

Abstract
A “branching–folding” synthetic strategy that affords a range of diverse cyclic benzo‐sulfonamide scaffolds is presented. Whereas different annulation reactions on common ketimine substrates build the branching phase of the scaffold synthesis, a common hydrogenative ring‐expansion method, facilitated by an increase of the ring‐strain during the branching phase, led to sulfonamides bearing medium‐sized rings in a folding pathway. Cell painting assay was successfully employed to identify tubulin targeting sulfonamides as novel mitotic inhibitors.
Keywords: branching pathway, cell painting, folding pathway, mitotic inhibitors, sulfonamides
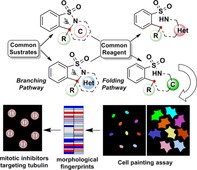
Branch, fold and paint! A “branching–folding” synthetic strategy delivers a range of diverse cyclic benzo‐sulfonamides and a cell painting analysis led to disclose novel tubulin targeting sulfonamide mitotic inhibitors.
The ability of small molecules to perturb biological systems in a temporal‐ and dose‐controlled manner endows them uniqueness to reveal insights of different biological functions. Advancement of chemical biology and drug discovery demands a regular supply of novel bioactive small molecules. The unmet medical needs and limited understanding of life at molecular level urgently call to interrogate biologically intriguing yet least explored areas of chemical space to deliver small molecules as probe and drug candidates.1 Unfortunately, organic synthesis had remained focused on a narrow range of molecular scaffolds despite the emergence of strategies, such as diversity‐oriented synthesis, which precisely aimed to deliver a range of diverse chemotypes.2
Sulfonamides, cyclic and acyclic, are a well‐known class of small molecules that represents several FDA‐approved drugs and potential candidates for a range of molecular targets and indications. Intriguingly, no natural product is yet known to embody a cyclic sulfonamide moiety. The primary sulfonamide and sulfamate groups are also rarely found in natural product structures (Scheme 1 a). Whereas the introduction of a sulfonamide moiety on small molecules is relatively easy and well‐established,3 the development of synthetic approaches affording a range of diverse biologically relevant cyclic sulfonamides remain underexplored.4 Herein, we disclose a scaffold‐diversity synthesis of cyclic benzo‐sulfonamides by employing a sequential “branching–folding” synthetic approach and using easily accessible saccharin‐derived cyclic N‐sulfonyl ketimines 1 as the precursors (Scheme 1 c).5 In this approach, the first branching pathway exploits different annulation and nucleophilic addition reactions on common substrates to form complex and cyclic sulfonamides with more sp 3 character and decorated with stereogenic centers. The next folding ring‐expansion strategy, driven by the ring‐strain in the scaffolds formed in branching phase, leads to medium ring‐sized benzo‐sulfonamides.
Scheme 1.
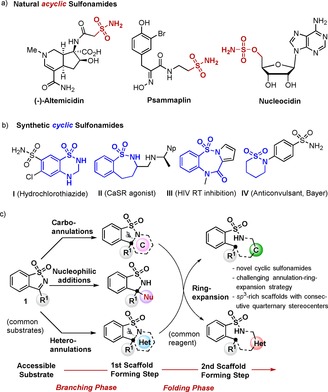
a) Some natural products with acyclic sulfonamides. b) Synthetic bioactive cyclic sulfonamides. c) A “branching–folding” synthesis planning to form diverse, complex and medium ring‐sized cyclic sulfonamides.
A branching synthesis pathway transforms a common substrate into structurally distinct scaffolds. The plan to employ saccharin‐derived ketimine 1 as the substrate for the sequential scaffold generating approach stemmed from both the reported as well as potential and unexplored reactivity of these cyclic ketimines in annulation and addition reactions. We focused on developing a range of different ring‐fusion transformations to the imine moiety, that is, from three‐membered aziridine to a six‐membered piperidine ring. This would offer possibilities for a subsequent and more challenging folding pathway, wherein common reaction conditions may induce ring‐expansion of different scaffolds formed in the first branching phase, delivering another set of cyclic sulfonamides (Scheme 1 c).
In our first reaction design in the branching phase, we planned to make annulation reactions with imine moiety of common substrate 1 employing 1‐ to 4‐carbon annulation partners to afford three to six‐membered aza‐heterocycle‐fused sulfonamides. To this end, the first scaffold target was the smallest stable aza‐ring system, that is, aziridines.6 Among different possible synthetic strategies to aziridines,7 the carbenium transfer approach through sulfonium ylides was employed affording different aziridine‐fused sulfonamides (2 a–h) with two different sites in acceptable to good yields (Scheme 2). Intriguingly, reactions of sulfonium ylides with N‐sulfonyl‐aldimine 1 (R1=H) did not afford the corresponding aziridines. Except for 2 c and 2 h that bear two adjacent aryl groups, and were formed in equimolar ratio of syn and anti isomers in high yields, aziridines were formed mostly as single syn diastereomers (2 b, 2 d–g) or as major isomer (2 a, d.r. 7:1). However, for ketimines with a carboethoxy moiety, in particular, 2 e–2 f, the formation of uncharacterizable by‐products (low molecular weight) was observed, which led to lower yields of desired products (Scheme 2).
Scheme 2.
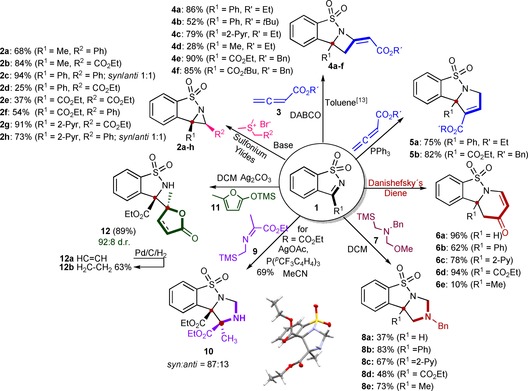
A branching pathway to cyclic sulfonamides by annulation and addition reactions of N‐sulfonyl ketimines. DABCO=1,4‐diazabicyclo[2.2.2]octane.
Towards 4‐ and 5‐membered ring‐fused benzo‐sulfonamides, Ye and co‐workers' approach8 of nucleophilic base‐catalyzed annulation reactions was exploited. Thus, the zwitterionic intermediate formed by addition of DABCO (cat.) and allenoates (3) added regioselectively from the terminal position of the allene to effectively result in a [2+2]‐annulation and formed benzosultam‐fused (E)‐azetidines (4 a–f). We observed that a reduced electrophilicity of imine moiety in 1 (R1=Me) negatively influenced the reaction progression and product yield for 4 d. Interestingly, aldimine 1 (R1 = H) did not react with allene‐derived zwitterions at all. Furthermore, five‐membered pyrroline‐based benzo‐sulfonamides were easily synthesized employing triphenylphosphine instead of DABCO as nucleophilic catalyst in the above methodology affording 5 a and 5 b in high yields (Scheme 2). The latter features two orthogonal esters for further selective functional group transformations.
For the six‐membered ring‐fused benzo‐sulfonamides, we used the Diels–Alder reaction.9 After some reaction conditions optimization, reaction of 1 with Danishefsky's diene under microwave irradiation in toluene furnished 1,4‐dihydro‐pyridones 6 a–d in good to excellent yields (Scheme 2). However, under the optimized microwave heating conditions, reaction of relatively electron‐rich ketimine 1‐Me, could afford only low yield of adduct 6 e.10
To introduce more heteroatoms as well as quaternary centers in more diverse and complex benzo‐sulfonamides, we employed two different azomethine ylides derived from 7 11 and α‐silyl imine 9 with ketimines 1 in dipolar annulation reactions and yielding imidazolidine‐based benzo‐sulfonamides 8 a–e and 10 in moderate to high yields (37–83 %).12 Interestingly, reactions of 7 derived dipole performed better with aryl or alkyl ketimine 1 than with aldimine (1, R1=H) or iminoester (1, R1=CO2Et, Scheme 2). Notably, α‐silyl imines (9) have rarely been explored in cycloaddition chemistry and are interesting substrates for generating small molecules embodying quaternary centers and complex stereochemical frameworks.13 Another reaction screening revealed that catalytic AgOAc and phosphine complex in acetonitrile could transform 1‐CO2Et with 9 into syn‐imidazolidine 10 (d.r.=87:13) as major adduct that bear two consecutive quaternary stereocenters (Table S1 in Supporting information).
In the last reaction of the branching pathway, a Mannich‐type addition of α‐angelica lactone14 as nucleophile to ketimine moiety in 1 was explored.15 In this way, we targeted molecular complexity both in terms of consecutive quaternary stereocenters16 in the product, as well as having a natural product fragment—the lactone ring—with a free rotation around the newly constructed bond. In our hands, silyl enol ether 11 worked better in the desired addition reaction than lactone itself. Variation of reaction temperature and Lewis acids identified AgOAc (Table S2) as the best catalyst for the vinylogous addition reaction, exhibiting high reactivity (quick reaction within 10 min) at −78 °C to form lactone 12 a in good yield (89 %) and diastereomeric ratio (25:2) in favor of syn‐diastereomer (based on nOe experiments). Hydrogenation of 12 a using 10 mol % Pd/C furnished a saturated γ‐lactone 12 b in 63 % yield.
With tricyclic scaffolds (2, 4–6, 8 and 10) in hand, we explored a common ring‐expansion approach in a'folding pathway' that could expand the range of distinct sulfonamides,17 in particular supporting medium‐ring‐sized frameworks.18 The key for the success of this approach was the selective cleavage of C−N bond in the annulation adducts formed in the branching phase. Although C−N bond cleavage does occur in the course of coupling19 and aziridine‐opening reactions,20 it has not been thoroughly utilized as a strategy to build larger cyclic systems.21 We hypothesized that, palladium on carbon could be used to cleave the N−C bond of the adducts from the branching phase in analogy to N‐debenzylation, but in intramolecular and stereoselective22 fashion to offer an additional set of novel cyclic sulfonamide scaffolds (Scheme 1). Interestingly, aziridines 2 a–h with one benzylic N−C bond (R2=CO2Et) under hydrogenation conditions (10 mol % Pd/C, H2), cleaved the quaternary benzylic C−N bond to deliver sultams 13 a–d (30–99 % yield) exclusively in syn configuration. However, aziridines with two benzylic N−C bonds (R1 and R2=Ar) were preferentially cleaved at the tertiary benzylic C−N bond to give sulfonamides 14 a–d (30–69 % yield). It seems that ring‐strain in aziridines with quaternary benzylic carbon indeed could drive the formation of sulfonamides 13 (for details see Supporting info). However, when possible, a tertiary benzylic C−N cleavage was preferred to form 14 a–d (Scheme 3).
Scheme 3.
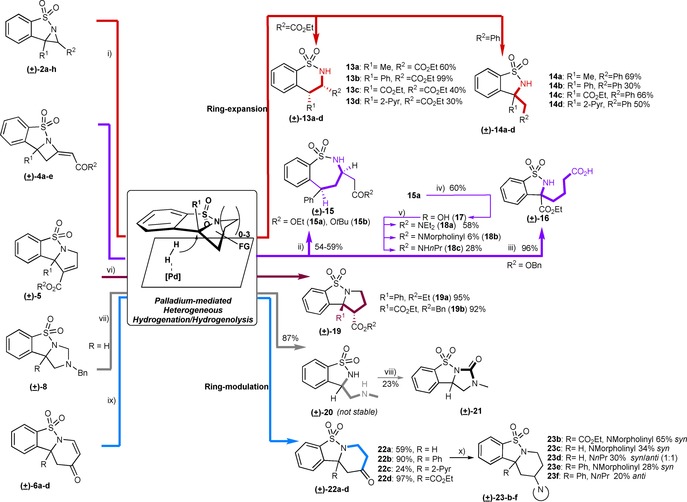
Ring‐expansion and modulation reactions through palladium‐mediated heterogeneous hydrogenation/hydrogenolysis. Conditions: i) 10 mol % Pd/C, EtOH/EtOAc,1 atm H2; ii) 14 mol % Pd/C, EtOH/EtOAc, 7.5 atm H2; iii) Pearlman′s catalyst (20 wt %., 9 mol %), MeOH/EtOAc, 2 h; iv) aq. NaOH/THF/MeCN, 19 h; v) first, (COCl)2 or SOCl2 in CHCl3, 60 °C; then, corresponding amine in THF at 0 °C; vi) Pd(OH)2/C, H2; vii) 10 mol % Pd/C, 7 atm H2, 16 h; viii) Et3N, CDI, MeCN, 16 h; ix) 10 mol % Pd/C H2, EtOH (+EtOAc for 22 d). For 22a: additional step of DMP/NaHCO3, DCM 0° C, 3 h; x) NaB(OAc)3H, corresponding amine, 4 Å MS, 1,2‐DCE, 16–48h.
Other substrates were also subjected to Pd‐mediated hydrogenation conditions. In particular, we expected to get medicinally important3g, 23 seven‐membered sulfonamides from azetidines 4. To our delight, under the same hydrogenative conditions, 4 a,b afforded the syn diastereomer of the corresponding seven‐membered benzo‐sulfonamides 15 a,b in moderate yields (Scheme 3). Isolation of a ring‐expanded intermediate 15 a' (49 % yield, see Supporting Information)12 suggests that hydrogenolysis of C−N bond precedes hydrogenation of exocyclic double bond (Scheme 2). We observed that a double benzylic substitution (R1=Ph) in azetidine 4 is a requisite for the ring expansion to occur. For instance, all our efforts for the desired ring expansion of ester‐substituted azetidine 4 e to the seven‐membered sulfonamide failed. In fact, the reaction led to extra‐cyclic opening of azetidine 4 e yielding 16 in 96 % yield. Nevertheless, ester hydrolysis and amide synthesis in 15 a and 15 b was performed to afford amides 18 a–c through carboxylic acid 17 (Scheme 3).
Pyrrolinyl sulfonamides 5 did not follow ring‐expansion. Instead, hydrogenation of the double bond occurred to give pyrrolidine syn‐19 in 95 % yield. Hydrogenation of aminal 8 a cleaved the ring at the expected aminal carbon to furnish a relatively unstable benzo‐sulfonamide 20 (87 % yield). The latter was quickly transformed into the stable tricyclic sulfonyl urea 21 that features a key motif of herbicidal24 and anti‐diabetic sulfonamides,25 by treatment with carbonyl diimidazole in presence of base.
Likewise, six‐membered dihydropyridones 6 a–d lacking ring‐strain were transformed into 1,4‐tetrahydropyridones 22 by heterogeneous catalytic hydrogenation. Furthermore, the ketone in 22 was used for reductive amination reactions using NaBH(OAc)3. It was observed that morpholine adducts displayed a higher degree of syn selectivity than n‐propylamine adducts (Scheme 3). The scaffold diversity generated in the branching and folding phases is clearly depicted in terms of three‐dimensional chemical space (PMI plot; Figure S1) and range of molecular properties (Figure S2) represented by the ensuing sulfonamide compound collection. To make an unbiased analysis of the novel sulfonamides formed, their biological activity was explored in a cell painting assay (CPA).26 CPA is an image‐based analysis that measures and quantifies a range of morphological changes in cells induced by a compound and condensed to generate a fingerprint for novel as well as reference compounds. Identification of reference compounds with a profile similar to the desired molecules may offer insights into the biological activity as well as modes of action of new compounds (Figure 1 a).27 To this goal, U2OS cells were treated with the compounds for 20 h prior to staining of cellular organelles and cellular components, that is, DNA, endoplasmic reticulum, nucleoli and cytoplasmic RNA, actin, mitochondria, Golgi and plasma membrane. Automated image analysis and data processing result in morphological fingerprints to display the change of 579 parameters in comparison to the DMSO‐treated cells (Figure S3). Two derivatives displayed activity in the CPA with induction of 51 % (4 a) and 35 % (5 b), i.e. percentage of the significantly changed parameters as compared to the DMSO control. These fingerprints were compared with fingerprints of biologically active reference compounds to generate target hypotheses. Interestingly, the morphological profile of 4 a displayed similarity to tubulin‐acting agents (fenbendazole29 −74 % similarity, tubulexin A30 −84 % similarity), (Figure 1 b and Figure S4a‐b), whereas the morphological profile of 5 b was similar to an inhibitor of the mitotic Polo‐like kinase 131 (PLK1‐56 % similarity, Figures S5 and S4c).
Figure 1.
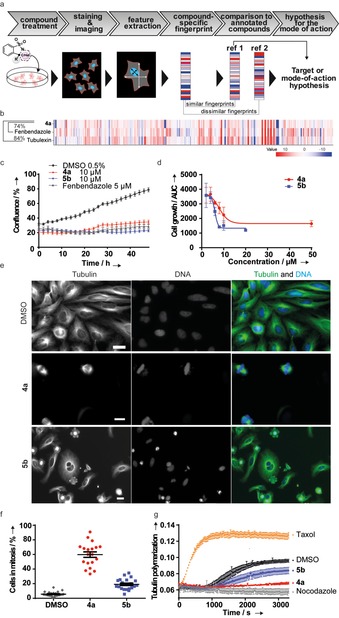
Influence of benzo‐sulfonamides 4 a and 5 b on cell growth, mitosis and tubulin polymerization. a) Schematic representation of morphological profiling, for example, the cell painting assay.28 b) Fingerprint comparison for 4 a (10 μm) with fenbendazole (3 μm) and tubulexin A (50 μm). c) The growth of U2OS cells was monitored for 48 h using kinetic live‐cell imaging in presence of the compounds or DMSO and fenbendazole as controls. Data are mean values (N=6)±SD and are representative of three biological replicates. d) Dose‐response analyses for cell growth inhibition were carried out as described in (a). The area under the curve was used to determine IC50 for cell growth inhibition. IC50 (4 a)=7.8±1.6 μm; IC50 (5 b)=6.0±0.4 μm. Data are mean values (N=3)±SD and are representative of two biological replicates. e) U2OS cells were treated with the compounds for 24 h prior to staining of DNA and tubulin using DAPI (blue) and anti‐alpha‐tubulin‐FITC antibody (green). Scale bar: 20 μm. f) U2OS cells were treated with the compounds for 24 h prior to staining for phospho‐histone 3 as a marker for mitotic arrest and DNA followed by automated image acquisition and analysis to quantify the percentage of cells in metaphase (i.e., phospho‐histone 3‐positive cells). Data are mean values (N=2)±SD and are representative of three biological replicates. g) In vitro tubulin polymerization assay. Tubulin polymerization was initiated in the presence of GTP and was monitored by means of turbidity measurement at 340 nm at 37 °C. Taxol and nocodazole were used as controls for tubulin stabilizing and destabilizing agents, respectively. Data are representative of three biological replicates.
Both, tubulin and PLK1 have an essential function during mitosis. Therefore, we monitored the growth of U2OS in presence of 4 a and 5 b cells by means of real‐time live‐cell analysis (Figure 1 a). Both compounds reduced cell growth dose‐dependently with IC50 values of 7.8±1.6 μm (4 a) and 6.0±0.4 μm (5 b) (Figure 1 b and Movies S1–S3). Inspection of cell morphology revealed the accumulation of round cells (Figure S6) with mitotic spindles (Figure 1 e) that is indicative of mitotic arrest. Arrest in mitosis was confirmed using phospho‐histone 3 staining that revealed increase in the fraction of mitotic cells from 5.2±2.4 % (DMSO‐treated cells) to 62.5±15.1 % and 19.8±6.9 % in the presence of 10 μm 4 a and 5 b, respectively (Figure 1 f and Figures S6–7). Tubulin polymerization is amenable to modulation by small molecules32 and interference with microtubule dynamics leads to mitotic arrest. Because the CPA revealed similarity of the fingerprints to the tubulin targeting agents fenbendazole and tubulexin A (Figure 1 b), we analyzed the influence of benzo‐sulfonamides 4 a and 5 b on in vitro tubulin polymerization. Compound 4 a strongly inhibited tubulin polymerization at 20 μm, whereas compound 5 b was less potent (Figure 1 g). These results confirm the CPA‐generated mode‐of‐action hypothesis and identified sulfonamides 4 a and 5 b as novel microtubule‐targeting agents. Notably, sulfonamide class of small molecules is not typically known for anti‐mitotic activities and therefore CPA was instrumental to offer an unexpected biological annotation to benzo‐sulfonamides.
In conclusion, a novel synthetic approach employing a “branching–folding” pathway strategy is presented to give access to biologically relevant yet underexplored cyclic benzo‐sulfonamides. Different annulation reactions of common substrates formed the core of branching pathway, and ring‐expansion strategy based on hydrogenolysis was key for the folding route. Cell painting assay was instrumental in identifying hits from this non‐natural class of small molecules and unravelling their unexpected anti‐mitotic activity and tubulin inhibition. We believe this approach will find further applications to reach out to novel chemical and biological space and to help advance discovery research.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
This research was funded partially by the Max Planck Society and the Innovative Medicines Initiative Joint Undertaking under the grant agreement (Nr. 115489), resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007‐2013) and EFPIA companies’ in‐kind contribution. Authors are grateful to Prof. H. Waldmann for his kind support and encouragement.
S. Zimmermann, M. Akbarzadeh, F. Otte, C. Strohmann, M. G. Sankar, S. Ziegler, A. Pahl, S. Sievers, K. Kumar, Chem. Eur. J. 2019, 25, 15498.
References
- 1.
- 1a. Scannell J. W., Blanckley A., Boldon H., Warrington B., Nat. Rev. Drug. Discovery 2012, 11, 191–200; [DOI] [PubMed] [Google Scholar]
- 1b. Ilardi E. A., Vitaku E., Njardarson J. T., J. Med. Chem. 2014, 57, 2832–2842; [DOI] [PubMed] [Google Scholar]
- 1c. Mullard A., Nat. Rev. Drug. Discovery 2016, 15, 669–669; [DOI] [PubMed] [Google Scholar]
- 1d. Garcia-Castro M., Zimmermann S., Sankar M. G., Kumar K., Angew. Chem. Int. Ed. 2016, 55, 7586–7605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7712–7732; [Google Scholar]
- 1e. Smietana K., Quigley D., Van de Vyver B., Møller M., Nat. Rev. Drug Discovery 2019, DOI: 10.1038/d41573-019-00046-3. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Schreiber S. L., Science 2000, 287, 1964–1969; [DOI] [PubMed] [Google Scholar]
- 2b. Burke M. D., Schreiber S. L., Angew. Chem. Int. Ed. 2004, 43, 46–58; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 48–60. [Google Scholar]
- 3.Selected reports:
- 3a. Hanson P. R., Probst D. A., Robinson R. E., Yau M., Tetrahedron Lett. 1999, 40, 4761–4764; [Google Scholar]
- 3b. Ji T., Wang Y., Wang M., Niu B., Xie P., Pittman C. U., Zhou A., ACS Comb. Sci. 2013, 15, 595–600; [DOI] [PubMed] [Google Scholar]
- 3c. Loh J. K., Asad N., Samarakoon T. B., Hanson P. R., J. Org. Chem. 2015, 80, 9926–9941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Lacour J., Kidd S. L., Guarnieri-Ibáñez A., Besnard C., Spring D. R., Medina F., Chem. Sci. 2017, 8, 5713–5720; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Laha J. K., Sharma S., Kirar S., Banerjee U. C., J. Org. Chem. 2017, 82, 9350–9359; [DOI] [PubMed] [Google Scholar]
- 3f. Liu R. R., Hu J. P., Hong J. J., Lu C. J., Gao J. R., Jia Y. X., Chem. Sci. 2017, 8, 2811–2815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Quan M., Wang X., Wu L., Gridnev I. D., Yang G., Zhang W., Nat. Commun. 2018, 9, 2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reviews:
- 4a. Rassadin V. A., Grosheva D. S., Tomashevskii A. A., Sokolov V. V., Chem. Heterocycl. Compd. 2013, 49, 39–65; [Google Scholar]
- 4b. Majumdar K. C., Mondal S., Chem. Rev. 2011, 111, 7749–7773; [DOI] [PubMed] [Google Scholar]
- 4c. Debnath S., Mondal S., Eur. J. Org. Chem. 2018, 933–956. [Google Scholar]
- 5.For some recent reports, see:
- 5a. Dauban P., Dodd R. H., Org. Lett. 2000, 2, 2327–2329; [DOI] [PubMed] [Google Scholar]
- 5b. Figueroa F. N., Heredia A. A., Peñéñory A. B., Sampedro D., Argüello J. E., Oksdath-Mansilla G., J. Org. Chem. 2019, 84, 3871–3880; [DOI] [PubMed] [Google Scholar]
- 5c. Wang H., Jiang T., Xu M. H., J. Am. Chem. Soc. 2013, 135, 971–974; [DOI] [PubMed] [Google Scholar]
- 5d. Zhong D. Y., Wu D., Zhang Y., Lu Z. W., Usman M., Liu W., Lu X. Q., Liu W. B., Org. Lett. 2019, 21, 5808–5812; [DOI] [PubMed] [Google Scholar]
- 5e. Kiefer L., Gorojankina T., Dauban P., Faure H., Ruat M., Dodd R. H., Bioorg. Med. Chem. Lett. 2010, 20, 7483–7487. [DOI] [PubMed] [Google Scholar]
- 6. Callebaut G., Meiresonne T., De Kimpe N., Mangelinckx S., Chem. Rev. 2014, 114, 7954–8015. [DOI] [PubMed] [Google Scholar]
- 7. Zhang S., Li L., Xin L., Liu W., Xu K., J. Org. Chem. 2017, 82, 2399–2406. [DOI] [PubMed] [Google Scholar]
- 8. Chen X. Y., Lin R. C., Ye S., Chem. Commun. 2012, 48, 1317–1319. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Heravi M. M., Ahmadi T., Ghavidel M., Heidari B., Hamidi H., RSC Adv. 2015, 5, 101999–102075; [Google Scholar]
- 9b. Nicolaou K. C., Snyder S. A., Montagnon T., Vassilikogiannakis G., Angew. Chem. Int. Ed. 2002, 41, 1668–1698; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1742–1773; [Google Scholar]
- 9c. Eschenbrenner-Lux V., Kumar K., Waldmann H., Angew. Chem. Int. Ed. 2014, 53, 11146–11157; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11326–11337. [Google Scholar]
- 10. Cao H.-w., Abramovich R. A., Stowers L. R., Heterocycles 1984, 22, 671. [Google Scholar]
- 11. Padwa A., Dent W., J. Org. Chem. 1987, 52, 235–244. [Google Scholar]
- 12.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201904175 (syn-10 and 15 a, respectively) contain the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
- 13. Kesava-Reddy N., Golz C., Strohmann C., Kumar K., Chem. Eur. J. 2016, 22, 18373–18377. [DOI] [PubMed] [Google Scholar]
- 14. Trost B. M., Hung C.-I. J., Scharf M. J., Angew. Chem. Int. Ed. 2018, 57, 11408–11412; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11578–11582. [Google Scholar]
- 15. Li X., Lu M., Dong Y., Wu W. B., Qian Q. Q., Ye J. X., Dixon D. J., Nat. Commun. 2014, 5, 4479. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Liu Y. Y., Han S. J., Liu W. B., Stoltz B. M., Acc. Chem. Res. 2015, 48, 740–751; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Trost B. M., Jiang C. H., Synthesis 2006, 369–396; [Google Scholar]
- 16c. Xu P. W., Yu J. S., Chen C., Cao Z. Y., Zhou F., Zhou J., ACS Catal. 2019, 9, 1820–1882. [Google Scholar]
- 17.
- 17a. Bauer R. A., Wenderski T. A., Tan D. S., Nat. Chem. Biol. 2013, 9, 21–29; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Rao B., Tang J., Wei Y., Zeng X., Chem. Asian J. 2016, 11, 991–995; [DOI] [PubMed] [Google Scholar]
- 17c. Guney T., Wenderski T. A., Boudreau M. W., Tan D. S., Chem. Eur. J. 2018, 24, 13150–13157; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Murai K., Kobayashi T., Miyoshi M., Fujioka H., Org. Lett. 2018, 20, 2333–2337; [DOI] [PubMed] [Google Scholar]
- 17e. Stephens T. C., Lawer A., French T., Unsworth W. P., Chem. Eur. J. 2018, 24, 13947–13953; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17f. Choi Y., Kim H., Park S. B., Chem. Sci. 2019, 10, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Burke M. D., Berger E. M., Schreiber S. L., J. Am. Chem. Soc. 2004, 126, 14095–14104; [DOI] [PubMed] [Google Scholar]
- 18b. Oguri H., Schreiber S. L., Org. Lett. 2005, 7, 47–50. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Esquivias J., Gómez Arrayás R., Carretero J. C., Angew. Chem. Int. Ed. 2006, 45, 629–633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 645–649; [Google Scholar]
- 19b. Liu C.-R., Li M.-B., Yang C.-F., Tian S.-K., Chem. Eur. J. 2009, 15, 793–797; [DOI] [PubMed] [Google Scholar]
- 19c. Liu C. R., Yang F. L., Jin Y. Z., Ma X. T., Cheng D. J., Li N., Tian S. K., Org. Lett. 2010, 12, 3832–3835; [DOI] [PubMed] [Google Scholar]
- 19d. Weng Z.-t., Li Y., Tian S.-k., J. Org. Chem. 2011, 76, 8095–8099; [DOI] [PubMed] [Google Scholar]
- 19e. Yang C. F., Wang J. Y., Tian S. K., Chem. Commun. 2011, 47, 8343–8345; [DOI] [PubMed] [Google Scholar]
- 19f. Wu X. S., Tian S. K., Chem. Commun. 2012, 48, 898–900. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Hu X. E., Tetrahedron 2004, 60, 2701–2743; [Google Scholar]
- 20b. Dauban P., Malik G., Angew. Chem. Int. Ed. 2009, 48, 9026–9029; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9188–9191; [Google Scholar]
- 20c. Schneider C., Angew. Chem. Int. Ed. 2009, 48, 2082–2084; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2116–2118; [Google Scholar]
- 20d. Lu P., Tetrahedron 2010, 66, 2549–2560; [Google Scholar]
- 20e. Stanković S., D′hooghe M., Catak S., Eum H., Waroquier M., Van Speybroeck V., De Kimpe N., Ha H.-J., Chem. Soc. Rev. 2012, 41, 643–665; [DOI] [PubMed] [Google Scholar]
- 20f. Yang P.-J., Qi L., Liu Z., Yang G., Chai Z., J. Am. Chem. Soc. 2018, 140, 17211–17217. [DOI] [PubMed] [Google Scholar]
- 21. Du H., Fairbridge C., Yang H., Ring Z., Appl. Catal. A 2005, 294, 1–21. [Google Scholar]
- 22. Kukula P., Prins R., Topics Catal. 2003, 25, 29–42. [Google Scholar]
- 23. Tollefson M. B., Kolodziej S. A., Fletcher T. R., Vernier W. F., Beaudry J. A., Keller B. T., Reitz D. B., Bioorg. Med. Chem. Lett. 2003, 13, 3727–3730. [DOI] [PubMed] [Google Scholar]
- 24. Hay J. V., Pestic. Sci. 1990, 29, 247–261. [Google Scholar]
- 25. Sola D., Rossi L., Schianca G. P. C., Maffioli P., Bigliocca M., Mella R., Corlianò F., Fra G. P., Bartoli E., Derosa G., Arch. Med. Sci. 2015, 4, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gustafsdottir S. M., Ljosa V., Sokolnicki K. L., Wilson J. A., Walpita D., Kemp M. M., Seiler K. P., Carrel H. A., Golub T. R., Schreiber S. L., Clemons P. A., Carpenter A. E., Shamji A. F., PLoS One 2013, 8, e80999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Christoforow A., Wilke J., Binici A., Pahl A., Ostermann C., Sievers S., Waldmann H., Angew. Chem. Int. Ed. 2019, 58, 14715–14723; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14857–14865. [Google Scholar]
- 28. Bray M. A., Singh S., Han H., Davis C. T., Borgeson B., Hartland C., Kost-Alimova M., Gustafsdottir S. M., Gibson C. C., Carpenter A. E., Nat. Protoc. 2016, 11, 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Friedman P. A., Platzer E. G., Biochim. Biophys. Acta Gen. Subj. 1978, 544, 605–614. [DOI] [PubMed] [Google Scholar]
- 30. Voigt T., Gerding-Reimers C., Tuyen T. N. T., Bergmann S., Lachance H., Scholermann B., Brockmeyer A., Janning P., Ziegler S., Waldmann H., Angew. Chem. Int. Ed. 2013, 52, 410–414; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 428–432. [Google Scholar]
- 31. Emmitte K. A., Andrews C. W., Badiang J. G., Davis-Ward R. G., Dickson H. D., Drewry D. H., Emerson H. K., Epperly A. H., Hassler D. F., Knick V. B., Kuntz K. W., Lansing T. J., Linn J. A., Mook R. A., Nailor K. E., Salovich J. M., Spehar G. M., Cheung M., Bioorg. Med. Chem. Lett. 2009, 19, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 32. Arnst K. E., Banerjee S., Chen H., Deng S. S., Hwang D. J., Li W., Miller D. D., Med. Res. Rev. 2019, 39, 1398–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary