

SUMMARY
Misregulation of alternative splicing is a hallmark of human tumors, yet to what extent and how it contributes to malignancy are only beginning to be unraveled. Here, we define which members of the splicing factor SR and SR-like families contribute to breast cancer and uncover differences and redundancies in their targets and biological functions. We identify splicing factors frequently altered in human breast tumors and assay their oncogenic functions using breast organoid models. We demonstrate that not all splicing factors affect mammary tumorigenesis in MCF-10A cells. Specifically, the upregulation of SRSF4, SRSF6, or TRA2β disrupts acinar morphogenesis and promotes cell proliferation and invasion in MCF-10A cells. By characterizing the targets of these oncogenic splicing factors, we identify shared spliced isoforms associated with well-established cancer hallmarks. Finally, we demonstrate that TRA2β is regulated by the MYC oncogene, plays a role in metastasis maintenance in vivo, and its levels correlate with breast cancer patient survival.
Graphical Abstract
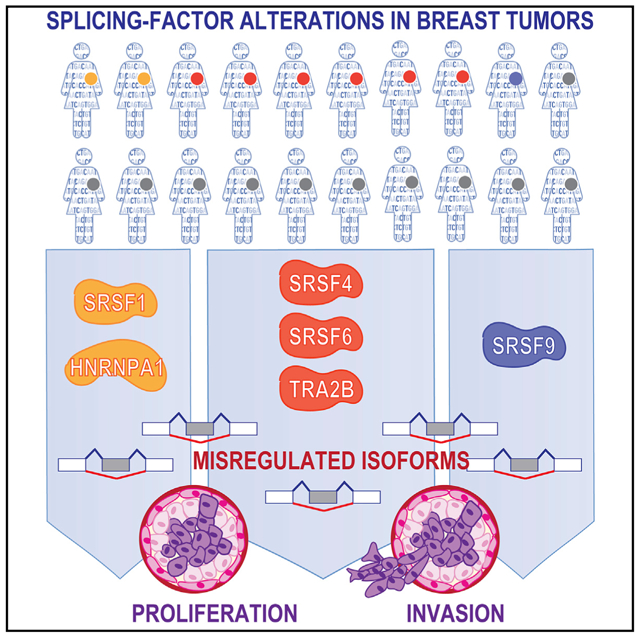
In Brief
Park et al. demonstrate that >50% of human breast tumors exhibit an alteration in one of the splicing factors from the SR protein family. Using in vitro and in vivo breast cancer models, they identify three splicing factors that promote cell proliferation and invasion by regulating isoforms associated with cancer hallmarks.
INTRODUCTION
Alternative RNA splicing is a key step in gene expression regulation and contributes to transcriptional diversity and plasticity by selecting which transcript isoforms are produced in a specific cell at a given time. Defects in alternative splicing (AS) are frequently found in human tumors, and RNA splicing regulators have recently emerged as a new class of oncoproteins or tumor suppressors (Dvinge et al., 2016). AS alterations can lead to malignancy by affecting the expression of oncogene and tumor-suppressor isoforms. Tumor-associated aberrant AS profiles result from mutations in AS regulatory elements of specific cancer genes or from changes in the splicing machinery (Urbanski et al., 2018). Recurrent somatic mutations in spliceosomal components frequently occur in myeloid tumors, suggesting that splicing factor (SF) alterations are a hallmark of cancer (Yoshida and Ogawa, 2014). In solid tumors, SFs exhibit copy-number variation (CNV) and/or changes in expression level, but they are rarely mutated (Urbanski et al., 2018).
One major class of SFs is the serine/arginine-rich (SR) protein family, which acts at multiple steps of spliceosome assembly and is involved in both constitutive splicing and AS (Black, 2003). The heterogeneous nuclear ribonucleoproteins (hnRNPs) are another group of SFs that are implicated in AS regulation. SR and hnRNP A/B proteins can exhibit antagonistic effects on particular exons. Both activator and repressor SFs bind directly to pre-mRNA and elicit changes in a concentration-dependent manner (Long and Caceres, 2009); thus, changes in SF levels would likely cause AS deregulation in cancer, even in the absence of mutations, and affect a network of downstream targets. Higher SF levels increase the availability of that SF, which can bind to additional exonic or intronic target sequences and directly affect the AS of target exons by recruiting or repelling the splicing machinery (Ge and Manley, 1990; Krainer et al., 1990; Wagner et al., 2016; Zahler et al., 1993; Zhu and Krainer, 2000). Alternatively, SF binding can limit the accessibility of another RNA-binding protein (RBP) that normally would repress or activate the AS of that specific exon and thus elicit the opposite effect. Conversely, decreased SF levels will also affect the AS of its targets by limiting its ability to bind target exons and by freeing SF-binding sites, which could now be occupied by other RBPs. Although SR proteins were initially described as activators promoting exon inclusion, and hnRNPs as AS repressors, transcriptomic studies suggest that members of both families can promote inclusion for some targets and skipping for others (Bradley et al., 2015; Huelga et al., 2012).
Changes in AS patterns are frequently detected in human breast tumors (Eswaran et al., 2013; Venables et al., 2008; Venables et al., 2009), yet the upstream regulators controlling these tumor-associated isoforms have not been extensively characterized. We demonstrated that the overexpression of SRSF1, an SF frequently upregulated in breast tumors (Karni et al., 2007), promotes the transformation of mammary cells in vitro and in vivo and acts by regulating spliced isoforms associated with proliferation and cell death (Anczuków et al., 2012, 2015). SRSF1 is a prototypical member of the SR protein family, which comprises 12 members (SRSF1–SRSF12) with structural similarities, containing 1 or 2 RNA recognition motifs (RRMs) and a C-terminal arginine-serine/rich (RS) domain (Long and Caceres, 2009). However, little is known about differences and redundancies in their targets and biological functions in human tissues. Multiple SR proteins may play roles in breast cancer pathogenesis, as follows: (1) SR protein levels increase during murine mammary tumorigenesis (Stickeler et al., 1999), and SRSF4, SRSF5, or SRSF6 are upregulated in human breast tumors or cell lines (Huang et al., 2007; Karni et al., 2007; Pind and Watson, 2003); (2) the increased expression of SRSF3 correlates with tumor grade in patients (Jia et al., 2010); and (3) the SR-like protein TRA2β is upregulated in breast tumors (Best et al., 2013; Watermann et al., 2006). Therefore, it is critical to characterize the differences and redundancies in SR protein targets in cancer and to assess their specificities as breast oncogenes. Here, we describe the molecular portraits of SF alterations in breast tumors, focusing on the SR protein family, and identify specific SFs that promote mammary cell transformation.
RESULTS
Comprehensive Molecular Portrait of SF Alterations in Human Breast Tumors
We systematically assessed SF mutations, CNVs, and expression changes in 960 human breast tumors from The Cancer Genome Atlas (TCGA) dataset (Figure 1). Based on previous studies reporting SF upregulation in smaller cohorts, we focused on the following: (1) 12 SR-protein family members, (2) the SR-like TRA2β, and (3) HNRNPA1, an SRSF1 antagonist, reported to be misregulated in breast tumors (Karni et al., 2007; Pelisch et al., 2012). No recurrent somatic mutations in these SFs were detected in breast tumors (Table S1A). This is in contrast to the recurrent SRSF2 mutations detected in patients with myelodysplastic syndromes or chronic myelomonocytic leukemia. These findings suggest that SF mutations are not likely to play a major role in breast cancer. Nonetheless, 57% of tumors had an alteration in at least one of the SFs analyzed, revealing frequent SF CNVs and/or expression changes in breast tumors (Figure 1). Recurrent SF alterations ranged from 2% to 15% in frequency; in particular, SRSF1, TRA2β, SRSF2, and SRFS6 are each altered in ≥10% of tumors, a substantial proportion of breast cancer patients, given that alterations in known cancer drivers ERBB2, MYC, or TP53 are detected in 17%, 27%, and 37% of tumors, respectively (Figure 1). For many SFs, the most frequent alterations are mRNA upregulation and DNA amplification, accounting for >85% of the total alterations detected for each SF, suggesting a selection for SF gain (Figure S1A). The two exceptions are SRSF4 and SRSF8, which are downregulated in 40% and 60%, respectively, of tumors with SF alteration, and upregulated in the remaining tumors (Figure S1A). Most SFs did not show a strong correlation between CNVs and expression changes (Table S1), suggesting additional layers of regulation (e.g., at the transcriptional or the post-transcriptional level). In addition, SRSF1, SRSF2, SRSF3, SRSF4, or TRA2β upregulation are enriched in basal breast tumors (Figure S1B), an aggressive cancer subtype. Finally, increased TRA2β levels are detected in 40% of triple-negative breast cancers (TNBCs) (Figure S1C), a tumor subtype associated with poor prognosis, high metastatic incidence, and lack of effective therapies.
Figure 1. SF Alterations Are Detected Frequently in Human Breast Tumors.

Graphical representation of SF alterations in TCGA human breast tumors (n = 960) sorted by frequency. CNVs and expression changes are assessed by DNA-and RNA-seq. Individual genes are represented as rows and patients as columns. Alterations in breast cancer genes BRCA1, BRCA2, TP53, MYC, and ERBB2 are in the bottom panel.
See also Figure S1.
We investigated whether alterations in SFs co-occur with or are mutually exclusive with alterations in known cancer genes, either considering any alteration type (Table S1C) or only tumors with upregulated SFs (Table S1D). We identified 23 SF alterations co-occurring with alterations in BRCA1, BRCA2, ERBB2, HRAS, MYC, PIK3CA, or TP53 genes (Table S1C). For example, co-occurring alterations include the following: (1) TP53 with SRSF3, SRSF12, and TRA2β; (2) HRAS with SRSF2; and (3) BRCA2 with SRSF12 and TRA2β (Table S1C). Considering only upregulated SFs, (1) amplified or upregulated MYC co-occurs with SRSF1, SRSF3, SRSF4, SRSF7, SRSF12, and TRA2β; (2) mutated TP53 with SRSF3, SRSF12, and TRA2β; (3) amplified or upregulated PIK3CA with TRA2β; (4) upregulated HRAS with SRSF2, SRSF3, SRSF4, SRSF7, and SRSF9; and (5) upregulated BRCA1 or BRCA2 with SRSF1, SRSF10, SRSF12, and TRA2β (Table S1D).
Furthermore, we asked whether SF alterations were correlated with alterations in other SFs, either considering any alteration type (Table S1E) or separating amplified and upregulated SFs (Table S1F and S1G). Of 94 possible pairs, 33 SF pairs had a tendency toward co-occurrence (Table S1E). SRSF1 and SRSF2 are located in a nearby region on chromosome 17, and amplifications in both genes co-occur in breast tumors (Table S1F). Other SFs located in close proximity (e.g., SRSF3 and SRSF12, or SRSF4, SRSF10 and SRSF11) do not exhibit this pattern. Focusing further on upregulated SFs (Table S1G), we identified 50 SF pairs with a tendency toward co-occurrence (e.g., SRSF1 or SRSF2 co-occurred with SRSF7, SRSF10, or TRA2β), whereas TRA2β co-occurred with SRFS3, SRSF7, SRSF10, and SRSF12 (Table S1G). Finally, for 31 co-occurring SF alteration pairs, we detected a correlation with tumor stage (Table S1E). The strongest correlation was found for alterations in both SRSF3 and SRSF7, which were enriched in T1c stage tumors.
To further determine whether defects in SF levels lead to AS alterations that could contribute to pathogenesis, we characterized the differentially spliced events (DSEs) in breast tumors with SF-high versus SF-low levels (Figure S2; Table S2A). AS changes were quantified using SpliceCore, commercial software that performs exon-centric AS analysis by mapping RNA sequencing (RNA-seq) data to a proprietary reference transcriptome. We combined SpliceCore results with an in-house bioinformatics pipeline based on rMATS (Shen et al., 2014) to filter and prioritize reproducible DSEs in terms of “differential percent spliced in” (ΔPSI) scores between SF-high versus SF-low tumors. We focused only on DSEs detected in ≥10 tumors from both groups. For SFs frequently altered, SRSF1, SRSF2, TRA2β, and SRFS6, we identified >1,000 DSEs in SF-high versus SF-low tumors (Figure S2B;Tables S2B-S2O), and 27%–47% of these were detected in ≥80 tumors (Figure S2C). The most frequent type of DSEs in SRSF1- and TRA2β-high tumors were cassette exon (CA) (Figure S2B), equally divided between inclusion and skipping events (Figure S2D). Conversely, SRSF6-high tumors were enriched in included events, including in retained introns (RIs) (Figure S2B and S2D). In addition, SRSF5- and SRSF11-high tumors each displayed >3,000 DSEs, but all DSEs were found in ≤80 tumors (Figure S2B and S2C), and these SF alterations were only detected in 3%−6% of tumors (Figure 1). Finally, SRSF3-, SRSF9-, SRSF12-, or HNRNPA1-high tumors displayed >1,000 DSEs, but most were detected in ≤80 tumors (Figure S2B and S2C).
We conducted a pairwise comparative analysis of the 14 SF tumor groups to identify shared and unique DSEs associated with SF-high levels; shared DSEs had the same ΔPSI direction (i.e., either both ΔPSI ≥10% or both ΔPSI ≤−10%). A total of 19 SF pairs shared between 20% and 45% of DSEs, while the remaining 72 SF pairs shared few DSEs (Table S2P). The largest overlap was detected between SRSF3-high and SRSF7-high tumors (Table S2P). For SFs frequently altered, SRSF1-high tumors shared 21% of DSEs with TRA2β-high tumors and 12% with SRSF2-high; TRA2β-high and SRSF2-high tumors shared 19% of DSEs (Table S2P). In addition, to determine whether SF-high tumors exhibit defects in specific gene sets, we searched for shared differentially spliced genes (DSGs), defined as any DSE within the same gene regardless of the ΔPSI direction or AS type. Here again, the largest overlap was found for SRSF3-high and SRSF7-high tumors (Table S2Q). Thus, although varying degrees of similarity and differences in AS are detected, the overall AS patterns are different in tumors with distinct SF alterations.
The finding that SFs are frequently altered in human breast tumors and are associated with the expression of distinct spliced isoforms prompted us to examine which of these SF alterations were likely to play a role in tumorigenesis.
SF Overexpression Differentially Affects Acinar Morphogenesis and Mammary Cell Proliferation
We selected eight SFs upregulated in breast tumors to systematically determine their individual roles in mammary cell transformation. Starting with non-transformed human mammary epithelial MCF-10A cells, we generated eight derivative lines, each stably overexpressing (OE) one SF using a retroviral construct containing a T7-tagged SF cDNA or an empty vector control. Expression was confirmed by qRT-PCR, western blotting, and immunofluorescence (Figures 2A, S3A, and S3B). All T7-tagged SFs were localized primarily in the nucleus, which is consistent with their role in AS. MCF-10A cells form organized growth-arrested three-dimensional (3D) structures when grown in Matrigel, a basement-membrane-like extracellular matrix, recapitulating acinar structures from the mammary gland (Debnath and Brugge, 2005). Oncogenes associated with breast cancer disrupt the organized architecture of MCF-10A acinar structures either by affecting both acinar size and organization, thus leading to the formation of enlarged dysmorphic structures (e.g., when ERBB2 or AKT are activated [Debnath et al., 2003b; Muthuswamy et al., 2001]) or by increasing acinar size only (e.g., following cyclin D1 or MYC expression [Debnath et al., 2002; Partanen et al., 2007]). We previously showed that SRSF1-OE increases cell proliferation and decreases apoptosis, leading to 1.5-fold larger acini, which is indicative of transformation in MCF-10A cells (Anczuków et al., 2012). SFs were classified into three categories according to their effects on acinar morphology compared to controls: (1) SRSF4-, SRSF6-, and TRA2β-OE promoted the formation of 3- to 4-fold larger and dysmorphic acini; (2) HNRNPA1-OE, similarly to SRSF1, increased acinar size by 2-fold, but did not disrupt acinar morphology; and (3) finally, SRSF2-, SRSF3-, and SRSF9-OE acini were not different from controls (Figures 2B and 2C). Moreover, SRSF6- and TRA2β-OE led to changes in acinar size that are detectable very early during morphogenesis and increased the number of proliferating acini (Figures S3C-S3E). Overall, our findings reveal that (1) not all SFs affect oncogenic hallmarks in this model; (2) only a subset of SFs affect acinar morphogenesis or proliferation, phenotypes indicative of cellular transformation in 3D-grown MCF-10A; and (3) distinct SFs affect different cellular phenotypes indicative of transformation in MCF-10A cells, suggesting underlying specificities and non-redundant functions of SFs, potentially mediated by their downstream targets.
Figure 2. Specificity of SF-Mediated Transformation.

(A) Expression of T7-tagged SFs in MCF-10A cells, detected by immunofluorescence using T7-tag antibody and DAPI nuclei co-stain (scale bar: 50 μm).
(B) Representative bright-field images of acinar size and morphology for control and SF-OE 3D MCF-10A cells on days 8 and 16 (scale bar: 100 μm).
(C) Quantification of acinar sizes in control and SF-OE 3D MCF-10A cells on day 16. The dot plot shows the size distribution of all of the structures and the median (horizontal line) for each condition (n = 4, >50 acini per experiment; t test, **p < 0.001, ***p < 0.0001; n.s., not significant).
See also Figure S3.
SRSF4-, SRSF6-, and TRA2β-Regulated AS Isoforms in Breast Cancer
We next characterized DSEs regulated by SFs in human mammary cells and tumors, focusing especially on the targets of SRSF4, SRSF6, and TRA2β, and defined the specificity and redundancy of these SFs. We performed RNA-seq on controls and SF-OE MCF-10A day 8 acini and identified DSEs in (1) highly proliferative and dysmorphic SRSF4-, SRSF6-, and TRA2β-OE acini and (2) SRSF2- and SRSF9-OE acini with no phenotypic difference from controls. SRSF1 targets were previously characterized by RNA-seq in MCF-10A cells (Anczuków et al., 2015) and were not investigated further here. We identified >1,500 DSEs in each condition, across multiple biological replicates (Figure 3A; Tables S3A-S3E). Almost half of these DSEs correspond to CA events, followed by RIs, alternative 5′ splice sites (A5′SSs), A3′SSs, and mutually exclusive exons (MXEs) (Figure 3A). SRSF4-, SRSF6-, and TRA2β-OE acini displayed the largest number of DSEs, while SRSF2- and SRSF9-OE acini had fewer DSEs (Figure 3A). All five SFs promoted both inclusion and skipping (Figure 3B), suggesting a dual role for SR proteins as AS activators and repressors, either directly through RNA binding or indirectly through secondary interactions; this is consistent with previous findings in mice, fruit flies, and humans (Bradley et al., 2015; Pandit et al., 2013). Analysis by AS event type revealed that all five SFs promoted relatively equal numbers of inclusion and skipping CA events (Figure S4A). SRSF2-, SRSF4-, SRSF6-, and SRSF9-OE primarily promoted RI events, whereas TRA2β-OE promoted a similar number of retained and skipped introns. The majority of A5′SS and A3′SS events promoted proximal SS usage (Figure S4A). Gene set enrichment analysis revealed that SRSF4-, SRSF6-, and TRA2β-OE affect the AS of genes that regulate key cellular processes associated with transformation (Figure S4B). We then examined the overlap across all SF-OE MCF-10A datasets, either at the AS event level (Table S3F) or the gene level (Table S3G). SRSF4-, SRSF6-, and TRA2β-OE acini, which had a similar phenotype in 3D, also exhibited the greatest overlap for both DSEs and DSGs, with 30%–40% shared DSEs (Figures 3C and S4B; Tables S3F and S3G).
Figure 3. SF Overexpression Is Associated with Differentially Spliced Events in MCF-10A.

(A) DSEs detected by RNA-seq in SF-OE versus control MCF-10A day 8 acini (n = 3; ∣ΔPSI∣ ≥ 10%, false discovery rate [FDR] < 5%, p < 0.01), sorted by AS event types. CA, cassette exon; MXE, mutually exclusive exon; RI, retained intron; A5′SS, alternative 5′SS; A3′SS, alternative 3′SS.
(B) Skipped (ΔPSI ≤ −10%) and included (ΔPSI R 10%) DSEs in SF-OE versus control are plotted by ΔPSI values for each SF for all AS event types.
(C) Overlap in DSEs for each SF pair. Bubble size is proportional to the number of shared DSEs and color indicates p values.
(D) RT-PCR validations of DSEs. A representative gel is shown, along with isoform structures. PSI for all samples and ΔPSI for significant SFs are calculated from RT-PCR (n = 3; mean ± SD; t test, *p < 0.05, n.s., not significant) and RNA-seq.
Next, we compared SF-driven DSEs identified in MCF-10A acini to DSEs detected in SF-high breast tumors (Tables S2B-S2O). A total of 173 DSEs were found in SRSF4-high MCF-10A acini and tumors, 96 DSEs in SRSF6-high, and 36 DSEs in TRA2β-high (Table S3I). Almost 40% of these DSEs affected regions with an annotated protein domain, suggesting that these AS alterations could produce novel protein isoforms in breast tumors.
We validated by RT-PCR 12 SF-regulated DSEs in SRSF4-, SRSF6-, and TRA2β-OE MCF-10A (Figure 3D) out of 21 randomly selected DSEs. Validated DSEs affect genes involved in proliferation (MKI67, APLP2), cell migration and adhesion (ARAP2, CD44, ERP29, LSR), signaling (ARHGEF11, MLPH, TCF7L2), or cytoskeleton organization (OSBPL3, RALGPS2, TLN1).
Since AS changes affecting protein domains are critical for protein function in cancer (Climente-González et al., 2017), we focused on DSEs that overlap protein domains and are therefore more likely to affect oncogenic phenotypes. DSEs co-occurring in SRSF4-, SRSF6-, and/or TRA2β-OE overlapped with annotated protein domains, compared to events unique to every SF (Figure S4D). In addition, to prioritize breast cancer-relevant biological processes, we selected genes associated with breast cancer based on human genetics and genomics studies in the Open Targets database (Carvalho-Silva et al., 2019). Gene set enrichment analysis on the subset of breast cancer-associated genes with alternatively spliced protein domains revealed that SRSF4-, SRSF6-, and TRA2β targets, but not SRSF2 and SRSF9 targets, are enriched in cancer-relevant biological processes (Table S3K). The first three SFs are therefore likely to affect the hallmarks that are indicative of mammary transformation through protein-altering AS. DSEs detected in SRSF4-, SRSF6-, and TRA2β-OE acini were enriched in genes associated with cell-cycle regulation, apoptosis, Golgi-vesicle transport, and cell adhesion (Table S3K). These results suggest a pivotal role for SRSF4, SRSF6, and TRA2β in the splicing-regulatory network that promotes mammary transformation in MCF-10A cells, which depends at least in part on discrete and effective co-regulatory interactions with one another.
SF-Regulated Pathways in Breast Cancer
We examined SF-induced gene expression changes in MCF-10A acini using Cuffdiff (Trapnell et al., 2013; Tables S4A-S4F). SRSF4-, SRSF6-, and TRA2β-OE acini exhibited the greatest number of gene expression changes, with >500 differentially expressed genes (DEGs) (Figure S4E; Tables S4B, S4C, and S4E). In contrast, SRSF2- and SRSF9-OE acini had ≤40 DEGs (Tables S4A-S4D). DEGs in SRSF4-, SRF6-, or TRA2β-OE acini were enriched in targets associated with cell proliferation, migration, cytoskeleton organization, polarity, cell signaling, or cholesterol metabolism (Tables S4H-S4J). In addition, SRSF4-, SRSF6-, and TRA2β-OE acini shared 15%–28% of DEGs (Figure S4F; Table S4F). Shared DEGs were associated with proliferation, cell cycle, cytokine-mediated signaling, cholesterol synthesis, migration, and extracellular matrix organization (Figure S4G; Tables S4K-S4M). Thus, SRSF4, SRSF6, and TRA2β not only share a similar phenotype in MCF-10A cells but also regulate the expression of shared genes and spliced isoforms that affect cellular processes associated with tumor initiation and progression.
TRA2β Cooperates with the MYC Breast Cancer Oncogene
Transformation often results from cooperation among several oncogenes. We previously demonstrated that SRSF1 cooperates with MYC (Anczuków et al., 2012), one of the most frequently altered genes in cancer. It remains unknown whether this cooperation extends to other SR proteins. Here, we generated MCF-10A cells OE selected SFs together with an inducible MYC transgene (Eilers et al., 1989). TRA2β-OE together with MYC promoted the formation of enlarged acinar structures compared to either alone (Figures 4A and 4B). In contrast, no significant differences in size or morphology were observed following OE of SRSF6 or SRSF4 with MYC (Figure 4B). Furthermore, MYC and TRA2β are co-expressed at high levels in human breast tumors and in other tumor types (Figure 4C). In addition, TRA2β mRNA levels increased 4 h after MYC induction in MCF-10A cells (Figure 4D), which is consistent with the time frame for the transcriptional activation of known MYC targets (Das et al., 2012; Jung et al., 2008; Oran et al., 2016; Yap et al., 2011). Increased TRA2β protein levels are detected at 24 h (Figure 4D), suggesting that TRA2β is likely a direct transcriptional target of MYC. Direct binding of MYC to the TRA2β proximal promoter region is further supported by genome-wide chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) data from mammary MCF-7 and MCF-10A cells (Figure 4E). Finally, MYC is amplified and/or upregulated in 51% of TRA2β-high versus 26% of TRA2β-low breast tumors (Figure 4F). Thus, our findings suggest that TRA2β-OE may be driven by MYC alterations in at least a subset of breast tumors.
Figure 4. Cooperation of TRA2β with the MYC Oncogene in Breast Cancer.

(A) Representative bright-field images of control or TRA2β-OE 3D MCF-10A acini expressing estrogen receptor ligand-binding domain (ER)-inducible MYC, activated by 4-hydroxytamoxifen (4-OHT) (scale bar: 100 μm).
(B) Acinar size distribution and median (horizontal line) of SRSF4-, SRSF6-, TRA2β-OE MYC.ER MCF-10A ± 4-OHT (n ≥ 3, >100 acini per condition; t test, *p < 0.01, n.s., not significant).
(C) Association plot showing the correlation between MYC and TRA2β expression in human tumors. Dataset identification numbers (IDs) and numbers of samples are indicated. MYC and TRA2β expression are grouped into four categories: (1) both high, (2) TRA2β high and MYC low, (3) TRA2β low and MYC high, and (4) both low.
(D) The TRA2β RNA and protein levels in MYC.ER MCF-10A +4-OHT at indicated time points are measured by qRT-PCR and western blotting (n = 3; mean ± SD; t test, *p < 0.05). RNA levels are normalized to GAPDH and HPRT; protein levels are normalized to tubulin.
(E) MYC binding to TRA2β genomic region as detected by MYC ChIP-seq experiments in MCF-7 and MCF-10A cells. Fold changes over control are calculated from pooled replicates; significant peaks are shown by rectangles.
(F) MYC amplification (AMP) or OE status inTRA2β-high versus TRA2β-low breast tumors.
Differential Role of SFs in Cell Migration and Invasion
To assess how SF-OE affects cell migration and invasion, we assayed several migratory or invasive phenotypes using 2D and 3D assays in non-invasive MCF-10A cells. In 3D collagen-Matrigel assays, the formation of intra-acinar bridges and multicellular protrusions, indicative of invasive behavior, was observed in 25%–50% of SRSF4-, TRA2β-, HNRNPA1-, and SRSF6-OE acini, but in <1% of control acini (Figures 5A and 5B). SRSF4-, TRA2β-, and SRSF6-OE also increased intra-acinar cell movement as detected by live-cell imaging (Figure S5A), and SRSF6-OE increased cell migration in 2D transwell assays (Figure S5C). Despite the lack of effect of SRSF9-OE on acinar morphology or size, SRSF9-OE cells migrated faster in wound-healing and transwell migration assays (Figure S5B and S5C) and exhibited invasive behavior in 3D assays (Figures 5A and 5B). These results suggest that SF-OE can affect cell invasion without affecting cell proliferation in MCF-10A cells.
Figure 5. Differential Roles of SFs in Cell Migration and Invasion.

(A) Representative bright-field images of acinar morphology for control or SF-OE 3D MCF-10A cells grown in Matrigel-collagen invasion assay at day 8. Multicellular protrusions and inter-acinar bridges are indicated by arrowheads (scale bar: 50 μm).
(B) Percentage of invasive versus non-invasive acini at day 8 (n = 3, >50 acini per condition).
(C) SF levels in MDA-MB231 expressing scrambled CTLsh or SF-targeting shRNAs are quantified 72 h after DOX treatment by western blotting using SF-specific antibodies and normalized to tubulin. Percentage of SF in +DOX is normalized to the corresponding −DOX (n = 3; mean ± SD).
(D and E) Migration of CTLsh or SFsh MDA-MB231 ± DOX in 2D wound-healing (D) or transwell assay
(E). Distribution and median (horizontal line) for each condition +DOX normalized to −DOX (n = 3; t test, *p < 0.05, **p < 0.005).
(F and G) Representative bright-field images of CTLsh or TRA2βsh MDA-MB231 cells, grown in 3D ± DOX (scale bar: 100 μm). DOX is added from either day 0 (F) or day 8 (G).
See also Figures S5 and S6.
To complement this analysis, which identified SFs that increase migratory properties in non-transformed MCF-10A cells, we asked whether SF levels are required to maintain migratory phenotypes in established breast cancer cells with metastatic potential (Figures 5C-5G). We focused on a TNBC model, as TRA2β is upregulated in 40% of TNBC tumors. We examined how SRSF4, SRSF6, SRSF9, and TRA2β, which increased migration and invasion in MCF-10A cells when overexpressed, affected the invasion of human MDA-MB231 TNBC cells. We established stable cell lines with a doxycycline (DOX)-inducible short hairpin RNA (shRNA) expression system that enables the tracking of retroviral transduction and shRNA induction through two fluorescent reporters—constitutively expressed GFP and DsRed expressed with the shRNA (Zuber et al., 2011). Six shRNAs were tested for each SF and the two shRNAs with the strongest knockdown (KD) were used further (Figure 5C). SF-KD did not decrease cell-doubling time (Figure S5D and S5E), allowing us to define the effect of SF-KD on cell invasion independently of its potential effect on proliferation. TRA2β-KD decreased cell migration in wound healing and transwell migration assays (Figures 5D, 5E, S5F, and S5G). A milder phenotype was observed for SRSF4-KD, which decreased migration in wound healing assays only (Figure 5D). The relatively modest KD efficiency for SRSF6 and SRSF9 (Figure 5C) may limit our ability to detect their role in cell migration and invasion. However, we noted that despite modest KD, both SRSF9 shRNAs decreased transwell migration to similar levels as TRA2β shRNAs (Figures 5E and S5G). SRSF9sh2+DOX cells, but not SRSF9sh1+DOX cells, migrated less in wound healing assays (Figure 5D), possibly because control SRSF9sh1−DOX cells migrated less on plastic than did SRSF9sh2−DOX cells (Figure S5F). Finally, we assessed SF-KD in MDA-MB231 cells grown in 3D culture in Matrigel, which form stellate structures representative of their invasive behavior. When shRNAs were induced from day 0, TRA2β-KD prevented the formation of stellate invasive structures and promoted the formation of round acinar-like structures lacking invasive behavior (Figures 5F and S5H-S5J). SRSF6-KD also prevented the formation of invasive structures, whereas SRSF4- and SRSF9-KD decreased the number of stellate structures and their size (Figure S5H and S5I). Finally, using the inducible system, we tested whether TRA2β is required to maintain cell invasion. TRA2β-shRNA induction on day 8, after the formation of invasive structures, was sufficient to reverse the phenotype and promote the formation of non-invasive acinar-like structures (Figure 5G). The effect was dose dependent, as the weaker shRNA, TRA2βsh1, had an intermediate phenotype compared to TRA2βsh2 (Figure 5G). We further demonstrate that TRA2β-KD prevented the formation of stellate invasive structures in another TNBC cell line, SUM159PT (Figures S6A and S6B). In summary, our findings suggest that specific SFs play a role in controlling cell migration in MCF-10A cells or in maintaining the invasive potential of breast cancer cells. In particular, TRA2β expression was sufficient to initiate invasion in mammary MCF-10A cells and was required to maintain invasiveness in MDA-MB231 and SUM159PT cells.
TRA2β Regulates the AS of Targets Implicated in Breast Cancer Metastasis
To uncover TRA2β-regulated targets controlling cell invasion, we performed RNA-seq and identified DSEs by comparing day 8 3D-grown MDA-MB231 cells across the following three conditions: (1) TRA2βsh1+DOX versus −DOX, (2) TRA2βsh2+DOX versus DOX, and (3) CTLsh+DOX versus −DOX. CTLsh+DOX versus −DOX cells were used to identify DOX-induced DSEs; in other words, significant DSEs from TRA2βsh+DOX versus −DOX were discarded if they were also significant and altered in the same direction in CTLsh+DOX versus −DOX (Table S5A). Following the exclusion of DOX-induced events, we identified >3,500 DSEs in TRA2βsh2+DOX, and >2,000 in TRA2βsh1+DOX (Figure 6A; Table S5B and S5C); the number of DSEs associated with the stronger shRNA TRA2βsh2 was higher than the number associated with the weaker shRNA. TRA2βsh1+DOX and TRA2βsh2+DOX cells shared 582 DSEs and 691 DSGs (Tables S5E and S5F). TRA2β-KD promoted primarily CA events, favoring skipping over inclusion (Figures 6A and 6B). TRA2β-KD affected the AS of genes that regulate key cellular processes associated with transformation (Figure 6C). Analysis of breast cancer-associated genes with alternatively spliced protein domains revealed that TRA2β-regulated targets are enriched in genes associated with G2/M regulation, protein deubiquitination, DNA repair, cell migration, and apoptosis (Table S5G and S5H). These biological processes were also highly ranked in TRA2β targets in MCF-10A cells, albeit represented by a different set of genes (Table S5H). These observations presumably reflect differences in the isoforms that are associated with TRA2β function in mammary transformation in MCF-10A cells versus its role in tumor maintenance and invasion in MDA-MB231 cells.
Figure 6. TRA2β-KD Promotes Changes in Spliced Isoforms in 3D MDA-MB231 Cells.

(A) DSEs detected by RNA-seq in TRA2βsh2 or TRA2βsh1 MDA-MB231 cells, grown in 3D ± DOX, at day 8 (n = 3; ∣ΔPSI∣ ≥ 10%, FDR < 5%, p < 0.01).
(B) Skipped (ΔPSI ≤−10%) and included (ΔPSI ≥ 10%) DSEs in TRA2βsh2 or TRA2βsh1 +DOX versus −DOX plotted by ΔPSI values.
(C) Gene set enrichment analysis for DSEs in TRA2βsh2 or TRA2βsh1 MDA-MB231 cells showing the top 10 hallmark gene sets.
(D) RT-PCR validations of selected DSEs. A representative gel is shown, along with isoform structures. PSI for all samples and ΔPSI for TRA2βsh2 are calculated from RT-PCR (n = 3; mean ± SD; t test, *p < 0.05, **p < 0.005, ***p < 0.0005, n.s., not significant) and RNA-seq.
(E) PSI for DSEs detected both in TRA2β-high versus TRA2β-low breast tumors (n = 146 and 449) and MDA-MB231 cells.
See also Table S5.
We validated 16 TRA2β-regulated DSEs by RT-PCR in 3D-grown MDA-MB231 cells (Figure 6D) out of 21 randomly selected DSEs. Validated DSEs affect genes associated with mitosis (ADAL), migration and adhesion (DHX32, IFI44, KMT5B, KIF23, TANK), signaling (FAM126A, CCDC88C, GNAS, NCOR2, TANK), AS (TRA2α), or metabolism (ADAL, FARSB, IDI1, UROS, PFKM). Furthermore, we demonstrated that 5 out of 6 TRA2β-regulated DSEs detected in MDA-MB231 cells are also differentially spliced in SUM159PT cells with TRA2β-sh2 (Figures S6A-S6C). ΔPSI magnitude is smaller in SUM159PT compared to MDA-MB231, which is consistent with a lower TRA2β-KD efficiency in SUM159PT.
TRA2β-KD also affected gene expression in 3D MDA-MB231 cells (Table S6), with >1,400 up- and >1,200 downregulated genes in TRA2βsh2 (Table S6B), whereas the weaker hairpin had an intermediate effect (Table S6C). DEGs were associated with Wnt signaling, cell polarity, cell cycle, and transcriptional control (Table S6D). Differential AS also led to changes in gene expression, with 312 genes affected both at the AS and the gene expression levels (Table S6E).
To further uncover spliced isoforms that are relevant to human breast tumors, we focused on DSEs detected both in MDA-MB231 cells and breast tumors with high TRA2β levels. We identified 32 shared DSEs, defined as DSEs regulated in opposite directions—in other words, ΔPSI ≥10% in TRA2βsh2+DOX versus −DOX MDA-MB231 and ΔPSI ≤−10% in TRA2β-high versus TRA2β-low tumors, and vice versa (Figure 6E; Table S5K). These DSEs affect genes associated with cell invasion and metastasis (ARVCF, DOCK7, TANK, TMPO, CCDC88C, SEC31A), cell cycle (TMPO), signaling (FAM126A, IRF3, NMU), or DNA replication and repair (BABAM1) (Table S5I). Finally, we assayed the function of two TRA2β-regulated isoforms, TANK+ex3 and CCDC88C+ex26, both included at higher levels in MDA-MB231 and breast tumors with TRA2β-high levels (Figures 6D-6F). Both TANK and CCDC88C genes were implicated in cell invasion (Ishida-Takagishi et al., 2012; Stellzig et al., 2013), although the function of these specific isoforms had yet to be characterized. MDA-MB231 cells expressing isoform-specific shRNAs targeting the TANK+ex3 isoform formed smaller and less invasive structures than control cells (TANK+ex3 sh +DOX versus −DOX), at least partially recapitulating the phenotype of TRA2β-KD (Figures S6E-S6G). Isoform-specific KD of CCDC88C+ex26 had a weaker phenotype, with only sh2 leading to smaller and less invasive structures (Figures S6F and S6G). Overall, our findings suggest that TRA2β regulates the AS of multiple genes linked with cancer and/or metastasis in both breast cancer cell lines and patients’ tumor tissues.
TRA2β Plays a Role in Breast Cancer Metastasis In Vivo, and Its Expression Correlates with Clinical Outcomes in Breast Cancer Patients
To assess the effect of TRA2β-KD on metastasis in vivo, TRA2βsh2 or CTLsh MDA-MB231 cells were orthotopically injected in the mammary gland of NOD.Cg-Prkdcscid Il2rgtm1Wjl/ SzJ (NSG) female mice (Figure 7A). Primary tumor and metastasis formations were monitored by luciferin bioluminescence imaging and histology. TRA2β-KD did not prevent the formation of primary mammary tumors (Figures 7B and S7A-S7C), nor did it affect their size, compared to non-induced (TRA2βsh2+DOX versus −DOX) and induced controls (TRA2βsh2+DOX versus CTLsh+DOX) (Figure S7B). However, TRA2β-KD animals had a decrease in lung and liver metastatic burden compared to non-induced and induced controls (Figures 7C and 7D), and in lung, liver, or lymph node macro-metastases (Figure S7D). TRA2β-KD reduced the number of metastatic foci, but had no effect on foci size (Figures S7B-S7J). In addition, TRA2β-KD also reduced lung metastasis formation in an experimental metastasis model in which cancer cells are injected in the tail vein (Figures S7K-S7N), modeling the latter steps of the metastatic cascade (e.g., survival in circulation, extravasation, colonization of a distant site). Finally, clinical data for human breast tumors revealed that TRA2β-high levels are associated with a decrease in overall survival (Figure 7E) and in distant metastasis-free survival (Figure 7F) in multiple patient cohorts. Overall, our data strongly suggest a role for TRA2β and its targets in the regulation of breast cancer metastasis initiation and maintenance.
Figure 7. TRA2β Plays a Role in Metastasis In Vivo in a TNBC Mouse Model and in Breast Cancer Patients.

(A) CTLsh or TRA2βsh2MDA-MB231 cells are injected into the mammary fat pad of NSG mice; primary tumors and metastasis are monitored by bioluminescence imaging.
(B) Bioluminescence detection of primary tumors and metastasis in mice injected with CTLsh or TRA2βsh2 MDA-MB231 cells ± DOX at 8 weeks post-injection.
(C) Representative H&E lung and liver sections of mice injected with TRA2βsh2 MDA-MB231 cells ± DOX (scale bar: 1 mm). Metastatic areas are circled in blue.
(D) Quantification of metastasis burden in mice injected with CTLsh or TRA2βsh2 MDA-MB231 cells ± DOX (n ≥ 4; t test, *p < 0.01, **p < 0.001, n.s., not significant).
(E and F) Correlation between TRA2β expression and overall survival (E) or distant metastasis-free survival (F) in cohorts of breast cancer patients stratified by TRA2β levels. Cohort size, dataset, and probe ID, and corrected p value (log-rank test) are indicated.
See also Figure S7.
DISCUSSION
SF Alterations in Mammary Cells Have Distinct Functional Consequences
Abnormal AS has emerged as a hallmark of human tumors (Urbanski et al., 2018). Defects in SFs are associated with many tumor types (Dvinge et al., 2016), yet the functional role of most SFs in tumorigenesis remains poorly characterized. Here, we identify alterations in the SR protein family and their frequency in human breast tumors. We then use breast cancer models to define the functional consequences of these SF alterations and identify their targets, uncovering distinct functions for each SR protein in mammary tissues. Our analysis of >900 human breast tumors revealed that SFs are frequently altered, but exhibit distinct alteration patterns and subtype specificity. In addition to the breast cancer oncogene SRSF1 (Anczuków et al., 2012), TRA2β, SRSF2, and SRSF6 are each overexpressed in ≥10% of tumors, all subtypes combined. TRA2β-, SRSF12-, SRSF3-, and SRSF2-high tumors represent >20%–40% of TNBCs, the most aggressive breast cancer subtype. Tumors with high levels of a given SF exhibit unique DSEs, and many DSEs are recurrently detected in >100 tumors. In addition, tumors with alterations in distinct SFs share relatively low numbers of DSEs, with the exception of SRSF3-high and SRSF7-high tumors, which share 46% of their DSEs. Our findings thus suggest that distinct SFs have different targets and biological functions in breast tumors.
Not all SFs confer oncogenic properties in our model. Although SRSF2 is upregulated in 13% of breast tumors, SRSF2-OE did not affect the phenotypes measured in MCF-10A cells. Recurrent mutations in SRSF2 are found in human hematologic malignancies, but not in other tumor types (Urbanski et al., 2018). Functionally, blood lineage-specific expression of Srsf2P95H causes defective hematopoiesis, and SRSF2 mutations alter its binding preference, thereby altering its target recognition (Kim et al., 2015). Our findings demonstrate that changes in SRSF2 levels trigger relatively few AS alterations in MCF-10A cells and that SRSF2 may not contribute to the early steps of mammary transformation. Our study also suggests a restricted role for SRSF9 in mammary tumorigenesis, which when overexpressed in MCF-10A cells increases cell migration and invasion, but does not affect cell proliferation, apoptosis, or acinar architecture. Consistently, SRSF9 plays a role in cell invasion in bladder cancer (Yoshino et al., 2012), and SRSF9-OE promotes β-catenin accumulation and activates Wnt signaling (Fu et al., 2013), thus possibly linking SRSF9 and cell invasion regulation. In summary, our findings reveal that SRSF4, SRSF6, and TRA2β play a role in both mammary transformation and metastasis, whereas SRSF9 affects only cell invasion in the models tested. Thus, in mammary cells, distinct SFs may be involved in each of the steps of tumorigenesis—some affecting cell proliferation, others invasion, and a subset promoting both, either directly or indirectly through downstream targets.
The AS Repertoire of SR Proteins in Mammary Cells Provides a Functional Link to Their Role in Breast Cancer
Our analysis reveals that in MCF-10A cells, SRSF4, SRSF6, and TRA2β regulate not only shared AS isoforms but also a set of shared genes associated with biological processes involved in transformation and metastasis. Several SF-induced AS isoforms detected in this study (ARHGEF11, RALGPS2, MLPH, CD44, and APLP2) were previously associated with the epithelial-mesenchymal transition (EMT) (Brown et al., 2011; Harvey et al., 2018; Shapiro et al., 2011; Warzecha et al., 2010), which is consistent with the invasive phenotype of SF-OE MCF-10A cells. In addition, several validated DSEs have been linked with cell invasion or proliferation: (1) ARHGEF11+ex38 is found in invasive basal breast cancer cells, but not in non-invasive luminal cells, and is required for cell migration and growth in vitro and in vivo (Itoh et al., 2017). The ARHGEF11+ex38 isoform is unable to interact with tight-junction protein ZO-1 and is localized away from cell-cell junctions (Itoh et al., 2017). (2) The AS switch from CD44v to CD44s isoforms is associated with tumor progression (Chen et al., 2018), and CD44s isoforms can potentiate Akt activation and promote cell survival (Brown et al., 2011). (3) Lipolysis-stimulated lipoprotein receptor (LSR) expression is linked with breast cancer cell migration, and differences in its cellular localization are associated with patient outcomes (Reaves et al., 2017). Skipping of exon 5, encoding the transmembrane domain, could affect LSR localization and activity. (4) TCF7L2 encodes a protein that interacts with β-catenin and plays a role in Wnt signaling. The TCF7L2+ex4 isoform displays a reduced ability to activate Wnt/β-catenin targets (Weise et al., 2010). (5) The MKI67Δex7 isoform lacks 360 amino acids compared to the full-length protein, and is associated with nutrient-starvation response (Chierico et al., 2017).
A subset of SR proteins can shuttle between the nucleus and the cytoplasm, including SRSF1, SRSF3, SRSF4, SRSF6, SRSF7, and SRSF10 (Cáceres et al., 1998; Cazalla et al., 2002; Sapra et al., 2009), and this ability may depend on the differentiation state of the cell (Botti et al., 2017). Moreover, several SR proteins have splicing-independent functions— for example, SRSF1, SRSF3, and SRSF7 regulate mRNA export (Huang et al., 2004; Müller-McNicoll et al., 2016); SRFS1 and SRSF2 are implicated in nonsense-mediated mRNA decay (Aznarez et al., 2018; Sato et al., 2008; Zhang and Krainer, 2004); SRSF1 regulates translation by interacting with components of the mammalian target of rapamycin (mTOR) pathway (Maslon et al., 2014; Michlewski et al., 2008; Sanford et al., 2004, 2008); and SRSF3, SRSF5, SRSF6, and SRSF7 affect the translation of viral RNA (Bedard et al., 2007; Fitzgerald and Semler, 2011,2013; Swanson et al., 2010; Swartz et al., 2007). In the present study, we cannot exclude the possibility that the described SF-associated phenotypes result from a dual role of the SF in AS and in mRNA export or translation, or other as yet to be defined functions. Potential non-splicing-related functions of SRSF4, SRSF6, and TRA2β in human cells warrant further investigation.
TRA2β Controls Mammary Cell Transformation and Breast Tumor Metastasis
TRA2β is overexpressed in many tumor types, and a role in tumorigenesis has been proposed (Best et al., 2013). Here, we provide a characterization of the function of TRA2β in promoting mammary cell transformation and metastasis in vitro and in vivo. TRA2β-OE tumors represent >40% of TNBCs, an aggressive breast cancer subtype with high metastasis incidence. Our results suggest that modulating TRA2β levels may represent a viable therapeutic option, even for patients with advanced metastatic disease.
In MDA-MB231 cells, TRA2β controls the AS of target genes associated with cell invasion, Wnt signaling, cell polarity, cell cycle, and transcriptional control. For example, high TRA2β levels in both TNBC cells and human tumors are associated with the AS of TANK, a member of the tumor necrosis factor receptor-associated factor protein family, which has been linked with cell proliferation and migration in brain tumors (Stellzig et al., 2013). The inclusion of TANK exon 3 encodes 58 additional amino acids in the N-terminal domain, which could affect the function of the protein. Using isoform-specific KD, we demonstrate that the exon 3-containing isoform is important for cell invasion in MDA-MB231 cells. Another TRA2β-regulated target, CCDC88C, encodes a signal transducer and Wnt signaling regulator, which can trigger cell invasion and drive metastasis (Ishida-Takagishi et al., 2012). CCDC88C has multiple spliced isoforms that differ in their ability to enhance cell invasiveness (Ear et al., 2019). The isoform detected here includes exon 26, which introduces a premature termination codon (PTC), truncating 493 amino acids and producing a transcript that could be degraded by nonsense-mediated mRNA decay (NMD). In addition, TRA2β-KD alters the AS of KMT5B, encoding the lysine methyltransferase SUV420H1, which targets histone H4. Reduced H4K20me3 levels are detected in breast tumors, and SUV420H1-OE can suppress cell invasion (Yokoyama et al., 2014). TRA2β also regulates the AS of KIF23, which encodes a kinesin-like protein involved in microtubule formation and movement. High KIF23 expression is associated with decreased overall survival and distant metastasis-free survival in breast cancer patients, and KIF23-KD suppresses the proliferation of TNBC cells (Wolter et al., 2017). Inclusion of exon 18 alters KIF23 cellular localization and is associated with longer survival in liver cancer patients (Sun et al., 2015). Other TRA2β targets associated with cell invasion include DOCK7, a gene regulating Rac activation and ErbB2 signaling (Yamauchi et al., 2008), and DPHS, which activates RhoA signaling, leading to increased cell motility and invasion in vitro and increased tumor growth in vivo (Muramatsu et al., 2016). In addition, several TRA2β-regulated targets are associated with metabolism and cell signaling: inclusion of FAM126A exon 11 leads to a C-terminally truncated protein that could have distinct biological functions and affect phosphatidylinositol 4-kinase (PI4K) localization (Baskin et al., 2016). PI4K is frequently amplified in breast tumors, and its expression promotes multi-acinar formation in MCF-10A cells (Pinke and Lee, 2011); AS of GNAS exon 3 regulates the expression of long and short GNAS isoforms, which display differences in localization and activities (Bastepe, 2007). GNAS amplification is found with breast cancer, and the expression of the long isoform leads to enhanced cyclic AMP (cAMP) signaling (Garcia-Murillas et al., 2014). The specific functions of each of the TRA2β-regulated isoforms remain to be determined. One may hypothesize that each TRA2β-regulated isoform contributes to a distinct aspect of TRA2β-mediated transformation, similarly to SRSF1 targets in breast cancer (Anczuków et al., 2015), and that SF-mediated transformation results from activating an oncogenic AS program, in the same way that a transcription factor activates hundreds of transcriptional targets.
The molecular mechanisms through which TRA2β levels are altered in tumors remain poorly understood. TRA2β is upregulated in 13% of breast tumors, but only 11% of these tumors show amplification at the gene locus. Our findings suggest that TRA2β is a direct transcriptional target of the MYC oncogene, that both genes are frequently co-expressed in breast tumors, and that their co-expression increases mammary acinar size. Furthermore, we demonstrate that 51% of TRA2β-high breast tumors exhibited MYC amplification or upregulation, suggesting that MYC transcriptional activation accounts for at least half of the TRA2β alterations. These findings, together with previous studies (Das et al., 2012; David et al., 2010; Hsu et al., 2015; Koh et al., 2015), show that MYC plays a critical role in regulating the expression of several SFs linked to tumorigenesis and provides a rationale for targeting AS in MYC-driven tumors.
RBPs as Breast Cancer Drivers
This study focused on the SR protein family and several related SFs. However, alterations in other SFs are reported in breast tumors and could contribute to transformation. In breast cancer, SF mutations remain rare; mutations in SF3B1 are detected in only 1.6% of breast tumors, all subtypes combined, whereas they are found in 20% of uveal melanomas and 70%–80% of myelodysplastic syndromes (Urbanski et al., 2018). Moreover, SF3B1 mutations are associated with luminal tumors, a breast cancer subtype that has a better prognosis than TNBC. Similarly, U2AF1, SRSF2, RBM10, QKI, and FUBP1 mutations are each found in <0.5% of TCGA breast tumors (Urbanski et al., 2018). Thus, in breast cancer, SF mutations may not represent the most valuable markers for tumor progression or therapeutics targets. Conversely, changes in SF levels are frequently detected and likely play a role in tumorigenesis. Beyond SR proteins, other SFs are dysregulated in breast tumors; hnRNPK, hnRNPM, PTBP1, and RBM10 are upregulated and RBM5 is both upregulated and downregulated (Urbanski et al., 2018). hnRNPM regulates the EMT in epithelial cells, in part by promoting the AS of CD44s and altering transforming growth factor β (TGF-β) signaling (Xu et al., 2014). PTBP1-OE increases cell proliferation, anchorage-independent growth, and invasion (He et al., 2014; Wang et al., 2008), whereas PTBP1-KD has the opposite effect (Wang et al., 2018). Furthermore, spliceosome inhibition with shRNAs or small molecules inhibits the growth of TNBC cells and patient-derived xenografts (Chan et al., 2017). Whether SF alterations trigger changes in shared DSEs or affect genes converging on pathways that lead to transformation remains to be determined. In addition, it is not yet known which SF alterations are drivers of tumor initiation and which result from a hyper-proliferative cell state. Our study reveals that specific SF alterations can contribute to mammary transformation and/or metastasis and also identifies two SFs upregulated in breast tumors but not sufficient by themselves to promote transformation in the models tested. These findings suggest that not all SF alterations are oncogenic in the mammary context or that they may require additional hits to cooperate in transformation. Given that tumors often exhibit alterations in multiple SFs, future studies should aim to understand how individual SFs regulate one another and whether they cooperate in transformation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Olga Anczukow (olga.anczukow@jax.org).
Plasmid and cell lines generated are available from the Lead Contact without restrictions, with reasonable compensation by requestor for shipping. Request for access to the SpliceCore software platform may be submitted to Envisagenics. Access to SpliceCore will be governed by the terms of a negotiated agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Cell Lines
Human mammary epithelial MCF-10A cells were obtained from the laboratory of Senthil K. Muthuswamy (Cold Spring Harbor Laboratory) and were maintained as described in DMEM/F12 supplemented with 5% horse serum, 20 ng/ml EGF (Peprotech), 0.5 mg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma), 10 μg/ml insulin (Sigma) and 1% penicillin streptomycin (Sigma) (Debnath et al., 2003a). Breast cancer cells lines, MDA-MB231-luciferase-GFP and SUM159PT were obtained from the laboratories of Min Yun (University of Southern California), and Steve Ethier (University of Southern California) respectively. MDA-MB231-luciferase-GFP cells were maintained as described (Yu et al., 2009). SUM-159PT were maintained in Ham’s F-12 (Sigma) supplemented with 5% FBS, 1% penicillin streptomycin (Sigma), 5 μg/ml insulin (Sigma) and 1 μg/ml hydrocortisone (Sigma). Cells were grown at 37°C under a humidified atmosphere with 5% CO2. Cells were routinely authenticated by STR profiling, tested negative for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza), and cell aliquots from early passages were used. All cell lines used here where established from female subjects.
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ female mice were obtained from The Jackson Laboratory and housed in Cold Spring Harbor Laboratory facilities. All mice used in these studies were female. Mice were group housed under a 12:12 light/dark cycle with access to food and water ad libitum. All animals were handled in accordance with Cold Spring Harbor Laboratory IACUC and AAALAC approved protocols and other ethics guidelines.
METHOD DETAILS
Plasmids
T7-tagged SRSF2, SRSF3, SRSF4, SRSF6, SRSF9, HNRNPA1 and TRA2β cDNA were subcloned from pcGT plasmids (Cáceres et al., 1997) into a pWZL-Hygro retroviral vector (a gift from S. Lowe, Cold Spring Harbor Laboratory) as described (Anczuków et al., 2012). PWZL-T7-SRSF1 (Anczuków et al., 2012) and pBabe-Puro-MYC.ER (Eilers et al., 1989) were described previously. SF-targeting shRNAs were designed using DSIR and sensor rules (Table S7A), and were cloned into TRMPV-Neo retroviral doxycycline-inducible shRNA expression vectors as described (Zuber et al., 2011). All vectors and inserts were verified and authenticated by Sanger sequencing.
Cell Culture and Cell Lines
Populations of MCF-10A cells expressing either T7-tagged SRSF1, SRSF2, SRSF3, SRSF4, SRSF6, SRSF9, HNRNPA1 or TRA2β cDNA, with or without MYC.ER overexpression, were generated by retroviral transduction and selection with 2μg/ml puromycin or 200 μg/ml hygromycin as described (Anczuków et al., 2012). Population of MDA-MB231-luciferase-GFP or SUM-159PT cells expressing rTTA3-puro and SF-shRNA-TRMPV-Neo were generated by retroviral transduction and selection with 2μg/ml puromycin or 1mg/ml G418 as described (Zuber et al., 2011).
Three-Dimensional Morphogenesis Assay
MCF-10A or MDA-MB231 stable cell lines were seeded on an 8-well glass chamber slide coated with Matrigel Growth Factor Reduced (BD Biosciences) as described (Debnath et al., 2003a) at a density of 5,000 cells per well and maintained in their respective media. At least 100 acini or structures were imaged at indicated time points using a Zeiss Axiovert 200M microscope and AxioVision 4.5 software (Zeiss). MYC.ER acini were stimulated with 1 μM 4-hydroxy-tamoxifen (Sigma) on day 3, and the medium with inducer was replaced every 3 days, as described (Zhan et al., 2008). For shRNA induction, MDA-MB231 or SUM159PT cell media was supplemented with 1-2 mg ml−1 doxycycline at the indicated time points, and the media was replaced every 3 days. For high-content imaging of 3D structures, 6,000 MDA-MB231 or 8,000 SUM159PT cells were plated per well in 48-well plates, and fluorescent images were acquired using a high-content imaging Opera Phoenix instrument (Perkin Elmer) and assembled using the Harmony software (Perkin Elmer) as a maximum projection image composed of 30-35 Z stack images taken every 55 μm. For downstream RNA or protein extraction from 3D cultured MCF-10A or MDA-MB231 cells were washed with PBS, and Matrigel was dissolved by incubating slides at 4°C in Cell Recovery Solution (BD).
Immunofluorescence
All procedures were performed as described (Anczuków et al., 2012). Microscopy was performed on a Zeiss Axiovert 200M instrument using the ApoTome imaging system (Zeiss) or the Dragonfly Spinning Disk system (Andor). T7 (CSHL antibody facility), cleaved caspase-3 (Cell Signaling) and ki67 (Zymed) primary antibodies were used at 1/50, 1/100 and 1/100 dilutions, respectively. Alexa Fluor 568 anti-mouse and 488 anti-rabbit secondary antibodies (Invitrogen) were used at 1/500 dilution. Acini were scored positive for ki67 when at least five cells within the acini were stained and positive for cleaved caspase-3 when at least one cell in the lumen was stained.
Quantitative RT-PCR Analysis
3D cultured MCF-10A cells were harvested as described above and RNA was extracted using an RNAeasy kit (QIAGEN) including DNase I treatment. 1 μg of RNA was reverse-transcribed with Superscript III reverse transcriptase (Invitrogen). QPCR was used to amplify endogenous transcripts with the SF-specific primers listed in Table S7B using cDNA corresponding to 5ng of RNA. QPCR was performed with iTaq Universal SYBR green Supermix (Bio-Rad) in 384-well plates (Life Technologies) using a ViiA7 Real-Time PCR system (Life Technologies) per manufacturer instructions. Results were analyzed with QuantStudio Real-Time PCR software, and SF expression was normalized to housekeeping genes GAPDH and HPRT.
Western Blot Analysis
Cells were lysed in Laemmli buffer (50 mM Tris-HCl pH 6.2, 5% (v/v) β-mercaptoethanol, 10% (v/v) glycerol, 3% (w/v) SDS). Equal amounts of total protein were loaded on a 12% SDS-polyacrylamide gel, transferred onto a nitrocellulose membrane (Millipore) and blocked in 5% (w/v) milk in Tween 20-TBST (50 mM Tris pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween 20). Blots were incubated with anti-T7 (EMD Millipore #69522-3), anti-SRSF4 (Bethyl Laboratories #A303-670A), anti SRSF6 (CSHL antibody facility AK9-156), anti-SRSF9 (CSHL antibody facility #AK251-24), anti-TRA2B (Abcam #ab31353), or anti-β Tubulin III (Genescript #A01203-40) primary antibodies. IR-Dye 680 anti-mouse or IR-Dye 800 anti-rabbit immunoglobulin G (IgG) secondary antibodies (LI-COR) were used for infrared detection and quantification with an Odyssey imaging system (LI-COR).
RNA-Sequencing Library Preparation
3D cultured MCF-10A or MDA-MB231 cells were harvested as described above and total RNA was extracted using an RNAeasy kit (QIAGEN) including DNase I treatment. RNA libraries were prepared and barcoded using a TrueSeq stranded mRNA kit with polyA selection (Illumina), and quantified using a Bioanalyzer DNA 1000 chip (Agilent). Equal amounts of libraries were pooled (3 libraries per lane) and sequenced as 101bp (MCF-10A) or 150bp (MDA-MB231) paired-ended reads on an Illumina HiSeq instrument at >80-100 million reads per library. At least 3 independent libraries were generated for each experimental condition.
RT-PCR Splicing Event Validation
MCF-10A, MDA-MB231 or SUM159PT cells were harvested as described above and RNA was extracted using an RNAeasy kit (QIAGEN) including DNase I treatment. 1 μg of total RNA was reverse-transcribed with Superscript III reverse transcriptase (Invitrogen). Semiquantitative PCR was used to amplify endogenous transcripts with the primers listed in Table S7C with cDNA from 5-20ng of RNA. Optimal PCR conditions were defined for each primer pair by testing amplification from 26-30 cycles to select semiquantitative conditions. PCR products were separated by 2% agarose gel stained with SYBRSafe (Invitrogen), and bands were quantified with a ChemiDoc MP Imaging System (Bio-Rad). The ratio of each isoform was first normalized to the sum of the different isoforms, and changes were then expressed as the fold increase compared to the levels obtained for cells or acini expressing the control vector.
Wound-Healing Assays
250,000 MCF-10A or 100,000 MDA-MB231 cells were grown in 6-well plates for 48 hours. For shRNA induction, the media was supplemented with 2μ ml−1 doxycycline at 24 hours before the start of the experiment and maintained until the end. A scratch was performed in the confluent monolayer with a P200 tip and the wells were washed twice with media. The same field was imaged with a Zeiss Observer microscope at 0h and 16h after induction of the wound. The size of the gap was measured using the Axiovision digital image processing software (Zeiss).
Transwell Migration Assays
150,000 MCF-10A or 25,000 MDA-MB231 cells in serum-free media were seeded on top of an 8-μm PET membrane transwell (BD Biosciences) in a 24-well format and allowed to migrate into the lower compartment containing complete media with serum and growth factors for 16 or 4 hours for MCF-10A or MDA-MB231 cells, respectively. For shRNA induction, the media was supplemented with 2μ ml−1 doxycycline at 24 hours before the start of the experiment and maintained until the end. After removal of the cells on top of the filter, the remaining cells were fixed with 5% formalin, permeabilized with 0.5% Triton X-100 and stained with DAPI. DAPI-positive nuclei were imaged using a Zeiss Observer microscope, and counted using the ImageJ digital image processing software.
3D Invasion Assays
Collagen-matrigel invasion assays were performed as described (Xiang and Muthuswamy, 2006). Briefly, 5,000 MCF-10A cells were seeded on a 1:2 mix of collagen:Matrigel in 8-well glass slide chamber. Media was replaced every 4 days, and acini were imaged at day-8 and day-16 using a Zeiss Observer microscope.
MTT Assay
Cell proliferation assays were performed with an ATCC MTT Assay kit (ATTC). Briefly 2,000 MDA-MB231 cells were plated into 96-well plates in triplicates in cell growth media with or without doxycycline. Cells were grown for 1-4 days and stained with the MTT reagent as recommended by the manufacturer. Wavelength was read on a SpectraMax M plate reader.
In Vivo Metastasis Modeling
Animal experiments were carried out in the Cold Spring Harbor Laboratory Animal Shared Resource in accordance with Cold Spring Harbor Laboratory Institutional Animal Care and Use Committee-approved procedures. 0.5-1×106 MDA-MB231-luciferase-GFP control or TRA2β shRNA inducible cells were injected either into the tail vein or into the mammary fat pad of 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (The Jackson Laboratory #5557) female mice. For shRNA induction, half of the mice were treated with doxycycline in both drinking water (1.5 mg ml–1 with 2% sucrose; RPI Corporation and Sigma-Aldrich) and food (625 mg kg–1, Harlan Laboratories). Doxycycline water was replaced every 3 days. Whole-body bioluminescent imaging was performed using an IVIS100 system (Caliper LifeSciences) as described (Zuber et al., 2011). Briefly, primary tumor growth and metastasis formation were monitored every 3-4 days by bioluminescence imaging following intraperitoneal injection of D-Luciferin (25 mg kg–1, Goldbio). Animal weight and primary tumor size was recorded bi-weekly. Animals were euthanized 50-60 days post-injection by cardiac perfusion of a 4% paraformaldehyde (PFA) solution. Primary tumors were collected and flash-frozen for RNA and protein extraction, or fixed in 4% PFA, washed with PBS, embedded in paraffin, sectioned, and stained with hematoxylin & eosin. Following a detailed necropsy, lung and liver tissues were fixed in 4% PFA, cryo-protected in sucrose gradient, embedded in OCT solution (Tissue-Tek) and frozen. Serial transversal sections of lung and liver were then performed through the whole tissue, every 2mm, and were re-embedded horizontally to obtain serial sections of the entire organ, and frozen in fresh OCT. Lung and liver sections were staining with hematoxylin & eosin and slides were imaged using an Aperio slide scanner (Leica). Micro- and macro-metastasis areas were then quantified relative to the whole organ area using an Aperio Scanscope (Leica).
Differential Splicing Analysis
We used the SpliceCore® software platform (https://www.envisagenics.com/platform/) to identify cancer-related differential splicing events (DSEs). SpliceCore is operated through a user interface built on the Microsoft Azure cloud to facilitate data trafficking, storage, HIPAA compliance and compute scalability. To identify DSEs changes co-occurring in cancer, SpliceCore utilizes a reference database called TXdb™, which incorporates over 5M exon-trio models (Wu et al., 2011) derived from the entire TCGA database, including ~1.5K breast cancer datasets. To prioritize reproducible DSEs, we complemented SpliceCore with additional in-house data analysis. Our in-house pipeline implemented STAR (v.2.5.1b) (Dobin et al., 2013), Cufflinks Suite (v.2.2.1) (Trapnell et al., 2012) and rMATS (v.3.2.5) (Shen et al., 2014). Paired-end reads were preprocessed for trimming of low-quality regions by Trimmomatic (v. 0.36) (Bolger et al., 2014) and mapped to the human reference genome using STAR in 2-pass mode with the Gencode GRCh37 v.25 reference transcript annotation. To include novel exons and introns in the analysis, we also performed an annotation-guided transcriptome reconstruction and merged the resulting transcriptome from each sample into a comprehensive transcript annotation (GTF) with Cufflinks and Cuffmerge (Cufflinks suites v.2.2.1) (Trapnell et al., 2012) using the “–multi-read-correct” and “–library-type fr-firststrand” (strand-specific library for dUTP protocol) parameters. Each RNA-seq replicate was processed independently in SpliceCore and in-house to produce individual “percent spliced in” (PSI) scores. In this manner we increased the sensitivity of the analysis by ensuring a larger number of AS events to be detected by at least one method. Next, we integrated both SpliceCore’s and in-house individual PSI scores to compute DSEs as “differential percent spliced in” (ΔPSI) scores. Global ΔPSI scores were estimated as the difference of the mean PSIs detected across RNA-seq replicates by at least one method: ΔPSIglobal = mean(PSIcase)-mean(PSIcontral). To eliminate inconsistent results, we performed a quality control whereby we estimated individual ΔPSIs for each “i”, where “i”s are the list of all case datasets analyzed by every method. For each “i” we estimated individual ΔPSIi = PSIi-mean(PSIcontrol). If the sign of the geometric mean of all “() did not equal sgn(ΔPSIglobal) then the splicing event was called inconsistent and filtered out. For each analyzed dataset (i.e., TCGA, MCF-10A and MDA-MB231) we applied further filtering criteria explained below.
TCGA Data Analysis
The RNA-seq data from TCGA breast tumors were retrieved and processed on the ISB Cancer Genomics Cloud Platform (https://isb-cgc.appspot.com/). Sample IDs are listed in Table S2A.
CNVs and expression changes in SFs were assessed by DNA-and RNA-seq, respectively, from a collection of human breast tumors from The Cancer Genome Atlas (TCGA) dataset (n = 960) (The Cancer Genome Atlas Network, 2012) as described previously (Cerami et al., 2012; Gao et al., 2013). Graphical Oncoprint representations were generated using the Cbio portal (http://www.cbioportal.org/) for all breast tumors, as well as for samples annotated as triple negative breast tumors. Pre-computed Z-score and clinical information, e.g., PAM50 signatures, were retrieved from Cbio for downstream analysis.
Human TCGA breast tumors (n = 960) (The Cancer Genome Atlas Network, 2012) were classified into SF-low (Z-score ≤ 0) or SF-high (Z-score ≥ 1.5) tumors according to their SF expression for SRSF1, SRSF2, SRSF3, SRSF4, SRSF5, SRSF6, SRSF7, SRSF8, SRSF9, SRSF10, SRSF11, SRSF12, TRA2β and HNRNPA1 using pre-computed Z-score values from cBioPortal (http://www.cbioportal.org/)(Cerami et al., 2012; Gao et al., 2013)(Table S2A). Splicing events were defined using both splice junction read counts and alternatively spliced exon body counts for each event to calculate a PSI score for each local event, as well as a ΔPSI in SFlow versus. SFhigh tumors using SpliceCore and in-house methods as described above. The in-house pipeline was implemented using corresponding dockers (https://www.docker.com/products/docker-hub) on a cloud instance. Significant DSEs were selected as follows: i) ΔPSI = ∣mean PSI SFhigh – mean PSI SFlow∣ ≥ 0.1; and ii) FDR ≤ 0.05; and iii) at least an average of 5 reads per dataset detected in both SFlow and SFhigh tumors that support either exon skipping or inclusion, i.e., (inclusion count > = 5 in either control OR case) AND (skipping count > = 5 in either control OR case). To correct for missing data due to sequencing depth below 100 million reads per sample in the TCGA dataset, we focused only on DSEs detected in at least 10 tumors from both groups.
Differential Splicing Analysis of MCF-10A and MDA-MB231 Cell Lines
DSEs between two groups, such as control versus SF-OE/KD, were determined using SpliceCore and in-house methods as described above. Significant DSEs were selected as follows: i) ΔPSI = ∣ mean PSISF-OE/KD – mean PSIcontrol ∣ ≥ 0.1; and ii) FDR ≤ 0.05; and iii) at least 5 reads (averaged across all biological replicates) detected in both the control and SF-OE/KD that support either exon skipping or inclusion, i.e., (inclusion count > = 5 in either control OR case) AND (skipping count > = 5 in either control OR case). For MDA-MB231 cells, the significant DSEs from TRA2βsh +DOX versus −DOX were discarded if they also appeared as significant and altered in the same direction in CTLsh +DOX versus −DOX cells.
Pairwise Comparison of DSEs Across Multiple Studies
To compare the overlap in DSEs between two studies, A and B, we performed a Fisher’s exact test (with ‘two-sided’ test to the null hypothesis) in which: i) DA and DB are all the detected AS events in study A or study B, respectively; ii) SA and SB are the statistically significant DSEs detected in study A or study B, respectively (∣ΔPSI∣ ≥ 0.1; FDR ≤ 0.05; ≥ 5 reads averaged across all replicates); iii) the intersection of significant DSEs with ΔPSI in the same direction (i.e., either both ≥ 0.1 or both ≤ −0.1) is defined as iS = SA & SB; and iv) the total splicing “universe” is defined by the union of splicing events as D = DA ∣ DB. Thus, the strength of overlap of DSEs in two studies is calculated using a Fisher’s exact test computed from the following 2×2 contingency table, along with odds ratio and P-values.
| inA | notA | |
|---|---|---|
| inB | iS | B-iS |
| notB | A - iS | D-iS |
To compare the similarity in DSEs between two studies, the Jaccard similarity index is calculated as J = SA & SB / SA ∣ SB. The analysis was conducted either on splicing events (DSEs) or one genes that exhibit at least one DSE (DSGs). P-values were corrected using Bonferroni’s method.
Differential Gene Expression Analysis
Preprocessing of reads and mapping steps was performed as mentioned for the differential splicing analysis. Using mapped files (BAM), differential expression of annotated genes (Gencode GRCh37) in control versus SF-OE/KD was determined with Cuffdiff (v.2.2.1) (Trapnell et al., 2012) using the “–multi-read-correct” and “–library-type fr-firststrand” (strand-specific library) parameters. Significant genes were selected as follows: i) FDR ≤ 0.05; ii) ∣log2 fold change∣ ≥ 0.3; and iii) test status = “OK.” For MDA-MB231 cells, genes with significant differential expression in TRA2βsh +DOX versus −DOX were discarded, if they also appeared as significant and altered in the same direction in CTLsh +DOX versus −DOX cells.
Protein Domain Annotation
Proteins domain were annotated using the InterProScan Pfam predictive module (Zdobnov and Apweiler, 2001). SpliceCore’s exon trios were translated to protein in three different reading frames and the optimal one was chosen for domain annotation. Optimal reading frames were selected based on alignment score to known protein sequences expressed from the coding locus.
Annotation of Genes with Breast Cancer Relevance
Genes associated with breast carcinoma based on GWAS, functional genomics, and literature data mining were retrieved from the Open Targets Platform (https://www.opentargets.org/).
Gene Enrichment Analysis
Gene enrichment analysis was performed using a Bioconductor package, enrichR (Chen et al., 2013; Kuleshov et al., 2016) for both i) genes that exhibit altered splicing (significant DSEs), ii) genes that exhibit differential splicing (DSGs), and iii) genes that exhibit changes in expression in SF-OE versus control MCF-10A cells or SF-KD versus control MDA-MB231 cells. We reported the top 100 enriched targets sorted based on FDR.
MYC and TRA2β Association Analysis
MYC and TRA2β amplification status and expression were calculated using the Cbio portal (http://www.cbioportal.org/) for human tumor RNA-seq data from The Cancer Genome Atlas Project as described (Cerami et al., 2012; Gao et al., 2013) or using microarray data from GEO GSE2109 (https://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE2109) as described (Anczuków et al., 2012). The association between MYC and TRA2β was computed using a two-tailed Fisher’s exact test and corrected for multiple testing.
MYC Binding Data
MYC ChIP-seq datasets from MCF-7 (ENCSR000DMJ) and MCF-10A (ENCSR000DOS) cells were analyzed as described on https://www.encodeproject.org/ and visualized using http://genome.ucsc.edu. Plotted tracks are fold changes over controls for pooled replicates, as well as representative ChIP-seq peaks called using conservative IDR thresholds.
TRA2β Expression and Clinical Outcome Analysis
Patients were stratified in TRA2β-high or -low expression groups using the minimum P-value approach as described (Mizuno et al., 2009). Correlations between TRA2β expression and overall survival or distant-metastasis-free-survival for multiple breast cancer patient cohorts were retrieved from the Prognoscan database (Mizuno et al., 2009) (http://gibk21.bse.kyutech.ac.jp/PrognoScan/index.html).
Graphs and Figures
Graphs were generated using GraphPad Prism 5 software. Figures were generated using Adobe CC 2018 Photoshop and Illustrator software in compliance with the Nature Publishing Group policy concerning image integrity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where appropriate, the data are presented as the mean ± s.d., as indicated. Data points were compared using an unpaired two-tailed Student t test or two-tailed Mann-Whitney test, as indicated in the legends. For quantification of proliferation and apoptosis markers, a two-tailed Fisher test was used. A Mantel-Cox test was used to compare tumor–free survival of mice in the transplantation experiment. P-values are indicated in the figure legends.
DATA AND CODE AVAILABILITY
The accession numbers for RNA-sequencing data are GEO: GSE137440 (RNA-seq MCF-10A) and GSE137408 (RNA-seq MDA-MB231).
RNA-sequencing data from TCGA breast tumors (The Cancer Genome Atlas Network, 2012) is available via ISB-CGC cloud. Sample IDs are listed in Table S2A. ChIP-seq datasets (ENCSR000DMJ and ENCSR000DOS) are available from https://www.encodeproject.org/.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-T7 mouse | EMD Millipore | #69522-3; RRID: AB_11211744 |
| Anti-cleaved caspase-3 rabbit | Cell Signaling | #9661L; RRID: AB_2341188 |
| Anti-TRA2β rabbit | Abcam | #AB31353; RRID: AB_778565 |
| Anti-β Tubulin III rabbit | Genescript | #A01203-40; RRID: AB_1289179 |
| anti-SRSF4 | Bethyl Labs | #A303-670A; RRID: AB_11204752 |
| anti SRSF6 | CSHL antibody Facility | AK9-156 |
| anti-SRSF9 | CSHL antibody Facility | #AK251-24 |
| Anti-ki67 rabbit | Zymed/ Invitrogen | #18-0191Z |
| Alexa Fluor 568 anti-mouse | Invitrogen | #A-11031; RRID: AB_144696 |
| Alexa Fluor 488 anti-rabbit | Invitrogen | #A-11034; RRID: AB_2576217 |
| IRDye 800CW Goat anti-Rabbit IgG (H + L) | Li-Cor | #926-32211; RRID: AB_621843 |
| IRDye 680 Goat anti-Mouse IgG (H + L) | Li-Cor | #926-3220 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Matrigel Growth Factor Reduced | BD /Corning | #354230 |
| Human Epithelial Growth Factor | Peprotech | #100-15 |
| 4-hydroxy-tamoxifen | Sigma | #H7904-5mg |
| Collagen | BD /Corning | #354236 |
| Cell Recovery Solution | BD /Corning | #354253 |
| Puromycin dihydrochloride | Sigma | #P8833 |
| Hygromycin B | Sigma | #400053 |
| G418 | Goldbio | #G-418 |
| Insulin | Sigma | #I-1882 |
| D-luciferin, potassium salt | Goldbio | #LUCK-10 g |
| Doxycycline | RPI corporation | #D43020 |
| Doxycycline food pellets | Harlan Teklad | #TD.05125 |
| Critical Commercial Assays | ||
| RNAeasy kit | QIAGEN | #74106 |
| TrueSeq stranded mRNA Kit | Illumina | #RS-122-2101 |
| iTaq Universal SYBR green Supermix | Bio-Rad | #172-5121 |
| MTT assay | ATCC | # 30-1010K |
| Deposited Data | ||
| RNA-seq MCF-10A | This paper | GSE137440 |
| RNA-seq MDA-MB231 | This paper | GSE137408 |
| Experimental Models: Cell Lines | ||
| MCF-10A | Laboratory of Senthil Muthuswamy | N/A |
| MDA-MB231 | Laboratory of Min Yu | N/A |
| SUM159PT | Laboratory of Steve Ethier | N/A |
| Recombinant DNA | ||
| PWZL-T7-SRSF1-Hygromycin | (Anczuków et al., 2012) | N/A |
| PWZL-T7-SRSF2-Hygromycin | This paper | N/A |
| PWZL-T7-SRSF3-Hygromycin | This paper | N/A |
| PWZL-T7-SRSF4-Hygromycin | This paper | N/A |
| PWZL-T7-SRSF6-Hygromycin | This paper | N/A |
| PWZL-T7-SRSF9-Hygromycin | This paper | N/A |
| PWZL-T7-HNRNPA1-Hygromycin | This paper | N/A |
| PWZL-T7-TRA2B-Hygromycin | This paper | N/A |
| pBABE-MYC.ER-Puromycin | (Eilers et al., 1989) | N/A |
| TRMPV-Neo-SRSF4-sh1 | This paper | N/A |
| TRMPV-Neo-SRSF4-sh2 | This paper | N/A |
| TRMPV-Neo-SRSF6-sh1 | This paper | N/A |
| TRMPV-Neo-SRSF6-sh2 | This paper | N/A |
| TRMPV-Neo-SRSF9-sh1 | This paper | N/A |
| TRMPV-Neo-SRSF9-sh2 | This paper | N/A |
| TRMPV-Neo-TRA2B -sh1 | This paper | N/A |
| TRMPV-Neo-TRA2B-sh2 | This paper | N/A |
| TRMPV-Neo-HNRNPA1-sh1 | This paper | N/A |
| TRMPV-Neo- HNRNPA1-sh2 | This paper | N/A |
| TRMPV-Neo- control-sh1 | This paper | N/A |
| rTTA3-Puro | (Zuber et al., 2011) | N/A |
| Sequence-Based Reagents | ||
| shRNA sequence | This paper Table S7A | N/A |
| QPCR primer sequences | This paper Table S7B | N/A |
| RT-PCR primer sequences | This paper Table S7C | N/A |
| Software and Algorithms | ||
| Cbio Portal | (Cerami et al., 2012) | www.cbioportal.org |
| ISB-CGC | (Reynolds et al., 2017) | https://isb-cgc.appspot.com/ |
| dockerhub | N/A | https://hub.docker.com/ |
| STAR v.2.5.1b | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Cufflinks 2.2.1 | (Trapnell et al., 2012) | http://cole-trapnell-lab.github.io/cufflinks/ |
| BedTools v.2.25.0 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| Trimmomatic v.0.36 | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| rMATS v.3.2.5 | (Shen et al., 2014) | http://rnaseq-mats.sourceforge.net/ |
| enrichR | (Chen et al., 2013; Kuleshov et al., 2016) | https://cran.r-project.org/web/packages/enrichR/vignettes/enrichR.html |
| SpliceCore® | Envisagenics | https://www.envisagenics.com/platform/ |
| Prognoscan database | (Mizuno et al., 2009) | http://gibk21.bse.kyutech.ac.jp/PrognoScan/index.html |
| Axiovision Digital Image Processing Software | Carl Zeiss | N/A |
| GraphPad Prism | GraphPad | N/A |
| ImageJ Digital Image Processing Software | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Photoshop and Illustrator CC2018 | Adobe | N/A |
Highlights.
52% of breast tumors have an alteration in at least one SR protein family member
SRSF4, SRSF6, or TRA2β promotes mammary cell proliferation and invasion
SRSF4, SRSF6, or TRA2β regulates shared spliced isoforms associated with cancer hallmarks
TRA2β is regulated by MYC and plays a role in breast cancer metastasis
ACKNOWLEDGMENTS
We thank J.E. Wilkinson for assistance with histopathology, S. Sampson and T. Helenius for comments on the manuscript, and M. Yurieva for assistance with GEO submission. We acknowledge assistance from The Jackson Laboratory (JAX) and Cold Spring Harbor Laboratory (CSHL) Microscopy and Sequencing Shared Resources, funded by the National Cancer Institute (NCI) Cancer Center Support grants P30CA034196 and P30CA045508. This work was supported by grants from the NCI R00CA1728206 to O.A. and P01CA13106 to A.R.K.; National Institute of General Medical Sciences (NIGMS) R43GM116478 to M.A.; and postdoctoral awards to O.A. from the Susan G. Komen Foundation for the Cure (KG091029), the Terri Brodeur Breast Cancer Foundation (66810101), and JAX start-up funds. We acknowledge the use of data generated by TCGA, managed by NCI and the National Human Genome Research Institute (NHGRI), and processed using the Institute for Systems Biology (ISB) Cancer Genomics Cloud. The content is solely the responsibility of the authors and does not necessarily represent NIH official views.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.110.
DECLARATION OF INTERESTS
M.A. is a founder and shareholder of Envisagenics, Inc., and A.R.K. is a member of its scientific advisory board.
REFERENCES
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, and Krainer AR (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol 19, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. (2015). SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 60, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznarez I, Nomakuchi TT, Tetenbaum-Novatt J, Rahman MA, Fregoso O, Rees H, and Krainer AR (2018). Mechanism of Nonsense-Mediated mRNA Decay Stimulation by Splicing Factor SRSF1. Cell Rep. 23, 2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin JM, Wu X, Christiano R, Oh MS, Schauder CM, Gazzerro E, Messa M, Baldassari S, Assereto S, Biancheri R, et al. (2016). The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol 18, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M (2007). The GNAS Locus: Quintessential Complex Gene Encoding Gsalpha, XLalphas, and other Imprinted Transcripts. Curr. Genomics 8, 398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Daijogo S, and Semler BL (2007). A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 26, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best A, Dagliesh C, Ehrmann I, Kheirollahi-Kouhestani M, Tyson-Capper A, and Elliott DJ (2013). Expression of Tra2 β in Cancer Cells as a Potential Contributory Factor to Neoplasia and Metastasis. Int. J. Cell Biol 2013, 843781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem 72, 291–336. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti V, McNicoll F, Steiner MC, Richter FM, Solovyeva A, Wegener M, Schwich OD, Poser I, Zarnack K, Wittig I, et al. (2017). Cellular differentiation state modulates the mRNA export activity of SR proteins. J. Cell Biol 216, 1993–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley T, Cook ME, and Blanchette M (2015). SR proteins control a complex network of RNA-processing events. RNA 21, 75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, and Cheng C (2011). CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Invest 121, 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Misteli T, Screaton GR, Spector DL, and Krainer AR (1997). Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J. Cell Biol 138, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Screaton GR, and Krainer AR (1998).A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 12, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E, et al. (2019). Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47 (D1), D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Zhu J, Manche L, Huber E, Krainer AR, and Cáceres JF (2002). Nuclear export and retention signals in the RS domain of SR proteins. Mol. Cell. Biol 22, 6871–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Sridhar P, Kirchner R, Lock YJ, Herbert Z, Buonamici S, Smith P, Lieberman J, and Petrocca F (2017). Basal-A Triple-Negative Breast Cancer Cells Selectively Rely on RNA Splicing for Survival. Mol. Cancer Ther 16, 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Zhao S, Karnad A, and Freeman JW (2018). The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chierico L, Rizzello L, Guan L, Joseph AS, Lewis A, and Battaglia G (2017). The role of the two splice variants and extranuclear pathway on Ki-67 regulation in non-cancer and cancer cells. PLoS One 12, e0171815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climente-González H, Porta-Pardo E, Godzik A, and Eyras E (2017). The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 20, 2215–2226. [DOI] [PubMed] [Google Scholar]
- Das S, Anczuków O, Akerman M, and Krainer AR (2012). Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 1, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, and Manley JL (2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, and Brugge JS (2005). Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5, 675–688. [DOI] [PubMed] [Google Scholar]
- Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, and Brugge JS (2002). The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003a). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Debnath J, Walker SJ, and Brugge JS (2003b). Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J. Cell Biol 163, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ear J, Dunkel Y, Mittal M, Lim BBC, Liu L, Holda M, Nitsche U, Barbazan J, Goel A, Janssen KP, et al. (2019). Two Isoforms of the Guanine Nucleotide Exchange Factor, Daple/CCDC88C Cooperate as Tumor Suppressors. Sci. Rep 9, 12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M, Picard D, Yamamoto KR, and Bishop JM (1989). Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature 340, 66–68. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Horvath A, Godbole S, Reddy SD, Mudvari P, Ohshiro K, Cyanam D, Nair S, Fuqua SA, Polyak K, et al. (2013). RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep 3, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KD, and Semler BL (2011). Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 7, e1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KD, and Semler BL (2013). Poliovirus infection induces the co-localization of cellular protein SRp20 with TIA-1, a cytoplasmic stress granule protein. Virus Res. 176, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Huang B, Shi Z, Han J, Wang Y, Huangfu J, and Wu W (2013). SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing β-catenin biosynthesis. EMBO Mol. Med 5, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Murillas I, Sharpe R, Pearson A, Campbell J, Natrajan R, Ashworth A, and Turner NC (2014). An siRNA screen identifies the GNAS locus as a driver in 20q amplified breast cancer. Oncogene 33, 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, and Manley JL (1990). A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 62, 25–34. [DOI] [PubMed] [Google Scholar]
- Harvey SE, Xu Y, Lin X, Gao XD, Qiu Y, Ahn J, Xiao X, and Cheng C (2018). Coregulation of alternative splicing by hnRNPM and ESRP1 during EMT. RNA 24, 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, and Beck WT (2014). Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis 3, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yario TA, and Steitz JA (2004). A molecular link between SR protein dephosphorylation and mRNA export. Proc. Natl. Acad. Sci. USA 101, 9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CS, Shen CY, Wang HW, Wu PE, and Cheng CW (2007). Increased expression of SRp40 affecting CD44 splicing is associated with the clinical outcome of lymph node metastasis in human breast cancer. Clin. Chim. Acta 384, 69–74. [DOI] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. (2012). Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 1, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida-Takagishi M, Enomoto A, Asai N, Ushida K, Watanabe T, Hashimoto T, Kato T, Weng L, Matsumoto S, Asai M, et al. (2012). The Dishevelled-associating protein Daple controls the non-canonical Wnt/Rac pathway and cell motility. Nat. Commun 3, 859. [DOI] [PubMed] [Google Scholar]
- Itoh M, Radisky DC, Hashiguchi M, and Sugimoto H (2017). The exon 38-containing ARHGEF11 splice isoform is differentially expressed and is required for migration and growth in invasive breast cancer cells. Oncotarget 8, 92157–92170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R, Li C, McCoy JP, Deng CX, and Zheng ZM (2010). SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci 6, 806–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung P, Menssen A, Mayr D, and Hermeking H (2008). AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 105, 15046–15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, and Krainer AR (2007). The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol 14, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabò A, et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. [DOI] [PubMed] [Google Scholar]
- Krainer AR, Conway GC, and Kozak D (1990). Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 4, 1158–1171. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, and Caceres JF (2009). The SR protein family of splicing factors: master regulators of gene expression. Biochem. J 417, 15–27. [DOI] [PubMed] [Google Scholar]
- Maslon MM, Heras SR, Bellora N, Eyras E, and Cáceres JF (2014). The translational landscape of the splicing factor SRSF1 and its role in mitosis. eLife May 6, e02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlewski G, Sanford JR, and Cáceres JF (2008). The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol.Cell 30, 179–189. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Kitada K, Nakai K, and Sarai A (2009). PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med. Genomics 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, and Neugebauer KM (2016). SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 30, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T, Kozaki KI, Imoto S, Yamaguchi R, Tsuda H, Kawano T, Fujiwara N, Morishita M, Miyano S, and Inazawa J (2016). The hypusine cascade promotes cancer progression and metastasis through the regulation of RhoA in squamous cell carcinoma. Oncogene 35, 5304–5316. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJand Brugge JS (2001). ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol 3, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oran AR, Adams CM, Zhang XY, Gennaro VJ, Pfeiffer HK, Mellert HS, Seidel HE, Mascioli K, Kaplan J, Gaballa MR, et al. (2016). Multifocal control of mitochondrial gene expression by oncogenic MYC provides potential therapeutic targets in cancer. Oncotarget 7, 72395–72414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr., and Fu XD (2013). Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 50, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen JI, Nieminen AI, Mäkelä TP, and Klefstrom J (2007). Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc. Natl. Acad. Sci. USA 104, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Khauv D, Risso G, Stallings-Mann M, Blaustein M, Quadrana L, Radisky DC, and Srebrow A (2012). Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J. Cell. Biochem 113, 2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pind MT, and Watson PH (2003). SR protein expression and CD44 splicing pattern in human breast tumours. Breast Cancer Res. Treat 79, 75–82. [DOI] [PubMed] [Google Scholar]
- Pinke DE, and Lee JM (2011). The lipid kinase PI4KIIIβ and the eEF1A2 oncogene co-operate to disrupt three-dimensional in vitro acinar morphogenesis. Exp. Cell Res 317, 2503–2511. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves DK, Hoadley KA, Fagan-Solis KD, Jima DD, Bereman M, Thorpe L, Hicks J, McDonald D, Troester MA, Perou CM, and Fleming JM (2017). Nuclear Localized LSR: A Novel Regulator of Breast Cancer Behavior and Tumorigenesis. Mol. Cancer Res 15, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SM, Miller M, Lee P, Leinonen K, Paquette SM, Rodebaugh Z, Hahn A, Gibbs DL, Slagel J, Longabaugh WJ, et al. (2017). The ISB Cancer Genomics Cloud: A Flexible Cloud-Based Platform for Cancer Genomics Research. Cancer Res. 77, e7–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, and Cáceres JF (2004). A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 18, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Coutinho P, Hackett JA, Wang X, Ranahan W, and Caceres JF (2008). Identification of nuclear and cytoplasmic mRNA targets for the shuttling protein SF2/ASF. PLoS One 3, e3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapra AK, Ankö ML, Grishina I, Lorenz M, Pabis M, Poser I, Rollins J, Weiland EM, and Neugebauer KM (2009). SR protein family members display diverse activities in the formation of nascent and mature mRNPs in vivo. Mol. Cell 34, 179–190. [DOI] [PubMed] [Google Scholar]
- Sato H, Hosoda N, and Maquat LE (2008). Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol. Cell 29, 255–262. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, and Gertler FB (2011). An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 7, e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellzig J, Chariot A, Shostak K, Ismail Göktuna S, Renner F, Acker T, Pagenstecher A, and Schmitz ML (2013). Deregulated expression of TANK in glioblastomas triggers pro-tumorigenic ERK1/2 and AKT signaling pathways. Oncogenesis 2, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickeler E, Kittrell F, Medina D, and Berget SM (1999). Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene 18, 3574–3582. [DOI] [PubMed] [Google Scholar]
- Sun X, Jin Z, Song X, Wang J, Li Y, Qian X, Zhang Y, and Yin Y (2015). Evaluation of KIF23 variant 1 expression and relevance as a novel prognostic factor in patients with hepatocellular carcinoma. BMC Cancer 15, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CM, Sherer NM, and Malim MH (2010). SRp40 and SRp55 promote the translation of unspliced human immunodeficiency virus type 1 RNA. J. Virol 84, 6748–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz JE, Bor YC, Misawa Y, Rekosh D, and Hammarskjold ML (2007). The shuttling SR protein 9G8 plays a role in translation of unspliced mRNA containing a constitutive transport element. J. Biol. Chem 282, 19844–19853. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, and Anczuków O (2018). Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 9, e1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, et al. (2008). Identification of alternative splicing markers for breast cancer. Cancer Res. 68, 9525–9531. [DOI] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. (2009). Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol 16, 670–676. [DOI] [PubMed] [Google Scholar]
- Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, Thornton CA, Wang ETand Berglund JA (2016). Dose-Dependent Regulation of Alternative Splicing by MBNL Proteins Reveals Biomarkers for Myotonic Dystrophy. PLoS Genet. 12, e1006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Norton JT, Ghosh S, Kim J, Fushimi K, Wu JY, Stack MS, and Huang S (2008). Polypyrimidine tract-binding protein (PTB) differentially affects malignancy in a cell line-dependent manner. J. Biol. Chem 283, 20277–20287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Fan Y, Yu X, Mao X, and Jin F (2018). PTBP1 promotes the growth of breast cancer cells through the PTEN/Akt pathway and auto-phagy. J. Cell. Physiol 233, 8930–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, and Carstens RP (2010). An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 29, 3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watermann DO, Tang Y, Zur Hausen A, Jäger M, Stamm S, and Stickeler E (2006). Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 66, 4774–4780. [DOI] [PubMed] [Google Scholar]
- Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S, and Hecht A (2010). Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 38, 1964–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter P, Hanselmann S, Pattschull G, Schruf E, and Gaubatz S (2017). Central spindle proteins and mitotic kinesins are direct transcriptional targets of MuvB, B-MYB and FOXM1 in breast cancer cell lines and are potential targets for therapy. Oncotarget 8, 11160–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Akerman M, Sun S, McCombie WR, Krainer AR, and Zhang MQ (2011). SpliceTrap: a method to quantify alternative splicing under single cellular conditions. Bioinformatics 27, 3010–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang B, and Muthuswamy SK (2006). Using three-dimensional acinar structures for molecular and cell biological assays. Methods Enzymol. 406, 692–701. [DOI] [PubMed] [Google Scholar]
- Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, et al. (2014). Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 28, 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Chan JR, and Tanoue A (2008). ErbB2 directly activates the exchange factor Dock7 to promote Schwann cell migration. J. Cell Biol 181, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CS, Peterson AL, Castellani G, Sedivy JM, and Neretti N (2011). Kinetic profiling of the c-Myc transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle 10, 2184–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y, Matsumoto A, Hieda M, Shinchi Y, Ogihara E, Hamada M, Nishioka Y, Kimura H, Yoshidome K, Tsujimoto M, and Matsuura N (2014). Loss of histone H4K20 trimethylation predicts poor prognosis in breast cancer and is associated with invasive activity. Breast Cancer Res. 16, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, and Ogawa S (2014). Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 5, 445–459. [DOI] [PubMed] [Google Scholar]
- Yoshino H, Enokida H, Chiyomaru T, Tatarano S, Hidaka H, Yamasaki T, Gotannda T, Tachiwada T, Nohata N, Yamane T, et al. (2012). Tumor suppressive microRNA-1 mediated novel apoptosis pathways through direct inhibition of splicing factor serine/arginine-rich 9 (SRSF9/SRp30c) in bladder cancer. Biochem. Biophys. Res. Commun 417, 588–593. [DOI] [PubMed] [Google Scholar]
- Yu M, Smolen GA, Zhang J, Wittner B, Schott BJ, Brachtel E, Ramaswamy S, Maheswaran S, and Haber DA (2009). A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 23, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM, Neugebauer KM, Lane WS, and Roth MB (1993). Distinct functions of SR proteins in alternative pre-mRNA splicing. Science 260, 219–222. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, and Apweiler R (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. [DOI] [PubMed] [Google Scholar]
- Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, and Muthuswamy SK (2008). Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Krainer AR (2004). Involvement of SR proteins in mRNA surveillance. Mol. Cell 16, 597–607. [DOI] [PubMed] [Google Scholar]
- Zhu J, and Krainer AR (2000). Pre-mRNA splicing in the absence of an SR protein RS domain. Genes Dev. 14, 3166–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, and Lowe SW (2011). Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat. Biotechnol 29, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for RNA-sequencing data are GEO: GSE137440 (RNA-seq MCF-10A) and GSE137408 (RNA-seq MDA-MB231).
RNA-sequencing data from TCGA breast tumors (The Cancer Genome Atlas Network, 2012) is available via ISB-CGC cloud. Sample IDs are listed in Table S2A. ChIP-seq datasets (ENCSR000DMJ and ENCSR000DOS) are available from https://www.encodeproject.org/.