

Thioredoxin system–mediated persulfidation of Cys residues controls protein function and protects them from oxidative stress.
Abstract
Irreversible oxidation of Cys residues to sulfinic/sulfonic forms typically impairs protein function. We found that persulfidation (CysSSH) protects Cys from irreversible oxidative loss of function by the formation of CysSSO1-3H derivatives that can subsequently be reduced back to native thiols. Reductive reactivation of oxidized persulfides by the thioredoxin system was demonstrated in albumin, Prx2, and PTP1B. In cells, this mechanism protects and regulates key proteins of signaling pathways, including Prx2, PTEN, PTP1B, HSP90, and KEAP1. Using quantitative mass spectrometry, we show that (i) CysSSH and CysSSO3H species are abundant in mouse liver and enzymatically regulated by the glutathione and thioredoxin systems and (ii) deletion of the thioredoxin-related protein TRP14 in mice altered CysSSH levels on a subset of proteins, predicting a role for TRP14 in persulfide signaling. Furthermore, selenium supplementation, polysulfide treatment, or knockdown of TRP14 mediated cellular responses to EGF, suggesting a role for TrxR1/TRP14-regulated oxidative persulfidation in growth factor responsiveness.
INTRODUCTION
The formation of hydropersulfide (─SSH) and hydropolysulfide (─SSnH) moieties on protein cysteine (Cys) residues has gained attention as a primary element in hydrogen sulfide (H2S) biology (1–4). Accumulating evidence indicates that per- or polysulfide species can be abundant protein modifications that may possibly act as regulators of enzymatic functions and signaling processes (4–7). In addition, on the basis of their chemical properties, it has been hypothesized that these modifications could serve as a means to protect critical Cys residues against irreversible oxidative inactivation under elevated oxidative stress (3, 8, 9). In equivalent conditions, persulfide groups are better nucleophiles than thiols because of the presence of a lone electron pair on the vicinal sulfur atom in the persulfide, resulting in the commonly known alpha effect, which increases reactivity toward oxidants and electrophiles. The stepwise oxidation of a persulfide group leads to the consecutive formation of perthiosulfenic acid, perthiosulfinic acid, and perthiosulfonic acid (─SSOH, ─SSO2H, and ─SSO3H, respectively; hereafter ─SSO1-3H); the equivalently oxidized forms of a thiol moiety are sulfenic, sulfinic, and sulfonic acids (─SOH, ─SO2H, and ─SO3H) (3). These two sets of modifications differ in the presence of an intrinsic disulfide bond in the persulfide derivatives, which renders them reducible by enzymatic or small inorganic disulfide reductants.
Early experimental support of the concept of regulated protein protection by persulfidation by Greiner et al. (8) showed that, upon H2O2 oxidation, the PTEN active-site mutant C71A could only be reactivated by dithiothreitol (DTT) if it had been preconditioned with NaHS before H2O2 exposure, causing persulfidation of C124 before oxidation. Furthermore, the prevalence of in vivo CysSSOH residues was recently reported, along with the important finding that these species react with dimedone (10). Dimedone labeling was hitherto considered specific for sulfenic acids and has been widely used for their detection (11). The relative contribution of CysSOH versus CysSSOH to dimedone labeling outcomes thus needs to be revisited.
In a cellular context, the important biological oxidant H2O2 reacts primarily with peroxiredoxins (Prxs) (12). On the basis of their high abundance and fast as well as specific reactivity with H2O2, Prx1 and Prx2 likely capture most cytosolic H2O2 (13). Prxs therefore serve as the cellular frontline antioxidant defense and also act as central hubs in catalytically transmitting oxidative redox signals to other proteins (14). Prx1 and Prx2 are so-called 2-Cys Prxs, having a highly reactive peroxidative (Cp) and a resolving (Cr) Cys residue. The extraordinary reactivity of Cp toward H2O2 stems from the unique juxtapositioning of proximal electron-donating and electron-withdrawing functional groups (15). Upon oxidation, the generated Cp sulfenic acid derivative reacts with the Cr on another Prx subunit within the multimeric enzyme to form an intermolecular disulfide link. The disulfide is recycled to the active reduced form by the thioredoxin (Trx) system. The reaction of Cp-SOH with Cr is, however, relatively slow; therefore, in the presence of excess peroxide, Cp can be further oxidized to the corresponding Cp-SO2H form. This process serves as a redox switch of the peroxidase activity, which can only be reactivated back to the active thiol form in a very slow ATP (adenosine 5′-triphosphate)– and cytosolic Trx1-consuming process catalyzed by sulfiredoxin (16).
Protein tyrosine phosphatase 1B (PTP1B) provides a classic example for redox regulation of a signaling protein (17, 18). As a member of the class I PTP superfamily of phosphatases, PTP1B features an HCX5R active-site motif with a highly nucleophilic Cys residue at position 215. Because of its low pKa (where Ka is the acid dissociation constant) (~5.4), Cys215 is susceptible to oxidation by H2O2, leading to enzymatic inactivation (17, 19, 20). Both the Trx and the glutathione (GSH) systems have been implicated in the reactivation of PTP1B (17, 18, 21–23). A unique redox property of PTP1B is the reported formation of a sulfenyl amide intermediate via the reaction of the primary sulfenic acid with an adjacent backbone nitrogen. The cyclic sulfenyl amide is readily converted back to the active thiol form by small thiols (DTT and GSH), thus providing a potential transient protection route for the enzyme from overoxidation (19). In addition, the active-site Cys can acquire a variety of modifications inside the cell, such as S-nitrosylation, S-glutathionylation, or S-persulfidation, which similarly inhibit the activity of PTP1B yet allow reactivation by the disulfide reductase systems (23–25). Of particular relevance to this study, Krishnan and coworkers (23) reported that persulfidation of Cys215 is a prevalent modification of PTP1B in response to endoplasmic reticulum stress, which reversibly inhibits the enzyme. Here, we hypothesized that reduction of the persulfidated Cys215and its oxidized states in PTP1B can provide a reversible regulatory mechanism of control also under oxidative conditions that would otherwise irreversibly destroy PTP1B function. It would also be possible that persulfidation states could thereby affect cellular responses to endogenous oxidative bursts such as those occurring during growth factor stimulation.
In the study presented here, we show that persulfidation of protein thiols is reversible by the action of disulfide reductase machineries and demonstrate using genetically modified mouse liver samples that this mechanism regulates the persulfidation status of low–molecular weight (LMW) Cys species and protein Cys residues. We show that persulfidation plays a universal role in protecting protein Cys under oxidative stress in cells. We developed a new mass spectrometry (MS) method, which allowed quantitative detection of CysSO3H and CysSSO3H modifications on protein Cys. Using this method, we demonstrated that protein CysSSO3H derivatives are abundant modifications in vivo and their levels are mediated by the Trx system in mouse liver samples. Our data also suggest that oxidation and reduction of persulfidated functional Cys thiols are likely to play a role in redox regulation of cellular signaling via reversible persulfidation and persulfide oxidation of Prx Cp residues and the active-site Cys of PTP1B. In line with this proposal, we present evidence that, in a cytosolic thioredoxin reductase 1 (TrxR1)– and thioredoxin-related protein of 14 kDa (TRP14)–dependent manner, persulfidation is involved in modulation of epidermal growth factor (EGF)–induced signaling processes in cells.
RESULTS
Persulfidation status is orchestrated by the disulfide reductase machineries in vivo
MS-based metabolomics experiments were carried out using mouse liver tissue samples to assess the relative abundance of Cys persulfide modifications and other sulfur species. The mice used in these experiments had livers that were genetically engineered so that all hepatocytes were either wild type (WT), homozygous glutathione disulfide reductase (Gsr)–null, double-homozygous TrxR1/Gsr-null, or triple-homozygous TrxR1/Gsr/Trx1–null (26, 27). Consistent with previous data indicating a regulatory role of the Trx and the GSH systems in protein [high–molecular weight (HMW)] persulfide homeostasis (28), we here used a more quantitative approach that can also assess persulfidation of metabolite (LMW) fractions (29). This showed that persulfidation levels of HMW and LMW molecules in the liver samples were critically and in a species-dependent manner responsive to disruptions of either the Trx or the GSH systems (Fig. 1). In the TrxR1-null livers, Cys persulfide (CysSSH) was significantly elevated (Fig. 1B), whereas in the Gsr-null livers, the concentrations of GSH-persulfide (GSSH) and sulfide anion (Bis-S) were significantly higher compared to the corresponding WT levels (Fig. 1, D and E). In the HMW pool, about two times larger protein CysSSH concentrations were measured in Gsr-null and TrxR1/Gsr-null compared to WT livers (Fig. 1H). Moreover, the levels of sulfide and inorganic disulfide (Bis-SS) were at least doubled in these samples, up to approximately fourfold higher levels of HMW Bis-SS in TrxR1/Gsr-null livers (Fig. 1, J and K). The levels of the sulfide oxidation product thiosulfate (HS2O3) also increased in Gsr- and TrxR1/Gsr-null livers as compared to WT livers (Fig. 1L). These latter LMW sulfur species that appeared in the HMW pool were most likely the result of Cys-polysulfide chain cleavage by the alkylating agent or other artifactual oxidation/hydrolysis liberating these species from proteins, as we demonstrated previously (30). The HMW Cys trisulfide (CysSSSH) pool was significantly smaller in all the mutant samples (Fig. 1I). This may possibly relate to elevated oxidative stress as a result of the disrupted reductase activities (27), because longer polysulfide chains are more prone to oxidation (3, 5). This explanation is also supported by the TrxR1/Gsr/Trx1-null samples, wherein the oxidative burden is highest (27), showing significantly lower Cys levels than in the WT samples (Fig. 1G) and lower CysSSH than in Gsr- or TrxR1/Gsr-null livers (Fig. 1H). These findings provided a foundation for our hypothesis that persulfidation of Cys residues under oxidative stress may play a role in protecting protein thiols from irreversible damage.
Fig. 1. Quantitative assessment for the roles of Trx and GSH systems in control of oxidized sulfur species in vivo.
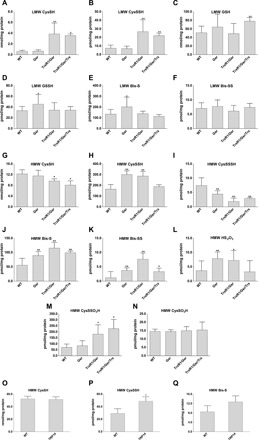
Concentrations of LMW (A to F) and protein-derived (HMW) (G to Q) thiol or persulfide and other oxidized sulfur species were determined using previously described high-sensitivity liquid chromatography–MS/MS protocols with appropriate modifications, as well as a newly developed LC-MS/MS method to detect protein CysSO3H and CysSSO3H (see Materials and Methods). Hepatocytes in the mouse liver tissue samples were WT, homozygous Gsr-null (Gsr), double-homozygous TrxR1/Gsr-null (TrxR1/Gsr), or triple-homozygous TrxR1/Gsr/Trx1-null (TrxR1/Gsr/Trx1). Livers of full-body knockout TRP14-null mice were used in (O) to (Q). The indicated LMW (A to F) and protein-derived (G to Q) sulfur species were extracted from deep-frozen tissue samples as described in Materials and Methods. Analyte levels were normalized to total protein concentrations. Data values and errors are means ± SD of measurements from n = 9 (WT), 8 (Gsr), 7 (TrxR1/Gsr), 5 (TrxR1/Gsr/Trx1), and 3 (TRP14) animals, all gender- and age-matched controls (young female adults) with similar feeding and lightning conditions. Significant differences in values compared to WT are indicated (*P < 0.05 and **P < 0.01).
To investigate the in vivo relevance of this model, we developed a new quantitative MS method to measure CysSO3H and CysSSO3H concentrations in liver proteins. The method uses internal standards that were quantified using authentic CysSO3H and CysSSO3H compounds (see Materials and Methods and fig. S1). Measurements with this method demonstrated that protein CysSSO3H (Fig. 1M) was comparable to Cys persulfidation levels (Fig. 1H) in the WT and different mutant mouse liver samples, which were 5 to 20 times higher compared to the corresponding CysSO3H concentrations (Fig. 1N). In addition, the elevated levels of CysSSO3H in TrxR1/Gsr and TrxR1/Gsr/Trx1 but not in Gsr compared to WT livers (Fig. 1M) indicate that the TrxR1-dependent Trx1 system plays a key role in regulating oxidized protein persulfide levels in vivo.
The role of TRP14 in regulation of protein persulfidation in vivo
TRP14 is a unique member of the Trx family because most protein disulfides are not substrates for this protein, in contrast to Trx1, while TRP14 likely plays roles in recovering thiols from protein CysSNO and Cys persulfide derivatives (28, 31). These observations led us to hypothesize that TRP14 may be specific to only a subset of potentially redox-regulated signaling proteins.
To assess whether TRP14 functions in protein persulfide reduction in vivo, we generated a conditional-null allele of the mouse Txndc17 gene encoding TRP14 (fig. S2). Preliminary phenotypic assessment of both full-body and liver-specific homozygous disruption of TRP14 revealed that, unlike the constitutive Trx1-null state, which is embryonic lethal (27, 32), both liver-specific and constitutively TRP14-null mice developed normally and showed no overt defects. Using livers from adult TRP14-null mice, we carried out quantitative liquid chromatography (LC)–MS measurements to assess the impacts of TRP14 on global protein persulfide levels. Moderate but significant elevation of the HMW protein-bound Cys persulfide level was observed in the TRP14-null livers compared to age- and sex-matched WT controls (Fig. 1P). These data suggest that TRP14 participates in reduction of specific persulfidated proteins. No significant differences were found either in LMW Cys persulfide levels or in other sulfide species that we measured (fig. S3), which supports the hypothesis that TRP14 is likely responsible for reducing only a subset of persulfidated proteins in vivo.
The protective effect of persulfidation illustrated on human serum albumin model protein
The chemical rationale of the hypothesized protective function of persulfidation is depicted in Fig. 2A, illustrating how previous persulfidation provides a possibility of functional reversibility in oxidation of Cys residues together with cellular reductase systems. We used human serum albumin (HSA) as a model protein to investigate the potential Cys-protecting role of per/polysulfidation under oxidizing conditions. The single surface–exposed and non–disulfide-linked Cys residue of HSA, Cys34, is readily polysulfidated upon inorganic polysulfide treatment (28). Here, polysulfide-treated HSA was subsequently further oxidized with a high concentration of H2O2 to generate the corresponding ─SxSOH, ─SxSO2H, and ─SxSO3H forms (x ≥ 1, referring to a mixture of different chain lengths). A nonpolysulfidated control was included to yield the expected ─SOH, ─SO2H, and ─SO3H oxidation products. The protein samples with these oxidized modifications on Cys34 were subsequently treated with DTT, and the reductive liberation of free HSA thiol form was finally monitored by labeling with the thiol-reactive fluorescent agent 5-iodoacetamido fluorescein (5-IAF), followed by nonreducing denaturing electrophoretic separation [SDS–polyacrylamide gel electrophoresis (SDS-PAGE)] and fluorimetric detection (Fig. 2B). DTT reduction led to an increased level of fluorescent labeling when the protein had been persulfidated in step 1 before H2O2 oxidation (Fig. 2C). This observation corroborates the hypothesis that, because of the presence of an intrinsic disulfide bond within the oxidative modification, precursor polysulfidation of Cys in proteins may yield reducible oxidation products, in contrast to the same oxidative challenge on the corresponding native Cys thiol.
Fig. 2. Reversible oxidation of HSA upon previous persulfidation.
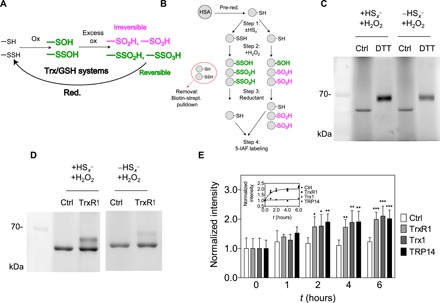
(A) Hypothesized role of the Trx or GSH system in reduction of excessively oxidized persulfide species, which become functionally reversible oxidation states (green), while nonpersulfidated Cys become irreversibly oxidized (magenta). (B) Experimental workflow for generation and detection of HSA-SSO1-3H species (for details, see Materials and Methods) (C and D) Fluorescent gels (n = 3 and 5, respectively) resulting from an experiment presented in (B) using DTT or TrxR1 as reducing agent. Excessively oxidized polysulfide species are more prone to DTT and NADPH (reduced form of nicotinamide adenine dinucleotide phosphate)/TrxR1 reduction compared to their thiol counterparts. (E) Addition of Trx1 or TRP14 does not increase the TrxR1 catalyzed reduction rate of HSA-SSO1-3H species. A representative time-resolved gel with WT TrxR1 is shown in fig. S4A (upper gel). Fluorescent signal intensities were determined densitometrically at each time point and normalized to the respective intensities at t = 0. Data points and error bars represent means ± SD of n = 5 experiments (*P < 0.05, **P < 0.01, and ***P < 0.001). The inset shows the derived kinetic curves, and the dashed lines represent a linear fit of the control (Ctrl) dataset and a single exponential least-square fit of all data points from the enzymatically reduced samples.
Enzymatic reduction of oxidized HSA polysulfide species
We previously reported that TrxR1 has polysulfide reductase capacity, further potentiated by the TrxR1 substrate Trx1 or TRP14 (28). On the basis of this, we here tested whether these members of the Trx system can also reduce oxidized HSA polysulfide species. Using the same workflow as shown in Fig. 2B, DTT was substituted in step 3 by NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) and TrxR1. The higher recovered thiol content in the polysulfide-treated samples compared to the untreated controls further illustrated the protective role of polysulfidation and, at the same time, demonstrated that TrxR1 can reduce HSA-SxSO1-3H species (Fig. 2D). Control experiments revealed that the recovery of the thiol groups from oxidized persulfide species by TrxR1 was approximately 60% under these conditions (fig. S4B). Next, we investigated whether the addition of Trx1 or TRP14 could further accelerate the reduction, which was not the case under these conditions, and all samples with TrxR1 were recovered at similar rates (Fig. 2E). Using mutant TrxR1 enzymes in which its active-site selenocysteine (Sec) residue was replaced with Ser or Cys, we demonstrated that the direct HSA-SxSO1-3H reductase activity of TrxR1 is Sec dependent (fig. S4A).
Persulfidation of the peroxidative cysteine protects Prx2 against overoxidation
Prxs are the first line of defense against H2O2 and other peroxides in cells. MS revealed that approximately 15% of Prx2 Cp was detected in its persulfidated form in normally cultured human embryonic kidney (HEK) 293 cells under the applied conditions (Fig. 3, A and D). The persulfidated fraction of Cp decreased upon treating cells with H2O2 (Fig. 3D) with a concomitant increase in the corresponding oxidized perthiosulfenic acid derivative (Cp-SSOH; Fig. 3E) and the perthiosulfinic form (Cp-SSO2H; Fig. 3F). Following knockdown (KD) of TrxR1, both the persulfidated (Fig. 3D) and perthiosulfinic acid (Fig. 3F) derivatives of Prx2 Cp were elevated compared to normal controls, suggesting a role for the Trx system in recovering the active Cp thiol from the persulfidated Prx Cp. Oxidation of the native thiol form of Cp followed a similar pattern (Fig. 3, B and C).
Fig. 3. Persulfidation of the peroxidative cysteine of Prx2 is modulated by the Trx system.
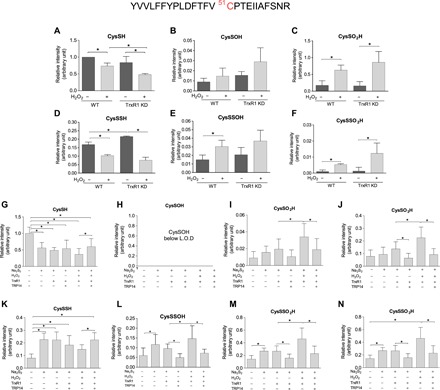
(A to F) WT and TrxR1 KD HEK293 cells were exposed to 600 μM H2O2 in Hanks’ balanced salt solution buffer at 37°C for 5 min and lysed in radioimmunoprecipitation assay (RIPA) buffer without SDS containing 5 mM iodoacetamide. The lysates were immunoprecipitated with anti-PRDX2 antibody. The eluted protein was subjected to tryptic digest in the presence of 5 mM iodoacetamide. (G to N) Recombinant human Prx2 (hPrx2) (12 μM) was incubated with or without 100 μM Na2S2 for 10 min, followed by 1 mM H2O2 for 10 min at 25°C. Samples were desalted and treated with or without TrxR1 (250 nM), TRP14 (4 μM), and NADPH (1 mM) for 1 hour at 37°C. The samples were alkylated with 1 mM iodoacetamide for 30 min, followed by tryptic digestion (37°C, 10 hours). Following LC–quadrupole time-of-flight (Q-TOF)–MS analyses, Mascot search refined the active-site Cys51-containing tryptic peptide (see at the top). Cys modifications were identified as shown in the panel headlines. The obtained intensities for modified peptides were normalized to the 93-EGGLGPLNIPLLADVTR-109 peptide fragment of Prx2 from the corresponding tryptic digest. Intensity values are plotted as a relative value compared to the CysSH peptide form from the untreated WT cells (A to F) or recombinant Prx2 (G to N). Data points and error bars represent means ± SD of n = 3 (A to F) and n = 9 experiments (G to N). *P < 0.05.
The reversal of the Prx2 oxidized persulfide species with or without exogenously added H2O2 display different, but not significantly different, steady-state levels between WT and TrxR1 KD cells (see Fig. 3, E and F). In addition, the role of TrxR1 in this system is rather complicated because, as detailed in Introduction, it is also an electron donor for Prxs via Trx1. Therefore, to investigate the Prx persulfide and oxidized persulfide reducing capacity of the Trx system, we performed MS-based experiments on isolated Prx2. As shown in Fig. 3 (G to N), when Prx2 was treated with Na2S2 followed by H2O2, TrxR1 alone (in the presence of NADPH) cannot reduce CysSSO2-3H modifications, but in the presence of TRP14, significantly less of these derivatives were detected. Despite this, it is well known that the Trx system cannot reduce CysSO2-3H species; significantly higher levels of CysSO2H and CysSO3H were detected after consecutive Na2S2 and H2O2 treatment followed by treatment with TrxR1 alone compared to when TRP14 was also added. Therefore, we checked the possibility whether the reason of the increased CysSO3H levels could be due to decomposition or reorganization of a fraction of the elevated levels of CysSSO3H species in these samples [this type of chemistry has been reported previously (33)]. Using the authentic CysSSO3H compound, we observed the formation of CysSO3H upon incubation in phosphate buffer or upon the addition of formic acid under oxidative conditions, which may serve as an explanation for the increased levels of sulfinic and sulfonic acid forms in samples that contain larger amounts of CysSSO2-3H derivatives.
Moreover, we observed a drop in the levels of the peroxidative Cys thiol upon consecutive treatment of Prx2 with Na2S2, H2O2, and NADPH/TrxR1/TRP14. This might be due to more Prx2 dimer formation upon polysulfide treatment (see fig. S5I), which cannot be reduced by TrxR1/TRP14 (34). The fact that the perthio- modifications were observed in samples, which had no polysulfide added to them, may be the result of noncomplete reduction of endogenous peroxidative Cys persulfides by the low amount (1 mM) of DTT during the protein purification phase.
On technical grounds, note that it is challenging to adequately measure the redox states of Prx Cp. Normally, large concentrations of fast-reacting alkylating agents must be used to outcompete cell lysis–induced artifactual oxidation of this extremely reactive thiol (35). However, because under these conditions protein Cys per- and polysulfides are destroyed via alkylating agent–induced polysulfur chain cleavage (30, 36), we here used mild alkylating conditions to capture the perthio- derivatives. Even milder electrophiles such as iodoacetamide have the potential to disrupt polysulfide chains on longer time scales (30). Figure S5 shows a case when tryptic digest was carried out in the absence of iodoacetamide (for 10 hours), in which case a high amount of ─SSO3H form was detected upon H2O2 treatment of HEK293 cells, which was significantly elevated in TrxR1 KD compared to WT cells (also in the absence of H2O2 treatment). In addition, the formation of CysSO3H was significantly impaired in TrxR1 KD compared to WT cells, further corroborating the role of TrxR1 in the persulfide-mediated protection of the Prx2 Cp thiol. However, it has to be noted that, using these mild alkylating conditions, the relative amounts of oxidation observed on the Cys residues of Prx compared to the reduced thiol form are likely to be overestimated. Nevertheless, our data indicate that persulfidation of the peroxidative Cys residue of Prx2 is a potential protecting mechanism against overoxidation and provide strong support that TRP14 coupled to TrxR1, which cannot reduce Prx2 dimers, is likely to be involved in the recovery of the active peroxidative thiol from its persulfide and oxidized persulfide derivatives. This model could potentially give another layer of regulation for Prx functions not only in cellular antioxidant protection but also in mediating redox signaling (see Discussion), which is worthy and require future comprehensive investigations to make firm mechanistic conclusions. These are outside the scope of the present study.
Persulfidation is a universal protective mechanism of protein Cys residues in oxidative stress
The reversible formation and physiological relevance of perthiosulfenic acid (─SSOH) species was recently reported by Heppner et al. (10). Historically, dimedone has been used to discriminate sulfenic (─SOH) derivatives from thiol (─SH) groups during oxidation mediated by reactive oxygen species (ROS) (11). However, we recently demonstrated that dimedone also reacts with perthiosulfenic acid (─SSOH) groups (10). In the study presented here, we subjected A431 cells to oxidative stress in the form of H2O2 treatment (Fig. 4), and oxidized protein modifications were captured by labeling with DCP-Bio1, a biotin-tagged dimedone-based probe. We found that about 30% of the DCP-Bio1–labeled untreated A431 cellular proteome was reducible by DTT, representing the fraction of the dimedone-labeled proteome that contained a disulfide bond and therefore was the portion corresponding to perthiosulfenic acid rather than sulfenic acid moieties (Fig. 4, A and B). As we reported earlier, protein labeling could result from either persulfide oxidation or polysulfide hydrolysis (30). To resolve the relative contributions of the oxidative effects, the samples were titrated with H2O2. The increase in dimedone-labeled proteins with increased H2O2 (up to 0.5 mM H2O2) indicated that a substantial proportion of the observed DTT-reducible perthiosulfenic acid pool arose from oxidative processes. Thus, the proposed mechanism in Fig. 2A might universally protect protein Cys residues during oxidative stress. The observed drop in Cys-dimedonylation at the highest H2O2 concentration (1 mM) is indicative of further oxidation to give, among others, perthiosulfinic or perthiosulfonic acid derivatives, as proposed in our model (Fig. 2A), which would not be labeled with dimedone. To identify specific protein targets under these experimental conditions, we affinity-captured the biotinylated fraction (representing dimedone-labeled protein Cys derivatives) and performed Western blot analyses against proteins well known to be subjected to redox regulation or have significant roles in antioxidant protection (Fig. 4, C and D). The protective effect of persulfidation against irreversible oxidation of Cys residues was apparent for all investigated proteins with redox-active thiols, namely, PTP1B, PTEN, KEAP1, and HSP90. The fact that PTP1B was affected by perthiosulfenylation was particularly interesting because previous findings suggested that Prx2 cannot transfer oxidative equivalents to PTP1B (22). Although redox regulation of PTP1B has major effects on growth factor signaling, the actual mechanisms of its oxidative inhibition are still unknown. Therefore, we further investigated how persulfidation and the Trx system can modulate PTP1B activities.
Fig. 4. H2O2-dependent formation of cellular protein SxSOH in A431 cells.
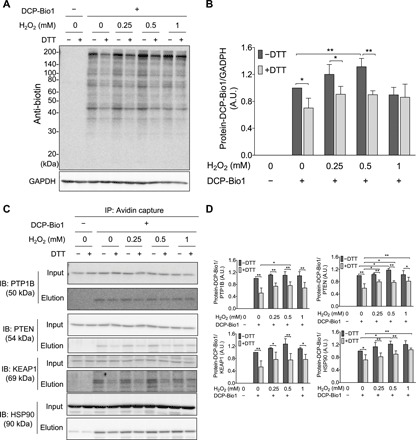
(A and B) DTT-reversible formation of oxidative modifications susceptive to dimedone labeling from whole-cell lysates. (A) A431 cells were exposed to increasing amount of H2O2 and disrupted by lysis buffer containing DCP-Bio1, a specific sulfenic acid probe containing a biotin tag. Control samples were included without the labeling agent. Cell lysates were then incubated with or without DTT, subjected to nonreducing SDS-PAGE, and immunoblotted (IB) against biotin. (B) Band intensities of the Western blot results (obtained by densitometric analyses) were plotted as a ratio of the untreated sample (─H2O2 and ─DTT). A.U., arbitrary units. (C and D) DCP-Bio1–labeled proteins were affinity-captured on avidin beads from whole-cell lysates generated as in (A), and specific proteins were identified from the eluates by Western blotting. (D) Band intensities of the membranes shown in (C). Western blots shown in (A) and (C) are representative of n = 3 independent experiments. The band intensities were assessed by densitometry using ImageJ software and normalized to (B) glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or (D) the respective protein band intensities from the input gels [whole-cell lysate before immunoprecipitation (IP)]. Each value is the mean ± SD of n = 3 independent experiments. *P < 0.05 and **P < 0.01.
Protective effect of persulfidation against irreversible oxidative inhibition of PTP1B
First, we tested whether polysulfide pretreatment could alleviate H2O2-induced irreversible inhibition of pure PTP1B in vitro. Consistent with a previous report (23), incubation of PTP1B with 50 μM Na2S2 for 10 min decreased its catalytic activity by about 70% (Fig. 5A). Subsequent treatment of this incubation mixture with 100 μM H2O2 resulted in further inhibition of PTP1B activity (Fig. 5A). DTT could partially restore PTP1B activity that was diminished by H2O2 alone (to about 25% of the initial activity after 30-min incubation) but more robustly recovered H2O2-dependent activity loss when the enzyme had been preconditioned with Na2S2 (Fig. 5A). However, when Na2S2 addition to the reaction mixture was subsequent to H2O2 treatment, it had no significant effect on the recovery of enzymatic activity by DTT. These data indicate that preemptive persulfidation can preserve reduction-mediated recovery of oxidation-induced PTP1B activity loss.
Fig. 5. Reversible inhibition of PTP1B and concomitant formation of active-site CysSSO1-3H derivatives upon inorganic polysulfide and H2O2 treatment.
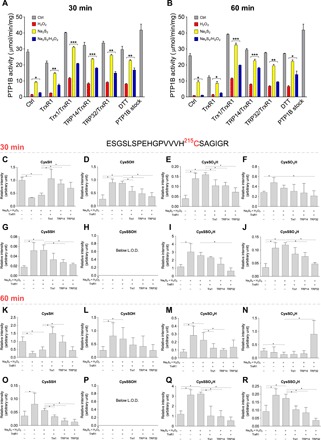
Recombinant human PTP1B [(A and B), 13.5 μM; (C to R), 20 μM] was incubated with or without 50 μM Na2S2 for 10 min followed by 100 μM H2O2 for 10 min at 25°C. Samples were then treated with or without Trx1 (15 μM), TRP14 (15 μM), or TRP32 (15 μM) with TrxR1 (250 nM) in NADPH [(A and B), 500 μM; (C to R), 1 mM] or 20 mM DTT for 30 min (A and C to J) or 60 min (B and K to R) at 37°C. (A and B) Phosphatase activity of PTP1B was measured in spectrophotometric assays using para-nitrophenyl phosphate (pNPP) as substrate. The control activity (PTP1B stock; last bar to the right) represents the untreated sample. (C to R) Protein samples were alkylated with 1 mM iodoacetamide for 30 min, digested with trypsin (37°C, 10 hours), and analyzed by LC-Q-TOF-MS. Peptides with modified active-site Cys derivatives were determined using Mascot; their levels were normalized to the intensities of the corresponding 157-QLELENLTTQETR-169 tryptic peptides and shown as relative values to the Cys thiol form in nontreated PTP1B set as 1.0. Data values are means ± SD of n = 3 (A to J) or n = 6 (K to R) experiments; *P < 0.05; **P < 0.01, and ***P < 0.001. L.O.D., limit of detection.
Reduction of oxidized persulfidated PTP1B by the Trx system
Previous findings show that the Trx system has prominent roles in reactivation of oxidized PTP1B (21, 22). On the basis of our results with HSA, we hypothesized that the Trx system would more efficiently restore oxidation-diminished PTP1B activity when the oxidative insult is preceded by persulfidation of the active-site Cys215. We found that Trx1, TRP14, and the thioredoxin-related protein of 32 kDa (TRP32) each potentiated reactivation of oxidized PTP1B-persulfide species as compared to the corresponding oxidized thiol form that had not been pretreated with polysulfide (Fig. 5, A and B). In contrast to its activity on HSA, TrxR1 alone was inefficient at reactivating PTP1B after consecutive treatment with Na2S2 and H2O2, but in the presence of Trx1, TRP14, or TRP32, it became highly efficient. In the presence of NADPH, TrxR1 seemingly catalyzed the inactivation of PTP1B compared to the control sample. This latter observation may be due to the previously reported NADPH oxidase activity of TrxR1, which is apparent when there is no reductase substrate around (37). Trx1 and TRP14 had the largest PTP1B reactivating capacity under the applied experimental conditions.
Formation of active-site Cys215 persulfide derivatives in PTP1B
To gain further insights into how polysulfide treatment aids reduction-mediated recovery of oxidatively inhibited PTP1B activity, we used ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) to investigate the posttranslational modifications of the active-site Cys215 residue upon polysulfide and/or H2O2 treatments. As shown in fig. S6 (A and B), incubation with Na2S2 lowered the amount of the CysSH form and induced concomitant formation of the persulfidated species. This modification was fully reversible by DTT. Corroborating our model on Fig. 2A, consecutive Na2S2 and H2O2 treatment converted the PTP1B Cys215-SH thiols to oxidized persulfide species, some of which were susceptible to dimedone labeling (fig. S6, A to D). These dimedone-labeled derivatives were entirely reducible by DTT (fig. S6D). DTT reduction should result in thiodimedone (dimedone-SH) release. To optimize detection of thiodimedone upon release from derivatized persulfide species, we used a well-characterized thiol-activated Sepharose 4B bead system, with which these oxidative modifications were previously demonstrated (9). The activated resin was persulfidated and then treated with H2O2 and dimedone to obtain R-SS-dimedone arms on the Sepharose beads. Next, DTT was added to the incubation mixture to cleave the generated disulfide bonds. The retention time, mass, and fragmentation pattern of the released thiodimedone product were finally characterized by UPLC-MS analyses (fig. S6, E to I). Using similar LC-MS/MS conditions, the dimedone-SH product could be detected using PTP1B samples pretreated with polysulfide and oxidized with H2O2 followed by reduction with DTT (fig. S6, E and J to L), thus corroborating the formation of PTP1B-SxSOH species during Na2S2 and H2O2 treatment.
To further characterize the generated oxidative posttranslational modifications on Cys215 and their relevance in the Trx system–mediated recovery of PTP1B activity, we used high-resolution LC-MS with quantitative quadrupole time-of-flight (LC-MS-Q-TOF) detection under similar conditions that were applied for the kinetic assays. On the basis of measured exact molecular masses, retention times, and fragmentation patterns of the Cys215-containing tryptic peptides, we validated the formation of Cys persulfide and oxidized perthio- derivatives at the active site of PTP1B and found additional evidence that these modifications are reduced by TrxR1 coupled to Trx1, TRP14, or TRP32 (Fig. 5, I, J, Q, and R). In line with the enzyme activity data (Fig. 5, A and B) at the 60-min time point after the addition of the Trx system, reduction of the oxidized persulfidated forms (Fig. 5, Q and R) was found to be more prominent compared to the 30-min time point (Fig. 5, I and J), which indicates that (similar to the HSA system) the enzymatic recovery of the PTP1B active-site Cys215 thiol from its CysSO2-3H forms is relatively slow. The slight but significantly lower sulfinic or sulfonic acid forms in some of the samples that were treated with different compositions of the Trx system may be due to similar effects that are discussed at the Prx2 section. The elevated levels of CysSO3H in TRP32-treated samples at the 60-min time point were unexpected, yet it occurred in all replicates and is worthy of further investigations.
The formation and reduction of Cys215 oxidized persulfide products corroborate that the recovery of PTP1B activity shown in Fig. 5 (A and B), which was measured under similar experimental conditions, is likely to be largely attributed to the model proposed in Fig. 2A, wherein the restoration of enzymatic activity is due to cleavage of polysulfide-induced disulfide bonds in ─SSH, ─SSOH, and ─SSO2,3H functional groups on Cys215. The fact that other LMW thiol–containing reducing agents such as Cys 2-mercaptoethanol (2-ME), and GSH functioned in recovery of PTP1B activity upon Na2S2 and H2O2 treatments further supports this proposal (fig. S7). In the case of these monothiols, some mixed disulfides can be generated; therefore, these small monothiols are slightly weaker at restoring activity to PTP1B as compared to DTT, for which thiols are the sole protein Cys–containing product (38).
Effect of TRP14-mediated persulfidation on EGF-induced protein phosphorylation
EGF stimulation of A431 cells is known to lead to PTP1B oxidation and inactivation, which enables EGF-triggered intracellular phosphorylation cascades (17). Using the A431 cell model, we found that pretreatment of A431 cells with polysulfide before EGF stimulation markedly increased both total EGF-dependent protein phosphorylation and specific EGF receptor (EGFR) phosphorylation (Fig. 6, A to C). As reported before and shown above, oxidized PTP1B can be reactivated by TrxR1 together with Trx1 or with TRP14 (21). Work by the Tonks laboratory identified a greater potency of the Trx system to recover enzymatic activity of persulfidated as compared to H2O2-inactivated recombinant PTP1B (23). In addition, our studies demonstrate that the TrxR1/TRP14 system potently reduces specific persulfidated proteins in cells (28) and in mouse liver (see above) and can reverse the activity of PTP1B by reducing its persulfidated and oxidized persulfide derivatives (see above). Moreover, the increases seen in phosphorylation upon polysulfide and EGF treatment suggest that EGFR and the involved protein kinases are more resistant to inhibition by persulfidation than PTP1B and potentially other phosphatases.
Fig. 6. Effect of persulfidation on EGF-induced phosphorylation cascades.
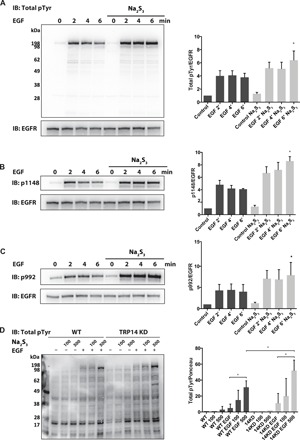
(A) Overnight starved A431 cells were pretreated with 500 μM sodium polysulfide (Na2S3) for 10 min and subsequently stimulated with EGF ligand (100 ng/ml) for the indicated times. (B and C) A431 cells were stimulated as in (A) and immunoblotted against EGFR phosphorylation sites, p1148 and p992. (D) Overnight starved WT and TRP14-deficient HEK293 (TRP14 KD) cells were pretreated with the indicated amounts of sodium polysulfide (Na2S3) for 10 min and subsequently stimulated with EGF ligand for 2 min. Total EGFR phosphorylation and EGFR expression were determined by immunoblotting using antibodies against phosphotyrosine (4G10) and EGFR, respectively (left). Equal protein loadings were confirmed by Ponceau S staining. Phosphorylation intensities were measured by densitometric analyses (right side of the panels) and normalized to respective EGFR expression levels. Western blots are representative of n = 3 (B to D) or n = 4 experiments (A). Data points are means ± SD of the indicated number of repeats; *P < 0.05. Significance refers to the untreated control samples (Control), unless otherwise specified.
Considering the importance of TRP14 and its dependence on the selenoprotein TrxR1, we hypothesized that either KD of TRP14 or lack of selenium supplementation (required to yield adequate activity of TrxR1) would potentiate the effect of polysulfide treatment on protein phosphorylation upon EGF stimulation. In addition, in HEK293 cells, we found that polysulfide pretreatment potentiated EGF-dependent phosphorylation, and in these cells, we could knock down TRP14, which also potentiated the effects of EGF stimulation (Fig. 6D). Moreover, fully supplementing the cells with selenium inhibited early responses to EGF in A431 cells, and more polysulfide treatment was needed to yield protein phosphorylation compared to cells that had not been saturated with selenium (fig. S8).
DISCUSSION
Here, we have shown that persulfidation can protect Cys residues from irreversible overoxidation and that the GSH and Trx system, particularly TrxR1 together with TRP14, can reduce oxidized persulfidated proteins. We furthermore found that persulfidation can modulate protein phosphorylation cascades in response to growth factor treatment, and we propose that persulfidation and redox regulation of PTPs such as PTP1B underpin these observations.
Our study should be considered in light of redox regulatory pathways in general, but perhaps especially observations related to H2S signaling. Soon after it was first proposed that H2S could play important mediatory functions in biology, a number of investigations reported that it participates in different pathophysiological conditions by alleviating oxidative stress–induced damage. This included protection against ROS-induced damage in brain, gastric mucosa and hepatic ischemia-reperfusion injury, as well as vascular endothelium in hypoglycemia [see (4) and references therein]. Initially, those effects were proposed to be due to direct scavenging of ROS. However, we and others argued that, because of the low physiological concentrations of free sulfide, even the fastest ROS scavenging reactions (e.g., the reduction of HOCl by sulfide, which is almost diffusion-controlled) would not be kinetically competitive in the presence of millimolar concentrations of GSH and protein thiols (39). We proposed that a potential mechanism to explain the protecting effects of sulfide, and of polysulfide species, could be associated with their reactions with metal centers of metalloproteins (4), as demonstrated with myeloperoxidase (40) or hemoglobin (41). Sulfide efficiently reduces the highly oxidizing and reactive ferryl heme intermediates of these enzymes, with concomitant production of polysulfide species (42) that are the most effective persulfidating agents of protein Cys side chains. Alternative redox-mediated events could also potentially induce protein per/polysulfidation under oxidative stress, as we reviewed previously (4). Thus, we proposed that per/polysulfidation contributes to protection against oxidative stress. In the present study, we have systematically addressed the physiological relevance of persulfidation-mediated protection of protein Cys residues.
Cys persulfidation and oxidation of persulfides
We previously introduced the ProPerDP method as a convenient tool for protein persulfide detection (28). In the present study, state-of-the-art LC-MS–based methods allowed a more quantitative determination of Cys persulfide and trisulfide species, which confirmed the elevated levels of both protein-derived and LMW CysSSH in liver tissues of mice with disrupted functions of the NADPH-dependent reductases glutathione reductase (Gsr) and/or TrxR1. As we measured lower steady-state concentrations of the more oxidant-sensitive CysSSSH species in livers in which the NADPH reductase machineries were disrupted, this may possibly suggest that these species play roles in scavenging a proportion of the elevated ROS in these livers. To corroborate this hypothesis, we developed a quantitative MS method to measure protein CysSO3H and CysSSO3H levels in tissue samples. Our method revealed that CysSSO3H is an abundant protein modification in mouse liver tissue. The fact that, compared to WT livers, CysSSO3H levels were significantly elevated in TrxR1/Gsr and TrxR1/Gsr/Trx1-null livers, in which cells are exposed to excessive oxidative stress, supports our model that persulfidation protects cellular protein Cys residues during oxidative stress in vivo. The fact that, compared to WT, CysSSO3H levels were not elevated in Gsr livers, in which only the GSH-based reduction was impaired, suggests that oxidized persulfide species are primarily reduced by the Trx system.
A large body of literature reports the intracellular formation of protein sulfenic acids under mild oxidative stress (11). Most of these studies are based on the reactivity of dimedone derivatives with CysSOH. However, Heppner et al. (10) recently showed that these conditions also generate abundant Cys-SSOH modifications, which are labeled by dimedone and thereby result in disulfide-linked Cys-SS-dimedone products. Moreover, we hypothesized that the presence of dimedone in a biological system is capable of shifting the hydrolysis equilibria of protein persulfides toward the formation of ─SSOH species, meaning that a fraction of the putative sulfenylated proteome originates from the protein persulfide pool (30). Here, we have further corroborated that considerable amounts of reducible dimedone-reactive protein modifications are generated by extracellular H2O2, which is attributable to the process of perthiosulfenylation. Note that CysSSO2H and CysSSO3H are unreactive toward dimedone, so the method does not account for those species. As we identified perthiosulfenylated species of protein targets connected to antioxidant protection (KEAP1), chaperone activity with various posttranslational modification (HSP90), and redox-regulated signaling proteins (PTP1B and PTEN), it seems clear that this reducible protein modification can have major biological roles especially in light that it is likely to be enzymatically regulated and controlled.
Roles of the Trx1 system in regulating persulfidation-mediated protein thiol protection
Components of the cytosolic Trx system (TrxR1 alone or coupled with Trx1 or TRP14) were previously shown to exhibit protein persulfide reductase activity (28). Here, we found that TrxR1 can directly reduce oxidized persulfide species into thiols. The classical TrxR1 substrates Trx1 and TRP14, generally involved in protein disulfide or persulfide reduction reactions (28, 31, 43), had no additional effects on the reduction rates of oxidized HSA polysulfide species. Although its biological relevance may be disputed considering that HSA is found in serum and TrxR1 is cytosolic, in case this activity is not unique to HSA, then it could potentially represent a previously unrecognized property of TrxR1.
The Trx system was previously found to be more effective in reactivating both H2O2-treated and persulfidated PTP1B compared to the GSH system (21–25). In mouse embryonic fibroblasts, we found that the alternative TrxR1 substrate TRP14 also has the capability to reactivate PTP1B (21, 31). Under similar conditions, the oxidized form of SHP2, a nonreceptor type PTP, remained unaffected by both Trx1 and TRP14. In addition, recently, we reported that TrxR1 alone protects PTP1B from excessive oxidation, most likely by directly reducing the sulfenic acid intermediate (22). These studies collectively suggest that the members of the Trx family contribute to the regulation of phosphorylation signaling in a substrate-specific targeted manner. Here, we found that TrxR1 has no significant reducing activity toward oxidized persulfide derivatives of Cys215 in enzymatic assays of PTP1B. We also found that, unlike the case for oxidized persulfidated HSA, TRP14 or Trx1 coupled to TrxR1 increased the reducing efficiency. Using the livers from TRP14-null mice generated for this study, we could assess quantitative information of the persulfide reductase activity of TRP14, noting only slightly elevated protein CysSSH levels in TRP14-null livers compared to WT, while no difference was detected in the LMW persulfide or related reactive sulfur species pools. These observations further corroborate the delicate substrate specificity of different components of the disulfide reductase systems. We hypothesize that the GSH system is primarily responsible for protein persulfide homeostasis, while the Trx system, perhaps especially TRP14, can target a specific subset of proteins. Here, note that TRP14 catalyzed the recovery of persulfidated Prx2 and PTP1B (Figs. 3, L to N, and 5, I, J, Q, and R) but not similarly modified HSA (Fig. 2E). A comprehensive proteomics approach will be necessary in the future to identify protein persulfide modifications specifically reduced by TRP14 or other members of the Trx or GSH systems.
Cellular implementation of the persulfide-based thiol protection mechanism
As a potential antioxidant defense mechanism, the relative importance of persulfidation was investigated on Prxs, the primary cellular peroxide-detoxification enzyme family. Note that cytosolic Prx2 reduces H2O2 with a rate constant of ~107 M−1 s−1 and can transmit redox signals to redox-regulated proteins, thus interlinking oxidative stress protection and signaling. Note, in this context, that a considerable fraction of the peroxidative Cys of Prx2 is persulfidated in HEK293 cells at steady-state levels and that its persulfidation is further increased upon TrxR1 KD. Our experiments also showed that H2O2 treatment generated overoxidized Cys51-SSO2H and Cys51-SSO3H species in normal cells. In vitro experiments on purified Prx2 confirmed that persulfidation of the peroxidative Cys and its oxidized persulfide forms are reduced back to the active thiol by TRP14 coupled to TrxR1 but not with TrxR1 alone. These findings suggest that persulfidation of Prx2 may protect the enzyme from permanent loss of activity due to overoxidation of Cys51 and that TRP14 is potentially involved in Prx2 functions by modulating its persulfidated states.
The current study shows that protein persulfidation normally exists in vivo and can, together with the cellular reducing systems, protect the proteins from irreversible overoxidation. However, the question arises: How is this protective mechanism executed in cells? Once an oxidative event has initiated, it is likely too late to begin persulfidating critical proteins, because the most critical thiols (e.g., in the active sites of Prxs, PTPs, and others) will also be the most reactive with H2O2 and will therefore become irreversibly oxidized before they could be protectively persulfidated. It is, in that context, important that even in unstressed states, we found that roughly 30% of protein thiols are persulfidated in vivo. It is thus possible that, rather than being a reactive defense, protein persulfidation is a preemptive mechanism that continuously invests ~30% of key proteins in inactive persulfidated state at the active site such that, following an oxidative event, these proteins can be rapidly reactivated to aid recovery. Depending on disulfide reductase–mediated on and off rates, this persulfidated pool could be achieved either by having a discrete subset of proteins on reserve or by having each protein spend 30% of its time in the persulfidated form and 70% of its time in the active thiol form. In this regard, it is interesting that we recently found that Cys persulfides are also cotranslationally inserted into proteins (29). This could ensure that even during events that would immediately overoxidize reactive Cys residues in newly synthesized proteins, these proteins have the ability to be activated at a later time by the cellular disulfide reductase systems.
Possible roles of thiol persulfidation beyond thiol protection
Protein thiol persulfidation, in particular of a catalytic active-site Cys, introduces a bulky extra sulfur atom into this site. This will alter the spatial characteristics of the active site and thereby likely interfere with normal catalysis. Persulfidation has been shown to disrupt normal enzymatic activity of various enzymes, such as PTP1B, KEAP1, or PTEN (4). However, note that persulfides are typically more acidic and more reactive than thiols. Some enzymes such as mitochondrial rhodanase, which function in cyanide and sulfide detoxification (44), have an active-site persulfide as an essential part of its catalytic mechanism (45). Although the reactivity of a given Cys persulfide will be dependent on how that persulfide interacts with other residues in the microenvironment of each active site, one should consider the possibility that, for some enzymes, the persulfidated versions might not be “inactivated” but rather “reprogrammed” for different substrate specificities. This possibility has not yet, to our knowledge, been explored.
Protein persulfidation in signaling
PTP1B is a well-characterized subject of redox regulation, and PTP1B has widespread involvement in growth factor signaling (17, 22, 46, 47). Reversible activity loss of PTP1B was reported in A431 squamous carcinoma cells after EGF stimulation, which was associated with elevated intracellular production of H2O2 (17). The requirement of increased H2O2 levels in the promotion of receptor tyrosine kinase–initiated phosphorylation pathways strongly supports a role of H2O2 as a second messenger molecule (17, 47). The molecular mechanism of EGF-induced H2O2 production, however, remained obscure until the recent description of the Ca2+-dependent activation of dual oxidase 1 (Duox1) from the NADPH oxidase family, triggered by EGF binding (48). The tyrosine kinase transmembrane receptor EGFR is the subject of redox regulation itself, with the formation of a Cys sulfenic acid modification detected on its Cys797 residue that promotes kinase activity (49). The nature of this modification has been revisited and suggested to also partially be polysulfide-derived perthiosulfenic acid (10). As a feedback loop in their interaction, phosphorylated EGFR was identified as a direct substrate of PTP1B, and it was suggested that a vital function of PTP1B is to prevent the nonspecific, non–ligand-induced phosphorylation of EGFR (46). This finding implies that inhibition of PTP1B results in sustained EGFR activity and coupled downstream signaling.
EGFR is the starting point of several signal transduction cascades, including the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/Akt, and c-Jun N-terminal kinase (JNK) pathways, and thereby, it regulates phenotypes including cellular growth, proliferation, migration, or adhesion. In the current study, we demonstrated that in A431 and HEK293 cells, general protein phosphorylation is not prominent in the absence of EGF, and polysulfide treatment alone had little effect under these conditions. Consistent with the fact that A431 cells have elevated EGFR levels, we observed substantially increased protein phosphorylation upon EGF treatment in these cells. Polysulfide treatment largely increased EGF-triggered protein phosphorylation. These observations suggest that kinases are less susceptible to polysulfides than phosphatases; therefore, EGFR, NOX, and involved kinase activities are likely not affected by polysulfide treatment. In HEK293 cells, we observed little effect on total protein phosphorylation when EGF or polysulfide was added alone, but a strong increase in phosphorylation signal was seen when they were used in combination. KD of TRP14 or lack of Se supplementation (which results in diminished TrxR1 activity) further enhanced polysulfide-induced elevated phosphorylation upon EGF treatment. These observations further support the hypothesis that endogenous persulfidation and/or oxidized polysulfidation-induced PTP1B inactivation is regulated, at least in part, by TrxR1/TRP14 (fig. S9). These cellular studies provide credence to the enzyme kinetic data (Fig. 5, A and B) and represent the first demonstration for the cooperative actions of the Trx system and protein persulfidation events in protecting functional protein Cys residues on PTP1B under EGF-induced elevated oxidant production. Thus, our observations and proposed mechanisms represent a novel concept of how sulfur species may orchestrate redox-mediated phosphorylation signaling events.
MATERIALS AND METHODS
Study design
Sample size: Each experiment was repeated at least three times. For mouse liver experiments, the number of replicates represents individual animals (biological replicates). Exact sample sizes are indicated for each figure in the figure legends. Data exclusions: No data were excluded from the analyses. Replication: All attempts at replication were successful. Randomization: Not applicable; mouse liver samples were allocated into groups according to their genotypes. Blinding: Not applicable.
Reagents
All reagents used were of analytical grade or higher purity. Chemicals were purchased from Sigma-Aldrich unless otherwise indicated. β-(4-Hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) was obtained from Molecular Biosciences (Boulder, CO, USA). Stock solution (100 mM) was prepared in dimethyl sulfoxide (DMSO), and aliquots were stored at −20°C. Stable isotope–labeled internal standards for LC–electrospray ionization (ESI)–MS/MS measurements were synthesized in Tohoku University, Japan. EZ-Link Iodoacetyl-PEG2-Biotin (IAB) and Zeba desalting spin column [molecular weight cutoff (MWCO), 7 kDa] were from Thermo Fisher Scientific (Waltham, MA, USA). Pronase, DCP-Bio1, and Amicon ultraconcentrator filters were ordered from Merck Millipore (Darmstadt, Germany). Thiol-activated Sepharose 4B, N-hydroxysuccinimide (NHS)–Sepharose, and PD MidiTrap G-25 columns are registered trademarks of GE Healthcare Life Sciences (Pittsburgh, PA, USA). para-Nitrophenyl phosphate (pNPP) was from Research Organics (Cleveland, OH, USA). Na2S2, Na2S3, and Na2S4 are products of Dojindo Laboratories (Kumamoto, Japan). EGF is distributed by R&D Systems (Minneapolis, MN, USA).
Cell culture
A431 cells [human squamous cell carcinoma, female, RRID:CVCL_0037; American Type Culture Collection (ATCC)] and HEK293 cells (female; ATCC) were grown in Dulbecco’s modified Eagle’s medium with high glucose (4.5 g/liter). Cell culture media were supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, penicillin, and streptomycin. Cells were kept in logarithmic growth phase at 37°C in humidified air containing 5% CO2. Stable TrxR1 or TRP14 KD HEK293 cell lines were generated as previously described (31).
Animal models and related procedures
All mice in this study were housed in the American Association for the Accreditation of Laboratory Animal Care (AAALAC)–accredited Animal Resources Center at Montana State University under specialized care conditions, including continuous-flow high-efficiency particulate air (HEPA)–filtered air cages (Tecniplast), sterilized bedding and enrichment, unrestricted access to sterilized feed (Picolabs 5058 or 5053) and sterilized water, and on a 14-hour light/10-hour dark cycle. Animal protocols were approved by the Montana State University Institutional Animal Care and Use Committee (protocol #2015-5). WT C57Bl/6J mice or B6.Cg-Tg(Alb-cre)21Mgn/J mice bearing the “AlbCre” transgene were obtained from The Jackson Laboratory (Bar Harbor, ME; stocks JAX#000664 and JAX#003574, respectively). Mice bearing the Gsr allele were provided by L. Rogers (50). Mice bearing the Txnrd1cond [B6.129(Cg)-Txnrd1tm1Ees/J, JAX#028283] or Txn1cond alleles (Txn1Tm1.1Ees/J, JAX#030221) (27) and Txnrd1cond/cond;AlbCre+ (AlbCre-driven liver-specific “TrxR1-null”), Txnrd1cond/cond;Gsr−/−;AlbCre+ (AlbCre-driven liver-specific “TrxR1/Gsr-null”), or Txn1cond/cond;Txnrd1cond/cond;Gsr−/−;AlbCre+ (AlbCre-driven liver-specific “Trx1/TrxR1/Gsr-null”) genotypes were described previously (26, 27). Mice were genotyped by polymerase chain reaction (PCR) using genomic DNA extracted from tail biopsies harvested between postnatal day 10 (P10) and P14.
All analyses shown were done on mice of both sexes between P60 and P90. For harvests, animals were euthanized with isoflurane. Thereafter, livers were perfused with sterile saline (cardiac portal), removed, divided into ~200-mg pieces, placed in 1.5-ml tubes, and snap-frozen in liquid nitrogen. Liver pieces were stored at −80°C and shipped to collaborating laboratories on dry ice for biochemical, molecular, and enzymatic analyses.
A conditional-null allele of the mouse Txndc17 gene, in which exons 1 to 4 are flanked by codirectional loxP sites (Txndc17fl) (fig. S2A), was generated and targeted into male mouse F1-C57Bl/6 × 129SvEvTac hybrid embryonic stem (ES) cells. All critical regions of the vector and all cloning junctions were sequence-verified. The targeting vector consisted of a region of chromosome 11 (Chr11) from positions 72,205,834 to 72,212,541, which was amplified from Fosmid WI1-1814P1 (BACPAC Resource Center) by PCR with Phusion polymerase (New England Biolabs, #M0531S). Restriction endonuclease sites Not I and Xho I were engineered onto the amplified fragment at the 5′ and 3′ ends, respectively. A loxP site flanked on its 3′ side by an engineered Spe I site was inserted into the 5′ untranslated region (5′UTR) of the Txndc17 gene, which replaced bases 72,207,602 to 72,207,604 of Chr11. Downstream of the Txndc17 3′ UTR, a construct consisting of frt-loxP-SV40 terminator-frt-Prm1pFlpe-Pol2pNeo-frt was inserted into Chr11 at position 72,210,526, which also replaced endogenous sequence of Chr11 from 72,210,526 to 72,210,542. This construct was flanked by engineered Asc I and Pac I sites at the 5′ and 3′ ends, respectively. The SV40 terminator and self-excising frt-Prm1pFlpe-Pol2pNeo-frt region were amplified from plasmid FnF11 (51). The completed Txndc17 targeting vector was inserted between Not I and Xho I sites in the TK1-TK2c plasmid (52) and linearized with Not I before electroporation into ES cells. Targeted ES cell clones were identified by PCR analysis of genomic DNA using primer pairs Txndc17 5′-forward1 (5′-gcactcctacaagctgtcccatg-3′) and Txndc17 Spe I-loxP-reverse (5′-gtacaacagtgcggagactagtat-3′) or Neo-forward (5′-tgctttacggtatcgccgct-3′) and Txndc17 3′-reverse1 (5′-gcctgtagatttgctcttcatgatgc-3′). One targeted ES cell clone was used to make chimeric mice in C57Bl/6J recipient blastocysts carrying the “Txndc17founder” allele. Phenotypically male chimeras were bred to C57Bl/6J females, and pups were screened for self-excision of the Prm1pFlpe-Pol2pNeo cassette.
Verification of Txndc17 alleles
Genotypes of mice carrying the unrecombined Txndc17 founder allele or the Flpe-recombined Txndc17fl allele were determined by PCR analysis of genomic DNA from tail biopsies of P15 mice using primer pairs Txndc17 3′-forward (primer i) (5′-gagaaccaggaatgcagtgagacat-3′) and Prm1p-reverse (5′-agggaagctgaaaggtggacagg-3′) or primer i and Txndc17 3′-reverse2 (primer ii) (5′-gttgtgagccactacatgatgctg-3′), respectively (fig. S2, A and B). Mice bearing the Txndc17fl allele were bred with Cre-deleter mice (53) to generate the Txndc17null allele. Genotyping of mice that carry the Txndc17null allele used three primers: primer ii, Txndc17 5′-forward2 (primer iii) (5′-atggctattcaaggctcccgtagc-3′), and Txndc17 exon1-reverse (primer iv) (5′-atccttagaaccgctgaagtaggc-3′). Inclusion of primer iv in this genotyping strategy is required only for detection of the WT allele of Txndc17 (fig. S2, A and C). General PCR thermocycle conditions for targeting vector construction, allelic confirmation, and genotyping were as follows: 94°C, 2 min; 94°C, 30 s; 61.5°C, 30 s; and 72°C, 1 min. Cycling was repeated 33 times with a final incubation at 72°C for 5 min.
Complementary DNA (cDNA) analysis of the Txndc17null and Txndc17 WT (+) allele was performed on oligo(dT)-primed cDNA samples using primers Txndc17 exon1-forward (5′-aggaccctagggctcctcccg-3′) and Txndc17 exon2-reverse (5′-cagtcttcggtaacatgcttcag-3′). β-Actin cDNA analysis used primer pair β-act exon5/6-forward (5′-ctcctagcaccatgaagatcaagatcat-3′) and β-act exon6-reverse (5′-ctcatcgtactcctgcttgctgat-3′) (fig. S2D).
LC-MS detection of LMW persulfide species
Mouse liver tissue samples were analyzed for LMW CysSH, CysSSH, GSH, GSSH, Bis-S (H2S), Bis-SS (HSSH), cystine, GSSG, GSSSG, HS2O3−, and homocysteine (HCysSH) analytes as described previously (29), with minor modifications. Briefly, deep-frozen tissue samples were cut to approximately one-third pieces and homogenized with a Physcotron homogenizer (Microtec Co. Ltd.) in 0.5 ml of ice-cold methanol in the presence of 5 mM HPE-IAM, which was recently shown to have a stabilizing effect on polysulfide chains (36). Homogenates were incubated at 37°C for 20 min and then centrifuged for 10 min at 4°C and 15,000g. Supernatants were separated and diluted 10 times with 0.1% formic acid. In the next step, samples were further diluted fivefold, and 25 nM final concentration of stable isotope–labeled internal standards was introduced and also dissolved in 0.1% formic acid solution. Ten microliters was injected to LC-ESI-MS/MS analysis. Cell pellets were reconstituted in 0.1% SDS in phosphate-buffered saline (PBS) buffer, homogenized by sonication, and centrifuged again for 10 min at 4°C at 15,000g. Bicinchoninic acid (BCA) protein assay was performed from the supernatants to determine total protein content of the samples. Each analyte concentration was normalized to the measured protein concentration. LC-ESI-MS/MS measurements were carried out using a Shimadzu LCMS-8060 triple quadrupole mass spectrometer connected to a Shimadzu Nexera UHPLC instrument, as reported previously (29). The HPE-IAM derivatized reactive sulfur species were separated on a YMC-Triart C18 reversed-phase chromatography column (50 × 2.0 mm inner diameter). Analytes were eluted with the following setup of mobile phases: 0.1% formic acid (mobile phase A) was mixed with mobile phase B (methanol and 0.1% formic acid) with a linear gradient from 5 to 90% within 15 min. The flow rate was set to 0.2 ml/min at T = 40°C. The indicated polysulfur and other oxidized species were identified and quantified by multiple reaction monitoring (MRM), with the MRM parameters published elsewhere (29).
LC-MS detection of HMW (protein-derived) persulfide species
Pronase digestion was performed on mouse liver tissue samples, similarly to previously described procedure, with adequate modifications (29). Briefly, deep-frozen tissue samples were homogenized in 0.5 ml of ice-cold radioimmunoprecipitation assay (RIPA) buffer [10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.1% NP-40, 0.1% sodium deoxycholate, and 0.1% SDS] containing 5 mM HPE-IAM. Lysates were centrifuged at 15,000g for 10 min at room temperature. Supernatant was recovered, and 150 μl was applied to PD SpinTrap G-25 desalting column (GE Healthcare) to remove the LMW fraction. HPE-IAM (5 mM) was immediately reintroduced to the filtrates. After BCA protein assay, protein levels were brought equal to 1 mg/ml and digested with pronase (3 mg/ml) in 50 mM sodium acetate buffer (pH 5.0) in the presence of 40 nM stable isotope–labeled internal standards for 7 hours at 37°C. After centrifugation, the supernatants were subjected to LC-ESI-MS/MS analysis as mentioned above.
Preparation of stable isotope–labeled CysSO3H, CysSSO3H
The stable isotope–labeled [13C1]CysSO3H and [13C1]CysS[34S1]O3H were synthesized according to the method described in a previous study (29) with modification. Briefly, 3 mM stable isotope–labeled [13C1]cysteine was reacted with 3 mM NaH[34S1] in the presence of 3 mM 1-hydroxy-2-oxo-3(N-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC7, Dojindo Laboratories) in 100 mM tris-HCl buffer (pH 7.4) at room temperature for 15 min, after which 2.5 mM peracetic acid (Sigma-Aldrich) was added to the reaction mixture and incubated at room temperature for 30 min to form CysSO3H and CysSSO3H. For stabilization of CysSO3H and CysSSO3H concentrations, 50 mM methionine was added to the reaction mixture to quench the residual oxidant. The concentrations of isotope-labeled CysSO3H and CysSSO3H were calculated with CysSO3H (Sigma-Aldrich) or CysSSO3H (Sigma-Aldrich) standards by using LC-MS/MS. These stable isotope–labeled CysSO3H and CysSSO3H reaction mixtures were stored at −80°C.
LC-MS/MS detection of protein CysSO3H and CysSSO3H in mouse liver
Pronase digestion was performed on mouse liver tissue samples, similarly to previously described procedure, with adequate modifications (29) . Briefly, deep-frozen tissue samples were homogenized in 0.5 ml of ice-cold RIPA buffer, containing 5 mM HPE-IAM. Lysates were centrifuged at 15,000g for 10 min at room temperature. Supernatants were recovered, and 150 μl was applied to PD SpinTrap G-25 desalting column to remove the LMW fraction. HPE-IAM (5 mM) was immediately reintroduced to the filtrates. After BCA protein assay, protein levels were brought equal to 1 mg/ml and digested with pronase (3 mg/ml) in 50 mM sodium acetate buffer (pH 5.0) for 7 hours at 37°C. After centrifugation, an aliquot of the stable isotope–labeled CysSO3H and CysSSO3H mixture was added to the supernatants, after which the samples were subjected to LC-ESI-MS/MS analysis. LC-ESI-MS/MS measurements were carried out using a Shimadzu LCMS-8060 triple quadrupole mass spectrometer connected to a Shimadzu Nexera UHPLC instrument. CysSO3H and CysSSO3H were separated on an Intrada Amino Acid column (100 × 3.0 mm inner diameter; Imtakt Corporation, Kyoto). Analytes were eluted with the following setup of mobile phases: Acetonitrile and 0.1% formic acid (mobile phase A) was mixed with mobile phase B (100 mM ammonium formate) with a linear gradient from 10 to 60% within 15 min. The flow rate was set to 0.3 ml/min at T = 40°C. CysSO3H and CysSSO3H were identified and quantified by MRM, using MRM parameters shown in table S1A.
Generation of overoxidized HSA polysulfide species
HSA (5 mg/ml) in 100 mM tris-HCl and 100 μM DTPA (diethylenetriamine pentaacetic acid) (pH 7.40) buffer was pre-reduced with 0.5 mM DTT overnight at 4°C to bring back the redox active Cys34 residue in the thiol form. HSA polysulfide was generated by inorganic polysulfide treatment, as described previously (28, 54). Briefly, polysulfide stock solution was prepared by mixing H2S and sodium hypochlorite stock solutions in 5:1 ratio, where the concentrations of the components were determined spectrophotometrically. Hypochlorite concentration was measured at 292 nm [ε(OCl−)292 nm = 350 M−1 cm−1]. Sulfide concentration was calculated from the average of two measurements, one being the direct absorbance of sulfide stock solution in water at 230 nm [ε(HS−)230 nm = 7700 M−1 cm−1] and the other one is 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) reduction by sulfide [(ε(TNB−)412 nm = 14,100 M−1 cm−1]. The reduced HSA samples were incubated with 2 mM polysulfide reagent at room temperature for 30 min in the dark, and an untreated sample was used as control. Excess polysulfides were removed by desalting spin columns (Thermo Zeba, 7 kDa MWCO), and 2 mM H2O2 was added to each sample for 1 hour at room temperature in the dark. H2O2 concentration was also determined spectrophotometrically (ε240 nm = 43.6 M−1 cm−1). Excess H2O2 was removed by desalting spin columns. Because HSA-SSH species are effectively reduced by the Trx system (28) and residual HSA-SH species would have interfered with our downstream detection method, potential remaining polysulfide and thiol species from incomplete oxidation were removed from the samples by alkylating with 1 mM IAB (EZ-Link Iodoacetyl-PEG2-Biotin, Thermo Fisher Scientific) for 1 hour at room temperature in the dark and subsequent affinity pulldown with streptavidin-coated magnetic beads (Sigma-Aldrich) for 1 hour at 4°C on a rotating wheel. Before the pulldown step, excess IAB was removed from the samples with desalting spin columns and Amicon ultraconcentrators (Merck, 30 kDa MWCO).
Reduction of overoxidized HSA polysulfide species
Overoxidized HSA polysulfide and thiol samples prepared as described above were first mixed with 1 mM DTT and reduced for 2 hours at 4°C. In the case of the DTT reduced samples (represented in Fig. 2C), the band position of HSA is higher than for the untreated samples, which is likely because of concomitant DTT-induced reduction of intramolecular disulfide bonds of the protein. Similar bands were previously identified as HSA by LC-MS proteomics, as discussed in (28).
Next, the different members of the Trx system were tested as reducing agents, in the following combinations: 500 μM NADPH and 200 nM TrxR1 WT or mutants (see Results and the Supplementary Materials) with or without the addition of 5 or 1 μM Trx1 or 5 or 1 μM TRP14. Rat TrxR1 WT and mutants, hTrx1, and hTRP14 were purified as described below. A control sample without any reducing equivalent was included into each group of samples. The time resolution of the reaction was monitored by fluorescent labeling of the reduced thiol product and subsequent SDS-PAGE. A sample of equal volume was taken from the reaction mixture at the indicated time points, and 0.5 mM 5-IAF (10 mM stock solution prepared in DMSO) was added to alkylate the thiol product and provide it with a fluorescent moiety. The proteins were heat-denatured at 100°C for 3 min and then separated on 10% nonreducing polyacrylamide gels.
Detection of Prx2 Cp modifications in HEK293 cells
Normal control (NC) and TrxR1 KD HEK293 cells were seeded to cell culture dishes at 3 × 106 cells per 100-mm2 density 48 hours before experiments. The cells were treated with or without 0.6 mM H2O2 for 5 min in Hanks’ balanced salt solution buffer at 37°C. Untreated samples were introduced as controls. After peroxide treatment, cells were washed in PBS three times and lysed in 1 ml of lysis buffer [10 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.1% NP-40, 0.1% sodium deoxycholate, deoxyribonuclease I (10 μg/ml), ribonuclease A (10 μg/ml), and protease inhibitor cocktail] without SDS containing 5 mM iodoacetamide (Fujifilm Wako Pure Chemical Corporation). Cells were scraped and disrupted by sonication. The lysates were incubated for 20 min at room temperature and then centrifuged for 20 min at 4°C at 15,000g. Supernatants were transferred into low protein–binding microcentrifuge tubes (LoBind Tubes, Eppendorf), mixed with 30 μl of protein G Mag beads (GE Healthcare; previously washed with lysis buffer), and agitated for 20 min at 4°C. After magnetic separation, cell lysates were transferred to low protein–binding tubes and reacted with 5 μl of anti-Prx2 (α-PRDX2; clone: EPR5155) overnight at 4°C. The samples were incubated with 50 μl of lysis buffer–washed protein G Mag beads for 2 hours at 4°C. The beads were washed five times with 1 ml of 200 mM tris-HCl (pH 8.0) and incubated with Trypsin Gold (20 μg/ml) (Promega) in 200 mM tris-HCl (pH 7.0) with or without 5 mM iodoacetamide (Fig. 3 and fig. S5, respectively) overnight at 4°C in a shaker. The digestion process was quenched with 1% final concentration of formic acid. After centrifugation, the supernatants were analyzed by LC-Q-TOF-MS and subsequent Mascot search as described below.
Preparation of recombinant human Prx2
To construct a plasmid for human Prx2 (hPrx2) expression, pET53-hPrx2 (N-terminal His tag and C-terminal StrepII tag) was constructed through Gateway technology (Thermo Fisher Scientific) with primers (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAGCCTCCGGTAACGCGCGCATCGGAA-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGATTGTGTTTGGAGAAATATTCCTTG-3′), pET53-DEST (Merck Millipore), and cDNA library generated from HEK293 cells. To prepare recombinant hPrx2, Escherichia coli strain T7 Express (New England Biolabs) transformed with pET53-hPrx2 was grown at 37°C in LB medium containing carbenicillin (100 μg/ml). When the OD600 (optical density at 600 nm) value was 0.6, 0.2 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to the culture medium to induce gene expression. After cultivation for 18 hours at 17°C, cells were harvested and suspended in ice-cold buffer A [50 mM phosphate, 300 mM NaCl, 10 mM imidazole, 1 mM DTT, and protease inhibitor (pH 8.0)] and disrupted by sonication under cooling. After centrifugation, the supernatant was loaded onto Ni–nitrilotriacetic acid agarose (Qiagen) and hPrx2 was eluted with ice-cold buffer A containing 250 mM imidazole. After a dilution of the sample three times with ice-cold buffer B [100 mM tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM DTT (pH 8.0)], the diluted sample was applied to Strep-Tactin Sepharose (IBA Lifesciences, Goettingen, Germany). hPrx2 was eluted with ice-cold buffer B containing 2.5 mM desthiobiotin (IBA Lifesciences). After desalting the sample by using PD10 column (GE Healthcare) with 50 mM tris-HCl buffer (pH 7.4), the purified hPrx2 was stored at −80°C before use. Protein concentration was determined by using the Bradford method with bovine serum albumin as a standard protein.
Identification of recombinant Prx2 and PTP1B perthiosulfonic acid species by LC-MS-Q-TOF experiments
Recombinant hPrx2 (12 μM) was incubated with or without 100 μM Na2S2 for 10 min and then 1 mM H2O2 for 10 min at 25°C. Samples were desalted with PD SpinTrap G-25 desalting column and then treated with or without TrxR1 (250 nM), TRP14 (4 μM), and NADPH (1 mM) for 1 hour at 37°C. Samples were alkylated with 1 mM iodoacetamide for 30 min, followed by digestion with Trypsin Gold at 37°C for 10 hours. The digested samples were subjected to LC-Q-TOF MS (Agilent Technologies).
Recombinant human PTP1B (hPTP1B) containing 10 mM DTT was desalted by PD SpinTrap G-25 desalting column to remove DTT. Recombinant DTT-free hPTP1B (20 μM) was incubated with or without 50 μM Na2S2 for 10 min and then 100 μM H2O2 for 10 min at 25°C. Samples were treated with or without TrxR1 (250 nM), Trx1 (15 μM), TRP14 (15 μM), TRP32 (15 μM), and NADPH (1 mM) for 30 or 60 min at 37°C. Samples were alkylated with 1 mM iodoacetamide for 30 min, followed by digestion with Trypsin Gold at 37°C for 10 hours. The digested samples were subjected to LC-Q-TOF MS (Agilent Technologies), and the data were narrowed to the active-site peptide by Mascot search.
LC-Q-TOF and Mascot search on recombinant PTP1B and Prx2 from HEK293 cells
The modification of Cys residue in PTP1B and Prx2 was identified by LC-Q-TOF-MS analysis combined with Mascot searches similarly to our previous report (29). Briefly, an Agilent 6510 Q-TOF mass spectrometer coupled to an Agilent HPLC chip/MS system was used. The latter consists of a nanopump with a four-channel microvacuum degasser and a microfluidic chip cube. Tryptic peptides were separated with a microfluidic reversed-phase HPLC chip (ZORBAX 300SB-C18; particle size, 5 μm; inner diameter, 75 μm; and length, 43 mm; Agilent Technologies). Samples were loaded using 0.1% formic acid and 5% acetonitrile in water as mobile phase and a flow rate of 4 μl/min. Peptide separation was carried out using the following elution conditions: 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) were used for a linear gradient elution. Percentage of mobile phase B was increased from the initial 5 to 90% within 15 min, keeping a flow rate of 400 nl/min. The ESI-Q-TOF instrument was set to positive ionization mode; the ionization voltage was 1750 V, and the fragmentor voltage was 175 V at a temperature of 300°C. The mass/charge ratio (m/z) values were monitored in the 500- to 1200-Da range with an MS scan rate of 4 s−1. Mascot MS/MS ion searches of the National Center for Biotechnology Information nonredundant database were used to obtain the exact mass numbers of the peptide containing the carbamidomethylated Cys residues via the Matrix Science Web server Mascot version 2.2. The following default search settings were applied: enzyme: trypsin; maximum missed cleavage: 1; variable modifications: Carbamidomethyl (C), Oxidation (C), Dioxidation (C), Trioxidation (C), SulfurDioxide (C), Sulfo (C); peptide tolerance: ±1.2 Da; MS/MS tolerance: ±1.2 Da. Peptide fragments containing persulfidated and/or oxidized Cys residues were identified on the basis of their calculated m/z values, adding the equivalent mass of sulfur and oxygen atoms to the unmodified peptide. The m/z values of the detected peptides that contain the active-site Cys residues of PTP1B (200-ESGSLSPEHGPVVVHCSAGIGR-221) or Prx2 (37-YVVLFFYPLDFTFVCPTEIIAFSNR-61) with the denoted modifications together with the corresponding PTP1B peptides (157-QLELENLTTQETR-169) and with the corresponding Prx2 peptides (93-EGGLGPLNIPLLADVTR-109), which were used for normalization of signal intensities, respectively (and were generated simultaneously in the same tryptic digest), are shown in table S1B.
Western blotting of Prx2
Prx2 dimerization was followed by Western blotting under nonreducing conditions. Human recombinant Prx2 (10 μM) was desalted with 10 mM DTT for 30 min at 4°C. Excess DTT was removed by desalting spin columns. The reduced protein was incubated with 100 μM Na2S2 (Dojindo Laboratories) for 10 min at room temperature, and the reaction was quenched by 1 mM iodoacetamide for 30 min at 37°C. After Bradford protein assay, equal amounts of protein were separated on 12% SDS-PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes. Immunoblotting was performed using anti-Prx2 (Sigma R8656; 1:10,000) and goat anti-rabbit horseradish peroxidase (HRP) (Dako P0448; 1:5000) antibodies and the Bio-Rad Clarity Western ECL Substrate.
Reversible intracellular protein oxidation monitored by dimedone labeling
A431 cells were cultured in 10-cm cell culture dishes and subjected to serum starvation for 24 hours before experiments. Cells were exposed to increasing concentrations of H2O2 (0 to 1 mM) for 15 min at 37°C, washed three times with cold PBS, and then collected in lysis buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1% protease inhibitor cocktail] containing 1 mM DCP-Bio1 [biotin-conjugated dimedone derivative (55), Millipore], catalase (200 U/ml), 10 mM iodoacetamide, and protease inhibitor (150 μl per plate). Lysates were incubated on ice for 1 hour and then centrifuged at 14,000g for 10 min. Supernatants were divided equally and treated with or without 1 mM DTT for 30 min at 25°C. Excess unreacted compounds were removed by Amicon ultraconcentrator filters and subjected to nonreducing SDS-PAGE. Immunoblotting was performed against biotin using an HRP-conjugated antibody, and membranes were evaluated by densitometry using the ImageJ software [National Institutes of Health (NIH), Bethesda]. Immunoprecipitation experiments were carried out in the DTT-reduced and untreated samples to identify target proteins. Biotinylated proteins were pulled down from the mixture by incubation with 40 μl of avidin agarose resin (Pierce) overnight at 4°C with gentle rotation. The beads were then washed with tris-buffered saline buffer [20 mM tris-HCl (pH 7.5) and 150 mM NaCl] six times, and proteins were eluted by boiling at 95°C for 10 min in reducing SDS-PAGE buffer. Eluates were analyzed by Western blotting using antibodies against PTP1B, PTEN, KEAP1, and HSP90.
Preparation of recombinant PTP1B protein (Kumagai laboratory)
The catalytic domain of recombinant hPTP1B was purified as described previously (56). E. coli BL21 cells, which were transformed with a plasmid containing the isolated catalytic domain of human 37-kDa PTP1B (NH2-terminal 321 residues) in a pET15b vector, were preincubated into LB containing ampicillin (100 μg/ml) at 37°C with shaking overnight at 120 rpm (TAITEC, Saitama, Japan). Then, the bacteria were large-scale cultured and grown to 0.6 to 0.8 absorbance at 600 nm. The protein was induced by the addition of 200 μM IPTG at 15°C, 120 rpm, for an additional 21 hours. The cells were harvested by centrifugation and then suspended in lysis buffer that consisted of 50 mM tris-HCl (pH 7.5), 100 mM NaCl, 10 mM 2-ME, and 5% glycerol. Cell lysates were sonicated on ice and then centrifuged at 105,398g for 1 hour to collect supernatant, which was next applied to a Ni-IDA ProBond column at 4°C. The column was extensively washed with buffer containing 50 mM tris-HCl (pH 7.5), 100 mM NaCl, and 10 mM 2-ME; then, PTP1B was eluted with imidazole in a linear gradient from 0 to 100 mM. The collected fractions were monitored by measuring the absorbance at 280 nm and SDS-PAGE. The His tag remained on the protein during further experimentation. Thiol groups oxidized during purification were reduced by incubation with 10 mM DTT for 1 hour on ice, and the LMW compounds were removed by Econo-Pac 10DG column. The protein concentration was determined by Bradford assay. The purified PTP1B was stored in 20 mM tris-HCl (pH 7.5) containing 10 mM DTT at −80°C, in which DTT was removed before each experiment by using Amicon ultraconcentrator filters. This preparation of PTP1B was used for experiments shown in Fig. 5 (A and B) and figs. S6 (A to D and J to L) and S7.
Preparation of recombinant PTP1B protein (Arnér laboratory)
The catalytic domain of PTP1B (residues 1 to 322) was expressed and purified as previously described (22). The His tag was cleaved off from the protein for further experiments. This preparation of PTP1B was used for experiments shown in Fig. 5 (C to R).
Preparation of Trx1, TRP14, and TRP32 (Kumagai laboratory)
Plasmid vectors for Trx1-, TRP14-, or TRP32-encoded pGEX-6p1, which were constructed as described previously (57, 58), were transformed to E. coli JM109. The E. coli cells were preincubated in LB containing ampicillin (100 μg/ml) at 37°C with shaking at 120 rpm overnight. Then, the bacteria were large-scale cultured and grown to ~0.6 to 0.8 absorbance at 600 nm. The proteins expressed as glutathione S-transferase (GST) fusion forms were then induced by the addition of 500 μM IPTG at 15°C, 120 rpm, for an additional 4 hours. The cells were harvested by centrifugation and then suspended in lysis buffer consisting of 20 mM tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride. The supernatant was retained following centrifugation at 105,398g for 1 hour of the sonicated cell lysates. GST-fusion proteins were purified using GSH Sepharose 4 fast flow (GE Healthcare) by batch method according to the manufacturer’s protocol. GST tag was cleaved off by digestion with PreScission protease (GE Healthcare). The proteins, which were eluted in the buffer containing 50 mM tris-HCl (pH 7.0), 150 mM NaCl, and 1 mM EDTA, were dialyzed against 50 mM Hepes-NaOH (pH 7.2) and stocked at −80°C before use. These preparations of Trx1, TRP14, and TRP32 were used for experiments shown in Fig. 5 (A and B).
Preparation of TrxR1 WT and mutants, Trx1, and TRP14 (Arnér laboratory)
WT and Sec mutated rat TrxR1, human Trx1, and TRP14 were expressed and purified as described previously (28, 31, 59). These preparations of TrxR1, Trx1, and TRP14 were used for experiments shown in Figs. 2 (D and E) and 5 (C to R).
Phosphatase assay for PTP1B activity
The phosphatase assay was performed as described previously (56) with a minor modification by using pNPP as a phosphatase substrate. Briefly, recombinant human 37-kDa PTP1B (13.5 μM), 50 mM tris-HCl (pH 7.5), and 0.1 mM EDTA were treated with or without 50 μM Na2S2 for 10 min and then 100 μM H2O2 for 10 min at 25°C. Samples were then treated with or without Trx1 (15 μM), TRP14 (15 μM), or TRP32 (15 μM) with TrxR1 (250 nM) in NADPH (500 μM) for the indicated times at 37°C. Then, PTP1B (13.5 nM) was incubated with the reaction mixture, which consisted of 4 mM pNPP, 100 mM acetate buffer (pH 5.5), and 0.1 mM EDTA for 10 min at 25°C. The phosphatase reaction was terminated by the addition of equal volume of 2 M NaOH. p-Nitrophenol (pNP) formation was measured at 405 nm.
Nano UPLC-MSE analysis
Recombinant hPTP1B (13.5 μM) after treatment with the indicated compounds (fig. S6, A to D) were analyzed by nanoACQUITY UPLC-MSE system (Waters, Milford, MA, USA), equipped with a BEH130 nanoACQUITY C18 column (100 mm by 75 μm internal diameter, 1.7 μm; Waters), and kept at 35°C. The total run time including conditioning the column at the initial conditions was 100 min. Mobile phases (A, 0.1% formic acid in water; B, 0.1% formic acid in acetonitrile) at a flow rate of 0.3 μl/min were linearly mixed in a gradient program as follows: 3% B for 1 min; linearly increasing over 74 min to 40% B, maintained for 4 min; then linearly increasing over 1 min to 95% B, maintained for 5 min before returning linearly to 3% B over 1 min. The eluted peptides were directly connected to the nanoelectrospray source of a mass spectrometer (SYNAPT high-definition Q-TOF, Waters) through a Teflon capillary union and a precut Picotip (Waters). The initial parameters of the mass spectrometer were set with a capillary voltage of 2.5 kV and a sampling cone voltage of 35 V. The source temperature was 100°C, and the detector was operated in positive ion mode. A low collision energy (6 eV) and an elevated collision energy (stepped from 15 to 30 eV) were used to generate intact peptide precursor ions and peptide product ions, respectively. MassLynx version 4.1 software (Waters) was used to control the system and to analyze the mass spectral data. The MS survey scan was m/z 300 to 2000 Da. The data were collected in centroid mode and acquired using an independent reference. Glu-1-fibrinopeptide B (m/z = 785.8426), which was used as an external mass calibration, was infused via the NanoLockSpray ion source and sampled every 10 s. BiopharmaLynx version 1.2 software (Waters) was used to perform baseline subtraction, smoothing, and deisotoping; to identify de novo peptide sequences; and to perform database searches.
Detection of thiodimedone by UPLC-MSE analysis
Preparation of thiodimedone from thiol-activated Sepharose 4B
Thiol-activated Sepharose 4B was reduced by 10 mM DTT for 30 min in the dark, and the unreacted DTT was removed by filtration (Durapore PVDF, Merck, Darmstadt, Germany). The gel was incubated with a polysulfide solution that was generated by the reaction of 3 mM NaOCl and 10 mM Na2S in 50 mM Hepes-HCl (pH 7.5) for 30 min at room temperature and washed three times with distilled deionized water (DDW) by filtration. H2O2 (5 mM) and dimedone (5 mM) were reacted with the persulfidated gel for 1 hour at room temperature. After washing the gel three times with DDW, R-SS-dimedone was reduced by 10 mM DTT for 30 min in the dark. The released 2-thiodimedone sample was collected by centrifugal filter following the addition of 30% acetonitrile–0.33% formic acid to the gel.
Preparation of thiodimedone from PTP1B bound to NHS-Sepharose
hPTP1B-bound NHS-Sepharose (Sigma-Aldrich, St. Louis, MO) was prepared following the manufacturer’s instruction. Briefly, hPTP1B was incubated with the Sepharose at 4°C overnight with gentle rotation. After the incubation, unbound hPTP1B was removed by centrifugation. Unreacted arms on the gel were blocked by 0.5 M ethanol amine–0.5 M NaCl (pH 8.3) for 30 min at room temperature, and the gel was washed three times by altering 100 mM tris-HCl (pH 8) and 100 mM acetic buffer–0.5 M NaCl (pH 4). Using this hPTP1B-bound NHS-Sepharose, hPTP1B-SS-dimedone–bound NHS-Sepharose was prepared as described above. hPTP1B-SS-dimedone was reduced by 10 mM DTT for 30 min in the dark, and the released 2-thiodimedone was collected using a centrifugal filter after the addition of 30% acetonitrile–0.33% formic acid on the gel.
The samples were analyzed using an UPLC-MSE system (Waters), equipped with a BEH130 nanoACQUITY C18 column (2.1 mm long, 50 μm inside diameter, and 1.7 μm particle size) that was maintained at 35°C. Mobile phases (A, 0.1% formic acid in water; B, 0.1% formic acid in acetonitrile) at a flow rate of 0.3 μl/min were linearly mixed in a gradient program as follows: 15% B for 3 min; linearly increase over 5 min to 85% B. The eluted compounds were transferred to the electrospray source of the SYNAPT High-Definition MS System (Waters), whose control and analyses were performed using MassLynx software version 4.1. The ESI was used with a capillary voltage of 2.8 kV, a sampling cone voltage of 35 V, a low collision energy of 6 eV (to detect precursor ions), and an elevated collision energy of 15 to 30 eV (to generate intact product ions). The data were collected in centroid mode and acquired using an independent reference. All analyses were acquired with an independent reference; leucine enkephalin [M − H]− ion as lock mass (m/z = 554.2615) was infused via the LockSpray ion source to ensure accuracy and reproducibility. Data were analyzed with MassLynx version 4.1 software and MassFragment version 1.1 software.
Western blotting for phosphotyrosine
At 90% confluence, WT and TRP14 KD HEK293 cells were starved overnight in Eagle’s Minimum Essential Medium (EMEM) supplemented with 0.1% (v/v) fetal calf serum. Cells were then treated with sodium polysulfide (Na2S3; Dojindo) for 10 min and subsequently stimulated with recombinant EGF ligand (100 ng/ml) (R&D Systems, 236-EG-200), as indicated in Fig. 6 and fig. S8. After stimulation, the medium was aspirated and the cells were washed in ice-cold 1× PBS and lysed with lysis buffer [0.5% Triton X-100, 0.5% deoxycholic acid, 150 mM NaCl, 20 mM tris (pH 7.4), 10 mM EDTA, and 30 mM sodium pyrophosphate] supplemented with 200 μM Na3VO4, 50 mM NaF (phosphatase inhibitors), and protease inhibitor cocktail. Lysates were cleared by centrifugation. Total EGFR phosphorylation was analyzed using immunoblotting with anti-phosphotyrosine (clone 4G10) (Millipore). EGFR expression and EGFR p1148 and p992 phosphorylation were monitored in parallel. Total protein loading was determined using Bradford assay and staining with Ponceau.
Statistical analysis
Densitometric analysis was carried out using the ImageJ software (NIH, Bethesda, MD). All data were shown by means ± SD from at least three independent experiments (exact sample sizes are indicated in the figure captions). The following statistical tests were used to assess significance: one-way or two-way analysis of variance (ANOVA) test, followed by a Dunnett’s multiple comparison test or Bonferoni post test and two-tailed unpaired Student’s t test. Tests were performed using GraphPad Prism version 6.0 software (San Diego, CA, USA) and Microsoft Excel 2013 (Redmond, WA, USA). Differences were considered significant if *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank J. Kundert, C. Miller, I. Cavigli, S. Tamowski, T. Frerk, and I. Pader for technical assistance with the preparation and maintenance of mouse models and R. Kudo and T. Sonobe for initial experiments on HSA-SxSO1-3H reduction. Funding: This work was supported by the 2019 Hungarian Thematic Excellence Program (TUDFO/51757/2019-ITM); the Hungarian National Research, Development and Innovation Office under grant nos. KH_126766 and K_129286 for N.P. and PD_132082 for É.D.; and the Japan Society for the Promotion of Science (Invitational Fellowship L19520) for P.N. This work was also supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Oxygen biology: A new criterion for integrated understanding of life” (no. 17H05519 to Y.K.) and a Grant-in-Aid (no. 17K15489 to Y.A.) for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Financial support is, in part, provided by Grants-in-Aid for Scientific Research (S), (C), for Young Scientists (B), and for Scientific Research on Innovative Areas from the Ministry of Education, Sciences, Sports, and Technology (MEXT), Japan, to T.A. (18H05277, 26111001, and 15K21759), to T.I. (17K08619), and to A.N. (17K15408). E.S.J.A. acknowledges funding from Karolinska Institutet, The Swedish Research Council (2013-765, 2014-2603, and 2017-01872), The Swedish Cancer Society (2015/238 and 2018/333), The Knut and Alice Wallenberg Foundations (KAW 2015.0063), and The Swedish Foundation for International Cooperation in Research and Higher Education (STINT, JA2018-7581 grant for Joint Japan-Sweden Research Collaboration). M.D. acknowledges support from the Swedish Research Council (no. 537-2014-360), The Swedish Society of Medicine (SLS-786841), and Åke Wiberg Stiftelse (nos. M17-0101 and M18-0141). E.E.S. was supported by grants from the U.S. NIH (OD026444, AG040020, GM110732, AG055022, and CA215784), the Montana/Interstate Agricultural Experiment Station (MONB00443 and W4171), and the Montana State University Department of Microbiology. Author contributions: P.N. conceived the study and oversaw the project, contributed to the design and interpretation of experimental results, and, with É.D., wrote the paper. T.A., Y.K., E.S.J.A., and E.E.S. supervised experiments in their individual laboratories, analyzed data, and contributed to manuscript writing. J.F. provided essential intellectual input and edited the manuscript. É.D. (Figs. 1 and 2 and figs. S3, S4, S5I, S6E, and S9), T.I. (Figs. 1, 3, and 5, C to R, and figs. S1, S3, and S5, A to H), Y.A. (Figs. 4 and 5 and figs. S6 and S7), M.D. (Fig. 6 and fig. S8), N.B. (Fig. 2 and figs. S4 and S5), T.T. (Fig. 5, A and B), B.E. (Fig. 6 and fig. S8), N.C.L. (Figs. 4 and 5 and figs. S6 and S7), J.R.P. (Fig. 1 and figs. S2 and S3), and A.N. (Figs. 3 and 5, C to R, and fig. S5, A to H) collected and analyzed data and edited figures and text in the manuscript. Q.C., Y.F., and H.M. provided tools. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The Gsr-mutant line of mice (Gsrnull allele) can be provided by W. Pretsch or E.E.S. pending a completed material transfer agreement. All other lines of mice used in this study, including the novel TRP14-mutant lines reported here (Txndc17null and Txndc17fl alleles), the TrxR1-mutant lines (Txnrd1null and Txnrd1cond alleles), and the Trx1-mutant lines (Txn1null and Txn1fl alleles), are available upon completion of a material transfer agreement from E.E.S. The following lines are also available from Jackson Labs upon completion of a material transfer agreement: Txndc17fl, available as line B6;129-Txndc17tm1Ees/J, JAX# 034607; Txnrd1cond allele, available as line Txnrd1tm1.1Ees/J, JAX# 028283; Txn1fl allele, available as line Txn1tm1.1Ees/J, JAX# 030221. Requests for these lines of mice can be submitted to Jackson Laboratories at https://www.jax.org/ or to E.E.S. at eschmidt@montana.edu.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaax8358/DC1
Table S1. MS settings for the analyses of Cys modifications.
Fig. S1. LC-ESI-MS/MS profiles for CysSO3H and CysSSO3H.
Fig. S2. Txndc17fl and Txndc17null alleles.
Fig. S3. MS-based detection of LMW sulfur species from WT and TRP14 knockout mouse liver samples.
Fig. S4. The partial reduction of oxidized HSA polysulfide species by TrxR1 is Sec dependent.
Fig. S5. The TrxR1-dependent persulfidation-mediated protection of the peroxidative cysteine of Prx2 against overoxidation is more prominently observed under mild alkylating condition.
Fig. S6. UPLC-MS analysis of perthiosulfenic acid modification in the active site of PTP1B and its derivatization with dimedone.
Fig. S7. Reactivation of PTP1B by LMW thiols after inorganic polysulfide and H2O2 treatment.
Fig. S8. A431 cells (cultured with or without 100 nM sodium selenite addition) treated with EGF ligand and 0 to 2000 μM polysulfides.
Fig. S9. Suggested mechanism of PTP1B-mediated EGFR signaling regulation by persulfidation and TRP14.
REFERENCES AND NOTES
- 1.Kabil O., Banerjee R., Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paul B. D., Snyder S. H., H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Ono K., Akaike T., Sawa T., Kumagai Y., Wink D. A., Tantillo D. J., Hobbs A. J., Nagy P., Xian M., Lin J., Fukuto J. M., Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free Radic. Biol. Med. 77, 82–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagy P., Mechanistic chemical perspective of hydrogen sulfide signaling. Methods Enzymol. 554, 3–29 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N. O., Xian M., Fukuto J. M., Akaike T., Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuevasanta E., Möller M. N., Alvarez B., Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. (2016). [DOI] [PubMed] [Google Scholar]
- 7.Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., Banerjee R., Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 138, 289–299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greiner R., Palinkas Z., Bäsell K., Becher D., Antelmann H., Nagy P., Dick T. P., Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 19, 1749–1765 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millikin R., Bianco C. L., White C., Saund S. S., Henriquez S., Sosa V., Akaike T., Kumagai Y., Soeda S., Toscano J. P., Lin J., Fukuto J. M., The chemical biology of protein hydropersulfides: Studies of a possible protective function of biological hydropersulfide generation. Free Radic. Biol. Med. 97, 136–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heppner D. E., Hristova M., Ida T., Mijuskovic A., Dustin C. M., Bogdándi V., Fukuto J. M., Dick T. P., Nagy P., Li J., Akaike T., van der Vliet A., Cysteine perthiosulfenic acid (Cys-SSOH): A novel intermediate in thiol-based redox signaling? Redox Biol. 14, 379–385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulsen C. E., Carroll K. S., Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 113, 4633–4679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood Z. A., Poole L. B., Karplus P. A., Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Winterbourn C. C., Hampton M. B., Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Stöcker S., Maurer M., Ruppert T., Dick T. P., A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14, 148–155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., Winterbourn C. C., Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: A kinetic and computational study. J. Biol. Chem. 286, 18048–18055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., Rhee S. G., Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653–656 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Lee S. R., Kwon K. S., Kim S. R., Rhee S. G., Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Tonks N. K., Redox redux: Revisiting PTPs and the control of cell signaling. Cell 121, 667–670 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Salmeen A., Andersen J. N., Myers M. P., Meng T.-C., Hinks J. A., Tonks N. K., Barford D., Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 (2003). [DOI] [PubMed] [Google Scholar]
- 20.van Montfort R. L., Congreve M., Tisi D., Carr R., Jhoti H., Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Dagnell M., Frijhoff J., Pader I., Augsten M., Boivin B., Xu J., Mandal P. K., Tonks N. K., Hellberg C., Conrad M., Arner E. S., Ostman A., Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-β receptor tyrosine kinase signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 13398–13403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dagnell M., Pace P. E., Cheng Q., Frijhoff J., Östman A., Arnér E. S. J., Hampton M. B., Winterbourn C. C., Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 292, 14371–14380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishnan N., Fu C., Pappin D. J., Tonks N. K., H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 4, ra86 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett W. C., DeGnore J. P., Konig S., Fales H. M., Keng Y. F., Zhang Z. Y., Yim M. B., Chock P. B., Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38, 6699–6705 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Chen Y. Y., Chu H. M., Pan K. T., Teng C. H., Wang D. L., Wang A. H., Khoo K. H., Meng T. C., Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 283, 35265–35272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eriksson S., Prigge J. R., Talago E. A., Arner E. S., Schmidt E. E., Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat. Commun. 6, 6479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prigge J. R., Coppo L., Martin S. S., Ogata F., Miller C. G., Bruschwein M. D., Orlicky D. J., Shearn C. T., Kundert J. A., Lytchier J., Herr A. E., Mattsson A., Taylor M. P., Gustafsson T. N., Arner E. S. J., Holmgren A., Schmidt E. E., Hepatocyte hyperproliferation upon liver-specific co-disruption of thioredoxin-1, thioredoxin reductase-1, and glutathione reductase. Cell Rep. 19, 2771–2781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doka E., Pader I., Biro A., Johansson K., Cheng Q., Ballago K., Prigge J. R., Pastor-Flores D., Dick T. P., Schmidt E. E., Arner E. S. J., Nagy P., A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2, e1500968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akaike T., Ida T., Wei F.-Y., Nishida M., Kumagai Y., Alam M. M., Ihara H., Sawa T., Matsunaga T., Kasamatsu S., Nishimura A., Morita M., Tomizawa K., Nishimura A., Watanabe S., Inaba K., Shima H., Tanuma N., Jung M., Fujii S., Watanabe Y., Ohmuraya M., Nagy P., Feelisch M., Fukuto J. M., Motohashi H., Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 8, 1177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogdándi V., Ida T., Sutton T. R., Bianco C., Ditrói T., Koster G., Henthorn H. A., Minnion M., Toscano J. P., Vliet A., Pluth M. D., Feelisch M., Fukuto J. M., Akaike T., Nagy P., Speciation of reactive sulfur species and their reactions with alkylating agents: Do we have any clue about what is present inside the cell? Br. J. Pharmacol. 176, 646–670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pader I., Sengupta R., Cebula M., Xu J., Lundberg J. O., Holmgren A., Johansson K., Arnér E. S., Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc. Natl. Acad. Sci. U.S.A. 111, 6964–6969 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsui M., Oshima M., Oshima H., Takaku K., Maruyama T., Yodoi J., Taketo M. M., Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 178, 179–185 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Segel I. H., Johnson M. J., Synthesis and characterization of sodium cysteine-S-sulfate monohydrate. Anal. Biochem. 5, 330–337 (1963). [DOI] [PubMed] [Google Scholar]
- 34.Jeong W., Yoon H. W., Lee S. R., Rhee S. G., Identification and characterization of TRP14, a thioredoxin-related protein of 14 kDa. New insights into the specificity of thioredoxin function. J. Biol. Chem. 279, 3142–3150 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Sobotta M. C., Barata A. G., Schmidt U., Mueller S., Millonig G., Dick T. P., Exposing cells to H2O2: A quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic. Biol. Med. 60, 325–335 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Hamid H. A., Tanaka A., Ida T., Nishimura A., Matsunaga T., Fujii S., Morita M., Sawa T., Fukuto J. M., Nagy P., Tsutsumi R., Motohashi H., Ihara H., Akaike T., Polysulfide stabilization by tyrosine and hydroxyphenyl-containing derivatives that is important for a reactive sulfur metabolomics analysis. Redox Biol. 21, 101096 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Q., Antholine W. E., Myers J. M., Kalyanaraman B., Arner E. S., Myers C. R., The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J. Biol. Chem. 285, 21708–21723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy P., Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 18, 1623–1641 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagy P., Winterbourn C. C., Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 23, 1541–1543 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Palinkas Z., Furtmüller P. G., Nagy A., Jakopitsch C., Pirker K. F., Magierowski M., Jasnos K., Wallace J. L., Obinger C., Nagy P., Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 172, 1516–1532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Potor L., Nagy P., Méhes G., Hendrik Z., Jeney V., Pethő D., Vasas A., Pálinkás Z., Balogh E., Gyetvai A., Whiteman M., Torregrossa R., Wood M. E., Olvasztó S., Nagy P., Balla G., Balla J., Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxid. Med. Cell. Longev. 2018, 3812568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garai D., Rios-Gonzalez B. B., Furtmuller P. G., Fukuto J. M., Xian M., Lopez-Garriga J., Obinger C. C., Nagy P., Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 113, 551–563 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Lu J., Holmgren A., The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Libiad M., Yadav P. K., Vitvitsky V., Martinov M., Banerjee R., Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdolrasulnia R., Wood J. L., Transfer of persulfide sulfur from thiocystine to rhodanese. Biochim. Biophys. Acta 567, 135–143 (1979). [DOI] [PubMed] [Google Scholar]
- 46.Flint A. J., Tiganis T., Barford D., Tonks N. K., Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 94, 1680–1685 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sundaresan M., Yu Z. X., Ferrans V. J., Irani K., Finkel T., Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Sirokmány G., Pató A., Zana M., Donkó A., Biró A., Nagy P., Geiszt M., Epidermal growth factor-induced hydrogen peroxide production is mediated by dual oxidase 1. Free Radic. Biol. Med. 97, 204–211 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., Carroll K. S., Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogers L. K., Tamura T., Rogers B. J., Welty S. E., Hansen T. N., Smith C. V., Analyses of glutathione reductase hypomorphic mice indicate a genetic knockout. Toxicol. Sci. 82, 367–373 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Wu S., Ying G., Wu Q., Capecchi M. R., A protocol for constructing gene targeting vectors: Generating knockout mice for the cadherin family and beyond. Nat. Protoc. 3, 1056–1076 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Thomas K. R., Capecchi M. R., Introduction of homologous DNA sequences into mammalian cells induces mutations in the cognate gene. Nature 324, 34–38 (1986). [DOI] [PubMed] [Google Scholar]
- 53.Gu H., Zou Y. R., Rajewsky K., Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73, 1155–1164 (1993). [DOI] [PubMed] [Google Scholar]
- 54.Doka E., Arner E. S. J., Schmidt E. E., Nagy P., ProPerDP: A Protein Persulfide Detection Protocol. Methods Mol. Biol. 2007, 51–77 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Poole L. B., Klomsiri C., Knaggs S. A., Furdui C. M., Nelson K. J., Thomas M. J., Fetrow J. S., Daniel L. W., King S. B., Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug. Chem. 18, 2004–2017 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwamoto N., Sumi D., Ishii T., Uchida K., Cho A. K., Froines J. R., Kumagai Y., Chemical knockdown of protein-tyrosine phosphatase 1B by 1,2-naphthoquinone through covalent modification causes persistent transactivation of epidermal growth factor receptor. J. Biol. Chem. 282, 33396–33404 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Ishii T., Funato Y., Miki H., Thioredoxin-related protein 32 (TRP32) specifically reduces oxidized phosphatase of regenerating liver (PRL). J. Biol. Chem. 288, 7263–7270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morinaka A., Yamada M., Itofusa R., Funato Y., Yoshimura Y., Nakamura F., Yoshimura T., Kaibuchi K., Goshima Y., Hoshino M., Kamiguchi H., Miki H., Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 4, ra26 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Xu J., Eriksson S. E., Cebula M., Sandalova T., Hedstrom E., Pader I., Cheng Q., Myers C. R., Antholine W. E., Nagy P., Hellman U., Selivanova G., Lindqvist Y., Arner E. S., The conserved Trp114 residue of thioredoxin reductase 1 has a redox sensor-like function triggering oligomerization and crosslinking upon oxidative stress related to cell death. Cell Death Dis. 6, e1616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaax8358/DC1
Table S1. MS settings for the analyses of Cys modifications.
Fig. S1. LC-ESI-MS/MS profiles for CysSO3H and CysSSO3H.
Fig. S2. Txndc17fl and Txndc17null alleles.
Fig. S3. MS-based detection of LMW sulfur species from WT and TRP14 knockout mouse liver samples.
Fig. S4. The partial reduction of oxidized HSA polysulfide species by TrxR1 is Sec dependent.
Fig. S5. The TrxR1-dependent persulfidation-mediated protection of the peroxidative cysteine of Prx2 against overoxidation is more prominently observed under mild alkylating condition.
Fig. S6. UPLC-MS analysis of perthiosulfenic acid modification in the active site of PTP1B and its derivatization with dimedone.
Fig. S7. Reactivation of PTP1B by LMW thiols after inorganic polysulfide and H2O2 treatment.
Fig. S8. A431 cells (cultured with or without 100 nM sodium selenite addition) treated with EGF ligand and 0 to 2000 μM polysulfides.
Fig. S9. Suggested mechanism of PTP1B-mediated EGFR signaling regulation by persulfidation and TRP14.