

Summary:
Ribosome assembly is an efficient, but complex and heterogeneous process during which ribosomal proteins assemble on the nascent ribosomal RNA (rRNA) during transcription. Understanding how the interplay between nascent RNA folding and protein binding determines the fate of transcripts remains a major challenge. Here, using single-molecule fluorescence-microscopy, we follow assembly of the entire 3’domain of the bacterial small ribosomal subunit in real-time. We find that co-transcriptional rRNA folding is complicated by the formation of long-range RNA interactions, and that r-proteins self-chaperone the rRNA folding process prior to stable incorporation into a ribonucleoprotein complex (RNP). Assembly is initiated by transient rather than stable protein binding, and the protein-RNA binding dynamics gradually decrease during assembly. This work questions the paradigm of strictly sequential and cooperative ribosome assembly and suggests that transient binding of RNA binding proteins to cellular RNAs could provide a general mechanism to shape nascent RNA folding during RNP assembly.
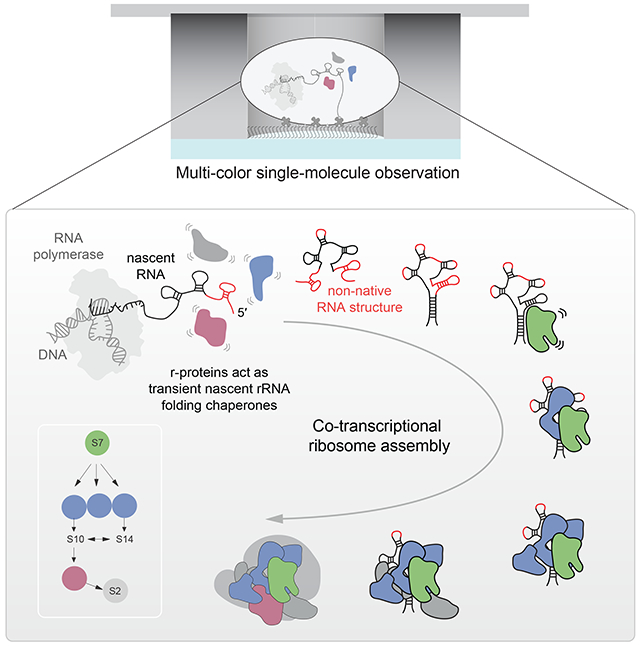
Graphical abstract
INTRODUCTION
Assembly of protein-RNA (RiboNucleoProtein; RNP) complexes is fundamental to all life forms, underlying transcription, translation and splicing, as well as the function of long noncoding RNAs. The formation of an RNP complex involves synthesis and correct folding of the individual protein and nucleic acid components, and the assembly of all the individual parts into a functional complex. In many cases, the choice of alternative RNA structures is guided by the interaction with specific proteins, thereby determining the fate of the RNA as an RNP (Ganser et al., 2019; Hentze et al., 2018). For example, specific RNA structural elements located in the 5’ untranslated region of mRNAs are modulated by the binding to RNA binding proteins and these interactions determine the translation efficiency of those mRNAs (Leppek et al., 2018). A major barrier to understanding RNP complex assembly is the challenge of tracking RNA folding and how this is correlated to protein binding. Furthermore, the bulk of knowledge is based on in vitro studies focusing on the interactions between pre-formed RNA with protein molecules, whereas in cells many such complexes form on the nascent RNA while it is emerging from the RNA polymerase (RNAP).
The best-studied noncoding RNP is the ribosome, which is the large and complex macromolecular machine that synthesizes cellular proteins. Assembly of the bacterial ribosome, which is composed of a small (30S) and large (50S) ribosomal subunit, is initiated by the synthesis of a ~4.5 kb long transcript. During transcription, the ribosomal RNA (rRNA) folds into secondary and tertiary structures, is modified at specific positions, binds with > 50 different ribosomal proteins (r-proteins), and is processed into the two ribosomal subunits (Shajani et al., 2011; Sykes and Williamson, 2009; Woodson, 2008, 2011). Because all those processes are coupled, this complicates studying ribosome assembly at the molecular level and requires methods that can directly and simultaneously track such processes in real-time.
The r-proteins are stably associated with the rRNA in mature ribosomes, with binding lifetimes of hours for the majority of the r-proteins (Robertson et al., 1977; Subramanian and van Duin, 1977). Early in vitro ribosome reconstitution experiments demonstrated that assembly is sequential and cooperative (Held et al., 1974; Shajani et al., 2011; Sykes and Williamson, 2009; Traub and Nomura, 1969; Woodson, 2008, 2011). Primary r-proteins bind directly to the rRNA and are required for the stable association of the secondary and tertiary binding proteins. The canonical model for binding cooperativity and stepwise assembly involves the binding of primary r-proteins that fold the rRNA in order to create the binding sites for the secondary r-proteins, thereby increasing their binding affinity and driving assembly forward. Despite sequential and cooperative assembly branches, primary r-proteins nucleate assembly at different positions along the rRNA (Adilakshmi et al., 2008) and assembly follows multiple parallel pathways (Adilakshmi et al., 2008; Davis et al., 2016; Mulder et al., 2010) that further complicates dissecting the assembly mechanism.
In vitro reconstitutions of 30S subunits from native 16S rRNA and r-proteins purified from cells demonstrate that assembly is 1-2 orders of magnitude slower than in vivo (Lindahl, 1975; Powers et al., 1993; Talkington et al., 2005), likely due to rRNA misfolding and slow refolding (Adilakshmi et al., 2008; Bunner et al., 2010; Powers et al., 1993). It has been postulated that vectorial (5’ to 3’) co-transcriptional RNA folding during in vivo assembly could be a key contributor to resolving such rRNA folding problems (Boyle et al., 1980; Heilman-Miller and Woodson, 2003; Incarnato et al., 2017; Isambert, 2009; Nussinov and Tinoco, 1981; Pan and Sosnick, 2006). For example, coupling transcription with protein binding is crucial for efficient assembly in vivo (de Narvaez and Schaup, 1979; Lewicki et al., 1993) and in vitro co-transcriptional assembly in the presence of whole-cell extract works efficiently under physiological conditions, (Jewett et al., 2013) in contrast to assembly from pre-synthesized components (Traub and Nomura, 1969). Yet, how transcription affects nascent rRNA folding and ribosome assembly and what is the nature for rRNA misfolding remain elusive.
We have recently developed (Duss et al., 2018) a single-molecule approach using zero-mode waveguide technology (Chen et al., 2014; Levene et al., 2003; Uemura et al., 2010; Zhu and Craighead, 2012) that directly tracks transcription and the binding of a protein to a nascent RNA transcript. Here, we extend this approach by tracking additionally the real-time folding of the nascent RNA. We focus on the assembly of the 3’domain of the E. coli 30S ribosomal subunit, which is initiated by the primary r-protein S7. Because the 3’-domain folds the slowest during in vitro reconstitutions (Adilakshmi et al., 2008; Soper et al., 2013; Talkington et al., 2005), this provides an ideal system to study nascent RNA folding and the influence of r-proteins on guiding RNA folding and assembly. Simultaneously and directly tracking transcription, nascent rRNA folding and assembly of the r-proteins on the nascent RNA transcript, we observe in real-time the assembly of the entire 3’-domain consisting of 8 r-proteins. We show that in presence of only primary r-protein S7, nascent RNA folding is very inefficient and S7 only binds transiently, demonstrating that vectorial rRNA synthesis alone does not solve the rRNA folding problem. We delineate the molecular basis for nascent rRNA misfolding by showing that proper formation of long-range helix 28 (H28) is a major barrier for initiating 3’domain assembly. In contrast to the current view of ribosome assembly whereby stable binding of primary r-proteins initiate assembly by reshaping the rRNA in order to create the binding site for the subsequent r-protein to drive assembly (Abeysirigunawardena et al., 2017; Ha et al., 1999; Kim et al., 2014; Orr et al., 1998; Ramaswamy and Woodson, 2009; Recht and Williamson, 2001), we find that later binding r-proteins also chaperone rRNA folding early in assembly before their stable incorporation and that their binding gradually decreases the protein-RNA binding dynamics during assembly. Overall, our findings challenge the paradigm of strictly sequential and cooperative ribosome assembly and provide an alternative role for integral RNA binding proteins acting as semi-specific RNA folding chaperones in cellular ribonucleoprotein (RNP) assembly.
RESULTS
Real-time binding of S7 to nascent 3’domain RNA
The 3’domain of the 16S rRNA is connected by its 5’ and 3’ termini to the rest of the 30S submit and forms the structurally isolated head of the 30S subunit (Figure 1A and 1B). Furthermore, the 3’domain can assemble independently in vitro forming a compact particle very similar in shape to the head domain of the 30S ribosomal subunit (Samaha et al., 1994). Those findings indicate that assembly of the isolated 3’-domain is a good model for assembly of the entire 30S subunit (Bunner et al., 2010; Kitahara and Suzuki, 2009). To reduce complexity further, we first investigated the binding dynamics of primary r-protein S7, which is the first protein to associate with the 3’domain during sequential 3’domain assembly in absence of the other 3’domain r-proteins (Held et al., 1974; Traub and Nomura, 1969). S7 binds simultaneously to a 4-way junction (4WJ) and a 3WJ, which are both connected by helix 29 (H29) and flanked by H30 and H28, respectively (Figures 1A and 1B). H28 is formed by the 5’- and 3’-termini of the 3’domain, connecting the 3’domain to the rest of the 30S ribosomal subunit. To observe real-time encounters of S7 with the nascent RNA, we applied our recently developed single-molecule approach (Figures 1C and 1D) (Duss et al., 2018). In short, we first immobilize single stalled transcription-elongation complexes through the nascent RNA to the surface of zero-mode waveguides (ZMWs) (Figure 1C). Then, co-transcriptional assembly is initiated by delivering a mixture containing all four NTPs, Cy5-labeled S7 and a Cy3-labeled DNA oligonucleotide that can specifically hybridize to either terminus of the nascent 3’domain RNA. The Cy3-labeled oligonucleotide was positioned to allow significant fluorescence energy transfer (FRET) to Cy5-S7 when bound at or near its specific binding site (Figures 1B and 1D), allowing discrimination of specific binding from non-specific surface absorption (Duss et al., 2018). Transcription elongation is visualized by labeling the DNA transcription template with two Cy3.5 dyes at the 3’-end resulting in a fluorescence intensity increase during transcription elongation due to movement of the dyes closer to the surface of the ZMWs (Figure 1C).
Figure 1. Real-time S7 encounters with nascent RNA.

(A) Secondary structure of E. coli S7 binding site. (B) 3-dimensional structure of E. coli 30S ribosomal subunit with labeling sites (PDB accession code: 4V9P); the Cy3 dye of the labeled DNA oligonucleotide binding to the 3’-end of the nascent RNA is modeled into the structure. (C and D) Single-molecule fluorescence approach (Duss et al., 2018). (E) Representative single-molecule trace with fluorescence intensity (top) and FRET efficiency (bottom); transcription (TC; orange), Cy5-S7 (red), Cy3-oligo (green). See also Figure S1.
To detect S7 binding during transcription, we used FRET to a Cy3-oligo that binds to the 5’-end of the nascent RNA. We did not observe S7 binding during transcription (Figure S1A) but binding occurs only post-transcriptionally to the nascent RNA. Unexpectedly for a primary binding protein, we see either no binding or a very dynamic S7 binding, with bursts of transient S7 binding interrupted by periods of tens of seconds to minutes that are devoid of S7 binding (Figures 1E and S1B). The distribution of S7 binding-event durations allows determination of S7 dissociation rates, koff,1 = (4.6 ± 0.4) x 10−2 s−1 (25 %) and koff,2 = 0.79 ± 0.03 s−1 (75 %) (Figure S1C), corresponding to two average S7-bound lifetime phases of ~20 s and ~1 s, respectively at 20 °C. We performed experiments at different S7 protein concentrations and found a fast concentration-dependent association rate of kon = (1.1 ± 0.3) x 108 M−1 s−1 (Figure S1D). We ascribe this fast rate to encounter complexes in agreement with previous findings of in vitro assembly of pre-transcribed 16S rRNA using hydroxyl radical foot-printing (Adilakshmi et al., 2008). In addition, we see a slow association rate phase that is not concentration-dependent, kon = 0.032 ± 0.019 s−1, suggesting that the nascent RNA is transitioning between S7 binding competent and incompetent conformations on a timescale of ~30 s.
Inefficient and slow 3’domain rRNA folding
We clustered all the single-molecule traces, each representing a different single transcribing RNA molecule, according to S7 binding behavior (Figure S2A and STAR Methods). We assigned RNA molecules based on their longest S7-bound lifetime per trace to either folded or misfolded states with a threshold of 2 s to separate the two populations (Figure S2B and STAR Methods). At 20 nM Cy5-S7, only ~30 % of the nascent full-length 3′-domain RNA molecules are able to bind S7 at least once for >2 s at 20 °C (Figure 2A) and the fraction of S7 binding competent RNA molecules does not significantly increase using 200 nM Cy5-S7 (Figure S2C). The other RNA molecules are either incompetent for S7 binding or show only short-lived S7 encounters with a dissociation rate of 2.3 ± 0.6 s−1. By increasing the temperature to 35 °C or pre-transcribing and pre-folding the RNA at 37 °C prior to protein binding, the fraction of RNA molecules that can bind S7 for >2 s increases to ~50 % (Figure 2A). This demonstrates that some kinetic RNA folding traps can be overcome by thermal energy in agreement with cold-sensitive ribosome assembly defects observed in vivo attributed to stable RNA misfolding (Kaczanowska and Ryden-Aulin, 2007; Shajani et al., 2011; Soper et al., 2013; Woodson, 2008). Having shown that S7 binding occurs after completion of transcription, we next determined when S7 stably associates with the nascent rRNA after transcription has completed. On average, we detect the first >2 s S7 binding event ~1-2 minutes after full transcription but ~40 % of the nascent RNA molecules require >5 minutes for engaging in S7 binding (Figure 2B).
Figure 2. Inefficient and slow nascent RNA folding.

(A and B) Nascent 3’domain rRNA folding is inefficient (A) and slow (B) in absence of other r-proteins. In (B), the time from full transcription till appearance of the first r-protein binding event with a duration of >2 s at 20 °C is represented. (C) Smaller nascent RNAs fold more efficiently. Δ147 stands for the truncation of 147 nt compared to the full-length 3’domain construct and other constructs are named accordingly. Concentrations: 20 nM Cy5-S7 or 25 nM Cy5-S15. Data with S15 in (A and B) is from reference (Duss et al., 2018). Number of molecules analyzed (n) = 172, 138, 64, 144, 92 (A), (n) = 46, 40 (B) and (n) = 138, 86, 119, 89 (C). See also Figure S2 and Data S2.
In comparison, for the central domain of the 16S rRNA nucleated by protein S15, ~80 % of the nascent RNA molecules stably engage with S15 at 35 °C in absence of other r-proteins (Duss et al., 2018), and stable association of S15 occurs within <10 s for >60 % of the nascent RNA molecules (Duss et al., 2018). The binding site for S15 consists of a simple 3-way junction, while the binding site for S7 is much more complex (Figure 1A). This may be one of the reasons why both the folding rate (Figure 2B) and efficiency (Figure 2A) of the 3’domain are much lower compared to the central domain rRNA. These results demonstrate that 3’domain nascent rRNA folding is slow and inefficient in the absence of other 3’domain r-proteins and that directional rRNA synthesis alone does not solve the problem of 3’domain rRNA misfolding.
Molecular basis for nascent RNA misfolding
A large portion of the S7 binding site consists of helices formed by long-range base-pairing interactions. For helices H28 and H29, the 5′- and 3′-strands forming those helices are separated by more than 460 or 400 nucleotides in primary sequence, respectively (Figure 1A). Thus, with an average transcription rate of ~20-25 nt s−1 at saturating 500 μM NTP concentrations at 20 °C (Figure S2E), in agreement with in vivo transcription rates (Ryals et al., 1982), the 5’-halves of H28 and H29 are emerging from the RNAP exit tunnel ~20 s before their complementary 3’-halves are completed. During this time, the 5′-halves of those helices could participate in non-native interactions, thereby trapping the RNA into stable non-native structures. To test this hypothesis, we deleted different internal regions of the 3’-domain RNA that are not part of the S7 binding site (Dragon and Brakier-Gingras, 1993; Dragon et al., 1994), thus shortening the number of nucleotides between the 5’ and 3’ ends of the transcript. Successive removal of RNA results in a steady increase in the fraction of nascent RNA molecules that can bind S7 for >2 s (Figure 2C) and an increased fraction of long-lived S7 binding events (Figure 4G). These findings are in agreement with a decreased probability for stable non-native structure formation and a shorter time window during which the nascent 5’-half of H28 is accessible for mispairing before its complementary 3’-half is transcribed to allow H28 formation. In support of this model, we observe decreasing RNA folding efficiency with decreasing transcription rate (Figures S2F–S2H).
Figure 4. 3’-proteins chaperone and stabilize rRNA.

(A) Temperature dependence of the long S7-bound lifetime phase shown for Δ270 truncation construct and the full 3’domain RNA in absence of any other r-proteins. The error bars represent the 95 % confidence intervals of the fit. (B) Nomura ribosome assembly map. (C) Experimental setup. (D) The probability of stable S7 incorporation increases with the presence of secondary binding r-proteins; temperature = 35 °C; [Cy5-S7] = 20 nM; [unlabeled r-proteins] = 400 nM, [S15] = 2 μM. (E and F) Simplified single-molecule traces for S7 binding at 35 °C in absence (E) or presence (F) of S9, S13 and S19. (G) Tertiary interactions between H41 and H42 are required for stable S7 binding. The area of the dots is proportional to the populations of the two lifetime phases. The error bars represent the 95 % confidence intervals of the fit. (H) Model on how RNA tertiary interactions and r-proteins progressively stabilize S7. Number of molecules analyzed in (D) (n) = 101, 120, 144, 115, 111, 141, 98, 149, 121, 57, 57. See also Figures S4, S5 and Data S2.
To track nascent RNA folding directly in real-time, we sought to correlate H28 formation with S7 binding (Figures 3 and S3). After immobilization of the stalled transcription complex, we initiated transcription with NTPs, 100 nM Cy3-oligo that binds 5’ to H28, 100 nM Cy3.5-oligo that binds 3’ to H28 and 5 nM Cy5.5-S7 (Figure 3A). Excitation with a single 532 nm laser allows direct detection of binding by both Cy3-oligo and Cy3.5-oligo and indirectly, via FRET, the binding of Cy5.5-S7 as in previous experiments. Upon successful formation of H28, the Cy3 and Cy3.5-labeled oligonucleotides are in close proximity, and we observe high FRET between them (Figure 3A). For transcription of the entire 3’domain at 20 °C, ~55 % of the nascent RNA molecules are unable to form H28 during the 10 minute experimental time (Figure 3B), while the other RNA molecules either have H28 formed upon binding of the two labeled oligos (~30 %) (Figures 3C, S3C and S3D) or we see real-time formation of H28 (~15 %) (Figure S3B). Repeating these experiments with the internally-truncated Δ270 construct, we find that H28 formation is much more efficient with ~97 % of the nascent RNA molecules able to form H28 (Figure 3D). This demonstrates that the formation of long-range helices is a main challenge for successful 5’-3’ directional co-transcriptional folding of the 3’domain RNA.
Figure 3. Correlation of nascent H28 formation with S7 binding.

(A) Experimental approach to detect nascent H28 formation and correlate it with S7 binding. (B and C) Representative single-molecule traces for nascent RNA molecules with H28 not formed (B) and H28 formed with subsequent Cy5.5-S7 binding (C). TC stands for transcription. (D and E) H28 formation efficiency (D) and S7 binding competency of H28-containing RNA molecules (E) for different constructs and conditions. (F) Model for nascent H28 formation and correlation to S7 protein binding. All the experiments were initiated by delivering 500 μM NTPs, 100 nM Cy3-oligo, 100 nM Cy3.5-oligo, 5 nM Cy5.5-S7 and different combinations of unlabeled 3’domain r-proteins (400 nM each) or non-cognate binder S15 (2 μM) to the stalled transcription complex. Number of molecules analyzed in (D and E) (n) = 100, 60, 72, 115, 147, 139, 115. See also Figure S3 and Data S2.
H28 formation is required for S7 binding (Figure 3B), consistent with the absence of S7 binding during transcription, since H28 can only form once the entire 3’domain RNA is synthesized. Yet, formation of H28 is not sufficient to ensure a proper conformation of the 3’ domain to bind S7. Only ~20 % or ~40 % of the nascent RNA molecules able to form H28 at 5 nM Cy5.5-S7 are competent to bind S7 at 20 °C and 35 °C, respectively, while this fraction is ~55 % for the internally truncated Δ270 construct (Figure 3E). Thus, a main reason for 3’domain nascent RNA misfolding is lack of H28 formation, but H28 formation alone is not sufficient for S7 binding. There are likely an ensemble of other misfolded nascent RNA conformations preventing successful S7 binding (Figure 3F).
3’-domain r-proteins stabilize rRNA
Primary binding r-proteins are believed to bind stably to the rRNA to initiate assembly and to organize or reorganize the RNA for the binding of the subsequent r-proteins. Our above observations showed relatively rapid S7 dissociation upon binding to a folding-competent RNA, with S7 bound for ~ 20 s at 20 °C. Notably, at 35 °C, the average S7-bound lifetime decreases to only ~ 6 s (Figures 4A and 4E), clearly not sufficient for formation of a stable ribosomal particle. Therefore, we investigated the effect of subsequently-binding r-proteins, on the stability of the S7-rRNA complex (Figures 4B and 4C). At 20 °C, S13 significantly stabilizes the S7-rRNA complex, while S9 or S19 have a smaller stabilizing effect (Figure S5A). In contrast, at 35 °C, we see a gradual increase in the fraction of RNA molecules that can stably incorporate S7 with the progressive addition of one, two, or all three secondary r-proteins, with S19 having the largest stabilizing effect (Figures 4D–F and S5B). Further addition of the tertiary binding r-proteins (S10, S14, S3, S2) does not further increase stable S7 incorporation, demonstrating that the combination of all three secondary binders is sufficient for stable S7 incorporation. We do not observe stabilization of S7 by the addition of only the tertiary binding r-proteins (S10, S14, S3, S2) in absence of the secondary r-proteins (S9, S13, S19), or at high concentrations of r-protein S15, which binds to the central domain and does not interact specifically with the 3’domain. Overall, these data support the conclusion that the stabilization of S7 is a property shared by the secondary binding proteins (S9, S13, S19) and is not a general property of all RNA binding proteins or r-proteins. In presence of all three secondary r-proteins, we see in ~30 % of the traces, short S7 encounters followed by a single S7 binding event that lasts for several minutes and is limited by photobleaching of the Cy5-S7 or the Cy3-oligo dyes (Figures 4F, S4A and S7G). We assign the initial S7 sampling as the binding to a rRNA that is attempting to fold into its native state, while the final S7 bound state represents a rRNA molecule with native binding sites for S7 and the secondary binding r-proteins.
In the fully assembled 30S ribosomal subunit, S13 and S19 do not directly contact the S7 r-protein but wrap around helices H30, H41 and H42 (Figure S4B). This suggests that S13 and S19 stabilize the relative orientation of these helices, thereby reducing the conformational dynamics of the underlying S7 binding site, stabilizing the S7-rRNA interaction. To test this hypothesis, we deleted the tip of H41 and H42 to abolish the tertiary interactions between these helices (Figures S4C and S4D), which abrogated stable S7 binding at 20 °C with only transient S7 binding events remaining (lifetime 3.6 ± 0.1 s) (Figure 4G). These experiments demonstrate RNA-mediated S7 cooperativity, whereby stable S7 binding requires a combination of remote RNA tertiary interactions and the association of the secondary binding r-proteins to the helices emerging from the 3WJ-4WJ S7 binding site (Figure 4H).
3’-domain r-proteins chaperone nascent rRNA folding
Since 3’-domain r-proteins increase the stability of S7 binding, we wondered if the 3’-domain r-proteins may also affect nascent RNA folding rate and efficiency. While the 3’domain r-proteins do not increase the S7 association rate following transcription of the S7-binding-competent 3’domain rRNA molecules (Figure S2D), in the presence of all the 3’domain r-proteins, we find a ~25 % increase in the fraction of nascent RNA molecules that can bind S7 (Figure 2A) at 35 °C. We next determined that the 3’domain r-proteins increase the fraction of RNA molecules that successfully form H28, from ~55 % to ~75 % at 35 °C (Figure 3D). We observed the same effect using only the secondary binding 3’domain r-proteins (S9, S13, S19) and as above H28 formation efficiency was unaffected by the addition of only the tertiary binders (S10, S14, S3, S2) or the addition of a non-cognate binding protein (r-protein S15 that binds specifically to the central domain). Notably, H28 formation is also not self-chaperoned by S7 as the H28 formation efficiency is unchanged at 0-400 nM S7 (Figure S3A). These findings demonstrate that H28 formation is specifically chaperoned by the secondary binding r-proteins.
We next investigated the effect of the 3’ domain r-proteins on the RNA folding efficiency for the subset of nascent RNA molecules with successfully formed H28. The population of RNA molecules with H28 formed that can bind S7 for >2 s increases from ~40 % to ~60 % in presence of all 3’domain proteins (Figure 3E). Notably, the RNA folding efficiency is increased to a similar extend both by the secondary or tertiary binding r-proteins while the non-cognate binding protein S15 has no effect on RNA folding efficiency. These data suggest that the 3’domain r-proteins both assist in nascent H28 formation and rescue other misfolded RNA structures, and that the secondary and tertiary binding r-proteins have different specific functions in nascent RNA folding of the 3’domain (Figure 3F).
According to the thermodynamic paradigm of sequential ribosome assembly, the secondary and tertiary binding r-proteins are not supposed to be associated with the rRNA prior to binding of the primary binding r-protein S7, but our data show that an interaction must occur in order to affect nascent RNA folding prior to S7 binding. We therefore sought to track binding of secondary binding r-protein S13 in the presence and absence of S7 (Figures S4E–S4M). In presence of S7, we observe two average S13-bound lifetime phases of ~10 s and ~1 s at 20 °C, while in absence of S7 we did not detect S13 binding (n = 139 molecules). We conclude that the interaction of S13 to chaperone the nascent RNA must be transient with interaction lifetimes below our detection limit of ~ 100 ms.
Overall, our data demonstrate that the secondary and tertiary binding r-proteins are not only thermodynamic stabilizers of S7 binding but are also acting as early RNA folding chaperones that promote efficient formation of the S7-rRNA interaction by transient binding.
Assembly of entire 3’domain
Finally, we monitored assembly of the entire 3’domain in real-time. Since S2 rapidly exchanges (Robertson et al., 1977; Subramanian and van Duin, 1977), S3 is the last protein to associate stably with the 3’domain (Samaha et al., 1994) and S3 binding serves as a reporter for the successful assembly of the entire 3’domain (Figure 5A). First, we measured the real-time binding of 100 nM Cy5-labeled S3 to pre-folded 3’domain rRNA at 35 °C in presence of 400 nM concentrations of different combinations of unlabeled 3’domain r-proteins (Figure 5B). The full repertoire of 3’domain r-proteins upstream of S3 are required for stable S3 incorporation (Figure S7D) (Held et al., 1974). Removal of individual r-proteins upstream of S3 almost completely abolished detectable S3 binding (single S3 binding event observed in ~1 % of the traces) (Figure 5C). Instead, in presence of all 3’domain proteins, we observed S3 binding (Figure S6A and S6B) to ~10-15 % of all the rRNA molecules, which is in agreement with assembly efficiencies found for the 3’-domain using in-vitro transcribed RNA (STAR Methods). S3 binds ~ 7.5 minutes after r-protein addition, kon(obs) = 0.135 ± 0.004 min−1 (Figure S6C), in agreement with pulse-chase quantitative mass-spectrometry measurements of in vitro reconstituted 30S subunits with kon(obs) = 0.055/0.23 min−1 at 30/40 °C, respectively (Talkington et al., 2005). ~40 % of the traces with S3 binding show only a few short-lived binding events (Figure S6B) with a lifetime of ~4 s, kof = 0.27 ± 0.01 s−1 (Figure S6D), suggesting a partially-formed S3 binding site. The other RNA molecules show occasional short-lived binding events, koff = 0.16 ± 0.01 s−1, followed by a single long-lived S3 binding event with koff = (6.0 ± 0.3) x 10−3 s−1 (Figure S6A, S6B and S6E). This corresponds to an average S3-bound lifetime of > 2.5 minutes that is likely limited by Cy5 dye photobleaching (Figures S7E and S7F). We interpret this final S3 binding event as successful assembly of the entire 3’domain.
Figure 5. Assembly of entire 3’domain.

(A) Nomura ribosome assembly map. (B) Experimental setup: 400 nM unlabeled S7, 100 nM Cy5-S3 and 400 nM all other unlabeled 3’domain r-proteins were injected; 35 °C. (C) All r-proteins binding upstream of S3 are required for stable S3 binding. Number of molecules analyzed in (C) (n) = 505, 1017, 924, 2699, 1453. See also Figures S6, S7 and Data S2.
Next, to correlate directly the binding of primary r-protein S7 with final r-protein S3, we repeated the experiments using Cy5.5-S7, Cy5-S3 with all the other 3’domain r-proteins unlabeled (Figures 6A, 6B and S7A). S3 binding events occur only after final stable S7 incorporation (Figure 6D), as expected for sequential ribosome assembly and the requirement for stable S7 binding prior to completion of 3’domain assembly. In ~60 % of those traces, we find S7 and S3 simultaneously bound (Figure S7B), while the remaining traces show S3 binding after final S7 signal disappearance (Figure 6C). We attribute the latter case to early S7 photobleaching. To estimate the time to finalize assembly of the 3’domain once the large S7 binding site has stably incorporated S7, we measured the dwell time from the start of the final stable S7 binding event, until the arrival of S3 (Figure 6E). These final steps take ~ 4 min, k(obs) = 0.24 ± 0.01 min−1 (Figure S7C). This slow incorporation suggests that RNA conformational changes are also required later in assembly of the 3’domain rRNA, subsequent to native folding of the large S7 binding site.
Figure 6. Correlating S7 with S3 binding.

(A) 3-dimensional structure of E. coli 30S ribosomal subunit with labeling sites for Cy5-S3 and Cy5.5-S7 (PDB accession code: 4V9P); the Cy3 dye of the labeled DNA oligonucleotide binding to the 3’-end of the nascent RNA is modeled into the structure. (B) Experimental setup: 100 nM Cy5-S3, 5 nM Cy5.5-S7 and 400 nM of all the other unlabeled 3’domain r-proteins were injected; 35 °C. (C) Representative single-molecule trace with Cy5-S3 (red), Cy5.5-S7 (purple) and Cy3-oligo (green). The trace was not corrected for fluorescence spectral bleed-through from the Cy5 into the Cy5.5 channel and the reverse. (D) The first Cy5-S3 binding event always follows the final Cy5.5-S7 binding event demonstrating sequential ribosome assembly. (E) The assembly process from stable S7 incorporation until 3’domain completion is slow, indicating that RNA conformational changes are also required later in assembly. Number of molecules analyzed in (D and E) (n) = 64. See also Figure S7 and Data S2.
DISCUSSION
Simultaneous and direct tracking of nascent RNA folding and RNP assembly
The formation of cellular RNP complexes involves synthesis and correct folding of the individual protein and nucleic acid components and specific intermolecular interactions between them. Recent studies have directly tracked RNA folding of a pre-synthesized RNA and correlated to protein binding in ribosome assembly (Abeysirigunawardena et al., 2017; Kim et al., 2014), but a major barrier to understanding RNP assembly is the reliance on in vitro studies investigating the interactions of protein molecules with a pre-formed structured RNA rather than RNA directly emerging from the RNA polymerase. Here, we have tracked the assembly of the entire 3’-domain of the 30S ribosomal subunit in real-time by directly correlating rRNA transcription, nascent RNA folding, binding of primary protein S7, and binding of tertiary protein S3 that completes 3’domain assembly (Figure 7). By providing real-time information on transient biochemically-unstable states, our dynamic data complement the molecular understanding of ribosome assembly that is heavily based on compositional (Chaker-Margot et al., 2015; Chen and Williamson, 2013) and structural knowledge of different assembly intermediates (Davis et al., 2016; Klinge and Woolford, 2018).
Figure 7. Model for nascent 3’domain assembly in absence of assembly factors.

Nascent RNA molecules can either misfold (shown in red) into stable non-native structures (deep valley on the left) or fold into RNA molecules that eventually transition into S7 binding competent conformations. The r-proteins chaperone the rRNA folding process early in assembly prior to their stable incorporation into the growing RNP particle. Nascent RNA folding and assembly are slow due to the rugged energy landscape. Progressive binding of r-proteins facilitates folding and stabilizes the nascent RNA. The single-molecule traces on the bottom demonstrate the decreasing protein binding dynamics during assembly.
Long-range interactions complicate nascent RNA folding
Nascent 3’-domain rRNA folding is slow and inefficient due to the challenge of long-range H28 formation. Supporting our data, the formation of non-native interactions of the 5’-half of H28 was also suggested in vivo using nascent RNA structural probing with DMS (Incarnato et al., 2017). Furthermore, x-ray hydroxyl radical foot-printing experiments in vivo demonstrated that the 3′-domain misfolds at low temperature in the absence of specific assembly factors (Soper et al., 2013). The binding occupancy of the S7 protein in the accumulating assembly intermediates was less than 40 %, indicating that a majority of those misfolded RNA conformations were unable to bind S7 stably at low temperature also in vivo (Soper et al., 2013). These prior experiments demonstrated the importance of assembly factors in guiding rRNA folding and assembly. The installation of rRNA modifications may also modulate RNA folding and ribosomal assembly, as suggested by the significantly lower in vitro reconstitution efficiencies of in vitro transcribed unmodified rRNA compared to natively purified rRNA (Jewett et al., 2013; Samaha et al., 1994). Our assay will allow investigation of the effects of specific assembly factors on nascent rRNA folding and assembly, especially in combination with recently developed transcription-translation coupled ribosome reconstitution systems (Jewett et al., 2013). While we do not find evidence for RNAP pausing during transcription of the 3’domain under our conditions, pausing would be expected to also decrease RNA folding efficiency as does decreased transcription rate (Figures S2F–S2H). In vivo, suppression of RNAP pausing during transcription of the rRNA is ensured by the rRNA transcription antitermination complex (rrnTAC) (Vogel and Jensen, 1995). Our findings suggest that besides preventing “traffic jam” during dense RNAP traffic (Klumpp and Hwa, 2008), the rrnTAC could also increase nascent rRNA folding efficiency by reducing the time window for RNA misfolding during formation of long-range interactions.
Recently, it has been demonstrated that many natural RNAs contain long-range interactions in vivo, spanning up to several thousands of nucleotides in sequence (Lu et al., 2016). Overall, this suggests that long-range RNA structure formation could be a general challenge in cellular nascent RNA structure formation and that mechanisms must exist to chaperone nascent RNA folding in vivo.
Late binding r-proteins act as early rRNA folding chaperones
Work from the last decade demonstrated that assembly proceeds through multiple parallel assembly pathways but with sequential and cooperative assembly within the individual pathways. Time-resolved hydroxyl radical footprinting showed that assembly nucleates at different positions along the rRNA (Adilakshmi et al., 2008) and, more recently, cryoEM structures of 13 different assembly intermediates of the large 50S ribosomal subunit revealed that individual cooperative folding blocks are incorporated into the growing ribosomal particle in different orders demonstrating that assembly occurs through parallel routes (Davis et al., 2016). In the present work, we zoom into one of those folding blocks or assembly routes and demonstrate that assembly is also not strictly sequential within those blocks revealing further complexity in assembly.
We find that r-proteins increase nascent H28 folding efficiency and help the folding of other parts of the extended S7 binding site. Earlier work on pre-folded RNA demonstrated the contribution of r-proteins as RNA folding aids in the context of sequential and cooperative ribosome assembly. Primary binding proteins were previously established to refold the rRNA to create the binding sites for the subsequent r-proteins to be recruited the complex. For example, primary r-protein S15, which binds to a 3-way junction in the central domain of the 16S rRNA, changes the conformation of the junction and creates the binding site for the secondary binding proteins S6/S18 (Ha et al., 1999; Orr et al., 1998; Recht and Williamson, 2001). Further work, using hydroxyl radical foot-printing, showed that r-protein S16, which binds to the 5’-domain of the 16S rRNA, suppresses a non-native folding intermediate smoothing the path to the native complex (Ramaswamy and Woodson, 2009) and recent single-molecule experiments demonstrated how r-protein S4 (Kim et al., 2014) or r-proteins S4, S17, S20 and S16 (Abeysirigunawardena et al., 2017) steer the rRNA towards the native state in order to facilitate the recruitment of the subsequent r-proteins to join the complex. These prior experiments provided a rationale for the cooperative and sequential paradigm of ribosome assembly where early binding r-proteins refold the rRNA to facilitate the binding of the subsequent r-proteins to be recruited to the assembling ribosomal complex.
Challenging and expanding on this current view of sequential ribosome assembly, we now find that later binding r-proteins, which should not yet be bound according to the sequential paradigm of ribosome assembly, act as nascent rRNA folding chaperones in early steps of assembly, presumably by transient and semi-specific binding. Transient binding must occur with dwell times <100 ms because we do not detect binding of secondary r-protein S13 (Figures S4E–S4M) nor tertiary binding r-protein S3 (Figure 5C) in absence of the upstream binding r-proteins. Using fluorescence triple-correlation spectroscopy, association of secondary binders with the 16S rRNA was detected in absence of primary binder S7 (Ridgeway et al., 2012), supporting our findings that later binding r-proteins assist in rRNA folding early in assembly before those r-proteins are stably incorporated and their binding sites entirely formed. One could therefore consider the secondary and tertiary binding r-proteins as transiently binding primary r-proteins.
Nascent RNA chaperone activity by late binding proteins could be a general mechanism contributing to efficient RNP maturation. For example, small nucleolar RNPs (snoRNPs) are composed of several proteins, which assemble in a specific order on the nascent snoRNA (Massenet et al., 2017; Reichow et al., 2007). Similarly, the signal recognition particle (Doudna and Batey, 2004) and the telomerase (Schmidt and Cech, 2015; Stone et al., 2007) require a stepwise assembly of several proteins on the corresponding RNAs. In those cases, late binding proteins could transiently interact with the RNA early in assembly to help RNA folding. Significantly more complex, spliceosome assembly requires the defined assembly of dozens of proteins onto the nascent mRNAs directly emerging from Pol II (Neugebauer, 2019; Will and Luhrmann, 2011). Several splicing factors consist of multi-domain RNA binding proteins (Lunde et al., 2007) and the transient interactions of individual protein domains with parts of the nascent RNA could help shape mRNA folding before their stable incorporation into the spliceosome complex. Finally, eukaryotic translation initiation requires the ordered recruitment of multiple RNA binding proteins to structured 5’ untranslated regions (Leppek et al., 2018). In light of our findings, it would not be surprising if proteins that stably recruit into the translation initiation machinery later in assembly could have RNA chaperone activity at an early stage of mRNA translation initiation.
RNA binding proteins do not interact with a single specific high affinity target, but with multiple RNA targets in a continuum between specific and non-specific binding (Buenrostro et al., 2014; Ferre-D’Amare, 2016; Jankowsky and Harris, 2015). Considering these recent findings, our new data and high local cellular concentrations of many RNA binding proteins, transient and semi-specific binding of RNA binding proteins to cellular RNAs could provide a general mechanism to actively facilitate nascent RNA folding during assembly of cellular RNPs.
Quality control mechanism for nascent rRNA folding
In contrast to the prevailing view that ribosome assembly consists of a series of stepwise events initiated by stable binding of primary binding r-proteins (Duss et al., 2018; Held et al., 1974; Kim et al., 2014; Shajani et al., 2011; Traub and Nomura, 1969), we find that each step in the assembly process involves significant dynamics, including multiple rounds of transient binding prior to formation of kinetically stable complexes. In the absence of other r-proteins, the primary binding r-protein S7 only transiently samples the nascent 3’-domain rRNA. However, during the course of assembly, the protein binding dynamics gradually decrease as additional components are installed. These findings are complemented by recent observations that RNA conformational dynamics decrease upon stable binding of the primary r-protein S4 to the 5’-domain (Kim et al., 2014). Transient sampling of primary binder S7 before the successful association of the secondary binders S9, S13 and S19 with their native binding sites, could prevent kinetic trapping of misfolded RNA conformations early in assembly by premature stable S7 association and thus, could provide a quality control mechanism for native rRNA folding.
Summary
In summary, by directly observing nascent rRNA folding and correlating it with ribosome assembly progression, we show that directional RNA synthesis alone does not fix the RNA misfolding problem as generally postulated. Instead, r-proteins cooperatively steer nascent RNA folding by progressively decreasing protein-RNA dynamics to obtain a stable native ribosome.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
The Lead Contact is James R. Williamson (jrwill@scripps.edu). Plasmids generated in this study have been deposited to Addgene (see Key Resource Table). All other materials are available without restriction. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21-Gold (DE3) | Agilent | Cat#230132 |
| E. coli Tuner™(DE3) | Novagen | Cat#70623-3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protocatechuic acid (PCA) | Pacific Biosciences | Cat#100-215-400 |
| Protocatechuate-3,4-dioxygenase (PCD) | Pacific Biosciences | Cat#001-028-310 |
| TSY | Pacific Biosciences | Cat#100-214-900 |
| Cy5 Maleimide Mono-Reactive Dye 5-Pack | GE Healthcare | Cat#PA25031 |
| Cy5.5 Sulfo-Cyanine5 maleimide | Lumiprobe | Cat#13380 |
| Cy3 Maleimide Mono-Reactive Dye 5-Pack | GE Healthcare | Cat#PA23031 |
| E. coli RNA polymerase holoenzyme | NEB | Cat#M0551S |
| Biolipidure-203 | NOF America Corporation | Cat#Biolipidure-203 |
| Biolipidure-206 | NOF America Corporation | Cat#Biolipidure-206 |
| Purified ribosomal proteins of 16S rRNA 3’domain | This paper | N/A |
| Oligonucleotides | ||
| Primers, geneBlocks-based DNA transcription templates, labeled DNA oligonucleotides | See Data S1 | N/A |
| ACU RNA trinucleotide | Dharmacon | N/A |
| Recombinant DNA | ||
| pRSF-1b | Novagen | Cat#71330-3 |
| pRSF-1b-S2 | This paper | Addgene Plasmid #128590 |
| pRSF-1b-S3 | This paper | Addgene Plasmid #128591 |
| pRSF-1b-S7-S83C/delta157-178 | This paper | Addgene Plasmid #128592 |
| pRSF-1b-S10 | This paper | Addgene Plasmid #128594 |
| pRSF-1b-S13-C85S/P112C | This paper | Addgene Plasmid #128595 |
| pRSF-1b-S14 | This paper | Addgene Plasmid #128596 |
| pRSF-1b-S19-T48C | This paper | Addgene Plasmid #128597 |
| pET-S9 | This paper | Addgene Plasmid #132948 |
| pRSF-1b-S3-M129C | This paper | Addgene Plasmid #132949 |
| pRSF-1b-S15-T79C | This paper | Addgene Plasmid #133048 |
| Software and Algorithms | ||
| MATLAB R2015a | Matworks Inc | http://matlab.com |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The genes for the E. coli 30S 3’domain r-proteins were cloned into a pRSF-1b expression vector (Novagen) and overexpressed in BL21-Gold (DE3) (Invitrogen) or Tuner™ (DE3) E. coli strains (Novagen). E. coli strains were grown in LB medium at 37 °C in culture flasks ranging in volume from 5 ml test tubes to 2 L baffled flasks.
METHOD DETAILS
Protein expression and purification
The seven expressed tag-free r-proteins (S2, S3, S7, S10, S13, S14 and S19) and one N-terminally 6xHis-tagged protein (S9) were purified from inclusion bodies as described in details elsewhere (Duss et al., 2018) and summarized in Table S1. In short, after expression at 37 °C in LB medium and subsequent cell lysis by sonication, the inclusion bodies were solubilized overnight in 8 M guanidine chloride followed by dialysis against 6 M urea or were solubilized in 8 M urea omitting subsequent dialysis. Purification of all proteins was performed under denaturing conditions in 6 M urea using either a SP HiTrap column (GE Healthcare), a Q column (for r-protein S2) or a Ni-NTA column followed by a SP column (for r-protein S9) as detailed in Table S1. The proteins were refolded by overnight dialysis, except for S10, which was refolded by 20-fold dilution into refolding buffer. S2, S3, S7, S14 and S19 were further purified with a Heparin HP column. All proteins were additionally purified by size-exclusion chromatography on a Superdex 75 26/600 HiLoad gel filtration column (GE Healthcare) equilibrated with the final sample buffers as summarized in Table S1. The monomeric fractions were concentrated using Vivaspin® 2 kDa MWCO (Sartorius) or Amicon® 3-10 kDa Ultra Centrifugal Filters (Millipore) depending on the size of the protein and finally frozen at −80 °C. The S10 protein was frozen immediately at −80 °C without concentrating.
Protein fluorescence-dye labeling
For S7, the S7(S83C) variant with a truncated C-terminus (157-178) was labeled with Cy5-maleimide or Cy5.5-maleimide, retaining full RNA binding activity, showing similar in vitro reconstitution efficiencies like unlabeled S7 (Figure S7D) and able to form active ribosomes (Hickerson et al., 2005; Ridgeway et al., 2012). For S3, the S3(M129C) variant was labeled with Cy5-maleimide, showing similar in vitro reconstitution efficiencies like unlabeled S3 (Figure S7D) and able to form active ribosomes (Hickerson et al., 2005). For S13, the P112C/C85S variant was labeled with Cy5-maleimide or Cy5.5-maleimide and showing similar in vitro reconstitution efficiencies like unlabeled S13 (Figure S7D) and able to form active ribosomes with unaffected kinetics of translocation (Cunha et al., 2013). For labeling, 1 mg of r-protein in the labeling buffer A (100 mM Na2HPO4/KH2PO4 (pH 7.0), 100 mM NaCl) was reduced by incubating the sample with 10 mM DTT for 2 hours on ice. Then, ammonium sulfate powder (70 % w/v) was added to the protein solution and incubated for 20 minutes at 4 °C with gentle agitation. The precipitated protein was separated by centrifugation at 14,000 rpm for 20 minutes, washed with 70 % ammonium sulfate in labeling buffer A and span down at 14,000 rpm. The washed pellet was dissolved in 475 μL of labeling buffer, containing 6 M urea (labeling buffer A1). 1 mg of Cy5-, or Cy3-maleimide dye (GE Healthcare), or Sulfo-Cyanine5.5 maleimide dye (Lumiprobe) was dissolved in 25 μL of ultra-pure DMSO (Sigma) and mixed with the protein solution in buffer A1. The conjugation reaction continued for 30 minutes with moderate shaking at room-temperature and was quenched by adding final 0.5 % (72 mM) β-mercaptoethanol to the reaction mixture.
To remove the excess of dye from the labeled protein, the reaction mixture was passed over a 5 ml Nap5 column (GE Healthcare) equilibrated with buffer A1. The Nap5 column elutes were analyzed with 4-20 % SDS-PAGE by visualization of fluorescence on a VersaDoc™ MP 4000 Imager (Bio-Rad) and Coomassie staining. To further remove free dye, the labeled protein fractions were pooled, refolded by 10-fold dilution with 20 mM TrisHCl (pH 7.6), 20 mM NaCl, 0.5 mM EDTA (buffer E) and passed over a 1-mL Heparin HP column (GE Healthcare) at 4 °C. The column was washed with 10 column volumes of buffer E and the labeled protein was eluted with 1 M NaCl in 20 mM TrisHCl (pH 7.6), 0.5 mM EDTA (buffer F), pooled, aliquoted and stored at −80 °C.
Protein concentration and labeling efficiency
The total protein concentration was quantified by amino acid hydrolysis in the UC Davis Molecular Structure Facility Core. The dye-coupled protein concentration was calculated based on the extinction coefficient of each dye: ϵ550(Cy3) = 150’000 M−1 cm−1; ϵ650(Cy5) = 250’000 M−1 cm−1; ϵ673(Sulfo-Cyanine5.5) = 195’000 M−1 cm−1. The labeling efficiency was determined using the ratio between dye-coupled and total protein concentration.
In vitro reconstitutions
In vitro reconstitutions (standard conditions) were performed by mixing 400 nM native 16S rRNA and 1.2 μM recombinantly expressed 3’domain proteins in 25 mM TrisHCl (pH 7.5), 330 mM KCl, 20 mM MgCl2, 2 mM DTT (reconstitution buffer) and 40 units/μL of RNAseOUT (Invitrogen) in a total volume of 1.2 ml and incubated at 42 °C for 20 minutes. In vitro reconstitutions in ZMW conditions were perform using the same concentrations of 16S RNA and r-proteins but in 50 mM TrisHCl (pH 7.5), 14 mM MgCl2, 20 mM NaCl, 0.04 mM EDTA, 40 μg/ml BSA, 0.01 % Triton X-100, 2 mM spermidine, 1 mM putrescine, 150 mM KCl, 0.25 % Biolipidure 203, 0.25 % Biolipidure 206, 0.5 mg/ ml yeast total tRNA, 0.5 mM DTT, 2.5 mM protocatechuic acid (PCA), 250 nM protocatechuate-3,4-dioxygenase (PCD), 2 mM TSY, 100 nM Cy3-oligo and 40 units/ μL of RNAseOUT (Invitrogen) in a total volume of 1.25 ml and incubated at 35 °C for 30 minutes. 16S rRNA was obtained as previously described (Talkington et al., 2005) and was heated for 5 minutes at 42 °C prior to reconstitutions. After reconstitution, the mixture was concentrated to 300 μL, washed twice with reconstitution buffer and purified with a 10-40 % sucrose gradient with ultracentrifugation at 32,000 rpm for 15 hours at 4 °C. After fractionation, the fractions containing rRNA were concentrated with an Amicon 100 kDa MWCO centrifugal filter to 30 μL and buffer-exchanged to remove excess of sucrose. The protein composition was analyzed on a 16 % SDS-PAGE gel (Invitrogen).
In vitro assembly of 30S ribosomal subunits with 16S rRNA and total proteins (TP30) that were purified from native ribosomes gives high reconstitution efficiencies approaching 80-90 % (Adilakshmi et al., 2008; Samaha et al., 1994; Talkington et al., 2005). However, reconstitutions of 30S subunits with in vitro transcribed 16S rRNA or 3’domain rRNA, which lack all the rRNA modifications, result in assembly efficiencies of only ~30 % (Samaha et al., 1994). Yet, those efficiencies can only be achieved using elevated temperature-gradients between 40-50 °C, requiring > 1 hour and using non-physiological buffer conditions (330 mM KCl, 20 mM MgCl2). Instead, we performed our single-molecule experiments at more physiologically relevant buffer compositions (150 mM KCl, 14 mM MgCl2) and at a single near-physiological temperature of 35 °C. We observe in our experiments ~12 % of the RNA molecules capable of binding Cy5-S3. Correcting for the labeling efficiency of Cy5-S3 of ~ 75 %, these are ~16 % of the RNA molecules, which are competent to complete 3’domain assembly. Overall, our observed assembly efficiencies for the entire 3’domain are expected using our more physiological in vitro reconstitution conditions.
DNA templates for transcription
The DNA transcription templates were labeled at the 3’-end by hybridizing a fluorescently-labeled DNA oligonucleotide, containing two Cy3.5 dyes, to a single-stranded overhang on the double-stranded DNA transcription template (see Data S1). The DNA oligonucleotide was obtained from IDT and was used without additional purification. The DNA template containing a single-stranded overhang was generated by autosticky PCR using a gBlocks gene fragment template available from IDT (Gal et al., 1999): The PCR primers contained a sequence complementary to the DNA template, an abasic site, and the sequence complementary to the labeled DNA oligonucleotide. PCR was performed with Phusion DNA polymerase according to the manufacturer’s instructions, using 35 cycles and optimizing the annealing temperature with a temperature gradient. The PCR product was purified on an agarose gel with subsequent QIAGEN gel extraction, followed by buffer exchange (10 mM TrisHCl (pH 7.5), 20 mM KCl) using a 30 kDa molecular weight cutoff centrifugal filter (Amicon). The template sequence was verified by Sanger sequencing. The DNA template was effectively labeled by duplex formation with a 20 % molar excess of labeled DNA oligonucleotide, heating to 68 °C for 5 minutes and slow cooling to room temperature. The labeled template DNA duplex was not further purified, and the labeling efficiency (~100 %) was quantified on a 4 % agarose gel.
Stalled transcription elongation complex
As described in detail previously (Duss et al., 2018), a stalled transcription elongation complex, composed of a DNA template, an RNA polymerase (RNAP) molecule and a nascent RNA of 50 nucleotides (Figure 1C), was formed by incubation of 25 nM labeled DNA transcription template, 100 μM ACU trinucleotide (Dharmacon), 10 μM ATP, CTP and UTP, 2 mM DTT, and 100 nM E. coli RNAP (NEB; holoenzyme) in a buffer containing 50 mM TrisHCl (pH 8.0), 14 mM MgCl2, 20 mM NaCl, 0.04 mM EDTA, 40 μg/ml BSA, 0.01 % Triton X-100, for 20 minutes at 37 °C. Then, transcription re-initiation was stopped by addition of 1 mg/ml heparin. Simultaneously, 20 nM of a biotinylated double-stranded DNA with a single-stranded overhang was added and the mixture was incubated for 20 minutes at 37 °C. During this time, the biotinylated dsDNA oligo was annealed to the 5’-end of the nascent RNA and efficient annealing was verified by native gel electrophoresis as described previously (Duss et al., 2018). The biotinylated DNA oligo was obtained by autosticky PCR similarly as the transcription template (see Data S2). The biotinylated stalled transcription complex was put on ice until used for the single-molecule experiments.
Pre-folded 3’domain RNA
For the single-molecule experiments with protein binding to pre-transcribed and pre-folded RNA, we performed a similar procedure as for the co-transcriptional experiments, except that rather than forming a stalled transcription complex by adding only 3 out of the 4 NTPs, we performed a transcription reaction using 1 mM of all 4 NTPs for 10 minutes at 37 °C, to generate the full-length RNA. Then, we incubated the RNA for 10 minutes at 37 °C with 1 mg/ml heparin and 20 nM biotinylated DNA as described above. The biotinylated full-length RNA was put on ice until used for the single-molecule experiments.
ZMW instrument and single-molecule imaging
Single-molecule experiments were conducted with a commercial RS II sequencer (Pacific Biosciences) that has been modified to allow the collection of single-molecule fluorescence intensities from individual ZMW wells in four different dye channels corresponding to Cy3, Cy3.5, Cy5, and Cy5.5 (Chen et al., 2014). The RS sequencer uses two lasers for dye excitation at 532 nm and 642 nm. Data were collected at ten frames per second for 10 minutes (co-transcriptional experiments) or three frames per seconds for 30 minutes (pre-transcribed RNA) if not otherwise stated. We used a single laser at 532 nm with an energy flux of 0.32 μW μm−2 for all the experiments. We note that Cy5 and Cy5.5 emission was observed due to a FRET from the excited Cy3 and /or Cy3.5 fluorophores as specified in the corresponding experiments.
For the co-transcriptional single-molecule experiments, biotinylated stalled transcription complexes were immobilized to the bottom of biotin-functionalized ZMWs using NeutrAvidin. First, 3-11 nM stalled complex was incubated with 200 nM NeutrAvidin in 50 mM TrisHCl (pH 7.5), 14 mM MgCl2, 20 mM NaCl, 0.04 mM EDTA, 40 μg/ml BSA, 0.01 % Triton X-100, 2 mM spermidine, 1 mM putrescine and 150-500 mM KCl for 10 minutes at room temperature. The stalled complex concentration was adjusted to obtain an optimal SMRT Cell loading efficiency of ~15 %. Larger constructs required higher concentrations of stalled complex for optimal immobilization. In parallel, a SMRT Cell V3 ZMW chip (Pacific Biosciences) was prepared by first wetting the chip with Tris buffer (20 mM TrisHCl (pH 8.0), 50 mM NaCl), then washing with the buffer used to incubate the stalled complex with NeutrAvidin (see above), and finally incubating the stalled complex/NeutrAvidin mixture on the chip. This incubation required 20 minutes to several hours, in proportion to template length, to obtain a satisfactory immobilization efficiency. After immobilization, non-immobilized complex was removed by washing with reaction buffer, composed of 50 mM TrisHCl (pH 7.5), 14 mM MgCl2, 20 mM NaCl, 0.04 mM EDTA, 40 μg/ml BSA, 0.01 % Triton X-100, 2 mM spermidine, 1 mM putrescine, 150 mM KCl, 0.25 % Biolipidure 203, 0.25 % Biolipidure 206 and 0.5 mg/ ml yeast total tRNA. The wash buffer also contained an oxygen-scavenging system consisting of 2.5 mM protocatechuic acid (PCA) with 250 nM protocatechuate-3,4-dioxygenase (PCD) (Aitken et al., 2008), and 2 mM TSY (Pacific Biosciences) to minimize fluorescence instability. After washing, 20 μl of this wash mix was left on the chip to keep the surface wet.
Before the experiment, the SMRT cell was loaded into the RS II sequencer. At the start of the experiment, the instrument illuminates the SMRT cell with a 532 nm laser and then automatically delivers 20 μl of the delivery mixture onto the cell surface at t ~10 s. The experiments were performed at the indicated temperature. The delivery mix consisted of reaction buffer supplemented with 0.5 mM NTPs, 100 nM labeled DNA oligonucleotide for hybridization to the 3′-end of nascent RNA (and/or the 5′-end if specifically stated), and labeled and unlabeled r-proteins at the indicated concentrations; if not otherwise stated, standard concentrations are 20 nM Cy5-S7, 5 nM Cy5.5-S7, 100 nM Cy5-S3 and/or 400 nM unlabeled r-proteins. DNA oligonucleotides (IDT) were labeled at their 3′-end and for some experiments, a quencher was attached to the other terminus (Zhang et al., 2014). We used labeled DNA oligos with and without quencher (Data S1) in our experiments, but we found a slightly reduced background fluorescence in presence of the quencher.
For the full-length RNA experiments, prior to immobilization, the biotinylated full-length RNA was diluted 4-fold and further incubated for 10 minutes at 37 °C with 200 nM NeutrAvidin in 50 mM Tris (pH 7.5), 14 mM MgCl2, 20 mM NaCl, 0.04 mM EDTA, 40 μg/ml BSA, 0.01 % Triton X-100, 2 mM spermidine, 1 mM putrescine, 150 mM KCl and 100 nM Cy3-oligo that binds to the 3′-end of the full-length RNA. Except for a shorter 10 minutes immobilization time, immobilization was identical to the co-transcriptional experiments as was delivery mix injection during the ZMW experiment, except that the Cy3-oligo and the NTPs were omitted in the delivery mix.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-molecule data analysis
Single-molecule data were analyzed with in-house–written MATLAB (MathWorks) scripts (Chen et al., 2014; Chen et al., 2013). For the co-transcriptional experiments, only traces showing a Cy3.5 fluorescence intensity increase, which is characteristic for transcription elongation (Figure 1C), and with the subsequent binding of a single Cy3-oligo (Figure 1D), which is binding to the 3′-end of the nascent RNA and indicates the presence of a single full-length RNA, were selected for further analysis. The exact start and end of transcription elongation were determined as detailed previously (Duss et al., 2018). For the experiments with pre-transcribed and pre-folded RNA, only traces showing a single Cy3-oligo were selected for further analysis.
Specific binding of dye-labeled protein (Cy5- or Cy5.5-labeled; denoted Cy5 in the following for simplicity) was detected by the presence of a high-FRET value between the Cy5-protein and a Cy3-oligo that is binding to the 5’-end or 3’-end of the nascent RNA (see e.g. Figure 1). The presence of FRET reports only on specific Cy5-S7 binding which was verified by following experiments: Using a binding incompetent Cy5-S7(delta 1-18) mutant (Robert et al., 2000), we observe no binding events except of a single transient event in ~5 % of the traces. Furthermore, we observe no binding for Cy5-S7 to an unspecific RNA target consisting of the 16S central domain rRNA. We calculated the FRET efficiency by EFRET = IA/(IA + ID) from background corrected traces, where ID and IA are the apparent fluorescence intensities of the donor (Cy3) and acceptor (Cy5), respectively. To assign the bound state, we defined the threshold in the middle of the two FRET states, with subsequent manual inspection for traces that showed substantial non-specific Cy5-protein binding and therefore a non-zero FRET efficiency baseline, as detailed previously (Duss et al., 2018). In short, for traces with substantial non-specific Cy5-protein binding, we used the presence of a clear anti-correlated change in Cy3 and Cy5 intensities, which can be readily distinguished from non-specific Cy5-protein binding due to the presence of a high-FRET value in the bound state by design (Figures 1B and 6A). In contrast, non-specific Cy5-protein binding results only in a Cy5 fluorescence increase in absence of a reciprocal decrease in Cy3 fluorescence (Figure S1A).
To calculate rate constants, all dwell times were fitted to single- or double-exponential distributions using maximum-likelihood parameter estimation in MATLAB.
The single-molecule traces were hierarchically clustered using the Weighted Pair Group Method with Arithmetic Mean (WPGMA) and the Spearman distance metric using MATLAB. First, we determined for each trace: i) the longest S7-bound lifetime, ii) the number of binding events, and iii) the time between end of transcription and the first stable binding event. Then, we clustered the traces according to those three parameters.
Defining RNA folding classes
For r-protein S15 binding to the central domain of the 16S rRNA, we observed three types of kinetic binding behaviors: i) RNA molecules without S15 binding, ii) RNA molecules with multiple ~ 1-1.5 s transient S15 binding events or iii) RNA molecules with mostly a single long-lived S15 binding event with a duration of more than several minutes (Duss et al., 2018). The very distinct kinetic binding fingerprints of the 3 RNA folding classes allowed their unambiguous assignment into one of the three folding classes. In contrast, for S7, descriptors for binding behavior such as number of binding events per trace or the longest binding event per trace do not directly permit identification of discrete RNA folding classes. Considering that nascent RNA folding is a time-dependent process, which ultimately leads to a folded or misfolded RNA molecule, a useful descriptor of the final folding state of a specific RNA molecule could be the longest S7-bound lifetime of that RNA molecules during the experimental time. Therefore, we plotted the longest S7-bound lifetime of every single-molecule trace as a cumulative distribution (Figure S2B). Remarkably, this data can be well fitted to a double exponential function. We interpret the “fast” phase as the misfolded RNA population and the “slow” phase as the natively folded RNA population. We obtain a threshold of 2 s for the longest lifetime per trace in order to assign a specific RNA molecule to either a folded or misfolded state.
Statistical details
Measurements from single-molecule fluorescence assays resulted from a specified number (n) of molecules from a single experiment. Statistical details of individual experiments, including number of analyzed molecules (n), the definition of error bars and confidence intervals of the fits are indicated in the corresponding figure legends.
DATA AND CODE AVAILABILITY
The published article includes all relevant data generated or analyzed during this study. Source data for all the figures are available in supplementary file Data S2. Additional data and codes are available from Lead Contact, James R. Williamson (jrwill@scripps.edu) on request.
Supplementary Material
Figure S7. Correlating S7 with S3 binding and ensemble experiments. Related to Figures 5 and 6.
(A-C) Correlating S7 with S3 binding. (A) Overview of the analyzed single-molecule traces. Every row represents a single pre-folded RNA molecule. (B) Representative single-molecule trace with Cy5-S3 (red), Cy5.5-S7 (purple) and Cy3-oligo (green). The trace was not corrected for fluorescence spectral bleed-through from the Cy5 into the Cy5.5 channel and the reverse. (C) To estimate the rate between stable S7 and initial S3 incorporation, we fitted the dwell times between final Cy5.5-S7 and first Cy5-S3 event to a single-exponential function. The error represents the 95 % confidence interval of the fit. Number of molecules analyzed (n) = 64 (C). Experimental setup in (A-C): 100 nM Cy5-S3, 5 nM Cy5.5-S7 and 400 nM of all the other unlabeled 3’domain r-proteins were injected at t = 0 seconds. All the experiments were performed at 35 °C. (D-G) Ensemble experiments. (D) In vitro reconstitutions of native 16S rRNA with individually recombinantly expressed 3’domain r-proteins (see STAR Methods). This data shows that: 1) all r-proteins upstream of S3 are required for S3 r-protein association, 2) S2 binding downstream of S3 is not required for stable S3 binding, 3) fluorescence labeling of S3, S7 and S13 give similar in vitro reconstitution efficiencies as unlabeled wild-type proteins, 4) using ZMW conditions (see STAR Methods) instead of established in vitro reconstitution conditions (Held et al., 1974; Traub and Nomura, 1969) has no effect on the assembly efficiency. (E-F) The actual S3-bound lifetime of the final S3 binding event is longer than what we can observe during our single-molecule experiments. Shown are competition experiments of 25 nM reconstituted 3’domain containing Cy5-S3 that was chased with unlabeled S3 at 16-fold excess for the indicated times and at 35 °C in presence of standard conditions or ZMW conditions. The Cy5 fluorescence was visualized with native agarose gel electrophoresis and was normalized by the amount of rRNA that was visualized with EtBr staining. Each dot represents the mean normalized fluorescence intensity remaining relative to the 5 min time point, and error bars represent standard deviation from different experiments. The line represents a fit to y = a * exp(−b*t) + c. The parameter c represents a slow phase which is too slow to be determined reliably during our experimental time of 1-2 days (c * exp(−d*t) = c if decay rate d is very small). We obtain two similarly populated lifetime phases of ~4-5 h and > several days, respectively. It is possible that the shorter lifetime phase of S3 represents a 3’domain particle which has not obtained its native fold yet, while the longer S3 lifetime phase represents a native 3’domain. However, while in vivo the majority of S3 is stably associated with the ribosome, ~ 25 % of S3 also exchanges rapidly (Robertson et al., 1977) suggesting that S3 could be associated with the native rRNA in two different states, one which exchanges faster than the other one. (G) Competition experiments of reconstituted 3’domain containing Cy5-S7 (25 nM) with unlabeled S7 at 16-fold excess for the indicated times and at 35 °C. The same experimental setup was used as in (E), except that the 3’domain was only reconstituted with the secondary binding proteins S9, S13 and S19 instead of all 3’domain proteins and Cy5-S7 was used instead of unlabeled S7. The data demonstrate that S7 does not significantly exchanges during the course of the experiment and shows that S9, S13 and S19 are sufficient to stabilize S7 on a relevant biological timescale. Error bars represent standard deviation from different experiments. Gel repetitions, n = 3 (E-G). See also Data S2.
Table S1. Related to STAR Methods.
Overview of r-protein expression and purification. a inclusion bodies.
Figure S1. Binding of S7. Related to Figure 1.
(A) Detection of co-transcriptional binding of S7. To detect specific binding of S7 during transcription, we used the experimental approach depicted in Figures 1C and 1D, and used a Cy3-oligo that hybridizes to the 5’-end of the nascent RNA. We did not find co-transcriptional S7 binding even using 200 nM Cy5-labeled S7. Shown is representative single-molecule fluorescence trace, which was background and fluorescence spectral bleed-through corrected (Duss et al., 2018). Due to unspecific Cy5-S7 binding to the surface of the ZMW at 200 nM concentrations, we used a different metric than FRET efficiency to detect anticorrelated Cy3-Cy5 intensity changes that are characteristic for specific Cy5-S7 binding (depicted on the lower panel) (Duss et al., 2018). We used: Ia(t) * [Id(max)- Id(t)], where Id and Ia are the apparent fluorescence intensities of the donor and acceptor, respectively and Id(max) is the intensity of the Cy3-oligo in absence of FRET. (B) Representative single-molecule traces for binding of S7 to nascent 3’domain rRNA in real-time. Shown are background corrected fluorescence intensities (top), FRET efficiency (bottom) and its probability distribution (bottom right). Cy5-S7 concentration: 1 nM. Temperature: 20 °C. (C) Single and double exponential fit of the Cy5-S7-bound lifetimes at 20 °C. The errors represent the 95 % confidence intervals of the fit. (D) S7 r-protein concentration titration. To obtain the on-rates, we fitted the arrival times between subsequent S7 binding events to a double-exponential function for the 3’domain and the Δ270 construct (see Data S1) and at different Cy5-S7 concentrations, at 20 °C. The dot sizes are proportional to the populations of the fast phase (blue) and the slow phase (red). The fast phase is protein concentration-dependent and was fitted to a linear equation up to the 5 nM point. At 20 nM, the arrival times are too fast to obtain an accurate on-rate. The slow phase is not concentration-dependent and the rate is presented as the average and standard deviation of all the concentration points. The error bars represent the 95 % confidence intervals of the fit. Number of dwells analyzed: (n) = 780 (C). See also Data S2.
Figure S2. Defining RNA folding classes and transcription rate dependence. Related to Figure 2.
(A) Clustering of the single-molecule traces with each row representing a single nascent 3’-domain rRNA molecule and synchronized post-transcriptionally. Cy5-S7 concentration: 20 nM. Temperature: 20 °C. (B) How to set the threshold for the longest S7-bound event per trace in order to assign that trace to either a folded or misfolded RNA molecule? For simplicity, we assume that there is a single natively folded population of RNA molecules that are competent to bind S7. In addition, we assume a single non-natively folded population of RNA molecules that shows transient S7 binding being either detectable or too short to be detected. To assign all the single-molecule traces to either of the two populations, we fitted the distribution of the longest S7-bound lifetime per trace to a double-exponential function with the fitting parameters providing the relative populations of folded versus misfolded RNA molecules. We note that this approach allows also to classify traces containing a single S7 binding event. The graph shows a cumulative probability distribution of durations of the longest S7 binding event per trace including the traces without any detectable binding events (set to 0 seconds and shown in red). The cumulative distribution was then fitted to a double exponential function, y = 1 – a*exp(−b*t) – (1-a)*exp(−c*t), in order to obtain the “dissociation-rates” and the corresponding populations a and (1-a). The threshold to assign a trace to either folded or misfolded RNA, corresponds to the population of the slow dissociation rate phase (blue horizontal line separates the two populations). The obtained threshold is shown as blue vertical line. See also STAR Methods. (C) The fraction of nascent 3’domain RNA molecules that are competent to bind S7 for > 2 seconds is similar using 20 nM or 200 nM Cy5-S7. (D) 3’domain r-proteins do not affect S7 on-rate. The time from full transcription till appearance of the first S7 binding event with a duration of >2 s at 35 °C is represented. Concentrations: 20 nM Cy5-S7 and 400 nM each unlabeled protein. (E) Distribution of durations to transcribe the entire 3’-domain RNA (517 nts) for individual RNA molecules at 500 μM NTPs at 20 °C. At 35 °C, our in vitro transcription rates increase to ~60 nt s−1 (Duss et al., 2018) similar to transcription rates in vivo (Ryals et al., 1982). The mean and standard deviation of the Gaussian fit are shown. (F-H) RNA folding efficiency is transcription-rate dependent. The fraction of RNA molecules, which can bind S7 at least once for >2 s was plotted against the average transcription rate (F) or the NTP concentration (G) for the Δ270 construct at 20 °C. (H) shows the dependence of the transcription rate on the NTP concentration. Number of molecules analyzed (n) = 138 (A and B), 138, 68, 92 (C), 28, 83, 60, 61 (D), 60 (E), 107, 122, 103, 86 (F-H). See also Data S2.
Figure S3. H28 formation of nascent RNA. Related to Figure 3.
(A) H28 formation efficiency is independent of the S7 protein concentration. (B-D) Representative single-molecule fluorescence traces showing real-time H28 formation (B) or RNA molecules, in which H28 is formed upon binding of both Cy3-oligo and Cy3.5 oligos that are required to detect H28 formation (C and D). Traces without (C) or with S7 binding (D) are shown. Note the spectral bleed-through from the Cy3 into the Cy3.5 and Cy3.5 into the Cy5.5 channels. For clarity, the Cy5.5 intensity trace (violet) is not shown in panels (B) and (C). Number of molecules analyzed in (A) (n) = 72, 114, 128. The experiments were initiated by delivering 500 μM NTPs, 100 nM Cy3-oligo, 100 nM Cy3.5-oligo and 5 nM Cy5.5-S7 (or the indicated S7 concentration in (A)) to the stalled transcription complex. See also Data S2.
Figure S4. Stabilization of S7 by r-proteins and RNA tertiary interactions and detection of S13 binding. Related to Figure 4.
(A) In presence of the secondary binding r-proteins S9, S13 and S19, S7 binds stably also at 35 °C. Single-molecule traces containing a long S7 binding event were clustered. Each row represents a single 3′-domain rRNA molecule. Cy5-S7 concentration: 20 nM. (B) S13 and S19 wrap around the RNA helices emerging from the S7 binding site, thereby stabilizing S7. Shown are r-protein S7 (green surface) and r-proteins S9, S13 and S19 (different cyan surfaces) bound to the 16S rRNA in the E. coli 30S ribosomal subunit (PDB accession code: 4V9P). For clarity, only the RNA portion present in the Δ270 construct is shown. (C and D) Long-range RNA tertiary interactions outside of the S7 binding site increase the S7 binding stability. Secondary structure (C), with long-range tertiary interactions shown as lines, and 3D structure (D) highlight the parts of H41 (red) and H42 (orange) that are missing in the smallest Δ318 construct. (E-M) Detecting S13 binding using FRET between Cy5-S13 and Cy3-S7 (E-I) or between Cy5.5-S13 and a Cy3-oligo binding to the 3’-end of the nascent RNA in presence (J-L) or absence (M) of unlabeled S7. (E and F) The dye labeling positions for the simultaneous detection of Cy3-S7 and Cy5-S13 are illustrated. Protein S7 (green surface) and protein S13 (cyan surface) bound to the 16S rRNA in the E. coli 30S ribosomal subunit (PDB accession code 4V9P). (G) Example traces for simultaneous binding of Cy3-S7 (20 nM) and Cy5-S13 (50 nM) to the nascent RNA. Note that no Cy3-oligo was used in this experiment. (H) Double exponential fit of the Cy5-S13-bound lifetimes. (I) Overview of single-molecule traces with each row representing a single nascent 3′-domain rRNA molecule. (J) Labeling scheme. (K) Example traces for detection of Cy5.5-S13 using a Cy3-oligo hybridized to the 3’-end of the nascent RNA. (L) Double exponential fit of the Cy5.5-S13-bound lifetimes. (M) No Cy5.5-S13 binding was detected in absence of S7. In (E-M), the co-transcriptional experiments were initiated by delivering 500 μM NTPs and the following reagents to the stalled transcription complex of the Δ270 construct at 20 °C: 20 nM Cy3-S7, 50 nM Cy5-S13 (E-I) or 200 nM Cy5.5-S13, 1.5 μM unlabeled S7 and 100 nM Cy3-oligo (J-L) or 200 nM Cy5.5-S13 and 100 nM Cy3-oligo (M). Number of dwells analyzed: (n) = 144 (H) and 35 (L). The errors in (H and L) represent 95 % confidence intervals of the fits. See also Data S2.
Figure S5. Stabilization of Cy5-S7 by combinations of secondary binding r-proteins. Related to Figure 4.
(A and B) The survival probability of Cy5-S7 is plotted at 20 °C (A) and 35 °C (B) in presence of different combinations of 3’domain r-proteins at 400 nM each (except of S15 at 2 μM). The survival probability, which is (1 – cumulative probability), is the probability that a trace has the longest Cy5-S7 binding event longer than time t (x-axis). The bar plots are representing the fraction of traces with the longest Cy5-S7 binding event longer than the time indicated. The data represented in the bar plots are equal to the y-values of the survival probability plots at the different thresholds (dotted lines). The bar plot at lifetime >100 s in (B) corresponds to Figure 4D. Number of molecules analyzed in (A) (n) = 114, 122, 118, 102 and (B) (n) = 101, 120, 144, 115, 111, 141, 98, 149, 121, 57, 57. See also Data S2.
Figure S6. Assembly of entire 3’domain detected by Cy5-labeled S3. Related to Figure 5.
100 nM Cy5-S3 and 400 nM of all the unlabeled 3’domain r-proteins were delivered at t = 0 seconds to a pre-folded 3’domain RNA to which a Cy3-oligo is hybridized to its 3’-end. (A) Representative single-molecule traces of Cy5-S3 binding in presence of all unlabeled 3’domain proteins; Cy5-S3 (red); Cy3-oligo (green). Dotted line indicates that S3 is likely still bound but the FRET signal has disappeared due to Cy3 or Cy5 dye photobleaching (see Figures S7E, S7F and STAR Methods). To detect anticorrelated Cy3-Cy5 intensity changes characteristic for specific Cy5-S3 binding, we used: Ia(t) * [Id(max)- Id(t)], where Id and Ia are the apparent fluorescence intensities of the donor and acceptor, respectively and Id(max) is the intensity of the Cy3-oligo in absence of FRET as explained in Figure S1A (Duss et al., 2018). (B) Overview of the analyzed single-molecule traces. Every row represents a single pre-folded RNA molecule. (C) Estimation of observed S3 binding rate. To estimate the observed on-rate for S3, we fitted the arrival times of Cy5-S3 to a single-exponential function. The arrival times are from protein delivery till appearance of the first S3 event of >10 seconds lifetime. (D and E) Single-exponential fit (D) or double-exponential fit (E) of the Cy5-S3-bound dwell-times in the subset of traces containing only Cy5-S3 binding events <10 s (D) or containing at least one Cy5-S3 binding event of >10 s (E). All the experiments were performed at 35 °C. The errors represent the 95 % confidence intervals of the fits. Number of molecules/ dwells analyzed (n) = 94/ 94 (C), (n) = 57/ 112 (D) and (n) = 97/ 205 (E). See also Data S2.
ACKNOWLEDGMENTS
The authors thank Edit Sperling, Rajan Lamichhane, Seán O’Leary, Alexey Petrov, Rosslyn Grosely, Alex Johnson, Junhong Choi, John Hammond and the Williamson, Puglisi and David Millar laboratories for helpful discussions and experimental advice. We thank Alex Johnson for critical reading of the manuscript. Funding: This work was supported by the Swiss National Science Foundation (SNSF) early postdoc.mobility grant no. P2EZP3-152131, advanced postdoc.mobility grant no. P300PA-160978 and the Human Frontier Science Program grant no. LT000628/2015-L to O.D., NIH R01 GM051266 and NIH R01 GM113078 to J.D.P and NIH R01 GM053757 to J.R.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures S1–S7 and Table S1
Data S1. Template and primer DNA sequences. Related to STAR Methods.
Data S2. Source data for all figures. Related to Figures 1–7 and S1–S7.
REFERENCES
- Abeysirigunawardena SC, Kim H, Lai J, Ragunathan K, Rappe MC, Luthey-Schulten Z, Ha T, and Woodson SA (2017). Evolution of protein-coupled RNA dynamics during hierarchical assembly of ribosomal complexes. Nature communications 8, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adilakshmi T, Bellur DL, and Woodson SA (2008). Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 455, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, Marshall RA, and Puglisi JD (2008). An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J 94, 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle J, Robillard GT, and Kim SH (1980). Sequential folding of transfer RNA. A nuclear magnetic resonance study of successively longer tRNA fragments with a common 5′ end. J Mol Biol 139, 601–625. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, and Greenleaf WJ (2014). Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nature biotechnology 32, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunner AE, Beck AH, and Williamson JR (2010). Kinetic cooperativity in Escherichia coli 30S ribosomal subunit reconstitution reveals additional complexity in the assembly landscape. Proc Natl Acad Sci U S A 107, 5417–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaker-Margot M, Hunziker M, Barandun J, Dill BD, and Klinge S (2015). Stage-specific assembly events of the 6-MDa small-subunit processome initiate eukaryotic ribosome biogenesis. Nat Struct Mol Biol 22, 920–923. [DOI] [PubMed] [Google Scholar]
- Chen J, Dalal RV, Petrov AN, Tsai A, O’Leary SE, Chapin K, Cheng J, Ewan M, Hsiung PL, Lundquist P, et al. (2014). High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc Natl Acad Sci U S A 111, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrov A, Tsai A, O’Leary SE, and Puglisi JD (2013). Coordinated conformational and compositional dynamics drive ribosome translocation. Nat Struct Mol Biol 20, 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SS, and Williamson JR (2013). Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J Mol Biol 425, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha CE, Belardinelli R, Peske F, Holtkamp W, Wintermeyer W, and Rodnina MV (2013). Dual use of GTP hydrolysis by elongation factor G on the ribosome. Translation (Austin) 1, e24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JH, Tan YZ, Carragher B, Potter CS, Lyumkis D, and Williamson JR (2016). Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell 167, 1610–1622 e1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Narvaez CC, and Schaup HW (1979). In vivo transcriptionally coupled assembly of Escherichia coli ribosomal subunits. J Mol Biol 134, 1–22. [DOI] [PubMed] [Google Scholar]
- Doudna JA, and Batey RT (2004). Structural insights into the signal recognition particle. Annu Rev Biochem 73, 539–557. [DOI] [PubMed] [Google Scholar]
- Dragon F, and Brakier-Gingras L (1993). Interaction of Escherichia coli ribosomal protein S7 with 16S rRNA. Nucleic Acids Res 21, 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon F, Payant C, and Brakier-Gingras L (1994). Mutational and structural analysis of the RNA binding site for Escherichia coli ribosomal protein S7. J Mol Biol 244, 74–85. [DOI] [PubMed] [Google Scholar]
- Duss O, Stepanyuk GA, Grot A, O’Leary SE, Puglisi JD, and Williamson JR (2018). Real-time assembly of ribonucleoprotein complexes on nascent RNA transcripts. Nature communications 9, 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-D’Amare AR (2016). RNA Binding: Getting Specific about Specificity. Cell Chem Biol 23, 1177–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Schnell R, Szekeres S, and Kalman M (1999). Directional cloning of native PCR products with preformed sticky ends (autosticky PCR). Molecular & general genetics : MGG 260, 569–573. [DOI] [PubMed] [Google Scholar]
- Ganser LR, Kelly ML, Herschlag D, and Al-Hashimi HM (2019). The roles of structural dynamics in the cellular functions of RNAs. Nature reviews Molecular cell biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Zhuang X, Kim HD, Orr JW, Williamson JR, and Chu S (1999). Ligand-induced conformational changes observed in single RNA molecules. Proc Natl Acad Sci U S A 96, 9077–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilman-Miller SL, and Woodson SA (2003). Effect of transcription on folding of the Tetrahymena ribozyme. RNA 9, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held WA, Ballou B, Mizushima S, and Nomura M (1974). Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J Biol Chem 249, 3103–3111. [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, and Preiss T (2018). A brave new world of RNA-binding proteins. Nature reviews Molecular cell biology 19, 327–341. [DOI] [PubMed] [Google Scholar]
- Hickerson R, Majumdar ZK, Baucom A, Clegg RM, and Noller HF (2005). Measurement of internal movements within the 30 S ribosomal subunit using Forster resonance energy transfer. J Mol Biol 354, 459–472. [DOI] [PubMed] [Google Scholar]
- Incarnato D, Morandi E, Anselmi F, Simon LM, Basile G, and Oliviero S (2017). In vivo probing of nascent RNA structures reveals principles of cotranscriptional folding. Nucleic Acids Res 45, 9716–9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isambert H (2009). The jerky and knotty dynamics of RNA. Methods 49, 189–196. [DOI] [PubMed] [Google Scholar]
- Jankowsky E, and Harris ME (2015). Specificity and nonspecificity in RNA-protein interactions. Nature reviews Molecular cell biology 16, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett MC, Fritz BR, Timmerman LE, and Church GM (2013). In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol Syst Biol 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczanowska M, and Ryden-Aulin M (2007). Ribosome biogenesis and the translation process in Escherichia coli. Microbiol Mol Biol Rev 71, 477–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Abeysirigunawarden SC, Chen K, Mayerle M, Ragunathan K, Luthey-Schulten Z, Ha T, and Woodson SA (2014). Protein-guided RNA dynamics during early ribosome assembly. Nature 506, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara K, and Suzuki T (2009). The ordered transcription of RNA domains is not essential for ribosome biogenesis in Escherichia coli. Mol Cell 34, 760–766. [DOI] [PubMed] [Google Scholar]
- Klinge S, and Woolford JL Jr. (2018). Ribosome assembly coming into focus. Nature reviews Molecular cell biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, and Hwa T (2008). Stochasticity and traffic jams in the transcription of ribosomal RNA: Intriguing role of termination and antitermination. Proc Natl Acad Sci U S A 105, 18159–18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Das R, and Barna M (2018). Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nature reviews Molecular cell biology 19, 158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, and Webb WW (2003). Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299, 682–686. [DOI] [PubMed] [Google Scholar]
- Lewicki BT, Margus T, Remme J, and Nierhaus KH (1993). Coupling of rRNA transcription and ribosomal assembly in vivo. Formation of active ribosomal subunits in Escherichia coli requires transcription of rRNA genes by host RNA polymerase which cannot be replaced by bacteriophage T7 RNA polymerase. J Mol Biol 231, 581–593. [DOI] [PubMed] [Google Scholar]
- Lindahl L (1975). Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J Mol Biol 92, 15–37. [DOI] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, and Varani G (2007). RNA-binding proteins: modular design for efficient function. Nature reviews Molecular cell biology 8, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenet S, Bertrand E, and Verheggen C (2017). Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA Biol 14, 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, Potter CS, Carragher B, and Williamson JR (2010). Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 330, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM (2019). Nascent RNA and the Coordination of Splicing with Transcription. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, and Tinoco I Jr. (1981). Sequential folding of a messenger RNA molecule. J Mol Biol 151, 519–533. [DOI] [PubMed] [Google Scholar]
- Orr JW, Hagerman PJ, and Williamson JR (1998). Protein and Mg(2+)-induced conformational changes in the S15 binding site of 16 S ribosomal RNA. J Mol Biol 275, 453–464. [DOI] [PubMed] [Google Scholar]
- Pan T, and Sosnick T (2006). RNA folding during transcription. Annu Rev Biophys Biomol Struct 35, 161–175. [DOI] [PubMed] [Google Scholar]
- Powers T, Daubresse G, and Noller HF (1993). Dynamics of in vitro assembly of 16 S rRNA into 30 S ribosomal subunits. J Mol Biol 232, 362–374. [DOI] [PubMed] [Google Scholar]
- Ramaswamy P, and Woodson SA (2009). S16 throws a conformational switch during assembly of 30S 5’ domain. Nat Struct Mol Biol 16, 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht MI, and Williamson JR (2001). Central domain assembly: thermodynamics and kinetics of S6 and S18 binding to an S15-RNA complex. J Mol Biol 313, 35–48. [DOI] [PubMed] [Google Scholar]
- Reichow SL, Hamma T, Ferre-D’Amare AR, and Varani G (2007). The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res 35, 1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgeway WK, Millar DP, and Williamson JR (2012). Quantitation of ten 30S ribosomal assembly intermediates using fluorescence triple correlation spectroscopy. Proc Natl Acad Sci U S A 109, 13614–13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert F, Gagnon M, Sans D, Michnick S, and Brakier-Gingras L (2000). Mapping of the RNA recognition site of Escherichia coli ribosomal protein S7. RNA 6, 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson WR, Dowsett SJ, and Hardy SJ (1977). Exchange of ribosomal proteins among the ribosomes of Escherichia coli. Molecular & general genetics : MGG 157, 205–214. [DOI] [PubMed] [Google Scholar]
- Ryals J, Little R, and Bremer H (1982). Temperature dependence of RNA synthesis parameters in Escherichia coli. J Bacteriol 151, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaha RR, O’Brien B, O’Brien TW, and Noller HF (1994). Independent in vitro assembly of a ribonucleoprotein particle containing the 3’ domain of 16S rRNA. Proc Natl Acad Sci U S A 91, 7884–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, and Cech TR (2015). Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev 29, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, and Williamson JR (2011). Assembly of bacterial ribosomes. Annu Rev Biochem 80, 501–526. [DOI] [PubMed] [Google Scholar]
- Soper SFC, Dator RP, Limbach PA, and Woodson SA (2013). In Vivo X-Ray Footprinting of Pre-30S Ribosomes Reveals Chaperone-Dependent Remodeling of Late Assembly Intermediates. Mol Cell 52, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MD, Mihalusova M, O’Connor CM, Prathapam R, Collins K, and Zhuang X (2007). Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature 446, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian AR, and van Duin J (1977). Exchange of individual ribosomal proteins between ribosomes as studied by heavy isotope-transfer experiments. Molecular & general genetics : MGG 158, 1–9. [DOI] [PubMed] [Google Scholar]
- Sykes MT, and Williamson JR (2009). A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys 38, 197–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkington MW, Siuzdak G, and Williamson JR (2005). An assembly landscape for the 30S ribosomal subunit. Nature 438, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub P, and Nomura M (1969). Structure and function of Escherichia coli ribosomes. VI. Mechanism of assembly of 30 s ribosomes studied in vitro. J Mol Biol 40, 391–413. [DOI] [PubMed] [Google Scholar]
- Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, and Puglisi JD (2010). Real-time tRNA transit on single translating ribosomes at codon resolution. Nature 464, 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel U, and Jensen KF (1995). Effects of the antiterminator BoxA on transcription elongation kinetics and ppGpp inhibition of transcription elongation in Escherichia coli. J Biol Chem 270, 18335–18340. [DOI] [PubMed] [Google Scholar]
- Will CL, and Luhrmann R (2011). Spliceosome structure and function. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson SA (2008). RNA folding and ribosome assembly. Curr Opin Chem Biol 12, 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson SA (2011). RNA folding pathways and the self-assembly of ribosomes. Acc Chem Res 44, 1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Revyakin A, Grimm JB, Lavis LD, and Tjian R (2014). Single-molecule tracking of the transcription cycle by sub-second RNA detection. eLife 3, e01775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, and Craighead HG (2012). Zero-mode waveguides for single-molecule analysis. Annu Rev Biophys 41, 269–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S7. Correlating S7 with S3 binding and ensemble experiments. Related to Figures 5 and 6.
(A-C) Correlating S7 with S3 binding. (A) Overview of the analyzed single-molecule traces. Every row represents a single pre-folded RNA molecule. (B) Representative single-molecule trace with Cy5-S3 (red), Cy5.5-S7 (purple) and Cy3-oligo (green). The trace was not corrected for fluorescence spectral bleed-through from the Cy5 into the Cy5.5 channel and the reverse. (C) To estimate the rate between stable S7 and initial S3 incorporation, we fitted the dwell times between final Cy5.5-S7 and first Cy5-S3 event to a single-exponential function. The error represents the 95 % confidence interval of the fit. Number of molecules analyzed (n) = 64 (C). Experimental setup in (A-C): 100 nM Cy5-S3, 5 nM Cy5.5-S7 and 400 nM of all the other unlabeled 3’domain r-proteins were injected at t = 0 seconds. All the experiments were performed at 35 °C. (D-G) Ensemble experiments. (D) In vitro reconstitutions of native 16S rRNA with individually recombinantly expressed 3’domain r-proteins (see STAR Methods). This data shows that: 1) all r-proteins upstream of S3 are required for S3 r-protein association, 2) S2 binding downstream of S3 is not required for stable S3 binding, 3) fluorescence labeling of S3, S7 and S13 give similar in vitro reconstitution efficiencies as unlabeled wild-type proteins, 4) using ZMW conditions (see STAR Methods) instead of established in vitro reconstitution conditions (Held et al., 1974; Traub and Nomura, 1969) has no effect on the assembly efficiency. (E-F) The actual S3-bound lifetime of the final S3 binding event is longer than what we can observe during our single-molecule experiments. Shown are competition experiments of 25 nM reconstituted 3’domain containing Cy5-S3 that was chased with unlabeled S3 at 16-fold excess for the indicated times and at 35 °C in presence of standard conditions or ZMW conditions. The Cy5 fluorescence was visualized with native agarose gel electrophoresis and was normalized by the amount of rRNA that was visualized with EtBr staining. Each dot represents the mean normalized fluorescence intensity remaining relative to the 5 min time point, and error bars represent standard deviation from different experiments. The line represents a fit to y = a * exp(−b*t) + c. The parameter c represents a slow phase which is too slow to be determined reliably during our experimental time of 1-2 days (c * exp(−d*t) = c if decay rate d is very small). We obtain two similarly populated lifetime phases of ~4-5 h and > several days, respectively. It is possible that the shorter lifetime phase of S3 represents a 3’domain particle which has not obtained its native fold yet, while the longer S3 lifetime phase represents a native 3’domain. However, while in vivo the majority of S3 is stably associated with the ribosome, ~ 25 % of S3 also exchanges rapidly (Robertson et al., 1977) suggesting that S3 could be associated with the native rRNA in two different states, one which exchanges faster than the other one. (G) Competition experiments of reconstituted 3’domain containing Cy5-S7 (25 nM) with unlabeled S7 at 16-fold excess for the indicated times and at 35 °C. The same experimental setup was used as in (E), except that the 3’domain was only reconstituted with the secondary binding proteins S9, S13 and S19 instead of all 3’domain proteins and Cy5-S7 was used instead of unlabeled S7. The data demonstrate that S7 does not significantly exchanges during the course of the experiment and shows that S9, S13 and S19 are sufficient to stabilize S7 on a relevant biological timescale. Error bars represent standard deviation from different experiments. Gel repetitions, n = 3 (E-G). See also Data S2.
Table S1. Related to STAR Methods.
Overview of r-protein expression and purification. a inclusion bodies.
Figure S1. Binding of S7. Related to Figure 1.
(A) Detection of co-transcriptional binding of S7. To detect specific binding of S7 during transcription, we used the experimental approach depicted in Figures 1C and 1D, and used a Cy3-oligo that hybridizes to the 5’-end of the nascent RNA. We did not find co-transcriptional S7 binding even using 200 nM Cy5-labeled S7. Shown is representative single-molecule fluorescence trace, which was background and fluorescence spectral bleed-through corrected (Duss et al., 2018). Due to unspecific Cy5-S7 binding to the surface of the ZMW at 200 nM concentrations, we used a different metric than FRET efficiency to detect anticorrelated Cy3-Cy5 intensity changes that are characteristic for specific Cy5-S7 binding (depicted on the lower panel) (Duss et al., 2018). We used: Ia(t) * [Id(max)- Id(t)], where Id and Ia are the apparent fluorescence intensities of the donor and acceptor, respectively and Id(max) is the intensity of the Cy3-oligo in absence of FRET. (B) Representative single-molecule traces for binding of S7 to nascent 3’domain rRNA in real-time. Shown are background corrected fluorescence intensities (top), FRET efficiency (bottom) and its probability distribution (bottom right). Cy5-S7 concentration: 1 nM. Temperature: 20 °C. (C) Single and double exponential fit of the Cy5-S7-bound lifetimes at 20 °C. The errors represent the 95 % confidence intervals of the fit. (D) S7 r-protein concentration titration. To obtain the on-rates, we fitted the arrival times between subsequent S7 binding events to a double-exponential function for the 3’domain and the Δ270 construct (see Data S1) and at different Cy5-S7 concentrations, at 20 °C. The dot sizes are proportional to the populations of the fast phase (blue) and the slow phase (red). The fast phase is protein concentration-dependent and was fitted to a linear equation up to the 5 nM point. At 20 nM, the arrival times are too fast to obtain an accurate on-rate. The slow phase is not concentration-dependent and the rate is presented as the average and standard deviation of all the concentration points. The error bars represent the 95 % confidence intervals of the fit. Number of dwells analyzed: (n) = 780 (C). See also Data S2.
Figure S2. Defining RNA folding classes and transcription rate dependence. Related to Figure 2.
(A) Clustering of the single-molecule traces with each row representing a single nascent 3’-domain rRNA molecule and synchronized post-transcriptionally. Cy5-S7 concentration: 20 nM. Temperature: 20 °C. (B) How to set the threshold for the longest S7-bound event per trace in order to assign that trace to either a folded or misfolded RNA molecule? For simplicity, we assume that there is a single natively folded population of RNA molecules that are competent to bind S7. In addition, we assume a single non-natively folded population of RNA molecules that shows transient S7 binding being either detectable or too short to be detected. To assign all the single-molecule traces to either of the two populations, we fitted the distribution of the longest S7-bound lifetime per trace to a double-exponential function with the fitting parameters providing the relative populations of folded versus misfolded RNA molecules. We note that this approach allows also to classify traces containing a single S7 binding event. The graph shows a cumulative probability distribution of durations of the longest S7 binding event per trace including the traces without any detectable binding events (set to 0 seconds and shown in red). The cumulative distribution was then fitted to a double exponential function, y = 1 – a*exp(−b*t) – (1-a)*exp(−c*t), in order to obtain the “dissociation-rates” and the corresponding populations a and (1-a). The threshold to assign a trace to either folded or misfolded RNA, corresponds to the population of the slow dissociation rate phase (blue horizontal line separates the two populations). The obtained threshold is shown as blue vertical line. See also STAR Methods. (C) The fraction of nascent 3’domain RNA molecules that are competent to bind S7 for > 2 seconds is similar using 20 nM or 200 nM Cy5-S7. (D) 3’domain r-proteins do not affect S7 on-rate. The time from full transcription till appearance of the first S7 binding event with a duration of >2 s at 35 °C is represented. Concentrations: 20 nM Cy5-S7 and 400 nM each unlabeled protein. (E) Distribution of durations to transcribe the entire 3’-domain RNA (517 nts) for individual RNA molecules at 500 μM NTPs at 20 °C. At 35 °C, our in vitro transcription rates increase to ~60 nt s−1 (Duss et al., 2018) similar to transcription rates in vivo (Ryals et al., 1982). The mean and standard deviation of the Gaussian fit are shown. (F-H) RNA folding efficiency is transcription-rate dependent. The fraction of RNA molecules, which can bind S7 at least once for >2 s was plotted against the average transcription rate (F) or the NTP concentration (G) for the Δ270 construct at 20 °C. (H) shows the dependence of the transcription rate on the NTP concentration. Number of molecules analyzed (n) = 138 (A and B), 138, 68, 92 (C), 28, 83, 60, 61 (D), 60 (E), 107, 122, 103, 86 (F-H). See also Data S2.
Figure S3. H28 formation of nascent RNA. Related to Figure 3.
(A) H28 formation efficiency is independent of the S7 protein concentration. (B-D) Representative single-molecule fluorescence traces showing real-time H28 formation (B) or RNA molecules, in which H28 is formed upon binding of both Cy3-oligo and Cy3.5 oligos that are required to detect H28 formation (C and D). Traces without (C) or with S7 binding (D) are shown. Note the spectral bleed-through from the Cy3 into the Cy3.5 and Cy3.5 into the Cy5.5 channels. For clarity, the Cy5.5 intensity trace (violet) is not shown in panels (B) and (C). Number of molecules analyzed in (A) (n) = 72, 114, 128. The experiments were initiated by delivering 500 μM NTPs, 100 nM Cy3-oligo, 100 nM Cy3.5-oligo and 5 nM Cy5.5-S7 (or the indicated S7 concentration in (A)) to the stalled transcription complex. See also Data S2.
Figure S4. Stabilization of S7 by r-proteins and RNA tertiary interactions and detection of S13 binding. Related to Figure 4.
(A) In presence of the secondary binding r-proteins S9, S13 and S19, S7 binds stably also at 35 °C. Single-molecule traces containing a long S7 binding event were clustered. Each row represents a single 3′-domain rRNA molecule. Cy5-S7 concentration: 20 nM. (B) S13 and S19 wrap around the RNA helices emerging from the S7 binding site, thereby stabilizing S7. Shown are r-protein S7 (green surface) and r-proteins S9, S13 and S19 (different cyan surfaces) bound to the 16S rRNA in the E. coli 30S ribosomal subunit (PDB accession code: 4V9P). For clarity, only the RNA portion present in the Δ270 construct is shown. (C and D) Long-range RNA tertiary interactions outside of the S7 binding site increase the S7 binding stability. Secondary structure (C), with long-range tertiary interactions shown as lines, and 3D structure (D) highlight the parts of H41 (red) and H42 (orange) that are missing in the smallest Δ318 construct. (E-M) Detecting S13 binding using FRET between Cy5-S13 and Cy3-S7 (E-I) or between Cy5.5-S13 and a Cy3-oligo binding to the 3’-end of the nascent RNA in presence (J-L) or absence (M) of unlabeled S7. (E and F) The dye labeling positions for the simultaneous detection of Cy3-S7 and Cy5-S13 are illustrated. Protein S7 (green surface) and protein S13 (cyan surface) bound to the 16S rRNA in the E. coli 30S ribosomal subunit (PDB accession code 4V9P). (G) Example traces for simultaneous binding of Cy3-S7 (20 nM) and Cy5-S13 (50 nM) to the nascent RNA. Note that no Cy3-oligo was used in this experiment. (H) Double exponential fit of the Cy5-S13-bound lifetimes. (I) Overview of single-molecule traces with each row representing a single nascent 3′-domain rRNA molecule. (J) Labeling scheme. (K) Example traces for detection of Cy5.5-S13 using a Cy3-oligo hybridized to the 3’-end of the nascent RNA. (L) Double exponential fit of the Cy5.5-S13-bound lifetimes. (M) No Cy5.5-S13 binding was detected in absence of S7. In (E-M), the co-transcriptional experiments were initiated by delivering 500 μM NTPs and the following reagents to the stalled transcription complex of the Δ270 construct at 20 °C: 20 nM Cy3-S7, 50 nM Cy5-S13 (E-I) or 200 nM Cy5.5-S13, 1.5 μM unlabeled S7 and 100 nM Cy3-oligo (J-L) or 200 nM Cy5.5-S13 and 100 nM Cy3-oligo (M). Number of dwells analyzed: (n) = 144 (H) and 35 (L). The errors in (H and L) represent 95 % confidence intervals of the fits. See also Data S2.
Figure S5. Stabilization of Cy5-S7 by combinations of secondary binding r-proteins. Related to Figure 4.
(A and B) The survival probability of Cy5-S7 is plotted at 20 °C (A) and 35 °C (B) in presence of different combinations of 3’domain r-proteins at 400 nM each (except of S15 at 2 μM). The survival probability, which is (1 – cumulative probability), is the probability that a trace has the longest Cy5-S7 binding event longer than time t (x-axis). The bar plots are representing the fraction of traces with the longest Cy5-S7 binding event longer than the time indicated. The data represented in the bar plots are equal to the y-values of the survival probability plots at the different thresholds (dotted lines). The bar plot at lifetime >100 s in (B) corresponds to Figure 4D. Number of molecules analyzed in (A) (n) = 114, 122, 118, 102 and (B) (n) = 101, 120, 144, 115, 111, 141, 98, 149, 121, 57, 57. See also Data S2.
Figure S6. Assembly of entire 3’domain detected by Cy5-labeled S3. Related to Figure 5.
100 nM Cy5-S3 and 400 nM of all the unlabeled 3’domain r-proteins were delivered at t = 0 seconds to a pre-folded 3’domain RNA to which a Cy3-oligo is hybridized to its 3’-end. (A) Representative single-molecule traces of Cy5-S3 binding in presence of all unlabeled 3’domain proteins; Cy5-S3 (red); Cy3-oligo (green). Dotted line indicates that S3 is likely still bound but the FRET signal has disappeared due to Cy3 or Cy5 dye photobleaching (see Figures S7E, S7F and STAR Methods). To detect anticorrelated Cy3-Cy5 intensity changes characteristic for specific Cy5-S3 binding, we used: Ia(t) * [Id(max)- Id(t)], where Id and Ia are the apparent fluorescence intensities of the donor and acceptor, respectively and Id(max) is the intensity of the Cy3-oligo in absence of FRET as explained in Figure S1A (Duss et al., 2018). (B) Overview of the analyzed single-molecule traces. Every row represents a single pre-folded RNA molecule. (C) Estimation of observed S3 binding rate. To estimate the observed on-rate for S3, we fitted the arrival times of Cy5-S3 to a single-exponential function. The arrival times are from protein delivery till appearance of the first S3 event of >10 seconds lifetime. (D and E) Single-exponential fit (D) or double-exponential fit (E) of the Cy5-S3-bound dwell-times in the subset of traces containing only Cy5-S3 binding events <10 s (D) or containing at least one Cy5-S3 binding event of >10 s (E). All the experiments were performed at 35 °C. The errors represent the 95 % confidence intervals of the fits. Number of molecules/ dwells analyzed (n) = 94/ 94 (C), (n) = 57/ 112 (D) and (n) = 97/ 205 (E). See also Data S2.
Data Availability Statement
The published article includes all relevant data generated or analyzed during this study. Source data for all the figures are available in supplementary file Data S2. Additional data and codes are available from Lead Contact, James R. Williamson (jrwill@scripps.edu) on request.