

Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII), regulated by inhibitor 1 of protein phosphatase 1 (I1PP1), is vital for maintaining cardiovascular homeostasis. However, the role and mechanism of I1PP1 against hypoxia-reoxygenation (H/R) injury in cardiomyocytes remain a question. In our study, after I1PP1 overexpression by adenovirus infection in the neonatal cardiomyocytes followed by hypoxia for 4 h and reoxygenation for 12 h, the CaMKIIδ alternative splicing subtype, ATP content, and lactate dehydrogenase (LDH) release were determined. CaMKII activity was evaluated by phosphoprotein phosphorylation at Thr17 (p-PLB Thr17), CaMKII phosphorylation (p-CaMKII), and CaMKII oxidation (ox-CaMKII). Reactive oxygen species (ROS), mitochondrial membrane potential, dynamin-related protein 1 (DRP1), and optic atrophy 1 (OPA1) expressions were assessed. Our study verified that I1PP1 overexpression attenuated the CaMKIIδ alternative splicing disorder; suppressed PLB phosphorylation at Thr17, p-CaMKII, and ox-CaMKII; decreased cell LDH release; increased ATP content; attenuated ROS production; increased mitochondrial membrane potential; and decreased DRP1 expression but increased OPA1 expression in the cardiomyocytes after H/R. Contrarily, CaMKIIδ alternative splicing disorder, LDH release, ATP reduction, and ROS accumulation were aggravated after H/R injury with the I1PP1 knockdown. Collectively, I1PP1 overexpression corrected disorders of CaMKIIδ alternative splicing, inhibited CaMKII phosphorylation, repressed CaMKII oxidation, suppressed ROS production, and attenuated cardiomyocyte H/R injury.
1. Introduction
Myocardial ischemia-reperfusion injury (MIRI) is a phenomenon wherein the myocardial function is not improved but aggravated immediately after blood perfusion is restored in the ischemic myocardium [1–4]. MIRI is often accompanied by cardiac and vascular adverse events such as arrhythmia, enlarged infarct size, persistent ventricular systolic dysfunction, or even no reflow, which seriously impair the prognosis of myocardial ischemia [5–7]. MIRI is a complex pathophysiological process involving multiple factors, such as oxygen-free radicals, calcium overload, inflammation, apoptosis, and endothelial cell homeostasis imbalance [8–10], in which excess of oxygen-free radicals is the critical factor for reperfusion injury [11]. Moreover, MIRI is a common cause of early cardiac dysfunction after cardiac surgery, which is a difficult problem to limit the treatment and prognosis of ischemic heart disease [12].
Calcium/calmodulin-dependent protein kinase II (CaMKII) is one serine-threonine protein kinase with multifunctions, which is abundant in the myocardium and other excitable tissues [13, 14]. Four isoforms of CaMKIIδ (α, β, γ, and δ) have been found as of now. CaMKIIδ is the most abundant subtype in the myocardium [15]. In the presence of alternative splicing, CaMKIIδ is capable of producing three splicing variants of δA, δB, and δC with the action of splicing factors [8–10]. Three different subtypes of CaMKIIδ play diverse roles in the cardiovascular system. CaMKIIδA was critical for cardiac systolic and diastolic function [16]. CaMKIIδB regulated myocardial hypertrophy and progenitor cell survival in the myocardium [17]. CaMKIIδC was closely related to cardiomyocyte apoptosis during myocardial remodeling [18].
Besides Ca2+/calmodulin, CaMKII could also be activated by phosphorylation and oxidation [19–21]. Sustained excessive CaMKII activation has adverse effects on a variety of heart diseases [22, 23]. Some studies have found that CaMKII activity bloomed at the beginning of MIRI, which promoted calcium extravasation of the sarcoplasmic reticulum and aggravated myocardial dysfunction and injury [24, 25]. Alleviation of CaMKII activity is beneficial to attenuate the degree of MIRI, reduce the excessive production of reactive oxygen species (ROS) [26], and inhibit apoptosis and necrosis, which is advantageous for the recovery of the myocardial function [27]. In addition, CaMKII is also one of the key mediators for myocardial necroptosis [28]. Therefore, CaMKII is commonly regarded as the core signal in cardiomyocytes [14]. Although there were several common CaMKII inhibitors such as KN93 or AIP, the specificity and potency of these compounds limit their application due to some other effects unrelated to CaMKII inhibition [29]. Moreover, as an inhibitor of CaMKII, KN93 competitively inhibits the binding of calmodulin to kinase, but not the autonomic activity of CaMKII [29]. Altogether, CaMKII activity regulation might be a potential method to alleviate MIRI.
Protein phosphatase 1 (PP1) is regulated by intracellular calcium fluctuations in the heart to change the phosphorylation level of multiple proteins [30–33]. Studies have shown that PP1 is upregulated in the heart of patients suffering from cardiac hypertrophy, cardiac dysfunction, and heart failure [34–36]. Lack of PP1 protected against arrhythmias and myocardial hypertrophy [37]. Moreover, PP1 promoted CaMKIIδ splicing. When PP1 increased, the ratio of CaMKIIδ alternative splicing products could be imbalanced, resulting in an enhancement of CaMKIIδC variants but a reduction of CaMKIIδB and CaMKIIδA variants [33]. Therefore, the pharmacological targeting of PP1 activity is believed to be useful in therapy and interventions for a variety of cardiomyopathies such as MIRI [38–41]. As far as we know, inhibitor 1 of protein phosphatase 1 (I1PP1) is an endogenous inhibitor to decrease PP1 activity or to inhibit PP1 expression [42]. I1PP1 upregulation accelerated Ca2+ cycling, improved cardiac function, and alleviated cell injury [43, 44].
Therefore, hypoxia-reoxygenation (H/R) of cardiomyocytes in vitro was used to verify the effect of I1PP1 overexpression on CaMKII activity and alternative splicing. The detailed mechanism of I1PP1 overexpression against H/R injury was also explored. It is beneficial to provide a new strategy for MIRI.
2. Methods and Materials
2.1. Cell Culture and Hypoxia-Reoxygenation Injury Model
Neonatal cardiomyocytes were extracted from Sprague-Dawley (SD) rats aging 1-3 days by trypsin (Beyotime, Shanghai, China) digestion. After culturing for 2-3 days, the cells were incubated with the DMEM medium (Gibco, Carlsbad, CA) containing 1 g/L glucose and 0.5% FBS (Gibco, Carlsbad, CA) and cultured in the incubator with 94% N2, 1% O2, and 5% CO2 to induce hypoxia. Four hours later, the medium was changed into 5.5 g/L glucose and 10% FBS and incubated in an incubator with 5% CO2 and normal air. After another 12 h for reoxygenation, the cardiomyocytes were collected for further experiments. CaMKII inhibitor KN93 (1 μM, MedChemExpress, Monmouth Junction, NJ) was preincubated for 1 h before hypoxia and reoxygenation.
The present experiment strictly complied with guidelines for the Care and Use of Laboratory Animals from the Institute for Laboratory Animal Research, National Research Council, Washington, D.C., National Academy Press, 2011, and any updates. The detailed protocols were also approved by a specific committee in Nantong University (NTU-20161210).
2.2. I1PP1 Adenovirus Infection
Rat full length of I1PP1 (Gene ID, 58977, 1 × 1011 PFU/mL) or vector (1 × 1011 PFU/mL) recombinant adenovirus solution (Hanbio Biotechnology Co., Ltd., Shanghai, China) with MOI value of 100 was infected into the cardiomyocytes. After 4 h, the adenovirus solution were washed out and DMEM medium with 10% FBS was replaced. Vector (1 × 1011 PFU/mL) recombinant adenovirus was applied as a negative control. Then, the cells were subjected to immunofluorescent staining, western blot, or hypoxia-reoxygenation injury.
2.3. RNA Interference
Three antisense oligodeoxynucleotides against rat I1PP1 mRNA (#1, 5′-AGACAATGGTTGAACATCA-3′; #2, 5′-GCAGAATCCAAACCCAAGA-3′; #3, 5′-TCAGCGTCAAGGCCAGATA′-3′) and nonspecific control siRNA (NC siRNA) were commercially obtained (RiboBio, Guangzhou, China).
After serum deprivation for 24 h, the above siRNA was transfected into the cardiomyocytes with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and subjected to hypoxia-reoxygenation injury.
2.4. Lactate Dehydrogenase (LDH) Measurement
The level of LDH in the medium was detected by a commercial LDH-Cytotoxic Assay Kit (Beyotime, Shanghai, China). The cardiomyocytes were cultured in a 24-well plate with a density about 80%. After I1PP1 recombinant adenovirus infection followed by hypoxia for 4 h and reoxygenation for 12 h, the supernatant of 120 μL was taken into a 96-well plate after centrifugation, and 60 μL of the test solution was mixed into each well. Then, the plate was placed without light for 30 min at 25°C. LDH release into the medium was calculated according to the absorbance at 490 nm, which was normalized by the value in the control group.
2.5. ATP Measurement
The content of ATP in the cardiomyocytes was detected by a commercial ATP Assay Kit (Beyotime, Shanghai, China). After treatment and equilibration, 100 μL of a CellTiter-Lumi™ luminescence assay reagent was completely mixed with the medium. Then, the luminescence intensity was obtained with a spectrophotometer (BioTek Instruments, Inc., USA). The relative ATP level was calculated and normalized by the value in the control group.
2.6. ROS Measurement
After incubation with dihydroethidium (DHE, 2 μM, Beyotime, Shanghai, China) at 37°C for 30 min, the superoxide production, considered as the DHE fluorescence intensity, in the cardiomyocytes was assessed with a laser confocal microscope (Leica, Wetzlar, Germany) and quantified using ImageJ software.
After incubation with MitoTracker Green (100 nM, Beyotime, Shanghai, China) and MitoSOX (5 μM, YEASEN, Shanghai, China) at 37°C for 20 min, mitochondrial ROS, considered as the MitoSOX fluorescence intensity, in the cardiomyocytes was assessed with a laser confocal microscope at 490/516 nm wavelengths for MitoTracker Green and 510/580 nm wavelengths for MitoSOX, respectively, which was quantified using ImageJ software.
2.7. Mitochondrial Membrane Potential (Δψm) Detection
After incubation with JC-1 staining solution (Beyotime, Shanghai, China) for 20 min, Δψm, considered as the fluorescence intensity, in the cardiomyocytes was assessed with a laser confocal microscope at 495/519 nm wavelengths for the J-monomer and 550/570 nm wavelengths for the J-aggregates, which was quantified using ImageJ software.
2.8. Immunofluorescent Staining
After incubation with primary anti-optic atrophy 1 (OPA1, 1 : 50) or anti-dynamin-related protein 1 (DRP1, 1 : 50) antibodies for 12 h at 4°C, the cardiomyocytes were incubated with IgG conjugated with Cy3 or Alexa Fluor 488 (1 : 500; Beyotime, Shanghai, China) for 2 h. The protein expression which is considered as the fluorescence intensity was assessed.
2.9. Real-Time PCR
After extraction of total RNA with TRIzol from the cardiomyocytes, 1 μL RNA sample, 2 μL 5x reaction buffer, and 7 μL DEPC water were mixed in the RNase-free tube. The procedure for reverse transcription was set up: incubation at 37°C for 15 minutes and 85°C for 5 s, and preservation at 4°C. Reversed cDNA was added into the SYBR Green qPCR mix for amplification (ABI, Carlsbad, CA, USA). The primer sequences (Table 1) of CaMKIIδ and housekeeping mRNA were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). Quantitative real-time PCR analyses were performed three times for each group of DNA. The relative mRNA level was calculated by the comparative delta-delta cycle threshold (CT) method.
Table 1.
The sequences of the primers for real-time PCR.
| Gene | Forward primers | Reverse primers |
|---|---|---|
| CaMKIIδA | 5′-CGAGAAATTTTTCAGCAGCC-3′ | 5′-ACAGTAGTTTGGGGCTCCAG-3′ |
| CaMKIIδB | 5′-CGAGAAATTTTTCAGCAGCC-3′ | 5′-GCTCTCAGTTGACTCCATCATC-3′ |
| CaMKIIδC | 5′-CGAGAAATTTTTCAGCAGCC-3′ | 5′-CTCAGTTGACTCCTTTACCCC-3′ |
| 18S | 5′-AGTCCCTGCCCTTTGTACACA-3′ | 5′-CGATCCGAGGGCCTCACTA-3′ |
2.10. Western Blot
The proteins of about 50 μg were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane. Next, the membrane was blocked by 5% milk without fat for 2 h followed by incubation with primary antibodies (Table 2) [45–47] at 4°C overnight. After washing, the membrane was incubated by horseradish peroxidase- (HRP-) conjugated IgG (Beyotime, Shanghai, China) for another 2 h. Enhanced chemiluminescence (ECL, Thermo Fisher Scientific Inc., Rockford, IL, USA) was dropped on the membrane to visualize the protein bands.
Table 2.
The primary antibodies for western blot.
| Protein | Dilution | Company |
|---|---|---|
| I1PP1 | 1 : 5000 | Abcam, Cambridge, UK |
| OPA1 | 1 : 1000 | Abcam, Cambridge, UK |
| CaMKII | 1 : 1000 | Abcam, Cambridge, UK |
| PP1 | 1 : 1000 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| PLB | 1 : 1000 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| p-PLB Thr17 | 1 : 1000 | Santa Cruz Biotechnology, Santa Cruz, CA, USA |
| DRP1 | 1 : 1000 | Cell Signaling Technology, Danvers, MA, USA |
| p-CaMKII | 1 : 1000 | Thermo Fisher Scientific, Rockford, IL, USA |
| ox-CaMKII | 1 : 1000 | Millipore, Billerica, MA, USA |
| GAPDH | 1 : 5000 | Sigma-Aldrich, St. Louis, MO, USA |
| β-Tubulin | 1 : 3000 | CMCTAG, Milwaukee, WI, USA |
2.11. Statistical Analysis
The data were expressed as mean ± Standard Error of the Mean (SEM), which were analyzed by one-way ANOVA followed by Bonferroni post hoc test. P values lower than 0.05 were regarded as a significant difference.
3. Results
3.1. CaMKIIδ Variants Disordered in Cardiomyocytes during Hypoxia-Reoxygenation Injury
Because antibodies for CaMKIIδ variants were unavailable, CaMKIIδA, CaMKIIδB, and CaMKIIδC expressions were measured by quantitative real-time PCR at the start of hypoxia and different times after reoxygenation. There was no significant change on CaMKIIδ variants after hypoxia for 4 h. Both CaMKIIδA and CaMKIIδB expressions were reduced, while the CaMKIIδC expression increased after reoxygenation for 6 h, which was most obvious at 12 h. The data suggested that cardiomyocyte H/R injury induced a significant disorder on CaMKIIδ variants (Figure 1).
Figure 1.
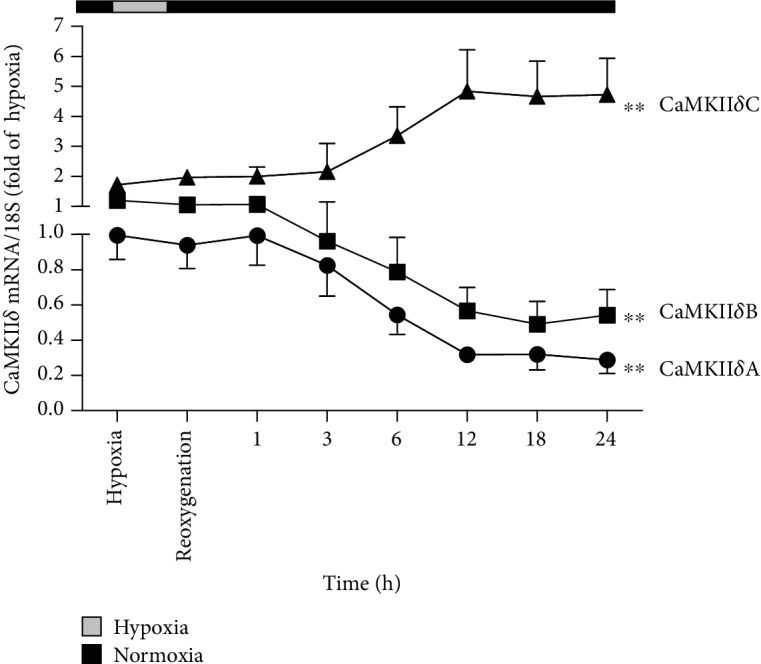
CaMKIIδ variants disordered in cardiomyocytes during H/R injury. After culture in 94% N2, 1% O2, and 5% CO2 for 4 h, the cardiomyocytes were changed into 95% air and 5% CO2. The mRNA levels of CaMKIIδA, CaMKIIδB, and CaMKIIδC of the cardiomyocytes at the start of the hypoxia and different times after reoxygenation were detected by quantitative real-time PCR. 18S was serviced as a housekeeping mRNA. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with “the start of the hypoxia.”
3.2. I1PP1 Overexpression Reduced LDH Release but Increased ATP Level in Cardiomyocytes after H/R Injury
However, whether the correction of the CaMKIIδ variant disorder was beneficial to attenuate H/R injury remains unknown. Next, the recombinant adenovirus technology was applied to induce I1PP1 overexpression in our study. PP1 antibody against PP1α was applied for detection of PP1 family catalytic subunits. We found that I1PP1 expression increased while PP1 expression decreased after recombinant adenovirus infection (Figure 2).
Figure 2.

Recombinant adenovirus infection increased I1PP1 but decreased PP1 expression in cardiomyocytes. (a) After the recombinant adenovirus solution carrying the I1PP1 gene or vector was infected into the cardiomyocytes, I1PP1 and PP1 were immunofluorescence stained using Alexa Fluor 488- (green) or Cy3- (red) conjugated IgG. The nuclei were stained using DAPI (blue). Bar = 100 μm. (b) Expression of the I1PP1 and PP1 protein was quantified in the cardiomyocytes by western blot. β-Tubulin was used as a loading control. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control.
After infection, hypoxia-reoxygenation was performed in the cardiomyocytes. There was more LDH in the medium after H/R, suggesting that H/R induced more serious injury. Moreover, I1PP1 overexpression in the cardiomyocytes significantly reduced LDH release (Figure 3(a)).
Figure 3.

I1PP1 overexpression reduced the LDH release but increased the ATP level in cardiomyocytes after H/R injury. After infection of I1PP1 recombinant adenovirus, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) LDH release in the culture medium was measured. (b) ATP level of the cardiomyocytes was measured. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; #P < 0.05 and ##P < 0.01 compared with H/R.
Dysfunction of energy metabolism is an important mechanism of myocardial ischemia-reperfusion injury [48]. ATP production was also detected to evaluate H/R injury in the cardiomyocytes. The data showed that ATP production was blocked after H/R in the cardiomyocytes, which was restored by I1PP1 overexpression (Figure 3(b)).
3.3. I1PP1 Overexpression Regulated CaMKII Activity and CaMKIIδ Alternative Splicing in Cardiomyocytes after H/R Injury
Previous researches verified that PLB phosphorylation at Thr17 was a robust marker for CaMKII activity, which was commonly used for CaMKII activity assessment [33]. Our present study confirmed that I1PP1 overexpression significantly inhibited PLB phosphorylation at Thr17 after H/R, suggesting that CaMKII activity was weakened due to a high level of I1PP1 (Figure 4(a)). It was also noted that CaMKIIδA and CaMKIIδB splicing variants significantly decreased, while CaMKIIδC significantly increased after H/R. Moreover, I1PP1 overexpression increased CaMKIIδA and CaMKIIδB but decreased CaMKIIδC expression (Figures 4(b)–4(d)). All these data suggested that both CaMKII activity and CaMKIIδ alternative splicing disorder were corrected by I1PP1 overexpression after H/R.
Figure 4.

I1PP1 overexpression regulated CaMKII activity and CaMKIIδ alternative splicing in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) Expression of p-PLB Thr17 and PLB in the cardiomyocytes was quantified by western blot. β-Tubulin was used as a loading control. (b–d) The mRNA levels of CaMKIIδA, CaMKIIδB, and CaMKIIδC in the cardiomyocytes were detected by quantitative real-time PCR. 18S was serviced as a housekeeping mRNA. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; ##P < 0.01 compared with H/R.
3.4. I1PP1 Overexpression Inhibited ROS but Elevated Mitochondrial Membrane Potential in Cardiomyocytes after H/R Injury
Excessive ROS production was considered as one critical mechanism for myocardial I/R injury [49, 50]. Therefore, ROS production in the global cell and the mitochondria was detected with DHE and MitoSOX staining, respectively. The results indicated that there was stronger fluorescence intensity of DHE and MitoSOX after H/R, which was significantly attenuated by I1PP1 overexpression (Figures 5(a) and 5(b)).
Figure 5.

I1PP1 overexpression inhibited ROS but elevated mitochondrial membrane potential in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) Superoxide production in the cardiomyocytes was detected using a fluorescence microscope with DHE fluorescent probe and quantified using ImageJ analysis software. Bar = 25 μm. (b) Mitochondrial ROS was measured using MitoSOX. Mitochondrial localization of MitoSOX signal was confirmed by colocalization with MitoTracker Green and quantified using ImageJ analysis software. Bar = 10 μm. (c) Mitochondrial membrane potential (Δψm) was measured by JC-1 staining and quantified using ImageJ analysis software. Bar = 25 μm. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; ##P < 0.01 compared with H/R.
Studies have shown that decrease of mitochondrial membrane potential (Δψm) might lead to ROS accumulation and cell damage. Our study found that the green fluorescence intensity of JC-1 monomers, suggesting the impaired mitochondrial membrane potential, increased after H/R. Meanwhile, red fluorescence intensity of JC-1 aggregates, suggesting the normal membrane potential, decreased. Moreover, I1PP1 overexpression inhibited green but enhanced red fluorescence intensity. The data demonstrated that I1PP1 overexpression in the cardiomyocytes elevated mitochondrial membrane potential after H/R (Figure 5(c)).
3.5. I1PP1 Overexpression Alleviated CaMKII Oxidation and Phosphorylation in Cardiomyocytes after H/R Injury
CaMKII oxidation and phosphorylation, as two main activation forms of CaMKII, are favorable to accelerate the deterioration of cardiac function after MIRI [28]. The study verified that both oxidation and phosphorylation of CaMKII were heightened in cardiomyocytes after H/R injury, while I1PP1 overexpression in the cardiomyocytes diminished CaMKII oxidation and phosphorylation after H/R injury (Figures 6(a) and 6(b)).
Figure 6.

I1PP1 overexpression alleviated CaMKII oxidation and phosphorylation in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) Expressions of CaMKII oxidation (ox-CaMKII) and total CaMKII in cardiomyocytes after H/R injury were quantified by western blot. GAPDH was used as a loading control. (b) Expression of CaMKII phosphorylation (p-CaMKII) and total CaMKII in cardiomyocytes after H/R injury was quantified by western blot. GAPDH was used as a loading control. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; ##P < 0.01 compared with H/R.
3.6. I1PP1 Overexpression Regulated Mitochondrial OPA1 and DRP1 Expression in Cardiomyocytes after H/R Injury
OPA1 and DRP1 are mitochondrial fusion- and fission-associated proteins, respectively [51, 52]. The dynamic balance of OPA1 and DRP1 maintains mitochondrial structures and functions [53, 54]. We found that H/R injury significantly inhibited OPA1 but elevated DRP1 expression. I1PP1 overexpression in the cardiomyocytes increased OPA1 but decreased DRP1 after H/R injury, which was beneficial to maintaining the balance of OPA1 and DRP1 (Figures 7(a)–7(c)).
Figure 7.

I1PP1 overexpression regulated mitochondrial OPA1 and DRP1 expression in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) OPA1 and DRP1 were immunofluorescence stained using Cy3- (red) or Alexa Fluor 488- (green) conjugated IgG and quantified using ImageJ analysis software. The nuclei were stained using DAPI (blue). Bar = 10 μm. (b, c) Expressions of OPA1 and DRP1 in the cardiomyocytes were quantified by western blot. GAPDH was used as a loading control. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; ##P < 0.01 compared with H/R.
3.7. I1PP1 Overexpression Combining with CaMKII Inhibitor KN93 Attenuated Oxidative Stress in Cardiomyocytes after H/R Injury
In order to confirm the above protective effect of I1PP1 overexpression against oxidative stress during H/R injury was ascribed to CaMKII inhibition, the global cellular ROS and mitochondrial ROS were further measured. The study found that CaMKII inhibitor KN93 preadministration significantly inhibited ROS accumulation after H/R injury (Figures 8(a)–8(d)).
Figure 8.

KN93 attenuated oxidative stress in cardiomyocytes after H/R injury. After CaMKII inhibitor KN93 (1 μM) preadministration for 1 h, the cardiomyocytes were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a, b) Superoxide production in the cardiomyocytes was detected using a fluorescence microscope with a DHE fluorescent probe and quantified using ImageJ analysis software. Bar = 100 μm. (c, d) Mitochondrial ROS was measured using MitoSOX. Mitochondrial localization of the MitoSOX signal was confirmed by colocalization with MitoTracker Green and quantified using ImageJ analysis software. Bar = 10 μm. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with the control; ##P < 0.01 compared with H/R.
Moreover, both I1PP1 overexpression and I1PP1 overexpression combining with KN93 similarly decreased LDH release (Figure 9(a)) and enhanced ATP production (Figure 9(b)), weakened ROS production (Figure 10), and increased mitochondrial membrane potential (Figure 11) in cardiomyocytes after H/R. Furthermore, no significant difference appeared between the above two groups. These data indicated that I1PP1 overexpression alleviated cell damage, inhibited ROS production, and elevated mitochondrial membrane potential in cardiomyocytes after H/R injury via inhibition of CaMKII activation.
Figure 9.

Effects of I1PP1 overexpression and KN93 on LDH release and ATP level in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes with or without CaMKII inhibitor KN93 incubation for 1 h were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) LDH release in the culture medium was measured. (b) ATP level of the cardiomyocytes was measured. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗P < 0.05 and ∗∗P < 0.01 compared with vector+H/R.
Figure 10.

Effects of I1PP1 overexpression and KN93 on oxidative stress in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes with or without CaMKII inhibitor KN93 incubation for 1 h were subjected to hypoxia for 4 h followed by 12 h reoxygenation. (a) Superoxide production in the cardiomyocytes was detected using a fluorescence microscope with DHE fluorescent probe and quantified using ImageJ analysis software. Bar = 25 μm. (b) Mitochondrial ROS was measured using MitoSOX. Mitochondrial localization of the MitoSOX signal was confirmed by colocalization with MitoTracker Green and quantified using ImageJ analysis software. Bar = 25 μm. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with vector+H/R.
Figure 11.

Effects of I1PP1 overexpression and KN93 on mitochondrial membrane potential in cardiomyocytes after H/R injury. After infection of the I1PP1 recombinant adenovirus, the cardiomyocytes with or without CaMKII inhibitor KN93 incubation for 1 h were subjected to hypoxia for 4 h followed by 12 h reoxygenation. Mitochondrial membrane potential (Δψm) was measured by JC-1 staining and quantified using ImageJ analysis software. Bar = 25 μm. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with vector+H/R.
3.8. I1PP1 Knockdown Exacerbated CaMKIIδ Alternative Splicing Disorder, Cell Injury, and Oxidative Stress in Cardiomyocytes after H/R Injury
Contrarily, we also assessed the effect of I1PP1 knockdown with siRNA technology in cardiomyocytes under H/R injury. We found that all three siRNA significantly reduced I1PP1 expression, and there was the least expression of I1PP1 after siRNA#2 transfection. Therefore, siRNA#2 was applied in a further study (Figure 12(a)). Next, we found that the CaMKIIδ alternative splicing disorder (Figure 12(b)), LDH release (Figure 12(c)), ATP reduction (Figure 12(d)), and ROS accumulation (Figure 12(e)) in cardiomyocytes were aggravated after H/R in the cardiomyocytes with I1PP1 knockdown. However, there was no evidence regarding CaMKII activation in neonatal cardiomyocytes after I1PP1was knocked down. Altogether, I1PP1 knockdown exacerbated the CaMKIIδ alternative splicing disorder, cell injury, and oxidative stress in neonatal cardiomyocytes after H/R injury.
Figure 12.

Effects of I1PP1 knockdown on CaMKIIδ alternative splicing disorder, cell injury, and oxidative stress in cardiomyocytes after H/R injury. (a) After I1PP1 siRNA or NC siRNA was transfected into neonatal rat cardiomyocytes, expressions of I1PP1 in the cardiomyocytes were quantified by western blot. GAPDH was used as a loading control. (b) After I1PP1 siRNA or NC siRNA was transfected into neonatal rat cardiomyocytes, cells were subjected to hypoxia for 4 h followed by 12 h reoxygenation. The mRNA levels of CaMKIIδA, CaMKIIδB, and CaMKIIδC of the cardiomyocytes after reoxygenation were detected by quantitative real-time PCR. 18S was serviced as a housekeeping mRNA. (c) LDH release in the culture medium was measured. (d) ATP level of the cardiomyocytes was measured. (e) Mitochondrial ROS was measured using MitoSOX. Mitochondrial localization of the MitoSOX signal was confirmed by colocalization with MitoTracker Green and quantified using ImageJ analysis software. Bar = 25 μm. Plots represent the mean ± SEM; n = 6. Statistical significance: ∗∗P < 0.01 compared with NC; #P < 0.05 and ##P < 0.01 compared with NC+H/R.
4. Discussion
Although reperfusion is essential for blood flow recovery in the myocardium, it might result in serious damage to the heart during myocardium ischemia and reperfusion. Thus, how to attenuate MIRI has been a very difficult problem in the clinic.
CaMKII is capable of integrating β-adrenergic, ROS, Gq-coupled receptors, hyperglycemia, and proapoptotic cytokine signals to induce oxidative stress in the myocardium [14]. The excessive activation of CaMKII exacerbates cardiomyocyte damage and accelerates the progression of cardiovascular disease. Previous research indicated that CaMKII was crucial in the pathogenesis of MIRI [55–59], and CaMKIIδ was the most vital isoform in the myocardium. Our present results showed that hypoxia-reoxygenation caused a disorder of CaMKIIδ alternative splicing in the cardiomyocytes, which was characterized by decreased expression of the δA and δB subtypes but increased expression of δC. On the other hand, levels of ox-CaMKII and p-CaMKII, as well as expression of p-PLB Thr17 representing CaMKII activity, also increased. Previous study found that PLB phosphorylation increased in the heart of I1PP1 transgenic mice in the basal state. I1PP1 elevated PLB phosphorylation after global ischemia for 40 min followed by reperfusion, whereas there were no differences at 60 min postreperfusion [43]. I1PP1 also increased PLB phosphorylation in the heart of pigs in the basal state [60]. In the present study, we found that I1PP1 significantly inhibited PLB phosphorylation after H/R in cardiomyocytes. The divergent effects on PLB phosphorylation may be explained by different types of cells or animals used in such studies, different ischemia or reperfusion times, different times for measurement, or a combination of these factors. To our knowledge, ROS oxidizes the methionine residue site of Met281/282 to promote CaMKII oxidation. CaMKII was able to be phosphorylated by itself. However, whether restoring the above abnormalities of CaMKII is able to alleviate H/R injury is not well known.
PP1 is able to alter the splicing factor phosphorylation and regulate CaMKII alternative splicing in the heart. A previous study found that hypoxia (but without reoxygenation) decreased the activity of PP1 in the cardiomyocytes [61], in the skeletal muscle of frogs [62], in the eukaryotic cells [63], and in the hippocampus [64]. A previous study found that enhanced PP1 activity was involved in Ca2+ cycling, augmenting during H/R in the adult heart [43]. Moreover, previous research found that inducible overexpression of I1PP1 enhanced basal cardiac function and protected against ischemia-reperfusion injury [43], which was consistent with our results that I1PP1 overexpression attenuated cardiomyocyte H/R injury. Our research also confirmed that I1PP1 corrected the disorder of CaMKIIδ splicing, reduced LDH release, but enhanced ATP production to alleviate cell injury. Contrarily, I1PP1 knockdown exacerbated the CaMKIIδ alternative splicing disorder and aggravated cell injury after H/R injury. The protective effect of I1PP1 overexpression may be ascribed to the regulation of CaMKII alternative splicing and inhibition of CaMKII activity and ox-CaMKII as well as p-CaMKII. These data indicated that I1PP1 had a negative regulatory effect on H/R injury of cardiomyocytes through CaMKII regulation.
The protective mechanism of the attenuating effect on H/R damage by I1PP1 overexpression might be related to ROS inhibition. As far as we know, excessive ROS usually resulted in several types of pathological damage including myocardial hypertrophy [65, 66], liver injury or fibrosis [67, 68], pulmonary damage [69], and neuronal apoptosis [70]. The unsaturated fatty acids were vital for maintaining mitochondrial integrity and function, which is extremely susceptible to oxidative injury. The increase of oxidized unsaturated fatty acids directly impaired the function of the mitochondrial electron transport chain, thereby further promoting more ROS generation in mitochondria [71]. During H/R, ROS also induced the transformation of mitochondrial permeability by opening several small pores in the mitochondria immediately at the beginning of reperfusion [72], which resulted in mitochondrial matrix swelling and outer membrane integrity destruction. Besides, mitochondrial damage also directly blocks the production and storage of cellular ATP. Therefore, maintaining normal structure and function of mitochondria is critical for cell survive [73, 74]. Our present results showed that H/R decreased the mitochondrial membrane potential and promoted ROS accumulation. After I1PP1 successfully inhibited PP1 expression, the CaMKII pathway, cell damage, ATP production, and mitochondrial membrane potential as well as ROS accumulation were restored significantly. Moreover, CaMKII-specific inhibitor KN93 was further applied to clarify the causal relationship between negative regulation of CaMKII and the protective effect on cardiomyocytes. Our data indicated that KN93 was capable of attenuating both global intracellular and mitochondrial ROS after H/R. We also found that I1PP1 overexpression with or without CaMKII inhibition by KN93 similarly decreased LDH release, weakened ROS production, and increased mitochondrial membrane potential in cardiomyocytes after H/R. Altogether, it suggested that I1PP1 overexpression attenuated cardiomyocyte injury under stress conditions via inhibiting CaMKII activation.
Dynamic fusion and fission is beneficial to maintaining mitochondrial structure and function to appropriately respond to frequent changing environmental conditions [75]. OPA1 localizes on the mitochondrial inner membrane (IMM) and membrane gap to control IMM fusion and sputum structure [76]. OPA1 deficiency promoted mitochondrial fragmentation and abnormal sputum remodeling to suppress respiratory ability and inhibit mitochondria-dependent cell metabolism [77, 78]. Besides, OPA1 also prevented cytochrome C redistribution and release to inhibit cell injury [54]. Contrarily, DRP1 has been verified to mediate mitochondrial fission [79]. In our present study, hypoxia-reoxygenation caused the imbalance of OPA1 and DRP1 in cardiomyocytes, which would cause fatal damage to the morphology and function of mitochondria. Overexpression of I1PP1 reversed the imbalance of proportions between OPA1 and DRP1 which is normal to maintain the mitochondrial homeostasis.
However, there are several limitations in our present study. Firstly, the use of human cardiomyocytes and measurement of CaMKII levels will greatly benefit extending the significance of the study, which will directly demonstrate the real impact of I1PP1 overexpression on cardiomyocytes for humans. However, human cardiomyocytes are unavailable in the present study. Secondly, due to the experimental strategy that mainly relies on the overexpression of I1PP1, it is unclear whether endogenous I1PP1 has a significant role under H/R injury in neonatal cardiomyocytes. The knockdown approach was performed in addition to overexpression of I1PP1. However, detection of CaMKII activity and CaMKII phosphorylation as well as PP1 levels was unavailable in the present study, which will be a great benefit to validate the effect and mechanism of I1PP1 on H/R injury in cardiomyocytes.
Collectively, I1PP1 overexpression corrected disorders of CaMKIIδ alternative splicing, inhibited CaMKII phosphorylation, repressed CaMKII oxidation, suppressed ROS production, and attenuated H/R injury in the cardiomyocytes (Figure 13). The present study is helpful to provide new biological targets for prevention and treatment of myocardial ischemia-reperfusion injury in the clinic.
Figure 13.
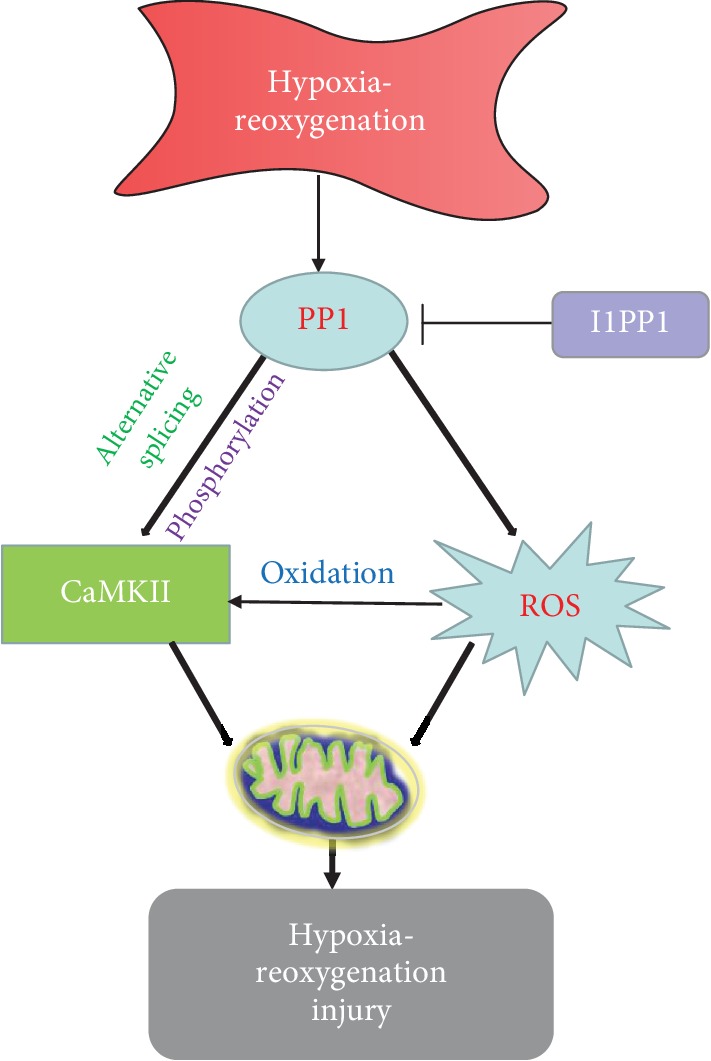
I1PP1 overexpression corrected CaMKIIδ alternative splicing disorders, inhibited CaMKII phosphorylation, repressed CaMKII oxidation, suppressed ROS production, and attenuated hypoxia-reoxygenation injury in the cardiomyocytes.
Acknowledgments
The work was funded by grants 81670243, 81873470, and 81770279 from the National Natural Science Foundation of China, grant BK20151276 from the Natural Science Foundation of Jiangsu Province, a major project of the Natural Science Research of Jiangsu Higher Education Institutions of China (18KJA310005), the Six Talent Peaks Project in Jiangsu Province (2018-WSN-062), grants from the China Postdoctoral Science Foundation (2017M610342 and 2019T120449), the Jiangsu Planned Projects for Postdoctoral Research Funds (1701050A), the Graduate Research and Innovation Projects of Jiangsu Province (KYCX18_2401), and the Nantong University Cooperative Innovation Program of Small Molecular Compound R&D (NTU2016-1).
Contributor Information
Guoliang Meng, Email: mengguoliang@ntu.edu.cn.
Wei Zhang, Email: zhangw@ntu.edu.cn.
Data Availability
The data used to support the findings of this study are available from the corresponding authors upon request.
Conflicts of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contributions
Huiqin Luo, Shu Song, and Yun Chen contributed equally to this study.
References
- 1.Hausenloy D. J., Yellon D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. Journal of Clinical Investigation. 2013;123(1):92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muntean D. M., Sturza A., Dănilă M. D., Borza C., Duicu O. M., Mornoș C. The role of mitochondrial reactive oxygen species in cardiovascular injury and protective strategies. Oxidative Medicine and Cellular Longevity. 2016;2016:19. doi: 10.1155/2016/8254942.8254942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalogeris T., Bao Y., Korthuis R. J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biology. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hausenloy D. J., Botker H. E., Engstrom T., et al. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. European Heart Journal. 2017;38(13):935–941. doi: 10.1093/eurheartj/ehw145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hausenloy D. J., Yellon D. M. Ischaemic conditioning and reperfusion injury. Nature Reviews Cardiology. 2016;13(4):193–209. doi: 10.1038/nrcardio.2016.5. [DOI] [PubMed] [Google Scholar]
- 6.Heusch G. The coronary circulation as a target of cardioprotection. Circulation Research. 2016;118(10):1643–1658. doi: 10.1161/CIRCRESAHA.116.308640. [DOI] [PubMed] [Google Scholar]
- 7.Ibanez B., Heusch G., Ovize M., Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. Journal of the American College of Cardiology. 2015;65(14):1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 8.Sun T., Zhang Y., Zhong S., et al. N-n-Butyl haloperidol iodide, a derivative of the anti-psychotic haloperidol, antagonizes hypoxia/reoxygenation injury by inhibiting an Egr-1/ROS positive feedback loop in h9c2 cells. Frontiers in Pharmacology. 2018;9:p. 19. doi: 10.3389/fphar.2018.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mokhtari-Zaer A., Marefati N., Atkin S. L., Butler A. E., Sahebkar A. The protective role of curcumin in myocardial ischemia-reperfusion injury. Journal of Cellular Physiology. 2018;234(1):214–222. doi: 10.1002/jcp.26848. [DOI] [PubMed] [Google Scholar]
- 10.Geng H. H., Li R., Su Y. M., et al. Curcumin protects cardiac myocyte against hypoxia-induced apoptosis through upregulating miR-7a/b expression. Biomedicine & Pharmacotherapy. 2016;81:258–264. doi: 10.1016/j.biopha.2016.04.020. [DOI] [PubMed] [Google Scholar]
- 11.Raedschelders K., Ansley D. M., Chen D. D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacology & Therapeutics. 2012;133(2):230–255. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Frank A., Bonney M., Bonney S., Weitzel L., Koeppen M., Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Seminars in Cardiothoracic and Vascular Anesthesia. 2012;16(3):123–132. doi: 10.1177/1089253211436350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westenbrink B. D., Ling H., Divakaruni A. S., et al. Mitochondrial reprogramming induced by CaMKIIδ mediates hypertrophy decompensation. Circulation Research. 2015;116(5):e28–e39. doi: 10.1161/CIRCRESAHA.116.304682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng N., Anderson M. E. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. Journal of Molecular and Cellular Cardiology. 2017;103:102–109. doi: 10.1016/j.yjmcc.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mollova M. Y., Katus H. A., Backs J. Regulation of CaMKII signaling in cardiovascular disease. Frontiers in Pharmacology. 2015;6:p. 178. doi: 10.3389/fphar.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luczak E. D., Anderson M. E. CaMKII oxidative activation and the pathogenesis of cardiac disease. Journal of Molecular and Cellular Cardiology. 2014;73:112–116. doi: 10.1016/j.yjmcc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quijada P., Hariharan N., Cubillo J. D., et al. Nuclear calcium/calmodulin-dependent protein kinase II signaling enhances cardiac progenitor cell survival and cardiac lineage commitment. Journal of Biological Chemistry. 2015;290(42):25411–25426. doi: 10.1074/jbc.M115.657775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao R. J., Tong L. J., Huang C., et al. Rescue of cardiac failing and remodelling by inhibition of protein phosphatase 1γ is associated with suppression of the alternative splicing factor-mediated splicing of Ca2+/calmodulin-dependent protein kinase δ. Clinical and Experimental Pharmacology and Physiology. 2014;41(12):976–985. doi: 10.1111/1440-1681.12308. [DOI] [PubMed] [Google Scholar]
- 19.Dewenter M., Neef S., Vettel C., et al. Calcium/Calmodulin-Dependent Protein Kinase II activity persists during chronic β-adrenoceptor blockade in experimental and human heart failure. Circulation-Heart Failure. 2017;10(5, article e003840) doi: 10.1161/CIRCHEARTFAILURE.117.003840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shioda N., Fukunaga K. Physiological and pathological roles of CaMKII-PP1 signaling in the brain. International Journal of Molecular Sciences. 2018;19(1):p. 20. doi: 10.3390/ijms19010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mustroph J., Neef S., Maier L. S. CaMKII as a target for arrhythmia suppression. Pharmacology & Therapeutics. 2017;176:22–31. doi: 10.1016/j.pharmthera.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Daniels L. J., Wallace R. S., Nicholson O. M., et al. Inhibition of calcium/calmodulin-dependent kinase II restores contraction and relaxation in isolated cardiac muscle from type 2 diabetic rats. Cardiovascular Diabetology. 2018;17(1):p. 89. doi: 10.1186/s12933-018-0732-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong P., Quan D., Peng J., et al. Role of CaMKII in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo. Journal of Molecular and Cellular Cardiology. 2017;109:1–16. doi: 10.1016/j.yjmcc.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Bell J. R., Vila-Petroff M., Delbridge L. M. D. CaMKII-dependent responses to ischemia and reperfusion challenges in the heart. Frontiers in Pharmacology. 2014;5:p. 96. doi: 10.3389/fphar.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell J. R., Erickson J. R., Delbridge L. M. Ca2+/calmodulin dependent kinase II: a critical mediator in determining reperfusion outcomes in the heart? Clinical and Experimental Pharmacology and Physiology. 2014;41(11):940–946. doi: 10.1111/1440-1681.12301. [DOI] [PubMed] [Google Scholar]
- 26.Sanders P. N., Koval O. M., Jaffer O. A., et al. CaMKII is essential for the proasthmatic effects of oxidation. Science Translational Medicine. 2013;5(195, article 195ra97) doi: 10.1126/scitranslmed.3006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szobi A., Rajtik T., Carnicka S., Ravingerova T., Adameova A. Mitigation of postischemic cardiac contractile dysfunction by CaMKII inhibition: effects on programmed necrotic and apoptotic cell death. Molecular and Cellular Biochemistry. 2014;388(1-2):269–276. doi: 10.1007/s11010-013-1918-x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang T., Zhang Y., Cui M., et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nature Medicine. 2016;22(2):175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 29.Pellicena P., Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Frontiers in Pharmacology. 2014;5:p. 21. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139(3):468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Fang H. Y., Hung M. Y., Lin Y. M., et al. 17β-Estradiol and/or estrogen receptor alpha signaling blocks protein phosphatase 1 mediated ISO induced cardiac hypertrophy. PLoS One. 2018;13(5, article e0196569) doi: 10.1371/journal.pone.0196569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebelo S., Santos M., Martins F., da Cruz e Silva E. F., da Cruz e Silva O. A. B. Protein phosphatase 1 is a key player in nuclear events. Cellular Signalling. 2015;27(12):2589–2598. doi: 10.1016/j.cellsig.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Huang C., Cao W., Liao R., et al. PP1γ functionally augments the alternative splicing of CaMKIIδ through interaction with ASF. American Journal of Physiology-Cell Physiology. 2014;306(2):C167–C177. doi: 10.1152/ajpcell.00145.2013. [DOI] [PubMed] [Google Scholar]
- 34.Brüchert N., Mavila N., Boknik P., et al. Inhibitor-2 prevents protein phosphatase 1-induced cardiac hypertrophy and mortality. American Journal of Physiology-Heart and Circulatory Physiology. 2008;295(4):H1539–H1546. doi: 10.1152/ajpheart.00515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiang D. Y., Li N., Wang Q., et al. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovascular Research. 2014;103(1):178–187. doi: 10.1093/cvr/cvu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoyama H., Ikeda Y., Miyazaki Y., et al. Isoform-specific roles of protein phosphatase 1 catalytic subunits in sarcoplasmic reticulum-mediated Ca2+ cycling. Cardiovascular Research. 2011;89(1):79–88. doi: 10.1093/cvr/cvq252. [DOI] [PubMed] [Google Scholar]
- 37.El-Armouche A., Wittköpper K., Degenhardt F., et al. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovascular Research. 2008;80(3):396–406. doi: 10.1093/cvr/cvn208. [DOI] [PubMed] [Google Scholar]
- 38.Miyazaki Y., Ikeda Y., Shiraishi K., et al. Heart failure-inducible gene therapy targeting protein phosphatase 1 prevents progressive left ventricular remodeling. PLoS One. 2012;7(4, article e35875) doi: 10.1371/journal.pone.0035875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr A. N., Schmidt A. G., Suzuki Y., et al. Type 1 phosphatase, a negative regulator of cardiac function. Molecular and Cellular Biology. 2002;22(12):4124–4135. doi: 10.1128/mcb.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sotoud H., Borgmeyer U., Schulze C., El-Armouche A., Eschenhagen T. Development of phosphatase inhibitor-1 peptides acting as indirect activators of phosphatase 1. Naunyn-Schmiedeberg′s Archives of Pharmacology. 2015;388(3):283–293. doi: 10.1007/s00210-014-1065-2. [DOI] [PubMed] [Google Scholar]
- 41.Liu C. L., He Y. Y., Li X., Li R. J., He K. L., Wang L. L. Inhibition of serine/threonine protein phosphatase PP1 protects cardiomyocytes from tunicamycin-induced apoptosis and I/R through the upregulation of p-eIF2α. International Journal of Molecular Medicine. 2014;33(3):499–506. doi: 10.3892/ijmm.2013.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber S., Meyer-Roxlau S., El-Armouche A. Role of protein phosphatase inhibitor-1 in cardiac beta adrenergic pathway. Journal of Molecular and Cellular Cardiology. 2016;101:116–126. doi: 10.1016/j.yjmcc.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 43.Nicolaou P., Rodriguez P., Ren X., et al. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circulation Research. 2009;104(8):1012–1020. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicolaou P., Kranias E. G. Role of PP1 in the regulation of Ca cycling in cardiac physiology and pathophysiology. Frontiers in Bioscience. 2009;14(14):3571–3585. doi: 10.2741/3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu M., Hua Y., Qi Y., Meng G., Yang S. Exogenous hydrogen sulphide supplement accelerates skin wound healing via oxidative stress inhibition and vascular endothelial growth factor enhancement. Experimental Dermatology. 2019;28(7):776–785. doi: 10.1111/exd.13930. [DOI] [PubMed] [Google Scholar]
- 46.Sun L., Chen Y., Luo H., Xu M., Meng G., Zhang W. Ca2+/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochemical Pharmacology. 2019;163:194–205. doi: 10.1016/j.bcp.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J., Yu J., Chen Y., et al. Exogenous hydrogen sulfide supplement attenuates isoproterenol-induced myocardial hypertrophy in a sirtuin 3-dependent manner. Oxidative Medicine and Cellular Longevity. 2018;2018:17. doi: 10.1155/2018/9396089.9396089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tu L., Pan C. S., Wei X. H., et al. Astragaloside IV protects heart from ischemia and reperfusion injury via energy regulation mechanisms. Microcirculation. 2013;20(8):736–747. doi: 10.1111/micc.12074. [DOI] [PubMed] [Google Scholar]
- 49.Meng G., Wang J., Xiao Y., et al. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. Journal of Biomedical Research. 2015;29(3):203–213. doi: 10.7555/JBR.28.20140037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Sun J., Liu C., Fang C. Protective effects of crocetin pretreatment on myocardial injury in an ischemia/reperfusion rat model. European Journal of Pharmacology. 2014;741:290–296. doi: 10.1016/j.ejphar.2014.07.052. [DOI] [PubMed] [Google Scholar]
- 51.Chu D. T., Tao Y., Tasken K. OPA1 in lipid metabolism: function of OPA1 in lipolysis and thermogenesis of adipocytes. Hormone and Metabolic Research. 2017;49(4):276–285. doi: 10.1055/s-0043-100384. [DOI] [PubMed] [Google Scholar]
- 52.Zhou H., Cheang T., Su F., et al. Melatonin inhibits rotenone-induced SH-SY5Y cell death via the downregulation of dynamin-related protein 1 expression. European Journal of Pharmacology. 2018;819:58–67. doi: 10.1016/j.ejphar.2017.11.040. [DOI] [PubMed] [Google Scholar]
- 53.Meng G., Liu J., Liu S., et al. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. British Journal of Pharmacology. 2018;175(8):1126–1145. doi: 10.1111/bph.13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ong S. B., Kalkhoran S. B., Cabrera-Fuentes H. A., Hausenloy D. J. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. European Journal of Pharmacology. 2015;763(Part A):104–114. doi: 10.1016/j.ejphar.2015.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong L. H., Gu X. M., Wu F., Jin Z. X., Zhou J. J. CaMKII inhibition mitigates ischemia/reperfusion-elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts. Biochemical and Biophysical Research Communications. 2017;491(3):687–692. doi: 10.1016/j.bbrc.2017.07.128. [DOI] [PubMed] [Google Scholar]
- 56.Salas M. A., Valverde C. A., Sánchez G., et al. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2010;48(6):1298–1306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Said M., Becerra R., Valverde C. A., et al. Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. Journal of Molecular and Cellular Cardiology. 2011;51(6):936–944. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajtik T., Carnicka S., Szobi A., et al. Oxidative activation of CaMKIIδ in acute myocardial ischemia/reperfusion injury: a role of angiotensin AT1 receptor-NOX2 signaling axis. European Journal of Pharmacology. 2016;771:114–122. doi: 10.1016/j.ejphar.2015.12.024. [DOI] [PubMed] [Google Scholar]
- 59.Gray C. B., Suetomi T., Xiang S., et al. CaMKIIδ subtypes differentially regulate infarct formation following ex vivo myocardial ischemia/reperfusion through NF-κB and TNF-α. Journal of Molecular and Cellular Cardiology. 2017;103:48–55. doi: 10.1016/j.yjmcc.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe S., Ishikawa K., Fish K., et al. Protein phosphatase inhibitor-1 gene therapy in a swine model of nonischemic heart failure. Journal of the American College of Cardiology. 2017;70(14):1744–1756. doi: 10.1016/j.jacc.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu G., Marin-Garcia J., Rogers T. B., Lakatta E. G., Long X. Phosphorylation and hypoxia-induced heme oxygenase-1 gene expression in cardiomyocytes. Journal of Cardiac Failure. 2004;10(6):519–526. doi: 10.1016/j.cardfail.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 62.MacDonald J. A., Storey K. B. Protein phosphatase type-1 from skeletal muscle of the freeze-tolerant wood frog. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology. 2002;131(1):27–36. doi: 10.1016/S1096-4959(01)00477-8. [DOI] [PubMed] [Google Scholar]
- 63.Comerford K. M., Leonard M. O., Cummins E. P., et al. Regulation of protein phosphatase 1γ activity in hypoxia through increased interaction with NIPP1: implications for cellular metabolism. Journal of Cellular Physiology. 2006;209(1):211–218. doi: 10.1002/jcp.20726. [DOI] [PubMed] [Google Scholar]
- 64.Zhang S., Zhang Y., Jiang S., et al. The effect of hypoxia preconditioning on DNA methyltransferase and PP1γ in hippocampus of hypoxia preconditioned mice. High Altitude Medicine & Biology. 2014;15(4):483–490. doi: 10.1089/ham.2014.1042. [DOI] [PubMed] [Google Scholar]
- 65.Zhao L., Wu D., Sang M., Xu Y., Liu Z., Wu Q. Stachydrine ameliorates isoproterenol-induced cardiac hypertrophy and fibrosis by suppressing inflammation and oxidative stress through inhibiting NF-κB and JAK/STAT signaling pathways in rats. International Immunopharmacology. 2017;48:102–109. doi: 10.1016/j.intimp.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 66.Wang P., Luo L., Shen Q., et al. Rosuvastatin improves myocardial hypertrophy after hemodynamic pressure overload via regulating the crosstalk of Nrf2/ARE and TGF-β/smads pathways in rat heart. European Journal of Pharmacology. 2018;820:173–182. doi: 10.1016/j.ejphar.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 67.Wang F., Liu J. C., Zhou R. J., et al. Apigenin protects against alcohol-induced liver injury in mice by regulating hepatic CYP2E1-mediated oxidative stress and PPARα-mediated lipogenic gene expression. Chemico-Biological Interactions. 2017;275:171–177. doi: 10.1016/j.cbi.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J., Yang A., Wu Y., et al. Stachydrine ameliorates carbon tetrachloride-induced hepatic fibrosis by inhibiting inflammation, oxidative stress and regulating MMPs/TIMPs system in rats. Biomedicine & Pharmacotherapy. 2018;97:1586–1594. doi: 10.1016/j.biopha.2017.11.117. [DOI] [PubMed] [Google Scholar]
- 69.Zhao X., Jin Y., Yang L., et al. Promotion of SIRT1 protein degradation and lower SIRT1 gene expression via reactive oxygen species is involved in Sb-induced apoptosis in BEAS-2b cells. Toxicology Letters. 2018;296:73–81. doi: 10.1016/j.toxlet.2018.07.047. [DOI] [PubMed] [Google Scholar]
- 70.Wang C., Nie X., Zhang Y., et al. Reactive oxygen species mediate nitric oxide production through ERK/JNK MAPK signaling in HAPI microglia after PFOS exposure. Toxicology and Applied Pharmacology. 2015;288(2):143–151. doi: 10.1016/j.taap.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 71.Birk A. V., Chao W. M., Bracken C., Warren J. D., Szeto H. H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. British Journal of Pharmacology. 2014;171(8):2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Halestrap A. P., Richardson A. P. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 73.Lu Y., Liu S., Wang Y., Wang D., Gao J., Zhu L. Asiatic acid uncouples respiration in isolated mouse liver mitochondria and induces HepG2 cells death. European Journal of Pharmacology. 2016;786:212–223. doi: 10.1016/j.ejphar.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 74.Wan C., Ma X., Shi S., et al. Pivotal roles of p53 transcription-dependent and -independent pathways in manganese-induced mitochondrial dysfunction and neuronal apoptosis. Toxicology and Applied Pharmacology. 2014;281(3):294–302. doi: 10.1016/j.taap.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 75.Nan J., Zhu W., Rahman M. S., et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochimica et Biophysica Acta-Molecular Cell Research. 2017;1864(7):1260–1273. doi: 10.1016/j.bbamcr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 76.Marin-Garcia J., Akhmedov A. T. Mitochondrial dynamics and cell death in heart failure. Heart Failure Reviews. 2016;21(2):123–136. doi: 10.1007/s10741-016-9530-2. [DOI] [PubMed] [Google Scholar]
- 77.MacVicar T., Langer T. OPA1 processing in cell death and disease – the long and short of it. Journal of Cell Science. 2016;129(12):2297–2306. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 78.Patten D. A., Wong J., Khacho M., et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO Journal. 2014;33(22):2676–2691. doi: 10.15252/embj.201488349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jin Q., Li R., Hu N., et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biology. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding authors upon request.