

Abstract
Paired two-component systems (TCSs), having a sensor kinase (SK) and a cognate response regulator (RR), enable the human pathogen Mycobacterium tuberculosis to respond to the external environment and to persist within its host. Here, we inactivated the SK gene of the TCS MtrAB, mtrB, generating the strain ΔmtrB. We show that mtrB loss reduces the bacterium's ability to survive in macrophages and increases its association with autophagosomes and autolysosomes. Notably, the ΔmtrB strain was markedly defective in establishing lung infection in mice, with no detectable lung pathology following aerosol challenge. ΔmtrB was less able to withstand hypoxic and acid stresses and to form biofilms and had decreased viability under hypoxia. Transcriptional profiling of ΔmtrB by gene microarray analysis, validated by quantitative RT-PCR, indicated down-regulation of the hypoxia-associated dosR regulon, as well as genes associated with other pathways linked to adaptation of M. tuberculosis to the host environment. Using in vitro biochemical assays, we demonstrate that MtrB interacts with DosR (a noncognate RR) in a phosphorylation-independent manner. Electrophoretic mobility shift assays revealed that MtrB enhances the binding of DosR to the hspX promoter, suggesting an unexpected role of MtrB in DosR-regulated gene expression in M. tuberculosis. Taken together, these findings indicate that MtrB functions as a regulator of DosR-dependent gene expression and in the adaptation of M. tuberculosis to hypoxia and the host environment. We propose that MtrB may be exploited as a chemotherapeutic target against tuberculosis.
Keywords: Mycobacterium tuberculosis, hypoxia, biofilm, bacterial signal transduction, infection, bacterial survival, dormancy, bacterial persistence, virulence, MtrB, sensor kinase, two component system
Introduction
The ability of Mycobacterium tuberculosis to remain dormant within its host for extended periods of time without eliciting any obvious symptoms of disease, its ability to relapse once drug treatment is withdrawn (1), and the lack of an efficacious vaccine pose a threat to worldwide efforts to control the disease, necessitating efforts to identify new therapeutic strategies. Two-component systems (TCSs)9 are involved in stimulus-dependent adaptation of bacteria to their environment. Canonical, paired TCSs comprise a membrane-bound sensor kinase (SK) and a cytosolic response regulator (RR). A phosphotransfer relay is activated when the SK senses a stimulus, culminating in phosphorylation of the cognate RR. TCSs regulate bacterial virulence, pathogenesis, biofilm formation, cell division, and metabolite production, to name a few functions. M. tuberculosis has 12 paired TCSs, six orphan RRs, and two orphan SKs (2). The mtrA–mtrB TCS is present in all mycobacterial species characterized (3). mtrA, the RR, is essential in M. tuberculosis. In Mycobacterium smegmatis, MtrB, the SK, localizes to the septa and the poles in a phosphorylation-dependent manner (4) to regulate the expression of the MtrA targets ripA, fbpB, and ftsI (5).
In high-G+C Gram-positive bacteria, MtrAB has been suggested to be the functional analog of the YycFG system in low GC–containing Gram-positive bacteria (6, 7). Similar to YycF in Bacillus subtilis, MtrA is the only response regulator of TCS, known to be essential in Mycobacterium (3). YycFG controls multiple cellular pathways linked to cell wall synthesis, cell growth, and cell division (8). Mutations in this TCS alter cell wall permeability, antibiotic resistance, biofilm formation, cellular morphology, osmotic stress, and virulence of pathogenic bacteria (9). Similar to YycFG, alterations in MtrAB expression also affect cell wall, cell division, and cellular morphology in Mycobacterium and Corynebacterium (4, 10, 11).
The foregoing preamble suggests that MtrB could be a regulator of cell wall synthesis, cellular morphology, biofilm formation, and survival of M. tuberculosis in vivo. To explore this, we inactivated mtrB in M. tuberculosis to generate the ΔmtrB strain. Inactivation of mtrB renders the bacilli unable to withstand hypoxia and acid stress. ΔmtrB is unable to form biofilms, possibly due to defective ketomycolic acid biosynthesis. The inability of ΔmtrB to survive under hypoxia is linked to dramatic down-regulation of the dosR–dosS regulon. Its response regulator DosR is induced in response to hypoxia and enables recovery from dormancy. Based on the relative transcription levels observed during progression of cells toward stationary phase in vitro, we hypothesized that the compromised transcription of the dosR regulon is directly associated with the inability of the bacteria to launch a response to hypoxia in the absence of mtrB. We confirmed direct interaction of MtrB with the noncognate response regulator DosR. MtrB enhanced binding of DosR with its targets, suggesting that in addition to its role as a sensor kinase, MtrB could exert MtrA-independent functions. Reduced transcription of dosR-regulated genes early during infection is likely to be linked to compromised survival of ΔmtrB in macrophages. The global changes in the bacterial transcriptome in the absence of mtrB could possibly explain the attenuated phenotype of ΔmtrB in a mouse model of infection with no visible granuloma formation. In summary, our results establish a previously unrecognized, central regulatory role of MtrB in the pathogenesis of M. tuberculosis. Therefore, MtrB could be exploited to develop novel therapeutics for the management of tuberculosis.
Results
M. tuberculosis MtrB is required to maintain growth, cell size, and surface architecture
To gain insight into the role of MtrB, we inactivated mtrB (ΔmtrB) of M. tuberculosis by disrupting the mtrB gene with a kanamycin cassette. PCR products of the expected size were obtained for the wildtype (WT) and ΔmtrB (Fig. S1, A–C). Southern blotting further confirmed the inactivation of mtrB (Fig. S1, D and E). PCR using genomic DNA as template confirmed the presence of mtrA in both the WT and ΔmtrB (Fig. S1F). The growth of ΔmtrB was retarded compared with the WT (Fig. 1A). This could be reversed upon complementation with mtrB (ΔmtrB::mtrB), suggesting that the growth retardation was specifically attributable to mtrB. Complementation of mtrB was confirmed by immunoblotting with MtrB antibody (Fig. S1G). ΔmtrB showed increased aggregation when grown in liquid medium containing Tween 80 for 10 min (Fig. 1B). The presence of Tween 80 did not affect the viability of the bacteria (Fig. S1H). This was reversed in ΔmtrB::mtrB. When grown on solid medium, ΔmtrB displayed altered colony morphology compared with the WT M. tuberculosis (Fig. 1C). This was reversed upon complementation with mtrB. Altered colony morphology of M. tuberculosis has been linked to alterations in cell wall synthesis (12).
Figure 1.

M. tuberculosis requires MtrB to maintain growth, cell size, surface integrity, biofilm formation, and survival under acid stress and hypoxia. A, growth curve of WT, ΔmtrB, and ΔmtrB::mtrB strains monitored by recording the cfu over a period of time in days (d). B, aggregation of WT, ΔmtrB, and ΔmtrB::mtrB strains observed after keeping growing cultures standing for 10 min. C, colony morphology of WT, ΔmtrB, and ΔmtrB::mtrB strains. D, scanning EM analyses of cell lengths of WT, ΔmtrB, and ΔmtrB::mtrB strains. Pole-to-pole lengths of 30 bacteria from at least five fields for each strain were measured. ***, p < 0.001. E, biofilm formation of WT, ΔmtrB, ΔmtrB::mtrB, and ΔmtrB::mtrA(Y102C) strains after 6 weeks of incubation. The right-hand panel indicates percentage of crystal violet (CV) taken up by biofilms of different strains compared with WT. Error bars represent S.D., n = 3. ***, p < 0.001. F, TLC of mycolic acid methyl esters. Apolar lipids were isolated from WT, ΔmtrB, and ΔmtrB::mtrB strains; derivatized as methyl esters; and resolved by TLC using petroleum ether:diethyl ether (95:5, v/v). ΔmtrB was deficient in ketomycolic acids compared with the WT, which was rescued by complementation with mtrB. G and H, percent reduction in growth of the M. tuberculosis strains at pH 5.5 (relative to growth at pH 7) in MB7H9 broth containing tyloxapol and ADC. Error bars represent S.D., n = 3. ***, p < 0.001; **, p < 0.01; ns, nonsignificant. I, survival of M. tuberculosis strains after recovery from hypoxia. After establishment of hypoxia, cfu were determined at the indicated time intervals. No colonies were obtained for ΔmtrB kept in hypoxic conditions for 14 or 28 days. Images in B, C, E, and F are representative of at least two independent experiments.
The pole-to-pole length of exponentially growing ΔmtrB was significantly greater than that of the WT M. tuberculosis. When evaluated by scanning EM, the average cell length of 30 bacteria was observed to be ∼2 μm for the WT, whereas it was ∼3 μm for ΔmtrB (Figs. 1D and S1I). These phenotypic changes suggested that MtrB possibly regulates cell division in M. tuberculosis. The length was restored in ΔmtrB::mtrB.
MtrB is required for biofilm formation
M. tuberculosis observed in infected guinea pigs after drug treatment or in certain caseating lesions in humans exhibits biofilm-like architecture (13). In view of this, we tested whether the absence of mtrB alters the ability of the bacterium to form biofilms. Strikingly, ΔmtrB was not able to form biofilms (Fig. 1E), whereas biofilm formation was restored in ΔmtrB::mtrB. MtrB is therefore required for biofilm formation in M. tuberculosis. We further tested whether the influence of MtrB on biofilm formation depends on MtrA. To do so, we introduced a gain-of-function mutant of MtrA, MtrA(Y102C), in ΔmtrB. This variant of MtrA functions even in the absence of MtrB (4, 14). The overexpression of His-tagged MtrA(Y102C) was confirmed by immunoblotting with His antibody (Fig. S1J). Biofilm formation could not be restored upon introduction of MtrA(Y102C) in ΔmtrB (Fig. 1E), suggesting that MtrB-dependent biofilm formation in M. tuberculosis probably occurs independently of MtrA. M. tuberculosis biofilms are rich in mycolic acids (15). We performed a qualitative analysis of mycolates in the WT strain and ΔmtrB. M. tuberculosis H37Rv is characterized by the presence of three major classes of mycolic acids, α, methoxy, and keto (Fig. 1F, second lane from left). ΔmtrB was compromised in ketomycolate formation compared with the WT (Fig. 1F, third lane from left), and this was rescued by complementation with mtrB (Fig. 1F, first lane from left). Pellicle biofilm formation in M. tuberculosis requires ketomycolates (16), suggesting that a deficiency of ketomycolates in ΔmtrB could be associated with compromised biofilm formation.
MtrB is required for survival under acid stress and hypoxia
Within the host, M. tuberculosis encounters a hostile environment to which it must respond appropriately to survive. Among the hostile conditions is the acidic pH of the phagosome or phagolysosome. We tested whether ΔmtrB is competent to withstand acid stress. The percent reduction of growth at pH 5.5 relative to growth at pH 7.0 was significantly higher for ΔmtrB compared with the WT after 4 or 6 days of exposure to pH 5.5 (Fig. 1G). This effect was reversed in the mtrB-complemented strain. Complementation of ΔmtrB with MtrA(Y102C) mutant did not reverse the acid sensitivity of the mutant (Fig. 1H), suggesting that the role of MtrB in aiding the bacterium to withstand acid stress is independent of MtrA. Wide-ranging remodeling of the transcriptome is associated with the entry of M. tuberculosis into a dormant state when it encounters hypoxia. In view of this, we tested the ability of ΔmtrB to withstand hypoxia. Hypoxic conditions were generated in sealed tubes, and the viability of the bacterium was evaluated by determination of cfu at different periods of time. Although WT and ΔmtrB::mtrB could grow when cells were replated on fresh medium after 14 or 28 days under hypoxia, no colonies were obtained for ΔmtrB at the same time points (Fig. 1I). These results strongly suggested that MtrB is required for mounting a response that enables bacteria to withstand hypoxia.
MtrB is required for survival of M. tuberculosis in macrophages
The observed changes in phenotypic behavior of ΔmtrB compared with the WT suggested that MtrB could be a likely regulator of the fate of M. tuberculosis during infection. To test whether MtrB has a role during infection ex vivo, we tested the ability of ΔmtrB to survive in bone marrow–derived macrophages (BMDMs). Loss of mtrB compromised the ability of the bacterium to survive in macrophages (Fig. 2A). This was reversed upon complementation of the mutant with mtrB. M. tuberculosis escapes lysosomal killing by subverting xenophagy (17). We tested whether attenuated survival of ΔmtrB within macrophages could be attributed to its increased association with autophagosomes and trafficking to lysosomes. Colocalization of the bacteria with LC3 was used as a method of analyzing association of bacteria with autophagosomes, whereas colocalization of the bacteria with LAMP1 was used to monitor trafficking of bacteria to lysosomes. FITC-labeled ΔmtrB colocalized at a significantly higher percentage than the WT with Alexa Fluor 546–labeled LC3B in BMDMs (Fig. 2B), suggesting that ΔmtrB was impaired in its ability to escape xenophagy compared with the WT. Furthermore, a larger percentage of FITC-labeled ΔmtrB colocalized with Alexa Fluor 546–labeled LAMP1 compared with the WT (Fig. 2C), suggesting increased trafficking to lysosomes and arguing in favor of a role of MtrB in blocking phagosome–lysosome fusion as well as bacterial survival in macrophages.
Figure 2.

MtrB is required for the survival of M. tuberculosis in macrophages and for trafficking to autophagosomes and lysosomes. A, survival of WT M. tuberculosis, ΔmtrB, and ΔmtrB::mtrB in BMDMs. Infected cells were lysed, and bacterial survival was determined by enumerating cfu. ΔmtrB was compromised for survival in macrophages compared with the WT. This was rescued by complementation with mtrB. B and C, BMDMs were infected with FITC-labeled M. tuberculosis (WT, ΔmtrB, or ΔmtrB::mtrB), fixed, and immunolabeled with anti-LC3 (B) or anti-LAMP1 (C) followed by Alexa Fluor 546–conjugated secondary antibody. Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Scale bars, 20 μm. B and C, representative images from three independent experiments. Percent colocalization of each of the three strains with LC3 (B) or LAMP1 (C) was calculated by counting at least 100 bacteria from five different fields. Error bars represent S.D. n = 3. ***, p < 0.001.
MtrB is required for establishing infection in mice
The reduced capability of ΔmtrB to survive in macrophages prompted us to test its ability to survive in vivo. Mice were infected with the WT or ΔmtrB via the aerosol route. Although the initial loads of both strains were the same (∼100 bacilli in the lungs), the cfu recovered from infected lungs decreased significantly over time in the case of ΔmtrB (Fig. 3A), suggesting a compromised ability of the mutant to replicate in the lungs. The WT M. tuberculosis disseminated to the spleens of infected mice (Fig. 3B), whereas for ΔmtrB, significantly less bacteria were recovered from the spleens after 12 and 14 weeks of infection (Fig. 3B). Splenomegaly was observed in mice infected with WT M. tuberculosis where the average length of the spleen was ∼2.8 cm, whereas spleens from mice infected with ΔmtrB were of an average length of ∼1.5 cm (Fig. 3C). During infection, M. tuberculosis persists within granulomas where it is shielded from the antimycobacterial immune effectors of the host. Histopathological evaluation showed that WT M. tuberculosis–infected lung sections displayed granulomas, whereas no granulomas were observed in ΔmtrB-infected lung sections (Fig. 3D). We conclude that the absence of mtrB is associated with an attenuated phenotype of the bacterium and an absence of immunopathology in the lung comparable with the WT strain in infected mice.
Figure 3.

MtrB is required to establish active infection in mice. A and B, survival of the WT M. tuberculosis or ΔmtrB strains in the lungs (A) or spleen (B) of mice after aerosol infection with ∼100 bacilli/lung. Error bars represent S.D. and the means of cfu obtained with four animals per strain from one experiment. ***, p < 0.001; *, p < 0.05. C, sizes of the spleens from two individual mice infected with either WT or ΔmtrB for 14 weeks. Scale, 3 cm. Images are representative of one experiment with four animals per strain. D, histopathology of lung sections collected after 12 weeks of infection with the indicated strains. Sections were stained with hematoxylin and eosin and observed under a light microscope at 40× magnification. G denotes granuloma (red arrow), and AS (black arrow) denotes alveolar space. Images are representative of one experiment with four animals per strain. Scale bar, 10 μm.
MtrB regulates the cytokine response during infection
Considering the attenuation of ΔmtrB as well as the absence of immunopathology in the mouse lung infected with ΔmtrB, we tested the expression of cytokines both ex vivo and in vivo. The release of proinflammatory cytokines TNFα and IL-6 were lower in the case of BMDMs infected with ΔmtrB compared with the WT (Fig. S2). This was reversed in ΔmtrB::mtrB. Similar to the results observed ex vivo, ΔmtrB elicited lower expression of Tnfα and Il6 in the lungs of infected mice (Fig. S2). These results were in harmony with the lack of immunopathology in the lungs of mice infected with ΔmtrB.
Transcriptome analysis shows perturbation of important pathways, processes, and nodes in the transcriptional network in ΔmtrB
To gain insights into the role of MtrB in regulating morphology, growth behavior, biofilm formation, and survival of M. tuberculosis within its host, we performed genome-wide transcriptional profiling of the WT and ΔmtrB. System-level analyses of the transcriptome showed that 1,014 genes were differentially expressed in ΔmtrB. 551 of these 1,014 genes were up-regulated in the mutant, whereas 463 genes were down-regulated. The differentially regulated processes were mapped according to their functions annotated in TubercuList https://mycobrowser.epfl.ch/ (57).10 Strikingly, a large number of genes down-regulated in ΔmtrB mapped to crucial functions/processes associated with cell wall biosynthesis/cell division, central carbon metabolism/respiration, and the response to hypoxia (Fig. 4, A and B). For the sake of clarity, we will focus on these down-regulated genes in this report.
Figure 4.

MtrB is a global regulator of the M. tuberculosis transcriptome. A, pie chart representing the distribution of genes down-regulated in ΔmtrB grown in vitro. B, heat maps of differentially regulated genes grouped according to their TubercuList functions or regulons. C, Venn diagrams representing the overlap between genes down-regulated in ΔmtrB and genes up-regulated in M. tuberculosis upon infection (left) or essential for growth of M. tuberculosis (middle) or up-regulated in M. tuberculosis under acid stress (right). The green arrow represents “down-regulated genes.”
Within its host, M. tuberculosis resides in acidic compartments such as the macrophage or the caseum of a necrotic granuloma. This requires sensing and responding to the acidic environment. A large number of genes are up-regulated when M. tuberculosis senses acidic pH or the phagosomal environment of macrophages. To obtain information on whether altered gene expression of ΔmtrB could possibly compromise its response to these environments, we tabulated genes that are up-regulated within macrophages (17) or under acid stress (18) but down-regulated in ΔmtrB (compared with the WT) grown in vitro. 101 genes (Table S1), which are induced upon infection of macrophages by M. tuberculosis, were down-regulated in ΔmtrB grown in vitro, and 32 genes (Table S2) that are part of the M. tuberculosis acid response were also down-regulated in the mutant (Fig. 4C). This was in line with our observation that ΔmtrB was compromised in its ability to respond appropriately to acidic pH. Furthermore, of the set of M. tuberculosis genes deemed essential for growth (19, 20), 71 were down-regulated in ΔmtrB compared with the WT (Table S3).
To better understand the effects of mtrB deletion at system level, we integrated an existing protein–DNA (pd) network of M. tuberculosis (21) with the protein–protein (pp) interaction network obtained from the STRING database to create a static network of M. tuberculosis pd and pp interactions. The microarray data available to us was mapped onto this network, enabling us to visualize the dysregulation of genes and interactomes linked to mtrB deletion. Dysregulation of genes likely of relevance to bacterial fitness under hypoxia and in macrophages is discussed below.
The transcriptional response to hypoxia and macrophage infection
Network analysis showed that a number of interacting partners of MtrB were down-regulated in ΔmtrB. The TCS DosR (RR)/DosS (SK) (also known as DevR/DevS) has been implicated in the initial response to hypoxia and in sensing NO, carbon monoxide, and acidic pH and for M. tuberculosis persistence in macaques (22, 23). A striking observation was the down-regulation of the dosR regulon in ΔmtrB (Figs. 4B and 5A). The majority of connections to DosR, including the pp connections, were down-regulated in ΔmtrB (Fig. S3A). We validated the down-regulation of several of these genes by qRT-PCR (Fig. 5B). qRT-PCR of genes representative of the dosR regulon, namely dosR, dosS, narX, narK2, pfkB, fdxA, Rv0081, and tgs1, confirmed their down-regulation in ΔmtrB compared with the WT (Fig. 5B). This was reversed in ΔmtrB::mtrB. These results were in conformity with the compromised survival of ΔmtrB under hypoxia. NarK2 is a H+/nitrate symporter, and narX is a part of the narK2X operon that is induced under hypoxia (24). pfkB, although encoding a putative phosphofructokinase, is reportedly devoid of phosphofructokinase activity (25). Its expression is enhanced upon hypoxia or growth arrest or in the intraphagosomal environment. fdxA encodes a ferredoxin and is induced at low pH (26) and in infected macrophages (27). Rv0081 is a transcriptional regulator that is part of an operonic locus under complex control (28). In silico modeling suggests that Rv0081 and Rv0082 could be responsible for communicating dormancy signals to the respiratory chain (29). Rv0081 is induced early during hypoxia and connects the early to the later hypoxic response by regulating the transcription factor Rv3334 (30). The biosynthesis of the major storage lipid triacylglycerol (TAG) depends on the TAG synthases tgs1 and tgs2 (31). tgs1 is the principal TAG-synthesizing enzyme. Shi et al. (32) have reported that during growth arrest, there is a shift of carbon metabolism from providing biosynthetic precursors and energy to the synthesis of storage compounds such as TAG, which depends on tgs1. tgs1 was down-regulated in ΔmtrB (Figs. 4B and 5B).
Figure 5.

MtrB regulates expression of genes of the DosR regulon. A, network representation of the dosR regulon showing a subset of genes differentially regulated in ΔmtrB. The blue edges denote protein–protein interactions; the pink edges denote protein–DNA interactions. Rectangular nodes represent transcription factors; circular nodes represent other proteins. The color scale of the nodes was assigned according to the log2 -fold change in gene expression in ΔmtrB compared with the WT (set as 1). B, relative expression of genes of the DosR regulon in ΔmtrB and ΔmtrB::mtrB compared with the WT under normoxia (set as 1). C, expression of DosR regulon genes after recovery from hypoxia analyzed at the indicated time intervals. D, expression of genes of the DosR regulon in M. tuberculosis strains after infection of macrophages. C and D, Error bars represent S.D., n = 3. *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ns, nonsignificant.
A large set of 230 genes is expressed at 4 and 7 days of hypoxia, constituting the “enduring hypoxic response” (33). Of these genes, 32 were down-regulated in ΔmtrB (Fig. S3B and Table S4), suggesting an inability of ΔmtrB to persist during long-term oxygen deprivation (Fig. 1H). 24 h after establishment of hypoxia, there was a striking increase in expression levels of dosR, narK2, and Rv0081 in the WT (Fig. 5C, left panel) (compared with the expression levels before hypoxia) but not in ΔmtrB (Fig. 5C, right panel), raising the possibility that the inability to turn on the DosR regulon contributes at least in part to the inability of ΔmtrB to survive under hypoxia. Although DosR has been linked to the survival of M. tuberculosis under hypoxia, we noted that the levels of transcription of dosR under normoxic conditions were lower in ΔmtrB compared with the WT (Fig. 5B).
Rohde et al. (18) and Peterson et al. (34) have reported that dosR regulon genes are up-regulated early during infection of macrophages with M. tuberculosis, although this is unlikely to be due to environmental cues such as hypoxia, nitric oxide, or carbon monoxide. To decipher the role of MtrB in regulating the expression of DosR regulon genes during in vitro infection, we tested the expression of selected genes in bacteria residing in macrophages by qRT-PCR. We confirmed that ΔmtrB is compromised with respect to transcription of genes associated with the dosR regulon (dosR, dosS, narK2, pfkB, fdxA, Rv0080, and Rv0081) compared with the WT (Fig. 5D). These results suggested that MtrB regulates transcription of DosR regulon genes that are possibly required for adaptation of the bacterium to the macrophage environment. These results were also in conformity with the compromised survival of ΔmtrB in macrophages (Fig. 2A).
In view of the above observations, we speculated that MtrB could potentially be involved in the transcription of basal levels of dosR under normoxia, possibly through physical interaction with DosR. To test whether DosR and MtrB interact physically, S-MtrB (cytosolic domain)-expressing Escherichia coli lysate was incubated with His-DosR (expressed in E. coli) immobilized on Ni2+-NTA-agarose. Bound proteins were positive for the presence of MtrB when analyzed by immunoblotting with S-antibody (Fig. 6A, last lane). As a negative control, lysates of E. coli expressing the cytosolic domain of another sensor kinase, KdpD (of the TCS KdpDE) (2), were incubated with immobilized His-DosR. No interaction was observed. We also tested for interaction between MtrB and DosR within mycobacteria using the mycobacterial protein fragment complementation (M-PFC) method. MtrB and DosR interacted when expressed in M. smegmatis (Fig. 6B). These results strengthened our view that MtrB interacts with DosR. To test whether these interactions regulate the DNA-binding ability of DosR, we performed electrophoretic mobility shift assays (EMSAs) to test the binding of DosR to an hspX promoter–derived DNA (35) in the absence or presence of MtrB. DosR (which had not been phosphorylated in vitro) did not show significant binding to hspX DNA at a concentration of 2.5 μm. However, a mobility shift was clearly visible when DosR was incubated with varying concentrations of MtrB prior to use in EMSAs (Fig. 6C). These results suggested that by virtue of interacting with DosR, MtrB enhances the DNA-binding ability of DosR. MtrB alone did not bind to the hspX DNA (Fig. S3C). DosR is autoregulated and binds to its own promoter. Similar to the hspX promoter, MtrB could enhance the binding of DosR with its own promoter (Fig. S3D). The mutation H305A rendered MtrB kinase-inactive (Fig. S3E). This kinase-inactive mutant was able to enhance the binding of DosR to the hspX or the dosR promoter DNA in the same manner as the WT (Fig. S3, F and G), suggesting that the ability of MtrB to enhance DosR activity does not require the kinase activity of MtrB. In vitro pulldown (Fig. S3H) and M-PFC assays in the mycobacterial environment (Fig. S3I) confirmed that both the WT and the kinase-inactive mutant of MtrB interact with DosR. We contend that interaction of the cytosolic domain of MtrB with DosR is required for binding of DosR to DNA under normoxia in a phosphorylation-independent manner and subsequent survival of the bacterium under hypoxia.
Figure 6.
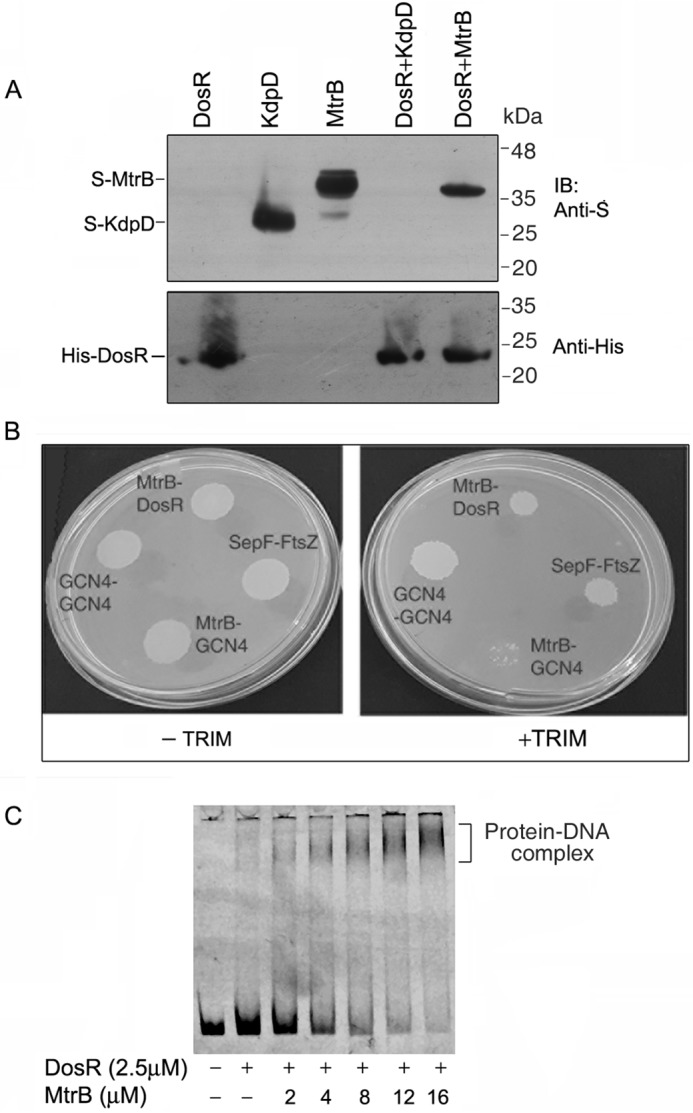
M. tuberculosis DosR interacts with MtrB and regulates DNA binding of DosR. A, S-MtrB (cytosolic domain)–expressing E. coli lysate (or KdpD (cytosolic domain)-expressing E. coli lysate as a negative control) was incubated with His-DosR (expressed in E. coli) immobilized on Ni2+-NTA-agarose. After washing the protein-bound resin, bound proteins were analyzed by immunoblotting (IB) with S-antibody and reprobed with His antibody. Lanes 1, 2, and 3, cell-free lysates of E. coli expressing His-DosR, S-KdpD, and S-MtrB (cytoplasmic domain), respectively; lanes 4 and 5, pulldowns of lysates expressing S-KdpD or S-MtrB, respectively, with DosR-bound Ni2+-NTA-agarose. B, M-PFC assay was used to determine association of DosR with MtrB. M. smegmatis expressing DosR fused to one half of murine DHFR and MtrB (cytosolic domain) fused to the other half of murine DHFR was grown in the absence or presence of TRIM. Plates were incubated at 37 °C for 7 days. Growth on TRIM indicated reconstitution of DHFR, which occurs when there is interaction between the protein pair. GCN4–GCN4 and SepF–FtsZ served as positive controls, and MtrB–GCN4 served as a negative control for protein–protein interaction. C, binding of DosR to the hspX promoter was analyzed by EMSA. EMSA was performed by incubating a Cy5-labeled PCR fragment derived from the hspX promoter with DosR in the absence or presence of different concentrations of MtrB. The reaction mixture was separated by PAGE, and the DNA–protein complex was visualized using a Typhoon biomolecular imager.
The division cell wall (DCW) cluster, cell division, and peptidoglycan synthesis
Considering that the cells of ΔmtrB were increased in length compared with the WT, we searched the transcriptomic data for changes in expression of genes associated with cell division. The DCW cluster in M. tuberculosis includes essential cell division genes such as ftsZ, ftsQ, ftsW, wag31, and sepF and the mur enzymes that are required for peptidoglycan synthesis (36). These genes are required for cell wall homeostasis, cell division, growth, and survival of M. tuberculosis. Microarray analysis (Fig. 4B) and qRT-PCR (Fig. 7A) showed that genes of the DCW cluster (ftsZ, ftsQ, and ftsW) were down-regulated in ΔmtrB. ilvE encodes an enzyme that catalyzes the conversion of valine to alanine for peptidoglycan synthesis. It was down-regulated in ΔmtrB (Fig. 7A). Furthermore, the mur enzymes that catalyze ligation and transferase reactions in peptidoglycan synthesis, namely murC, murF, murG, and murX, were down-regulated, as evidenced in the microarray data (Fig. 4B). The down-regulation of murG, the final enzyme in the peptidoglycan synthesis pathway, was validated by qRT-PCR (Fig. 7A). The slower growth rate and increased cell length of ΔmtrB are likely consequences of decreased expression of cell division and peptidoglycan-synthesizing genes. Previous studies have shown that transposon insertions in genes linked to cell wall synthesis render M. tuberculosis hypersusceptible to acid stress (37). The increased susceptibility of ΔmtrB to acid stress (Fig. 1G) was in harmony with the compromised expression of genes linked to cell wall synthesis. This effect was reversed in the mtrB-complemented strain.
Figure 7.
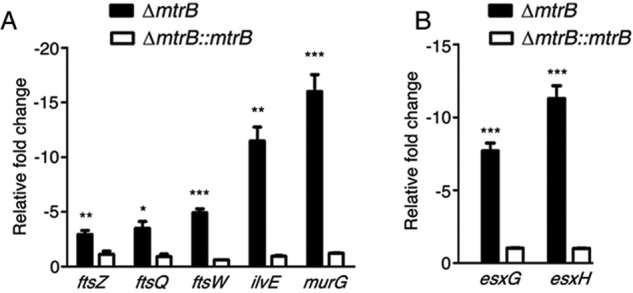
MtrB is required for expression of genes associated with peptidoglycan biosynthesis and lysosomal trafficking. A and B, relative expression of genes associated with peptidoglycan biosynthesis and cell division (A) or regulation of lysosomal trafficking (B) from ΔmtrB (filled bars) and ΔmtrB::mtrB (open bars) was quantitated by qRT-PCR. Gene expression is shown relative to that of the WT (set as 1). Error bars represent S.D., n = 3. ***, p < 0.001; **, p < 0.01; *, p < 0.05.
Genes regulating lysosomal trafficking
M. tuberculosis harbors five chromosomal esx clusters that encode specialized type VII secretion systems (38). esxG and esxH are components of the ESX-3 secretion system. The ESCRT machinery is required to deliver M. tuberculosis to the lysosome. The EsxH–EsxG complex disrupts ESCRT function and impairs phagosome maturation (39). We observed from the microarray data that esxG and esxH were down-regulated (Fig. 4B) in ΔmtrB compared with the WT. This was confirmed by qRT-PCR (Fig. 7B). The attenuated expression of esxG and esxH likely accounts in part for the greater trafficking of ΔmtrB to the lysosome compared with the WT (Fig. 2C).
Discussion
One of the major challenges in the management of tuberculosis is the lack of detailed understanding of the players that enable the bacterium to evade immune surveillance mechanisms and to withstand the harsh environment prevailing within its host. In a recent study, Gorla et al. (40) reported the knockout of mtrB in M. tuberculosis only in the MtrA(Y102C) genetic background. In this study, we conclusively demonstrate that mtrB can be inactivated (even in the absence of MtrA(Y102C)) and that MtrB is not essential in M. tuberculosis. However, ΔmtrB was compromised in terms of its ability to survive in macrophages compared with the WT. It is known that M. tuberculosis inhibits phagosome maturation and prevents acidification of phagosomes harboring live bacilli as well as their fusion with lysosomes. Recent reports suggest that M. tuberculosis survival and immune evasion are at least in part due to the suppression of autophagy and lysosomal trafficking. In line with this, a greater proportion of ΔmtrB colocalized with LC3 and LAMP1, markers of autophagosomes and lysosomes, respectively (Fig. 2). EsxH and EsxG in concert disrupt the function of the ESCRT machinery and hinder phagosome maturation (41). The down-regulation of esxG and esxH in ΔmtrB (Fig. 7B) is possibly linked to its diminished ability to subvert the host phagolysosomal fusion pathway and to survive in macrophages. We have therefore established that ex vivo, MtrB is required for subversion of the innate immune response, in terms of suppression of phagolysosomal trafficking of the bacterium. In vivo, bacterial cfu were much lower in the organs of mice infected with ΔmtrB compared with the WT (Fig. 3). These results suggest that the absence of MtrB attenuates the bacteria during in vivo infection. The inability to survive under acid stress and prevent phagolysosomal trafficking correlates directly with the attenuated phenotype of ΔmtrB. This is in concordance with previous studies reporting the attenuated phenotype of mutants that are susceptible to acid stress (37). Furthermore, infection with ΔmtrB was characterized by a lack of immunopathology in the lungs of mice. This was in harmony with the decreased expression of Tnfα and Il6 in the infected tissues. TNFα and IL-6 have been reported to be required for the initiation and maintenance of granuloma formation (42). To test whether the phenotype of ΔmtrB is associated with proinflammatory cytokine release from macrophages, we measured TNFα and IL-6 release from infected RAW264.7 cells. The release of the aforesaid cytokines was expectedly attenuated in the ΔmtrB-infected cells and restored upon complementation with mtrB (Fig. S2), strengthening the link between MtrB and the ability of M. tuberculosis to release cytokines involved in granuloma formation and the inflammatory response.
Considering that M. tuberculosis is exposed to a variety of stresses in its host, we tested the ability of ΔmtrB to withstand stress. We used the static culture hypoxia model described by McGillivray et al. (43) to test the role of MtrB in hypoxic survival. ΔmtrB was unable to survive under hypoxia, a characteristic that is partially restored in ΔmtrB::mtrB (Fig. 1H). DosR assists in maintaining metabolic homeostasis during hypoxia and enables recovery from dormancy (44, 45). The transition of M. tuberculosis to nonreplicating persistence is marked by induction of DosR genes and rerouting of carbon flux toward the synthesis of storage triacylglycerol. We observed by transcriptional profiling that MtrB is required for transcription of the dosR regulon under hypoxia, under normoxia, and in macrophages. This study therefore brings to light a hitherto unanticipated function of MtrB as a regulator of the DosR network. Interestingly, we demonstrated using pulldowns of recombinant MtrB from E. coli lysates with immobilized DosR that MtrB interacts directly with DosR (Fig. 6). This interaction was confirmed in the mycobacterial system as well. A phosphorylation-incompetent mutant of MtrB, H305A, was also able to interact with DosR in vitro. Vasisht et al. (46) have reported that phosphorylated DosR binds to the hspX promoter at a concentration of 0.5 μm, whereas nonphosphorylated DosR shows weak binding even at a concentration of 3 μm. We have demonstrated that nonphosphorylated DosR did not show binding to the hspX promoter at a concentration of 2.5 μm. However, binding was clearly enhanced in the presence of MtrB (Fig. 6). In addition, we have demonstrated that the phosphorylation-incompetent MtrB mutant H305A was also capable of enhancing DosR binding to the hspX promoter, suggesting that the DosR–MtrB interaction enhances the DNA-binding ability of DosR in a phosphorylation-independent manner. We hypothesize that this interaction is possibly required for maintaining basal levels of DosR under normoxic conditions in a signal-independent manner. The underlying mechanism of MtrB-dependent binding of DosR to specific promoter regions remains to be elucidated. The dosR–dosS operon is autoregulated (47). DosR binds to its own promoter in a sequence-specific manner and regulates dosR promoter activity. Our results demonstrated that MtrB could augment DNA-binding ability of DosR to its own promoter in a phosphorylation-independent manner (Fig. S3). This binding is likely linked to the activation of the dosR regulon. It is plausible that binding of MtrB with DosR induces conformational changes in DosR that could enhance its DNA-binding ability, allowing maintenance of basal levels of DosR under normoxia when DosS is not active and DosR is not phosphorylated. In harmony with this, ΔmtrB was unable to launch a transcriptional response similar to the WT under hypoxia (Fig. 5C) and is compromised in its ability to activate the DosR regulon when grown in macrophages (Fig. 5D).
Another important observation is the inability of ΔmtrB to form biofilms (Fig. 1E). The growth of pathogens in organized biofilm-like structures in tissues promotes persistence. In the case of M. tuberculosis, biofilms are associated with a drug-tolerant phenotype (16, 48), rendering the inability of ΔmtrB to form biofilms particularly significant. The compromised production of ketomycolic acids in ΔmtrB possibly accounts, at least in part, to its inability to form biofilms. How MtrB regulates the synthesis of ketomycolic acids awaits further investigation. It has earlier been reported that the pknG mutant of M. tuberculosis is compromised in terms of its ability to survive under hypoxia or to form biofilms and at the same time shows an attenuated phenotype in an animal model and a compromised ability to elicit granuloma formation (49). Therefore, the inability of ΔmtrB to form biofilms or to survive under hypoxia is likely linked to the attenuated phenotype of ΔmtrB. MtrA(Y102C) is a gain-of-function mutant of MtrA, which functions in the absence of MtrB. We observed that complementation of ΔmtrB with mtrA(Y102C) did not restore biofilm formation. Based on these observations, we suggest that besides its role as the cognate sensor kinase of MtrA, MtrB also exerts phosphorylation-independent functions of significance to global regulation of the M. tuberculosis transcriptome. Along somewhat similar lines, it has previously been reported that MtrB regulates the cellular localization of Wag31 in a phosphorylation-independent manner (5).
Cell wall homeostasis is impaired in mtrB-inactivated mutants of Mycobacterium avium (10). In Corynebacterium glutamicum, MtrB plays an important role in maintaining cell shape and morphology (11). Here, we show compromised expression of genes linked to peptidoglycan synthesis and cell division in ΔmtrB (Fig. 7A), which is likely linked to altered cell length of ΔmtrB compared with the WT. These are likely to reduce bacterial fitness under stress.
In conclusion, our study highlights a novel role of MtrB as an important regulatory hub in the M. tuberculosis gene expression network that controls the ability of M. tuberculosis to (a) respond to hypoxia, (b) subvert phagolysosomal trafficking, (c) form biofilms, (d) maintain cell wall homeostasis, and (e) regulate mycolate synthesis. We suggest that the regulation of these processes by MtrB is intimately linked to the ability of M. tuberculosis to infect mice because ΔmtrB was compromised in its ability to establish infection in vivo. The inability of ΔmtrB to survive under hypoxia suggests that MtrB is likely to be critical for persistence of M. tuberculosis within the host. Therefore, our studies bring to light the possibility of targeting MtrB for the development of novel therapeutics for the management of tuberculosis.
Materials and methods
Strains and growth conditions
M. tuberculosis H37Rv was grown in Middlebrook 7H9 medium containing 0.05% Tween 80 and 10% bovine albumin, dextrose, and catalase (ADC) (Difco) unless specified otherwise. The mtrB deletion strain (ΔmtrB) and the complemented strain (ΔmtrB::mtrB) were grown in the same medium containing 20 μg/ml kanamycin or 20 μg/ml kanamycin plus 50 μg/ml hygromycin B, respectively.
Inactivation of mtrB
The inactivation of mtrB was carried out in M. tuberculosis H37Rv using specialized transducing mycobacteriophages for allelic exchange. We inactivated mtrB by cloning 800-bp regions upstream (primers P1 and P2) and downstream (primers P3 and P4) of the gene into pYUB854Kan, ligating the construct to phAE159 to generate the allelic exchange substrate, and delivered the substrate by specialized transducing mycobacteriophages as described previously (50). The hygromycin cassette in pYUB854 was replaced with a kanamycin cassette from pMV261. Double crossovers were selected on 7H11 plates containing kanamycin. Positive colonies were verified by PCR amplifying a region 200 bp upstream and downstream of mtrB (primers P5 (A1) and P6 (A2)) and the kanamycin cassette (primers P7 (B1) and P8 (B2)). Southern blotting confirmed the deletion. The mtrB-complemented strain (ΔmtrB::mtrB) was generated by cloning the WT full-length mtrB in pMV306 (Hyg+) and introducing the construct into the ΔmtrB strain. The list of primers is provided in Table S5.
His-tagged mtrA(Y102C) was generated as described previously (51). Control vector pLAM12 or mtrA(Y102C)::pLAM12 was electroporated separately into ΔmtrB. WT, ΔmtrB, or ΔmtrB::mtrB was grown to midexponential phase at 37 °C in Middlebrook 7H9 broth with appropriate antibiotics followed by induction with acetamide. Cultures were harvested and lysed in PBS containing protease inhibitor mixture (Cell Signaling Technology) in a mini bead beater. Proteins were solubilized in buffer containing 1% (v/v) Triton X-100 for 4 h at 4 °C with shaking. Expression of His-MtrA(Y102C) or MtrB was confirmed by Western blotting bacterial lysates with anti-His (Abcam) or anti-MtrB (raised in our laboratory) antibody, respectively.
Aggregation and colony morphology
To test the aggregation phenotype, M. tuberculosis was grown in MB 7H9 containing 0.1% Tween 80 and 10% ADC to late log phase and left standing for 10 min at room temperature. For colony morphology, log-phase cultures were spotted on Dubos agar and allowed to grow for 4 weeks.
Biofilm formation
Log-phase cultures of M. tuberculosis strains were diluted 1:100 in 5 ml of Sauton's medium without detergent in T25 nonvented tissue culture flasks. The caps were wrapped in Parafilm, and flasks were incubated at 37 °C for 6 weeks as described by Ojha et al. (52). Caps were loosened after 4 weeks of incubation to facilitate biofilm growth. After 6 weeks, media below the biofilms were aspirated with a Pasteur pipette, and the film was allowed to dry. Crystal violet was added to the films and washed with water followed by reconstitution in alcohol after 15 min. Absorbance at 595 nm was measured to quantify the stain in the supernatant of each sample.
Mycolic acid isolation and analysis
Log-phase cultures were harvested and dried, and cells (50 mg) were treated with 40% tetrabutylammonium hydroxide (Merck) at 100 °C overnight. The mixture was cooled to room temperature. 2 ml of dichloromethane and 300 μl of methyl iodide (Merck) were added, and the mixture was rotated at room temperature for 1 h. Phase separation was carried out by centrifugation at 1,400 × g for 2 min. The aqueous phase was removed, and the organic phase was washed first with 2 ml of 3 n HCl followed by two washes with 2 ml of water. The organic phase was removed and dried followed by reconstitution in dichloromethane. Mycolic acid methyl esters were analyzed by TLC using petroleum ether:diethyl ether (95:5, v/v) six times. TLC plates were developed by charring after spraying with 20% (v/v) methanolic H2SO4.
Macrophage infections
BMDMs were prepared from the bone marrow obtained from the femora of BALB/c mice by culturing in Iscove's modified Dulbecco's medium containing 10% fetal bovine serum and macrophage colony-stimulating factor at 37 °C with 5% CO2 until differentiation was complete. Infections were carried out at an m.o.i. of 10 for 4 h followed by treatment with gentamycin for 2 h.
Immunofluorescence microscopy
For colocalization experiments, bacteria were labeled with 100 μg/ml FITC at 4 °C overnight. BMDMs were infected with FITC-labeled bacteria at an m.o.i. of 10 on coverslips. At the end of each infection, cells were fixed with 4% (v/v) paraformaldehyde for 10 min at 25 °C followed by permeabilization with 0.01% Triton X-100 in PBS for 10 min. The cells were then treated with 2% BSA in PBS for 90 min followed by treatment with either anti-LC3 antibody (MBL International) or anti-LAMP1 antibody (Abcam) overnight at 4 °C. Cells were treated with Alexa Fluor 546–conjugated goat anti-rabbit antibody (Abcam), and coverslips were mounted with Slowfade (Thermo Scientific). Slides were imaged by confocal microscopy.
Animal infections
All mouse experiments were approved by the institutional Animal Ethics Committees of the National JALMA Institute, Agra, India, and the Bose Institute, Kolkata, India. BALB/c mice were purchased from Central Drug Research Institute, Lucknow, India and infected with aerosolized M. tuberculosis at the National JALMA Institute for Leprosy and Other Mycobacterial Diseases, Agra, India. Successful infection was confirmed by determination of cfu from the lungs of mice on Day 1. Each dose was ∼100 cfu. Mice were sacrificed after 0, 4, 12, and 14 weeks of infection, and cfu were determined from the lungs and spleen. Lungs, after 4 weeks of infection, were homogenized in TRIzol, and RNA was isolated using the miRVANA RNA isolation kit (Ambion) following the manufacturer's protocol. For histopathology, infected lungs after 12 weeks were fixed in formalin, embedded in paraffin, and stained with hematoxylin and eosin. Sections were analyzed by a certified pathologist having no knowledge of the sample sets.
Microarray analysis
RNA was isolated from M. tuberculosis using the Qiagen RNA isolation kit. Labeling was carried out using Agilent's Quick Amp kit (cDNA synthesis and in vitro transcription) for 500 ng of RNA. Microarray-based gene expression profiling was carried out on an Agilent platform (8 × 15,000 format). Microarray data analysis, normalization of data, and statistical analysis were carried by Genotypic Technology Pvt. Ltd., Bangalore, India using Agilent Genespring GX and Genotypic Biointerpretor database. Log2 -fold change cutoff for up- or down-regulated genes was set to 0.8. Gene expression changes in the mutant greater than or equal to 0.8-fold of the WT were considered up-regulation, and changes less than or equal to −0.8-fold of WT were considered down-regulation.
Networks and heat maps
The M. tuberculosis transcription factor network was obtained from Minch et al. (21). The pp interaction data were obtained from the STRING v10 (53) database online. These two data sets were merged to form a larger M. tuberculosis interaction network containing both protein–DNA and protein–protein interactions. The microarray data were integrated into this network, and regulons that displayed marked dysregulation were illustrated with Cytoscape v2.8.3 (54). The blue connections represent pp interactions, whereas the red connections show pd interactions. Diamond-shaped nodes indicate transcription factors, and elliptical nodes represent other proteins. The color of each node defines the expression level of that gene in the microarray data and ranges from red (high) to green (low). Heat maps were generated using the “heatmap.2” function of the “gplots” package in R, with the color gradient from green (low) to red (high).
RNA isolation from intracellular bacteria
RNA was prepared from intracellular M. tuberculosis following the method of Rohde et al. (18). Briefly, RAW264.7 cells were seeded at 107 cells/T75 flask in four flasks for each strain of M. tuberculosis. The cells were infected at an m.o.i. of 10 for 4 h followed by treatment with gentamycin for another 2 h to remove extracellular bacteria. The cells were then washed with PBS and lysed in guanidinium thiocyanate buffer containing N-lauryl sarcosine, sodium citrate, and β-mercaptoethanol. Lysates were centrifuged, and pelleted bacteria were washed with PBS containing 0.1% Tween 80. The pellet was lysed by the addition of lysozyme and TRIzol followed by bead beating. RNA was prepared by chloroform extraction followed by application to an RNeasy kit column (Qiagen). RNA was treated with DNase and quantified.
RNA isolation and qRT-PCR
Cell pellets were harvested from growing cultures, reconstituted in a lysis buffer containing guanidinium thiocyanate and β-mercaptoethanol, and lysed using a bead beater. RNA was prepared following the method of Rustad et al. (55). The lysate was centrifuged at high speed for 15 min followed by purification of RNA from the supernatant using the Qiagen RNeasy kit following the manufacturer's protocol. RNA was treated with Turbo DNase (Ambion) following the manufacturer's protocol. SYBR Green–based real-time PCR was carried out using the primers provided in Table S5. The relative expression of the target gene was normalized to the endogenous reference gene (16S rRNA). -Fold change was determined by the comparative CT method.
Measurement of cytokines
Cytokine kits were purchased from Peprotech or Thermo Fischer Scientific. Cytokines were measured in the cell-free supernatants according to the manufacturers' instructions.
Cloning, expression, and purification of recombinant proteins
The cytosolic domain of MtrB encompassing amino acid residues Ser-234 to Gly-567 was cloned between EcoRI and HindIII in pET29a+ in-frame with an N-terminal S-tag using gene-specific primer pairs P15 and P16. The construct was verified by sequencing, transformed into E. coli BL21 (DE3), and then expressed with an S-tag by induction at 25 °C for 4 h with 250 μm isopropyl 1-thio-β-d-galactopyranoside. Cells were harvested and lysed by freeze/thaw cycles, and cell-free lysates were used for further experiments. The recombinant His-tagged construct of the cytoplasmic domain of MtrB was a gift from Dr. Deepak Saini, Indian Institute of Science, Bengaluru, India, and the His-DevR (DosR)–expressing construct was a gift from Dr. Jaya Tyagi, All India Institute of Medical Sciences, New Delhi, India. MtrB and DosR were expressed in E. coli Origami (DE3) and E. coli C43 (DE3), respectively. Cells were harvested and lysed by freeze/thaw cycles, and His-tagged proteins were purified from cell-free supernatants chromatography on Ni2+-NTA-agarose.
In vitro pulldown assay
The interaction between the cytosolic domain of MtrB and DosR was tested as follows. E. coli lysate expressing His-DosR was incubated with Ni2+-NTA-agarose at 4 °C for 30 min. The slurry containing bound His-DosR was washed thoroughly followed by incubation with E. coli lysate expressing S-tagged cytoplasmic domain or MtrB or with S-tagged KdpD (a nonspecific negative control) at 22 °C for 1 h. The slurry was washed thoroughly with wash buffer containing 30 mm imidazole, boiled in SDS gel denaturing buffer, and loaded on SDS-polyacrylamide gels. The separated proteins were electroblotted onto polyvinylidene difluoride membranes, immunoblotted with horseradish peroxidase–conjugated S-tag antibody (Novagen) followed by development with LumiGLO (Cell Signaling Technology) to detect agarose-bound S-tagged protein. Blots were reprobed with His antibody.
M-PFC assay
Protein interactions were analyzed in M. smegmatis using the M-PFC assay (56). The GCN4 homodimerization domain (fused to the N terminus of dihydrofolate reductase (DHFR) was excised from the episomal plasmid pUAB100 or the integrative plasmid pUAB200 and replaced with DosR or MtrB (cytoplasmic domain), respectively. DosR was cloned between the BamHI and ClaI sites of pUAB100 using primers P17 and P18. MtrB was cloned between the MunI and ClaI sites of pUAB200 using primers P19 and P20. All constructs were verified by sequencing. Recombinant and control plasmids were separately electroporated in M. smegmatis and grown in Middlebrook 7H9 medium supplemented with 0.2% glycerol, 0.05%Tween 80, kanamycin, and hygromycin. Interacting clones were selected by spotting transformants on Middlebrook 7H11 medium containing 0.5% (w/v) glucose, kanamycin, hygromycin, and trimethoprim (TRIM) at a concentration of 20 μg/ml.
EMSAs
The binding of DosR to the promoter of hspX or dosR was analyzed by EMSA. DNA fragments encompassing the upstream regions of hspX or dosR were PCR-amplified using appropriate primer pairs (Table S6) of which one was Cy5-labeled and genomic DNA of M. tuberculosis as a template. Binding of DosR with gel-purified PCR product (10 nm) was carried out in binding buffer (20 mm Tris/HCl, pH 8, 20 mm NaCl, 50 mm CaCl2, 10 mm MgCl2, 10 mm KCl, 5% glycerol, 0.05 mg of salmon sperm DNA/μl) for 30 min at 30 °C in a final volume of 10 μl. Where mentioned, recombinant DosR was incubated with recombinant MtrB (cytosolic domain) at different concentrations in binding buffer for 30 min at 25 °C in a final volume of 10 μl. Postincubation, protein mixtures were further incubated with gel-purified PCR product (10 nm) in binding buffer for another 30 min at 25 °C in a final volume of 20 μl. The reaction mixtures were separated on 7% native polyacrylamide gels run in 0.5× Tris acetate/EDTA buffer at 4 °C and 120 V for 120 min. The protein–DNA complex was scanned on a Typhoon Trio Plus scanner (GE Healthcare).
In vitro kinase assay
Recombinant MtrB (WT or the mutant H305A) (15 μm) was incubated with 1 μCi of [γ32P]ATP (3250 Ci/mmol; Board of Radiation and Isotope Technology, India) in kinase buffer (50 mm Tris/HCl, pH 8.0, 50 mm KCl, 10 mm MgCl2) containing 50 μm ATP at 30 °C for 60 min. Reactions were terminated by adding 1× SDS-PAGE loading dye (2% SDS, 50 mm Tris/HCl, pH 6.8, 0.02% bromphenol blue, 1% β-mercaptoethanol, 10% glycerol). The samples were resolved by SDS-PAGE (12.5%). After electrophoresis, the gel was washed with distilled water and exposed to X-ray film for 1 h.
Hypoxia and acid stress
Hypoxia was established as described by McGillivray et al. (39). Briefly, M. tuberculosis strains were grown to log phase and transferred to T12.5 nonvented tissue culture flasks such that they were filled to the neck leaving ∼⅛ volume free. The flasks were closed tightly, and the mouths were secured by Parafilm. One control flask containing methylene blue for each strain was also set up as an indicator of hypoxia. The flasks were incubated at 37 °C for 4–5 days before the methylene blue was completely discolored, marking day 0 of hypoxia, and cfu were assessed at the required time points. For acid stress, M. tuberculosis strains were grown in MB 7H9 containing 0.02% tyloxapol, pH 5.5, buffered with MES. cfu of all three strains were followed over a period of 6 days, and the percent reduction in cfu of cultures grown at pH 5.5 from that of cultures grown at pH 7.0 was plotted.
Statistical analysis
All statistical analyses were performed using GraphPad Prism v5. All data have been represented as means ± S.D., and Student's t test (two-tailed) was used for comparison of two groups.
Data availability
Data sets can be found in the NCBI Gene Expression Omnibus (GEO) under accession number GSE97958.
Ethics statement
Animal experiments were approved by the Institutional Animal Ethics Committee of National JALMA Institute for Leprosy and Other Mycobacterial Diseases, Agra India (IAEC/JALMA/55/2015) and Bose Institute, Kolkata, India (IAEC/BI/34/2015). All protocols adhered to the guidelines provided by the “Committee for the Purpose of Control and Supervision of Experiments on Animals” (CPCSEA), a statutory body of the Government of India, Ministry of Social Justice and Empowerment.
Author contributions
S. K. B., J. B., and M. Kundu conceptualization; S. K. B., D. S., S. S., J. B., and M. Kundu formal analysis; J. B. and M. Kundu supervision; J. B. and M. Kundu funding acquisition; S. K. B., S. L., A. K. S., and S. B. validation; S. K. B., S. L., A. K. S., S. B., M. Kumar, S. K. S., D. S., J. B., and M. Kundu investigation; S. K. B., S. L., A. K. S., S. B., P. G., and R. S. methodology; S. K. B., J. B., and M. Kundu writing-original draft; J. B. and M. Kundu project administration; K. J., U. D. G., P. G., and R. S. resources; R. S., M. Kundu, and J. B. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Dr. Ashok Mukherjee for histopathological analysis.
This work was supported in part by Science and Engineering Research Board Grant EMR/2015/001154 (to J. B. and M. Kundu) and J. C. Bose Fellowship SB/S2/JCB-049/2016 (to J. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–3 and Tables S1–S6.
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE97958.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- TCS
- two-component system
- SK
- sensor kinase
- RR
- response regulator
- cfu
- colony-forming unit(s)
- TAG
- triacylglycerol
- EMSA
- electrophoretic mobility shift assay
- DCW
- division cell wall
- BMDM
- bone marrow–derived macrophage
- TNF
- tumor necrosis factor
- IL
- interleukin
- pd
- protein–DNA
- pp
- protein–protein
- qRT-PCR
- quantitative RT-PCR
- NTA
- nitrilotriacetic acid
- M-PFC
- mycobacterial protein fragment complementation
- ESCRT
- endosomal sorting complex required for transport
- ADC
- bovine albumin, dextrose, and catalase
- m.o.i.
- multiplicity of infection
- DHFR
- dihydrofolate reductase
- TRIM
- trimethoprim
- MB
- Middlebrook.
References
- 1. Dorman S. E., and Chaisson R. E. (2007) From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat. Med. 13, 295–298 10.1038/nm0307-295 [DOI] [PubMed] [Google Scholar]
- 2. Parish T. (2014) Two-component regulatory systems of mycobacteria. Microbiol. Spectr. 2, MGM2-0010-2013 10.1128/microbiolspec.MGM2-0010-2013 [DOI] [PubMed] [Google Scholar]
- 3. Zahrt T. C., and Deretic V. (2000) An essential two-component signal transduction system in Mycobacterium tuberculosis. J. Bacteriol. 182, 3832–3838 10.1128/JB.182.13.3832-3838.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Plocinska R., Purushotham G., Sarva K., Vadrevu I. S., Pandeeti E. V., Arora N., Plocinski P., Madiraju M. V., and Rajagopalan M. (2012) Septal localization of the Mycobacterium tuberculosis MtrB sensor kinase promotes MtrA regulon expression. J. Biol. Chem. 287, 23887–23899 10.1074/jbc.M112.346544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plocinska R., Martinez L., Gorla P., Pandeeti E., Sarva K., Blaszczyk E., Dziadek J., Madiraju M. V., and Rajagopalan M. (2014) Mycobacterium tuberculosis MtrB sensor kinase interactions with FtsI and Wag31 proteins reveal a role for MtrB distinct from that regulating MtrA activities. J. Bacteriol. 196, 4120–4129 10.1128/JB.01795-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dubrac S., Boneca I. G., Poupel O., and Msadek T. (2007) New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism, cell division and biofilm formation in Staphylococcus aureus. J. Bacteriol. 189, 8257–8269 10.1128/JB.00645-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winkler M. E., and Hoch J. A. (2008) Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in Gram-positive bacteria. J. Bacteriol. 190, 2645–2648 10.1128/JB.01682-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukushima T., Szurmant H., Kim E. J., Perego M., and Hoch J. A. (2008) A sensor histidine kinase co-ordinates cell wall architecture with cell division in Bacillus subtilis. Mol. Microbiol. 69, 621–632 10.1111/j.1365-2958.2008.06308.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Senadheera M. D., Guggenheim B., Spatafora G. A., Huang Y. C., Choi J., Hung D. C., Treglown J. S., Goodman S. D., Ellen R. P., and Cvitkovitch D. G. (2005) A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB, and ftf expression, biofilm formation, and genetic competence development. J. Bacteriol. 187, 4064–4076 10.1128/JB.187.12.4064-4076.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cangelosi G. A., Do J. S., Freeman R., Bennett J. G., Semret M., and Behr M. A. (2006) The two-component regulatory system mtrAB is required for morphotypic multidrug resistance in Mycobacterium avium. Antimicrob. Agents Chemother. 50, 461–468 10.1128/AAC.50.2.461-468.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Möker N., Brocker M., Schaffer S., Krämer R., Morbach S., and Bott M. (2004) Deletion of the genes encoding the MtrA-MtrB two-component system of Corynebacterium glutamicum has a strong influence on cell morphology, antibiotics susceptibility and expression of genes involved in osmoprotection. Mol. Microbiol. 54, 420–438 10.1111/j.1365-2958.2004.04249.x [DOI] [PubMed] [Google Scholar]
- 12. Bhatt A., Fujiwara N., Bhatt K., Gurcha S. S., Kremer L., Chen B., Chan J., Porcelli S. A., Kobayashi K., Besra G. S., and Jacobs W. R. Jr. (2007) Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc. Natl. Acad. Sci. U.S.A. 104, 5157–5162 10.1073/pnas.0608654104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lenaerts A. J., Hoff D., Aly S., Ehlers S., Andries K., Cantarero L., Orme I. M., and Basaraba R. J. (2007) Location of persisting mycobacteria in a guinea pig model of tuberculosis revealed by r207910. Antimicrob. Agents Chemother. 51, 3338–3345 10.1128/AAC.00276-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Satsangi A. T., Pandeeti E. P., Sarva K., Rajagopalan M., and Madiraju M. V. (2013) Mycobacterium tuberculosis MtrAY102C is a gain-of-function mutant that potentially acts as a constitutively active protein. Tuberculosis 93, S28–S32 10.1016/S1472-9792(13)70007-6 [DOI] [PubMed] [Google Scholar]
- 15. Ojha A. K., Baughn A. D., Sambandan D., Hsu T., Trivelli X., Guerardel Y., Alahari A., Kremer L., Jacobs W. R. Jr., and Hatfull G. F. (2008) Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69, 164–174 10.1111/j.1365-2958.2008.06274.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sambandan D., Dao D. N., Weinrick B. C., Vilchèze C., Gurcha S. S., Ojha A., Kremer L., Besra G. S., Hatfull G. F., and Jacobs W. R. Jr. (2013) Keto-mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. MBio 4, e00222–13 10.1128/mBio.00222-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kimmey J. M., and Stallings C. L. (2016) Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol. Med. 22, 1060–1076 10.1016/j.molmed.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rohde K. H., Veiga D. F., Caldwell S., Balázsi G., and Russell D. G. (2012) Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 8, e1002769 10.1371/journal.ppat.1002769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rohde K. H., Abramovitch R. B., and Russell D. G. (2007) Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2, 352–364 10.1016/j.chom.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 20. Sassetti C. M., Boyd D. H., and Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 21. Minch K. J., Rustad T. R., Peterson E. J., Winkler J., Reiss D. J., Ma S., Hickey M., Brabant W., Morrison B., Turkarslan S., Mawhinney C., Galagan J. E., Price N. D., Baliga N. S., and Sherman D. R. (2015) The DNA-binding network of Mycobacterium tuberculosis. Nat. Commun. 6, 5829 10.1038/ncomms6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mehra S., Foreman T. W., Didier P. J., Ahsan M. H., Hudock T. A., Kissee R., Golden N. A., Gautam U. S., Johnson A. M., Alvarez X., Russell-Lodrigue K. E., Doyle L. A., Roy C. J., Niu T., Blanchard J. L., et al. (2015) The DosR regulon modulate adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am. J. Respir. Crit. Care Med. 191, 1185–1196 10.1164/rccm.201408-1502OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voskuil M. I., Schnappinger D., Visconti K. C., Harrell M. I., Dolganov G. M., Sherman D. R., and Schoolnik G. K. (2003) Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713 10.1084/jem.20030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sohaskey C. D., and Wayne L. G. (2003) Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J. Bacteriol. 185, 7247–7256 10.1128/JB.185.24.7247-7256.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Phong W. Y., Lin W., Rao S. P., Dick T., Alonso S., and Pethe K. (2013) Characterization of phosphofructokinase activity in Mycobacterium tuberculosis reveals that a functional glycolytic carbon flow is necessary to limit the accumulation of toxic metabolic intermediates under hypoxia. PLoS One 8, e56037 10.1371/journal.pone.0056037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fisher M. A., Plikaytis B. B., and Shinnick T. M. (2002) Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184, 4025–4032 10.1128/JB.184.14.4025-4032.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D., Nathan C., and Schoolnik G. K. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704 10.1084/jem.20030846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He H., Bretl D. J., Penoske R. M., Anderson D. M., and Zahrt T. C. (2011) Components of the Rv0081-Rv0088 locus, which encodes a predicted formate hydrogenlyase complex, are coregulated by Rv0081, MprA, and DosR in Mycobacterium tuberculosis. J. Bacteriol. 193, 5105–5118 10.1128/JB.05562-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hegde S. R., Rajasingh H., Das C., Mande S. S., and Mande S. C. (2012) Understanding communication signals during mycobacterial latency through predicted genome-wide protein interactions and Boolean modeling. PLoS One 7, e33893 10.1371/journal.pone.0033893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun X., Zhang L., Jiang J., Ng M., Cui Z., Mai J., Ahn S. K., Liu J., Zhang J., Liu J., and Li Y. (2018) Transcription factors Rv0081 and Rv3334 connect the early and the enduring hypoxic response of Mycobacterium tuberculosis. Virulence 9, 1468–1482 10.1080/21505594.2018.1514237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daniel J., Deb C., Dubey V. S., Sirakova T. D., Abomoelak B., Morbidoni H. R., and Kolattukudy P. E. (2004) Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 186, 5017–5030 10.1128/JB.186.15.5017-5030.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi L., Sohaskey C. D., Pheiffer C., Datta P., Parks M., McFadden J., North R. J., and Gennaro M. L. (2010) Carbon flux rerouting during Mycobacterium tuberculosis growth arrest. Mol. Microbiol. 78, 1199–1215 10.1111/j.1365-2958.2010.07399.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rustad T. R., Harrell M. I., Liao R., and Sherman D. R. (2008) The enduring hypoxic response to Mycobacterium tuberculosis. PLoS One 3, e1502 10.1371/journal.pone.0001502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peterson E. J., Bailo R., Rothchild A. C., Arrieta-Ortiz M. L., Kaur A., Pan M., Mai D., Abidi A. A., Cooper C., Aderem A., Bhatt A., and Baliga N. S. (2019) Path-seq identifies an essential mycolate remodelling program for mycobacterial host adaptation. Mol. Syst. Biol. 15, e8584 10.15252/msb.20188584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chauhan S., and Tyagi J. S. (2008) Cooperative binding of phosphorylated DevR to upstream sites is necessary and sufficient for activation of the Rv3134c-devRS operon in Mycobacterium tuberculosis: implication in the induction of DevR target genes. J. Bacteriol. 190, 4301–4312 10.1128/JB.01308-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hett E. C., and Rubin E. J. (2008) Bacterial growth and cell division: a mycobacterial perspective. Microbiol. Mol. Biol. Rev. 72, 126–156 10.1128/MMBR.00028-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vandal O. H., Roberts J. A., Odaira T., Schnappinger D., Nathan C. F., and Ehrt S. (2009) Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J. Bacteriol. 191, 625–631 10.1128/JB.00932-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simeone R., Bottai D., and Brosch R. (2009) ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 12, 4–10 10.1016/j.mib.2008.11.003 [DOI] [PubMed] [Google Scholar]
- 39. Mehra A., Zahra A., Thompson V., Sirisaengtaksin N., Wells A., Porto M., Köster S., Penberthy K., Kubota Y., Dricot A., Rogan D., Vidal M., Hill D. E., Bean A. J., and Philips J. A. (2013) Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog. 9, e1003734 10.1371/journal.ppat.1003734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gorla P., Plocinska R., Sarva K., Satsangi A. T., Pandeeti E., Donnelly R., Dziadek J., Rajagopalan M., and Madiraju M. V. (2018) MtrA response regulator controls cell division and cell wall metabolism and affects susceptibility of mycobacteria to the first line antituberculosis drugs. Front. Microbiol. 9, 2839 10.3389/fmicb.2018.02839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tinaztepe E., Wei J. R., Raynowska J., Portal-Celhay C., Thompson V., and Philips J. A. (2016) Role of metal-dependent regulation of ESX-3 secretion in intracellular survival of Mycobacterium tuberculosis. Infect. Immun. 84, 2255–2263 10.1128/IAI.00197-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Welsh K. J., Abbott A. N., Hwang S.-A., Indrigo J., Armitige L. Y., Blackburn M. R., Hunter R. L. Jr., and Actor J. K. (2008) A role for tumour necrosis factor-α, complement C5 and interleukin-6 in the initiation and development of the mycobacterial cord factor trehalose 6′,6-dimycolate induced granulomatous response. Microbiology 154, 1813–1824 10.1099/mic.0.2008/016923-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGillivray A., Golden N. A., and Kaushal D. (2015) The Mycobacterium tuberculosis Clp gene regulator is required for in vitro reactivation from hypoxia-induced dormancy. J. Biol. Chem. 290, 2351–2367 10.1074/jbc.M114.615534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leistikow R. L., Morton R. A., Bartek I. L., Frimpong I., Wagner K., and Voskuil M. (2010) The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 192, 1662–1670 10.1128/JB.00926-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schubert O. T., Ludwig C., Kogadeeva M., Zimmermann M., Rosenberger G., Gengenbacher M., Gillet L. C., Collins B. C., Röst H. L., Kaufmann S. H., Sauer U., and Aebersold R. (2015) Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe 18, 96–108 10.1016/j.chom.2015.06.001 [DOI] [PubMed] [Google Scholar]
- 46. Vasisht A., Prithvi Raj D., Gupta U. D., Bhat R., and Tyagi J. S. (2016) The α10 helix of DevR, the Mycobacterium tuberculosis dormancy regulator, regulates its DNA binding and activity. FEBS J. 283, 1286–1299 10.1111/febs.13664 [DOI] [PubMed] [Google Scholar]
- 47. Bagchi G., Chauhan S., Sharma D., and Tyagi J. S. (2005) Transcription and autoregulation of the Rv3134-devR-devS operon of Mycobacterium tuberculosis. Microbiology 151, 4045–4053 10.1099/mic.0.28333-0 [DOI] [PubMed] [Google Scholar]
- 48. Marrakchi H., Lanéele M. A., and Daffé M. (2014) Mycolic acids: structures, biosynthesis and beyond. Chem. Biol. 21, 67–85 10.1016/j.chembiol.2013.11.011 [DOI] [PubMed] [Google Scholar]
- 49. Khan M. Z., Bhaskar A., Upadhyay S., Kumari P., Rajmani R. S., Jain P., Singh A., Kumar D., Bhavesh N. S., and Nandicoori V. K. (2017) Protein kinase G confers survival advantage to Mycobacterium tuberculosis during latency-like conditions. J. Biol. Chem. 292, 16093–16108 10.1074/jbc.M117.797563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bardarov S., Bardarov S. Jr, Pavelka M. S. Jr., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., and Jacobs W. R. Jr. (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017 10.1099/00221287-148-10-3007 [DOI] [PubMed] [Google Scholar]
- 51. Chatterjee A., Sharma A. K., Mahatha A. C., Banerjee S. K., Kumar M., Saha S., Basu J., and Kundu M. (2018) Global mapping of MtrA-binding sites links MtrA to regulation of its targets in Mycobacterium tuberculosis. Microbiology 164, 99–110 10.1099/mic.0.000585 [DOI] [PubMed] [Google Scholar]
- 52. Ojha A. K., Jacobs W. R. Jr, and Hatfull G. F. (2015) Genetic dissection of mycobacterial biofilms. Methods Mol. Biol. 1285, 215–226 10.1007/978-1-4939-2450-9_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., and Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rustad T. R., Roberts D. M., Liao R. P., and Sherman D. R. (2008) Isolation of mycobacterial RNA in Mycobacteria Protocols (Parish T., and Brown A. C., eds) pp. 13–21, Humana Press, New York [Google Scholar]
- 56. Singh A., Mai D., Kumar A., and Steyn A. J. (2006) Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc. Natl. Acad. Sci. U.S.A. 103, 11346–11351 10.1073/pnas.0602817103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kapopoulou A., Lew J. M., Cole S. T. (2011) The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 91, 8–13 10.1016/j.tube.2010.09.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sets can be found in the NCBI Gene Expression Omnibus (GEO) under accession number GSE97958.