

Supplemental Digital Content is available in the text.
Keywords: arrhythmias, cardiac; genotype; heart; heart rate; long QT syndrome
Background:
Long QT syndromes (LQTS) arise from many genetic and nongenetic causes with certain characteristic ECG features preceding polymorphic ventricular tachyarrhythmias (PVTs). However, how the many molecular causes result in these characteristic ECG patterns and how these patterns are mechanistically linked to the spontaneous initiation of PVT remain poorly understood.
Methods:
Anatomic human ventricle and simplified tissue models were used to investigate the mechanisms of spontaneous initiation of PVT in LQTS.
Results:
Spontaneous initiation of PVT was elicited by gradually ramping up ICa,L to simulate the initial phase of a sympathetic surge or by changing the heart rate, reproducing the different genotype-dependent clinical ECG features. In LQTS type 2 (LQT2) and LQTS type 3 (LQT3), T-wave alternans was observed followed by premature ventricular complexes (PVCs). Compensatory pauses occurred resulting in short-long-short sequences. As ICa,L increased further, PVT episodes occurred, always preceded by a short-long-short sequence. However, in LQTS type 1 (LQT1), once a PVC occurred, it always immediately led to an episode of PVT. Arrhythmias in LQT2 and LQT3 were bradycardia dependent, whereas those in LQT1 were not. In all 3 genotypes, PVCs always originated spontaneously from the steep repolarization gradient region and manifested on ECG as R-on-T. We call this mechanism R-from-T, to distinguish it from the classic explanation of R-on-T arrhythmogenesis in which an exogenous PVC coincidentally encounters a repolarizing region. In R-from-T, the PVC and the T wave are causally related, where steep repolarization gradients combined with enhanced ICa,L lead to PVCs emerging from the T wave. Since enhanced ICa,L was required for R-from-T to occur, suppressing window ICa,L effectively prevented arrhythmias in all 3 genotypes.
Conclusions:
Despite the complex molecular causes, these results suggest that R-from-T is likely a common mechanism for PVT initiation in LQTS. Targeting ICa,L properties, such as suppressing window ICa,L or preventing excessive ICa,L increase, could be an effective unified therapy for arrhythmia prevention in LQTS.
WHAT IS KNOWN?
Spontaneous initiation of arrhythmias in long QT syndromes exhibits genotype-dependent ECG features including T-wave alternans, short-long-short sequence (or a pause).
R-on-T is the most common ECG phenomenon during ventricular arrhythmia initiation in long QT syndromes (LQTS).
WHAT THE STUDY ADDS?
Computer simulations well capture the diverse ECG features and genotype dependence of arrhythmia initiation in LQTS.
Despite the many different underlying molecular causes of LQTS, a common mechanism that we call R-from-T is likely responsible for arrhythmia initiation in LQTS.
Based on these new mechanistic insights, targeting L-type calcium current properties such as suppressing window calcium current or preventing excessive calcium current increase could be an effective unified therapy for arrhythmia prevention in patients with LQTS.
QT prolongation is a major risk factor of ventricular arrhythmias and sudden cardiac death in congenital and acquired long QT syndromes (LQTS),1–6 heart failure,7 and ischemia.8 During the last 2 decades, genetic sequencing and molecular studies have revealed a diverse taxonomy of congenital LQTS subtypes, classified by both the specific genetic mutations and the ion channels they affect. Since the discovery of LQTS type 1 (LQT1), 16 distinct subtypes of LQTS have been classified,9 with the major subtypes being LQT1, LQTS type 2 (LQT2), and LQTS type 3 (LQT3). In addition, many drugs have been identified to prolong QT interval resulting in acquired LQTS.10
While the molecular causes of LQTS are complex, clinical studies have shown that polymorphic ventricular tachyarrhythmias (PVTs) or Torsade de Pointes in LQTS patients are usually preceded by several characteristic ECG features (Figure 1):
Figure 1.
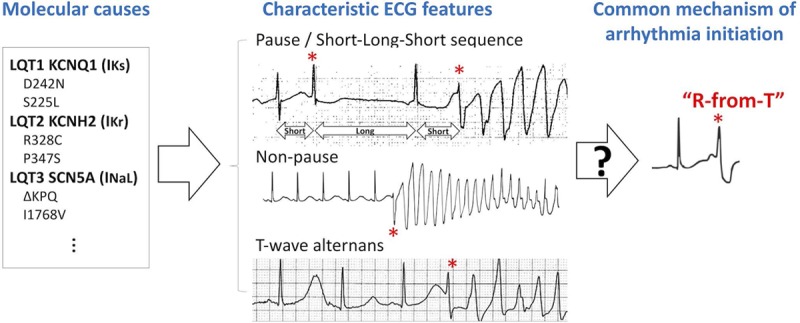
Schematic diagram linking the different molecular causes of long QT syndrome (LQTS) to characteristic ECG features, and a hypothetical common mechanism of spontaneous arrhythmia initiation. Left, Genetic and nongenetic causes of LQTS at the molecular scale. Middle, Characteristic ECG features of spontaneous arrhythmogenesis in LQTS patients. Top ECG: Pause-dependent initiation of polymorphic ventricular tachyarrhythmia (PVT). An ECG from a patient with acquired LQTS showing short-long-short (SLS) sequence preceding PVT, in which a first premature ventricular complex (PVC; *) occurring on the downslope of the T wave causes a compensatory pause, resulting in a second PVC (*) leading to PVT. Modified from Figure 3 in Chiang and Roden2 with permission. Copyright © 2000, American College of Cardiology. Middle ECG: Nonpause-dependent initiation of PVT. A representative ECG from an LQTS type 1 (LQT1) patient with ischemia showing PVT initiation without a preceding pause. Modified from Figure 8B in the study by Morita et al3 with permission. Copyright © 2008 Elsevier. Bottom ECG: T-wave alternans (TWA)-dependent initiation of PVT. An ECG recording from a patient with acquired LQTS. The sinus beats show prolonged QT intervals (>600 ms) and TWA. A PVC (*) then occurred on the downslope of the larger T wave, which then immediately initiated ventricular tachycardia. Modified from Figure 1 in Badri et al6 with permission. Copyright © 2015, American College of Cardiology Foundation. Right, A hypothetical common mechanism of arrhythmia initiation in LQTS investigated in the current study, termed R-from-T.
Pause-dependent and nonpause-dependent initiation of PVT: the onset of PVT in LQTS patients is mainly (≈70%) pause dependent,11,12 that is, PVT occurs after a prolonged RR interval. A ubiquitous ECG pattern preceding PVT is the so-called short-long-short (SLS) sequence (center top in Figure 1).2,4,10 A smaller portion (≈30%) is nonpause dependent,11,12 that is, PVT occurs spontaneously without a preceding pause or SLS sequence (center middle in Figure 1). Tan et al13 showed that the onset of PVT is mainly pause dependent in LQT2 and LQT3 but nonpause dependent in LQT1.
T-wave alternans (TWA): macro-volt TWA occurs frequently in LQTS.4,14–16 TWA can either directly precede PVT without a pause (center bottom in Figure 1)6 or occur much earlier with the onset of PVT preceded by a pause or SLS sequence.4,16
R-on-T: in all modes of PVT initiation above, the premature ventricular complexes (PVCs) or first beat of PVT (marked by * in Figure 1) occur on the downslope of the T wave—a well-known ECG phenomenon called R-on-T.
Reducing the many complex molecular causes to these several characteristic ECG features considerably simplifies our understanding of arrhythmogenesis in LQTS. However, how these molecular causes result in these characteristic ECG features and how these features are then mechanistically linked to the initiation of PVT remain incompletely understood.
In this study, we seek to bridge the gaps between molecular cause, ECG features, and arrhythmia initiation using in silico models of human LQTS. The ECG patterns from our anatomic ventricle simulations well captured the clinical ECG patterns and their genotype specificities. From our anatomic ventricle and simplified tissue simulations of LQT1, LQT2, and LQT3, we obtained the following mechanistic insight: regional action potential duration (APD) prolongation (and thus QT prolongation) increases repolarization gradients, which when combined with enhanced L-type calcium (Ca2+) current (ICa,L) causes the spontaneous genesis of PVCs. Because these PVCs emerge from the repolarization gradient that gives rise to the T wave, we call this mechanism R-from-T, in contrast with the classic explanation of R-on-T mechanism in which an exogenous PVC coincidentally encounters a vulnerable repolarizing region during the T wave, which we call R-to-T. In other words, in R-from-T, the PVC and the T wave are not coincidental but causally related. We further distinguish these 2 mechanisms in detail in Discussion. Because enhanced ICa,L is required for the R-from-T mechanism, targeting the properties of ICa,L could prevent PVT regardless of the specific subtype of LQTS.
Methods
The authors declare that all supporting data are available within the article and its Data Supplement.
Computer Models
The human anatomic ventricle model was adapted from the one previously developed by Ten Tusscher et al.17,18 We added a Purkinje network to the ventricle model (Figure I in the Data Supplement) generated using a method developed by Sahli Costabal et al.19 One-dimensional (1D) cable and 2-dimensional (2D) tissue models were used for mechanistic investigations. All computer simulations were performed on Tesla and GeForce GPUs (NVIDIA Corporation) with software written in the CUDA programing language.
The human ventricular action potential model by O’Hara et al20 was used for the ventricular myocytes. The ICa,L steady-state activation and inactivation curves were taken from either the study by O’Hara et al20 or Li et al.21 The human Purkinje action potential model by Stewart et al22 was used for the Purkinje network cells. Instead of using detailed ion channel models simulating specific mutations,23 we modeled LQT1 by removing the slow component of the delayed rectifier potassium current (IKs), LQT2 by removing the fast component of the delayed rectifier potassium (IKr), and LQT3 by increasing late sodium current (INaL) in the ventricles. Parameters are detailed in the Data Supplement for each type of LQTS. A bulk heterogeneity in the right ventricle was created by adjusting a nonmutated current for each LQTS subtype (Figure II in the Data Supplement), resulting in an APD map agreeing with the ECG-imaging studies of LQTS patients from Vijayakumar et al.24
Pseudo-ECGs (Figure III in the Data Supplement) were computed25,26 with the V5 lead shown unless otherwise specified.
Further details of the mathematical models, the fiber structure in the ventricles, the Purkinje network, Purkinje cell-myocyte coupling, and ECG computation are presented in the Data Supplement.
Arrhythmia Initiation Protocols
We used 3 protocols to elicit spontaneous initiation of arrhythmias:
ICa,L ramp protocol: constant heart rate with a 20-second ICa,L conductance (PCa) ramp (first panel, Figure 2A). The ramp begins at the control value (PCa,control=0.0001 cm/s=1 μm/s) to a specified high value (PCa,H). The heart rate is fixed. This protocol simulates the initial phase of a β-adrenergic surge, when ICa,L is quickly activated but IKs is not yet.27
Pause protocol (LQT2 and LQT3): constant PCa with a pause in the heart rate by a sudden change from 120 to 60 beats per minute.
Increasing heart rate protocol (LQT1): constant PCa with the RR interval gradually decreasing from 1000 (60 beats per minute) to 500 ms (120 beats per minute) in increments of 10 ms per beat.
Figure 2.
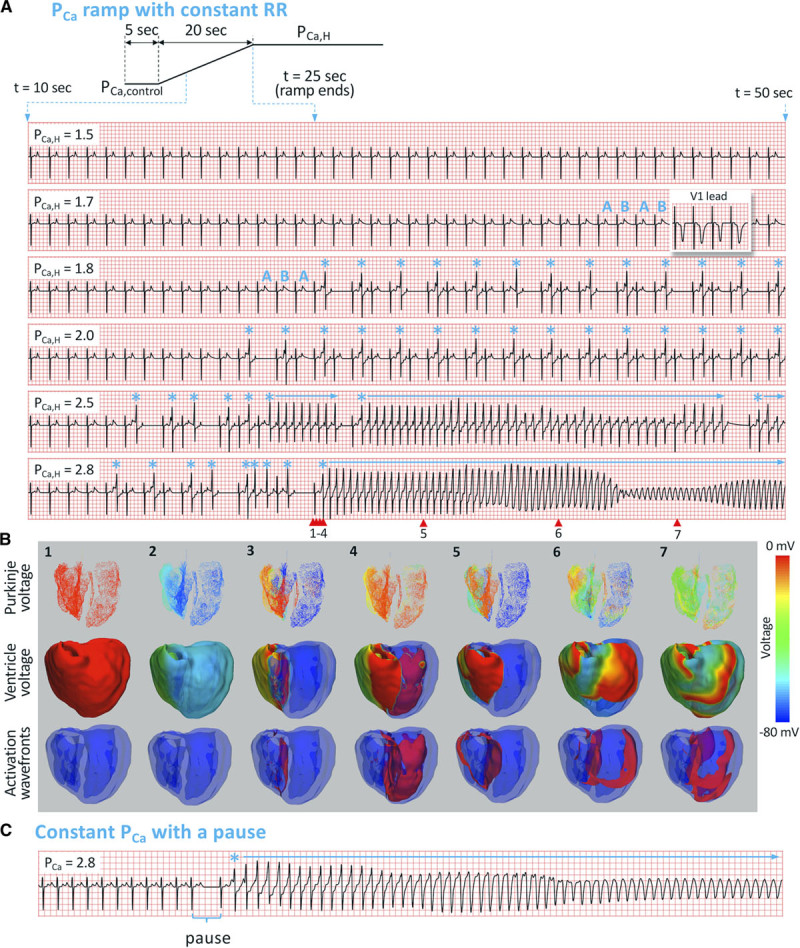
Spontaneous initiation of arrhythmias in LQT2 (long QT syndrome type 2). A, Top, Schematic of the PCa ramp protocol. The ramp starts at t=5 s and ends at t=25 s. ECG traces: ECG traces from t=10 to 50 s for 6 PCa,H values (in units of µm/s) as indicated on each ECG. Heart rate was 60 beats per minute. ABAB marks T-wave alternans (TWA). * marks the premature ventricular complexes, and horizontal arrows indicate episodes of polymorphic ventricular tachyarrhythmia (PVT). In the second ECG, we also show the V1 lead (inset) for TWA. B, Numbered snapshots of voltage maps (first and second rows) and wave fronts (colored red, third row) from the time points marked by corresponding red arrows on the last ECG in A (PCa,H=2.8 µm/s). See Movie I in the Data Supplement for full episode. C, PVT induced by the pause protocol, changing the heart rate from 120 to 60 beats per minute with a constant PCa=2.8 µm/s.
Results
Initiation of Arrhythmias in LQT2
Simulation of the Anatomic Ventricle Model
We first used the PCa ramp protocol (Figure 2A and 2B) to elicit spontaneous initiation of PVT. Figure 2A shows ECGs in the time interval from 10 to 50 s for different PCa,H values, and Figure 2B shows voltage snapshots for PCa,H=2.8 μm/s. Figure 2C shows the ECGs for the pause protocol in which PCa was held at 2.8 μm/s for the whole simulation period. The ECG dynamics from these simulations are summarized as follows:
TWA and PVCs: up to PCa,H=1.5 μm/s, the ECG remained in normal sinus rhythm (first ECG). When PCa,H was increased to 1.7 μm/s, TWA appeared (marked as ABAB on the second ECG). When PCa,H was increased to 1.8 μm/s, PVCs occurred (marked by * on the third ECG). These PVCs were always superimposed on the T wave, manifesting as R-on-T. TWA still preceded the appearance of PVCs (marked as ABA on the third ECG), with the first PVC occurring on the larger of the alternating T waves.
SLS sequence: at PCa,H=1.8 μm/s, the first PVC blocks next sinus beat, resulting in a compensatory pause. After this pause and the next sinus beat, a second PVC occurs resulting in a classic SLS sequence. This ECG pattern repeated as PCa was held at 1.8 μm/s. When PCa,H was increased to 2.0 μm/s (fourth ECG), after the first compensatory pause, an alternating interpolated PVC pattern formed where a PVC occurred after every other sinus beat without compensatory pauses—a behavior we call PVC alternans.
PVT preceding SLS sequence: when PCa,H was raised to above 2.5 μm/s, multiple episodes of nonsustained PVT (fifth ECG) and sustained PVT (sixth ECG) occurred. These episodes of PVT were always preceded by an SLS sequence. To reveal how PVCs and PVT occur spontaneously in the heart, we show selected snapshots in a 3-view sequence (Purkinje network voltage, ventricular myocardium voltage, and ventricular excitation wave fronts) for the initiation episode in the PCa,H=2.8 μm/s case (Figure 2B). Focal excitations repeatedly originate from the right ventricle heterogeneity and eventually evolve into a focal-reentrant mixture (see Movie I in the Data Supplement for the entire episode).
PVT preceding a pause: we were also able to initiate arrhythmias using the pause protocol. Figure 2C shows an ECG for PCa held at 2.8 μm/s, in which the heart rate was suddenly decreased from 120 to 60 beats per minute. When the heart rate was 120 beats per minute, no TWA or PVCs occurred. After the heart rate was changed to 60 beats per minute, PVT was initiated immediately.
Mechanistic Insights of Arrhythmogenesis From 1D and 2D Tissue Simulations
To better understand the underlying mechanism of arrhythmia initiation in the anatomic ventricle, we carried out additional simulations using 1D cable and 2D tissue models.
In the 1D cable, a region of longer APD was placed in the center by using a smaller IKs conductance (GKs), paced from the upper end. When PCa=1 μm/s (Figure 3A), the APD was stable in the entire pacing cycle length (PCL) range from 400 to 1600 ms. For PCa=2.0 μm/s (Figure 3B), when PCL was increased to 1000 ms, APD alternans occurred in the center region of the cable but did not generate PVCs (corresponding to TWA in the second ECG in Figure 2A). At PCLs >1150 ms, APD dynamics became more complex and PVCs occurred. For PCa=2.3 μm/s (Figure 3C), when PCL was increased to a value where APD alternans occurred, PVCs also occurred. These APD alternans lead to PVC alternans (corresponding to the PVC pattern seen in the fourth ECG in Figure 2A). At slower PCLs, PVCs occur on every beat. Importantly, these PVCs emerge spontaneously from the repolarization gradient region and propagate only in one direction—a process we call spontaneous unidirectional propagation.
Figure 3.
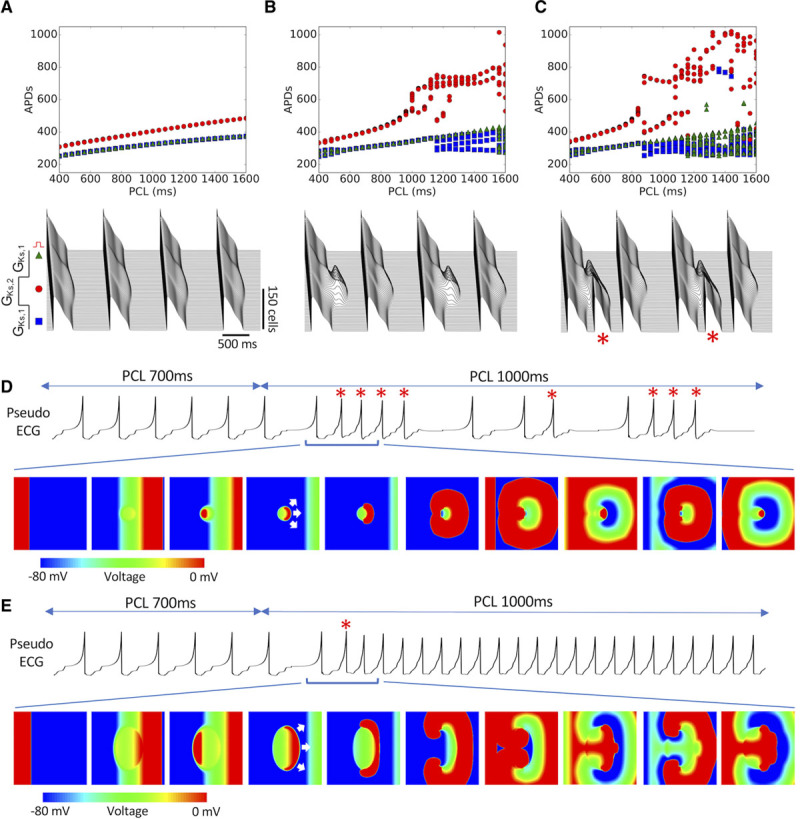
Mechanistic insights of arrhythmogenesis from 1-dimensional (1D) cable and 2-dimensional (2D) tissue simulations. A, Top, Action potential duration (APD) vs pacing cycle length (PCL) recorded from the 3 locations indicated in bottom. Bottom, Space-time plot of voltage in the 1D cable (300 cells) for 4 beats at PCL=1000 ms. PCa=1.0 µm/s. GKs,1=0.05 mS/μF, GKs,2=0.03 mS/μF. B, Same as A but for PCa=2.0 µm/s. APD alternans without premature ventricular complexes (PVCs) occurred from PCL=1000 to 1150 ms. C, Same as A but for PCa=2.3 µm/s. APD alternans occurred from PCL=900 to 1050 ms, in which PVC alternans (marked with * in bottom) occurred simultaneously. D, Pseudo-ECG and voltage snapshots from a 2D tissue (500×500 cells) simulation with a circular region of lower GKs. GKs=0.03 mS/μF in the center circular region (diameter, 100 cells), GKs=0.05 mS/μF elsewhere. The tissue was paced from the left side with 20 beats at PCL=700 ms and then changed to 1000 ms. White arrows in the fourth voltage panel indicate the direction of spontaneous unidirectional propagation of the PVC. See Movie II in the Data Supplement for full episode. E, Same as D, but the long APD region was changed to an oval (long axis, 300 cells; short axis, 200 cells). See Movie III in the Data Supplement for full episode.
In the 2D tissue, a longer APD region is similarly placed in the center of the tissue (Figure 3D and 3E). The tissue was paced from the left edge, and the PCL was maintained at 700 ms for 20 beats and then suddenly increased to 1000 ms. In the first case, the long APD region is circular (Figure 3D; Movie II in the Data Supplement). At PCL=700 ms, no PVCs occurred. After PCL was increased to 1000 ms, focal excitations began to emerge from the APD gradient region. The excitations propagated unidirectionally (indicated by the white arrows), first to the right, then around the longer APD region before finally colliding and annihilating at the left side, forming a target-like pattern. This repeated to generate a focal train of multiple PVCs. As the PCL was maintained at 1000 ms, multiple such episodes occurred. However, when we changed the shape of the longer APD region into a larger and more elongated geometry (Figure 3E; Movie III in the Data Supplement), the initial focal excitation directly evolved into sustained reentry instead, forming a figure-of-eight reentry since the region was large enough that the tips did not collide and annihilate. This reentry arose directly from spontaneous unidirectional propagation of the PVC itself and was not the result of another exogenous PVC encountering conduction block.
Initiation of Arrhythmias in LQT3
We used the same 2 protocols as in LQT2 to elicit arrhythmias. Figure 4A shows the ECGs for the PCa ramp protocol. Similar to LQT2, TWA preceded R-on-T PVCs (marked by *), and the first PVC always occurred on the larger of the alternating T wave. Most of the time, episodes of PVT followed SLS sequences (ie, a pause), but in some cases, PVT also occurred immediately following TWA without a pause (third ECG). This case of TWA immediately preceding PVT is similar to the clinical example shown in Figure 1. Sustained ventricular tachycardia occurred at PCa,H=3.2 μm/s (sixth ECG). In Figure 4B, we show selected snapshots in a 3-view mode for the initiation episode in the PCa,H=3.2 μm/s case (see Movie IV in the Data Supplement for the entire episode). The PVCs again emerged spontaneously and propagated out unidirectionally from the repolarization gradient region. In this case, the focal excitation traveled around the longer APD region and was able to quickly evolve into a stable reentry, manifesting as a monomorphic ventricular tachycardia. Using other parameters, the resulting arrhythmia could be either monomorphic or polymorphic.
Figure 4.
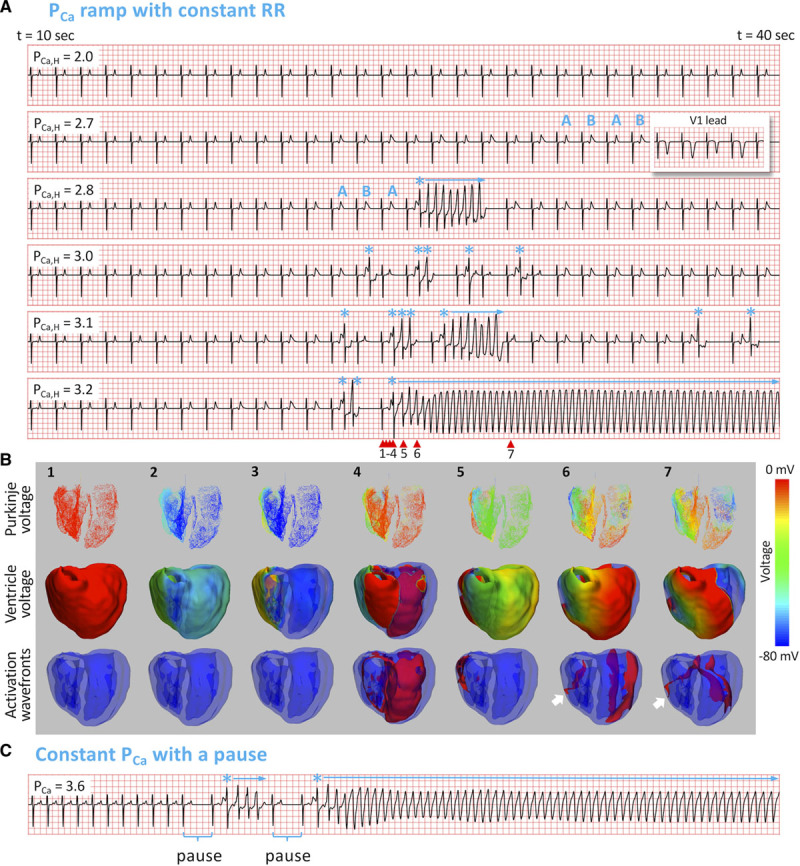
Spontaneous initiation of arrhythmias in long QT syndrome (LQTS) type 3. A, ECG traces from t=10 to 50 s for 6 PCa,H values as indicated on each ECG, using the same PCa ramp protocol in LQTS type 2 (Figure 2A, top). Heart rate was 60 beats per minute. ABAB marks T-wave alternans (TWA). * marks the premature ventricular complexes, and horizontal arrows indicate episodes of polymorphic ventricular tachyarrhythmia (PVT). In the second ECG, we also show the V1 lead (inset) for TWA. B, Numbered snapshots of voltage maps (first and second rows) and wave fronts (colored red, third row) from the time points marked by corresponding red arrows on the last ECG in A (PCa,H=3.2 µm/s). See Movie IV in the Data Supplement for full episode. C, PVT induced by the pause protocol, changing the heart rate from 120 to 60 beats per minute with a constant PCa=3.6 µm/s.
We also elicited arrhythmias using the pause protocol (Figure 4C). With a heart rate of 120 beats per minute, no PVCs or arrhythmias occurred even at PCa,H=3.6 μm/s, but arrhythmias occurred immediately after a sudden decrease in heart rate to 60 beats per minute.
Initiation of Arrhythmias in LQT1
Spontaneous initiation of PVT in LQT1 manifests differently than in LQT2 and LQT3. We first used the PCa ramp protocol with heart rates of 60 and 120 beats per minute. Figure 5A shows ECGs at 3 PCa,H values for both heart rates. At PCa,H=2.1 μm/s, both ECGs show normal sinus rhythm. When PCa,H was increased to 3.1 μm/s, arrhythmias occurred at 120 beats per minute but not at 60 beats per minute. When PCa,H was increased further to 4.1 μm/s, arrhythmias occurred at both heart rates. Note that in either heart rate, arrhythmias always occurred without a preceding pause or SLS sequence. In addition, no TWA was observed at any PCa,H. Despite these differences from LQT2 and LQT3, the arrhythmias were still initiated with an R-on-T (marked by *). The 3-view snapshots also show focal excitations emerging from the repolarization gradient region, resulting in PVT (Figure 5B; Movie V in the Data Supplement). PVT was also able to be elicited using the increasing heart rate protocol for certain PCa,H values between 3.1 and 4.1 μm/s, with an example shown at PCa,H=4.0 μm/s (Figure 5C).
Figure 5.
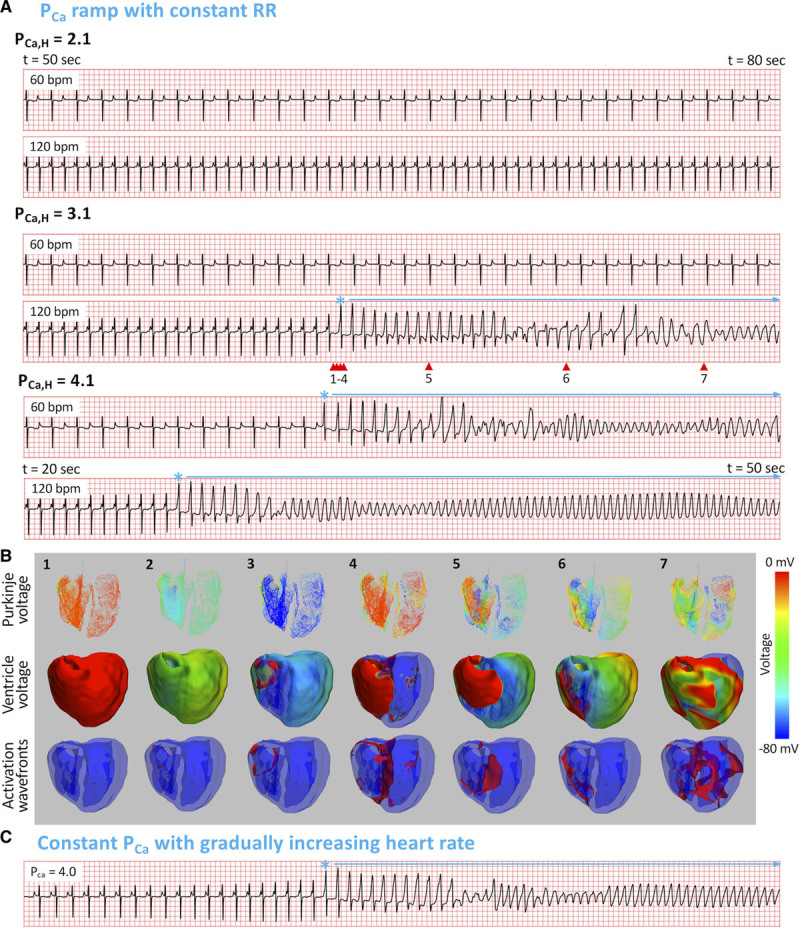
Spontaneous initiation of arrhythmias in long QT syndrome (LQTS) type 1. A, ECG traces at 2 heart rates (60 and 120 beats per minute) for 3 PCa,H values, using the same PCa ramp protocol in LQTS type 2 (Figure 2A, top). * marks the premature ventricular complexes, and horizontal arrows indicate the polymorphic ventricular tachyarrhythmia (PVT) episodes. B, Numbered snapshots of voltage maps (first and second rows) and wave fronts (colored red, third row) from the time points marked by corresponding red arrows on the fourth ECG in A (120 beats per minute, PCa,H=3.1 µm/s). See Movie V in the Data Supplement for full episode. C, PVT induced by the increasing heart rate protocol. Heart rate was gradually increased from 60 to 120 beats per minute for a constant PCa=4.0 µm/s.
Although the presence of IKs can explain why arrhythmias tend to occur at slower heart rates in LQT2 and LQT3,28 it is not clear why a unique SLS sequence occurs in LQT2 and LQT3 but not in LQT1. To understand the underlying mechanism, we carried out single-cell and 1D cable simulations comparing LQT1 and LQT2. Figure 6A shows APD versus PCa for LQT1 and LQT2. In the case of LQT1, increasing PCa causes APD to increase suddenly to repolarization failure. However, in the case of LQT2, APD increases as a staircase with incremental transitions. The sudden repolarization failure in LQT1 is due to the fact that peak IKr does not change with increasing APD since IKr has already saturated (Figure 6B, top), and, therefore, there is not enough outward current to oppose the increase of ICa,L. On the contrary, when IKs is present as in LQT2, it does increase with increasing APD because of its slow activation (Figure 6B, bottom), supplying enough outward current to oppose ICa,L for repolarization. At the tissue scale, repolarization failure in the long APD region causes repetitive firings at both fast and slow heart rates (Figure 6C). Therefore, in LQT1, the sudden transition from a normal action potential to repolarization failure causes the transition from sinus rhythm to repetitive focal firings. Single PVCs with a compensatory pause or SLS sequences do not have a chance to develop because full-blown arrhythmias occur immediately from the first PVC onward. Adding a small amount of IKs back into the LQT1 cell causes a more gradual APD change before reaching repolarization failure, and with enough IKs added back, the response is similar to LQT2 (Figure IV in the Data Supplement). Similarly, in the LQT1 1D cable, after adding a certain level of IKs back, the behavior changed from repolarization failure to distinct PVC alternans. These results imply that when IKs conductance is below a certain threshold, one will observe the typical LQT1 pattern of arrhythmia initiation, otherwise, one will observe the LQT2 behavior.
Figure 6.
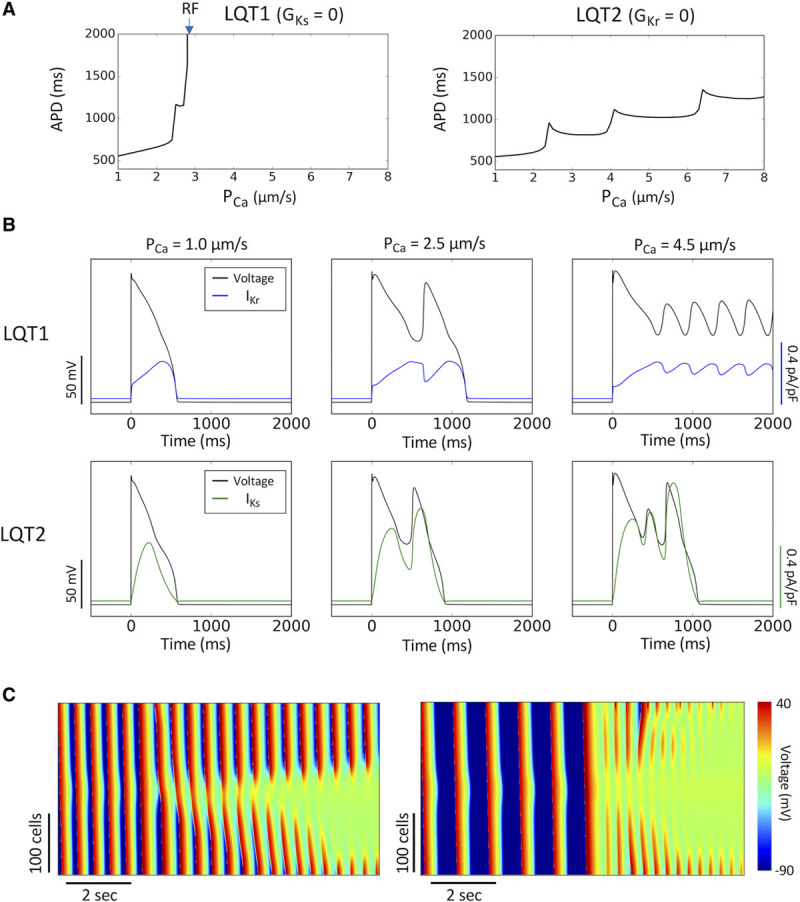
Cellular and tissue mechanisms of arrhythmogenesis in long QT syndrome (LQTS) type 1 (LQT1). A, Single-cell action potential duration (APD) vs PCa for LQT1 (left) and LQTS type 2 (LQT2; right). Arrow marks PCa where repolarization failure (RF) occurs. B, Single-cell voltage, IKr, and IKs vs time for LQT1 (top) and LQT2 (bottom) at 3 PCa values. C, Line scans of voltage in a 1-dimensional cable for PCa=3.1 µm/s at pacing cycle length (PCL)=500 ms (left, corresponding to the fourth ECG in Figure 5A) and PCa=4.1 µm/s at PCL=1000 ms (right, corresponding to the fifth ECG in Figure 5A) in LQT1, showing repolarization failure in the center of the cable generating new premature ventricular complexes.
Reducing Window ICa,L Prevents Arrhythmogenesis in LQTS
Although the modes of spontaneous arrhythmia initiation are genotype dependent, one common requirement is the enhancement of ICa,L. Moreover, the R-on-T focal excitations that initiate the arrhythmias always originate from the repolarization gradient regions, resulting in focal, reentrant, or mixed focal-reentrant arrhythmias. Based on our understanding of the role window ICa,L plays in both early afterdepolarization genesis in single cells29,30 and PVC formation in heterogeneous tissue,31 we hypothesized that reducing the window ICa,L can prevent arrhythmias in LQTS. We reduced the window ICa,L by shifting the steady-state inactivation curve (f∞) as indicated in Figure 7A. The advantage of shifting f∞ (in contrast to ICa,L blockade) is that it does not affect ICa,L in the normal action potential (Figure 7B, left) but can effectively suppress early afterdepolarizations (Figure 7B, right) and PVCs.31 Left shifting f∞ by 5 mV prevented the arrhythmias that were induced by the PCa ramp protocol in LQT1, LQT2, and LQT3 (Figure 7C). To systematically evaluate the efficacy of this therapeutic strategy, we carried out 1D cable simulations to identify the PCa threshold for PVCs versus the amount of f∞ shift (Figure 7D). In all 3 subtypes of LQTS, a 5-mV shift almost doubled the threshold for arrhythmias. At 8-mV shift, no PVCs were seen even at 6× the control PCa for any LQTS subtype.
Figure 7.
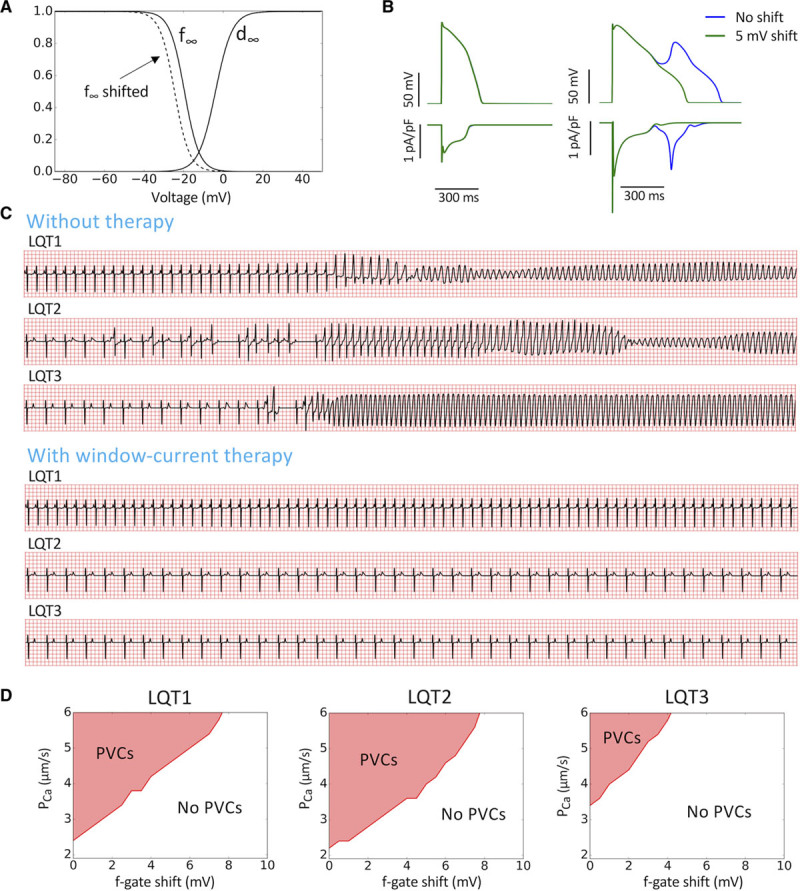
A unified genotype-independent therapy for long QT syndrome (LQTS) type 1 (LQT1), LQTS type 2 (LQT2), and LQTS type 3 (LQT3). A, Steady-state activation (d∞) and inactivation (f∞) curves for ICa,L. Dashed curve is f∞ with a 5-mV shift. B, Action potential and ICa,L before and after 5-mV shift of f∞. Left, Normal action potential in which the shift causes no change (the green and blue curves are superimposed). Right, The early afterdepolarization was suppressed by the 5-mV shift but with no change in peak ICa,L. C, ECG before (reproduced from Figures 2, 4, and 5) and after a 5-mV f∞ shift in LQT1, LQT2, and LQT3. D, PCa threshold for premature ventricular complexes (PVCs) in 1-dimensional cable simulations vs voltage shift of f∞ for LQT1, LQT2, and LQT3.
Discussion
In this study, we used anatomic human ventricle and simplified tissue models to systematically investigate the mechanisms of spontaneous arrhythmia initiation in LQTS. Our simulations reproduced the characteristic ECG features commonly seen in LQTS patients, including TWA, R-on-T, and SLS sequences leading to PVT in LQT2 and LQT3 and nonpause-dependent initiation of PVT in LQT1. 1D and 2D models further explored the detailed mechanisms of TWA, PVC formation, and initiation of arrhythmias. We showed that although the characteristic ECG features preceding the onset of PVT may vary between the different LQTS genotypes, a common mechanism for PVT initiation may exist, which we call R-from-T. In this R-from-T mechanism, PVCs are generated from increased repolarization gradients (as a result of QT prolongation) with an enhanced ICa,L (such as during a β-adrenergic surge), which then propagate unidirectionally away from the repolarization gradient region. This can generate either target-like excitation patterns resulting in focal arrhythmias or evolve into reentrant arrhythmias depending on the geometry of the tissue heterogeneity. This new understanding leads to a unified therapeutic strategy in which a single intervention of suppressing window ICa,L effectively prevented arrhythmias in multiple LQTS subtypes. Detailed mechanistic insights and implications for clinical arrhythmia prevention are discussed in detail below.
R-From-T as a Common Mechanism of Arrhythmia Initiation in LQTS: R-on-T Revisited
R-on-T is a descriptive term denoting the ECG appearance of an R wave superimposed on a T wave. R waves that occur during the downslope of a T wave have been widely associated with increased arrhythmia risk in a variety of conditions.32 Here, we discuss our current understanding of the different possible mechanisms by which arrhythmias can spontaneously initiate with the R-on-T phenomenon (Figure 8; Movie VI in the Data Supplement).
Figure 8.
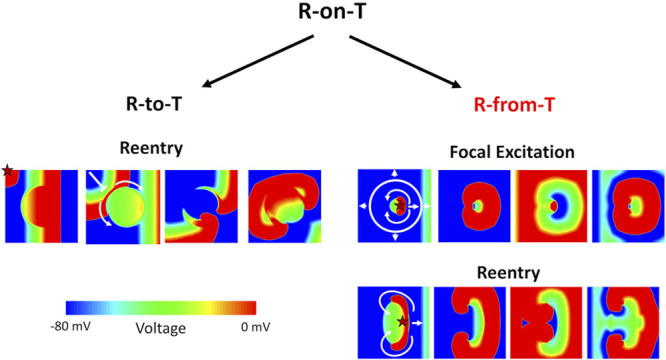
Schematic diagram distinguishing the R-to-T and R-from-T mechanisms of arrhythmia initiation. Spontaneous arrhythmias that initiate with an R-on-T phenomenon on ECG can arise from 2 different underlying mechanisms, which we call R-to-T and R-from-T. A heterogenous long action potential duration (APD) region is placed in the center of the tissue. The star marks the origin of the ectopic premature ventricular complex (PVC) in each scenario. Left, R-to-T is the mechanism in which an ectopic PVC encounters a repolarizing region, undergoing unidirectional conduction block and causing figure-of-eight reentry. The R wave and T wave are 2 independent events, and the arrhythmias are only reentry. Right, R-from-T is the mechanism investigated in this study, where a PVC itself emerges from a repolarization gradient resulting in spontaneous unidirectional propagation. The R wave is caused by the T wave, and the arrhythmias can be either focal excitations (top) or reentry (bottom) depending on the properties of the heterogeneity. An animated version of this figure is shown in Movie VI in the Data Supplement.
The classical mechanistic explanation for arrhythmias initiating as an R-on-T is as follows: when an ectopic PVC from somewhere either in the Purkinje network or ventricular myocardium encounters a refractory region with right timing, unidirectional conduction block can occur, which may then develop into figure-of-eight reentry (Figure 8, left). Because in this scenario the PVC travels and encounters a repolarizing T wave by chance, we call this classic mechanism the R-to-T mechanism.
In the current and our previous studies,18,31,33,34 we have shown that PVCs can also spontaneously arise from a repolarization gradient via a dynamical instability when ICa,L is significantly enhanced. The PVCs from this mechanism will always appear and coincide during the T wave, also manifesting as R-on-T on ECG. Because these PVCs arise from the repolarization gradient and thus are causally linked with the T wave, we call this new mechanism the R-from-T mechanism (Figure 8, right). Note that in R-to-T, the R wave and T wave are 2 independent events, while in R-from-T, the R wave is caused by the T wave.
In R-from-T, after the PVCs emerge spontaneously from the repolarization gradient region, they propagate unidirectionally away from the gradient—a behavior we call spontaneous unidirectional propagation. Evidence of this type of PVC behavior has been shown in optical mapping experiments of rabbit and canine hearts,31,35–37 where spontaneous PVCs would emerge from the steep repolarization gradient region and propagate unidirectionally. This behavior results in distinctly different mechanisms of arrhythmogenesis compared with the classical R-to-T mechanism. First, repeated PVCs can occur by R-from-T,31 manifesting as singlets, couplets, or a long train of PVCs, resulting in sustained or nonsustained focal arrhythmias. Second, since the PVC only propagates unidirectionally, it can also evolve directly into reentrant arrhythmias without requiring an additional region of heterogeneity to cause conduction block. Thus, depending on the properties of the heterogeneity, these PVCs can result in focal, reentrant, or mixed focal/reentrant arrhythmias all from the same R-from-T mechanism.
Since the R-from-T PVCs and the subsequent arrhythmias are caused by the same mechanism, a single PVC in this case can be considered as just the shortest possible run of an arrhythmia. Couplets, triplets, trains of PVCs, and the subsequent PVT are just longer runs of arrhythmias from the same underlying instability, which depends on the cellular and tissue properties, as well as the interactions of PVCs with the sinus beats. In this sense, the R-from-T mechanism blurs the line between the usual notions of a distinct trigger and substrate in arrhythmia initiation.
While the R-to-T mechanism is widely considered as the prototypical mechanism of arrhythmogenesis in cardiac diseases in general,38,39 it is unknown whether such a mechanism is indeed responsible for the spontaneous initiation of arrhythmias in LQTS. Although the R-to-T mechanism has been reproduced in computer simulations of LQTS using an externally applied premature stimulus (so-called S1S2 protocol),40,41 to our knowledge, there is no clear experimental evidence of spontaneous arrhythmias caused by R-to-T in LQTS. On the contrary, several optical mapping experiments of LQTS do show spontaneous unidirectional propagation from the steep repolarization gradient region,31,35–37 consistent with the R-from-T mechanism. Other recent simulation studies18,31,33,42,43 have also shown spontaneous initiation of arrhythmias without an external stimulus, while our current study is able to reproduce the characteristic ECG features seen in patients with LQTS and reveals how R-from-T serves as a common mechanism of arrhythmia initiation in different LQTS subtypes.
Mechanistic Links Between the Characteristic ECG Features and Spontaneous PVT Initiation in LQTS
TWA and SLS sequences are the major characteristic ECG features known to precede PVT in LQTS, but their detailed relationship is not well understood. Our results from this study offer a deeper explanation through the R-from-T mechanism.
TWA has been widely observed in LQTS4,14–16 and often precedes the initiation of PVT, either immediately (eg, center bottom in Figure 1)6 or many hours before a pause-induced arrhythmia.4,16 In our ICa,L ramp protocol simulations of LQT2 (Figure 2) and LQT3 (Figure 4), TWA was observed just before the appearance of PVCs and PVT. The first PVC during the ICa,L ramp always occurred on the larger of the alternating T waves. In most of the cases, PVT initiation was still preceded by an SLS sequence but could also occur immediately following TWA without an SLS sequence (third ECG in Figure 4A). These simulated ECG behaviors agree well with the clinical ECG features seen in patients with LQTS.4,6,16 The role of TWA in the genesis of PVCs and arrhythmias has been investigated in our recent study.33 We demonstrated that TWA can further exacerbate the repolarization gradient causing PVCs and PVT. However, TWA itself is an intermediate behavior, also caused by enhanced ICa,L (Figures 2 through 4) and slow heart rates (Figure 3) or simply by QT prolongation.33 PVCs and initiation of PVT also result from enhanced ICa,L and slow heart rates but do not necessarily require TWA to occur. The ICa,L threshold or the heart rate threshold for TWA is lower than that for PVCs and arrhythmias, which may explain why TWA occurs frequently in LQTS without arrhythmias or can occur long before arrhythmia onset. These insights agree well with the clinical observation that LQTS patients with TWA have a higher incidence of arrhythmias, but QT prolongation is still the primary risk factor.15
The SLS sequence or a pause immediately preceding PVT initiation is a unique ECG feature in LQTS, accounting for the majority of PVT initiation patterns.4,10–13,44 In our anatomic ventricle simulations, almost all PVT episodes were preceded by an SLS sequence in LQT2 and LQT3, but not in LQT1, agreeing with clinical observations.13 In our ICa,L ramp protocol simulations, the SLS sequence appeared naturally as ICa,L was ramped up. When the first PVC occurs (short), it propagates retrogradely through the Purkinje network, blocking the next sinus beat and resulting in a longer RR interval (long). This increased RR interval further exacerbates the repolarization gradient, leading to another PVC on the next beat (short). Since this pause is longer than the normal sinus RR interval, the repolarization gradient becomes more severe, which can lead to repetitive PVCs or reentrant PVT. The role of the first PVC, whether it arises from R-from-T or any additional mechanisms including delayed afterdepolarizations, Purkinje automaticity, or even heart rate variability (eg, in Figures 2C and 4C), is to simply set up the long compensatory pause.
Note that our anatomic ventricle simulations capture the wide range of ECG phenomenon in LQTS, including TWA, PVCs, SLS, R-on-T, and PVTs, with only a single intervention, that is, ramping up ICa,L. This suggests that the R-from-T mechanism is a likely candidate for spontaneous arrhythmogenesis in LQTS.
Genotype-Dependent Onset of PVT: Role of IKs
Different LQTS subtypes are known to exhibit different genotype-dependent triggers45 and characteristic ECG features preceding PVT.13 For example, LQT2 and LQT3 arrhythmias tend to be more bradycardia dependent, whereas in LQT1, this rate dependence is much less sensitive and can even be tachycardia dependent—a feature well captured by our simulations. This can be explained by the well-known effect of the slow transition to the deeper closed states of IKs28: a longer diastolic interval will cause fewer channels to be available for immediate opening in the next action potential and thus results in a lower repolarization reserve. However, it is not clearly understood why SLS sequences occur frequently in LQT2 and LQT3 but not in LQT1. This study offers further mechanistic insight.
The effects of slow activation of IKs on APD have been investigated in general in our previous study46 and in a recent study by Varshneya et al.47 As shown in our single-cell and 1D cable results (Figure 6), in the absence of IKs, the action potential tends to exhibit an all-or-none response to increasing ICa,L, that is, the action potential either repolarizes normally or fails to repolarize. But in the presence of IKs, the APD exhibits a more graded response to increasing ICa,L because of the slow activation of IKs. Therefore, in LQT1, repolarization failure will occur abruptly in the longer APD region once ICa,L reaches the threshold, directly generating repetitive focal PVCs and causing a sudden transition from sinus rhythm to PVT. In LQT2 and LQT3, however, the APD increases gradually in response to the ICa,L ramp. When ICa,L reaches the threshold, only a single PVC is generated, which can result in a compensatory pause and potentiates R-from-T arrhythmogenesis in a pause-dependent manner with an SLS sequence.
Although these genotype-dependent features leading up to PVT appear different, the mechanism of arrhythmia initiation is still the same, that is, R-from-T. Therefore, this study provides important insights that mechanistically link the molecular causes of LQTS to the characteristic ECG features and then these ECG features to a common R-from-T mechanism for spontaneous PVT initiation.
Implications for Developing a Unified Therapy for PVT Prevention in LQTS
A major insight from our study is that the R-from-T mechanism may be the final common pathway leading to PVT initiation, regardless of the LQTS genotype. One critical factor for R-from-T was an enhanced ICa,L. It is well known that blocking Ca2+ channels has adverse effects of weakening contraction,48 and thus, Ca2+ channel blockers are typically not appropriate for clinical use. In this study, we showed that R-from-T arrhythmogenesis does not occur until ICa,L is significantly increased above baseline (a 2- to 4-fold increase). Therefore, instead of blocking ICa,L overall, preventing excessive increase of ICa,L over baseline can still be effective in preventing R-from-T arrhythmias. This observation agrees with current clinical practice where β-blockers are used as the mainstay arrhythmia prevention therapy in LQTS,45 since β-blockers can prevent the excessive ICa,L increase during a sympathetic event, albeit still with notable side effects on heart rate, exercise tolerance, and blood pressure.
In this study, we demonstrated another potential therapy, treating different LQTS subtypes using only a single intervention by reducing the ICa,L window current with a simple shift in the steady-state inactivation curve (f∞). In other words, we were able to prevent arrhythmias in LQT1, LQT2, and LQT3, only by targeting the ICa,L window current. The advantages of this therapeutic strategy include the following: (1) it avoids affecting the normal calcium transient required for contractility as calcium channel blockers would; (2) it does not affect the APD (and thus QT interval) during sinus rhythm, differing from many of the ion channel blockers; and (3) it avoids affecting the heart rate as with β-blockers. Our results demonstrate that this approach was effective, as only a small 5-mV shift in f∞ doubled the arrhythmia threshold for all LQTS subtypes.
It has been shown that spontaneous Ca2+ release can also be a potential mechanism for arrhythmias in LQTS,49–51 which does not rely on the ICa,L window current. However, as demonstrated in the study by Wilson et al,49 blocking ICa,L can effectively abolish the spontaneous Ca2+ oscillations because of a reduction of the sarcoplasmic reticulum Ca2+ load. Therefore, we predict that blocking window ICa,L may still indirectly have an effect on arrhythmia suppression, since blocking window ICa,L in LQTS can also reduce the overall sarcoplasmic reticulum Ca2+ load. This effect needs to be explored in future modeling studies.
Limitations
For the sake of mechanistic clarity and data availability, some aspects of our models were simplified in as reasonable a manner as possible. The anatomic ventricle model tissue heterogeneity was simulated as a simplified single bulk region to elicit a clearly interpretable initiation of arrhythmias, which may result in T-wave morphologies that differ from real patients.52 More complicated heterogeneities may result in more mixed cases with different or multiple initiation sites. In addition, interpatient ionic current variability can play important roles in the mode of arrhythmia initiation, which may require population modeling approaches.53–55 We also simulated the β-adrenergic surge by only ramping up ICa,L without any other effects, focusing on the initial phase of sympathetic simulation. In this study, the most extreme congenital LQT1 and LQT2 mutations of zero IKs and IKr were simulated, but there are many other possible mutations with partial reduction or trafficking defects instead of complete deficiency, which may result in further mixed or intermediate behaviors. Finally, this study focused on congenital LQT1, LQT2, and LQT3, but the insights into PVT initiation and prevention by targeting ICa,L could also be applicable to other congenital LQTS and acquired LQTS since the R-from-T mechanism seems invariant to the particular cause of QT prolongation, as long as a repolarization gradient and sufficiently elevated ICa,L exists. This needs to be verified in other types of congenital LQTS and acquired LQTS in future modeling studies and eventually verified in future experimental and clinical studies.
Conclusions
Despite the complex genetic and nongenetic causes of LQTS and genotype-dependent clinical features, R-from-T offers a promising common mechanism for PVT initiation in LQTS. Because R-from-T arrhythmogenesis is promoted by a steep repolarization gradient and requires an enhanced ICa,L to occur, targeting ICa,L properties, such as suppressing window ICa,L or preventing excessive increase of ICa,L, can be an effective unified therapy for arrhythmia prevention in LQTS.
Acknowledgments
We thank the arrhythmia group at the University of California, Los Angeles for their useful comments and discussions.
Sources of Funding
This work was supported by National Institutes of Health grants R01 HL134709, R01 HL139829, T32 GM008042, and F30 HL132449.
Disclosures
None.
Supplementary Material
Footnotes
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCEP.119.007571.
References
- 1.Napolitano C, Bloise R, Monteforte N, Priori SG. Sudden cardiac death and genetic ion channelopathies: long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation. 2012;125:2027–2034. doi: 10.1161/CIRCULATIONAHA.111.055947. doi: 10.1161/CIRCULATIONAHA.111.055947. [DOI] [PubMed] [Google Scholar]
- 2.Chiang CE, Roden DM. The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol. 2000;36:1–12. doi: 10.1016/s0735-1097(00)00716-6. doi: 10.1016/s0735-1097(00)00716-6. [DOI] [PubMed] [Google Scholar]
- 3.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 4.Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, Philippides GJ, Roden DM, Zareba W American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology, the Council on Cardiovascular Nursing, and the American College of Cardiology Foundation. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation. 2010;121:1047–1060. doi: 10.1161/CIRCULATIONAHA.109.192704. doi: 10.1161/CIRCULATIONAHA.109.192704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knollmann BC, Roden DM. A genetic framework for improving arrhythmia therapy. Nature. 2008;451:929–936. doi: 10.1038/nature06799. doi: 10.1038/nature06799. [DOI] [PubMed] [Google Scholar]
- 6.Badri M, Patel A, Patel C, Liu G, Goldstein M, Robinson VM, Xue X, Yang L, Kowey PR, Yan GX. Mexiletine prevents recurrent Torsades de Pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin Electrophysiol. 2015;1:315–322. doi: 10.1016/j.jacep.2015.05.008. doi: 10.1016/j.jacep.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 8.Halkin A, Roth A, Lurie I, Fish R, Belhassen B, Viskin S. Pause-dependent torsade de pointes following acute myocardial infarction: a variant of the acquired long QT syndrome. J Am Coll Cardiol. 2001;38:1168–1174. doi: 10.1016/s0735-1097(01)01468-1. doi: 10.1016/s0735-1097(01)01468-1. [DOI] [PubMed] [Google Scholar]
- 9.Giudicessi JR, Wilde AAM, Ackerman MJ. The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc Med. 2018;28:453–464. doi: 10.1016/j.tcm.2018.03.003. doi: 10.1016/j.tcm.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 11.Viskin S, Fish R, Zeltser D, Belhassen B, Heller K, Brosh D, Laniado S, Barron HV. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart. 2000;83:661–666. doi: 10.1136/heart.83.6.661. doi: 10.1136/heart.83.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noda T, Shimizu W, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S. Classification and mechanism of Torsade de Pointes initiation in patients with congenital long QT syndrome. Eur Heart J. 2004;25:2149–2154. doi: 10.1016/j.ehj.2004.08.020. doi: 10.1016/j.ehj.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 13.Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze-Bahr E, Noda T, Wilde AA. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation. 2006;114:2096–2103. doi: 10.1161/CIRCULATIONAHA.106.642694. doi: 10.1161/CIRCULATIONAHA.106.642694. [DOI] [PubMed] [Google Scholar]
- 14.Narayan SM. T-wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol. 2006;47:269–281. doi: 10.1016/j.jacc.2005.08.066. doi: 10.1016/j.jacc.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W, Moss AJ, le Cessie S, Hall WJ. T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:1541–1546. doi: 10.1016/0735-1097(94)90653-x. doi: 10.1016/0735-1097(94)90653-x. [DOI] [PubMed] [Google Scholar]
- 16.Wegener FT, Ehrlich JR, Hohnloser SH. Amiodarone-associated macroscopic T-wave alternans and torsade de pointes unmasking the inherited long QT syndrome. Europace. 2008;10:112–113. doi: 10.1093/europace/eum252. doi: 10.1093/europace/eum252. [DOI] [PubMed] [Google Scholar]
- 17.Ten Tusscher KH, Hren R, Panfilov AV. Organization of ventricular fibrillation in the human heart. Circ Res. 2007;100:e87–101. doi: 10.1161/CIRCRESAHA.107.150730. doi: 10.1161/CIRCRESAHA.107.150730. [DOI] [PubMed] [Google Scholar]
- 18.Vandersickel N, de Boer TP, Vos MA, Panfilov AV. Perpetuation of torsade de pointes in heterogeneous hearts: competing foci or re-entry? J Physiol. 2016;594:6865–6878. doi: 10.1113/JP271728. doi: 10.1113/JP271728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahli Costabal F, Hurtado DE, Kuhl E. Generating Purkinje networks in the human heart. J Biomech. 2016;49:2455–2465. doi: 10.1016/j.jbiomech.2015.12.025. doi: 10.1016/j.jbiomech.2015.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li GR, Yang B, Feng J, Bosch RF, Carrier M, Nattel S. Transmembrane ICa contributes to rate-dependent changes of action potentials in human ventricular myocytes. Am J Physiol. 1999;276:H98–H106. doi: 10.1152/ajpheart.1999.276.1.H98. doi: 10.1152/ajpheart.1999.276.1.H98. [DOI] [PubMed] [Google Scholar]
- 22.Stewart P, Aslanidi OV, Noble D, Noble PJ, Boyett MR, Zhang H. Mathematical models of the electrical action potential of Purkinje fibre cells. Philos Trans A Math Phys Eng Sci. 2009;367:2225–2255. doi: 10.1098/rsta.2008.0283. doi: 10.1098/rsta.2008.0283. [DOI] [PubMed] [Google Scholar]
- 23.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–569. doi: 10.1038/23034. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 24.Vijayakumar R, Silva JNA, Desouza KA, Abraham RL, Strom M, Sacher F, Van Hare GF, Haïssaguerre M, Roden DM, Rudy Y. Electrophysiologic substrate in congenital Long QT syndrome: noninvasive mapping with electrocardiographic imaging (ECGI). Circulation. 2014;130:1936–1943. doi: 10.1161/CIRCULATIONAHA.114.011359. doi: 10.1161/CIRCULATIONAHA.114.011359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie F, Qu Z, Yang J, Baher A, Weiss JN, Garfinkel A. A simulation study of the effects of cardiac anatomy in ventricular fibrillation. J Clin Invest. 2004;113:686–693. doi: 10.1172/JCI17341. doi: 10.1172/JCI17341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sadrieh A, Domanski L, Pitt-Francis J, Mann SA, Hodkinson EC, Ng CA, Perry MD, Taylor JA, Gavaghan D, Subbiah RN, Vandenberg JI, Hill AP. Multiscale cardiac modelling reveals the origins of notched T waves in long QT syndrome type 2. Nat Commun. 2014;5:5069. doi: 10.1038/ncomms6069. doi: 10.1038/ncomms6069. [DOI] [PubMed] [Google Scholar]
- 27.Liu GX, Choi BR, Ziv O, Li W, de Lange E, Qu Z, Koren G. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol. 2012;590:1171–1180. doi: 10.1113/jphysiol.2011.218164. doi: 10.1113/jphysiol.2011.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qu Z, Xie LH, Olcese R, Karagueuzian HS, Chen PS, Garfinkel A, Weiss JN. Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res. 2013;99:6–15. doi: 10.1093/cvr/cvt104. doi: 10.1093/cvr/cvt104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madhvani RV, Angelini M, Xie Y, Pantazis A, Suriany S, Borgstrom NP, Garfinkel A, Qu Z, Weiss JN, Olcese R. Targeting the late component of the cardiac L-type Ca2+ current to suppress early afterdepolarizations. J Gen Physiol. 2015;145:395–404. doi: 10.1085/jgp.201411288. doi: 10.1085/jgp.201411288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang X, Kim TY, Koren G, Choi BR, Qu Z. Spontaneous initiation of premature ventricular complexes and arrhythmias in type 2 long QT syndrome. Am J Physiol Heart Circ Physiol. 2016;311:H1470–H1484. doi: 10.1152/ajpheart.00500.2016. doi: 10.1152/ajpheart.00500.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engel TR, Meister SG, Frankl WS. The “R-on-T” phenomenon: an update and critical review. Ann Intern Med. 1978;88:221–225. doi: 10.7326/0003-4819-88-2-221. doi: 10.7326/0003-4819-88-2-221. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, Kim TY, Huang X, Liu MB, Koren G, Choi BR, Qu Z. Mechanisms linking T-wave alternans to spontaneous initiation of ventricular arrhythmias in rabbit models of long QT syndrome. J Physiol. 2018;596:1341–1355. doi: 10.1113/JP275492. doi: 10.1113/JP275492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teplenin AS, Dierckx H, de Vries AAF, Pijnappels DA, Panfilov AV. Paradoxical onset of arrhythmic waves from depolarized areas in cardiac tissue due to curvature-dependent instability. Phys Rev X. 2018;8:021077. doi: 10.1103/PhysRevX.8.021077. doi: 10.1103/PhysRevX.8.021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Laurita KR. The mechanism of pause-induced torsade de pointes in long QT syndrome. J Cardiovasc Electrophysiol. 2005;16:981–987. doi: 10.1111/j.1540-8167.2005.40677.x. doi: 10.1111/j.1540-8167.2005.40677.x. [DOI] [PubMed] [Google Scholar]
- 36.Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN, Chen PS. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim TY, Kunitomo Y, Pfeiffer Z, Patel D, Hwang J, Harrison K, Patel B, Jeng P, Ziv O, Lu Y, Peng X, Qu Z, Koren G, Choi BR. Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm. 2015;12:220–228. doi: 10.1016/j.hrthm.2014.10.003. doi: 10.1016/j.hrthm.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waldo AL, Wit AL. Mechanisms of cardiac arrhythmias. Lancet. 1993;341:1189–1193. doi: 10.1016/0140-6736(93)91012-b. doi: 10.1016/0140-6736(93)91012-b. [DOI] [PubMed] [Google Scholar]
- 39.Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol. 2015;77:29–55. doi: 10.1146/annurev-physiol-021014-071622. doi: 10.1146/annurev-physiol-021014-071622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahrens-Nicklas RC, Clancy CE, Christini DJ. Re-evaluating the efficacy of beta-adrenergic agonists and antagonists in long QT-3 syndrome through computational modelling. Cardiovasc Res. 2009;82:439–447. doi: 10.1093/cvr/cvp083. doi: 10.1093/cvr/cvp083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai J, Wang K, Li Q, Yuan Y, Zhang H. Pro-arrhythmogenic effects of CACNA1C G1911R mutation in human ventricular tachycardia: insights from cardiac multi-scale models. Sci Rep. 2016;6:31262. doi: 10.1038/srep31262. doi: 10.1038/srep31262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang PC, Perissinotti LL, López-Redondo F, Wang Y, DeMarco KR, Jeng MT, Vorobyov I, Harvey RD, Kurokawa J, Noskov SY, Clancy CE. A multiscale computational modelling approach predicts mechanisms of female sex risk in the setting of arousal-induced arrhythmias. J Physiol. 2017;595:4695–4723. doi: 10.1113/JP273142. doi: 10.1113/JP273142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutta S, Mincholé A, Zacur E, Quinn TA, Taggart P, Rodriguez B. Early afterdepolarizations promote transmural reentry in ischemic human ventricles with reduced repolarization reserve. Prog Biophys Mol Biol. 2016;120:236–248. doi: 10.1016/j.pbiomolbio.2016.01.008. doi: 10.1016/j.pbiomolbio.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neal Kay G, Vance JP, Joaquin GA, Richard WH, Albert LW. Torsade de pointes: the long-short initiating sequence and other clinical features: observations in 32 patients. J Am Coll Cardiol. 1983;2:806–817. doi: 10.1016/s0735-1097(83)80226-5. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–877. doi: 10.1161/CIRCEP.111.962019. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qu Z, Chung D. Mechanisms and determinants of ultralong action potential duration and slow rate-dependence in cardiac myocytes. PLoS One. 2012;7:e43587. doi: 10.1371/journal.pone.0043587. doi: 10.1371/journal.pone.0043587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varshneya M, Devenyi RA, Sobie EA. Slow delayed rectifier current protects ventricular myocytes from arrhythmic dynamics across multiple species. Circ Arrhythm Electrophysiol. 2018;11:e006558. doi: 10.1161/CIRCEP.118.006558. doi: 10.1161/CIRCEP.118.006558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourgonje VJ, Vos MA, Ozdemir S, Doisne N, Acsai K, Varro A, Sztojkov-Ivanov A, Zupko I, Rauch E, Kattner L, Bito V, Houtman M, van der Nagel R, Beekman JD, van Veen TA, Sipido KR, Antoons G. Combined Na(+)/Ca(2+) exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ Arrhythm Electrophysiol. 2013;6:371–379. doi: 10.1161/CIRCEP.113.000322. doi: 10.1161/CIRCEP.113.000322. [DOI] [PubMed] [Google Scholar]
- 49.Wilson D, Ermentrout B, Němec J, Salama G. A model of cardiac ryanodine receptor gating predicts experimental Ca2+-dynamics and Ca2+-triggered arrhythmia in the long QT syndrome. Chaos. 2017;27:093940. doi: 10.1063/1.5000711. doi: 10.1063/1.5000711. [DOI] [PubMed] [Google Scholar]
- 50.Kim JJ, Němec J, Li Q, Salama G. Synchronous systolic subcellular Ca2+-elevations underlie ventricular arrhythmia in drug-induced long QT type 2. Circ Arrhythm Electrophysiol. 2015;8:703–712. doi: 10.1161/CIRCEP.114.002214. doi: 10.1161/CIRCEP.114.002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burashnikov A, Antzelevitch C. Block of I(Ks) does not induce early afterdepolarization activity but promotes beta-adrenergic agonist-induced delayed afterdepolarization activity. J Cardiovasc Electrophysiol. 2000;11:458–465. doi: 10.1111/j.1540-8167.2000.tb00342.x. doi: 10.1111/j.1540-8167.2000.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 52.Immanuel SA, Sadrieh A, Baumert M, Couderc JP, Zareba W, Hill AP, Vandenberg JI. T-wave morphology can distinguish healthy controls from LQTS patients. Physiol Meas. 2016;37:1456–1473. doi: 10.1088/0967-3334/37/9/1456. doi: 10.1088/0967-3334/37/9/1456. [DOI] [PubMed] [Google Scholar]
- 53.Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ, Rodriguez B. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci U S A. 2013;110:E2098–E2105. doi: 10.1073/pnas.1304382110. doi: 10.1073/pnas.1304382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ni H, Morotti S, Grandi E. A Heart for diversity: simulating variability in cardiac arrhythmia research. Front Physiol. 2018;9:958. doi: 10.3389/fphys.2018.00958. doi: 10.3389/fphys.2018.00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarkar AX, Christini DJ, Sobie EA. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J Physiol. 2012;590:2555–2567. doi: 10.1113/jphysiol.2011.223313. doi: 10.1113/jphysiol.2011.223313. [DOI] [PMC free article] [PubMed] [Google Scholar]