

Abstract
Glutamate and dopamine systems play distinct roles in terms of neuronal signalling, yet both have been proposed to contribute significantly to the pathophysiology of schizophrenia. In this paper we assess research that has implicated both systems in the aetiology of this disorder. We examine evidence from post‐mortem, preclinical, pharmacological and in vivo neuroimaging studies. Pharmacological and preclinical studies implicate both systems, and in vivo imaging of the dopamine system has consistently identified elevated striatal dopamine synthesis and release capacity in schizophrenia. Imaging of the glutamate system and other aspects of research on the dopamine system have produced less consistent findings, potentially due to methodological limitations and the heterogeneity of the disorder. Converging evidence indicates that genetic and environmental risk factors for schizophrenia underlie disruption of glutamatergic and dopaminergic function. However, while genetic influences may directly underlie glutamatergic dysfunction, few genetic risk variants directly implicate the dopamine system, indicating that aberrant dopamine signalling is likely to be predominantly due to other factors. We discuss the neural circuits through which the two systems interact, and how their disruption may cause psychotic symptoms. We also discuss mechanisms through which existing treatments operate, and how recent research has highlighted opportunities for the development of novel pharmacological therapies. Finally, we consider outstanding questions for the field, including what remains unknown regarding the nature of glutamate and dopamine function in schizophrenia, and what needs to be achieved to make progress in developing new treatments.
Keywords: Psychosis, schizophrenia, dopamine, glutamate, antipsychotics, striatum, NMDA receptors, D2 receptors, D1 receptors, dorsolateral prefrontal cortex, GABA interneurons, amphetamine, ketamine, cognitive symptoms
Schizophrenia is a severe mental disorder characterized by positive symptoms such as delusions and hallucinations, negative symptoms including amotivation and social withdrawal, and cognitive symptoms such as deficits in working memory and cognitive flexibility1. The disorder accounts for significant health care costs, and is associated with a reduced life expectancy of about 15 years on average2.
Antipsychotics were serendipitously discovered over fifty years ago, but it took another decade or so until dopamine antagonism was demonstrated as central to their clinical effectiveness3. Further evidence implicating the dopamine system in the pathophysiology of schizophrenia has subsequently accumulated, and it remains the case that all licensed first‐line treatments for schizophrenia operate primarily via antagonism of the dopamine D2 receptor4.
However, despite the central role that dopamine plays in our understanding of schizophrenia, it has also become increasingly clear that dysfunction of this system may not be sufficient to explain several phenomena. In particular, dopamine blockade is not an effective treatment for negative and cognitive symptoms and, in a significant proportion of patients, it does not improve positive symptoms either. As a result, attention has turned to additional neurochemical targets. Glutamate is the major excitatory neurotransmitter of the central nervous system. The finding that antagonists of a specific glutamate receptor, the N‐methyl‐D‐aspartate (NMDA) receptor, induce psychotic symptoms has led to a wealth of research implicating the glutamate system in the pathophysiology of schizophrenia.
In this paper we review the evidence regarding dopaminergic and glutamatergic functioning in schizophrenia. We survey indirect findings from preclinical, genetic and pharmacological studies, evidence from post‐mortem research, and results of neuroimaging studies that characterize functioning in living patients. We discuss how dysregulation of these systems may lead to the symptoms of the disorder, and the therapeutic possibilities associated with their pharmacological modulation. We then explore what may underlie this dysregulation, and the interaction between the two systems, before concluding by considering outstanding questions for the field.
DOPAMINE
Dopamine was initially thought to be a biologically inactive intermediary compound on the synthetic pathway between tyrosine and noradrenaline. Work by A. Carlsson and others, however, demonstrated that dopamine depletion inhibited movement, and that this effect could be reversed following the administration of the dopamine precursor L‐DOPA. This established that the molecule was of major biological importance in its own right5, and discrete dopaminergic projections were subsequently identified.
That dopaminergic dysfunction might play a role in the development of psychotic symptoms is one of the longest standing hypotheses regarding the pathophysiology of schizophrenia. Below, we discuss the evidence for dopamine dysfunction in schizophrenia, before considering how this may lead to psychotic symptoms, and the mechanisms through which dopamine modulating treatments exert their effects.
Indirect evidence for dopamine dysfunction in schizophrenia
Animal models
Rodent models of schizophrenia are useful for investigating molecular mechanisms that may be of pathophysiological relevance, and for testing novel therapeutic interventions.
One well characterized model of dopaminergic hyperactivity involves administering repeated doses of amphetamine. This has been shown to induce events that are also observed in individuals with schizophrenia, such as reduced prepulse inhibition, stereotyped behaviours, and impaired cognitive flexibility and attention6. Given that amphetamine results in dopamine release, and that the above effects can be ameliorated with the administration of dopamine antagonists, this provides indirect evidence for a role of dopamine in behaviour thought to be a proxy for psychotic symptoms.
Another example is that of mice genetically modified to overexpress dopamine D2 receptors in the striatum, which also display a wide range of schizophrenia‐like behaviours7. Similarly, transgenic insertion of tyrosine hydroxylase and guanosine triphosphate (GTP) cyclohydrase 1 into the substantia nigra in early adolescence increases dopamine synthesis capacity, and has been associated with a schizophrenia‐like behavioural phenotype8.
Other examples do not target the dopamine system directly, but are still associated with dopaminergic abnormalities. The methylazoxymethanol acetate (MAM) model involves inducing neurodevelopmental disruption of the hippocampus via the administration of MAM to pregnant rats, and is accompanied by increased firing rates of mesostriatal dopamine neurons9. A model of environmental risk factors in which rats were socially isolated post weaning has also been associated with increased striatal presynaptic dopamine function10.
In summary, multiple methods have been used to induce increased striatal dopamine signalling in animal models, and these consistently produce behaviours analogous to those observed in individuals with schizophrenia.
Cerebrospinal fluid and post‐mortem studies
Studies examining levels of dopamine and its metabolites – 3,4‐dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) – in schizophrenia, both peripherally and in cerebrospinal fluid (CSF), have given inconsistent results11, 12, 13. This may be due to the fact that these levels are a state dependent marker, and to the effects of antipsychotic treatment. Studies have found that levels of dopamine, HVA and DOPAC in CSF are only increased in those receiving antipsychotic treatment13, 14, and that reductions occur following the withdrawal of antipsychotics15, 16.
Some17, 18, 19, but not all20, studies of HVA have found higher levels in both CSF and plasma of acutely relapsed patients compared to stable patients. There have also been suggestions that baseline plasma HVA levels may predict subsequent response to antipsychotics, which shows some parallels with imaging findings considered below21.
This approach to studying the dopamine system, however, has declined in popularity over recent years. A major weakness is that the measurement occurs distal from the dopamine neurons of interest. Since both hypo‐ and hyperdopaminergic function may exist within an individual simultaneously, a technique that allows for anatomical specificity is required to understand the nature and localization of changes.
Early post‐mortem investigations suggested that striatal D2 receptor levels might be raised in individuals with psychosis22, and a meta‐analysis of seven post‐mortem studies suggested that receptor levels were increased with a large effect size23. However, no studies of antipsychotic naïve individuals exist, and the majority are of individuals chronically treated with antipsychotic medications, which have been found to lead to D2 receptor upregulation24, 25.
Post‐mortem studies have also examined the substantia nigra. In these studies evidence regarding dopamine function is inconsistent, with some studies suggesting an increase in tyrosine hydroxylase levels in patients26, but others finding no difference27, 28. Other studies have found abnormal nuclear morphology of substantia nigra neurons29, reduced dopamine transporter (DAT) and vesicular monoamine transporter (VMAT) gene expression, and increased monoamine oxidase A expression28.
Recent collaborative efforts in amassing significantly larger post‐mortem sample sizes, and applying more sophisticated methods of analysis, may improve our understanding in the near future30. However, even with these developments, the drawbacks of post‐mortem studies include heterogenous tissue quality, the fact that the majority of samples are from older patients with a long history of antipsychotic use, limited information regarding clinical phenotype, and that death itself leads to a wide range of neurobiological changes that may obscure important differences.
Studies in living participants have greater potential to include younger individuals, drug‐free subjects, and also the ability to look at within‐individual changes in symptoms and how these relate to pharmacological manipulation.
Psychopharmacology of dopaminergic agonists and antagonists
The discovery that chlorpromazine and reserpine were effective in the treatment of schizophrenia occurred prior to the identification of dopamine as a neurotransmitter. It was not until the 1970s that the clinical potency of antipsychotics was incontrovertibly linked to blockade of the dopamine D2 receptor31, 32. In addition, selective D2 antagonists show equivalent efficacy to drugs with a broad spectrum of activity33, indicating that D2 antagonism is sufficient for antipsychotic efficacy.
It was also noted that drugs such as amphetamine that increase dopaminergic neurotransmission could induce psychotic symptoms in healthy individuals, and exacerbate psychotic symptoms in individuals with schizophrenia34, 35. Similarly, L‐DOPA treatment in Parkinson's disease has been found to induce psychotic symptoms in some individuals36. However, while amphetamine‐induced psychosis is marked by hallucinations, delusions, paranoia, and conceptual disorganization, it is not typically associated with negative and cognitive symptoms of the same form as those observed in schizophrenia37. This relative specificity to positive psychotic symptoms contrasts with glutamatergic models of schizophrenia (see later).
Summary of indirect findings
The findings discussed above provide evidence that aberrant function of the dopamine system contributes to psychotic symptoms (see Table 1). However, these methods are unable to identify where within the brain this dysfunction is localized to and, for the most part, cannot provide a direct link to symptoms. We next discuss methods for in vivo imaging of the dopamine system, which has the potential to overcome these obstacles.
Table 1.
Summary of indirect evidence for dysfunction of dopamine and glutamate systems in schizophrenia
| Dopamine | Glutamate | |
|---|---|---|
| Animal models | Amphetamine administration, striatal D2 overexpression, and transgenically increased dopamine synthesis capacity are associated with schizophrenia‐like behaviours. Models of neurodevelopmental and social risk factors are associated with increased striatal dopamine function. | Administration of NMDA antagonists induces a wide variety of schizophrenia‐like behaviours. Genetic models that disrupt NMDA signalling (by reducing levels of D‐serine, inactivating D‐amino oxidase or decreasing dysbindin) show behavioural and neurobiological changes similar to those observed in schizophrenia. |
| Cerebrospinal fluid (CSF) | Studies of DOPAC and HVA both peripherally and in CSF have been inconsistent. | Studies of glutamate levels are inconsistent, but kynurenic acid (an NMDA antagonist) levels appear consistently raised. |
| Post‐mortem studies | Increased D2 receptor densities have been observed, but may result from medication use. | Glutamate neurons show reduced dendrite arborization, spine density and synaptophysin expression. Glutamate transporter EAAT2 protein and mRNA levels appear reduced in frontal and temporal areas. There is some evidence that glutaminase expression is increased in patients, and also that GRIN1 abnormalities exist. |
| Pharmacological studies | Clinical potency of antipsychotics is strongly linked to their affinity for the D2 receptor. Amphetamines can induce positive psychotic symptoms in healthy controls and worsen symptoms in patients. | NMDA antagonists induce positive, negative and cognitive psychotic symptoms in healthy controls. Chronic ketamine users show subthreshold psychotic symptoms. |
NMDA – N‐methyl‐D‐aspartate, DOPAC – 3,4‐dihydroxyphenylacetic acid, HVA – homovanillic acid, EAAT – excitatory amino acid transporter
Imaging dopamine in vivo
Both magnetic resonance imaging (MRI) and positron emission tomography (PET) have been used to characterize the dopamine system in vivo (Table 2). PET provides molecular specificity to the dopamine system, but this comes at the cost of lower temporal and spatial resolution compared to MRI.
Table 2.
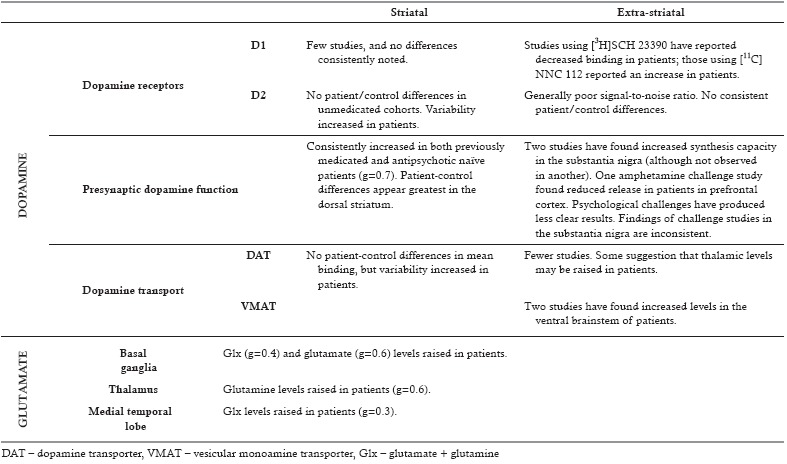
Summary of imaging studies of the dopamine and glutamate systems in schizophrenia
MRI
Although MRI lacks the ability to directly image the dopamine system, recent work imaging neuromelanin has shown some promise in quantifying the dopamine system in vivo. Neuromelanin is synthesized via iron dependent oxidation of cytosolic dopamine, and accumulates in dopamine neurons of the substantia nigra. It has been demonstrated that the neuromelanin MRI signal is associated with integrity of dopamine neurons, with dopamine release capacity in the striatum, and with the severity of psychosis in schizophrenia49.
Functional MRI (fMRI) has also been used in attempts to infer functioning of the dopamine system. Task‐based fMRI has been adopted to quantify the striatal response to reward, and this has been linked to dopamine function, although the precise relationship is complex50. There is consistent evidence of reduced ventral striatal activation to reward in schizophrenia51. We consider how this is consistent with the hypothesis of an overactive dopamine system in the section discussing psychotic symptoms below.
PET: dopamine receptors
Dopamine receptors have been studied using a wide range of radioligands. The majority of studies have used ligands specific for D2‐type (i.e., D2, D3 and D4) dopamine receptors, although several studies have also examined D1‐type (i.e., D1 and D5) receptors.
Striatum
It has been proposed that excessive dopaminergic neurotransmission in schizophrenia results from upregulation of striatal postsynaptic D2‐type receptors. However, meta‐analyses of studies using PET show only a small increase in receptor density at most in schizophrenia, and there is no significant difference between patients and controls in analyses restricted to medication naïve patients52. When combined with evidence that antipsychotic treatment appears to lead to D2 receptor upregulation24, 25, it appears possible that any patient‐control differences may be secondary to confounding by treatment.
There are caveats, however, to the above inference. First, the majority of studies are unable to measure the absolute density of receptors, because a proportion of receptors will be occupied by endogenous dopamine. If schizophrenia is associated with increased synaptic dopamine levels, this could mask a concurrent increase in receptor densities. Indeed, one study where dopamine depletion was undertaken prior to PET scanning showed significantly increased dopamine receptor availability in patients, although this increase was not significant in another study using this approach53, 54.
Second, the majority of ligands are selective for D2 over D3 and D4 receptors. The studies that have employed butyrophenone tracers (that have an affinity for D4 receptors in addition to D2 and D3 receptors) have tended to show raised receptor densities compared to those studies employing ligands that do not have D4 affinity52. In addition to potential differences in D2/3/4 subtype proportions, D2 receptors exist in both high and low affinity states, and some evidence suggests that schizophrenia may be associated with an increased proportion of receptors in the high affinity state55, 56, 57, 58.
Furthermore, following receptor internalization, some tracers remain bound, while others dissociate. So, if receptor internalization is increased in one group, this would register as reduced ligand binding if using a tracer that dissociates on internalization, but not if using a tracer that remains bound59, 60.
Finally, it has recently been shown that the variability of striatal D2 receptor levels is greater in patients than controls61, suggesting that differences in D2 receptor density may exist, but only within a subgroup of patients, although whether this reflects a primary pathology or an effect of prior antipsychotic treatment in some patients remains unclear.
D1‐type receptors have not been studied frequently in the striatum, and the studies that have been undertaken do not show any clear patient‐control differences52, 62.
Extra‐striatal regions
The measurement of dopamine receptors in extra‐striatal regions is complicated by the lower receptor densities, which means that the signal‐to‐noise ratio is much lower than in the striatum. Studies of thalamic, temporal cortex and substantia nigra D2/3 receptor availability have not consistently shown patient‐control differences63. Other cortical regions have rarely been studied, and have not shown consistent changes63.
D1 receptors have been more thoroughly examined in cortical regions than in the striatum. Two studies using [11C]NNC 112 reported an increase in patients64, 65, while one reported a decrease66. Four studies using [3H]SCH 23390 have reported a decrease62, 66, 67, 68, while two found no significant differences69, 70. The interpretation of these findings is complicated by the fact that dopamine depletion paradoxically decreases the binding of [3H]SCH 23390, while it has no effect upon [11C]NNC 112 binding. Furthermore, antipsychotic exposure decreases D1 receptor expression, and both the above ligands also show affinity for the 5‐HT2A receptor71, 72, 73.
PET: dopamine transport mechanisms
DAT is involved in reuptake of dopamine from the synaptic cleft, and is often interpreted in PET studies as a measure of the density of dopamine neurons. Studies examining DAT density in the striatum have found no consistent differences between patients and controls52, although, as with D2 receptors, variability is increased in schizophrenia, suggesting that differences may exist within a subgroup61. A more recent study did find significantly raised striatal DAT levels in patients, but this was observed in those with a chronic illness with long‐term antipsychotic exposure74.
There have been fewer studies examining extra‐striatal regions, although the ones that have been undertaken do suggest that thalamic DAT levels may be raised in patients74, 75.
VMAT2 transports intracellular monoamines into synaptic vesicles. Two PET studies have found that its levels were increased in the ventral brainstem of individuals with schizophrenia, but found no differences compared to controls in the striatum or thalamus76, 77. This is in contrast to the post‐mortem studies discussed above28, but in keeping with a study showing increased VMAT2 density within platelets from individuals with schizophrenia78.
PET: presynaptic dopamine function
Multiple methods exist for quantifying aspects of presynaptic dopamine function.
Several studies have investigated dopamine release capacity by studying the reaction of the dopamine system to an acute challenge, be that pharmacological such as amphetamine, or psychological such as a reward or stress task79. Animal studies have shown that comparing ligand binding during the challenge to binding at baseline allows one to infer the amount of dopamine release induced by the task80.
Alternatively, one can obtain a measure of baseline synaptic dopamine levels by comparing a baseline scan with a scan obtained following the administration of a dopamine depleting agent such as alpha‐methylparatyrosine.
Finally, radiolabelled L‐DOPA can be used to quantify dopamine synthesis capacity. Radiolabeled L‐DOPA is taken up by dopamine neurons, where it is converted by aromatic L‐amino acid decarboxylase to dopamine, which is then sequestered in vesicles within nerve terminals81. The rate of uptake provides an index of dopamine synthesis capacity.
Striatum
Studies have consistently demonstrated raised presynaptic dopamine function in schizophrenia, with Hedges’ g=0.7 (Hedges’ g is a measure of effect size, and values of 0.2 are typically considered small, those of 0.5 medium, and those of 0.8 large82). The studies using a challenge paradigm show larger effect sizes (g=1.0) compared to those quantifying dopamine synthesis capacity (g=0.5)83. The hyperdopaminergic state associated with schizophrenia appears greatest within the dorsal striatum83.
Further evidence for pathophysiological relevance comes from studies showing a direct association between synthesis capacity and the severity of positive psychotic symptoms84, 85, 86. The relationship with other symptom domains is less clear: an inverse relationship with depressive symptoms87 and a lower synthesis capacity associated with worse cognitive performance88 have been reported.
Extra‐striatal regions
Outside of the striatum, dopamine synthesis capacity can only be reliably measured in a limited number of brain regions, such as the substantia nigra and the amygdala, using current techniques89. Two studies have found increased dopamine synthesis capacity in the substantia nigra90, 91, although this was not observed in another92. One study also found raised dopamine turnover in the amygdala91.
Although cortical dopamine receptors are predominantly D1‐type, D1 receptor ligands are not reliably displaceable, and therefore not suitable for challenge or displacement studies. Cortical D2 receptors do exist, but studies are complicated by their sparsity93. Furthermore, although challenge paradigms have demonstrated validity in the striatum, the results of cortical studies are harder to interpret, with displacement not always observed94.
One study using amphetamine challenge in combination with the high‐affinity ligand FLB‐457 found reduced dopamine release in the prefrontal cortex in individuals with schizophrenia95. Two other recent FLB‐457 studies adopted psychological challenges. One of these used a psychological stressor, which did not induce cortical tracer displacement in either patients or controls96. The other used a cognitive test of executive function, which did show lower tracer displacement in patients, but interpretation was complicated by the fact that, again, the task did not consistently induce dopamine release97. A study using 18F‐fallypride found no differences between patients and controls in terms of stress‐induced cortical dopamine release98.
Two studies have examined dopamine release in the substantia nigra. One used a stress challenge and found an increased release in patients99; the other adopted an amphetamine challenge and found a non‐significant reduction95.
PET: dopamine across the psychosis spectrum
Several studies have investigated dopamine function in subjects at clinical high risk for psychosis. Initial studies showed evidence of raised presynaptic dopamine function in these individuals100, 101, 102. However, this was not seen in the largest study to date103. This may potentially result from the fact that raised presynaptic striatal dopamine function appears to be limited to those subjects who subsequently develop psychosis104, and transition rates have declined in recent years.
A study of healthy individuals that experience auditory hallucinations also found no difference in striatal dopamine synthesis capacity compared to healthy controls without hallucinations105. Studies in individuals at increased genetic risk for schizophrenia, such as patients’ relatives and individuals with 22q11.2 deletion syndrome, have also not shown consistent differences from controls in terms of presynaptic dopamine function106, 107, 108.
Studies in psychotic individuals with diagnoses other than schizophrenia, such as bipolar disorder and temporal lobe epilepsy86, 109, have found raised striatal dopamine synthesis capacity. This finding, along with the inconsistent evidence in people at increased clinical or genetic risk, may suggest that increased striatal dopamine synthesis capacity is associated with psychosis across psychiatric diagnoses, rather than being an underlying risk factor for schizophrenia.
Studies of dopamine receptor densities in individuals at both clinical99, 100, 110 and genetic107, 110, 111, 112, 113 high risk are similar to those in individuals with schizophrenia, in that they have shown no clear differences from controls.
Summary of PET findings
The studies reviewed above provide consistent evidence of a striatal presynaptic hyperdopaminergic state in schizophrenia (see Table 2), and little consistent evidence of altered D2/3 receptor levels. It remains uncertain as to whether abnormalities exist with regard to other dopamine receptors, or with cortical dopamine function.
Consequences of dopaminergic dysfunction
Prediction errors, salience and positive symptoms
After its role in movement was established, preclinical findings suggested that dopamine also played a role in signalling reward114. Later work demonstrated that signalling more specifically related to the discrepancy between expected and received reward – a reward prediction error115. More recently, it has been demonstrated that firing is not exclusively tied to reward prediction, but rather can occur in response to a wide range of salient stimuli116, 117, 118, 119, 120, and that in more dorsal regions of the striatum dopamine signalling is particularly associated with threat‐related stimuli118, 119.
Several related theories have proposed how disruption to normal dopamine function could underlie positive psychotic symptoms such as delusions and hallucinations121, 122, 123. Dysregulated dopamine neuron firing will aberrantly signal that irrelevant stimuli are of importance, thereby imbuing percepts and thoughts with abnormal salience, in turn leading to inappropriate associations and causal attributions124. There are also mechanisms through which uncoordinated dopamine signalling may contribute not only to the generation of delusional beliefs, but also to the imperviously rigid form of delusional thought123, 125.
Recent work has attempted to identify more precisely the mechanisms through which dopaminergic dysfunction may contribute to symptoms. The experience of a stimulus depends not only on the sensory inputs resulting from that stimulus, but also on prior expectations regarding the probability of a percept. Auditory hallucinations appear to result from a stronger influence of prior expectations upon sensory percepts126, and this increased weighting of priors has been associated with greater levels of amphetamine‐induced dopamine release in the striatum127.
In terms of understanding the development of delusions, a combined PET and MRI experiment found that dopamine release was related to neural signalling of belief updates rather than just sensory surprise128. This suggests that aberrant dopamine signalling may lead to irrelevant stimuli being understood as meaningful, the clinical relevance of which is supported by the finding that participants who displayed more aberrant belief updating showed greater subclinical paranoid ideation128.
In addition to mesostriatal dopamine signalling, several cortical regions have also been implicated in salience processing129, 130. The salience network comprises the anterior cingulate cortex and bilateral insula, and abnormalities of this network have been proposed as a core feature of schizophrenia pathophysiology131. The network has a key role in orchestrating dynamic switching between brain states, for example between a resting state and states associated with performing cognitively demanding tasks132. It is of relevance that dopamine signalling also plays a role in dynamic reorganization of brain states133, 134. Recent work has demonstrated that mesostriatal dopamine signalling and salience network connectivity are tightly linked135, although whether this relationship is disrupted in schizophrenia is not known.
Reward, motivation and negative symptoms
Reward and punishment are fundamental drivers of behaviour, and reinforcement learning models formalize the relationship between reward, states and behaviour. Prediction errors allow the value of states and actions to be learnt, and are a key signal in many reinforcement learning models. Given the central role of dopamine in both coding prediction errors and in the cortical representation of environmental states, several studies have used this framework to explore the behavioural consequences of disrupted dopamine signalling136.
Cortical D1 receptors play a central role in shaping the accurate neural representation of environmental states, by allowing precise inhibition of neural activity137. Reduced cortical dopamine signalling means that stimuli associated with reward cannot be accurately encoded, effectively foreclosing their ability to guide behaviour137. Furthermore, reduced cortical dopamine signalling may mean that reward‐related representations are short‐lived, with the consequence that, even if correctly represented, the motivational properties of reward‐associated stimuli have a briefer impact137.
Dopamine neurons fire in response to stimuli that have been previously associated with reward, and guide behaviour towards actions associated with previous reward138. A striatal hyperdopaminergic state may mean that reward‐associated stimuli have reduced motivational influence, as aberrantly high background levels of dopamine signalling reduce the signal‐to‐noise ratio of adaptive phasic signalling139. This mechanism also has the potential to reduce the appetitive properties of a given reward, thereby reducing its impact to shape future behaviour, and accounting for negative symptoms such as anhedonia and amotivation140, 141, 142. This reduced signal‐to‐noise ratio may account for the reduced striatal activation to reward observed with fMRI in individuals with schizophrenia51.
Cortical dopamine and cognitive symptoms
Cognitive symptoms of schizophrenia include deficits in working memory, executive function, and information processing. They occur prior to the onset of frank psychosis and account for a significant proportion of the morbidity associated with the illness143, 144, 145.
The dorsolateral prefrontal cortex is central to many cognitive processes, and both functional and structural pathology of the region has been linked to the deficits seen in schizophrenia146. The molecular changes underlying cognitive symptoms, however, are unknown. Given the importance of D1 receptor signalling for cognition147, reduced cortical dopamine signalling has long been hypothesized to contribute to the cognitive symptoms observed in schizophrenia. As discussed above, however, evidence regarding D1 receptors in schizophrenia is inconclusive, and there has been only a single study demonstrating reduced cortical dopamine release95.
On the basis of preclinical work using a model of striatal D2 overexpression, it has also been proposed that excessive striatal dopamine signalling may lead to reductions in cortical dopamine and associated cognitive symptoms7. Therefore, it appears that both reduced and excessive dopamine signalling can have deleterious effects on cognition, which may contribute to the fact that there has been minimal success in developing dopamine modulating treatments for cognitive symptoms of schizophrenia4, 145.
GLUTAMATE
As reviewed above, there is significant evidence that dysfunction of the dopamine system is involved in the pathogenesis of schizophrenia. This may, however, occur downstream of other pathophysiological processes. Furthermore, schizophrenia is a heterogenous disorder, and dopaminergic dysfunction may not play a significant role in some individuals, while in others it may be only one of several pathological mechanisms.
Substantial evidence has accumulated implicating the glutamate system in the pathogenesis of schizophrenia. Glutamate is the major excitatory neurotransmitter in the central nervous system, and, in contrast to the anatomically localized cell bodies of dopamine neurons, glutamatergic neurons are widespread throughout the brain.
Glutamate receptors show considerable variety, and are classified as either ionotropic or metabotropic. Ionotropic receptors include the NMDA and the non‐NMDA receptors – kainate and alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA). NMDA and non‐NMDA receptors are normally co‐localized on neurons, and act synergistically in that the NMDA receptor has slower kinetics and tends to enhance depolarization initiated by non‐NMDA receptors.
Following the discovery of the psychotomimetic effects of NMDA antagonists, the NMDA receptor has become a primary focus when considering potential glutamatergic dysfunction in schizophrenia. Below we examine the evidence associating schizophrenia with glutamatergic abnormalities, consider how these abnormalities may lead to symptoms, and review the potential of glutamate modulating agents as treatments.
Indirect evidence for glutamatergic dysfunction in schizophrenia
Animal models
The administration of NMDA antagonists to non‐human primates and rodents has been shown to induce a variety of schizophrenia‐like behaviours, such as sensorimotor gating impairments, increased locomotion, abnormal repetitive movements, and cognitive and social deficits38. NMDA antagonism has also been shown to lead to hippocampal hypermetabolism, similar to that which has been observed in schizophrenia148, 149.
A genetic animal model that reduced levels of the NMDA co‐agonist D‐serine was associated with neurobiological changes similar to those observed in schizophrenia, such as reduced dendritic spine density and hippocampal volume39. Several other genetic mice models, such as those involving inactivation of D‐amino oxidase40 and reduction of the synaptic protein dysbindin41, also show NMDA receptor hypofunction accompanied by behavioural and neurobiological changes analogous to those observed in schizophrenia.
CSF and post‐mortem studies
Initial small studies of CSF found reduced glutamate levels in patients, but these findings were not replicated42, 43. However, CSF and post‐mortem brain levels of kynurenic acid, an NMDA receptor antagonist, have consistently found to be raised, although not plasma levels44.
Post‐mortem studies investigating structural alterations of glutamate neurons have generally found reductions in dendrite arborization, spine density, and synaptophysin expression across frontal and temporal regions45.
mRNA expression of specific glutamate receptors and their subunits has also been investigated across multiple brain regions. Although several studies have found reduced expression of NMDA receptor subunits such as GRIN1, GRIN2A and GRIN2C, these have not been consistently replicated across brain regions45, although – when regions are examined individually – there is evidence of reduced GRIN1 expression in the hippocampus45. A recent study examining samples from over 500 individuals with schizophrenia found increased exon skipping in GRIN1, that would affect the extracellular ligand binding site30. Fewer studies have examined protein expression of these subunits, and these are also inconsistent in their findings45.
There have also been a small number of studies investigating enzymes involved in glutamate metabolism. Excitatory amino acid transporters (EAAT) remove glutamate from the synaptic cleft. After reuptake from the synapse, glutamine synthetase converts glutamate to glutamine. When glutamine is delivered to neurons, it is converted back to glutamate by glutaminase. There is some preliminary evidence that both mRNA and protein levels of EAAT2, the transporter responsible for the majority of glutamate uptake, are reduced in frontal and temporal areas of patients with schizophrenia, while glutaminase mRNA levels and enzymatic activity have been found to be raised in two studies examining, respectively, the thalamus and dorsolateral prefrontal cortex45.
Although the caveats regarding post‐mortem studies discussed above remain, several of the findings covered here are consistent with impaired functioning of the NMDA receptor. In addition to the direct pathology suggested by the findings relating to GRIN1, the reduced expression of EAAT2 and the increased expression of glutaminase may be understood as compensatory responses attempting to increase synaptic glutamate levels.
Psychopharmacology of NMDA antagonists
The administration of NMDA receptor antagonists, such as phencyclidine and ketamine, to healthy controls has been repeatedly shown to induce manifestations similar to the positive, negative and cognitive symptoms of schizophrenia46. It has been demonstrated that NMDA receptor blockade by these compounds is both necessary and sufficient for their psychotomimetic effects150.
These drugs have also been shown to exacerbate a similarly wide spectrum of symptoms in individuals with schizophrenia, in contrast to amphetamines, which predominantly worsen positive symptoms151. Furthermore, it has been shown that anti‐NMDA receptor encephalitis may be associated with psychiatric presentations that resemble schizophrenia in some individuals152. NMDA antagonism has, therefore, been proposed to be a superior pharmacological model of schizophrenia, compared to amphetamines, given its ability to more reliably induce negative as well as positive symptoms153, 154.
Acute NMDA antagonist administration has typically been employed in experimental settings, since chronic administration to healthy controls cannot be ethically undertaken. However, chronic ketamine users show subthreshold psychotic symptoms across positive, negative and cognitive domains47, 48, and a subgroup of these chronic users develop a persisting psychosis that is more similar to schizophrenia than that induced by acute single dose ketamine administration155.
Imaging glutamate in vivo
Proton magnetic resonance spectroscopy
Proton magnetic resonance spectroscopy (1H‐MRS) is the most frequently used technique for investigating the glutamate system in vivo. It does not require the use of ionizing radiation, and is significantly cheaper than PET.
1H‐MRS does, however, have several drawbacks, including the fact that it is unable to distinguish between intra‐ and extracellular compartments156. Glutamate does not act solely as a neurotransmitter, but is involved in protein synthesis and nitrogen metabolism, and is a precursor to GABA157. This means that it is not possible to infer whether differences between patients and controls relate to synaptic glutamate concentrations, as opposed to alterations in these other functions of glutamate. Even if one assumes that detectable abnormalities relate to differences in synaptic neurotransmission, it is not possible to determine whether this is secondary to differences in synaptic levels, as opposed to presynaptic dysfunction, or altered reuptake of glutamate.
At higher field strengths, glutamate and its metabolite glutamine can be distinguished, while at lower strengths the concentration of both, often abbreviated to Glx, is all that can be accurately quantified. Glutamine is synthesized from glutamate following the uptake of synaptic glutamate by astrocytes, and glutamine levels have been taken to be a marker of glutamate neurotransmission. However, as with glutamate, glutamine takes part in multiple cell process, which complicates interpretation158.
There have been over fifty 1H‐MRS studies of the glutamate system in schizophrenia. A synthesis of their findings is complicated by methodological differences, notably in the imaging sequences used, the brain regions studied, and the patient cohorts enrolled. Notwithstanding these issues, a meta‐analysis of these studies has found several relatively consistent findings159. There is evidence that Glx (g=0.4) and glutamate (g=0.6) levels are raised in the basal ganglia, that glutamine concentration is increased in the thalamus (g=0.6), and that Glx levels are raised in the medial temporal lobe (g=0.3) in patients with schizophrenia (Table 2).
Although these effect sizes are in some instances comparable to those observed for presynaptic dopamine function, they represent considerably less studies. In the case of both glutamate in the basal ganglia and glutamine in the thalamus, only three studies have been performed, and as such these findings should be regarded as preliminary. The increased Glx levels observed in the medial temporal lobe represent 18 studies (13 in schizophrenia and 5 in clinical high‐risk individuals).
If taken to reflect synaptic glutamate levels, the temporal lobe findings are consistent with the post‐mortem findings discussed above, and the PET study discussed below, which suggest that receptor dysfunction is accompanied by compensatory changes that increase synaptic glutamate levels. This is also consistent with studies that have found hippocampal hyperactivity in psychosis, potentially resulting from dysfunction of NMDA receptors located on GABAergic interneurons9, 148, 149.
In recent years, several studies have been performed using higher magnetic field strengths of up to 7 Tesla. Higher field strength has greater sensitivity, enabling greater separation of glutamate and glutamine peaks, and allowing for more accurate quantification. Two studies in first‐episode patients have shown reduced glutamate concentrations in the anterior cingulate cortex160, 161, while another only found this in a subset of patients with predominantly negative symptoms162. No difference has been observed in several other investigations163, 164, 165, 166. Overall this suggests that, even at high field strengths, group differences are inconsistent.
Another recent development is functional MRS. This involves acquiring multiple measures during a task or other stimulus, to investigate dynamic changes in metabolite measures. Changes in 1H‐MRS measures during a stimulus are proposed to result from compartmental shifts, as glutamate located extracellularly may contribute more to the signal. This results from the fact that, within presynaptic vesicles, metabolite movement is restricted, and therefore may have a faster T2 relaxation rate167.
A study using a heat pain stress found a reduced anterior cingulate cortex glutamate response in individuals with schizophrenia compared to healthy controls168, although interpretation is complicated by the fact that baseline glutamate levels were higher in patients. A similar pattern was seen in a study using a cognitive task in which patients showed reduced anterior cingulate glutamate response compared to controls166.
Other neuroimaging techniques
Given the limitations of 1H‐MRS in determining the precise nature of glutamatergic abnormalities, several attempts have been made to develop radioligands capable of directly measuring glutamate receptors.
One study found reduced NMDA receptor binding in the left hippocampus of patients with schizophrenia, but no further studies have been attempted, in part due to concerns regarding a lack of specificity of the tracer. A recent study using a tracer for the metabotropic glutamate receptor 5 found no patient‐control differences169. New tracers are under development, but lack of specificity remains an ongoing problem when attempting to image glutamate receptors170.
13C‐MRS is another non‐invasive imaging technique. It has lower sensitivity compared to 1H‐MRS, but, when combined with a 13C labelled infusion, it has the potential to overcome some of the limitations associated with 1H‐MRS, specifically as regards to characterizing the glutamate‐glutamine cycle171. This technique has been adopted to show that the majority of energy production in the brain supports glutamatergic activity and, although studies are yet to be performed in schizophrenia, it has recently been used in humans to show that ketamine increases glutamate‐glutamine cycling172, 173.
Glutamate chemical exchange saturation transfer (GluCEST) is another novel technique for measuring glutamate in vivo. In addition to improved sensitivity, it allows for whole brain imaging without the need to specify a single voxel174. It has so far been used in a single investigation, which reported reduced glutamate levels in individuals with schizophrenia and those at clinical high risk, compared to healthy controls175.
Glutamate across the psychosis spectrum
As with imaging of the dopamine system, efforts have been made to characterize glutamatergic function in individuals at clinical and genetic high risk for psychosis. Similarly, although a meta‐analysis suggested that increased Glx levels might exist in the medial frontal cortex in individuals at clinical high risk of psychosis165, recent studies have been negative176, 177, 178, and there appear to be no clear differences between at‐risk individuals and controls.
Given that 1H‐MRS measures of glutamate have been consistently shown to decline with age across the frontal cortex, anterior cingulate, hippocampus and basal ganglia179, 180, 181, it may be that schizophrenia is associated with a distinct trajectory of change, and so differences only become detectable later in the time course of the illness.
Consequences of glutamatergic dysfunction
Unlike dopamine neurons, which are restricted to relatively well circumscribed anatomical pathways, glutamate signalling occurs ubiquitously throughout the brain and, as a result, dysfunction of this system has the potential to account for a wide range of impairments.
However, given the limitations regarding techniques for directly quantifying the glutamate system in vivo, there is a paucity of direct evidence regarding the precise nature of glutamatergic dysfunction in schizophrenia, and studies looking at the relationship between 1H‐MRS measures of glutamate and symptom severity have produced inconsistent findings182. Indeed, both increased and decreased level of glutamate as measured by 1H‐MRS have been proposed to support a hypothesis of NMDA hypofunction in schizophrenia183, 184.
NMDA receptors, sparse coding and memory
NMDA receptors play a vital role in orchestrating several cog‐nitive processes, including working memory185. One of the mechanisms involved in the efficient cortical representation of information is that of sparse coding.
Different cortical cell classes encode information differently. More superficial cell layers code “sparsely”. This means that only a small proportion of cells within a region will be spiking, and information is thus encoded spatially186. This is in contrast to deeper layers, where the majority of cells may be firing, and information is encoded by variations in the rate of spiking186. Sparse coding involves great spatial precision in terms of the area of excitation, and this is mediated by strong lateral inhibition secondary to dense GABAergic interneuron networks within the superficial layers187.
Sparse coding allows the maintenance of multiple mnemonic networks, and also the protection of encoded memories from distractors137. Disruption to sparse coding mechanisms has been shown to lead to the development of false memories in animal models188. Both individuals with schizophrenia and healthy controls administered NMDA antagonists display phenomena that may result from impaired sparse coding – a smaller working memory buffer, decreased mnemonic precision, and false alarms in working memory137, 189. Decreased inhibition secondary to hypofunction of NMDA receptors on GABAergic interneurons could account for a disruption to the spatial precision of superficial layer firing, but this remains to be definitively tested.
Excitatory/inhibitory balance and neuronal oscillations
Synchronized neuronal oscillations are associated with a wide range of cognitive processes, such as working memory. These oscillations result from a tightly maintained balance between excitatory and inhibitory populations of neurons, and can be measured in vivo using electroencephalogram (EEG).
The balance between excitation and inhibition is crucial for normal physiological function, and NMDA receptors play a critical role here. Disruption to this balance has been proposed to result in the EEG abnormalities observed in schizophrenia. This disorder is associated with increased resting gamma oscillations, that have been linked to cognitive symptoms190. This disruption of normal oscillatory activity mirrors what has been observed with ketamine administration in healthy volunteers.
A range of molecular alterations have been proposed to potentially lead to excitatory/inhibitory imbalance in schizophrenia, including excessive pruning of dendritic spines, and intrinsic GABAergic abnormalities191. Another candidate mechanism is that of NMDA hypofunction. NMDA antagonists preferentially act on GABA interneurons, because these neurons have a more depolarized membrane potential. It has also been proposed that, in schizophrenia, NMDA hypofunction may preferentially affect interneurons192, which would in turn lead to greater activity of pyramidal neurons. This uncoordinated increased activity may underlie disruptions to normal oscillatory activity mentioned above, and act as noise, impairing the ability of coordinated activity to be passed down to subcortical regions150.
FACTORS UNDERLYING GLUTAMATERGIC AND DOPAMINERGIC DYSFUNCTION
Genetic factors
Over a hundred risk loci have been associated with schizophrenia by large scale genome‐wide association studies (GWAS)195. Given the evidence implicating dopamine in the disorder, it is surprising that only one of the loci was found to be associated with the dopamine system, specifically the dopamine D2 receptor.
Analyses specifically looking at genes involved in dopamine synthesis, signalling and metabolism have also been unable to find a signal other than for D2 receptors, and this accounts for a negligible proportion of the overall genetic risk196. However, other loci strongly linked to schizophrenia, such as 10q24.32, have shown associations with dopamine synthesis capacity197, as have polymorphisms of the gene DISC1198. Although relatively small sample sizes were used, these findings suggest that genetic factors related to schizophrenia may indirectly affect the dopamine system.
In the case of glutamate, many genes involved in the development and maintenance of glutamatergic synapses are not only implicated by loci significantly associated with schizophrenia in GWAS193, 195, but also by studies examining rarer genetic risk factors199, 200. This includes genes that directly code for components of glutamate receptors, such as GRIN2A, GRIA1 and GRM3, and genes involved in facilitating glutamatergic neurotransmission through other means, such as that coding for serine racemase (SRR). These results provide some of the strongest support that disruption of glutamatergic signalling is a fundamental component of schizophrenia pathophysiology.
Induced pluripotent stem cells (iPSCs) allow for the generation of live neurons, in vitro, from somatic cells taken from patients. These neurons are best conceptualized as modelling fetal brain tissue, primarily reflecting an underlying genetic architecture, distilled from environmental exposures201. The use of iPSCs can be particularly valuable in elucidating the effects of genetics in a polygenic disorder such as schizophrenia, where disease risk is encoded by complex networks of genes, the functional consequences of which are not easily intuited.
Several studies using this technique have investigated how the dopamine system may be affected. One study found reduced DAT expression, indicating immaturity in dopaminergic neurons derived from individuals with schizophrenia202. Another study, however, found that dopamine neurons derived from patients tended to develop more rapidly, and showed increased dopamine release203. Small sample sizes and differences in experimental protocols likely contributed to these discrepancies204.
iPSCs have also been used to study the glutamate system in neurons derived from individuals with schizophrenia. Two studies have demonstrated deficits in glutamate receptor signalling in patient‐derived cells205, 206, and a follow‐up study identified specific gene modules associated with these deficits in glutamatergic signalling201. One investigation found reduced glutamate release in schizophrenia‐derived cells differentiated to hippocampal dentate gyrus cells207, while another found delayed maturation of both dopaminergic and glutamatergic neurons, and that glutamatergic neurons displayed a reduced ability to form synaptic contacts202.
Environmental factors
Acute psychosocial stress has been shown to induce striatal dopamine release208, 209. Measures of presynaptic dopamine function are raised in migrants, and those that have experienced childhood trauma, both of which are risk factors associated with schizophrenia210, 211, 212, 213. However, another risk factor for schizophrenia, heavy cannabis use, is associated with blunted dopamine synthesis capacity and release214, 215.
Although acute stress has been shown to increase cortical glutamate level in preclinical models, this has not been demonstrated in humans using 1H‐MRS216, 217. 1H‐MRS studies in cannabis users generally report reduced glutamate levels in both cortical and subcortical areas, which is in keeping with animal work218. Of note, a recent iPSC study showed that, in neurons derived from individuals with schizophrenia, tetrahydrocannabinol administration led to depressed glutamate signalling219.
Neural circuits and dopamine‐glutamate interactions
The evidence discussed above suggests that, while the dopamine hypothesis can account for the positive symptoms of psychosis, it is less clear whether it can fully account for negative and cognitive symptoms. Similarly, while glutamatergic models of psychosis are able to replicate a wide range of symptoms of psychosis, they do not directly account for the finding of increased presynaptic striatal dopamine function, nor the clinical effectiveness of dopamine antagonists. This suggests that dysfunction in both systems contributes to the pathophysiology of schizophrenia, and highlights the need to understand how these two systems may interact.
Much research has investigated dopamine‐glutamate relationships in humans using pharmacological challenges. Amphetamine administration has been shown to increase cortical glutamate levels, as measured using 1H‐MRS220, but dopamine antagonists do not have consistent effects on glutamate levels as measured using 1H‐MRS221. Several, but not all, PET studies have found that ketamine administration is associated with striatal dopamine release222. While glutamatergic dysfunction may encourage dopaminergic disinhibition, it is clear that this is not the only route to symptoms, given that dopamine antagonists do not entirely ameliorate the effects of NMDA antagonists223.
Recent studies have combined PET and MRI measurements in the same individuals to investigate this relationship without the use of pharmacological modulation. One study in healthy individuals found that increased dopamine synthesis capacity in the ventral striatum was associated with both reduced cortical and increased striatal levels of glutamate224. This is in keeping with the imaging studies discussed above, in which both increased dopamine and glutamate measures were observed in the striatum in schizophrenia, which could potentially result from increased activity of glutamatergic projection to the striatum. Other studies found the same relationship between cortical glutamate and striatal dopamine in clinical high‐risk and first‐episode psychosis patients, but not in controls225, 226.
Theories linking glutamate and dopamine have proposed that defective NMDA receptors on cortical GABA interneurons result in inadequate inhibition of glutamatergic projections to the midbrain. This, in turn, may overstimulate mesostriatal dopamine neurons. In order to account for purported cortical dopamine deficits, it has also been proposed that overactive glutamatergic projections might overstimulate GABA interneurons in the ventral tegmental area, and thereby overinhibit mesocortical projection neurons227.
The first of these proposed mechanisms has support from studies demonstrating ketamine‐induced release of striatal dopamine. It is also in line with the 1H‐MRS studies discussed above, if the increased basal ganglia glutamate levels observed in schizophrenia are taken to represent increased activity of cortical projections. The second mechanism remains speculative222.
It is likely, however, that the relationship between glutamate and dopamine is not one‐way but bidirectional. As mentioned above, human studies have suggested that modulation of the dopamine system affects cortical glutamate levels. Preclinical studies have shown that mice genetically modified to have upregulated dopaminergic signalling display disrupted glutamatergic signalling of thalamocortical neurons228. Furthermore, cortical dopamine receptors have been shown to influence local glutamate release229.
The wide range of pathways potentially linking the two systems, and the potential for opposing effects depending on the number of interneurons within a circuit, means that it is not possible to disentangle precise mechanisms with currently available neuroimaging methods.
TREATMENT
Dopamine modulating treatments
Antipsychotics are effective treatments for positive symptoms in the majority of patients with a diagnosis of schizophrenia. However, about one third of patients show persistent positive symptoms despite treatment, and this has been termed treatment‐resistant schizophrenia230.
Recent studies have suggested that dopamine synthesis capacity may predict which patients respond to treatment. An initial study found that dopamine synthesis capacity was only raised in those with a treatment‐responsive illness84, which is consistent with a more recent prospective study231.
Even when effective in treating positive symptoms, dopamine antagonists do not typically show significant benefit for negative and cognitive symptoms. This is expected if these symptoms do not primarily relate to the hyperdopaminergic state, and particularly so if they result from deficits in dopaminergic signalling.
All currently licensed antipsychotics exert their dopaminergic effects primarily at postsynaptic D2 receptors, which is downstream of the presynaptic hyperdopaminergic state that has been observed in molecular imaging studies. There are currently a number of treatments in development which attempt to correct dysregulated dopamine function further upstream.
Apomorphine was shown to be an efficacious treatment in an early clinical trial232. Although later studies did not consistently replicate this finding4, a recent investigation suggested that this drug may normalize dopaminergic activity, potentially via agonism of presynaptic autoreceptors233. Another upstream approach involves agonism of trace amine type 1 receptors. This has been shown preclinically to both reduce midbrain dopamine neuron activity, and reduce the locomotor response to amphetamine234.
Finally, the PET studies discussed above have demonstrated that presynaptic dopaminergic dysfunction is greatest in the dorsal striatum83, and muscarinic receptor 4 positive allosteric modulators specifically inhibit dorsal striatal dopamine release, with efficacy demonstrated in some clinical trials235, 236.
The dopamine system may also be more indirectly regulated via upstream circuits. The potential of glutamate signalling to modulate dopamine neurotransmission has been discussed above. Reduced functioning of GABAergic interneurons has also been suggested to contribute to disinhibition of dopamine neurons, and alpha 5 selective GABA agonists have been proposed as means of addressing this. Although neuroimaging and preclinical work provides conceptual support, efficacy in patients is yet to be demonstrated4.
There is also the chance of intervening downstream of postsynaptic receptors. Stimulation of D2 receptors inhibits cyclic adenosine monophosphate (cAMP) production, while phosphodiesterase inhibitors have an opposing effect by preventing cAMP breakdown. Phosphodiesterase inhibitors may therefore have the potential to block the downstream effects of excessive D2 signalling, and also the potential benefit of boosting cortical D1 signalling237. Although clinical trials testing these compounds have not yet been successful4, there is a significant variability in the regional expression of phosphodiesterase inhibitor subtypes, and those showing the greatest cortical expression remain to be tested238.
Glutamate modulating treatments
Glutamate modulating treatments for schizophrenia fall into two camps: those that aim to augment NMDA signalling, and those that aim to reduce levels of synaptic glutamate proposed to be pathologically raised as a result of NMDA hypofunction. A general challenge for the development of glutamatergic treatments is that they will typically have relatively global effects, whereas pathology may be confined to discrete cell types such as NMDA receptors on specific GABAergic interneurons239, 240.
Since directly augmenting synaptic glutamate levels could have pathologically excitotoxic effects, efforts at augmenting NMDA signalling have focused on the receptor's glycine modulatory site. For activation of the NMDA receptor, glycine or D‐serine has to bind to the glycine modulatory site on GluN1 subunit, in addition to glutamate binding at the GluN2 subunit. Agonists of the glycine modulatory site – including glycine, D‐serine and D‐cycloserine – have demonstrated the ability to attenuate the psychotogenic effects of NMDA antagonists in preclinical studies, although this has not been clearly demonstrated in humans150.
A meta‐analysis of clinical trials in individuals with schizophrenia suggested that D‐serine may be effective in the treatment of negative symptoms241, but a relatively large trial was subsequently negative242. The same meta‐analysis also suggested that glycine may have a benefit for overall symptoms as well, but large scale trials are needed for clear confirmation of this effect241. A meta‐analysis of glutamate positive modulators in the treatment of cognitive symptoms, including D‐serine and glycine, did not demonstrate benefit over placebo243.
Poor blood‐brain barrier penetration by glycine and some of the other co‐agonists means that occupancy of the glycine modulatory site may be insufficient to exert clinically measurable effects. To address this, an alternative approach has involved attempts to increase synaptic glycine levels by blocking the glycine type 1 transporter, thereby inhibiting removal of glycine from the synapse.
Bitopertin, a glycine type I transporter inhibitor, appeared to be a promising compound in this regard, showing good blood‐brain barrier penetration, with encouraging results in early clinical trials244. However, later trials were not successful, perhaps due to the unusually high placebo responses, or the use of chronic patient populations, given that there is some evidence that intervention is likely to be more effective in early illness stages245. More recently, another glycine type I transporter inhibitor was shown to increase long‐term potentiation (a marker of neuroplasticity) in individuals with schizophrenia, and the compound awaits testing in clinical trials246.
Another potential approach may be to lower levels of kynurenic acid, which is an endogenous antagonist of the glycine modulatory site247. Cyclooxygenase‐2 inhibitors reduce kynurenic acid levels, and celecoxib has shown benefit as an adjunctive treatment in early psychosis, but not chronic illness248. However, a recent in vitro study found that celecoxib did not significantly reduce kynurenic acid levels, while parecoxib and niflumic acid did249. So, it may be that other cyclooxygenase‐2 inhibitors have the potential for greater efficacy.
Based on findings that NMDA antagonism increases synaptic glutamate levels, the second general approach has focused on hypotheses that NMDA hypofunction may result in pathologically raised levels of glutamate, and therefore inhibiting the release of glutamate from terminals may be therapeutic150, 250.
The metabotropic glutamate 2 receptor (mGluR2) is located presynaptically on glutamate neurons, where it acts as an autoreceptor to regulate glutamate release251. Positive allosteric modulators of mGluR2 have been found to be effective in reducing the cognitive impairments induced by ketamine252. However, efficacy in clinical trials has not been consistently shown253. One complication in these trials was the high rate of placebo response.
Riluzole (2‐amino‐6‐trifluormethoxy benzothiazole) has also been shown to reduce synaptic glutamate levels through a wide range of mechanisms, and an initial trial found it to be effective in treating negative symptoms in schizophrenia, potentially by altering striatocortical connectivity254, 255. Similarly, lamotrigine inhibits glutamate release via inhibition of several ion channels, and attenuates the psychotomimetic effects of ketamine256. Lamotrigine has also shown efficacy as an adjunctive medication for clozapine‐resistant schizophrenia, although studies to date are small and findings inconsistent257.
Neuroimaging studies have suggested that treatment‐resistant schizophrenia may not show the dopaminergic dysfunction seen in treatment‐responsive schizophrenia84, 231, 258, and that glutamatergic abnormalities may be of greater pathophysiological relevance in those cases of schizophrenia which do not respond to antipsychotic medications259, 260. Supporting this view, there is evidence that cortical glutamate levels are higher in patients with treatment‐resistant schizophrenia relative to responsive patients261. One of the reasons for unsuccessful clinical trials may therefore be that glutamate modulating treatments are only of significant benefit in a subgroup of patients.
OUTSTANDING QUESTIONS AND FUTURE DIRECTIONS
There are a number of outstanding issues when it comes to understanding the role of dopamine and glutamate in schizophrenia. In the case of glutamate, it is not possible to separate extra‐ and intracellular compartments using MRS, and we cannot accurately probe receptors and synaptic glutamate levels in vivo. As a result, it is not currently possible to precisely characterize the nature of glutamate dysfunction in schizophrenia. It remains unclear whether synaptic glutamate levels are abnormal, whether receptors are altered, and where any alterations might be localized within the brain.
Because of this, it is not clear whether treatments should aim to reduce synaptic glutamate levels or augment glutamatergic neurotransmission150. This likely contributes to the fact that to date no glutamate modulating agents exist that demonstrate unequivocal efficacy in schizophrenia.
There is a need for radioligands with reliable binding at the NMDA receptor to allow for investigation of receptor abnormalities in schizophrenia. PET ligands for other proteins involved in glutamatergic signalling, such as AMPA receptors, enzymes involved in glutamate synthesis and metabolism, and the kynurenine pathway, would also represent a considerable advance. In the meantime, other methods, such as functional MRS, 11C‐MRS, GluCEST and 7T 1H‐MRS, may advance our understanding by allowing more precise inferences regarding the nature of glutamatergic abnormalities in schizophrenia.
Imaging studies have provided more information when it comes to the dopamine system. However, several questions remain unanswered, such as the nature of cortical dopamine function in schizophrenia, whether a cortical hypodopaminergic state co‐exists with the striatal hyperdopaminergic condition, and how dopaminergic dysfunction evolves across illness course.
The improved resolution of PET cameras has allowed for greater anatomical precision in identifying the locus of dopaminergic dysfunction in schizophrenia. Early hypotheses had suggested that this dysfunction might be characterized by aberrant mesolimbic function. However, the use of new PET cameras has demonstrated that the greatest patient‐control differences are in the dorsal striatum83. The resolution of PET is still relatively coarse, however, and this limits the precision with which inferences can be made about which specific neuron groups are affected.
In addition to advances in hardware, further progress may be made by employing novel methods, such as super‐resolution techniques, in which multiple low‐resolution images are combined to create a high‐resolution image, or deep learning methods, where anatomical information from an MRI scan is used to help improve the resolution of the PET image262, 263. The limited resolution of in vivo imaging techniques means that it is difficult to directly test circuit level hypotheses regarding the nature of disrupted glutamate‐dopamine interactions in schizophrenia. Instead, hypotheses regarding circuit interactions are largely based on preclinical, post‐mortem and pharmacological studies, but await direct testing in patients.
Translation of research findings to clinically useful applications is not straightforward, and compounds acting though mechanisms other than dopamine receptor antagonism have not consistently shown efficacy in clinical trials. However, there exists a range of novel mechanisms for manipulation of both glutamate and dopamine signalling that show potential and await clinical testing. As discussed above, it may be that certain treatments are only of benefit in specific subgroups of patients, and clinical benefit may therefore be optimized by stratifying participants on the basis of underlying neurobiology231, 259. Given the coarseness of current clinical measures264, the development of imaging biomarkers to evaluate treatment effects at a neurobiological level may assist in moving the field forward265.
The non‐linearity inherent to neural signalling within a complex network means that, even if one disregards potential inconsistencies between studies, a coherent integration of existing findings is challenging. The combination of large datasets across illness phases, biophysical networks models to link molecular pathology to the macroscale dysfunction observed with neuroimaging, and carefully designed experiments to test and finesse these models is one route to integrating what at times appears to be a disparate collection of findings266, 267.
CONCLUSIONS
The hypothesis that dopamine signalling is altered in schizophrenia is supported by animal studies, post‐mortem research, and the clinical effects of drugs that either block or accentuate dopaminergic neurotransmission. In addition, over the past 25 years, substantial evidence has accumulated from PET studies that there is increased dopamine synthesis and release capacity in schizophrenia, that is greatest within the dorsal striatum.
Genetic findings do not provide strong support for the idea that dopaminergic dysregulation is a primary abnormality. Rather, it appears that the dopaminergic dysfunction is more likely to develop downstream of abnormalities in other systems, including the glutamatergic system. It also appears that environmental factors may play a significant role in the development of dopaminergic dysregulation. Dopamine antagonists remain the mainstay for pharmacological treatment of schizophrenia, but there is increasing evidence that these are not effective for all patients.
Evidence for glutamate playing a role in the pathophysiology of schizophrenia initially came from the psychotomimetic effects of NMDA antagonists. While preclinical and post‐mortem findings are consistent with this hypothesis, there is limited support from imaging studies. However, in contrast to dopamine, recent genetic findings do provide support for the view that glutamatergic abnormalities may play a major role in schizophrenia pathophysiology. However, progress is hampered by the challenges involved in precisely characterizing the system in vivo, and, while a wide range of glutamate modulating agents have been investigated, none have clear clinical efficacy.
Despite the limitations described, as regards both treatment efficacy and direct evidence for dysfunction, the dopamine and glutamate hypotheses of schizophrenia remain influential and relevant. This is not least because, as recent data demonstrate, they possess the flexibility to accommodate new findings, and to provide ongoing potential avenues for the development of novel treatments.
ACKNOWLEDGEMENTS
R.A. McCutcheon's work is funded by a Wellcome trust grant (no. 200102/Z/15/Z) and UK National Institute for Health Research (NIHR) fellowships.
REFERENCES
- 1. McCutcheon RA, Marques TR, Howes OD. Schizophrenia: an overview. JAMA Psychiatry (in press). [DOI] [PubMed] [Google Scholar]
- 2. Hjorthøj C, Stürup AE, McGrath JJ et al. Years of potential life lost and life expectancy in schizophrenia: a systematic review and meta‐analysis. Lancet Psychiatry 2017;4:295‐301. [DOI] [PubMed] [Google Scholar]
- 3. van Rossum JM. The significance of dopamine‐receptor blockade for the mechanism of action of neuroleptic drugs. Arch Int Pharmacodyn Thérapie 1966;160:492‐4. [PubMed] [Google Scholar]
- 4. Kaar SJ, Natesan S, McCutcheon R et al. Antipsychotics: mechanisms underlying clinical response and side‐effects and novel treatment approaches based on pathophysiology. Neuropharmacology (in press). [DOI] [PubMed] [Google Scholar]
- 5. Carlsson A, Lindqvist M, Magnusson T. 3,4‐Dihydroxyphenylalanine and 5‐hydroxytryptophan as reserpine antagonists. Nature 1957;180:1200. [DOI] [PubMed] [Google Scholar]
- 6. Featherstone RE, Kapur S, Fletcher PJ. The amphetamine‐induced sensitized state as a model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2007;31:1556‐71. [DOI] [PubMed] [Google Scholar]
- 7. Kellendonk C, Simpson EH, Polan HJ et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 2006;49:603‐15. [DOI] [PubMed] [Google Scholar]
- 8. Petty A, Cui X, Tesiram Y et al. Enhanced dopamine in prodromal schizophrenia (EDiPS): a new animal model of relevance to schizophrenia. NPJ Schizophr 2019;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grace AA. Dopamine system dysregulation and the pathophysiology of schizophrenia: insights from the methylazoxymethanol acetate model. Biol Psychiatry 2017;81:5‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lapiz MDS, Fulford A, Muchimapura S et al. Influence of postweaning social isolation in the rat on brain development, conditioned behavior, and neurotransmission. Neurosci Behav Physiol 2003;33:13‐29. [DOI] [PubMed] [Google Scholar]
- 11. Bowers MB. Central dopamine turnover in schizophrenic syndromes. Arch Gen Psychiatry 1974;31:50‐4. [DOI] [PubMed] [Google Scholar]
- 12. Kahn RS, Harvey PD, Davidson M et al. Neuropsychological correlates of central monoamine function in chronic schizophrenia: relationship between CSF metabolites and cognitive function. Schizophr Res 1994;11:217‐24. [DOI] [PubMed] [Google Scholar]
- 13. Gattaz WF, Riederer P, Reynolds GP et al. Dopamine and noradrenalin in the cerebrospinal fluid of schizophrenic patients. Psychiatry Res 1983;8:243‐50. [DOI] [PubMed] [Google Scholar]
- 14. Gattaz WF, Gasser T, Beckmann H. Multidimensional analysis of the concentrations of 17 substances in the CSF of schizophrenics and controls. Biol Psychiatry 1985;20:360‐6. [DOI] [PubMed] [Google Scholar]
- 15. Frecska E, Perényi A, Bagdy G et al. CSF dopamine turnover and positive schizophrenic symptoms after withdrawal of long‐term neuroleptic treatment. Psychiatry Res 1985;16:221‐6. [DOI] [PubMed] [Google Scholar]
- 16. Bagdy G, Perényi A, Frecska E et al. Decrease in dopamine, its metabolites and noradrenaline in cerebrospinal fluid of schizophrenic patients after withdrawal of long‐term neuroleptic treatment. Psychopharmacology 1985;85:62‐4. [DOI] [PubMed] [Google Scholar]
- 17. Maas JW, Bowden CL, Miller AL et al. Schizophrenia, psychosis, and cerebral spinal fluid homovanillic acid concentrations. Schizophr Bull 1997;23:147‐54. [DOI] [PubMed] [Google Scholar]
- 18. van Kammen DP, Kelley M. Dopamine and norepinephrine activity in schizophrenia. Schizophr Res 1991;4:173‐91. [DOI] [PubMed] [Google Scholar]
- 19. Post RM, Fink E, Carpenter WT et al. Cerebrospinal fluid amine metabolites in acute schizophrenia. Arch Gen Psychiatry 1975;32:1063‐9. [DOI] [PubMed] [Google Scholar]
- 20. Nybäck H, Berggren BM, Hindmarsh T et al. Cerebroventricular size and cerebrospinal fluid monoamine metabolites in schizophrenic patients and healthy volunteers. Psychiatry Res 1983;9:301‐8. [DOI] [PubMed] [Google Scholar]
- 21. Robinson DG, Woerner MG, Alvir JM et al. Predictors of treatment response from a first episode of schizophrenia or schizoaffective disorder. Am J Psychiatry 1999;156:544‐9. [DOI] [PubMed] [Google Scholar]
- 22. Lee T, Seeman P. Elevation of brain neuroleptic/dopamine receptors in schizophrenia. Am J Psychiatry 1980;137:191‐7. [DOI] [PubMed] [Google Scholar]
- 23. Zakzanis KK, Hansen KT. Dopamine D2 densities and the schizophrenic brain. Schizophr Res 1998;32:201‐6. [DOI] [PubMed] [Google Scholar]
- 24. Silvestri S, Seeman MV, Negrete J‐C et al. Increased dopamine D2 receptor binding after long‐term treatment with antipsychotics in humans: a clinical PET study. Psychopharmacology 2000;152:174‐80. [DOI] [PubMed] [Google Scholar]
- 25. Samaha A‐N, Seeman P, Stewart J et al. “Breakthrough” dopamine supersensitivity during ongoing antipsychotic treatment leads to treatment failure over time. J Neurosci 2007;27:2979‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mueller HT, Haroutunian V, Davis KL et al. Expression of the ionotropic glutamate receptor subunits and NMDA receptor‐associated intracellular proteins in the substantia nigra in schizophrenia. Mol Brain Res 2004;121:60‐9. [DOI] [PubMed] [Google Scholar]
- 27. Ichinose H, Ohye T, Fujita K et al. Quantification of mRNA of tyrosine hydroxylase and aromatic L‐amino acid decarboxylase in the substantia nigra in Parkinson's disease and schizophrenia. J Neural Transm ‐ Park Dis Dement Sect 1994;8:149‐58. [DOI] [PubMed] [Google Scholar]
- 28. Purves‐Tyson TD, Owens SJ, Rothmond DA et al. Putative presynaptic dopamine dysregulation in schizophrenia is supported by molecular evidence from post‐mortem human midbrain. Transl Psychiatry 2017;7:e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams MR, Galvin K, O'Sullivan B et al. Neuropathological changes in the substantia nigra in schizophrenia but not depression. Eur Arch Psychiatry Clin Neurosci 2014;264:285‐96. [DOI] [PubMed] [Google Scholar]
- 30. Gandal MJ, Zhang P, Hadjimichael E et al. Transcriptome‐wide isoform‐level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018;362:6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975;188:1217‐9. [DOI] [PubMed] [Google Scholar]
- 32. Creese I, Burt D, Snyder S. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976;192:481‐3. [DOI] [PubMed] [Google Scholar]
- 33. Huhn M, Nikolakopoulou A, Schneider‐Thoma J et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi‐episode schizophrenia: a systematic review and network meta‐analysis. Lancet 2019;394:939‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Connell P. Amphetamine psychosis. BMJ 1957;5018:582. [Google Scholar]
- 35. Bell DS. The experimental reproduction of amphetamine psychosis. Arch Gen Psychiatry 1973;29:35‐40. [DOI] [PubMed] [Google Scholar]
- 36. Moskovitz C, Moses H, Klawans HL. Levodopa‐induced psychosis: a kindling phenomenon. Am J Psychiatry 1978;135:669‐75. [DOI] [PubMed] [Google Scholar]
- 37. Voce A, Calabria B, Burns R et al. A systematic review of the symptom profile and course of methamphetamine‐associated psychosis: substance use and misuse. Subst Use Misuse 2019;54:549‐59. [DOI] [PubMed] [Google Scholar]
- 38. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 2001;158:1367‐77. [DOI] [PubMed] [Google Scholar]
- 39. Balu DT, Li Y, Puhl MD et al. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci USA 2013;110:E2400‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Labrie V, Duffy S, Wang W et al. Genetic inactivation of D‐amino acid oxidase enhances extinction and reversal learning in mice. Learn Mem 2009;16:28‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carlson GC, Talbot K, Halene TB et al. Dysbindin‐1 mutant mice implicate reduced fast‐phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci USA 2011;108:E962‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim JS, Kornhuber HH, Schmid‐Burgk W et al. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett 1980;20:379‐82. [DOI] [PubMed] [Google Scholar]
- 43. Goff DC, Wine L. Glutamate in schizophrenia: clinical and research implications. Schizophr Res 1997;27:157‐68. [DOI] [PubMed] [Google Scholar]
- 44. Plitman E, Iwata Y, Caravaggio F et al. Kynurenic acid in schizophrenia: a systematic review and meta‐analysis. Schizophr Bull 2017;43:764‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu W, Macdonald ML, Elswick DE et al. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann NY Acad Sci 2015;1338:38‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Javitt D, Zukin S. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 1991;148:1301‐8. [DOI] [PubMed] [Google Scholar]
- 47. Stone JM, Pepper F, Fam J et al. Glutamate, N‐acetyl aspartate and psychotic symptoms in chronic ketamine users. Psychopharmacology 2014;231:2107‐16. [DOI] [PubMed] [Google Scholar]
- 48. Fan N, Xu K, Ning Y et al. Profiling the psychotic, depressive and anxiety symptoms in chronic ketamine users. Psychiatry Res 2016;237:311‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cassidy CM, Zucca FA, Girgis RR et al. Neuromelanin‐sensitive MRI as a noninvasive proxy measure of dopamine function in the human brain. Proc Natl Acad Sci USA 2019;116:5108‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Urban NBL, Slifstein M, Meda S et al. Imaging human reward processing with positron emission tomography and functional magnetic resonance imaging. Psychopharmacology 2012;221:67‐77. [DOI] [PubMed] [Google Scholar]
- 51. Radua J, Schmidt A, Borgwardt SJ et al. Ventral striatal activation during reward processing in psychosis: a neurofunctional meta‐analysis. JAMA Psychiatry 2016;72:1243‐51. [DOI] [PubMed] [Google Scholar]
- 52. Howes OD, Kambeitz J, Stahl D et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 2012;69:776‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abi‐Dargham A, van de Giessen E, Slifstein M et al. Baseline and amphetamine‐stimulated dopamine activity are related in drug‐naïve schizophrenic subjects. Biol Psychiatry 2009;65:1091‐3. [DOI] [PubMed] [Google Scholar]
- 54. Kegeles LS, Abi‐Dargham A, Frankle WG et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 2010;67:231‐9. [DOI] [PubMed] [Google Scholar]
- 55. Kubota M, Nagashima T, Takano H et al. Affinity states of striatal dopamine D2 receptors in antipsychotic‐free patients with schizophrenia. Int J Neuropsychopharmacol 2017;20:928‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mizrahi R, Addington J, Rusjan PM et al. Increased stress‐induced dopamine release in psychosis. Biol Psychiatry 2012;71:561‐7. [DOI] [PubMed] [Google Scholar]
- 57. Frankle WG, Paris J, Himes M et al. Amphetamine‐induced striatal dopamine release measured with an agonist radiotracer in schizophrenia. Biol Psychiatry 2018;83:707‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Slifstein M, Abi‐Dargham A. Is it pre‐ or postsynaptic? Imaging striatal dopamine excess in schizophrenia. Biol Psychiatry 2018;83:635‐7. [DOI] [PubMed] [Google Scholar]
- 59. Guo N, Guo W, Kralikova M et al. Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology 2010;35:806‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sun W, Ginovart N, Ko F et al. In vivo evidence for dopamine‐mediated internalization of D2‐receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol 2003;63:456‐62. [DOI] [PubMed] [Google Scholar]
- 61. Brugger SP, Angelescu I, Abi‐Dargham A et al. Heterogeneity of striatal dopamine function in schizophrenia: meta‐analysis of variance. Biol Psychiatry (in press). [DOI] [PubMed] [Google Scholar]
- 62. Stenkrona P, Matheson GJ, Halldin C et al. D1‐dopamine receptor availability in first‐episode neuroleptic naive psychosis patients. Int J Neuropsychopharmacol 2019;22:415‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kambeitz J, Abi‐Dargham A, Kapur S et al. Alterations in cortical and extrastriatal subcortical dopamine function in schizophrenia: systematic review and meta‐analysis of imaging studies. Br J Psychiatry 2014;204:420‐9. [DOI] [PubMed] [Google Scholar]
- 64. Abi‐Dargham A, Mawlawi O, Lombardo I et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 2002;22:3708‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abi‐Dargham A, Xu X, Thompson JL et al. Increased prefrontal cortical D₁ receptors in drug naive patients with schizophrenia: a PET study with [11C]NNC112. J Psychopharmacol 2012;26:794‐805. [DOI] [PubMed] [Google Scholar]
- 66. Kosaka J, Takahashi H, Ito H et al. Decreased binding of [11C]NNC112 and [11C]SCH23390 in patients with chronic schizophrenia. Life Sci 2010;86:814‐8. [DOI] [PubMed] [Google Scholar]
- 67. Hirvonen J, Van Erp TGM, Huttunen J et al. Brain dopamine D1 receptors in twins discordant for schizophrenia. Am J Psychiatry 2006;163:1747‐53. [DOI] [PubMed] [Google Scholar]
- 68. Okubo Y, Suhara T, Suzuki K et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997;385:634‐6. [DOI] [PubMed] [Google Scholar]
- 69. Karlsson P, Farde L, Halldin C et al. PET study of D(1) dopamine receptor binding in neuroleptic‐naive patients with schizophrenia. Am J Psychiatry 2002;159:761‐7. [DOI] [PubMed] [Google Scholar]
- 70. Poels EMP, Girgis RR, Thompson JL et al. In vivo binding of the dopamine‐1 receptor PET tracers [11C]NNC112 and [11C]SCH23390: a comparison study in individuals with schizophrenia. Psychopharmacology 2013;228:167‐74. [DOI] [PubMed] [Google Scholar]
- 71. Guo N, Hwang DR, Lo ES et al. Dopamine depletion and in vivo binding of PET D1 receptor radioligands: implications for imaging studies in schizophrenia. Neuropsychopharmacology 2003;28:1703‐11. [DOI] [PubMed] [Google Scholar]
- 72. Ekelund J, Slifstein M, Narendran R et al. In vivo DA D(1) receptor selectivity of NNC 112 and SCH 23390. Mol Imaging Biol 2007;9:117‐25. [DOI] [PubMed] [Google Scholar]
- 73. Lidow MS, Elsworth JD, Goldman‐Rakic PS. Down‐regulation of the D1 and D5 dopamine receptors in the primate prefrontal cortex by chronic treatment with antipsychotic drugs. J Pharmacol Exp Ther 1997;281:597‐603. [PubMed] [Google Scholar]
- 74. Artiges E, Leroy C, Dubol M et al. Striatal and extrastriatal dopamine transporter availability in schizophrenia and its clinical correlates: a voxel‐based and high‐resolution PET study. Schizophr Bull 2017;43:1134‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arakawa R, Ichimiya T, Ito H et al. Increase in thalamic binding of [11C]PE2I in patients with schizophrenia: a positron emission tomography study of dopamine transporter. J Psychiatr Res 2009;43:1219‐23. [DOI] [PubMed] [Google Scholar]
- 76. Taylor SF, Koeppe RA, Tandon R et al. In vivo measurement of the vesicular monoamine transporter in schizophrenia. Neuropsychopharmacology 2000;23:667‐75. [DOI] [PubMed] [Google Scholar]
- 77. Zubieta JK, Taylor SF, Huguelet P et al. Vesicular monoamine transporter concentrations in bipolar disorder type I, schizophrenia, and healthy subjects. Biol Psychiatry 2001;49:110‐6. [DOI] [PubMed] [Google Scholar]
- 78. Zucker M, Valevski A, Weizman A et al. Increased platelet vesicular monoamine transporter density in adult schizophrenia patients. Eur Neuropsychopharmacol 2002;12:343‐7. [DOI] [PubMed] [Google Scholar]
- 79. Egerton A, Mehta MA, Montgomery AJ et al. The dopaminergic basis of human behaviors: a review of molecular imaging studies. Neurosci Biobehav Rev 2009;33:1109‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dewey SL, Logan J, Wolf AP et al. Amphetamine induced decreases in (18F)‐N‐methylspiroperidol binding in the baboon brain using positron emission tomography (PET). Synapse 1991;7:324‐7. [DOI] [PubMed] [Google Scholar]
- 81. Kumakura Y, Cumming P. PET studies of cerebral levodopa metabolism: a review of clinical findings and modeling approaches. Neuroscientist 2009;15:635‐50. [DOI] [PubMed] [Google Scholar]
- 82. Sawilowsky SS. New effect size rules of thumb. J Mod Appl Stat Methods 2009;8:597‐9. [Google Scholar]
- 83. McCutcheon R, Beck K, Jauhar S et al. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta‐analysis and test of the mesolimbic hypothesis. Schizophr Bull 2018;44:1301‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Demjaha A, Murray RM, McGuire PK et al. Dopamine synthesis capacity in patients with treatment‐resistant schizophrenia. Am J Psychiatry 2012;169:1203‐10. [DOI] [PubMed] [Google Scholar]
- 85. Abi‐Dargham A, Gil R, Krystal J et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry 1998;155:761‐7. [DOI] [PubMed] [Google Scholar]
- 86. Jauhar S, Nour MM, Veronese M et al. A test of the transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiatry 2017;74:1206‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hietala J, Syvälahti E, Vilkman H et al. Depressive symptoms and presynaptic dopamine function in neuroleptic‐naive schizophrenia. Schizophr Res 1999;35:41‐50. [DOI] [PubMed] [Google Scholar]
- 88. Avram M, Brandl F, Cabello J et al. Reduced striatal dopamine synthesis capacity in patients with schizophrenia during remission of positive symptoms. Brain 2019;142:1813‐26. [DOI] [PubMed] [Google Scholar]
- 89. Egerton A, Demjaha A, McGuire P et al. The test‐retest reliability of 18F‐DOPA PET in assessing striatal and extrastriatal presynaptic dopaminergic function. Neuroimage 2010;50:524‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Howes OD, Williams M, Ibrahim K et al. Midbrain dopamine function in schizophrenia and depression: a post‐mortem and positron emission tomographic imaging study. Brain 2013;136:3242‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kumakura Y, Cumming P, Vernaleken I et al. Elevated [18F]fluorodopamine turnover in brain of patients with schizophrenia: an [18F]fluorodopa/positron emission tomography study. J Neurosci 2007;27:8080‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Elkashef A, Doudet D, Bryant T. 6‐(18)F‐DOPA PET study in patients with schizophrenia. Psychiatry Res Neuroimaging 2000;100:1‐11. [DOI] [PubMed] [Google Scholar]
- 93. Hall H, Sedvall G, Magnusson O et al. Distribution of D1‐ and D2‐dopamine receptors, and dopamine and its metabolites in the human brain. Neuropsychopharmacology 1994;11:245‐56. [DOI] [PubMed] [Google Scholar]
- 94. Hernaus D, Mehta MA. Prefrontal cortex dopamine release measured in vivo with positron emission tomography: implications for the stimulant paradigm. Neuroimage 2016;142:663‐7. [DOI] [PubMed] [Google Scholar]
- 95. Slifstein M, Van De Giessen E, Van Snellenberg J et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 2015;72:316‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schifani C, Tseng H‐H, Kenk M et al. Cortical stress regulation is disrupted in schizophrenia but not in clinical high risk for psychosis. Brain 2018;141:2213‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rao N, Northoff G, Tagore A et al. Impaired prefrontal cortical dopamine release in schizophrenia during a cognitive task : a [11C]FLB 457 positron emission tomography study. Schizophr Bull 2019;45:670‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hernaus D, Collip D, Kasanova Z et al. No evidence for attenuated stress‐induced extrastriatal dopamine signaling in psychotic disorder. Transl Psychiatry 2015;5:e547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tseng H‐H, Watts JJ, Kiang M et al. Nigral stress‐induced dopamine release in clinical high risk and antipsychotic‐naïve schizophrenia. Schizophr Bull 2018;44:542‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Abi‐Dargham A, Kegeles LS, Zea‐Ponce Y et al. Striatal amphetamine‐induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I]iodobenzamide. Biol Psychiatry 2004;55:1001‐06. [DOI] [PubMed] [Google Scholar]
- 101. Howes OD, Montgomery AJ, Asselin M‐C et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 2009;66:13‐20. [DOI] [PubMed] [Google Scholar]
- 102. Egerton A, Chaddock CA, Winton‐Brown TT et al. Presynaptic striatal dopamine dysfunction in people at ultra‐high risk for psychosis: findings in a second cohort. Biol Psychiatry 2013;74:106‐12. [DOI] [PubMed] [Google Scholar]
- 103. Howes OD, Bonoldi I, McCutcheon RA et al. Glutamatergic and dopaminergic function and the relationship to outcome in people at clinical high risk of psychosis: a multi‐modal PET‐magnetic resonance brain imaging study. Neuropsychopharmacology (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Howes O, Bose S, Turkheimer FE et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]‐DOPA PET imaging study. Am J Psychiatry 2011;168:1311‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Howes OD, Shotbolt P, Bloomfield M et al. Dopaminergic function in the psychosis spectrum: an [18F]‐DOPA imaging study in healthy individuals with auditory hallucinations. Schizophr Bull 2013;39:807‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shotbolt P, Stokes PR, Owens SF et al. Striatal dopamine synthesis capacity in twins discordant for schizophrenia. Psychol Med 2011;41:2331‐8. [DOI] [PubMed] [Google Scholar]
- 107. van Duin EDA, Kasanova Z, Hernaus D et al. Striatal dopamine release and impaired reinforcement learning in adults with 22q11.2 deletion syndrome. Eur Neuropsychopharmacol 2018;28:732‐42. [DOI] [PubMed] [Google Scholar]
- 108. Huttunen J, Heinimaa M, Svirskis T et al. Striatal dopamine synthesis in first‐degree relatives of patients with schizophrenia. Biol Psychiatry 2008;63:114‐7. [DOI] [PubMed] [Google Scholar]
- 109. Reith J, Benkelfat C, Sherwin A et al. Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci USA 1994;91:11651‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vingerhoets C, Bloemen OJN, Boot E et al. Dopamine in high‐risk populations: a comparison of subjects with 22q11.2 deletion syndrome and subjects at ultra high‐risk for psychosis. Psychiatry Res Neuroimaging 2018;272:65‐70. [DOI] [PubMed] [Google Scholar]
- 111. Brunelin J, d'Amato T, van Os J et al. Increased left striatal dopamine transmission in unaffected siblings of schizophrenia patients in response to acute metabolic stress. Psychiatry Res 2010;181:130‐5. [DOI] [PubMed] [Google Scholar]
- 112. Lee KJ, Lee JS, Kim SJ et al. Loss of asymmetry in D2 receptors of putamen in unaffected family members at increased genetic risk for schizophrenia. Acta Psychiatr Scand 2008;118:200‐8. [DOI] [PubMed] [Google Scholar]
- 113. Hirvonen J, van Erp TGM, Huttunen J et al. Striatal dopamine D1 and D2 receptor balance in twins at increased genetic risk for schizophrenia. Psychiatry Res 2006;146:13‐20. [DOI] [PubMed] [Google Scholar]
- 114. Fouriezos G, Hansson P, Wise RA. Neuroleptic‐induced attenuation of brain stimulation reward in rats. J Comp Physiol Psychol 1978;92:661‐71. [DOI] [PubMed] [Google Scholar]
- 115. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science 1997;275:1593‐9. [DOI] [PubMed] [Google Scholar]
- 116. Lerner TN, Shilyansky C, Davidson TJ et al. Intact‐brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 2015;162:635‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Menegas W, Babayan BM, Uchida N et al. Opposite initialization to novel cues in dopamine signaling in ventral and posterior striatum in mice. Elife 2017;6:e21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Menegas W, Akiti K, Amo R et al. Dopamine neurons projecting to the posterior striatum reinforce avoidance of threatening stimuli. Nat Neurosci 2018;21:1421‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Groessl F, Munsch T, Meis S et al. Dorsal tegmental dopamine neurons gate associative learning of fear. Nat Neurosci 2018;21:952‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature 2009;459:837‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Miller R. Schizophrenic psychology, associative learning and the role of forebrain dopamine. Med Hypotheses 1976;2:203‐11. [DOI] [PubMed] [Google Scholar]
- 122. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 2003;160:13‐23. [DOI] [PubMed] [Google Scholar]
- 123. McCutcheon RA, Abi‐Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci 2019;42:205‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sterzer P, Adams RA, Fletcher P et al. The predictive coding account of psychosis. Biol Psychiatry 2018;84:634‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Corlett PR, Krystal JH, Taylor JR et al. Why do delusions persist? Front Hum Neurosci 2009;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Powers AR, Mathys C, Corlett PR. Pavlovian conditioning‐induced hallucinations result from overweighting of perceptual priors. Science 2017;357:596‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cassidy CM, Balsam PD, Weinstein JJ et al. A perceptual inference mechanism for hallucinations linked to striatal dopamine. Curr Biol 2018;28:503‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nour MM, Dahoun T, Schwartenbeck P et al. Dopaminergic basis for signaling belief updates, but not surprise, and the link to paranoia. Proc Natl Acad Sci USA 2018;115:E10167‐76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. McCutcheon RA, Bloomfield MAP, Dahoun T et al. Chronic psychosocial stressors are associated with alterations in salience processing and corticostriatal connectivity. Schizophr Res 2019;213:56‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Uddin LQ. Salience processing and insular cortical function and dysfunction. Nat Rev Neurosci 2015;16:55‐61. [DOI] [PubMed] [Google Scholar]
- 131. Palaniyappan L, Simmonite M, White TP et al. Neural primacy of the salience processing system in schizophrenia. Neuron 2013;79:814‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Menon V, Uddin LQ. Saliency, switching, attention and control: a network model of insula function. Brain Struct Funct 2010;214:655‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Roffman JL, Tanner AS, Eryilmaz H et al. Dopamine D1 signaling organizes network dynamics underlying working memory. Sci Adv 2016;2:e1501672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Braun U, Harneit A, Pergola G et al. Brain state stability during working memory is explained by network control theory, modulated by dopamine D1/D2 receptor function, and diminished in schizophrenia. bioRxiv 2019:679670. [Google Scholar]
- 135. McCutcheon R, Nour MM, Dahoun T et al. Mesolimbic dopamine function is related to salience network connectivity: an integrative positron emission tomography and magnetic resonance study. Biol Psychiatry 2019;85:368‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Maia TV, Frank MJ. From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci 2011;14:154‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Krystal JH, Anticevic A, Yang GJ et al. Impaired tuning of neural ensembles and the pathophysiology of schizophrenia: a translational and computational neuroscience perspective. Biol Psychiatry 2017;81:874‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol 1998;80:1‐27. [DOI] [PubMed] [Google Scholar]
- 139. Maia TV, Frank MJ. An integrative perspective on the role of dopamine in schizophrenia. Biol Psychiatry 2017;81:52‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Strauss GP, Frank MJ, Waltz JA et al. Deficits in positive reinforcement learning and uncertainty‐driven exploration are associated with distinct aspects of negative symptoms in schizophrenia. Biol Psychiatry 2011;69:424‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Gold JM, Waltz JA, Matveeva TM et al. Negative symptoms in schizophrenia result from a failure to represent the expected value of rewards: behavioral and computational modeling evidence. Arch Gen Psychiatry 2012;69:129‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Krystal JH, D'Souza DC, Gallinat J et al. The vulnerability to alcohol and substance abuse in individuals diagnosed with schizophrenia. Neurotox Res 2006;10:235‐52. [DOI] [PubMed] [Google Scholar]
- 143. MacCabe JH, Wicks S, Löfving S et al. Decline in cognitive performance between ages 13 and 18 years and the risk for psychosis in adulthood: a Swedish longitudinal cohort study in males. JAMA Psychiatry 2013;70:261‐70. [DOI] [PubMed] [Google Scholar]
- 144. Keefe RSE, Fenton WS. How should DSM‐V criteria for schizophrenia include cognitive impairment? Schizophr Bull 2007;33:912‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Green MF, Horan PW, Lee J. Nonsocial and social cognition in schizophrenia: current evidence and future directions. World Psychiatry 2019;18:146‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kelly S, Guimond S, Lyall A et al. Neural correlates of cognitive deficits across developmental phases of schizophrenia. Neurobiol Dis 2019;131:104353. [DOI] [PubMed] [Google Scholar]
- 147. Arnsten AFT. Catecholamine modulation of prefrontal cortical cognitive function. Trends Cogn Sci 1998;2:436‐47. [DOI] [PubMed] [Google Scholar]
- 148. Schobel SA, Chaudhury NH, Khan UA et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 2013;78:81‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lieberman JA, Girgis RR, Brucato G et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry 2018;23:1764‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012;37:4‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Malhotra AK, Pinals DA, Adler CM et al. Ketamine‐induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic‐free schizophrenics. Neuropsychopharmacology 1997;17:141‐50. [DOI] [PubMed] [Google Scholar]
- 152. Al‐Diwani A, Handel A, Townsend L et al. The psychopathology of NMDAR‐antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry 2019;6:235‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Krystal JH, Karper LP, Seibyl JP et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 1994;51:199‐214. [DOI] [PubMed] [Google Scholar]
- 154. Carhart‐Harris RL, Brugger S, Nutt DJ et al. Psychiatry's next top model: cause for a re‐think on drug models of psychosis and other psychiatric disorders. J Psychopharmacol 2013;27:771‐8. [DOI] [PubMed] [Google Scholar]
- 155. Cheng WJ, Chen CH, Chen CK et al. Similar psychotic and cognitive profile between ketamine dependence with persistent psychosis and schizophrenia. Schizophr Res 2018;199:313‐8. [DOI] [PubMed] [Google Scholar]
- 156. Poels EMP, Kegeles LS, Kantrowitz JT et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry 2014;19:20‐9. [DOI] [PubMed] [Google Scholar]
- 157. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA‐glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 2006;98:641‐53. [DOI] [PubMed] [Google Scholar]
- 158. McKenna MC. The glutamate‐glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 2007;85:3347‐58. [DOI] [PubMed] [Google Scholar]
- 159. Merritt K, Egerton A, Kempton MJ et al. Nature of glutamate alterations in schizophrenia. JAMA Psychiatry 2016;52:998‐1007. [DOI] [PubMed] [Google Scholar]
- 160. Reid MA, Salibi N, White DM et al. 7T proton magnetic resonance spectroscopy of the anterior cingulate cortex in first‐episode schizophrenia. Schizophr Bull 2019;45:180‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang AM, Pradhan S, Coughlin JM et al. Assessing brain metabolism with 7‐T proton magnetic resonance spectroscopy in patients with first‐episode psychosis. JAMA Psychiatry 2019;76:314‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kumar J, Liddle EB, Fernandes CC et al. Glutathione and glutamate in schizophrenia: a 7T MRS study. Mol Psychiatry (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Marsman A, Mandl RCW, Klomp DWJ et al. GABA and glutamate in schizophrenia: a 7T 1H‐MRS study. Neuroimage Clin 2014;6:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Posporelis S, Coughlin JM, Marsman A et al. Decoupling of brain temperature and glutamate in recent onset of schizophrenia: a 7T proton magnetic resonance spectroscopy study. Biol Psychiatry Cogn Neurosci Neuroimaging 2018;3:248‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Brandt AS, Unschuld PG, Pradhan S et al. Age‐related changes in anterior cingulate cortex glutamate in schizophrenia: a 1H MRS study at 7 Tesla. Schizophr Res 2016;172:101‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Taylor R, Neufeld RWJ, Schaefer B et al. Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color‐word Stroop task. NPJ Schizophr 2015;1:15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jelen LA, King S, Mullins PG et al. Beyond static measures: a review of functional magnetic resonance spectroscopy and its potential to investigate dynamic glutamatergic abnormalities in schizophrenia. J Psychopharmacol 2018;32:497‐508. [DOI] [PubMed] [Google Scholar]
- 168. Chiappelli J, Shi Q, Wijtenburg SA et al. Glutamatergic response to heat pain stress in schizophrenia. Schizophr Bull 2018;44:886‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Akkus F, Treyer V, Ametamey SM et al. Metabotropic glutamate receptor 5 neuroimaging in schizophrenia. Schizophr Res 2017;183:95‐101. [DOI] [PubMed] [Google Scholar]
- 170. Fu H, Chen Z, Josephson L et al. Positron emission tomography (PET) ligand development for ionotropic glutamate receptors: challenges and opportunities for radiotracer targeting N‐methyl‐d‐aspartate (NMDA), α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA), and kainate receptors. J Med Chem 2019;62:403‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mason G, Petersen K, de Graaf RA et al. Measurements of the anaplerotic rate in the human cerebral cortex using 13C magnetic resonance spectroscopy and [1‐13 C] and [2‐13C] glucose. J Neurochem 2007;100:73‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Abdallah CG, De Feyter HM, Averill LA et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 2018;43:2154‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sibson NR, Dhankhar A, Mason GF et al. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA 1998;95:316‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Cai K, Haris M, Singh A et al. Magnetic resonance imaging of glutamate. Nat Med 2012;18:302‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Roalf DR, Nanga RPR, Rupert PE et al. Glutamate imaging (GluCEST) reveals lower brain GluCEST contrast in patients on the psychosis spectrum. Mol Psychiatry 2017;22:1298‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Modinos G, S¸ ims¸ek F, Azis M et al. Prefrontal GABA levels, hippocampal resting perfusion and the risk of psychosis. Neuropsychopharmacology 2018;43:262‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Liemburg E, Sibeijn‐Kuiper A, Bais L et al. Prefrontal NAA and Glx levels in different stages of psychotic disorders: a 3T 1H‐MRS study. Sci Rep 2016;6:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Da Silva T, Hafizi S, Rusjan PM et al. GABA levels and TSPO expression in people at clinical high risk for psychosis and healthy volunteers: a PET‐MRS study. J Psychiatry Neurosci 2019;44:111‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hädel S, Wirth C, Rapp M et al. Effects of age and sex on the concentrations of glutamate and glutamine in the human brain. J Magn Reson Imaging 2013;38:1480‐7. [DOI] [PubMed] [Google Scholar]
- 180. Tayoshi S, Sumitani S, Taniguchi K et al. Metabolite changes and gender differences in schizophrenia using 3‐Tesla proton magnetic resonance spectroscopy (1H‐MRS). Schizophr Res 2009;108:69‐77. [DOI] [PubMed] [Google Scholar]
- 181. Grachev I, Apkarian A. Aging alters regional multichemical profile of the human brain: an in vivo 1H‐MRS study of young versus middle‐aged subjects. J Neurochem 2001;76:582‐93. [DOI] [PubMed] [Google Scholar]
- 182. Merritt K, McGuire P, Egerton A. Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front Psychiatry 2013;4:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lutkenhoff ES, van Erp TG, Thomas MA et al. Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry 2010;15:308‐18. [DOI] [PubMed] [Google Scholar]
- 184. Tebartz Van Elst L, Valerius G, Büchert M et al. Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry 2005;58:724‐30. [DOI] [PubMed] [Google Scholar]
- 185. Wang M, Yang Y, Wang CJ et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron 2013;77:736‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Harris KD, Mrsic‐Flogel TD. Cortical connectivity and sensory coding. Nature 2013;503:51‐8. [DOI] [PubMed] [Google Scholar]
- 187. Buzsáki G, Watson BO. Brain rhythms and neural syntax: implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin Neurosci 2012;14:345‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Lin AC, Bygrave AM, De Calignon A et al. Sparse, decorrelated odor coding in the mushroom body enhances learned odor discrimination. Nat Neurosci 2014;17:559‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Murray JD, Anticevic A, Gancsos M et al. Linking microcircuit dysfunction to cognitive impairment: effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex 2014;24:859‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 2010;11:100‐13. [DOI] [PubMed] [Google Scholar]
- 191. Lewis DA, Curley AA, Glausier JR et al. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 2012;35:57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Coyle J. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 2006;26:365‐84. [DOI] [PubMed] [Google Scholar]
- 193. Pers TH, Timshel P, Ripke S et al. Comprehensive analysis of schizophrenia‐associated loci highlights ion channel pathways and biologically plausible candidate causal genes. Hum Mol Genet 2015;25:1247‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Ma C, Gu C, Huo Y et al. The integrated landscape of causal genes and pathways in schizophrenia. Transl Psychiatry 2018;8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Schizophrenia Working Group of the Psychiatric Genomics Consortium . Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014;511:421‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Edwards AC, Bacanu S, Bigdeli TB et al. Evaluating the dopamine hypothesis of schizophrenia in a large‐scale genome‐wide association study. Schizophr Res 2016;176:136‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. D'Ambrosio E, Dahoun T, Pardiñas AF et al. The effect of a genetic variant at the schizophrenia associated AS3MT/BORCS7 locus on striatal dopamine function: a PET imaging study. Psychiatry Res Neuroimaging 2019;291:34‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Dahoun T, Pardiñas AF, Veronese M et al. The effect of the DISC1 Ser704Cys polymorphism on striatal dopamine synthesis capacity: an [18F]‐DOPA PET study. Hum Mol Genet 2018;27:3498‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Pocklington AJ, Rees E, Walters JTR et al. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron 2015;86:1203‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Timms AE, Dorschner MO, Wechsler J et al. Support for the N‐methyl‐D‐aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry 2013;70:582‐90. [DOI] [PubMed] [Google Scholar]
- 201. Brennand K, Savas JN, Kim Y et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry 2015;20:361‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Robicsek O, Karry R, Petit I et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle‐derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry 2013;18:1067‐76. [DOI] [PubMed] [Google Scholar]
- 203. Hook V, Brennand KJ, Kim Y et al. Human iPSC neurons display activity‐dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Reports 2014;3:531‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hartley BJ, Tran N, Ladran I et al. Dopaminergic differentiation of schizophrenia hiPSCs. Mol Psychiatry 2015;20:549‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Brennand KJ, Simone A, Jou J et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011;473:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Wen Z, Nguyen HN, Guo Z et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014;515:414‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yu DX, Di Giorgio FP, Yao J et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports 2014;2:295‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Pruessner JC, Champagne F, Meaney MJ et al. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci 2004;24:2825‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Vaessen T, Hernaus D, Myin‐Germeys I et al. The dopaminergic response to acute stress in health and psychopathology: a systematic review. Neurosci Biobehav Rev 2015;56:241‐51. [DOI] [PubMed] [Google Scholar]
- 210. Egerton A, Howes OD, Houle S et al. Elevated striatal dopamine function in immigrants and their children: a risk mechanism for psychosis. Schizophr Bull 2017;43:293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Egerton A, Valmaggia LR, Howes OD et al. Adversity in childhood linked to elevated striatal dopamine function in adulthood. Schizophr Res 2016;176:171‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Radua J, Ramella‐Cravaro V, Ioannidis JPA et al. What causes psychosis? An umbrella review of risk and protective factors. World Psychiatry 2018;17:49‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Morgan C, Knowles G, Hutchinson G. Migration, ethnicity and psychoses: evidence, models and future directions. World Psychiatry 2019;18:247‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Bloomfield MA, Morgan CJ, Egerton A et al. Dopaminergic function in cannabis users and its relationship to cannabis‐induced psychotic symptoms. Biol Psychiatry 2014;75:470‐8. [DOI] [PubMed] [Google Scholar]
- 215. Mizrahi R, Suridjan I, Kenk M et al. Dopamine response to psychosocial stress in chronic cannabis users: a PET study with [11C]‐+‐PHNO. Neuropsychopharmacology 2013;38:673‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Popoli M, Yan Z, McEwen BS et al. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci 2012;13:22‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Houtepen LC, Schür RR, Wijnen JP et al. Acute stress effects on GABA and glutamate levels in the prefrontal cortex: a 7T 1H‐magnetic resonance spectroscopy study. Neuroimage Clin 2017;14:195‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Colizzi M, McGuire P, Pertwee RG et al. Effect of cannabis on glutamate signalling in the brain: a systematic review of human and animal evidence. Neurosci Biobehav Rev 2016;64:359‐81. [DOI] [PubMed] [Google Scholar]
- 219. Guennewig B, Bitar M, Obiorah I et al. THC exposure of human iPSC neurons impacts genes associated with neuropsychiatric disorders. Transl Psychiatry 2018;8:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. White TL, Monnig MA, Walsh EG et al. Psychostimulant drug effects on glutamate, Glx, and creatine in the anterior cingulate cortex and subjective response in healthy humans. Neuropsychopharmacology 2018;43:1498‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Egerton A, Bhachu A, Merritt K et al. Effects of antipsychotic administration on brain glutamate in schizophrenia: a systematic review of longitudinal (1)H‐MRS studies. Front Psychiatry 2017;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kokkinou M, Ashok A, Howes O. The effects of ketamine on dopaminergic function: meta‐analysis and review of the implications for neuropsychiatric disorders. Mol Psychiatry 2018;23:59‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Krystal JH, D'Souza DC, Karper LP et al. Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology 1999;145:193‐204. [DOI] [PubMed] [Google Scholar]
- 224. Gleich T, Deserno L, Lorenz RC et al. Prefrontal and striatal glutamate differently relate to striatal dopamine: potential regulatory mechanisms of striatal presynaptic dopamine function? J Neurosci 2015;35:9615‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Jauhar S, McCutcheon R, Borgan F et al. The relationship between cortical glutamate and striatal dopamine in first‐episode psychosis: a cross‐sectional multimodal PET and magnetic resonance spectroscopy imaging study. Lancet Psychiatry 2018;5:816‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Stone JM, Howes OD, Egerton A et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry 2010;68:599‐602. [DOI] [PubMed] [Google Scholar]
- 227. Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol 2012;3:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Chun S, Westmoreland JJ, Bayazitov IT et al. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014;344:1178‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Gao WJ, Krimer LS, Goldman‐Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci USA 2001;98:295‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Howes OD, McCutcheon R, Agid O et al. Treatment‐resistant schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group consensus guidelines on diagnosis and terminology. Am J Psychiatry 2017;174:216‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Jauhar S, Veronese M, Nour MM et al. Determinants of treatment response in first‐episode psychosis: an 18F‐DOPA PET study. Mol Psychiatry 2019;24:1502‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Tamminga CA, Schaffer MH, Smith RC et al. Schizophrenic symptoms improve with apomorphine. Science 1978;200:567‐8. [DOI] [PubMed] [Google Scholar]
- 233. Jauhar S, Veronese M, Rogdaki M et al. Regulation of dopaminergic function: an [18F]‐DOPA PET apomorphine challenge study in humans. Transl Psychiatry 2017;7:e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Revel FG, Moreau J‐L, Gainetdinov RR et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc Natl Acad Sci USA 2011;108:8485‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Foster DJ, Wilson JM, Remke DH et al. Antipsychotic‐like effects of M4 positive allosteric modulators are mediated by CB2 receptor‐dependent inhibition of dopamine release. Neuron 2016;91:1244‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Shekhar A, Potter WZ, Lightfoot J et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry 2008;165:1033‐9. [DOI] [PubMed] [Google Scholar]
- 237. Heckman PRA, Blokland A, Bollen EPP et al. Phosphodiesterase inhibition and modulation of corticostriatal and hippocampal circuits: clinical overview and translational considerations. Neurosci Biobehav Rev 2018;87:233‐54. [DOI] [PubMed] [Google Scholar]
- 238. Kuroiwa M, Snyder GL, Shuto T et al. Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP‐32 signaling cascade in frontal cortex. Psychopharmacology 2012;219:1065‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Cohen SM, Tsien RW, Goff DC et al. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res 2015;167:98‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Alherz F, Alherz M, Almusawi H. NMDAR hypofunction and somatostatin‐expressing GABAergic interneurons and receptors: a newly identified correlation and its effects in schizophrenia. Schizophr Res Cogn 2017;8:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Singh SP, Singh V. Meta‐analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 2011;25:859‐85. [DOI] [PubMed] [Google Scholar]
- 242. Weiser M, Heresco‐Levy U, Davidson M et al. A multicenter, add‐on randomized controlled trial of low‐dose d‐serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry 2012;73:e728‐34. [DOI] [PubMed] [Google Scholar]
- 243. Iwata Y, Nakajima S, Suzuki T et al. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta‐analysis of double‐blind randomized controlled trials. Mol Psychiatry 2015;20:1151‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Beck K, Javitt DC, Howes OD. Targeting glutamate to treat schizophrenia: lessons from recent clinical studies. Psychopharmacology 2016;233:2425‐28. [DOI] [PubMed] [Google Scholar]
- 245. Goff D. Bitopertin: the good news and bad news. JAMA Psychiatry 2014;71:621‐2. [DOI] [PubMed] [Google Scholar]
- 246. D'Souza DC, Carson RE, Driesen N et al. Dose‐related target occupancy and effects on circuitry, behavior, and neuroplasticity of the glycine transporter‐1 inhibitor PF‐03463275 in healthy and schizophrenia subjects. Biol Psychiatry 2018;84:413‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Balu DT, Coyle JT. The NMDA receptor “glycine modulatory site” in schizophrenia: D‐serine, glycine, and beyond. Curr Opin Pharmacol 2015;20:109‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Zheng W, Cai DB, Yang XH et al. Adjunctive celecoxib for schizophrenia: a meta‐analysis of randomized, double‐blind, placebo‐controlled trials. J Psychiatr Res 2017;92:139‐46. [DOI] [PubMed] [Google Scholar]
- 249. Zakrocka I, Targowska‐Duda KM, Wnorowski A et al. Influence of cyclooxygenase‐2 inhibitors on kynurenic acid production in rat brain in vitro. Neurotox Res 2019;35:244‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Anticevic A, Cole MW, Repovs G et al. Connectivity, pharmacology, and computation: toward a mechanistic understanding of neural system dysfunction in schizophrenia. Front Psychiatry 2013;4:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Ellaithy A, Younkin J, González‐Maeso J et al. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci 2015;38:506‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Krystal JH, Abi‐Saab W, Perry E et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology 2005;179:303‐9. [DOI] [PubMed] [Google Scholar]
- 253. Uno Y, Coyle JT. Glutamate hypothesis in schizophrenia. Psychiatry Clin Neurosci 2019;73:204‐15. [DOI] [PubMed] [Google Scholar]
- 254. Pillinger T, Rogdaki M, Mccutcheon RA et al. Altered glutamatergic response and functional connectivity in treatment resistant schizophrenia: the effect of riluzole and therapeutic implications. Psychopharmacology 2019;236:1985‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Farokhnia M, Sabzabadi M, Pourmahmoud H et al. A double‐blind, placebo controlled, randomized trial of riluzole as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia. Psychopharmacology 2014;231:533‐42. [DOI] [PubMed] [Google Scholar]
- 256. Anand A, Charney DS, Oren DA et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N‐methyl‐D‐aspartate receptor antagonists. Arch Gen Psychiatry 2000;57:270‐6. [DOI] [PubMed] [Google Scholar]
- 257. Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine‐resistant schizophrenia: a systematic review and meta‐analysis. Schizophr Res 2009;109:10‐4. [DOI] [PubMed] [Google Scholar]
- 258. Mouchlianitis E, McCutcheon R, Howes OD. Brain‐imaging studies of treatment‐resistant schizophrenia: a systematic review. Lancet Psychiatry 2016;3:451‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Egerton A, Broberg BV, Van Haren N et al. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: a multicentre 1H‐MRS study (OPTiMiSE). Mol Psychiatry 2018;23:2145‐55. [DOI] [PubMed] [Google Scholar]
- 260. Demjaha A, Egerton A, Murray RM et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry 2014;75:e11‐3. [DOI] [PubMed] [Google Scholar]
- 261. Mouchlianitis E, Bloomfield MAP, Law V et al. Treatment‐resistant schizophrenia patients show elevated anterior cingulate cortex glutamate compared to treatment‐responsive. Schizophr Bull 2016;42:744‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Kennedy JA, Israel O, Frenkel A et al. Super‐resolution in PET imaging. IEEE Trans Med Imaging 2006;25:137‐47. [DOI] [PubMed] [Google Scholar]
- 263. Song T‐A, Chowdhury SR, Yang F et al. Super‐resolution PET imaging using convolutional neural networks. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 264. McCutcheon R, Pillinger T, Mizuno Y et al. The efficacy and heterogeneity of antipsychotic response in schizophrenia: a meta‐analysis. Mol Psychiatry (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Javitt DC, Carter CS, Krystal JH et al. Utility of imaging‐based biomarkers for glutamate‐targeted drug development in psychotic disorders. JAMA Psychiatry 2018;75:11‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. van den Heuvel MP, Scholtens LH, Kahn RS. Multiscale neuroscience of psychiatric disorders. Biol Psychiatry 2019;86:512‐22. [DOI] [PubMed] [Google Scholar]
- 267. Breakspear M. Dynamic models of large‐scale brain activity. Nat Neurosci 2017;20:340‐52. [DOI] [PubMed] [Google Scholar]