

New theoretical and experimental work suggests that 10-nm chromatin fiber is folded into tree-like domains in single cells.
Abstract
With the textbook view of chromatin folding based on the 30-nm fiber being challenged, it has been proposed that interphase DNA has an irregular 10-nm nucleosome polymer structure whose folding philosophy is unknown. Nevertheless, experimental advances suggest that this irregular packing is associated with many nontrivial physical properties that are puzzling from a polymer physics point of view. Here, we show that the reconciliation of these exotic properties necessitates modularizing three-dimensional genome into tree data structures on top of, and in striking contrast to, the linear topology of DNA double helix. These functional modules need to be connected and isolated by an open backbone that results in porous and heterogeneous packing in a quasi–self-similar manner, as revealed by our electron and optical imaging. Our multiscale theoretical and experimental results suggest the existence of higher-order universal folding principles for a disordered chromatin fiber to avoid entanglement and fulfill its biological functions.
INTRODUCTION
The intimate connection between three-dimensional (3D) interphase DNA structure and gene expression in eukaryotic cells has made chromatin folding a rapidly developing field. In the past decade, previously well-accepted concepts have been continuously challenged by new experimental discoveries. For example, it had previously been widely believed that chromatin is folded into 30-nm fibers (1), which are assembled into discrete higher-order structures. However, this regular folding hierarchy has been highly debated [see recent review (2) and references therein] and was not observed in the state-of-the-art electron microscopy tomography (ChromEMT) experiment (3), which instead revealed a highly disordered chromatin polymer that is heterogeneously packed even down to the level of single nucleosomes in situ. This structural heterogeneity of chromatin has been suggested by partial wave spectroscopy (PWS) (4), a label-free technique, to have a profound regulatory impact on the global transcriptional profile of live single cells (5). At the population-average level, because of the development of chromosome conformation capture (3C) (6) and related techniques, it is becoming well established that chromatin has frequent genomic contacts inside topologically associating domains (TADs) (7). A loop extrusion hypothesis based on CTCF (CCCTC-binding factor) and cohesin proteins was proposed to explain many TAD features at the ensemble level (8, 9). However, the importance of this putative mechanism at the single-cell level has been questioned by recent superresolution fluorescence microscopy experiment (10), which found chromatin segments to be rich in TAD-like clusters (11) that are persistent even after cohesin knockout. Together, these new experimental findings remind us of the complexity of chromatin folding, which requires the cooperation of a large family of architectural chromatin-regulatory proteins, as well as the interplay between multiple folding mechanisms such as supercoiling (12–15), phase separation (16–18), molecular binding (19), crowding effects (20), and loop extrusion (8), all under the feedback control of transcription to be responsive to external stimuli. They also raise many important questions such as (i) what are the functional units of chromatin, (ii) what is the hierarchy of chromatin folding hidden in the disordered morphology, and (iii) what are the inner workings of TADs at the single-cell level. Among these questions, perhaps the most fundamental one is whether there are abstract yet universal folding principles of our genomic code independent of the known molecular and mechanistic complexity.
To investigate the existence of these principles (mathematical rules) without the burden of accounting for all the physical interactions and biological mechanisms that are far from fully understood, one could search for an abstract folding algorithm that aims to recapitulate the major experimental observations with minimal adjustable parameters and computational complexity, the success of which would suggest a positive answer. Among the chromatin features known to date from experimental results, the most important two are (i) the frequent self-interactions (21) that link promoters and enhancers for transcriptional regulation and (ii) the heterogeneous packing (3, 22–24) that disperses local DNA accessibilities, making room for transcription and nuclear transport. However, there is an apparent conflict between these two major chromatin properties from a polymer physics point of view. For example, it has been hypothesized that chromatin resembles a fractal globule (FG) (25, 26), which is a self-similar polymer in a fully collapsed state. While the FG model predicts the observed high contact frequency, it cannot explain the spatial heterogeneity of chromatin packing. One can prove that it is impossible for any 1D chain to be simultaneous self-similar, rich in self-contacts, and diverse in packing density (see the Supplementary Materials for more details). This trilemma rules out the possibility of chromatin being any kind of fractal chain (Fig. 1A and fig. S1) and means that chromatin must have distinct folding modes at different length scales. Despite the unknown folding details, this scale dependence has manifested itself in the mass scaling measured by small-angle neutron scattering (27) and small-angle x-ray scattering (28), which undergoes a transition from slow to fast as the length scale increases (Fig. 1B). Because slow mass scaling is associated with polymer decondensation and therefore less intrapolymer contacts, one would expect chromatin contact frequency to decay faster at a smaller scale. However, high-throughput 3C (Hi-C) experiments indicate a contact scaling transition from slow to fast as the genomic scale increases (Fig. 1C), opposite to the expected result from mass scaling. Moreover, at the small length scale where mass scaling is not even close to 3 (i.e., space filling), chromatin exhibits a remarkably slow power-law decay of contact frequency with a scaling exponent around −0.75 inside TADs (8). These abnormal behaviors make chromatin folding not only an intriguing biological question but also a fascinating puzzle for polymer physics.
Fig. 1. The paradox of chromatin folding and the basic ideas of SRRW.
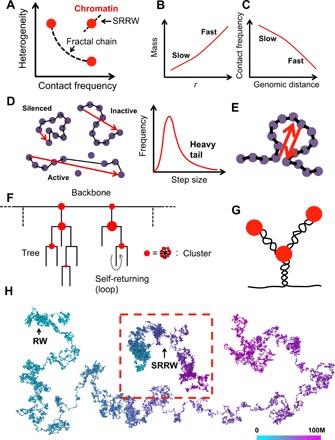
(A) Chromatin structure cannot be explained by a fractal chain that has anticorrelated contact frequency and density heterogeneity. SRRW is developed to unify these two properties. (B) Schematic representation of the mass scaling behavior of chromatin. (C) Schematic representation of the contact scaling behavior of chromatin (<10 Mb), which is counterintuitive given the mass scaling behavior. (D) Coarse-graining diverse epigenetic states at the nanoscale into a wide distribution of step sizes. One step approximately maps to 10 nucleosomes or 2-kb DNA. The balls represent histones, and the lines represent DNA. The arrows represent the coarse-grained steps in SRRW. (E) Self-returning as a coarse-grained representation of looping (in a broad sense including supercoiling and clustering). (F) SRRW’s topological architecture featuring random trees connected by a backbone. Tree nodes are formed by frequent self-returning of short steps. (G) One possible realization of tree structures is by combining nanoclusters with nested supercoils or loops. (H) Comparison between a free SRRW (highlighted in the red box) and a free RW, both of 50,000 steps (100 Mb)
In this paper, we address the above contact-structure paradox by introducing a self-returning random walk (SRRW) as a mathematical model that effectively breaks the nonbranching topology of the 10-nm chromatin fiber and generates tree-like topological domains connected by an open chromatin backbone. The decondensed backbone segments isolate the topological domains and allow them to form larger compartments with high folding variation. We carry out nanoscopic imaging using chromatin scanning transmission electron microscopy (ChromSTEM) and live-cell PWS microscopy to demonstrate DNA packing heterogeneity across different scales, visualizing chromatin domains and compartments at the single-cell level as predicted by our model. Our results support the hypothesis of local DNA density being an important transcriptional regulator and provide a new picture of genomic organization in which chromatin is folded into a variety of minimally entangled hierarchical organizations across length scales ranging between tens of nanometers to micrometers, without requiring a 30-nm fiber. We discuss the quantitative predictions of this folding picture and how they explain a wide range of existing experimental observations. Using heat shock as a model system, we further reveal couplings between chromatin properties during stress response. The strong agreement between our model and experiments on this structural perturbation sheds lights into the structure-function relationship of interphase DNA and suggests the existence of higher-order folding principles and substantial reduction of dimension during genomic landscape exploration. We end the paper by discussing possible molecular mechanisms and biological consequences of this novel organizational paradigm.
Results
Self-returning random walk
Given the enormous size of our genome, coarse graining is necessary in chromatin modeling. Typically, polymer models coarse-grain at least a few kilobases of DNA into one monomer bead to study chromatin structures above 10 Mb. This implicitly restricts the basic repeated unit of the coarse-grained chromatin to be of 30 nm or larger in diameter, incompatible with the recent ChromEMT observation where chromatin is predominantly a heterogeneous assembly of 5- to 24-nm fibers (3). Therefore, instead of large beads of one size, we use steps with a continuous spectrum of step sizes to cover the conformational freedom (29) of a 10-nm fiber at the kilobase level (see Fig. 1D). We choose each single step to correspond with 2 kb of DNA, roughly an average of 10 nucleosomes. To capture the frequent genomic contacts, we introduce stochastic, self-returning events (Fig. 1E). The return probability is assumed to decay with the length of the current step size U0 by a power law
| (1) |
where α > 1 is the folding parameter for the modeled chromatin. Here, a smaller α leads to a higher return frequency and vice versa. According to Eq. 1, large steps have a lower probability of returning than small steps do. Therefore, the decondensed and more accessible DNA segments are more likely to be found outside of loops. Once a step is returned, a further return is possible based on the same probability function. With a probability of 1 − Preturn, a jump from the current position can be issued to explore a new point. The jump takes an isotropic direction and a step size U1 that follows a power-law distribution
| (2) |
where u is larger than 1, with the resulting smallest step size in reduced units corresponding to roughly 30 nm in the end-to-end distance of the 2-kb nucleosome segment. Likewise, we eliminate unrealistically long step sizes, which correspond to 0.1% of the jumps. To model the confinement effect inside nucleus, we also introduce a global cutoff of 2α times the local cutoff during the conformation generation, which results in a DNA density of ~0.015 bp/nm3, comparable to that of an average diploid human eukaryotic nucleus. An accumulated stack of steps (both jumps and returns) tracks the overall conformation of chromatin, while a subset of steps (only the unreturned jumps that are, on average, more stretched) results in the formation of a chromatin “backbone.” With respect to the model, the stochastic jumps and returns thus biologically represent the conformations of 2-kb DNA segments in 3D space where (i) large steps without returns are akin to elongated segments of DNA, (ii) large steps with large returns are branches of loops and supercoiled plectonemes, and (iii) small steps with small returns are compacted clusters. By adding returning to jumping, the model thus turns a nonbranching topology into a branching one, with the degree of structural hierarchy controlled by the folding parameter α. As schematically shown in Fig. 1F, the overall topological architecture of SRRW is a string of random trees, with the branches formed by the low-frequency returning of long steps and the nodes formed by the clustering of high-frequency returning of short steps. Isolated by the unreturned long backbone segments, the trees integrate nested loops and clusters into domains for co-regulation. One possible realization of these hierarchical structures can be the combination of passive nanoscale phase separation with active supercoiling driven by DNA transcription, as shown in Fig. 1G. Nested loops formed by molecular binding or extrusion could also contribute to the effective branching of chromatin. In the rest of the paper, we use 50,000 steps to model 100 Mb of DNA, roughly the average genomic size of one entire human chromosome. We use an α around 1.15 to generate structures that resemble interphase chromatin, and we will discuss the implication of this parameter on higher-order chromatin folding.
Chromatin structure and scaling at the single-cell level predicted by SRRW
At a negligible computational cost, SRRW is able to stochastically generate chromatin-like conformations at 2-kb resolution of high case-to-case variations in spatial organization, but with consistent topological and statistical characteristics controlled by the global folding parameter α. Because of the hierarchical folding, a typical conformation generated by a free SRRW is much more compacted than that generated by a free random walk (RW), as shown in Fig. 1H. Because we are modeling one single interphase chromosome confined by the surrounding genome, we will focus on a confined SRRW (we keep the generic term SRRW for simplicity) as our chromatin model in the rest of the paper. As shown in Fig. 2A and fig. S2, the overall structure of our modeled chromatin is porous, nonglobular (30), with a rough surface, which is in stark contrast to the predictions of many polymer simulations where the modeled chromatins collapse into globules. The irregular shape of SRRW with large surface area would naturally facilitate accessibility of DNA to transcription inside interchromosomal domains between chromosome territories (31), in line with experimental observations (32). The rich porosity and sponge-like structure of SRRW also allow transcription factors to efficiently search the interior of chromatin and could explain the fractal-like nuclear diffusion of various biomolecules (33).
Fig. 2. Typical single-cell level chromatin structure and its scaling behaviors predicted by our model.
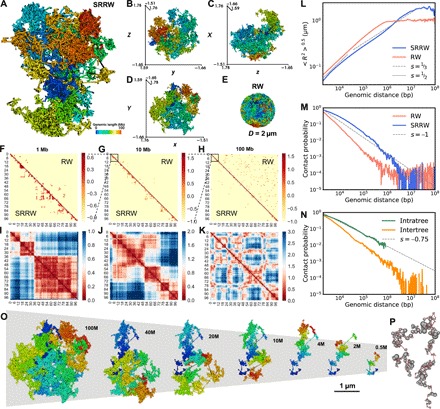
(A to D) Folded structure of our modeled chromatin and its xyz projections. (E) Equilibrium globule (confined RW) as a reference system. (F to H) Predicted single-cell level contact maps from local (1 Mb) to global (100 Mb) based on SRRW and RW. (I to K) Physical distance maps of SRRW from local to global. (L) Root mean square end-to-end distance (R) scaling of SRRW and RW. As guide to the eye, the dashed and dotted lines show power-law scaling, with exponents being 1/3 and 1/2, respectively. (M) Contact scaling of SRRW and RW. As guide to the eye, the dashed line shows power-law scaling, with exponent being −1. (N) Contact scaling of SRRW for intra- and intertree contacts. The intratree contacts are richer than the intertree ones and follow a power-law scaling with exponent s = −0.75, akin to the contact scaling within TADs. (O) Structures of the modeled chromatin at different genomic scales. (P) Beads-on-string representation of the secondary structure of chromatin modeled by a free SRRW. Tree domains are represented in beads, with the size of the bead corresponding to the genomic size of the domain. The backbone is represented in string.
Colored based on the 1D genomic sequence, the modeled chromatin is folded into domains and compartments reflected by the unmixed color. As a reference, an equilibrium globule (a confined RW, or RW for simplicity) shows no sign of territorial organization (Fig. 2E). The disparate organizations between SRRW and RW lead to strikingly different contact maps, as shown in Fig. 2 (F to H), where our modeled chromatin exhibits enriched contacts of TAD-like patterns (30, 34, 35) in the most regulation-relevant ranges (≪10 Mb) and is relatively depleted of random contacts above 10 Mb. To better illustrate the spatial organization of the modeled chromatin, we calculated the physical distance matrices at different scales as shown in Fig. 2 (I to K). The TAD-like domains at the kilobase-to-megabase scale are seen without requiring ensemble averaging, which is in consistence with recent superresolution observations of heterogeneous domains in single cells (10). To allow a more direct comparison between our predicted structures and the state-of-the-art imaging results (10), we have adapted the experimental resolution of 30 kb by coarse-graining our chromatin model 15-fold. A collection of typical structures of 2-Mb modeled chromatin segments in both 2- and 30-kb resolutions and their physical distance maps are shown in Fig. 3, which demonstrates how 3D chromatin clustering leads to various TAD-like 2D patterns. Our model predicts that these clustered contacts at the sub-megabase scale transition into plaid-like patterns at the multi-megabase scale (Fig. 2K), reminiscent of the AB-compartment patterns (6) found in Hi-C contact maps. Our distance matrices imply that the 3D genome modeled at the single-cell level is compartmentalized across many scales, which is visualized in Fig. 2O. The compacted yet minimally intermixed 3D packing below 10 Mb is reflected in the root mean square end-to-end distance (R) scaling with slope between 1/3 and 1/2 in the corresponding regime (Fig. 2L). At the genomic scale above 10 Mb, the slope drops to below 1/3, in line with experimental observation (36) and signifying a strong intermixture of large compartments. Nevertheless, at this scale, the modeled chromatin intermixes as clumps rather than wires, which would be expected to result in strong internal friction rather than entanglement.
Fig. 3. Typical local chromatin structure predictions.
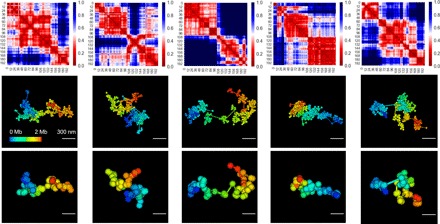
Five examples of 2 Mb of DNA at 2- and 30-kb resolutions and their physical distance maps (in micrometers) at the single-cell level predicted by the SRRW model. These examples suggest that structured domains exist at the single-cell level with high domain-to-domain variability.
Figure 2M shows the contact probability (Pc) curve of our modeled chromatin. Compared to the RW reference, SRRW features a slow decay of Pc at the sub-megabase scale, which is again in consistence with experimental findings (21), and as revealed in Fig. 2N, the contact frequency is higher inside the tree domains. The decay exponent, −0.75, of intratree Pc coincides with the value reported by Hi-C experiments for contacts inside TADs (8). Comparing intra- and intertree contacts, our model predicts that, in single cells, the genomic isolation across tree domains can be as large as 10-fold near the 1-Mb range, much higher than the puzzlingly small twofold isolation across TADs at the population-average level observed in Hi-C experiments (37, 38). The sufficient isolation at the single-cell level predicted by our model is provided by the elongated intertree backbone segments (~5% of the modeled chromatin), which are expected to be openly accessible and transcriptionally active. This prediction naturally explains the experimental finding that TAD boundaries are active in transcription (39). Because tree domains can further cluster and a large tree domain itself has a structural hierarchy, the contact pattern at the TAD (megabase) scale is therefore hierarchical (fig. S3). Whether this hierarchy extends to large scales is a matter of debate (35, 40, 41). While SRRW predicts compartmentalized organizations at different scales, the arrangement of the compartments at the largest scale (>10 Mb) (Fig. 2K) is nonhierarchical and can be unraveled to a polymeric string of tree domains (represented by beads), as shown in Fig. 2P. This “secondary” structure of our modeled chromatin highlights the functional importance of the tree domains, which exert strong topological constraints on the genomic organization at the single-cell level.
Tree domains and larger compartments in a 3D forest
The SRRW model predicts that the topological domains at the single-cell level have random tree structures. These tree domains are amalgams of chromatin nanoclusters and loops (likely supercoiled) at the kilobase-to-megabase scale and serve as building blocks of larger packing domains, i.e., the compartments of tree domains at the single-cell level. Collectively, these tree domains form a “3D forest” within a chromosome territory. A 3D map of tree domains of a typical chromatin structure predicted by SRRW is shown in Fig. 4A, with the color corresponding to the genomic size of each tree domain. The physical sizes of the tree domains measured by their radius of gyrations (Rg) are positively correlated with their genomic sizes but with considerable dispersion (Fig. 4B). The peak Rg of tree domains is around 70 nm, as shown in Fig. 4C. The genomic size distribution of the tree domains is shown in Fig. 4D, featuring a broad spectrum and an abundance of small domains. The average genomic size is around 50 kb, while most of the DNA is packed in tree domains around 300 kb, resembling the typical size of TAD at the Hi-C level (37). Recent high-resolution TAD analyses revealed a large population of small TADs of tens of kilobases (42, 43), which aligns well with our tree-domain analysis at the single-cell level. Note that the genomic size distribution of tree domains is predicted to be strongly non-Gaussian with a heavy tail. As a result, the median size is much smaller than the average size. It is interesting that significant median-average discrepancy has also been reported for TADs whose median size is about 185 kb (21) and average size is about 800 kb (7). While the values would depend on the Hi-C resolution, the heavy-tailed domain statistics has been shown in recent sub-kilobase Hi-C analysis (43). Our model suggests that this non-Gaussian statistics is a general feature of chromatin, in contrast to normal polymer behavior (fig. S4).
Fig. 4. A 3D map of tree domains and their structural statistics.
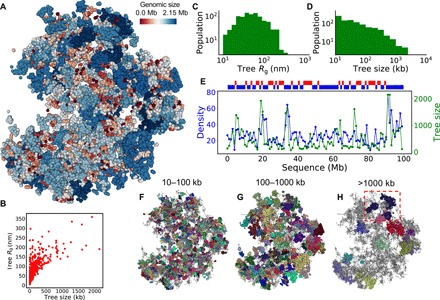
(A) Particle representation of the modeled chromatin with tree domains colored according to their genomic sizes in logarithm scale. (B) Scatter plot of the physical size (Rg) of tree domains versus their genomic sizes, with each domain being one point. (C) Physical size (Rg) distribution of tree domains. (D) Genomic size distribution of tree domains. (E) Local DNA density and averaged tree domain size walking along the sequence of the modeled chromatin, with each step being 1 Mb. Above the panel, the local density is divided into two groups, showing in red (low density) and blue (high density). (F to H) Tree domains marked by random colors in three size groups (10 to 100 kb, 100 to 1000 kb, and above 1000 kb). The unmarked structures are shown in gray. An example of subdomains inside a tree domain is highlighted in (H).
To reveal the regulatory effect of tree domains on local DNA accessibility, we investigated the relationship between tree domain size and packing density. We have observed a positive correlation between the genomic size of tree domains and the local packing density walking along the modeled chromatin sequence (Fig. 4E), suggestive of a size-dependent domain activity that is in line with recent finding that active domains are small in genomic size (42). The more condensed packing of larger tree domains is reflected in a decrease of the R scaling factor in the sub-megabase regime (Fig. 2L). Using a dichotomy of low (red) and high (blue) packing densities (low density defined as below 80% of mean), we found a quasi–self-similar density pattern (Fig. 4E, top) along the sequence of our modeled chromatin. Resembling the experimentally observed AB-compartment activity alternation (30), this pattern reflects an emergence of higher-order compartmentalization above tree domains as implied by the plaid-like matrix in Fig. 2K. These multitree packing domains emerge because of the Lévy flight–like statistics of the secondary structure of SRRW (Fig. 2P), which is non-Gaussian and inhomogeneous at large length scale. The higher-order compartmentalization, however, does not compromise the physical identities of individual tree domains. As shown in Fig. 4 (F to H), the tree domains (randomly colored) are physical entities with minimal intermixing. Their hierarchical inner structure also lowers the risk of self-entanglement and knotting at the sub-megabase level. These structural properties make tree domains good candidates for the functional modules of chromatin at the single-cell level. The similarities between our single-cell predictions and the population-level Hi-C observations suggest that TADs are the statistical consequence of chromatin folding into tree-like topological domains (fig. S3).
A quasi–self-similar heterogeneous packing picture of chromatin
One natural yet important structural consequence of SRRW is that the packing density is highly nonuniform across many length scales. To highlight the multi-megabase compartments, we calculated a 3D density map with the nanoscale density fluctuations filtered out. In Fig. 5A, the compartments are demonstrated in a colored particle representation, with larger and darker particles corresponding to higher compartmental DNA density. The cross sections of the filtered 3D density map in all three directions are displayed in density heat maps (Fig. 5A, top). Zooming into a local fraction of the modeled chromatin (without an applied filter) reveals nanoscale domains that are interconnected by physically extended backbone segments (Fig. 5A, bottom left) and rich in high-density tree nodes, as highlighted by golden particles in the second bottom panel of Fig. 5A. Our results are consistent with the experimental observation of widespread chromatin nanoclusters or clutches interspersed by nucleosome-depleted regions (22) and further suggest that large clutches as large tree nodes host higher-order interactions. Our structural prediction of the spatial separation of open and condensed chromatin domains, with the latter being larger clumps, is also remarkably in line with recent superresolution observation where active and repressive histone marks have little colocalization and correlate respectively with small low-density and large high-density DNA regions (44). Our prediction of the ubiquitous alternation between open and condensed chromatin domains suggests that transcriptional activation and repression are highly coupled, similar to the two sides of a coin. In particular, the model predicts that segments with higher transcriptional propensity tend to serve as stronger isolators between neighboring domains (fig. S3). The granular packing structure depicted by SRRW registers large surface area and rich porosity, resulting in a similarly heterogeneous encompassing media that could serve as a nuclear channel system (45). Such a unique space-filling property of SRRW sets it apart from normal disordered polymers, by greatly widening the local DNA density spectrum as shown in Fig. 5B. This broad density spectrum echoes the recent ChromEMT observation (3) and emphasizes the fundamental role of disordered chromatin packing in transcriptional regulation and nuclear transport.
Fig. 5. Granular and porous chromatin packing across scales demonstrated by model and experiments.
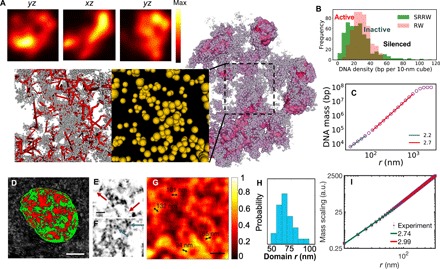
(A) Density field representation (with nanoscale fluctuation filtered) of the modeled chromatin (shown in purple) and its cross sections in three orthogonal directions (shown in the top panels). Zoomed-in bottom panels show the nanoscale packing of the modeled chromatin (without filter). Backbone segments are represented in red lines, and nanocluster tree nodes are represented as yellow particles, with the volume being proportional to the genomic size. (B) Local DNA density spectrum sampled by walking along the modeled chromatin with a probe of 150-nm radius (green). A reference is sampled from an RW (red). (C) DNA mass scaling of the modeled chromatin (sampled over 1000 SRRW trajectories). (D) Typical live cell PWS image of an A549 cell showing chromatin packing domains of mass fractal–like structure. Scale bar, 5 μm. High D regions are scaled in red, while low D regions are shown in green. D of 2.3 was used as a cutoff to separate high and low D regions. (E to G) ChromSTEM images of A549 cells. (E and F) Linker DNA (red arrows) and individual nucleosomes (blue arrows) captured by ChromSTEM. Scale bar, 30 nm. (G) Chromatin volume concentration based on the convolution of ChromSTEM image. Scale bar, 200 nm. (H) Histogram of chromatin domain size distribution based on the chromatin volume concentration. (I) DNA mass scaling based on ChromSTEM. a.u., arbitrary units.
The existence of two modes of chromatin organization (tree domains and chromatin compartments) leads to an increase in the mass scaling factor from small to large length scales, as shown in Fig. 5C. Such a trend is concordant with the biphasic finding from small-angle neutron scattering (27) and small-angle x-ray scattering (28). Because the scattering experiments do not distinguish nuclear molecules, we carried out a ChromSTEM method that focuses on the DNA mass distribution. Combining electron microscopy and DNA staining, ChromSTEM captures nucleosome-level chromatin nanostructure (Fig. 5, E and F). Consistent with recent ChromEMT observations, no 30-nm fibers were observed. The DNA mass scaling readout of ChromSTEM (Fig. 5I) confirmed the biphasic behavior predicted by the SRRW model. Despite the slow mass scaling at the small length scale, the tree topology predicted by our model enables high contact frequency within the nanoscale chromatin domains (Fig. 2N), reconciling the contact-structure conundrum present in nonbranching polymer models of the higher-order chromatin organization. A DNA density map based on ChromSTEM reveals a granular and porous packing of DNA as shown in Fig. 5G, in line with our prediction. Notably, the peak radius of the chromatin domains revealed by ChromSTEM is around 70 nm (Fig. 5H), which is consistent with recent single-cell fluorescence microscopy (23) and coincides with the peak Rg of tree domains predicted by SRRW (Fig. 4C). The excellent agreement between our model and nanoscopic imaging strongly suggests the existence of functional chromatin modules with minimal intermixing at the sub-megabase level in single cells. From the nanoclusters (clutches) to the tree domains, and to the larger-scale compartments (packing domains), our model predicts that chromatin has persistent packing heterogeneity across many length scales, hence resembling mass fractals (granular and porous in a quasi–self-similar way) at the single-cell level. To further investigate chromatin packing in live cells, we next performed optical imaging using PWS microscopy (4). As a label-free technique, PWS microscopy noninvasively measures the intracellular macromolecular arrangement and detects mass fractal–like structures. As shown in Fig. 5D, PWS microscopy reveals that chromatin is rich in packing domains of heterogeneous and mass fractal–like structure in the live-cell nucleus.
Emergence of a universal folding principle
As a minimal model, SRRW uses a single folding parameter α to describe the collective conformational freedom of chromatin. Here, we show that tuning α leads to structural alternations across many scales. Increasing α changes the chromatin architecture from network-like to chain-like. At the tree domain level, a small α promotes the formation of large tree domains and concomitantly more nanoclusters or clutches (22) as tree nodes, whereas a large α reduces the branching and clustering of the modeled chromatin. This is shown in Fig. 6A, where we marked the five largest tree domains in red and the tree nodes larger than 40 kb in yellow, for typical architectures at α = 1.1 and α = 1.3. At the compartment level, small α results in a small population of large compartments, while large α favors a large collection of small ones. This can be seen in typical physical distance matrices (Fig. 6, B and C), where fine plaid-like pattern appears when large packing domains are repressed at large α. Recall that from Fig. 2 (I to K), we have predicted the existence of TAD-like and AB compartment–like organizations at the single-cell level. Here, our structural analysis further suggests a coupling between the repression of packing domains and the finer compartmentalization in single cells. Our prediction finds an interesting analog with the Hi-C observation, where weakening TADs unmask a finer compartment structure at the population level (41). This counteraction between the two modes of chromatin organization can be intuitively understood in the framework of SRRW because the formation of tree domains driven by higher-order interactions imposes hierarchical topological constraints on heterogeneous chromatin segments and suppresses large-scale phase separation.
Fig. 6. Coupling between chromatin properties during structural alternation.
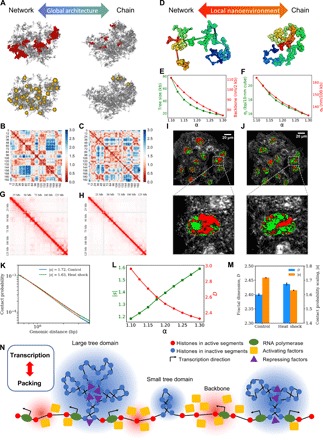
(A) Global architectural alternation as α changes. Left: Network-like (small α). Right: Chain-like (large α). For each structure, the five largest tree domains are shown in red in the first row, and the tree nodes larger than 40 kb are shown in yellow in the second row. (B and C) Physical distance maps at small (left) and large (right) α values. (D) Two different nanoenvironments at small (left) and large (right) α values. (E) Tree domain sizes and backbone openness as functions of α. (F) SDs of local DNA density and end-to-end distance as functions of α. (G and H) Contact matrices of hESCs exposed to heat shock and at normal temperature. (I and J) PWS live-cell images of HCT116 cells under heat shock and normal temperature conditions. (K) Contact probability curves for hESCs under normal temperature and heat shock conditions for the 100-kb to 1-Mb range. (L) How contact scaling and effective mass scaling for 100-kb to 1-Mb modeled chromatin segment depend on α. (M) Trends of contact probability scaling change and heterogeneity change after heat shock based on Hi-C analysis and PWS measurements. (N) Schematic presentation of the interplay between transcription and chromatin packing. High DNA density (inactive) regions are highlighted in blue, and low DNA density (active) regions are highlighted in red. For a simple demonstration, we scaled down all the domains, which are expected to contain more nucleosomes in reality.
The interplay between the two layers of chromatin organization allows a global architectural change to affect local nanoenvironments where transcription happens. As schematically shown in Fig. 6D, a global architecture rich in large tree domains (small α) has more polarized local DNA accessibility and hence transcription activity. While there is more DNA densely packed in large tree domains with more condensed tree nodes (Fig. 5A and fig. S5), the backbone chromatin segments connecting the tree domains are expected to become more open when the domain sizes increase. Numerical analyses of the average tree domain size and backbone openness (physical extent) as functions of α have confirmed this picture and are shown in Fig. 6E. Our analysis also shows that polarization of chromatin accessibility is coupled with DNA packing heterogeneity (Fig. 6F). To study the local contact-structure relation, we analyzed the contact frequency scaling and the R scaling for the modeled chromatin segments of 100 kb to 1 Mb. We fitted the scaling curves to power laws and extracted scaling factors (fig. S6). An effective mass scaling factor D defined as the inverse of R scaling factor measures the mass scaling based on consecutive DNA mass with definite segment length. Figure 6L shows that D decreases as α goes up, anticorrelated with the contact scaling s, but correlates with the local packing heterogeneity. The coupling between mass scaling and structural heterogeneity suggests, again, that chromatin compartments or packing domains can be approximated as a mass fractal with its structural heterogeneity quantified by a fractal dimension D. Note that our prediction of higher chromatin heterogeneity fostering more genomic contacts contrasts the behavior of a fractal chain (Fig. 1A). The reason why chromatin resembles 3D mass fractal rather than 1D fractal chain is due to the prevailing clusters and branches that lead to a heterogeneous and porous structure.
The above theoretical analysis suggests that during chromatin structural alternation, which could happen when cell responses to stress, the perturbations of many chromatin properties are coupled. Here, we test this prediction using heat shock as model experimental system. We analyze the contact maps (Fig. 6, G and H) and contact probability curves (Fig. 6K) at normal temperature and exposed to heat shock using the publicly available Hi-C data on hESCs (human embryonic stem cells) [Gene Expression Omnibus (GEO) access code GSE105028] (46). We noticed a decrease in contact probability scaling with the heat shock compared to the control condition, which signifies an increase in long-range contacts in hESCs subjected to heat shock. A similar trend has been observed in previous literature for heat shock experiments on Drosophila cells (47), signifying that this increase in long-range contacts may be an evolutionarily conserved part of the heat shock response. We have carried out live-cell PWS measurements on HCT116 cells to study the effect of heat shock on the packing heterogeneity of chromatin. As shown in Fig. 6 (I and J), we observed that temperature stress led to a higher PWS signal that means more heterogeneous packing of live-cell chromatin. Together, we found that the contact probability scaling factor s and the average mass fractal dimension D are anticorrelated, as shown in Fig. 6M, which is consistent with the prediction of our model (Fig. 6L). We performed additional PWS experiments on A549 lung adenocarcinoma and differentiated BJ fibroblast cell lines and observed the same trend of increased fractal dimension upon heat shock stimulation (fig. S9). Our model also suggests that the formation of long-range genomic contacts is coupled with promotion of high-density chromatin clusters, in line with the Hi-C report that chromatin forms more polycomb-silenced heterochromatin while increasing its self-interaction frequencies during heat shock (47). Moreover, upon treatment of both A2780 and A2780 m248 ovarian cancer cell lines with 100 μM valproic acid, a histone deacetylase inhibitor, we observe a significant decrease in mass fractal dimension D, which we would expect to result in an increase in contact probability scaling s, demonstrating the importance of histone markers in determining chromatin-chromatin interactions and altering global structural properties of chromatin (fig. S10). Together, our theoretical and experimental results suggest that structural hierarchy, packing heterogeneity, genomic interaction, and transcriptional polarization are intimately coupled during epigenetic reconfiguration.
As schematically shown in Fig. 6N, transcription could play an important role in these couplings. The divergent transcriptions are expected to generate overwound DNA flanked by promoters, facilitating nucleosome deposition and plectonemic supercoils. The merging of small plectonemes and the branching of large plectonemes can lead to tree-like structures. On the other hand, convergent transcriptions between tree domains can generate underwound DNA, which tends to evict histones and stiffen the chromatin fiber, resulting in open backbones. In this case, part of the torsion can be stored in small tree domains, which can be easily activated as transcription proceeds. The activating and repressing factors are expected to concentrate at different domains and facilitate their phase separation at larger scales. In this picture, transcription shapes the packing of remote DNA, while chromatin packing, in return, regulates the local activity of transcription. Because chromatin packing can be globally affected by physiochemical factors such as temperature, pH, and ionic strength, macrogenomic engineering (5) that regulates the global gene expression pattern through chromatin packing control can be useful to treat systems biology diseases such as cancer.
DISCUSSION AND CONCLUSION
In this paper, we asked the question of how chromatin folds to achieve simultaneously high self-interacting frequency and high space-filling heterogeneity. While there is no lack of 3D models with conflicting single-cell level predictions developed to explain the population-level Hi-C contact maps, the packing heterogeneity has received less attention and remained poorly understood. Nevertheless, a few lines of recent experimental evidences (3, 5, 24, 45) have converged to suggest that chromatin packing heterogeneity determines the functional accessibility and activity of interphase DNA. Here, we integrated modeling and imaging to understand 3D genome at the single-cell level. We have carried out ChromSTEM and PWS measurements to quantify the heterogeneity of chromatin packing across different length scales. At the nanoscale, concurring with recent study (3), we did not observe the 30-nm fiber and instead revealed a granular packing structure with prevailing nanodomains of a peak radius around 70 nm (Fig. 5H). To characterize chromatin packing at larger scales and in live cells, we have conducted PWS microscopy to reveal that heterogeneous packing domains exist throughout the nucleus (Fig. 5D). Our multiscale imaging suggests a quasi–self-similar picture of chromatin packing. Using heat shock as an example, we demonstrated that the average heterogeneity characterized by a mass fractal dimension is sensitive to environmental change and could serve as an indicator of chromatin folding state (Fig. 6, I and J).
There is a subtle yet important topological difference between a fractal chain and a mass fractal, as the former does not branch. For 1D chain, there is a trilemma between high packing heterogeneity, high contact frequency, and self-similarity. This means that the presumed nonbranching topology and the strict scale invariance of chromatin must be broken to unify high packing heterogeneity and contact frequency, hence necessitating distinct folding modes at different scales. In this light, we carried out ChromSTEM to find a transition in the mass scaling near 100 nm (Fig. 5I), close to the peak domain radius. Note that the scale-dependent power laws have also been reported in literature for chromatin contact frequency (8) and mean square distance (48). To understand the scale-dependent packing modes, we introduced an SRRW model, which generates tree-like topological domains with hierarchical structures (Fig. 1). The tree domains pack local genomic DNA into nested loops and nanoclusters with high contact frequency at the sub-megabase scale and constitute mass fractal–like compartments at the multi-megabase scale (Figs. 2 and 4). We predict that an open chromatin backbone connects and isolates the tree domains, rendering highly porous chromatin structure, flexible higher-order folding, and irregularly shaped chromosome territory (Fig. 5, A to C). Our model agrees well with our experiments demonstrating a heterogeneous chromatin structure across many length scales (Fig. 5, D to I). Moreover, the model predicts an anticorrelation between contract frequency scaling and packing heterogeneity, consistent with our experimental observation of heat shock response (Fig. 6).
It is interesting that our model predicts TAD-like and AB compartment–like patterns on a distance matrix at the single-cell level (Fig. 2, I to K), resembling the Hi-C contact patterns (21) at the population level. This resemblance becomes more intriguing, because the model further predicts a counteraction between the two single-cell organizational modes (Fig. 6, B and C), reminiscent of the similar phenomenon at the population level reported in Hi-C experiments (41). This suggests that well-organized 3D structures such as domains and compartments are intrinsic chromatin characteristics at the single-cell level rather than merely statistical patterns of large population (fig. S3). Furthermore, our model predicts that the 3D genome is more structured than that suggested by population-level contact patterns. Beyond the average twofold isolation associated with TADs (37, 38), tree domains allow single-cell chromatin structure to realize 5- to 10-fold probability difference between the intra- and interdomain contacts at the 100-kb to 1-Mb level (Fig. 2N). This apparent contradiction between our single-cell level prediction and the population-level observation of genomic contacts can be resolved by noting that the cell-to-cell variability of genomic organization is high (34). Compared to the diffuse and intermixing loops predicted by the loop extrusion hypothesis (9), our model depicts more compacted and isolated physical entities in single cells (Fig. 3), rich in spontaneous higher-order interactions as revealed by recent superresolution experiment (10). In line with recent Hi-C reports (42, 43), statistical analysis of our modeled chromatin suggests that there are prevailing small domains at the single-cell level, followed by a heavy tail of larger ones (Fig. 4D). Our model further predicts a correlation between local DNA density and domain size (Fig. 4E), which lends explanation to the experimental finding that small domains are, on average, more active than large ones (42). Last, our prediction that tree domains are isolated by easily accessible chromatin backbone segments (Figs. 2P and 3) is consistent with the observation that TAD boundaries are enriched in active genes (39).
It is remarkable that by resolving the contact-structure paradox (Fig. 1), a minimal model is able to pivot a wide array of chromatin features including untangled compaction (25), disordered morphology (3), nonglobular territory (30), porous nuclear medium (45), biphasic chromatin mass scaling (27, 28), abundance of nanoclusters or clutches (22), non-Gaussian domain size distribution (42, 43), size-dependent domain activity (42), active domain boundaries (39), higher-order interactions (10), and multiple layers of chromatin organization (41). This suggests an emerging picture that, on top of the first-order genomic structure, namely, the linear DNA double helix for data reproduction, our genome has evolved to physically adopt virtual tree data structure (Fig. 1F) for its higher-order functional modules to better organize the enormous genomic information. In this regard, functional optimization may be a better perspective to understand chromatin folding than polymer physics. In particular, we argue that forming a string of tree-like topological domains connected by open backbone segments has functional advantages in (i) proper organization of genomic contacts, (ii) packing-based regulation of transcription, (iii) transport and accommodation of nuclear proteins, and (iv) transition between interphase and mitosis.
Proper genomic contacts based on chromatin folding are critical to cell function. In stark contrast to the equilibrium globule reference (Fig. 2, F to H), we have shown that our model promotes short-range contacts and suppresses long-range (multi-megabase and above) contacts. Because longer-range contacts have more combinatorial possibilities, this suggests that chromatin folding can significantly lower the information entropy of genomic interaction, despite the observed “disordered” polymer structure (3). This ordered genomic organization not only enhances the local regulatory contacts within hierarchical domains but also restricts random long-range contacts that could lead to aberrant functions.
The identity of cell often depends on the coordination of highly active and strictly silenced genes, which requires a polarized transcription pattern. As shown in Fig. 5B, compared to a confined RW globule, our chromatin model has a broader spectrum of local DNA density with both boosted low-density and high-density regimes. In line with the emerging hypothesis that local chromatin packing density regulates the transcriptional activity, this packing heterogeneity predicted by our model provides a prerequisite physical environment for polarized gene expression. On the other hand, the quasi–self-similar packing picture revealed by our experiments and model (Fig. 5 and fig. S2) is advantageous in supporting fast transport of nuclear proteins of varying sizes. This porosity across many length scales can also accommodate the experimentally observed clusters and condensates from tens of nanometer to micrometer scale that are composed of different transcription factors, DNA-processing enzymes, and chromatin architectural proteins.
It is worth noting that, in our model, most chromatin is packed into tree domains, whereas only about 5% of interphase DNA is exposed in the backbone segments that are physically extended and of high transcriptional competence. It is this minority of the heterogeneous biopolymer that creates most of the low DNA density regions in the porous chromatin structure. Recent in vitro experiment (49) confirmed a condensin loop extrusion mechanism (50) on naked DNA, which is strictly one-sided and has been argued (51) to be far from sufficient for mitotic condensation. Nevertheless, on the basis of our prediction that only about 5% of the DNA swells the porous chromosome territory, the one-sided condensin extrusion on this extended backbone of chromatin could have a considerable effect of collapsing interphase DNA into mitotic form. This backbone extrusion could preserve the folding memory of chromatin into the topological information of tree domains, although most of the Hi-C detectable spatial proximity information between loci is inevitably lost during the transition from interphase to mitosis. In this manner, our folding picture allows reconciliation between mitotic condensation and folding memory inheritance. Consistently, recent superresolution single-nucleosome tracking suggested the existence of chromatin domains throughout the cell cycle (23). The reported peak radius of the domains is about 80 nm, similar to our ChromSTEM measurement and model’s prediction.
The formation of tree domains predicted by our model allows the functional modules of chromatin to have topological identities (a robust way to store information). However, these topological domains cannot be self-assembled without energy input (note that SRRW as a folding algorithm is not a memory-less Markov process and cannot happen at thermodynamic equilibrium). Nevertheless, a tree-like topology of chromatin segment is not totally unexpected because looping and supercoiling can effectively branch the biopolymer. Given the structural complexity and heterogeneity of tree domains predicted by SRRW, we expect the folding process to involve a diversity of molecular mechanisms to act in concert under the guidance of genetic (52) and epigenetic (53) codes. These could include transcription-induced supercoiling (12–15), phase separation (16–18), long noncoding RNA (54), DNA-mediated charge transfer (55), and the putative cohesive-CTCF loop extrusion (9). In particular, DNA-mediated charge transfer could enable fast long-range communication between responsible architectural proteins in a transcription-sensitive manner, whereas long noncoding RNA could allow slow but more specific loci communication and recruitment of folding agents. Given the hierarchical nature of the tree topology, we expect both the assembly and disassembly of the tree domains to be hierarchical processes, meaning that small tree domains can group into large ones where they become subdomains and vice versa similar to chemical reactions. During these grouping and ungrouping processes, backbone segments can be absorbed into new tree domains, and new backbone segments can be exposed. For a steady state when the cell is not preparing for division, we expect the overall statistics (Fig. 4), i.e., the tree size distribution, the degree of structural hierarchy, and the portion of backbone segments, to be stable. Similar to the inheritance of noncoding DNA (>90% of genome), maintaining physically extended backbone segments (~5% of genome) and their low DNA density local environment inside the highly crowded nucleus may seem uneconomical, because energy needs to be consumed against the conformational entropy of chromatin that tends to homogenize the DNA concentration. This puzzle could be explained by recent experimental finding that droplet of nuclear proteins initialized by DNA binding mechanically dispels heterochromatin and promotes the formation of low DNA density region (17). This picture is consistent with another recent observation that the total euchromatin density including proteins is only 1.5-fold lower than that of the surrounding heterochromatin, despite the 5.5- to 7.5-fold DNA density difference (56). Together, the formation of both tree domains and chromatin backbone in vivo is not only functionally desirable but also mechanistically possible.
The fact that many major chromatin features can be explained by an abstract folding algorithm indicates the existence of universal principles for chromatin to functionally fold and efficiently explore the genomic landscape. Our results suggest a global coupling between different chromatin properties including domain hierarchy, nanocluster size distribution, backbone openness, packing heterogeneity, genomic interaction, and transcriptional polarization, which means a substantial dimensionality reduction during chromatin folding due to the emergence of collective folding parameters or susceptibilities (for example, α in SRRW). This folding picture sheds new lights into the physics of epigenetics and stresses the importance of understanding 3D genome from a data structure point of view (Fig. 1F) on top of polymer physics, because chromatin folding is optimizing biological functions rather than minimizing thermodynamic free energy. The unexpected positive correlation between genomic contact scaling and DNA packing heterogeneity of living chromatin under heat shock is one example.
In summary, we have combined theoretical and experimental efforts to understand chromatin topology, statistics, scaling, and their couplings at the single-cell level. Our multiscale results, from kilobase-level nanoclusters to 100 Mb–level chromosome territories, provide an integrative view of higher-order chromatin folding as an alternative thinking to the classical 30-nm fiber–based picture. Our prediction that chromatin folds into tree-like topological domains connected by an active backbone explains and reconciles a wealth of exotic properties of this living biopolymer that are alien to the common sense of polymer physics. It is remarkable that despite the distinct folding modes at different scales, chromatin is able to maintain alternation between active and inactive states across many scales both in space and on DNA sequence, in a quasi–self-similar manner. The non-Gaussian folding statistics and global coupling between chromatin properties suggest that interphase DNA explores the great genomic landscape as a complex network rather than a simple polymer. The possibility of chromatin having tree data structures and universal folding principles opens an exciting new paradigm to understand genomic organization and presents many new questions, the answering of which would require collaborations between experimentalists and theorists from different fields. We hope that our insights in this paper could inspire future interdisciplinary efforts on this grand challenge of life science.
MATERIALS AND METHODS
Model
The folding algorithm of SRRW is given in the main text. Here, we summarized some parameters we used in the model and data analysis. The smallest step size in the model is 30 nm. For local DNA density analysis, we used a probe of 150-nm radius. For contact frequency calculation, we chose a contact criterion to be two loci being within 45 nm. The single-cell level modeling and analyses were performed at α = 1.15. The local and global cutoffs in this case are 24.85 and 57.15 in reduced units. The local cutoff was chosen to cover the length of a fully stretched 2-kb DNA double helix. The global cutoff was chosen to render a reasonable size of chromosome territory with an average DNA density that is comparable to that of diploid human nucleus. Population-level modeling and analyses were performed over 1000 independent samples.
Cell culture
Human lung carcinoma cells (A549) and human skin fibroblast (BJ) cells were purchased from the American Type Culture Collection (ATCC). A549 cells were cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific). BJ cells were cultured in minimum essential media (Thermo Fisher Scientific). All culture media were supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) and penicillin-streptomycin (100 μg/ml; Thermo Fisher Scientific). Ovarian (A2780 and A2780.m248) cell lines were a gift from C.-P. H. Yang and obtained from the laboratory of E. de Vries at Albert Einstein College of Medicine. A2780 and A2780.m248 cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific). All culture media were supplemented with 10% FBS (Thermo Fisher Scientific). HCT116 cells were purchased from ATCC and grown to confluence in McCoy’s 5A medium (Lonza) supplemented with 10% FBS and 1× penicillin-streptomycin at 37°C. All cells were cultured on 35-mm MatTek dishes (MatTek Corp.) in 5% CO2 and at the physiological oxygen level, and all experiments were performed on cells from passage 5 to passage 20.
ChromEM staining with STEM tomography and TEM imaging
ChromEM staining was used to label the DNA in the A549 cell and BJ cell nucleus, as previously described (3). The cells were fixed in 2.5% EM grade glutaraldehyde and 2% paraformaldehyde (EMS, USA) in 1× sodium cacodylate buffer for 20 min at room temperature and 1 hour on ice. All the following steps were conducted on ice or a customized cold stage with temperature control, and the reagents used in the protocol were prechilled in the fridge or on ice. After fixation, the cells were stained with DRAQ5 (Thermo Fisher Scientific, USA) that intercalates double-stranded DNA and bathed in 3,3′-diaminobenzidine solution (Sigma-Aldrich, USA). An inverted microscopy (Eclipse, Nikon, Japan) was used for photobleaching. Each spot was photobleached for 7 min by epi-illumination with a 100× objective. Osmium ferrocyanide was used to further enhance contrast of the DNA, and standard Durcupan resin (EMS, USA) embedding was carried out. After curing for 48 hours at 60°C, ultrathin sections were prepared by an ultramicrotome (UC7, Leica, USA) and deposited onto plasma-treated TEM grid with formar-carbon film (EMS, USA). To investigate the CVC (chromatin volume concentration) and radius of the chromatin nanodomains, dual-axis STEM high-angle annular dark-field tomography was performed on the 100-nm ultrathin section of A549 cell nucleus with 10-nm colloidal gold fiducial markers. The sample was tilted from −60° to 60° with 2° step along the two perpendicular axes. IMOD (57) was used to align the tilt series, and the tomography reconstruction was conducted in TomoPy (58) using a penalized maximum likelihood algorithm (PLM-hybrid) for each axis. The combination of tomograms was performed using IMOD to further suppress the influence of missing cone. Furthermore, to estimate the average cluster size for the whole nucleus across multiple cells experimentally, mass scaling analysis of TEM projection images on ultrathin sections of BJ cells was performed. The TEM contrast was converted to mass thickness by the Beer’s law before the mass scaling analysis. For more details, see section S6.
Live-cell PWS microscopy
Live-cell PWS microscopy measurements were performed as previously (4). Cells were imaged using a broad-spectrum light-emitting diode with illumination focused onto the sample plane using a 63× or 100× objective as indicated. Backscattered light was collected with spectral filtration performed using a liquid-crystal tunable filter (CRi, Woburn, MA; spectral resolution, 7 nm), collecting sequential monochromatic images between 500 and 700 nm. These monochromatic images were projected onto a CMOS (complementary metal-oxide semiconductor) camera (ORCA-Flash 4.0 v2, Hamamatsu City, Japan) producing a 3D image cube. Variations in the backscattered intensity were analyzed at each pixel and used to calculate mass fractal dimension D.
Heat shock experiments
Cells were first grown to confluence and then imaged with PWS microscopy. Briefly, cells were seeded onto size-0 glass-bottom 35-mm dishes at least 48 hours before imaging at a concentration of 50,000 cells/ml and cultured at 37°C with 21% O2 and 5% CO2. Cells incubated at 37°C were treated as controls. For heat shock, each dish was imaged using PWS microscopy, incubated at 42°C for 1 hour, and then immediately imaged afterward. All the experiments were blinded.
Hi-C analysis
Analysis was conducted on publicly available Hi-C data on hESCs (GEO accession code GSE105028) before and after heat shock exposure (46). The Juicer analysis tool was used to perform read alignment to the hg38 human reference genome, read pairing, and deduplication for each Hi-C replicate (59). Reads with a low mapping quality score [MAPQ (mapping quality score) < 30] were removed. Reads across replicates for each condition were merged. Matrix normalization was performed to normalize to a target coverage vector using Juicebox, which was used for visualization (60). Hi-C maps were plotted with 250-kb resolution. Contact probability was calculated from the merged Hi-C contact maps for each condition with a 5-kb resolution, and a linear regression fit to contact probability versus genomic distance (in base pairs) was performed to calculate contact probability scaling for long-range contacts.
Supplementary Material
Acknowledgments
We thank members of the Szleifer and Backman laboratories for their insights and comments. We thank Y. Q. Gao for useful discussion. Funding: We acknowledge funding from the NSF (Biol & Envir Inter of Nano Mat 1833214, EFRI research project 1830961) and the NIH (National Cancer Institute R01 CA228272 and R01 CA225002). This research was supported, in part, through the computational resources and staff contributions provided by the Genomics Compute Cluster, which is jointly supported by the Feinberg School of Medicine, the Center for Genetic Medicine, and Feinberg’s Department of Biochemistry and Molecular Genetics, the Office of the Provost, the Office for Research, and Northwestern Information Technology. The Genomics Compute Cluster is part of Quest, Northwestern University’s high-performance computing facility, with the purpose to advance research in genomics. Author contributions: K.H., V.B., and I.S. conceived the project. K.H. developed the theory and model. Y.L. performed the ChromSTEM experiments and analyzed the data. A.R.S. and R.J.N. participated in the modeling. V.A. and A.E. performed PWS experiments and analyzed the data. R.K.A.V. analyzed the publicly available Hi-C data on heat shock. K.H. wrote the original draft, and all authors reviewed and edited the manuscript. V.B. and I.S. secured the funding and supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaay4055/DC1
Section S1. Contact scaling of random fractal chain
Section S2. Granular and porous packing of SRRW
Section S3. Tree domains and TADs
Section S4. Non-Gaussian statistics
Section S5. Effect of α on the statistics of tree domains
Section S6. Chromatin scanning transmission electron microscopy
Section S7. Heat shock experiments across multiple cell lines
Section S8. Histone modification affects fractal dimension D
Fig. S1. Contact probabilities of RWs and LFs in 2D and 3D space.
Fig. S2. Comparing packing cross sections of confined SRRW and RW.
Fig. S3. Structure of local SRRW segment and ensemble-averaged contact map.
Fig. S4. End-to-end distance distributions of 100-kb SRRW segment (green) and of 100-kb RW segment (red).
Fig. S5. Effect of α on the tree domains.
Fig. S6. D and s from the modeled chromatin at varying α.
Fig. S7. ChromEM sample preparation and STEM imaging on A549 cells.
Fig. S8. ChromEM sample preparation and TEM imaging on BJ cells.
Fig. S9. Heat shock increases fractal dimension D across multiple cell lines.
Fig. S10. Histone deacetylase inhibitor valproic acid decreases fractal dimension D.
REFERENCES AND NOTES
- 1.Finch J., Klug A., Solenoidal model for superstructure in chromatin. Proc. Natl. Acad. Sci. U.S.A. 73, 1897–1901 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeshima K., Ide S., Babokhov M., Dynamic chromatin organization without the 30-Nm fiber. Curr. Opin. Cell Biol. 58, 95–104 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Ou H. D., Phan S., Deerinck T. J., Thor A., Ellisman M. H., O’Shea C. C., ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357, eaag0025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almassalha L. M., Bauer G. M., Chandler J. E., Gladstein S., Cherkezyan L., Stypula-Cyrus Y., Weinberg S., Zhang D., Ruhoff P. T., Roy H. K., Subramanian H., Chandel N. S., Szleifer I., Backman V., Label-free imaging of the native, living cellular nanoarchitecture using partial-wave spectroscopic microscopy. Proc. Natl. Acad. Sci. U.S.A. 113, E6372–E6381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almassalha L. M., Bauer G. M., Wu W., Cherkezyan L., Zhang D., Kendra A., Gladstein S., Chandler J. E., VanDerway D., Seagle B.-L. L., Ugolkov A., Billadeau D. D., O’Halloran T. V., Mazar A. P., Roy H. K., Szleifer I., Shahabi S., Backman V., Macrogenomic engineering via modulation of the scaling of chromatin packing density. Nat. Biomed. Eng. 1, 902–913 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O., Sandstrom R., Bernstein B., Bender M. A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L. A., Lander E. S., Dekker J., Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dekker J., Heard E., Structural and functional diversity of topologically associating domains. FEBS Lett. 589, 2877–2884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanborn A. L., Rao S. S. P., Huang S.-C., Durand N. C., Huntley M. H., Jewett A. I., Bochkov I. D., Chinnappan D., Cutkosky A., Li J., Geeting K. P., Gnirke A., Melnikov A., McKenna D., Stamenova E. K., Lander E. S., Aiden E. L., Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 112, E6456–E6465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L. A., Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bintu B., Mateo L. J., Su J.-H., Sinnott-Armstrong N. A., Parker M., Kinrot S., Yamaya K., Boettiger A. N., Zhuang X., Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabo Q., Jost D., Chang J.-M., Cattoni D. I., Papadopoulos G. L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M., Bantignies F., Cavalli G., TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 4, eaar8082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kouzine F., Gupta A., Baranello L., Wojtowicz D., Ben-Aissa K., Liu J., Przytycka T. M., Levens D., Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 20, 396–403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naughton C., Avlonitis N., Corless S., Prendergast J. G., Mati I. K., Eijk P. P., Cockroft S. L., Bradley M., Ylstra B., Gilbert N., Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 20, 387–395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corless S., Gilbert N., Effects of DNA supercoiling on chromatin architecture. Biophys. Rev. 8, 245–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S. H., Ganji M., Kim E., van der Torre J., Abbondanzieri E., Dekker C., DNA sequence encodes the position of DNA supercoils. eLife 7, e36557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabari B. R., Dall’Agnese A., Boija A., Klein I. A., Coffey E. L., Shrinivas K., Abraham B. J., Hannett N. M., Zamudio A. V., Manteiga J. C., Li C. H., Guo Y. E., Day D. S., Schuijers J., Vasile E., Malik S., Hnisz D., Lee T. I., Cisse I. I., Roeder R. G., Sharp P. A., Chakraborty A. K., Young R. A., Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin Y., Chang Y.-C., Lee D. S. W., Berry J., Sanders D. W., Ronceray P., Wingreen N. S., Haataja M., Brangwynne C. P., Liquid nuclear condensates mechanically sense and restructure the genome. Cell 175, 1481–1491.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Pierro M., Cheng R. R., Aiden E. L., Wolynes P. G., Onuchic J. N., De Novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc. Natl. Acad. Sci. U.S.A. 114, 12126–12131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianco S., Chiariello A. M., Annunziatella C., Esposito A., Nicodemi M., Predicting chromatin architecture from models of polymer physics. Chromosome Res. 25, 25–34 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Kim J. S., Backman V., Szleifer I., Crowding-induced structural alterations of random-loop chromosome model. Phys. Rev. Lett. 106, 168102 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Rao S. S. P., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A. L., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricci M. A., Manzo C., García-Parajo M. F., Lakadamyali M., Cosma M. P., Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M. T., Wendt K. S., Okada Y., Nagai T., Maeshima K., Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Schwartz U., Németh A., Diermeier S., Exler J. H., Hansch S., Maldonado R., Heizinger L., Merkl R., Längst G., Characterizing the nuclease accessibility of DNA in human cells to map higher order structures of chromatin. Nucleic Acids Res. 47, 1239–1254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grosberg A., Rabin Y., Havlin S., Neer A., Crumpled globule model of the three-dimensional structure of DNA. Europhys. Lett. 23, 373–378 (1993). [Google Scholar]
- 26.Mirny L. A., The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 19, 37–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebedev D. V., Filatov M. V., Kuklin A. I., Islamov A. K., Kentzinger E., Pantina R., Toperverg B. P., Isaev-Ivanov V. V., Fractal nature of chromatin organization in interphase chicken erythrocyte nuclei: DNA structure exhibits biphasic fractal properties. FEBS Lett. 579, 1465–1468 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Joti Y., Hikima T., Nishino Y., Kamada F., Hihara S., Takata H., Ishikawa T., Maeshima K., Chromosomes without a 30-Nm chromatin fiber. Nucleus 3, 404–410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A., Field Y., Moore I. K., Wang J.-P. Z., Widom J., A genomic code for nucleosome positioning. Nature 442, 772–778 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens T. J., Lando D., Basu S., Atkinson L. P., Cao Y., Lee S. F., Leeb M., Wohlfahrt K. J., Boucher W., O’Shaughnessy-Kirwan A., Cramard J., Faure A. J., Ralser M., Blanco E., Morey L., Sansó M., Palayret M. G. S., Lehner B., di Croce L., Wutz A., Hendrich B., Klenerman D., Laue E. D., 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cremer T., Cremer M., Dietzel S., Müller S., Solovei I., Fakan S., Chromosome territories–A functional nuclear landscape. Curr. Opin. Cell Biol. 18, 307–316 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Sutherland H., Bickmore W. A., Transcription factories: Gene expression in unions? Nat. Rev. Genet. 10, 457–466 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Bancaud A., Huet S., Daigle N., Mozziconacci J., Beaudouin J., Ellenberg J., Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 28, 3785–3798 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagano T., Lubling Y., Stevens T. J., Schoenfelder S., Yaffe E., Dean W., Laue E. D., Tanay A., Fraser P., Single-cell Hi-c reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan L., Xing D., Chang C.-H., Li H., Xie X. S., Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S., Su J.-H., Beliveau B. J., Bintu B., Moffitt J. R., Wu C., Zhuang X., Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen A. S., Cattoglio C., Darzacq X., Tjian R., Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dekker J., Mirny L., The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ulianov S. V., Khrameeva E. E., Gavrilov A. A., Flyamer I. M., Kos P., Mikhaleva E. A., Penin A. A., Logacheva M. D., Imakaev M. V., Chertovich A., Gelfand M. S., Shevelyov Y. Y., Razin S. V., Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 26, 70–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser J., Ferrai C., Chiariello A. M., Schueler M., Rito T., Laudanno G., Barbieri M., Moore B. L., Kraemer D. C., Aitken S., Xie S. Q., Morris K. J., Itoh M., Kawaji H., Jaeger I., Hayashizaki Y., Carninci P., Forrest A. R. R.; The FANTOM Consortium, Semple C. A., Dostie J., Pombo A., Nicodemi M., Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol. Syst. Biol. 11, 852 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N. A., Huber W., Haering C., Mirny L., Spitz F., Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramírez F., Bhardwaj V., Arrigoni L., Lam K. C., Grüning B. A., Villaveces J., Habermann B., Akhtar A., Manke T., High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q., Sun Q., Czajkowsky D. M., Shao Z., Sub-kb Hi-C in D. Melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun. 9, 188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J., Ma H., Jin J., Uttam S., Fu R., Huang Y., Liu Y., Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 24, 873–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cremer T., Cremer M., Hübner B., Strickfaden H., Smeets D., Popken J., Sterr M., Markaki Y., Rippe K., Cremer C., The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 589, 2931–2943 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Lyu X., Rowley M. J., Corces V. G., Architectural proteins and pluripotency factors cooperate to orchestrate the transcriptional response of HESCs to temperature stress. Mol. Cell 71, 940–955.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L., Lyu X., Hou C., Takenaka N., Nguyen H. Q., Ong C.-T., Cubeñas-Potts C., Hu M., Lei E. P., Bosco G., Qin Z. S., Corces V. G., Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol. Cell 58, 216–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sachs R. K., van den Engh G., Trask B., Yokota H., Hearst J. E., A random-walk/giant-loop model for interphase chromosomes. Proc. Natl. Acad. Sci. U.S.A. 92, 2710–2714 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ganji M., Shaltiel I. A., Bisht S., Kim E., Kalichava A., Haering C. H., Dekker C., Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alipour E., Marko J. F., Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 40, 11202–11212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banigan E. J., Mirny L. A., Limits of chromosome compaction by loop-extruding motors. Phys. Rev. X 9, 031007 (2018). [Google Scholar]
- 52.Liu S., Zhang L., Quan H., Hao T., Meng L., Yang L., Feng H., Gao Y. Q., From 1D sequence to 3D chromatin dynamics and cellular functions: A phase separation perspective. Nucleic Acids Res. 46, 9367–9383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Pierro M., Zhang B., Aiden E. L., Wolynes P. G., Onuchic J. N., Transferable model for chromosome architecture. Proc. Natl. Acad. Sci. U.S.A. 113, 12168–12173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang K. C., Yang Y. W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B. R., Protacio A., Flynn R. A., Gupta R. A., Wysocka J., Lei M., Dekker J., Helms J. A., Chang H. Y., A long noncoding rna maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Genereux J. C., Boal A. K., Barton J. K., DNA-mediated charge transport in redox sensing and signaling. J. Am. Chem. Soc. 132, 891–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Imai R., Nozaki T., Tani T., Kaizu K., Hibino K., Ide S., Tamura S., Takahashi K., Shribak M., Maeshima K., Density imaging of heterochromatin in live cells using orientation-independent-DIC microscopy. Mol. Biol. Cell 28, 3349–3359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Gürsoy D., De Carlo F., Xiao X., Jacobsen C., TomoPy: A framework for the analysis of synchrotron tomographic data. J. Synchrotron Radiat. 21, 1188–1193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Durand N. C., Shamim M. S., Machol I., Rao S. S. P., Huntley M. H., Lander E. S., Aiden E. L., Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durand N. C., Robinson J. T., Shamim M. S., Machol I., Mesirov J. P., Lander E. S., Aiden E. L., Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/2/eaay4055/DC1
Section S1. Contact scaling of random fractal chain
Section S2. Granular and porous packing of SRRW
Section S3. Tree domains and TADs
Section S4. Non-Gaussian statistics
Section S5. Effect of α on the statistics of tree domains
Section S6. Chromatin scanning transmission electron microscopy
Section S7. Heat shock experiments across multiple cell lines
Section S8. Histone modification affects fractal dimension D
Fig. S1. Contact probabilities of RWs and LFs in 2D and 3D space.
Fig. S2. Comparing packing cross sections of confined SRRW and RW.
Fig. S3. Structure of local SRRW segment and ensemble-averaged contact map.
Fig. S4. End-to-end distance distributions of 100-kb SRRW segment (green) and of 100-kb RW segment (red).
Fig. S5. Effect of α on the tree domains.
Fig. S6. D and s from the modeled chromatin at varying α.
Fig. S7. ChromEM sample preparation and STEM imaging on A549 cells.
Fig. S8. ChromEM sample preparation and TEM imaging on BJ cells.
Fig. S9. Heat shock increases fractal dimension D across multiple cell lines.
Fig. S10. Histone deacetylase inhibitor valproic acid decreases fractal dimension D.