hajj GN, et al. Prion protein: orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 2010;12(2):63‐86. [PubMed] [Google Scholar]
- 105. Guillot‐Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The α‐secretase‐derived N‐terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009;284(51):35973‐35986. 10.1074/jbc.M109.051086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax‐mediated apoptosis. J. Biol. Chem. 2001;276(42):39145‐39149. 10.1074/jbc.C100443200 [DOI] [PubMed] [Google Scholar]
- 107. Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J. Neurosci. Res. 2004;75(2):153‐161. 10.1002/jnr.10864 [DOI] [PubMed] [Google Scholar]
- 108. Tobler I, Gaus SE, Deboer T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380(6575):639‐642. 10.1038/380639a0 [DOI] [PubMed] [Google Scholar]
- 109. Nuvolone M, Hermann M, Sorce S, et al. Strictly co‐isogenic C57BL/6J‐Prnp −/− mice: a rigorous resource for prion science. J. Exp. Med. 2016;213(3):313‐327. 10.1084/jem.20151610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Baumann F, Tolnay M, Brabeck C, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26(2):538‐547. 10.1038/sj.emboj.7601510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bremer J, Baumann F, Tiberi C, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010;13(3):310‐318. 10.1038/nn.2483 [DOI] [PubMed] [Google Scholar]
- 112. Küffer A, Lakkaraju AKK, Mogha A, et al. The prion protein is an agonistic ligand of the G protein‐coupled receptor Adgrg6. Nature. 2016;536(7617):464‐468. 10.1038/nature19312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Brown DR, Qin K, Herms JW, et al. The cellular prion protein binds copper in vivo. Nature. 1997;390(6661):684‐687. 10.1038/37783 [DOI] [PubMed] [Google Scholar]
- 114. Watt NT, Taylor DR, Kerrigan TL, et al. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012;3(1):1134 10.1038/ncomms2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Haigh CL, Marom SY, Collins SJ. Copper, endoproteolytic processing of the prion protein and cell signalling. Front. Biosci. Landmark Ed. 2010;15(1):1086‐1104. [DOI] [PubMed] [Google Scholar]
- 116. Mouillet‐Richard S, Ermonval M, Chebassier C, et al. Signal transduction through prion protein. Science. 2000;289(5486):1925‐1928. 10.1126/science.289.5486.1925 [DOI] [PubMed] [Google Scholar]
- 117. Chiarini LB, Freitas ARO, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21(13):3317‐3326. 10.1093/emboj/cdf324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Spielhaupter C, Schatzl HM. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001;276(48):44604‐44612. 10.1074/jbc.M103289200 [DOI] [PubMed] [Google Scholar]
- 119. Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15(1):34 10.1186/s12915-017-0375-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐β oligomers. Nature. 2009;457(7233):1128‐1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Barry AE, Klyubin I, Mc Donald JM, et al. Alzheimer's disease brain‐derived amyloid‐β‐mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011;31(20):7259‐7263. 10.1523/jneurosci.6500-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kong C, Xie H, Gao Z, et al. Binding between prion protein and aβ oligomers contributes to the pathogenesis of Alzheimer's disease. Virol. Sin. 2019. 10.1007/s12250-019-00124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Aulić S, Masperone L, Narkiewicz J, et al. α‐Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell‐to‐cell spreading and block prion replication. Sci. Rep. 2017;7(1):10050 10.1038/s41598-017-10236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Linsenmeier L, Altmeppen HC, Wetzel S, et al. Diverse functions of the prion protein—does proteolytic processing hold the key? Biochim. Biophys. Acta Mol. Cell Res. 2017;1864(11, Part B):2128‐2137. 10.1016/j.bbamcr.2017.06.022 [DOI] [PubMed] [Google Scholar]
- 125. Linsenmeier L, Mohammadi B, Wetzel S, et al. Structural and mechanistic aspects influencing the ADAM10‐mediated shedding of the prion protein. Mol. Neurodegener. 2018;13(1):18 10.1186/s13024-018-0248-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Altmeppen HC, Puig B, Dohler F, et al. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012;1(1):15‐31. [PMC free article] [PubMed] [Google Scholar]
- 127. McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A new paradigm for enzymatic control of α‐cleavage and β‐cleavage of the prion protein. J. Biol. Chem. 2014;289(2):803‐813. 10.1074/jbc.M113.502351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mays CE, Coomaraswamy J, Watts JC, et al. Endoproteolytic processing of the mammalian prion glycoprotein family. FEBS J. 2014;281(3):862‐876. 10.1111/febs.12654 [DOI] [PubMed] [Google Scholar]
- 129. Shmerling D, Hegyi I, Fischer M, et al. Expression of amino‐terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93(2):203‐214. 10.1016/S0092-8674(00)81572-X [DOI] [PubMed] [Google Scholar]
- 130. Altmeppen HC, Prox J, Puig B, et al. Lack of a‐disintegrin‐and‐metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol. Neurodegener. 2011;6(1):36 10.1186/1750-1326-6-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Jarosz‐Griffiths HH, Corbett NJ, Rowland HA, et al. Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid‐β oligomers. J. Biol. Chem. 2019. 10.1074/jbc.RA118.005364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Haddon DJ, Hughes MR, Antignano F, Westaway D, Cashman NR, McNagny KM. Prion protein expression and release by mast cells after activation. J. Infect. Dis. 2009;200(5):827‐831. 10.1086/605022 [DOI] [PubMed] [Google Scholar]
- 133. Jimenez‐Huete A, Lievens PM, Vidal R, et al. Endogenous proteolytic cleavage of normal and disease‐associated isoforms of the human prion protein in neural and non‐neural tissues. Am. J. Pathol. 1998;153(5):1561‐1572. 10.1016/s0002-9440(10)65744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio‐Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995;270(32):19173‐19180. [DOI] [PubMed] [Google Scholar]
- 135. Lewis V, Johanssen VA, Crouch PJ, Klug GM, Hooper NM, Collins SJ. Prion protein “gamma‐cleavage”: characterizing a novel endoproteolytic processing event. Cell. Mol. Life Sci. 2016;73(3):667‐683. 10.1007/s00018-015-2022-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Linden R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017;10:77 10.3389/fnmol.2017.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993;268(21):15922‐15928. [PubMed] [Google Scholar]
- 138. Sunyach C, Jen A, Deng J, et al. The mechanism of internalization of glycosylphosphatidylinositol‐anchored prion protein. EMBO J. 2003;22(14):3591‐3601. 10.1093/emboj/cdg344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shyng SL, Moulder KL, Lesko A, Harris DA. The N‐terminal domain of a glycolipid‐anchored prion protein is essential for its endocytosis via clathrin‐coated pits. J. Biol. Chem. 1995;270(24):14793‐14800. [DOI] [PubMed] [Google Scholar]
- 140. Nunziante M, Gilch S, Schatzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half‐life of cellular prion protein. J. Biol. Chem. 2003;278(6):3726‐3734. 10.1074/jbc.M206313200 [DOI] [PubMed] [Google Scholar]
- 141. Shyng SL, Heuser JE, Harris DA. A glycolipid‐anchored prion protein is endocytosed via clathrin‐coated pits. J. Cell Biol. 1994;125(6):1239‐1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Taylor DR, Hooper NM. The low‐density lipoprotein receptor‐related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007;402(1):17‐23. 10.1042/bj20061736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Peters PJ, Mironov A, Peretz D, et al. Trafficking of prion proteins through a caveolae‐mediated endosomal pathway. J. Cell Biol. 2003;162(4):703‐717. 10.1083/jcb.200304140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mayor S, Riezman H. Sorting GPI‐anchored proteins. Nat. Rev. Mol. Cell Biol. 2004;5(2):110‐120. 10.1038/nrm1309 [DOI] [PubMed] [Google Scholar]
- 145. Galvan C, Camoletto PG, Dotti CG, Aguzzi A, Ledesma MD. Proper axonal distribution of PrPC depends on cholesterol‐sphingomyelin‐enriched membrane domains and is developmentally regulated in hippocampal neurons. Mol. Cell. Neurosci. 2005;30(3):304‐315. 10.1016/j.mcn.2005.07.003 [DOI] [PubMed] [Google Scholar]
- 146. Sarnataro D, Caputo A, Casanova P, et al. Lipid rafts and clathrin cooperate in the internalization of PrPC in epithelial FRT cells. PLoS One. 2009;4(6):e5829 10.1371/journal.pone.0005829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schmid SL. Clathrin‐coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 1997;66(1):511‐548. 10.1146/annurev.biochem.66.1.511 [DOI] [PubMed] [Google Scholar]
- 148. Parkyn CJ, Vermeulen EGM, Mootoosamy RC, et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 2008;121(6):773‐783. 10.1242/jcs.021816 [DOI] [PubMed] [Google Scholar]
- 149. Willnow TE. The low‐density lipoprotein receptor gene family: multiple roles in lipid metabolism. J. Mol. Med. 1999;77(3):306‐315. [DOI] [PubMed] [Google Scholar]
- 150. Strickland DK, Gonias SL, Argraves WS. Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 2002;13(2):66‐74. [DOI] [PubMed] [Google Scholar]
- 151. Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4(11):724‐738. [DOI] [PubMed] [Google Scholar]
- 152. Parton RG, Simons K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007;8(3):185‐194. 10.1038/nrm2122 [DOI] [PubMed] [Google Scholar]
- 153. Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J. Cell Biol. 1994;127(5):1199‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vey M, Pilkuhn S, Wille H, et al. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae‐like membranous domains. Proc. Natl. Acad. Sci. U. S. A. 1996;93(25):14945‐14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB. COOH‐terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl. Acad. Sci. U. S. A. 1997;94(6):2333‐2338. 10.1073/pnas.94.6.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 2002;277(28):25457‐25464. 10.1074/jbc.M203248200 [DOI] [PubMed] [Google Scholar]
- 157. Sarnataro D, Caputo A, Casanova P, et al. Lipid rafts and clathrin cooperate in the internalization of PrP in epithelial FRT cells. PLoS One. 2009;4(6):e5829 10.1371/journal.pone.0005829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI‐anchored proteins are delivered to recycling endosomes via a distinct cdc42‐regulated, clathrin‐independent pinocytic pathway. Dev. Cell. 2002;2(4):411‐423. [DOI] [PubMed] [Google Scholar]
- 159. Kim JI, Surewicz K, Gambetti P, Surewicz WK. The role of glycophosphatidylinositol anchor in the amplification of the scrapie isoform of prion protein in vitro. FEBS Lett. 2009;583(22):3671‐3675. 10.1016/j.febslet.2009.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Priola SA, McNally KL. The role of the prion protein membrane anchor in prion infection. Prion. 2009;3(3):134‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810‐813. [DOI] [PubMed] [Google Scholar]
- 162. Prusiner SB, Scott M, Foster D, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63(4):673‐686. [DOI] [PubMed] [Google Scholar]
- 163. Meier P, Genoud N, Prinz M, et al. Soluble dimeric prion protein binds PrPSc in vivo and antagonizes prion disease. Cell. 2003;113(1):49‐60. [DOI] [PubMed] [Google Scholar]
- 164. Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell. 1994;77(7):967‐968. [DOI] [PubMed] [Google Scholar]
- 165. Bueler H, Raeber A, Sailer A, et al. High prion and PrPSc levels but delayed onset of disease in scrapie‐inoculated mice heterozygous for a disrupted PrP gene. Mol. Med. 1994;1(1):19‐30. [PMC free article] [PubMed] [Google Scholar]
- 166. Manson JC, Clarke AR, McBride PA, McConnell I, Hope J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration. 1994;3(4):331‐340. [PubMed] [Google Scholar]
- 167. Prusiner SB. Molecular biology of prion diseases. Science. 1991;252(5012):1515‐1522. [DOI] [PubMed] [Google Scholar]
- 168. Zhou Z, Xiao G. Conformational conversion of prion protein in prion diseases. Acta Biochim. Biophys. Sin. 2013;45(6):465‐476. 10.1093/abbs/gmt027 [DOI] [PubMed] [Google Scholar]
- 169. Jarrett JT, Lansbury PT Jr. Seeding “one‐dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73(6):1055‐1058. [DOI] [PubMed] [Google Scholar]
- 170. Combet S, Cousin F, Rezaei H, Noinville S. Membrane interaction of off‐pathway prion oligomers and lipid‐induced on‐pathway intermediates during prion conversion: A clue for neurotoxicity. Biochim. Biophys. Acta Biomembr. 2018;1861(2):514‐523. 10.1016/j.bbamem.2018.12.001 [DOI] [PubMed] [Google Scholar]
- 171. Sandberg MK, Al‐Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470(7335):540‐542. 10.1038/nature09768 [DOI] [PubMed] [Google Scholar]
- 172. Sandberg MK, Al‐Doujaily H, Sharps B, et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014;5(1):4347 10.1038/ncomms5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kocisko DA, Come JH, Priola SA, et al. Cell‐free formation of protease‐resistant prion protein. Nature. 1994;370(6489):471‐474. 10.1038/370471a0 [DOI] [PubMed] [Google Scholar]
- 174. Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non‐genetic propagation of strain‐specific properties of scrapie prion protein. Nature. 1995;375(6533):698‐700. 10.1038/375698a0 [DOI] [PubMed] [Google Scholar]
- 175. Priola SA, Chabry J, Chan K. Efficient conversion of normal prion protein (PrP) by abnormal hamster PrP is determined by homology at amino acid residue 155. J. Virol. 2001;75(10):4673‐4680. 10.1128/jvi.75.10.4673-4680.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease‐resistant state. EMBO J. 1999;18(12):3193‐3203. 10.1093/emboj/18.12.3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kirby L, Birkett CR, Rudyk H, Gilbert IH, Hope J. In vitro cell‐free conversion of bacterial recombinant PrP to PrPres as a model for conversion. J. Gen. Virol. 2003;84(Pt 4):1013‐1020. 10.1099/vir.0.18903-0 [DOI] [PubMed] [Google Scholar]
- 178. Caughey WS, Raymond LD, Horiuchi M, Caughey B. Inhibition of protease‐resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. U. S. A. 1998;95(21):12117‐12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Maxson L, Wong C, Herrmann LM, Caughey B, Baron GS. A solid‐phase assay for identification of modulators of prion protein interactions. Anal. Biochem. 2003;323(1):54‐64. [DOI] [PubMed] [Google Scholar]
- 180. Hill AF, Antoniou M, Collinge J. Protease‐resistant prion protein produced in vitro lacks detectable infectivity. J. Gen. Virol. 1999;80(1):11‐14. [DOI] [PubMed] [Google Scholar]
- 181. Castilla J, Gonzalez‐Romero D, Saa P, et al. Crossing the Species Barrier by PrPSc Replication In Vitro Generates Unique Infectious Prions. Cell. 2008;134(5):757‐768. 10.1016/j.cell.2008.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Castilla J, Morales R, Saa P, et al. Cell‐free propagation of prion strains. EMBO J. 2008;27(19):2557‐2566. 10.1038/emboj.2008.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Green KM, Castilla J, Seward TS, et al. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008;4(8):e1000139 10.1371/journal.ppat.1000139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Castilla J, Saa P, Soto C. Detection of prions in blood. Nat. Med. 2005;11(9):982‐985. 10.1038/nm1286 [DOI] [PubMed] [Google Scholar]
- 185. Concha‐Marambio L, Pritzkow S, Moda F, et al. Detection of prions in blood from patients with variant Creutzfeldt‐Jakob disease. Sci. Transl. Med. 2016;8(370):370ra183 10.1126/scitranslmed.aaf6188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Bieschke J, Weber P, Sarafoff N, Beekes M, Giese A, Kretzschmar H. Autocatalytic self‐propagation of misfolded prion protein. Proc. Natl. Acad. Sci. U. S. A. 2004;101(33):12207‐12211. 10.1073/pnas.0404650101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease‐resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 2005;280(29):26873‐26879. 10.1074/jbc.M503973200 [DOI] [PubMed] [Google Scholar]
- 188. Orru CD, Wilham JM, Vascellari S, Hughson AG, Caughey B. New generation QuIC assays for prion seeding activity. Prion. 2012;6(2):147‐152. 10.4161/pri.19430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Atarashi R, Sano K, Satoh K, Nishida N. Real‐time quaking‐induced conversion: a highly sensitive assay for prion detection. Prion. 2011;5(3):150‐153. 10.4161/pri.5.3.16893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wilham JM, Orru CD, Bessen RA, et al. Rapid end‐point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010;6(12):e1001217 10.1371/journal.ppat.1001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Orru CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt‐Jakob disease using nasal brushings. N. Engl. J. Med. 2014;371(6):519‐529. 10.1056/NEJMoa1315200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Orru CD, Groveman BR, Hughson AG, et al. Rapid and sensitive RT‐QuIC detection of human Creutzfeldt‐Jakob disease using cerebrospinal fluid. mBio. 2015;6(1). 10.1128/mBio.02451-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kraus A, Raymond GJ, Race B, et al. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J. Virol. 2017;91(21):e01276‐e01217. 10.1128/JVI.01276-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Colby DW, Wain R, Baskakov IV, et al. Protease‐sensitive synthetic prions. PLoS Pathog. 2010;6(1):e1000736 10.1371/journal.ppat.1000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Li Q, Wang F, Xiao X, et al. Structural attributes of mammalian prion infectivity: insights from studies with synthetic prions. J. Biol. Chem. 2018;293(48):18494‐18503. 10.1074/jbc.RA118.005622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Legname G, Baskakov IV, Nguyen HO, et al. Synthetic mammalian prions. Science. 2004;305(5684):673‐676. 10.1126/science.1100195 [DOI] [PubMed] [Google Scholar]
- 197. Supattapone S. What makes a prion infectious? Science. 2010;327(5969):1091‐1092. 10.1126/science.1187790 [DOI] [PubMed] [Google Scholar]
- 198. Wang F, Yang F, Hu Y, et al. Lipid interaction converts prion protein to a PrPSc‐like proteinase K‐resistant conformation under physiological conditions. Biochemistry. 2007;46(23):7045‐7053. 10.1021/bi700299h [DOI] [PubMed] [Google Scholar]
- 199. Wang F, Yin S, Wang X, Zha L, Sy MS, Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP‐lipid interaction. Biochemistry. 2010;49(37):8169‐8176. 10.1021/bi101146v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005;15(2):102‐111. 10.1016/j.tcb.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 201. Goold R, Rabbanian S, Sutton L, et al. Rapid cell‐surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011;2(1):281 10.1038/ncomms1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Kanu N, Imokawa Y, Drechsel DN, et al. Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 2002;12(7):523‐530. [DOI] [PubMed] [Google Scholar]
- 203. Jeffrey M, McGovern G, Goodsir CM, Brown KL, Bruce ME. Sites of prion protein accumulation in scrapie‐infected mouse spleen revealed by immuno‐electron microscopy. J. Pathol. 2000;191(3):323‐332. [DOI] [PubMed] [Google Scholar]
- 204. Veith NM, Plattner H, Stuermer CA, Schulz‐Schaeffer WJ, Burkle A. Immunolocalisation of PrPSc in scrapie‐infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 2009;88(1):45‐63. 10.1016/j.ejcb.2008.08.001 [DOI] [PubMed] [Google Scholar]
- 205. Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992;267(23):16188‐16199. [PubMed] [Google Scholar]
- 206. Caughey B, Raymond GJ. The scrapie‐associated form of PrP is made from a cell surface precursor that is both protease‐ and phospholipase‐sensitive. J. Biol. Chem. 1991;266(27):18217‐18223. [PubMed] [Google Scholar]
- 207. Gilch S, Winklhofer KF, Groschup MH, et al. Intracellular re‐routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 2001;20(15):3957‐3966. 10.1093/emboj/20.15.3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent‐insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 1997;272(10):6324‐6331. [DOI] [PubMed] [Google Scholar]
- 209. Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease‐resistant state requires insertion of PrP‐res (PrP (Sc)) into contiguous membranes. EMBO J. 2002;21(5):1031‐1040. 10.1093/emboj/21.5.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor‐dependent and ‐independent prion protein association with model raft membranes on conversion to the protease‐resistant isoform. J. Biol. Chem. 2003;278(17):14883‐14892. 10.1074/jbc.M210840200 [DOI] [PubMed] [Google Scholar]
- 211. Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell. 1992;3(8):851‐863. 10.1091/mbc.3.8.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zurzolo C, van Meer G, Mayor S. The order of rafts. Conference on microdomains, lipid rafts and caveolae. EMBO Rep. 2003;4(12):1117‐1121. 10.1038/sj.embor.7400032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Taraboulos A, Scott M, Semenov A, et al. Cholesterol depletion and modification of COOH‐terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995;129(1):121‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Critchley P, Kazlauskaite J, Eason R, Pinheiro TJ. Binding of prion proteins to lipid membranes. Biochem. Biophys. Res. Commun. 2004;313(3):559‐567. [DOI] [PubMed] [Google Scholar]
- 215. Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J. Mol. Biol. 2002;315(5):1241‐1256. 10.1006/jmbi.2001.5322 [DOI] [PubMed] [Google Scholar]
- 216. Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB. COOH‐terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl. Acad. Sci. U. S. A. 1997;94(6):2333‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Walmsley AR, Zeng F, Hooper NM. The N‐terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J. Biol. Chem. 2003;278(39):37241‐37248. 10.1074/jbc.M302036200 [DOI] [PubMed] [Google Scholar]
- 218. Sarnataro D, Campana V, Paladino S, Stornaiuolo M, Nitsch L, Zurzolo C. PrPC association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell. 2004;15(9):4031‐4042. 10.1091/mbc.e03-05-0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Sarnataro D, Paladino S, Campana V, Grassi J, Nitsch L, Zurzolo C. PrPC is sorted to the basolateral membrane of epithelial cells independently of its association with rafts. Traffic. 2002;3(11):810‐821. [DOI] [PubMed] [Google Scholar]
- 220. Harris DA. Trafficking, turnover and membrane topology of PrP: protein function in prion disease. Br. Med. Bull. 2003;66(1):71‐85. 10.1093/bmb/66.1.71 [DOI] [PubMed] [Google Scholar]
- 221. Zavodszky E, Hegde RS. Misfolded GPI‐anchored proteins are escorted through the secretory pathway by ER‐derived factors. eLife. 2019;8 10.7554/eLife.46740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Beranger F, Mange A, Goud B, Lehmann S. Stimulation of PrPCRetrograde Transport toward the Endoplasmic Reticulum Increases Accumulation of PrPScin Prion‐infected Cells. J. Biol. Chem. 2002;277(41):38972‐38977. 10.1074/jbc.M205110200 [DOI] [PubMed] [Google Scholar]
- 223. Hegde RS, Rane NS. Prion protein trafficking and the development of neurodegeneration. Trends Neurosci. 2003;26(7):337‐339. 10.1016/S0166-2236(03)00143-7 [DOI] [PubMed] [Google Scholar]
- 224. Arnold JE, Tipler C, Laszlo L, Hope J, Landon M, Mayer RJ. The abnormal isoform of the prion protein accumulates in late‐endosome‐like organelles in scrapie‐infected mouse brain. J. Pathol. 1995;176(4):403‐411. 10.1002/path.1711760412 [DOI] [PubMed] [Google Scholar]
- 225. McKinley MP, Taraboulos A, Kenaga L, et al. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 1991;65(6):622‐630. [PubMed] [Google Scholar]
- 226. Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: a complex membrane‐anchoring structure for proteins. Biochemistry. 2008;47(27):6991‐7000. 10.1021/bi8006324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Tsai Y‐H, Gotze S, Vilotijevic I, et al. A general and convergent synthesis of diverse glycosylphosphatidylinositol glycolipids. Chem. Sci. 2013;4(1):468‐481. 10.1039/C2SC21515B [DOI] [Google Scholar]
- 228. Olschewski D, Seidel R, Miesbauer M, et al. Semisynthetic murine prion protein equipped with a GPI anchor mimic incorporates into cellular membranes. Chem. Biol. 2007;14(9):994‐1006. 10.1016/j.chembiol.2007.08.007 [DOI] [PubMed] [Google Scholar]
- 229. Chesebro B, Wehrly K, Caughey B, Nishio J, Ernst D, Race R. Foreign PrP expression and scrapie infection in tissue culture cell lines. Dev. Biol. Stand. 1993;80:131‐140. [PubMed] [Google Scholar]
- 230. Lawson VA, Priola SA, Wehrly K, Chesebro B. N‐terminal truncation of prion protein affects both formation and conformation of abnormal protease‐resistant prion protein generated in vitro. J. Biol. Chem. 2001;276(38):35265‐35271. 10.1074/jbc.M103799200 [DOI] [PubMed] [Google Scholar]
- 231. Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443(7113):803‐810. 10.1038/nature05294 [DOI] [PubMed] [Google Scholar]
- 232. Kundel F, Tosatto L, Whiten DR, Wirthensohn DC, Horrocks MH, Klenerman D. Shedding light on aberrant interactions ‐ a review of modern tools for studying protein aggregates. FEBS J. 2018;285(19):3604‐3630. 10.1111/febs.14409 [DOI] [PubMed] [Google Scholar]
- 233. Sang JC, Meisl G, Thackray AM, et al. Direct observation of murine prion protein replication in vitro. J. Am. Chem. Soc. 2018;140(44):14789‐14798. 10.1021/jacs.8b08311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Sang JC, Lee J‐E, Dear AJ, et al. Direct observation of prion protein oligomer formation reveals an aggregation mechanism with multiple conformationally distinct species. Chem. Sci. 2019;10(17):4588‐4597. 10.1039/C8SC05627G [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437(7056):257‐261. 10.1038/nature03989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Rutishauser D, Mertz KD, Moos R, et al. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One. 2009;4(2):e4446 10.1371/journal.pone.0004446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Barmada SJ, Harris DA. Visualization of prion infection in transgenic mice expressing green fluorescent protein‐tagged prion protein. J. Neurosci. 2005;25(24):5824‐5832. 10.1523/jneurosci.1192-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Moore RA, Vorberg I, Priola SA. Species barriers in prion diseases—brief review. Arch. Virol. Suppl. 2005;19:187‐202. [DOI] [PubMed] [Google Scholar]
- 239. Caughey B. Interactions between prion protein isoforms: the kiss of death? Trends Biochem. Sci. 2001;26(4):235‐242. [DOI] [PubMed] [Google Scholar]
- 240. McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J. Virol. 2009;83(9):4469‐4475. 10.1128/jvi.02412-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Chu NK, Shabbir W, Bove‐Fenderson E, et al. A C‐terminal membrane anchor affects the interactions of prion proteins with lipid membranes. J. Biol. Chem. 2014;289(43):30144‐30160. 10.1074/jbc.M114.587345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Charco JM, Erana H, Venegas V, et al. Recombinant PrP and its contribution to research on transmissible spongiform encephalopathies. Pathogens. 2017;6(4). 10.3390/pathogens6040067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Merrifield RB. Solid Phase Peptide Synthesis. I. The synthesis of a tetrapeptide. J. Am . Chem. Soc. 1963;85(14):2149‐2154. [Google Scholar]
- 244. Dawson PE, Muir TW, Clark‐Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266(5186):776‐779. [DOI] [PubMed] [Google Scholar]
- 245. Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U. S. A. 1998;95(12):6705‐6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Evans TC Jr, Benner J, Xu MQ. The in vitro ligation of bacterially expressed proteins using an intein from Methanobacterium thermoautotrophicum. J. Biol. Chem. 1999;274(7):3923‐3926. [DOI] [PubMed] [Google Scholar]
- 247. Gorodinsky A, Harris DA. Glycolipid‐anchored proteins in neuroblastoma cells form detergent‐resistant complexes without caveolin. J. Cell Biol. 1995;129(3):619‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Aguzzi A, Heppner FL. Pathogenesis of prion diseases: a progress report. Cell Death Differ. 2000;7(10):889‐902. 10.1038/sj.cdd.4400737 [DOI] [PubMed] [Google Scholar]
- 249. Weissmann C. Molecular biology of prion diseases. Trends Cell Biol. 1994;4(1):10‐14. [DOI] [PubMed] [Google Scholar]
- 250. Eberl H, Tittmann P, Glockshuber R. Characterization of Recombinant, Membrane‐attached Full‐length Prion Protein. J. Biol. Chem. 2004;279(24):25058‐25065. 10.1074/jbc.M400952200 [DOI] [PubMed] [Google Scholar]
- 251. Hicks MR, Gill AC, Bath IK, et al. Synthesis and structural characterization of a mimetic membrane‐anchored prion protein. FEBS J. 2006;273(6):1285‐1299. 10.1111/j.1742-4658.2006.05152.x [DOI] [PubMed] [Google Scholar]
- 252. Breydo L, Sun Y, Makarava N, et al. Nonpolar substitution at the C‐terminus of the prion protein, a mimic of the glycosylphosphatidylinositol anchor, partially impairs amyloid fibril formation. Biochemistry. 2007;46(3):852‐861. 10.1021/bi061923v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Ball HL, King DS, Cohen FE, Prusiner SB, Baldwin MA. Engineering the prion protein using chemical synthesis. J. Pept. Res. 2001;58(5):357‐374. [DOI] [PubMed] [Google Scholar]
- 254. Musiol H‐J, Dong S, Kaiser M, et al. Toward semisynthetic lipoproteins by convergent strategies based on click and ligation chemistry. ChemBioChem. 2005;6(4):625‐628. 10.1002/cbic.200400351 [DOI] [PubMed] [Google Scholar]
- 255. Araman C, Thompson RE, Wang S, Hackl S, Payne RJ, Becker CFW. Semisynthetic prion protein (PrP) variants carrying glycan mimics at position 181 and 197 do not form fibrils. Chem. Sci. 2017;8(9):6626‐6632. 10.1039/C7SC02719B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Matveenko M, Hackl S, Becker CFW. Utility of the phenacyl protecting group in traceless protein semisynthesis through ligation‐desulfurization chemistry. ChemistryOpen. 2018;7(1):106‐110. 10.1002/open.201700180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Becker CF, Liu X, Olschewski D, et al. Semisynthesis of a glycosylphosphatidylinositol‐anchored prion protein. Angew. Chem. Int. Ed. Engl. 2008;47(43):8215‐8219. [DOI] [PubMed] [Google Scholar]
- 258. Olschewski D, Becker CF. Chemical synthesis and semisynthesis of membrane proteins. Mol. Biosyst. 2008;4(7):733‐740. 10.1039/b803248c [DOI] [PubMed] [Google Scholar]
- 259. Chu NK, Becker CF. Semisynthesis of membrane‐attached prion proteins. Methods Enzymol. 2009;462:177‐193. 10.1016/s0076-6879(09)62009-7 [DOI] [PubMed] [Google Scholar]
- 260. Chu NK, Olschewski D, Seidel R, et al. Protein immobilization on liposomes and lipid‐coated nanoparticles by protein trans‐splicing. J. Pept. Sci. 2010;16(10):582‐588. 10.1002/psc.1227 [DOI] [PubMed] [Google Scholar]
- 261. Chu NK, Becker CF. Recombinant expression of soluble murine prion protein for C‐terminal modification. FEBS Lett. 2013;587(5):430‐435. 10.1016/j.febslet.2012.12.026 [DOI] [PubMed] [Google Scholar]
- 262. Volkmann G, Mootz H. Recent progress in intein research: from mechanism to directed evolution and applications. Cell. Mol. Life Sci. 2013;70(7):1185‐1206. 10.1007/s00018-012-1120-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Shah NH, Muir TW. Inteins: nature's gift to protein chemists. Chem. Sci. 2014;5(2):446‐461. 10.1039/C3SC52951G [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Xu MQ, Perler FB. The mechanism of protein splicing and its modulation by mutation. EMBO J. 1996;15(19):5146‐5153. [PMC free article] [PubMed] [Google Scholar]
- 265. Wu H, Hu Z, Liu XQ. Protein trans‐splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U. S. A. 1998;95(16):9226‐9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid‐modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913‐916. 10.1126/science.1068539 [DOI] [PubMed] [Google Scholar]
- 267. Rose K, Vizzavona J. Stepwise solid‐phase synthesis of polyamides as linkers. J. Am. Chem. Soc. 1999;121(30):7034‐7038. 10.1021/ja9909879 [DOI] [Google Scholar]
- 268. Martin DD, Xu MQ, Evans TC Jr. Characterization of a naturally occurring trans‐splicing intein from Synechocystis sp. PCC6803. Biochemistry. 2001;40(5):1393‐1402. [DOI] [PubMed] [Google Scholar]
- 269. Kaneko K, Wille H, Mehlhorn I, et al. Molecular properties of complexes formed between the prion protein and synthetic peptides. J. Mol. Biol. 1997;270(4):574‐586. 10.1006/jmbi.1997.1135 [DOI] [PubMed] [Google Scholar]
- 270. Baldwin MA. Analysis of glycosylphosphatidylinositol protein anchors: the prion protein. Methods Enzymol., (Academic Press. 2005;405:172‐187. 10.1016/S0076-6879(05)05008-1 [DOI] [PubMed] [Google Scholar]
- 271. Schumacher MC, Resenberger U, Seidel RP, et al. Synthesis of a GPI anchor module suitable for protein post‐translational modification. Biopolymers. 2010;94(4):457‐464. 10.1002/bip.21380 [DOI] [PubMed] [Google Scholar]
- 272. Vilotijevic I, Götze S, Seeberger PH, & Silva DV (2013) Chemical synthesis of GPI anchors and GPI‐anchored molecules Modern Synthetic Methods in Carbohydrate Chemistry, eds Werz D. B. & Vidal S. (Wiley‐VCH; ), pp 335‐372. 10.1002/9783527658947.ch12 [DOI] [Google Scholar]
- 273. Roller R, Vilotijevic I, Michel D, Seeberger HP, Silva DV. Semi‐synthesis of glycosylphosphatidylinositol anchored glycopeptides and glycoproteins. J. Pept. Sci. 2014;20:S47‐S48. [Google Scholar]
- 274. Grube M, Lee B‐Y, Garg M, et al. Synthesis of galactosylated glycosylphosphatidylinositol derivatives from trypanosoma brucei. Chem. Eur. J. 2018;24(13):3271‐3282. 10.1002/chem.201705511 [DOI] [PubMed] [Google Scholar]
- 275. Makarava N, Baskakov IV. Expression and purification of full‐length recombinant PrP of high purity. Methods Mol. Biol. 2008;459:131‐143. 10.1007/978-1-59745-234-210 [DOI] [PubMed] [Google Scholar]
- 276. Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105‐125. EMBO J. 2007;26(2):548‐558. 10.1038/sj.emboj.7601507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J. Neurochem. 2009;108(1):44‐56. 10.1111/j.1471-4159.2008.05719.x [DOI] [PubMed] [Google Scholar]
- 278. Otvos L Jr, Cudic M. Post‐translational modifications in prion proteins. Curr. Protein Pept. Sci. 2002;3(6):643‐652. [DOI] [PubMed] [Google Scholar]
- 279. Stimson E, Hope J, Chong A, Burlingame AL. Site‐specific characterization of the N‐linked glycans of murine prion protein by high‐performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry. 1999;38(15):4885‐4895. 10.1021/bi982330q [DOI] [PubMed] [Google Scholar]
- 280. Shi L, Chen H, Zhang SY, et al. Semi‐synthesis of murine prion protein by native chemical ligation and chemical activation for preparation of polypeptide‐α‐thioester. J. Pept. Sci. 2017;23(6):438‐444. 10.1002/psc.3008 [DOI] [PubMed] [Google Scholar]
- 281. Mishra R, Elgland M, Begum A, et al. Impact of N‐glycosylation site variants during human PrP aggregation and fibril nucleation. Biochim. Biophys. Acta Proteins Proteom. 2019;1867(10):909‐921. 10.1016/j.bbapap.2019.03.010 [DOI] [PubMed] [Google Scholar]